

Abstract
Background
Mitochondrial uncouplers are well-known for their ability to treat a myriad of metabolic diseases, including obesity and fatty liver diseases. However, for many years now, mitochondrial uncouplers have also been evaluated in diverse models of cancer in vitro and in vivo. Furthermore, some mitochondrial uncouplers are now in clinical trials for cancer, although none have yet been approved for the treatment of cancer.
Scope of Review
In this review we summarise published studies in which mitochondrial uncouplers have been investigated as an anti-cancer therapy in preclinical models. In many cases, mitochondrial uncouplers show strong anti-cancer effects both as single agents, and in combination therapies, and some are more toxic to cancer cells than normal cells. Furthermore, the mitochondrial uncoupling mechanism of action in cancer cells has been described in detail, with consistencies and inconsistencies between different structural classes of uncouplers. For example, many mitochondrial uncouplers decrease ATP levels and disrupt key metabolic signalling pathways such as AMPK/mTOR but have different effects on reactive oxygen species (ROS) production. Many of these effects oppose aberrant phenotypes common in cancer cells that ultimately result in cell death. We also highlight several gaps in knowledge that need to be addressed before we have a clear direction and strategy for applying mitochondrial uncouplers as anti-cancer agents.
Major conclusions
There is a large body of evidence supporting the therapeutic use of mitochondrial uncouplers to treat cancer. However, the long-term safety of some uncouplers remains in question and it will be critical to identify which patients and cancer types would benefit most from these agents.
Keywords: Mitochondrial uncouplers, Cancer therapy, Cytotoxicity, Mouse models of cancer
Abbreviations
- Δψm
mitochondrial membrane potential
- AMPK
5′ adenosine monophosphate-activated protein kinase
- ANT
adenine nucleotide translocase
- ATP
adenosine triphosphate
- CSCs
cancer stem cells
- CDK
cyclin-dependent kinase
- DNP
2,4-dinitrophenol
- ECAR
extracellular acidification rate
- ETC
electron transport chain
- GSH
reduced glutathione
- HCC
hepatocellular carcinoma
- IMM
inner mitochondrial membrane
- mitoROS
mitochondrial reactive oxygen species
- mPTP
mitochondrial permeability transition pore
- mTOR
mammalian target of rapamycin
- NADPH
nicotinamide adenine dinucleotide phosphate
- NTZ
nitazoxanide
- OCR
oxygen consumption rate
- OXPHOS
oxidative phosphorylation
- PMF
proton motive force
- ROS
reactive oxygen species
- TNBC
triple negative breast cancer
- UCP
uncoupling protein
- μM
micromolar
1. Introduction
Cancer poses severe health, economic, and societal burdens on the world, being the second leading cause of death globally and a recognised public health priority [1]. In 2018, an estimated 18.1 million people globally received a cancer diagnosis, with a further 9.6 million people succumbing to the disease [2]. Lung (2,093,876 new cases) and breast cancers (2,088,849 new cases) were the most commonly diagnosed malignancies, and lung (1,761,007 deaths), stomach (782,685 deaths), and liver cancers (781,631 deaths) were the deadliest [2]. These staggering numbers demonstrate the importance of identifying new ways to eradicate cancer cells and increase treatment options that improve survival rates of cancer patients.
Cancer is delineated as a cellular disease whereby healthy cells undergo genetic damage that enables their spread and growth in different parts of the body. Cancer cells continually divide and evade programmed death and other mechanisms such as immunosurveillance that ensure cellular stability and homeostasis within the body. Indeed, each neoplasm is genetically different, rendering cancer a heterogeneous disease that is difficult to treat [4]. However, despite the nearly unlimited possibilities of genetic changes, there is a much smaller set of potential phenotypic changes that occur in cancer. In the 1920s, Otto Warburg made the seminal discovery that tumour tissues have increased rates of glycolysis compared to non-malignant tissues despite sufficient oxygen availability, which is now called “aerobic glycolysis” or the Warburg effect (as reviewed in [5]). Warburg initially postulated that this may be caused by dysfunctional mitochondria in tumour cells. However, we now know that most cancer cells have functional mitochondria and readily use aerobic glycolysis and OXPHOS to support the production of ATP, macromolecules, ROS-regulators (for example, GSH and NADPH), and oncometabolites depending on the microenvironment, nutrient and oxygen availability, and disease stage [5,6]. The role that mitochondrial metabolism plays in cancer development, including metastatic lung cancer [7], and opportunities to target mitochondria for cancer therapy [6] has been highlighted. However, the therapeutic potential of mitochondrial uncouplers as anti-cancer agents has not yet been discussed and is therefore the focus of this review.
2. Oxidative phosphorylation and mitochondrial uncoupling
The mitochondrial oxidative phosphorylation (OXPHOS) process is governed by the chemiosmotic theory whereby nutrient oxidation is coupled to ATP production via a series of redox reactions in the electron transport chain (ETC) complex that ultimately results in the reduction of molecular oxygen to water at complex IV (outlined in Figure 1). Electron transfer through complexes I, III, and IV provides energy to pump protons (H+) from the mitochondrial matrix into the intermembrane space (IMS) to create a proton concentration gradient (ΔpH) and membrane potential (Δψm) that collectively form the proton motive force (PMF) (Δp). The electrochemical proton gradient of the PMF drives H+ along the concentration gradient from the intermembrane space (low pH and high protons) through complex V (ATP synthase) in the inner mitochondrial membrane (IMM) into the mitochondrial matrix (high pH and low protons) [8]. ATP is produced as protons re-enter the matrix via the ATP synthase enzyme complex (Figure 1). However, protons can re-enter the mitochondrial matrix independent of ATP synthase via a process that is “uncoupled” from ATP production [9,10] (Figure 1). Mitochondrial uncoupling is a normal metabolic process, and approximately 35% of basal respiration in perfused rat muscle is uncoupled [11]. It is estimated that up to 25% of the whole-body basal metabolic rate in rats is due to mitochondrial uncoupling [11]. Examination of the oxygen consumption rate (OCR) in mitochondrial stress tests shows that uncoupling (“proton leak”) accounts for ∼13–25% of basal respiration in different cancer cell lines in vitro [7,[12], [13], [14], [15]]. However, few studies have directly assessed uncoupled respiration between cancer cells and their non-cancerous cells of origin. One example is a study that showed that ∼50% of basal respiration was uncoupled in mouse-derived Myc-dependent osteosarcoma cells, whereas ∼40% of basal respiration was uncoupled in differentiated mouse osteocytes [16].
Figure 1.

Mitochondrial uncouplers increase mitochondrial respiration. Mitochondrial uncouplers mediate proton (H+) transport into the mitochondrial matrix via a pathway independent of ATP synthase [9,10]. Uncouplers bind to protons in the intermembrane space proceeding to cross the inner mitochondrial membrane (IMM). Mitochondrial protonophore uncouplers are typically lipophilic weak acids that can penetrate membranes [18]. Once an uncoupler has crossed the IMM, it is deprotonated and cycles out of the mitochondrial matrix, where it becomes protonated again to continue the uncoupling cycle. This figure was created with BioRender.com and adapted from Demine et al. [17].
Protons cannot freely penetrate the mitochondrial inner membrane, so intermediate proteins and lipids are required including uncoupling proteins (UCPs), fatty acids, and other protein-dependent processes, as reported [17]. Apart from endogenous mitochondrial uncoupling mechanisms, pharmacological mitochondrial uncoupling can be mediated by natural products and small molecules that become deprotonated when they enter the alkaline pH of the mitochondrial matrix. The mitochondrial uncoupling process is illustrated in Figure 1.
2.1. Endogenous uncoupling
Endogenous mitochondrial uncoupling mechanisms are mediated in part by proteins present in the IMM [19]. Uncoupling proteins, a transmembrane group of 5 homologues (UCP1-5) in the IMM, catalyse the transport of protons across the mitochondrial membrane [20]. Each UCP is found in different regions in the body and has various roles in thermogenesis [21], fatty acid metabolism, macrophage activation [22], anti-oxidant effects, and calcium regulation [23]. There are wide variations in the expression levels and effects UCPs have on cancer phenotypes. UCPs are generally upregulated in many cancers including leukaemia, breast, ovarian, bladder, oesophageal, pancreatic, kidney, testicles, lung, prostate, and skin cancers, and their expression is associated with tumorigenicity, metastasis, and chemoresistance [3,5]. UCP2 is the most broadly upregulated UCP in a range of cancer types. UCP2 is best known for its role in decreasing reactive oxygen species (ROS) and is overexpressed or more “active” (deglutathionylated) in drug-resistant cancer cells [24]. Knocking down UCP2 in cancer cells (or inhibiting glutathionylation of UCP2) restores sensitivity to chemotherapy agents [24], reverses the Warburg effect [25], and increases ROS, ROS-dependent nuclear translocation of GAPDH, and autophagy [26]. In contrast, overexpression of UCP2 increased tumour growth and blocked the anti-tumour effects of camptothecin (a topoisomerase inhibitor) in colon cancer xenografts [27]. In human lung cancers, a high expression of UCP3 was observed in large cell carcinomas that was associated with a poor prognosis in squamous cell carcinomas [28]. However, overexpression of UCP3 in the basal epidermis of mice inhibited Akt activation and prevented carcinogen-induced skin tumour formation in v-Ha-Ras transgenic mice [29]. Interestingly, histological staining of prostate tissues showed stronger UCP1 expression in androgen receptor (AR)-positive and AR-negative tumours and metastases compared with normal/benign prostate tissue [30] despite UCP1 being considered a marker of brown and beige adipocytes. These studies demonstrated that UCPs are deregulated in different types of cancer and may have pro- or anti-tumorigenic roles that are likely linked to their ability to regulate oxidative stress.
The anion carrier complex adenosine nucleoside translocase (ANT) has 4 isoforms and is responsible for ADP/ATP exchange across the IMM and regulation of the mitochondrial permeability transition pore and mitochondrial apoptosis pathways through an undetermined method [31]. Both UCP and ANT complexes require free fatty acids for proton leak. The “fatty acid cycling” hypothesis suggests both complexes act as a carrier translocating the anionic form of fatty acid (FA) into the IMM. The fatty acid carboxylate group becomes deprotonated to form a carboxylic acid group in the matrix [32]. Endogenous uncoupling is activated by ROS [33], alkylsulfonate [34], adrenaline and noradrenaline [35], leptin [36], thyroid hormone [37], and peroxisome proliferator-activated receptor α (PPAR-α) agonist [38] and inhibited by purine nucleotides such as guanosine diphosphate (UDP), guanosine triphosphate (GTP), and ATP [39]. UCPs and ANTs can also be induced by oxidised lipid products (for example, 4-hydroxy-2-nonenal (HNE)) [40] and upregulated by high-fat diets [41], ketogenic diets [42,43], or fasting [44,45] as a potential mechanism to combat oxidative stress.
2.2. Exogenous uncoupling
Exogenous uncoupling mechanisms are modulated by chemically synthesised or naturally derived small molecules and divided into two broad categories: protonophores or non-protonophores. Protonophores have a molecular structure consisting of a bulky lipophilic moiety to binds to mitochondria and resides within the bilipid layer of the IMM, a weak acidic group with a pKa value between 3 and 8 to translocate protons [46], and a strong electron-withdrawing group for expulsion of the proton using negative charge delocalisation [47]. Protonophores exert uncoupling via H+ translocation across the IMM [47]; some common protonophores are 2,4-dinitrophenol (DNP), carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), carbonyl cyanide m-chlorophenyl hydrazone (CCCP), and BAM15. Non-protonophore uncouplers exhibit uncoupling properties through other mechanisms such as formononetin as a PPARγ agonist [48] and UCP1 activator [49], aspartate-glutamate carrier (AGC)-mediated uncoupling of arylamino-NBD analogues [50], and ATPase modification by 2,6-diisopropylphenol [51].
The first pharmacological application of exogenous uncouplers in humans was 2,4-dinitrophenol (DNP) in the 1930s [52]. While DNP treatment proved beneficial as a weight loss treatment, it had a narrow therapeutic index between efficacy and toxicity, non-specific membrane uncoupling, and off-target side effects that resulted in it being banned by the FDA in 1938 [52]. Since then, there has been a large number of DNP derivatives [[53], [54], [55], [56], [57]] and structurally unrelated uncouplers [[58], [59], [60], [61]] developed with the goal of attaining a better therapeutic window than DNP. Prodrugs of DNP include DNPME [55] and MP201 [56], and controlled-release formulations of DNP include CRMP [62]. Structurally unrelated molecules include BAM15 [58], OPC-163493 [60] CZ5 [63], niclosamide ethanolamine [64], nemorosone [65], and others are listed in Table 1. Many of these uncouplers have been evaluated for treating metabolic diseases (obesity/diabetes), hepatic steatosis, ischemia-reperfusion injury, and neurodegenerative diseases such as Alzheimer's and Huntington's disease [63]. A broad range of mitochondrial uncouplers has been evaluated for their efficacy in diverse cancer models in vitro and in vivo (Table 1).
Table 1.
The anti-cancer efficacy of mitochondrial uncouplers in cancer models.
| Uncoupler name, logP, and structure | Cancer cell types/cell lines | Mechanisms associated with cytostatic/cytotoxic effects in cancer cells | IC50 concentrations (assay type) | Other comments |
|---|---|---|---|---|
| Classical uncouplers | ||||
|
2,4-DNP LogP = 1.7 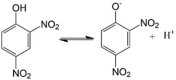 2,4-Dinitrophenol |
In vitro: Human T-cell leukaemia (Jurkat), human lymphoblastic leukaemia (CEM), and human cervical cancer (HeLa) cells [71], CEM and CEM-Crma cells [70], human pulmonary adenocarcinoma (Calu-6) [72], and mouse juxtaglomerular carcinoma cells (As4.1) [73] |
Activation of Fas surface receptor (CD95/APO-1) in CEM, CEM-Crma, HeLa, Jurkat, and CEM cells Downregulation of cyclin D1 and cyclin E, induction of p27 or p53 levels, elevation of the Bax to Bcl-2 ratio, loss of Δψm, activation of caspase-3 or -9, increment in intracellular ROS production, and reduction in GSH levels in Jurkat and As4.1 cells |
IC50 (μM) = 200 in Calu-6 cells with 72 h of treatment (MTT assay) | Off-target depolarisation of plasma membrane Sensitivity toward normal, healthy peripheral blood lymphocytes |
|
FCCP LogP = 3.7 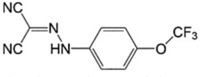 Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone |
In vitro: Human pulmonary adenocarcinoma (Calu-6 cells) [76,77] and mouse juxtaglomerular carcinoma cells (As4.1 cells) [74,75] |
Dissipation of Δψm, activation of caspase-3, induction of PARP-1 cleavage, increment in ROS production, and reduction in GSH levels in As4.1 cells Upregulation of CDKI and p27, activation of caspase-3, -8, and -9, downregulation of CDK2, CDK4, CDK6, cyclin D1, cyclin E, and phosphorylated forms of Rb, reduction in GSH levels, increase in ROS levels, decreased GSH levels, and dissipation of Δψm |
IC50 (μM) = 6.6 in Calu-6 cells with 72 h of treatment, 10 in As4.1 cells with 48 h of treatment (MTT assay) | |
|
CCCP LogP = 3.4 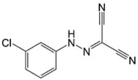 Carbonyl cyanide m-chlorophenyl hydrazone |
In vitro: Burkitt's lymphoma (ST486) [166], Human promyelocytic leukaemia (HL-60) [167], human adenocarcinoma (HeLa cells) [168], human neuroblastoma (SH-SY5Y) [80], human osteosarcoma (143B TK−) [78], human breast adenocarcinoma (MCF-7) [79], lymphoblastic leukaemia cells (CEM and CEM-Crma cells) [70], human B-lymphoblastoid (SKW6) [71], and murine pro-B lymphoid (FL5.12) cells [169] |
Reduction in Δψm and ATP levels, induction of PGAM5 expression, induction of PINK1/Parkin-mediated mitophagy, and inhibition of DRP1 translocation to mitochondria in SH-SY5Y cells Generation of ROS and Bax translocation to mitochondria in a dose-dependent manner; mitochondrial permeability transition (MPT) and mitochondrial membrane depolarisation without cytochrome c release in CCCP-treated osteosarcoma cells (as determined by confocal microscopy) Caspase-3 activation is responsible (but not solely) for CCCP-induced apoptosis (fluorometric assay) |
CCCP alone did not induce apoptosis in 143B TK−, SKW6, MCF-7, Jurkat, and CEM cells but induced apoptosis in FL5.12 and Jurkat cells following longer treatment | |
| Repurposed drugs | ||||
|
Niclosamide LogP = 4 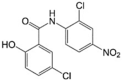 5-Chloro-N-(2-chloro-4-nitrophenyl)-2-hydroxybenzamide |
In vitro: Human glioblastoma (pGBM, LN229, T98G, U87(MG),U138, and U373(MG)) [85], adrenocortical carcinoma [91] (BD140A, SW-13, and NCI-H295R) [90], human colon carcinoma (HCT116) [92], human colon cancer (LoVo, SW620, HCT116, HT29, LS174T, SW480, and DLD-1) [86,170,171], human breast cancer (MDA-MB-468, MCF-7) [87,88,172], human ovarian cancer (A2780ip2, A2780cp20, and SKOV3Trip2) [89], human head and neck squamous cell carcinoma (WSU-HN6 and CNE-2Z) [173], human oesophageal cancer (ESO26, FLO-1, KYAE-1, OE33, SK-GT-4, and OE19) [94], human liver carcinoma (HepG2, QGY7701, QGY-7703, and SMMC-7721) [64,174,175] cells In vivo: Human glioblastoma (pGBMs) xenografts [85], oesophageal cancer xenografts (ESO26 and ESO26-PAC) [176], adrenocortical carcinoma xenografts (NCI-H295R) [90,91], non-small lung cancer xenografts (CL1-5) [93], breast cancer C57BL/6 model (E00771) [95], B16F0 mouse melanoma syngeneic model [96], MC38 allografts in NSG mice [92], and WSU-HN6 xenografts [173] |
Inhibition of intracellular Wnt/β-catenin, NOTCH-, mTOR-, and NF-kB signalling cascades in glioblastoma cells Activation of caspase-dependent apoptosis and G1 cell-cycle arrest and decreases in cellular migration by silencing the Wnt/β-catenin pathway in adrenocortical carcinoma cells Inhibition of Wnt/β-catenin, mTOR, and STAT3 signalling in ovarian cancer Induction of apoptosis via upregulation of ATF3 and activation of PERK in HCC cells Downregulation of anti-apoptotic protein Mcl-1 and survivin by inhibiting phosphorylation of STAT3 at Y705 in HCC cells Inhibition of S100A4-induced migration, invasion, tumorigenesis, and proliferation in colon cancer Inhibition of mTORC1 via acidification of cytoplasm due to an influx of protons through the plasma membrane and dissipation of proton gradients in lysosomes in breast cancer cells |
IC50 (μM) = 31.91 in HepG2 cells, 10.24 in QGY-7703 cells, and 13.46 in SMMC-7721 cells after 48h treatment (MTT assay). IC50 (μM) = 120 in BD140A cells, 150 in SW-13 cells, and 530 in NCI-H295R cells (qHTS screening). |
Poor water solubility 0.23 μg/mL and oral bioavailability (∼10%) [177] Synergistic anti-cancer activity with temozolomide for glioblastoma, cisplatin for hepatocellular carcinoma, erlotinib in colon cancer, and reversal of paclitaxel resistance in oesophageal cancer cells mainly through inhibiting Wnt/β-catenin Treatment for 26 days reduced liver metastasis [mean metastasis score: 34.9% (control 100%); median survival: 46.5 days (control 24 days)] in mice bearing HCT116 tumour xenografts (in vivo luminescence imaging) Reversal of adipocyte-induced epithelial mesenchymal transition (EMT) through inhibition of IL-6/STAT3 activation and downregulation of EMT-TFs TWIST and SNAIL in breast cancer |
|
Nitazoxanide (NTZ) LogP = 1.58 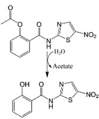 2-(Acetyloxy)-N-(5-nitro-2-thiazolyl) benzamide |
In vitro: 3D cultures of human epithelial colon carcinoma cells (HCT116 and HT-29) [106] Human breast adenocarcinoma cells (SKBR3), osteosarcoma-derived cells 1325 [107], colorectal cancer (Caco2, LS174T, SW403, SW480, DLD-1, and HT-29) [178], human glioblastoma cells (LN229, A172, U87), and human umbilical vein endothelial cells (HUVEC) [108] In vivo: Human colon cancer HCT116 xenografts [106], SKBR3 adenocarcinoma xenografts [107], and glioma LN229 xenografts [108] |
Activation of AMPK pathway and inhibition of c-Myc, mTOR, and Wnt signalling in HCT116 spheroids resulting in energetic stress Inhibition of c-Myc (IC50 of protein inhibition = 100 nM) in SKBR3 cells |
IC50 (μM) = 3 in HCT116 spheroids with hypoxic core vs 10–20 in HCT116 cells cultured in 2D (fluorometric microculture cytotoxicity assay and GFP fluorescence imaging) IC50 (μM) = 1 in SKBR3 cells after 24 h of treatment (MTS assay) |
NTZ more cytotoxic toward HCT116 cells grown in 3D vs 2D NTZ-induced cytotoxicity was reversible and required continuous high plasma concentrations in vivo Combination with Irinotecan (topoisomerase I inhibitor) had a greater effect on tumour growth than NTZ or irinotecan alone NTZ did not induce weight loss, diarrhoea, or skin rash in xenografted mice, whereas irinotecan caused weight loss |
|
Oxyclozanide LogP = 4.4 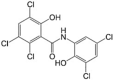 2,3,5-Trichloro-N-(3,5-dichloro-2-hydroxyphenyl)-6-hydroxybenzamide |
In vitro: Mouse colon adenocarcinoma (MC38) and human colorectal carcinoma (HCT116) cells [92] In vivo: Human colon adenocarcinoma (MC38) xenografts [92] |
Induction of AMPK activation and downregulation of mTOR activity, decreased phosphorylation of p70S6K, and upregulation of AMPK | Less potent than niclosamide; minimal efficacious concentration: 20–40 μM but is more stable in animals Reduced hepatic formation and metastases in vivo in NSG mice with colon adenocarcinoma (MC38) allografts |
|
| Chemical library | ||||
|
BAM15 LogP = 3.7 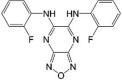 (2-Fluorophenyl) 6-[(2-fluorophenyl)amino](1,2,5-oxadiazolo[3,4-e]pyrazin-5-yl) amine |
In vitro: Human breast cancer (MCF-7) [113], and human melanoma (A375, SK-Mel28, MeWo) [114] and normal murine liver (NmuLi), rat myoblasts (L6), mouse myoblasts (C2C12), and primary rat left ventricular cardiomyocytes (NRVC) cells [58] |
Induced OCR and mitochondrial fragmentation and decreased Δψm but did not alter ERK phosphorylation in melanoma cell lines | ∗IC50 (μM) => 50 in NmuLi cells, 26.6 in L6 cells, 17.8 in C2C12 cells, and 29.1 in NRVC cells after 48 h of treatment (crystal violet or MTT assays) ∗IC50 values were not calculated for any cancer cell lines |
No off-target depolarisation of plasma membrane and no change in colony formation of melanoma cells at a 10 μM dose Apoptosis enhanced and colony formation inhibited in melanoma cells treated with BAM15 combined with PLX4032 (BRAFV600E inhibitor) or GSK1120212 (MEK inhibitor) and ABT737 (Bcl-2 and Bcl-xL inhibitor) |
|
SR4 LogP = 5.53 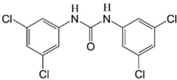 1, 3-Bis(3,5-dichlorophenyl) urea |
In vitro: Human amyloid leukaemia (HL-60) [118], human lung cancer (H1417, H1618, H520, and H358) [123], human hepatocellular carcinoma (HepG2), human lung cancer (H358), human melanoma cell (A2058) [122], human breast cancer (MCF-7 and MDA-MB-23) [118], and human/mouse melanoma cells (B16-F0, A2058, Hs600T A101D, A375, A2058, MeWo, SK-MEL-2, SK-MEL-5, and SK-MEL-28) [119,120] In vivo: B16-F0 mouse melanoma syngeneic model, human melanoma A2058 xenografts [120], human lung cancer H358 xenografts [123], and vemurafenib-sensitive and -resistant human A375 melanoma xenograft mouse models [119] |
Activation of AMPK and inhibition of mTORC1 signalling pathway in HepG2 cells Induction of G0/G1 cell cycle arrest through reduction in cyclin D1, cyclin E2, and cyclin-dependent kinases (CDK 2 and CDK 4) and upregulation of CDK inhibitors p21WAF1/Cip1 and p27Kip1 in HL-60 cells DNA fragmentation, activation of caspase 3, 7, and 9, release of cytochrome c to cytosol, PARP cleavage, and reduction in Δψm in HL-60 cells |
IC50 (μM) = 3.5 in HepG2 cells after 48 h of treatment (MTT assay) IC50 (μM) = > 20 in non-cancerous primary mouse hepatocytes after 48 h of treatment (MTT assay) |
In vitro cytotoxicity assay results suggest that SR4 may be more toxic to liver cancer cells (lower IC50 values) than normal hepatocytes |
|
FR58P1a LogP = 5.8 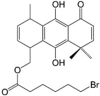 (9,10-Dihydroxy-4,8,8-trimethyl-5-oxo-1,4,5,8-tetrahydroanthracene-1-yl)methyl 6-bromohexanoate (9,10-Dihydroxy-4,8,8-trimethyl-5-oxo-1,4,5,8-tetrahydroanthracene-1-yl)methyl 6-bromohexanoate |
In vitro: Human (triple negative) breast cancer cells (MDA-MB-231) [124] |
Upregulation of AMPK via sirtuin 1 (Sirt1) activation, upregulation of glucose transporter 4 (GLUT4) and mitochondrial ADP/ATP transport genes (ANT3), downregulation of oxidative phosphorylation-related genes (COX-IV isoform 1, Cyt c, and ATP5FA1), and reduced expression of respiratory complexes (II, IV, and V) in MDA-MB-231 cells | No off-target depolarisation of the plasma membrane FR58P1a is mild uncoupler that promotes survival and proliferation in MDA-MB-231 cells but inhibits their fibronectin-dependent migratory ability |
|
|
FH535 LogP = 5.83 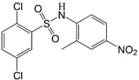 2,5-Dichloro-N-(2-methyl-4-nitrophenyl)benzenesulfonamide |
In vitro: Human pancreatic cancer (PANC-1 and BxPC-3) [126], human breast cancer (MDA-MB-231, HCC38) [128], human hepatocellular carcinoma cells (LCSC, Huh7, Hep3B, and PLC) [133], human colorectal cancer (HT29 and SW480) [130], and human osteosarcoma cells (143b, 143b-DxR, U2OS, SaOS-2, HOS, and K7M2) [131]. In vivo: Pancreatic PANC-1 xenografts [126], hepatocarcinoma HuH7 xenografts [134], and colon cancer HT29 xenografts [130] |
Inhibition of Wnt/β-catenin signalling pathway, inhibition of PPARδ and PPARγ receptors, and activation of AMPK (independent of the Wnt pathway) | IC50 (μM) = 13.8 in LCSC cells, 10.9 in HuH7 cells, 9.25 in Hep3B cells, and 6.6 in PLC cells after 72 h of treatment (3H-thymidine incorporation assay) | In vivo, FH535 (15–25 mg/kg) inhibits the growth of pancreatic (PANC-1), colon (HT29), and hepatocellular (HuH7) cancer cell xenograft tumours FH535 also suppresses metastasis of pancreatic cancer cells by inhibiting migration, invasion, and adhesion of detached cells and inhibits angiogenesis |
|
F16 and 5BMF LogP = 3.6 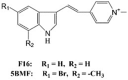 F16: 3-[2-(1-Methylpyridin-1-ium-4-yl)ethenyl]-1H-indole 5BMF: 5-Bromo-7-methyl-3-[(E)-2-(1-methylpyridin-1-ium-4-yl)ethenyl]-1H-indole |
In vitro: Human gastric carcinoma (SGC-7901), human breast adenocarcinoma (MCF-7) [137], human bladder cancer (T24), human non-small cell lung cancer cells (H838, HCC4006, HCC827, H1693, H2030, H2228, A549, H1437, and H1944) [136], human breast cancer (MD-MBA-231), human glioblastoma (U87MG), and mouse embryonic fibroblast cells (NIH/3T3) [140] In vivo: Glioblastoma U87MG cell xenografts [140] and non-small cell lung cancer (HCC827) xenografts [136] |
Inhibition of erb-2/neu protooncogene, selective accumulation in mitochondria leading to its swelling, increased ROS production, and outer membrane rupturing; elevation in mitochondrial inner membrane permeabilization to H+/K+, alteration in mitochondrial membrane fluidity, OCR increase, reduction in ATP levels, and Δψm, release of cytochrome c causing mitochondrial permeability transition mPTP-mediated cytotoxicity | F16 IC50 (μM) = 18.88 in T24 cells and 46.6 in H838 cells after 4 days of treatment 5BMF IC50 (μM) = 0.82 in T24 cells, 0.36 in H838 cells, 0.11 in HCC4006 cells, 1.99 in HCC827 cells, 1.18 in H1693 cells, 0.05 in H2228 cells, 0.73 in A549 cells, 1.43 in H1437 cells, 0.26 in H1944 cells, and 11 in NIH-3T3 cells after 96 h of treatment (direct cell counting) 5BMF IC50 = 4.15 μM in MD-MBA-231 cells, 0.36 μM in U87MG cells, and 10.23 μM in NIH/3T3 cells after 72 h of treatment (direct cell counting). |
F16 preferentially accumulates in cancer cell mitochondrial matrix with >100-fold selectivity over normal cell mitochondria In vitro cytotoxicity assay results suggest that 5BMF may be more toxic to cancer cells (lower IC50 values) than normal mouse embryonic fibroblasts In vivo, 5BMF (and 5BMF complexed with serum albumin) inhibits the growth of U87MG subcutaneous xenograft tumours without effecting mouse body weights |
| Polycyclic polyprenylated acylphloroglucinol | ||||
|
Nemorosone LogP = 8.3 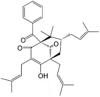 (1R,5R,7S)-1-Benzoyl-4-hydroxy-8,8-dimethyl-3,5,7-tris(3-methyl-2-buten-1-yl)bicyclo[3.3.1]non-3-ene-2,9-dione |
In vitro: Human hepatocellular carcinoma (HepG2) [65], human pancreatic cancer (MIA-PaCa-2, AsPC-1, and Capan-1) [141], human neuroblastoma (LAN-1 and NB69) [142], human breast adenocarcinoma (MCF-7) [144], human prostate cancer (PC-3), and human metastatic colorectal carcinoma cells (LoVo WT) [146] |
Dissipation of Δψm, depletion of ATP, release of mitochondrial Ca2+, and reduction in Ca2+ uptake leading to Ca2+-induced mPTP opening in HepG2, MIA-PaCa-2, AsPC-1, and Capan-1 cells | IC50 (μM) = 16 (24 h) and 4.5 (72 h) in MIA-PaCa-2 cells, 16 (24 h) and 5 (72 h) in AsPC-1 cells, 25 (24 h) and 5 (72 h) in Capan-1 cells, 80 (24 h) and 43 (72 h) in human dermal fibroblasts (HDF), and 73 (24 h) and 35 (72 h) in human foreskin fibroblast (HFF) cells (resazurin cell viability assay) | In vitro results suggest that nemorosone is more cytotoxic to cancer cells than normal cells |
|
Guttiferone A LogP = 10.4 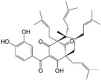 (1R,5R,7R,8S)-3-(3,4-Dihydroxybenzoyl)-4-hydroxy-8-methyl-1,5,7-tris(3-methyl-2-buten-1-yl)-8-(4-methyl-3-penten-1-yl)bicyclo[3.3.1]non-3-en-2,9-dione |
In vitro: Human hepatocellular carcinoma (HepG2) [147] and breast adenocarcinoma cells (MCF-7) [148] |
Dissipation of Δψm, depletion of ATP and increase in intracellular ATP levels in HepG2 cells Induction of Ca2+ release, prevention of Ca2+ uptake, induction of membrane permeabilization, induction of ROS levels, and depletion of NADPH in HepG2 mitochondria |
IC50 (μM) = 15 in MCF-7 cells after 48 h of incubation (MTT assay) | |
TPP-linked polyhydroxybenzoates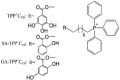 TPP + C10: Triphenyl(10-((3,4,5-trihydroxybenzoyl)oxy) decyl)phosphonium bromide SA-TPP + C10: (10-((2-Hydroxybenzoyl)oxy) decyl)triphenylphosphonium bromide GA-TPP + C10: (10-((2,5-Dihydroxybenzoyl)oxy) decyl)triphenylphosphonium bromide |
In vitro: TPP + C10: Mouse mammary adenocarcinoma (TA3/Ha cells), lab-generated multi-resistant variant of TA3/Ha cells (TA3-MTX-R), and human acute lymphoblastic leukaemia cells (CCRF-CEM) [153] TPP + C10, GA-TPP + C10, and SA-TPP + C10: human breast cancer cells (MCF7, AU565, BT549, MDA-MB-361, and MDA-MB-231 cells) [156], and human colon cancer cells (HCT-15 and COLO 205) [157] GA-TPP + C10: human breast cancer cells (MCF7, ZR-75-1, BT-474, BT-549, MDA-MB-231, AU565, and MDA-MB-361) [154] In vivo: TPP + C10: TA3/Ha mouse mammary adenocarcinoma syngeneic CAF1-Jax mouse model [153] TPP + C10: TA3/Ha syngeneic AJ mouse model [155] |
Dissipation of Δψm, induction of OCR, reduction in ATP levels, promotion of NAD(P)H oxidation, reduction in the NAD(P)H/NAD(P)+ ratio, and mitochondrial swelling indicating mPTP opening in TA3/Ha cells and their isolated mitochondria | TPP + C10 IC50 (μM) = 0.4 in TA3/Ha cells, 0.38 in TA3-MTX-R cells, 1.65 in CEM cells, and 7.1 in non-tumoral mammary gland cells (MM3MG) after 48 h of treatment (neutral red assay) TPP + C10 IC50 (μM) = 6.17 in MCF-7, 5.01 in AU565, 5.04 in BT549, and 11.99 in MDA-MB-231 cells after 48 h of treatment (MTT assay) SA-TPP + C10 IC50 (μM) = 2.33 in MCF-7 cells, 2.88 in AU565 cells, 1.67 in BT549 cells, and 8.34 in MDA-MB-231 cells after 48 h of treatment (MTT assay) GA-TPP + C10 IC50 (μM) = = 5.69 in MDA-MB-361 cells, 2.35 in MCF7 cells, 3.44 in AU565 cells, 1.2 in BT549 cells, and 8 in MDA-MB-231 cells after 48 h of treatment (MTT assay) |
In vitro results suggest that TPP + C10 is more cytotoxic to cancer cells than normal cells GA-TPP + C10 is more cytotoxic than SA-TPP + C10 and SA-TPP + C10 is more cytotoxic than TPP + C10 in cancer cells |
CTU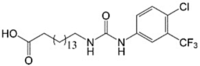 16-(([4-Chloro-3-(trifluoromethyl) phenyl]carbamoyl)amino)hexadecanoic acid |
In vitro: Human breast cancer cells (MDA-MB-231, T47D, MDA-MB-468, and MCF-7) [161] In vivo: Human breast cancer MDA-MB-231 xenografts [161] |
Dimeric self-assembly of CTU induced depolarisation of mitochondrial membrane, decreased mitochondrial cardiolipin and its precursor phosphatidylglycerol, increased caspase-3/7 activity, and depleted ATP levels | This is the chemically distinct and only protonophoric uncoupler that acts by forming lipophilic dimeric head-to-tail complex | |
|
VE-3N LogP = 3.1 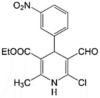 Ethyl 6-chloro-5-formyl-2-methyl-4-(3-nitrophenyl)-1,4-dihydrpyridine-3-carboxylate Ethyl 6-chloro-5-formyl-2-methyl-4-(3-nitrophenyl)-1,4-dihydrpyridine-3-carboxylate |
In vitro: Human hepatocellular carcinoma cells (HepG2) [165] |
Dissipation of Δψm and ATP production, suppression of respiration rate, reduction in ATP levels and Ca2+ uptake, induction of Ca2+ release, and increase in mitochondrial membrane fluidity and mitochondrial swelling in HepG2 cells | No IC50 values, although the viability of HepG2 cells at 10 μM was 43% (flow cytometry) | 40 and 50 μM VE-3N prevented Ca2+ uptake and enhanced Ca2+ release from mitochondria while lower concentrations did not affect calcium homeostasis (spectrophotometry) |
Abbreviations (not covered at the start of the review): inhibitory concentration (concentration that inhibits cell viability by 50%): IC50, RNA-dependent protein kinase-like kinase: PERK, activating transcription factor 3: ATF3, adenine nucleotide translocase 3: ANT3, NOD-scid gamma: NSG, calcium ion: Ca2+.
3. Mitochondrial uncouplers in cancer
3.1. 2,4-DNP
2,4-DNP (DNP) is one of the earliest known protonophoric mitochondrial uncouplers. It was first introduced in the US in 1933 as a weight-reducing agent. However, it was removed from the market by the FDA in 1938 due to significant adverse effects, such as severe hyperthermia, hyperlactacidaemia, hepatotoxicity, possible acute cataract, and agranulocytosis resulting in several fatalities [66,67]. These fatalities were exacerbated by negligent self-medication, rapid oral/dermal absorption, a narrow therapeutic window, steep dose responses, and variable interpatient sensitivity. Current therapeutic uses of DNP are being reconsidered for cancer therapy and neurodegenerative and metabolic diseases either in conjunction with other agents that can modify its toxicity profile [67], after physicochemical modification for mitochondrial targeting [54,68], or as a prodrug [56,69].
Studies have shown that DNP induces apoptosis in diverse human cancer cell lines in a dose-dependent manner, although the inhibitory concentration which that 50% of cells died (IC50) was shown to be as high as 200 μM in lung cancer cells (Table 1). DNP's mechanism of cytotoxicity has been linked to activation of CD95/APO-1/Fas receptors [70,71], G1 cell cycle arrest, downregulation of cell cycle proteins, increased p27 expression, elevated Bax:Bcl-2 ratios, activation of caspase-3 and -9, dissipated mitochondrial membrane potential (Δψm), decreased GSH levels, and increased intracellular ROS production [72,73]. However, DNP also induced cytotoxicity in peripheral blood lymphocytes (PBL), suggesting that it may not have selective toxicity toward cancer cells [72] (Table 1).
3.2. FCCP
FCCP, a fluorinated hydrazonomalononitrile, is a classical ionophoric mitochondrial uncoupler used as a model compound for studying uncoupling effects in vitro. FCCP reduced cell growth in mouse juxtaglomerular carcinoma cells (As4.1) and human lung cancer cells (Calu-6) in a dose- and time-dependent manner, with IC50 values less than 10 μM in both cell lines (Table 1). In As4.1 cells, FCCP-induced cell death (apoptosis) was associated with dissipated Δψm, activation of caspase-3, PARP-1 cleavage, and increased ROS coupled with decreased GSH levels [74,75]. In Calu-6 cells, FCCP inhibited cell growth via upregulation of CDKI and p27, activation of caspase-3, -8, and -9, and downregulation of CDK2, CDK4, CDK6, cyclin D1, cyclin E, and phosphorylated forms of Rb, increasing ROS/decreasing GSH levels and altering Δψm [76,77] (Table 1).
3.3. CCCP
CCCP, a chlorinated hydrazonomalononitrile, is another classical protonophore. Similar to FCCP, CCCP is lipophilic weak acid that is widely used to investigate mitochondrial functions in cells. The cytotoxicity of CCCP has been reported in many cancer cells (Table 1). Lim et al. reported that CCCP induced reversible mPTP opening and mitochondrial swelling in human osteosarcoma cells (143B TK-) without cytochrome c release or significant cell death [78]. Chaudhari et al. reported that CCCP enhanced TRAIL-induced apoptosis in human breast adenocarcinoma cells (MCF-7) in dose-dependent manner via ROS generation and dissipation of Δψm [79]. Similar to DNP, it induced Fas-triggered cell death in Jurkat, CEM, and CEM-Crma cells in a dose-dependent manner [70]. In human neuroblastoma cells (SH-SY5Y), CCCP induced cell death by reducing ΔΨm and ATP levels, increasing PGAM5 expression, inducing PINK1/Parkin-mediated mitophagy, and inhibiting DRP1 translocation to mitochondria [80].
3.4. Niclosamide
Niclosamide is a mitochondrial uncoupler that has been approved by the FDA to treat tapeworms for over 50 years [81]. Studies have shown that niclosamide has anti-neoplastic activity against leukaemia, renal, ovarian, colon, melanoma, non-small cell lung, prostate, and breast cancers [82]. Niclosamide has been demonstrated to alter Wnt/β-catenin [[83], [84], [85]], Notch [86], mTOR [87,88], NF-kB [85], and STAT3 signalling pathways [89]. Niclosamide has also been shown to decrease cell proliferation in a dose-dependent manner and induce G1 cell cycle arrest and apoptosis [90]. In vivo studies showed no toxicity at doses as high as 200 mg/kg/day [90], with reductions in the growth of tumours from adrenocortical carcinoma [90,91], colon adenocarcinoma [92], non-small lung cancer [93], oesophageal cancer [94], breast cancer [95], and melanoma [96] (Table 1). Some potential drawbacks that may limit the use of this uncoupler include both poor water solubility [97] and oral bioavailability [98]. Indeed, the maximum plasma concentration in humans following oral dosing with 500 mg 3 times per day (maximum tolerated dose) was 182 ng/mL, lower than the effective dose in preclinical studies for castration-resistant prostate cancer [99]. Nevertheless, niclosamide is currently in other phase I/II clinical trials for prostate [NCT03123978 and NCT02807805] and colon cancer [NCT02687009].
3.5. Nitazoxanide
Nitazoxanide (NTZ), a prodrug of tizoxanide, is a broad-spectrum anti-parasitic drug approved by the FDA in 2002 to treat giardiasis and cryptosporidiosis [100,101]. It is also active against several bacteria including Clostridium difficile [102] and Helicobacter pylori [103] and hepatitis B and C viruses [104]. NTZ has been shown to induce concentration-dependent collapse of Δψm in Mycobacterium tuberculosis comparable to FCCP [105]. The anti-cancer efficacy of NTZ has also been explored in diverse cancer cell types (Table 1). Of particular interest, Senkowski et al. identified NTZ, niclosamide, and closantel as mitochondrial uncouplers with selective activity toward multicellular tumour spheroids (MCTS) of human epithelial colon carcinoma cells (HCT116 and HT-29) via high-throughput drug screening of 1,600 clinically tested compounds [106]. The IC50 concentration of NTZ in 2D-cultured HCT116 cells was 10–20 μM compared with 3 μM in 3D-cultured HCT116 MCTS, suggesting NTZ's selectivity toward dormant, hypoxic, and nutrient-deprived cancer cells in the acidic interior of MCTS [106] (Table 1). When individually tested in dissociated spheroids (comprising both quiescent and proliferating cells), NTZ and the topoisomerase 1 inhibitor irinotecan did not completely inhibit colony formation. However, when both agents were used in combination, colony formation was strongly inhibited. Then when tested in vivo, NTZ combined with irinotecan caused a significant reduction in the growth of HCT116 colon xenograft tumours compared with irinotecan alone [106]. Similarly, NTZ treatment of nu/nu mice bearing human breast cancer cell (SKBR3) subcutaneous xenografts led to lower tumour volumes [107]. Wang et al. also revealed that high concentrations of NTZ (100–1600 μM) dose- and time-dependently inhibited proliferation of human glioblastoma cell lines (LN229, A172, U87, and HUVEC) in vitro, and NTZ penetrated the blood-brain barrier to reach orthotopic LN229 glioma xenografts in the caudate nucleus of the BALB/C nude mice model in vivo (after oral administration), prolonging the survival time [108].
NTZ has also been shown to activate AMPK and inhibit the mTOR pathway, indicating energetic deficit, and indirectly downregulate other oncogenic pathways including decreasing c-Myc levels and Wnt signalling [106] (Table 1). Notably, the inhibitory effect of NTZ on c-Myc has also been demonstrated in human breast cancer, osteosarcoma, and lymphoma cells in vitro [107]. In other studies, NTZ decreased ATP levels and strongly increased the extracellular acidification rate (ECAR) and OCR at low concentrations, leading to G1 cell cycle arrest [109]. NTZ also suppressed glioma cell viability and induced cell cycle arrest in the G0/G1 phase by increasing the transcription of inhibitor of growth 1 (ING1), a tumour suppressor gene, and by inhibiting late-stage autophagosome-induced ING1 degradation [108]. In colorectal cancer cells (Caco2 and LS174T cells), NTZ-induced G1 cell cycle arrest was associated with mTOR inhibition and c-Myc depletion-induced p27 stabilisation [109] (Table 1).
Given that NTZ has a remarkable pharmacokinetic safety profile with an average maximal plasma concentration exceeding 6 μM 2–6 h after a single (500 mg) dose without severe adverse effects and is also shown to be well tolerated in a single dose up to 4 g [110], NTZ may represent a promising anti-cancer therapy and is currently in a phase II clinical trial for advanced cancers [NCT02366884] alongside diverse anti-fungal/anti-bacterial/anti-protozoal agents.
3.6. Oxyclozanide
Oxyclozanide is a mitochondrial uncoupler that is approved for veterinary use as an anthelmintic drug [111]. There is limited literature in which oxyclozanide has been trialled as a potential cancer therapeutic. Alsadia et al. confirmed that oxyclozanide exhibited mitochondrial uncoupling properties at 20 μM and had no effect on the plasma membrane until much higher concentrations [92]. Cell profiling revealed that treatment with oxyclozanide increased cell accumulation in the G0/G1 phase and decreased cells in the S phase in MC38 and HCT116 colon cancer cell lines [92] (Table 1). Interestingly, oxyclozanide (20 μM) showed limited effects on MC38 cell viability over a 24-h period, but long-term treatment (2 weeks) showed anti-proliferative activity in clonogenic assays [92]. A 3-week study conducted in immune-deficient NOD-scid gamma (NSG) mice bearing MC38 cell xenografts revealed that oxyclozanide (that reached plasma concentrations of 20–40 μM) reduced metastasis to the spleen compared with control mice [92]. NMR metabolomics data showed a dose-dependent increase in AMPK activation and decrease in mTOR in MC38 cells treated with oxyclozanide for 2 h in vitro [92]. In vivo treatment of mice with oxyclozanide (via oral gavage) revealed an increase in AMPK activation in hepatic tissue 6 h after administration [92]. Prolonged oral treatment also showed a reduction in mTOR activity [92].
3.7. BAM15
BAM15 is a novel mitochondrial protonophore uncoupler that was first identified from a small molecule chemical library screen performed in L6 rat myoblasts [58]. In this study, Kenwood et al. showed that over a concentration range from 100 nM to 1 μM, BAM15 increased OCR comparable to FCCP and also reduced ROS [58]. At higher doses (between 1 μM and 50 μM), BAM15 maintained uncoupled respiration at a higher rate than FCCP, and this phenotype was observed in multiple cell types including L6 myoblasts, primary rat neonatal ventricular cardiomyocytes, mouse C2C12 myoblasts, normal murine hepatocytes, and human primary fibroblasts [58]. Furthermore, BAM15 increased the cellular extracellular acidification rate (ECAR), which can indicate higher rates of glycolysis. BAM15 was also determined to be a protonophore due to its ability to cause proton-dependent mitochondrial swelling and was not reliant on ANT [58]. Membrane electrophysiology experiments showed that BAM15 did not change current or plasma membrane depolarisation compared with FCCP. This indicates that unlike FCCP, BAM15 does not cause off-target depolarisation of the plasma membrane [58]. Pre-treatment of rats with BAM15 (bolus of 1 or 5 mg/kg) was also shown to protect rat kidneys from ischaemic reperfusion injury [58].
Since its discovery, BAM15 has been tested in AML12 hepatocytes, 3T3-L1 adipocytes, C2C12 mouse myotubes [112], breast cancer cells (MCF-7) [113], melanoma cells (A375, SK-Mel28, and MeWo) [114], and normal murine liver (NMuLi) cells [115] and was shown to activate AMPK in rat aorta myoblasts (A10 cells) [116], consistent with findings in C2C12 cells [112]. However, BAM15's uncoupling activity has predominantly been explored in mouse models of obesity. Findings from Axelrod et al. using obese C57BL/6 mice treated with 85 mg/kg/day of BAM15 were similar to those of Alexopoulos et al. using obese C57BL/6J mice treated with 100 mg/kg/day of BAM15 [112,115]. Both studies showed a reduction in fat mass and improved glucose homeostasis with BAM15 treatment [112,115]. BAM15 treatment also increased the levels of reduced glutathione (GSH) by 2.3-fold (oxidised glutathione levels were unaffected), decreased bioactive oxidised lipids (for example, 4-HNE), decreased pro-inflammatory lipid mediators, and did not alter ATP levels or induce AMPK activation in the livers of Western diet-fed mice (which is the primary tissue target of BAM15) [115]. These results indicated that BAM15 did not induce energetic stress and may improve anti-oxidant defence and decrease inflammation in vivo.
Only one study to date has reported the anti-cancer activity of BAM15 in vitro. Serasinghe et al. showed that BAM15 (10 μM) induced mitochondrial fragmentation, but on its own did not induce apoptosis or inhibit colony formation in melanoma cell lines (A375, SK-Mel28, and MeWo) [114]. However, when BAM15 was combined with minimally toxic doses of PLX4032 (a BRAFV600E inhibitor) or GSK1120212 (a MEK inhibitor), apoptotic cell death was markedly enhanced and colony formation was inhibited compared with any of the drugs alone [114]. These results suggest an additive or synergistic effect between BAM15 and inhibitors of MAPK signalling, a phenotype attributed to activation of the Bcl-2 family-mediated mitochondrial pathway of apoptosis [114]. Moreover, apoptosis was further enhanced and colony formation ablated in A375 cells when BAM15 was combined with the BH3 mimetic ABT737 (that inhibits Bcl-2 and Bcl-xL) and PLX4032 or GSK1120212 [114]. These studies reveal that therapies targeting mitochondria (such as with BAM15) may be useful to overcome resistance to MAPK inhibitors, which is a major obstacle to successful melanoma treatment.
While studies in cancer models are lacking, comprehensive studies in mice have shown that BAM15 is orally bioavailable, well-tolerated, not toxic at high doses (200 mg/kg), and does not alter food intake or lean mass [115]. However, BAM15 has a low aqueous solubility and half-life of 1.7 h, which may limit some applications [115]. Nevertheless, if BAM15 shows strong anti-cancer activity in future in vitro and in vivo studies either alone or in combination with other cancer drugs, this novel uncoupler has drug-like features that may be desirable for early-phase clinical trials.
3.8. SR4
Figarola et al. designed and synthesised SR4 based on a chemical library of aryl/heterocyclic ureido and aryl/heterocyclic carboxaminido phenoxyisobutyric acid derivatives that were originally established to find an advanced glycation inhibitor [117,118]. SR4 has demonstrated cytotoxicity against diverse cancer cell types in vitro [[118], [119], [120]] (Table 1), and its anti-cancer activity has been attributed to G0/G1 cell cycle arrest, reduced expression of cyclins D1 and E2, cyclin-dependent kinases (CDK 2 and CDK 4), upregulation of CDK inhibitors p21WAF1/Cip1 and p27Kip1, mitochondrial-dependent apoptosis, DNA fragmentation, activation of caspase 3, 7, and 9, release of cytochrome c to the cytosol, PARP cleavage, and a reduction in Δψm [118] (Table 1). In 3T3-L1 preadipocyte cells, SR4 exhibited no cytotoxicity but inhibited adipogenesis by activating AMPK via an increase in the AMP:ATP ratio and inhibiting mTORC1 (mammalian target of rapamycin complex 1) activity. Various cancer cells such as HL-60, HeLa, MCF-7, A549, H-358, and H-520 also showed dose-dependent activation of AMPK with SR4 treatment [121]. SR4 also induced mitochondrial membrane swelling in non-respiring mouse liver mitochondria, increased ROS production, and decreased cellular ATP production within 1 h in HepG2 cells [122]. ATP depletion triggered activation of AMPK and inhibition of the mTORC1 signalling pathway in a dose- and time-dependent manner, which ultimately led to G0/G1 cell cycle arrest and apoptosis [122].
The anti-cancer activity of SR4 has also been evaluated in several mouse models of cancer (Table 1). SR4 significantly inhibited melanoma tumour growth, prolonged the survival rate of mice, and was well tolerated without overt toxicity as indicated by no weight loss [120]. SR4 also significantly reduced tumour burden in lung cancer cell (H358) xenografts [123]. The in vivo anti-cancer efficacy of orally administered SR4 was also assessed in vemurafenib-sensitive and -resistant human melanoma (A375) xenografts and compared with niclosamide. Both uncouplers inhibited the growth of vemurafenib-sensitive and -resistant melanoma tumours in mice without evidence of toxicity to major organs [119]. However, SR4 demonstrated better potency than niclosamide in reducing tumour growth, which might have been attributed to the better pharmacokinetic profile of SR4 (longer half-life, greater maximum blood concentration, and higher bioavailability) [119].
3.9. FR58P1a
Urra et al. discovered the mild protonophoric uncoupler FR58P1a (a bromoalkyl ester of hydroquinone) in a drug screen of phenolic compounds in MDA-MB-231 triple-negative breast cancer (TNBC) cells [124] (Table 1). FR58P1a (5–30 μM) induced mild loss in Δψm in MDA-MB-231 cells compared to FCCP without off-target depolarisation of the plasma membrane [124]. FR58P1a (30 μM) induced ATP depletion and a decrease in intracellular NADPH levels, thereby modulating AMPK phosphorylation via sirtuin 1 (Sirt1) activation. Interestingly, activation of Sirt1/AMPK by FR58P1a promoted survival and proliferation in MDA-MB-231 cells but inhibited their fibronectin-dependent migratory ability, a phenotype not observed in normal mammary epithelial cells (MCF10A) [124] (Table 1). Prolonged FR58P1a-induced uncoupling promoted metabolic rewiring toward glycolysis and mitochondrial clearance. FR58P1a-induced metabolic rewiring was associated with upregulation of glucose transporter 4 (GLUT4) and ANT3 and downregulation of oxidative phosphorylation-related genes (COX-IV isoform 1, cytochrome c, and ATP5FA1) and respiratory complexes (II, IV, and V) (Table 1). FR58P1a also decreased mitochondrial mass in MDA-MB-231 as indicated by reduced cardiolipin content and proteins of the outer mitochondrial membrane (VDAC and TOMM20) as well as an increased PINK1 levels, an initiator of mitophagy [124].
3.10. FH535
Handeli and Simon identified FH535 via high-throughput chemical library screening as a potent inhibitor of Wnt/β-catenin and peroxisome proliferator-activated receptor (PPAR) signalling [125] (Table 1). Since aberrant Wnt/β-catenin pathway activation is closely related to carcinogenesis, the anti-cancer activity of FH535 has been tested in pancreatic cancer [126,127], TNBC [128], hepatocellular carcinoma [129], colorectal cancer [130], and osteosarcoma cells [131] (Table 1). Similar to FCCP, FH535 is sufficiently lipophilic (logP 5.83) to reach the mitochondrial membrane and possesses an ionisable nitrogen-hydrogen bond that can participate in proton translocation [132]. When the hydrogen in nitrogen-hydrogen bonds was replaced with a methyl group, the methyl analogue displayed no uncoupling activity and did not activate AMPK or inhibit Wnt [132]. In HuH7 and PLC/PRF/5 cells, FH535 specifically targeted the electron transport chain complexes I and II. The biological activity of FH535 was found to be due to interlinkage between inhibition of Wnt/β-catenin, OXPHOS, the ETC, and AMPK activation [133].
In vivo, FH535 treatment of mice bearing PANC-1 pancreatic tumour xenografts reduced tumour growth and angiogenesis as determined by immunostaining CD34 (a vascular endothelial cell marker) and microvessel density measurements [126]. FH535 also significantly reduced the growth of HuH7 hepatocellular xenograft tumours compared to controls without major signs of toxicity, such as weight loss and decreased ambulation [134]. FH535 also reduced the size and weight of HT29 colon adenocarcinoma xenograft tumours compared to controls without significant effects on weight loss, indicating good tolerability to FH535 [130] (Table 1).
3.11. F16 and 5BMF
F16, a delocalised lipophilic cation (DLC), was identified through a high-throughput chemical library screen to find a selective inhibitor of the ErbB-2/Neu protooncogene [135] (Table 1). DLCs easily penetrate the hydrophobic plasma and mitochondrial membrane and preferentially accumulate in the cancer cell mitochondrial matrix with >100-fold selectivity over normal cell mitochondria because of higher negative transmembrane potential difference in cancer cells (∼-220 mV) compared to normal cells (∼−140 mV) [136].
F16 displayed strong cytotoxicity against human gastric carcinoma (SGC-7901) and human breast cancer (MCF-7) cells in vitro [137]. F16 specifically localised in mitochondria, increased ROS production, induced swelling and outer membrane rupturing, increased inner membrane permeabilization to H+/K+, and changed mitochondrial membrane fluidity in isolated rat liver mitochondria in a time- and dose-dependent manner [137]. F16 showed strong uncoupling activity in mitochondria as indicated by increased OCR, decreased intracellular ATP levels, decreased Δψm, and the release of cytochrome c, suggesting mPTP-mediated cytotoxicity [137]. Due to its small size, fluorescence (absorption at 420 nm and emission at 520 nm), and mitochondrial selectivity, F16 was also used as a targeting moiety for anti-cancer agents (such as fluorouracil [138], chlorambucil [139], and boron-dipyrromethene [135]) for theragnostic purposes, bioimaging, and combinational chemotherapy.
Based on its structure-activity relationship, F16 was substituted at its indole position to increase cytotoxicity and cancer selectivity. 5BMF, a fluorescent 5-bromo-7-methyl substituted F16, showed relatively high selectivity and stronger anti-tumour activity (IC50: 0.82 μM in T24 human bladder cancer cells and 0.36 μM in H838 non-small cell lung cancer cells) compared to F16 (IC50: 18.88 μM in T24 cells and 46.6 μM in H838 cells) (Table 1). In athymic nude mice (nu/nu) bearing human lung adenocarcinoma HCC827 tumour xenografts, 5BMF significantly suppressed tumour growth compared to a control group. Insignificant changes in body weight and the histological examination of major organs of the treated group indicated 5BMF's low in vivo toxicity [136]. The same group non-covalently complexed 5BMF to human serum albumin to increase its solubility and consequently bioavailability for in vivo application and investigated its efficacy in athymic nude mice (nu/nu) bearing human glioblastoma U87MG xenografts. Ex vivo fluorescence imaging demonstrated 5BMF accumulated in tumours (high fluorescent intensity) and strongly inhibited tumour growth compared to controls (Table 1). Histological sections of major organs of the 5BMF-treated group showed no obvious tissue damage, indicating no or minimal in vivo toxicity [140].
3.12. Nemorosone
Pardo-Andreu et al. reported that nemorosone, a polycyclic polyprenylated acylphloroglucinol (PPAP) extracted from the floral resin of Clusia rosea, displayed protonophoric uncoupling activity (50–500 nM) with potency comparable to that of CCCP in isolated rat liver mitochondria [65]. It elicited concentration-dependent in vitro cytotoxicity against hepatocellular carcinoma HepG2 cells, but minimal toxicity against non-cancer human embryonic kidney HEK293T cells (Table 1). It also induced mitochondrial membrane potential dissipation, ATP depletion, Ca2+ release from Ca2+-loaded mitochondria, and Ca2+ uptake reduction in HepG2 cells in a dose-dependent manner [65]. The authors suggested that nemorosone's toxicity might be explained by sensitisation of mitochondria to Ca2+-induced permeability transition pore opening [65]. Nemorosone (5–20 μM) was also found to induce a dose-dependent reduction in mitochondrial membrane potential (Δψm) with concomitant Ca2+ release in pancreatic cancer cells (MIA-PaCa-2, AsPC-1, and Capan-1 cells) with complete abolition of Δψm at 50 μM as demonstrated by voltage-dependent fluorescent dye JC-1 staining [141]. Interestingly, nemorosone-induced ΔΨm abolition in non-cancerous HDF cells required a higher dose of 160 μM [141]. Diaz-Carballo et al. also noted that nemorosone exerted potent cytotoxic activity in vitro against neuroblastoma cell lines (LAN-1 and NB69 cells) at concentrations below 6.5 μM [142]. However, mouse fibroblast (NIH-3T3) and human embryonic fibroblast cells (MRC-5) displayed high resistance to nemorosone [142]. Nemorosone is also cytotoxic against oestrogen receptor-positive human breast cancer cells (MCF-7) [143], prostate cancer cells (PC-3) [144], leukaemia [145], and colorectal carcinoma cells (LoVo WT) in vitro [146] but has not yet been evaluated in in vivo cancer models (Table 1).
3.13. Guttiferone A
Another structurally related PPAP, guttiferone A, which was isolated from Symphonia globulifera roots and Garcinia aristata fruits, displayed uncoupling action in HepG2 cells and rat liver isolated mitochondria [147] (Table 1). Guttiferone decreased cell viability, promoted Δψm dissipation, and depleted ATP in HepG2 cells in a dose-dependent manner [147]. In isolated rat liver mitochondria, guttiferone A decreased Δψm, induced mitochondrial Ca2+ release, prevented mitochondrial Ca2+ uptake, induced cyclosporine A/EGTA-insensitive mitochondrial membrane permeabilization in isotonic sucrose-based medium, increased ROS levels, and depleted NADPH [147] (Table 1). Similar effects of guttiferone were reported in breast cancer (MCF-7) cells [148]. In addition to anti-cancer activity, anti-microbial [149], anti-malarial [150], anti-trypanosomal [151], and anti-leishmanial [152] activities of guttiferone and nemorosone have also been reported.
3.14. TPP-linked polyhydroxybenzoates
As previously noted, delocalised lipophilic cations (DLCs) such as triphenylphosphonium (TPP) compounds, rhodamine-123, rhodacyanine MKT-077, guanidine, F16, and dequalinium can easily enter and accumulate in the highly negatively charged mitochondrial matrix by virtue of their lipophilicity and delocalised positive charge and as such have been used as mitochondria-targeting ligands. Some of these conjugates have mitochondrial uncoupling activity, which is thought to play a major role in their cytotoxicity.
Jara et al. synthesised a series of conjugates of gallic acid ester (3,4,5-trihydroxy-benzoate) with TPP linked by alkyl chains of 8, 10, 11, and 12 carbons and showed their dose-dependent cytotoxicity in mouse mammary adenocarcinoma (TA3/Ha cells), lab-generated multi-resistant variants of TA3/Ha cells (TA3-MTX-R), and human acute lymphoblastic leukaemia cells (CCRF-CEM) [153]. Compared to other alkyl linkers, TPP + C10 in which gallic acid ester is conjugated to TPP via decyl linker showed the best compromise between cytotoxicity, cancer selectivity, and safety. TPP + C10 exhibited 17-fold selective cytotoxicity in TA3/Ha cells (IC50 0.4 μM) compared to non-cancerous mouse epithelial mammary gland MM3MG cells (7.1 μM). TPP + C10 showed uncoupling activity by decreasing Δψm in TA3/Ha cells and their isolated mitochondria, increasing OCR, decreasing ATP levels, and promoting the oxidation of NAD(P)H and reduction in the NAD(P)H/NAD(P)+ ratio in TA3/Ha cells. It also induced mitochondrial swelling in TA3/Ha and TA3-MTX-R cells, indicating MPT pore opening. In a TA3/Ha syngeneic CAF1-Jax mouse model, TPP + C10 significantly inhibited tumour growth compared with the control group [153]. TPP + C10 also exhibited dose-dependent cytotoxicity in breast cancer cells [154]. In another study, the combination of TPP + C10 and doxycycline, a tetracycline antibiotic, inhibited tumour growth in a TA3/Ha syngeneic AJ mouse model and improved the mouse survival rate [155].
Sandoval-Acuña et al. (from the same group) synthesised a series of polyhydroxybenzoates (2-hydroxy-, 2,5-dihydroxy-, 2,3-dihydroxy-, 3,4-dihydroxy-, and 3,4,5-trihydroxy-benzoate) conjugated to TPP via decyl linker and investigated their activity in human breast cancer cells with different oestrogen and HER2/neu expression [156] (Table 1). All five conjugates exerted a dose-dependent cytotoxic effect (IC50 6–16 μM after 24 h of incubation) on all five cell lines studied, although MDA-MB-231 cells required a higher dose. They also dose-dependently increased the OCR and mitochondrial superoxide anion production (at higher doses) and rapidly decreased Δψm. These effects were minimal in non-cancerous breast epithelial cells (MCF 10F cells), indicating these conjugates' cancer-selective action [156]. As single agents, they had minimal effects on ATP levels but markedly decreased ATP levels in the presence of AMPK inhibitors (compound C and diadenylate-pentaphosphate) [156]. Sublethal doses of conjugates also diminished the migratory capacity of MDA-MB-231 cells [156]. In another study, these conjugates' cytotoxicity was investigated in chemo-resistant human colon cancer cell lines (HCT-15 and COLO 205 cells) [157]. 3,4,5 tri-hydroxybenzoate (TPP + C10), 2,5-di-hydroxybenzoate (GA-TPP + C10), and 2-hydroxybenzoate (SA-TPP + C10) conjugated to TPP via a decyl unit showed cytotoxicity (IC50 5–16 μM after 24 h of treatment) in a concentration- and time-dependent manner in both cells [157] (Table 1). They increased the OCR, decreased Δψm, decreased ATP levels, activated AMPK, and induced caspase 3 activation [157]. Among these, GA-TPP + C10, which showed higher toxicity toward tumour cells (compared to normal colon epithelial cells CCD 841 CoN) was investigated in breast cancer cells in combination with doxycycline [154] (Table 1). Mitochondrial uncoupling of OXPHOS by GA-TPP + C10 induced a pro-survival adaptive response toward glycolysis, which was mediated by AMPK as indicated by increased transcription of nuclear and mitochondrial genes related to the ETC and upregulation of the mitochondrial transporter genes [154]. Doxycycline can inhibit the 28S subunit of the human mitochondrial ribosome and block the adaptive response induced by GA-TPP + C10, thereby promoting synergistic cytotoxicity [154].
3.15. CTU
CTU, an aryl-urea derived from ω-3-17,18-epoxyeicosanoic acid (ω-3-EEA), is a new protonophore mitochondrial uncoupler that acts via dimeric self-assembly [158] (Table 1). ω-3-EEA induced apoptosis in human MDA-MB-231 cells in vitro and suppressed tumour growth in MDA-MB-231 xenografted Balb/c nu/nu mice in vivo when co-administered with a soluble epoxide hydrolase inhibitor 2-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA) [159] (Table 1). Soluble epoxide hydrolase inhibitors prevented the hydrolysis of labile epoxide groups by endogenous epoxide hydrolase [160]. Similarly, CTU decreased the viability of MDA-MB-231 cells in vitro, which was coupled with decreased ATP levels and increased cells in the Sub-G1 phase of the cell cycle [160]. CTU also exhibited anti-cancer activity against breast cancer cells in vitro after prolonged treatment [161] and depolarised the mitochondrial membrane, decreased mitochondrial cardiolipin (and its precursor phosphatidylglycerol), and increased caspase-3/7 activity in MDA-MB-231 cells [161]. In female nu/nu mice xenografted with MDA-MB-231 cells in their mammary fat pads, CTU without co-administration of epoxide hydrolase inhibitor produced a dose-dependent decrease in tumour volume [161] (Table 1). Histology of tumour sections showed that CTU treatment increased TUNEL staining (a marker of apoptosis) and decreased Ki67 staining (a marker of proliferation) compared with control tumours [161].
Rawling et al. recently reported the molecular mechanism of the mitochondrial uncoupling action of CTU and analogues that had high pKa values (11–14) [158]. They established the uncoupling activity of CTU and analogues in TNBC cells using JC-1 assays, OCR tests, and non-cell-based assays that measured uncoupler-induced changes in the ionic membrane conductance of lipid bilayers of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) tethered to a thin film gold electrode using electrical impedance spectroscopy [158]. Density functional theory (DFT) calculations and molecular dynamics simulations in DOPC bilayers indicated that diffusion of the deprotonated uncoupler across the MIM proceeded via self-assembly of aryl ureas into dimeric complexes formed by intermolecular hydrogen bonds between carboxylate and urea groups [158]. Furthermore, it was reported that meta- and para-aryl substitution and dimerization delocalised the negative charge to promote overall complex lipophilicity and membrane permeability [158].
3.16. VE-3N
Several 1,4-dihydropyridine derivatives are known to exert potent pharmacological activities, such as calcium channel antagonist effect [162], reversal of multi-drug resistance in tumours [163], and anti-cancer activities [164]. VE-3N, a 1,4-dihydropyridine derivative, exerted potent protonophoric uncoupling activity in HepG2 cells and isolated rat liver mitochondria [165] (Table 1). VE-3N (1–10 μM) induced concentration-dependent apoptosis and decreased Δψm and ATP production in HepG2 cells (Table 1). In isolated mitochondria, VE-3N reduced the respiration rate, Δψm, ATP levels, and Ca2+ uptake and promoted Ca2+ release, membrane fluidity, and mitochondrial swelling in a dose-dependent manner [165]. However, VE-3N has not (to the best of our knowledge) been evaluated in in vivo cancer models.
4. Common phenotypes exhibited by mitochondrial uncouplers in cancer cells
4.1. Mitochondrial mechanisms associated with cytotoxicity
At least 16 structurally diverse mitochondrial uncouplers have been evaluated for their cytotoxicity against numerous cancer cell types and normal (non-cancerous) cells in vitro (Table 1). Furthermore, several of these uncouplers demonstrated anti-cancer activity in mouse models, but there was little consistency with respect to cancer types and models (allograft, xenograft, and syngeneic) (Table 1). Overall, these uncouplers display several common phenotypes that are associated with their mechanism of action and potentially underlie their cytotoxicity to cancer cells (Table 2). The relationship between these phenotypes and their links to cytotoxicity, cell cycle changes, and proliferation are shown in Figure 2. The most common phenotypes are expected for mitochondrial uncouplers, namely loss of mitochondrial membrane potential (Δψm) and increased OCR (Table 2). Other reported mechanisms include decreased cardiolipin (an important component of the IMM), increased expression of mitochondrial serine/threonine protein phosphatase family member 5 (PGAM5) and PTEN-induced kinase 1 (PINK1), dynamin-related protein 1 (DRP1) translocation to mitochondria, mitophagy, and subsequent loss of mitochondrial mass (Table 1, Table 2). Importantly, phenotypes relating to mitophagy may serve as one clue to the potential of uncouplers as anti-cancer agents, since dysfunctions in mitophagy are associated with cancer progression [179].
Table 2.
Common phenotypes associated with the anti-cancer effects of mitochondrial uncouplers.
| Common phenotypes associated with uncoupler-induced cytotoxicity | Mitochondrial uncouplers shown to induce these phenotypes |
|---|---|
| ↓ Δψm | DNP, FCCP, CCCP, SR4, FR58P1a, F16, nemorosone, guttiferone A, TPP + C10, GA-TPP + C10, SA-TPP + C10, CTU, VE-3N |
| Mitochondrial swelling | CCCP, SR4, F16, TPP + C10 |
| ↓ Mitochondrial cardiolipin | FR58P1a, CTU |
| Ca2+ release from mitochondria | Nemorosone, VE-3N |
| ↓ ATP | CCCP, NTZ, SR4, FR58P1a, FH535, F16, nemorosone, guttiferone A, TPP + C10, GA-TPP + C10, SA-TPP + C10, CTU, VE-3N |
| Activation of AMPK | NTZ, oxyclozanide, SR4, FR58P1a, FH535, TPP + C10, GA-TPP + C10, SA-TPP + C10 |
| Inhibition of mTOR signalling | Niclosamide, NTZ, oxyclozanide |
| ↓ Wnt signalling | NTZ, FH535 |
| ↑ ROS | DNP, FCCP, CCCP, SR4, F16, guttiferone A |
| ↓ NADPH | FR58P1a, guttiferone A, TPP + C10 |
| Apoptosis | DNP, FCCP, CCCP, niclosamide, SR4, F16, TPP + C10, GA-TPP + C10, SA-TPP + C10, VE-3N, CTU |
| Changes in cell cycle proteins | DNP, FCCP, SR4 |
| Cell cycle arrest (G0/G1 and G2/M) | DNP, niclosamide, NTZ, oxyclozanide, SR4, CTU |
Note: BAM15 is not listed in this table because it has not been reported to show cytotoxicity as a single agent in cancer cell lines.
Figure 2.

Relationships between mitochondrial uncoupler-induced phenotypes in cancer cells. Mitochondrial uncouplers induce mitochondrial membrane depolarisation, decrease ATP levels, increase ROS, and decrease anti-oxidant defence by reducing levels of GSH. These phenotypes are linked to changes in signalling pathways including Wnt/β-catenin, AMPK, mTOR, and HIF1α, many of which inhibit cancer cell proliferation and viability (apoptosis). Other phenotypes include arrest in the Go/G1 phase of the cell cycle, altered expression of cell cycle-regulated proteins including cyclin-dependent kinases (CDK), and upregulation of CDK inhibitors such as p21 and p27. Mitochondrial uncouplers also increase opening the mitochondrial permeability transition pore (mPTP), mitochondrial swelling, and rupture of mitochondrial membranes, which leads to calcium and cytochrome c release and subsequent apoptosis (or necrosis) when given in higher doses and longer durations (denoted by ∗). Phenotypes elevated by uncouplers are shown in green boxes, phenotypes decreased by uncouplers are shown in red boxes, and those in the blue boxes show differences in phenotypes (increased or decreased) that are closely related.
Many chemo-resistant and metastatic cancers and cancer stem cells (CSCs) have been found to rely on OXPHOS for ATP production [[180], [181], [182]]. Therefore, it is possible that some cancer cells are more sensitive to uncoupler-induced disruptions in mitochondrial metabolism and OXPHOS. Cancer cell mitochondria are also generally hyperpolarised (ΔΨc, ∼−220 mV) compared to normal cells (ΔΨn, ∼−140 mV) [136,183], but it remains unclear whether mitochondrial hyperpolarisation confers a selective vulnerability for cancer cells to pharmacologic mitochondrial uncouplers. One possibility is that hyperpolarised mitochondria will be more sensitive to mitochondrial uncouplers and result in higher rates of mitochondrial uncoupling than non-cancer cells. Another possibility is that mitochondrial uncouplers could be designed to target cancer cells in part by a high pKa, whereby the molecules can be deprotonated in the more alkaline mitochondrial matrix of cancer cells and may have less activity in non-cancerous cells. The studies discussed in this review have shown that several uncouplers display greater cytotoxicity toward cancer cells than normal cells, including SR4, 5BMF, nemorosone, and TPP + C10, and that F16 preferentially accumulates in the cancer cell mitochondrial matrix with >100-fold selectivity over normal cell mitochondria (Table 1). Thus, the inherent differences in mitochondria between cancer and normal cells may represent an opportunity to selectively kill cancer cells, which is chemotherapy's major goal.
4.2. Energetic stress and signalling pathways
Mitochondrial uncoupling decreases metabolic efficiency in the context of the amount of ATP derived per nutrient oxidised. Mild mitochondrial uncoupling increases OCR but has little effect on mitochondrial membrane potential or ATP production because mitochondria increase their nutrient oxidation rate to maintain a normal proton gradient and normal ATP production. However, high rates of mitochondrial uncoupling that exceed cellular respiratory capacity lower the supply of ATP, limiting available energy, impairing cell proliferation, and/or inducing cytotoxicity [184]. Indeed, a decrease in ATP (and/or an increase in the AMP/ATP ratio) and activation of AMP-activated protein kinase (AMPK) were reported in many studies (Table 2), suggesting that the doses of uncouplers used may have promoted high rates of uncoupling that exceeded cellular respiratory capacity and therefore induced energetic stress and cytotoxicity (Table 2).
The AMPK signalling pathway plays an important role in cancer cell proliferation, tumorigenesis, and cell survival and is imperative for cellular homeostasis with activation modulated by oxidative stress and changes in AMP/ATP ratios [185]. AMPK activation increases catabolic reactions including lipid oxidation, autophagy, and mitophagy as well as increasing glucose uptake to replenish ATP levels [185,186]. In particular, activation of AMPK induces phosphorylation and inhibition of mTORC1, a key mediator of anabolic growth [186]. The method by which uncouplers activate AMPK and whether this activation influences mTOR signalling or other pathways is possibly dose- or cell-type dependent. Hsu et al. found that 5 μM of CCCP suppresses HIF-1α in HepG2 cells by deregulating mTOR via AMPK signalling [187]. However, Kwon et al. reported that CCCP inhibits mTORC1 independently of AMPK [188]. Lee et al. found that wogonin, a natural compound from Scutellaria baicalensis, may act as an uncoupler and induce apoptosis in human glioblastoma cells by p53-mediated caspase signalling that is activated downstream of AMPK [189] (Figure 2). Interestingly, niclosamide has been shown to inhibit mTORC1 signalling via its ability to induce cytoplasmic acidification by inducing an influx of protons through the plasma membrane and dissipating proton gradients of other organelles such as lysosomes [88].
Mitochondrial uncouplers have also been shown to inhibit Wnt or downregulate Wnt signalling [132] (Table 1, Table 2), which is modulated by cellular ATP supplies (Figure 2) [132,190]. Increased Wnt signalling drives cancer cell proliferation, maintaining populations of tumour-initiating cells, preventing cell senescence, and promoting metastasis [191]. Thus, mitochondrial uncouplers may show efficacy against cancer types in which elevated Wnt signalling, both β-catenin-dependent and -independent, is a strong driver of malignancy and progression, including colon cancer [132].
4.3. ROS, calcium homeostasis, and cell death
ROS are chemical species that contain oxygen, such as superoxide anions, hydrogen peroxide, and hydroxyl radicals. ROS can cause cellular damage to DNA, proteins, and lipids, but are also important signalling molecules [192]. In cancer cells, it is thought that low levels of ROS are cytostatic, while elevated levels of ROS are tumour-promoting [192]. However, excessive ROS can be detrimental to cancer cells and cause cytotoxicity [192]. In early stage cancers, ROS promotes angiogenesis and tumour progression through an increase in both the genomic mutation rate and stabilisation of hypoxia-inducible factor 1-alpha (HIF-1α) [193]. As mitochondrial uncouplers can lower ROS, for example, BAM15 [58], mitochondrial uncouplers could prevent cancer cell proliferation and progression, mitigating ROS-driven genetic mutations and HIF-1α activation (Figure 2). However, the majority of uncouplers reported in this review were shown to increase ROS in cancer cells and often decreased anti-oxidant defence by lowering GSH and NADPH levels (Table 2). These features may be related to the dosing and length of exposure to the uncoupler, the anti-oxidant defence capacity of the cells (levels of ROS scavengers), the basal levels of ROS (cancer cells were shown to have higher basal levels of ROS [192]), or a combination of these parameters. Nevertheless, the induction of ROS by uncouplers in cancer may be responsible for tipping the balance in ROS levels toward cytotoxicity.
In many of the studies listed in Table 1, it was not always clear whether the uncouplers that induced ROS did so by increasing mitochondrial ROS (mitoROS) or non-mitochondrial ROS. One study showed that 5 μM of SR4 increased MitoSOX Red (a mitochondrial-specific superoxide probe) fluorescence in HepG2 cells [122]. Furthermore, SR4-induced mitoROS was rescued by the anti-oxidants MitoTempo and N-acetylcysteine and the recoupler 6-ketocholestanol, but not the mPTP blocker cyclosporin A, indicating that SR4-induced mitoROS was due to uncoupling and not opening of the mPTP [122]. In another study, 4.5 μM of FCCP increased MitoSOX Red fluorescence in human pro-myelocytic leukaemia cells (HL-60), whereas 293 μM was required to inhibit HL-60 cell viability by 50% after 6 h of treatment (IC50 = 18 μM for 24 h of treatment) [194]. These results demonstrated that FCCP increased mitoROS at doses lower than those required to induce significant cytotoxicity. However, other studies showed that 1–5 μM of FCCP did not induce mitoROS in HepG2 cells [122,195]. An off-target effect of FCCP is its ability to depolarise the plasma membrane (as reviewed in [17]) and events such as this can promote activation of NADPH oxidase 2 (NOX2, which is localised at the plasma membrane) and induce ROS [196]. As such, it is likely that FCCP and other uncouplers such as niclosamide [88] that dissipate other proton gradients outside of mitochondria may stimulate ROS production in other cellular locations in addition to mitochondria.
High levels of mitochondrial calcium (Ca2+) coupled with increased levels of mitoROS induce opening of the mPTP, swelling of the mitochondrial matrix, calcium efflux, and subsequent cell death [197]. Calcium overload and the mPTP play critical roles in the pathogenesis of cardiac [198] and neuronal injury as well as neurodegenerative diseases such as Alzheimer's [199] and Parkinson's disease [200]. Interestingly, DNP's therapeutic benefit against ischaemic injury in neurodegenerative diseases has been attributed to its ability to prevent mitochondrial calcium overload [201]. In this review, it appears that many uncouplers disrupt calcium homeostasis and induce ROS, leading to mitochondrial swelling, calcium efflux, and subsequent apoptosis in cancer cells (Table 1). However, one study showed that the mPTP is not required for calcium efflux in HeLa cells [202], suggesting that the phenotypes exhibited by uncouplers may be cell-type specific.
5. Conclusions and future considerations
In the past few years, several clinical trials have emerged that investigated the anti-cancer potential of mitochondrial-targeted drugs including metformin, an ETC complex I inhibitor, and CPI-613, a lipoate analogue that inhibits α-ketoglutarate dehydrogenase and pyruvate dehydrogenase [6]. Phase II and phase III clinical trials were also conducted to examine the anti-cancer effects of curcumin [NCT02944578, NCT02782949, and NCT02064673], which in some studies was shown to induce mitochondrial uncoupling [[203], [204], [205]]. Similarly, low doses of the multi-tyrosine kinase inhibitor sorafenib were recently reported to induce mild uncoupling and reduce HCC formation in mice and non-alcoholic steatohepatitis in mice and monkeys [206]. The uncoupler niclosamide is in phase I/II clinical trials for prostate [NCT03123978 and NCT02807805] and colon cancer [NCT02687009], and the uncoupler nitazoxanide (NTZ) is in a phase II clinical trial for advanced cancers [NCT02366884]. As this review illustrated, many other mitochondrial uncouplers have also shown promising anti-cancer effects in vitro and in vivo (Table 1) and may progress to clinical trials in the future.
In this review, we reported that some uncouplers showed increased anti-cancer efficacy when used in combination with irinotecan (NTZ), doxycycline (TPP + C10 and GA-TPP + C10), temozolomide (niclosamide), cisplatin (niclosamide), erlotinib (niclosamide), and MAPK inhibitors (BAM15) in various cancer types in vitro and/or in vivo (Table 1). Of particular interest, doses of BAM15 that were sufficient to induce uncoupling were not toxic to melanoma cells, but when combined with MAPK inhibitors, cell death was enhanced [114]. These results suggest that it is possible that low doses of uncouplers (that induce mild uncoupling) may be effective to elicit an anti-cancer response when used in combination with other chemotherapy agents. However, it is important to note that the opposite effect was also observed in some cases. For example, as a single agent, FCCP induced apoptosis in lymphoid CCRF-CEM cells and myeloid HL60 cells in a dose-dependent manner, but pre-treatment of cells with FCCP also delayed apoptosis induced by the protein kinase inhibitors staurosporine and chelerythrine in CCRF-CEM cells [13]. Similarly, low micromolar FCCP doses decreased apoptosis and ROS induced by the topoisomerase inhibitor camptothecin in HCT116 colon cancer cells [27] and prevented cisplatin-induced cytotoxicity in HT-29 and SW-620 colon cancer cells [207]. It is thought that mild uncoupling may be cytoprotective due to the anti-oxidant/ROS-inhibiting effects at low concentrations. In one instance, it was shown that mild uncoupling induced by FCCP, BAM15, and DNP protected cells against radiation therapy that stimulates ROS production [208].
Momcilovic et al. showed differences in mitochondrial membrane potential between lung tumours in vivo using a voltage-sensitive, positron emission tomography (PET) radiotracer 4-[18F]fluorobenzyl-triphenylphosphonium (18F-BnTP), which is a positively charged lipophilic cation that localises to the negatively charged IMM [209]. Using a genetically engineered mouse model of lung cancer combined with imaging with [18F]fluoro-2-deoxyglucose (18F-FDG) and 18FBnTP showed that lung tumours that were positive for 18F-FDG but negative for 18F-BnTP were squamous cell carcinomas (SCC), whereas tumours positive for 18FBnTP but negative for 18F-FDG were lung adenocarcinomas (ADC) [209]. They also showed that treating mice with complex I inhibitors phenformin or metformin decreased 18FBnTP uptake, suggesting that this tracer can detect loss of Δψm in vivo [209]. Importantly, this study highlights these tracers' potential to not only identify tumour histology but also reveal which tumours may be more responsive to treatment with mitochondrial-targeted agents. Furthermore, some cancer stem cells that rely on OXPHOS (as reviewed in [182]) may also show increased sensitivity to agents that disrupt mitochondrial metabolism. Indeed, the uncoupler FH535 inhibited the proliferation of liver cancer stem cells in vitro [129].
In summary, a number of important factors require attention as mitochondrial uncouplers progress toward the clinic, including 1) which mitochondrial uncouplers are most effective against tumours in vivo and are well-tolerated, non-toxic to normal cells, and ideally orally bioavailable; 2) what cancer types/stages and patients would benefit most from treatment with these uncouplers; 3) what mediates sensitivity to these agents, for example, cancer cells that rely on OXPHOS and/or have high Δψm; 4) potential mechanisms of resistance, for example, prolonged treatment with uncouplers may upregulate glycolytic pathways in cancer cells; 5) whether the systemic effects of mitochondrial uncouplers overshadow the direct effects on cancer cells; and 6) what standard-of-care drugs they should be combined with for synergistic or additive effects. Collectively, this review highlights published evidence supporting uncouplers' potential as anti-cancer agents, and future studies addressing the remaining gaps in knowledge will help progress more of these agents into cancer clinical trials.
Funding
R.S. is funded by a Scientia PhD Scholarship from UNSW, and F.L.B. is funded by a Cancer Institute NSW Early Career Fellowship (2018/ECF003).
Author contributions
FLB: Conceptualisation, supervision, writing original draft, and review. RS: Visualisation, writing original draft, review, and editing. EJ: Visualisation, writing original draft, review, and editing.
Conflict of interest
F.L.B. has a commercial interest in mitochondrial uncoupling through Life Biosciences, Inc.
References
- 1.Prager G.W., Braga S., Bystricky B., Qvortrup C., Criscitiello C., Esin E. Global cancer control: responding to the growing burden, rising costs and inequalities in access. ESMO Open. 2018;3(2) doi: 10.1136/esmoopen-2017-000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.Faubert B., Solmonson A., DeBerardinis R.J. Metabolic reprogramming and cancer progression. Science. 2020;368(6487) doi: 10.1126/science.aaw5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.DeBerardinis R.J., Chandel N.S. We need to talk about the Warburg effect. Nature Metabolism. 2020;2(2):127–129. doi: 10.1038/s42255-020-0172-2. [DOI] [PubMed] [Google Scholar]
- 6.Vasan K., Werner M., Chandel N.S. Mitochondrial metabolism as a target for cancer therapy. Cell Metabolism. 2020;32(3):341–352. doi: 10.1016/j.cmet.2020.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chuang C.-H., Dorsch M., Dujardin P., Silas S., Ueffing K., Hölken J.M. Altered mitochondria functionality defines a metastatic cell state in lung cancer and creates an exploitable vulnerability. Cancer Research. 2021;81(3):567–579. doi: 10.1158/0008-5472.CAN-20-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cadenas S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2018;1859(9):940–950. doi: 10.1016/j.bbabio.2018.05.019. [DOI] [PubMed] [Google Scholar]
- 9.Weinbach E.C., Garbus J. Mechanism of action of reagents that uncouple oxidative phosphorylation. Nature. 1969;221(5185):1016–1018. doi: 10.1038/2211016a0. [DOI] [PubMed] [Google Scholar]
- 10.Loomis W.F., Lipmann F. Reversible inhibition of the coupling between phosphorylation and oxidation. Journal of Biological Chemistry. 1948;173(2):807–808. [PubMed] [Google Scholar]
- 11.Rolfe D.F., Newman J.M., Buckingham J.A., Clark M.G., Brand M.D. Contribution of mitochondrial proton leak to respiration rate in working skeletal muscle and liver and to SMR. American Journal of Physiology. 1999;276(3):C692–C699. doi: 10.1152/ajpcell.1999.276.3.C692. [DOI] [PubMed] [Google Scholar]
- 12.Curley S.A., Palalon F., Lu X., Koshkina N.V. Noninvasive radiofrequency treatment effect on mitochondria in pancreatic cancer cells. Cancer. 2014;120(21):3418–3425. doi: 10.1002/cncr.28895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isono T., Chano T., Yonese J., Yuasa T. Therapeutic inhibition of mitochondrial function induces cell death in starvation-resistant renal cell carcinomas. Scientific Reports. 2016;6(1):25669. doi: 10.1038/srep25669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu J., Yang S., Cao B., Zhou G., Zhang F., Wang Y. Targeting B7-H3 via chimeric antigen receptor T cells and bispecific killer cell engagers augments antitumor response of cytotoxic lymphocytes. Journal of Hematology & Oncology. 2021;14(1):21. doi: 10.1186/s13045-020-01024-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schcolnik-Cabrera A., Chavez-Blanco A., Dominguez-Gomez G., Juarez M., Vargas-Castillo A., Ponce-Toledo R.I. Pharmacological inhibition of tumor anabolism and host catabolism as a cancer therapy. Scientific Reports. 2021:5222. doi: 10.1038/s41598-021-84538-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anso E., Mullen A.R., Felsher D.W., Matés J.M., DeBerardinis R.J., Chandel N.S. Metabolic changes in cancer cells upon suppression of MYC. Cancer & Metabolism. 2013;1(1):7. doi: 10.1186/2049-3002-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demine S., Renard P., Arnould T. Mitochondrial uncoupling: a key controller of biological processes in physiology and diseases. Cells. 2019;8(8):795. doi: 10.3390/cells8080795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garlid K.D., Nakashima R. Studies on the mechanism of uncoupling by amine local anesthetics. Evidence for mitochondrial proton transport mediated by lipophilic ion pairs. Journal of Biological Chemistry. 1983;258(13):7974–7980. [PubMed] [Google Scholar]
- 19.Zhao R.Z., Jiang S., Zhang L., Yu Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling. International Journal of Molecular Medicine. 2019;44(1):3–15. doi: 10.3892/ijmm.2019.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rousset S., Alves-Guerra M.-C., Mozo J., Miroux B., Cassard-Doulcier A.-M., Bouillaud F. The biology of mitochondrial uncoupling proteins. Diabetes. 2004;53(suppl 1):S130–S135. doi: 10.2337/diabetes.53.2007.s130. [DOI] [PubMed] [Google Scholar]
- 21.Riley C.L., Dao C., Kenaston M.A., Muto L., Kohno S., Nowinski S.M. The complementary and divergent roles of uncoupling proteins 1 and 3 in thermoregulation. The Journal of Physiology. 2016;594(24):7455–7464. doi: 10.1113/JP272971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emre Y., Nübel T. Uncoupling protein UCP2: when mitochondrial activity meets immunity. FEBS Letters. 2010;584(8):1437–1442. doi: 10.1016/j.febslet.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 23.Andrews Z.B., Diano S., Horvath T.L. Mitochondrial uncoupling proteins in the CNS: in support of function and survival. Nature Reviews Neuroscience. 2005;6(11):829–840. doi: 10.1038/nrn1767. [DOI] [PubMed] [Google Scholar]
- 24.Pfefferle A., Mailloux R.J., Adjeitey C.N.-K., Harper M.-E. Glutathionylation of UCP2 sensitizes drug resistant leukemia cells to chemotherapeutics. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research. 2013;1833(1):80–89. doi: 10.1016/j.bbamcr.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Baffy G., Derdak Z., Robson S.C. Mitochondrial recoupling: a novel therapeutic strategy for cancer? British Journal of Cancer. 2011;105(4):469–474. doi: 10.1038/bjc.2011.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dando I., Fiorini C., Pozza E.D., Padroni C., Costanzo C., Palmieri M. UCP2 inhibition triggers ROS-dependent nuclear translocation of GAPDH and autophagic cell death in pancreatic adenocarcinoma cells. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2013;1833(3):672–679. doi: 10.1016/j.bbamcr.2012.10.028. [DOI] [PubMed] [Google Scholar]
- 27.Derdak Z., Mark N.M., Beldi G., Robson S.C., Wands J.R., Baffy G. The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Research. 2008;68(8):2813–2819. doi: 10.1158/0008-5472.CAN-08-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giatromanolaki A., Balaska K., Kalamida D., Kakouratos C., Sivridis E., Koukourakis M.I. Thermogenic protein UCP1 and UCP3 expression in non-small cell lung cancer: relation with glycolysis and anaerobic metabolism. Cancer Biology & Medicine. 2017;14(4):396–404. doi: 10.20892/j.issn.2095-3941.2017.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nowinski S.M., Solmonson A., Rundhaug J.E., Rho O., Cho J., Lago C.U. Mitochondrial uncoupling links lipid catabolism to Akt inhibition and resistance to tumorigenesis. Nature Communications. 2015;6(1):8137. doi: 10.1038/ncomms9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhau H.E., He H., Wang C.Y., Zayzafoon M., Morrissey C., Vessella R.L. Human prostate cancer harbors the stem cell properties of bone marrow mesenchymal stem cells. Clinical Cancer Research. 2011;17(8):2159–2169. doi: 10.1158/1078-0432.CCR-10-2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brand M.D., Pakay J.L., Ocloo A., Kokoszka J., Wallace D.C., Brookes P.S. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochemical Journal. 2005;392(2):353–362. doi: 10.1042/BJ20050890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garlid K.D., Orosz D.E., Modrianský M., Vassanelli S., Jezek P. On the mechanism of fatty acid-induced proton transport by mitochondrial uncoupling protein. Journal of Biological Chemistry. 1996;271(5):2615–2620. doi: 10.1074/jbc.271.5.2615. [DOI] [PubMed] [Google Scholar]
- 33.Chouchani E.T., Kazak L., Jedrychowski M.P., Lu G.Z., Erickson B.K., Szpyt J. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature. 2016;532(7597):112–116. doi: 10.1038/nature17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jabůrek M., Vařecha M., Ježek P., Garlid K.D. Alkylsulfonates as probes of uncoupling protein transport mechanism ion pair transport demonstrates that direct H+ translocation by UCP1 is not necessary for uncoupling. Journal of Biological Chemistry. 2001;276(34):31897–31905. doi: 10.1074/jbc.M103507200. [DOI] [PubMed] [Google Scholar]
- 35.Kozak U.C., Held W., Kreutter D., Kozak L.P. Adrenergic regulation of the mitochondrial uncoupling protein gene in brown fat tumor cells. Molecular Endocrinology. 1992;6(5):763–772. doi: 10.1210/mend.6.5.1603085. [DOI] [PubMed] [Google Scholar]
- 36.Henry B.A., Andrews Z.B., Rao A., Clarke I.J. Central leptin activates mitochondrial function and increases heat production in skeletal muscle. Endocrinology. 2011;152(7):2609–2618. doi: 10.1210/en.2011-0143. [DOI] [PubMed] [Google Scholar]
- 37.Solmonson A., Mills E. Uncoupling proteins and the molecular mechanisms of thyroid thermogenesis. Endocrinology. 2016;157(2):455–462. doi: 10.1210/en.2015-1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barberá M.J., Schlüter A., Pedraza N., Iglesias R., Villarroya F., Giralt M. Peroxisome proliferator-activated receptor α activates transcription of the brown fat uncoupling protein-1 gene a link between regulation of the thermogenic and lipid oxidation pathways in the brown fat cell. Journal of Biological Chemistry. 2001;276(2):1486–1493. doi: 10.1074/jbc.M006246200. [DOI] [PubMed] [Google Scholar]
- 39.Modrianský M., Murdza-Inglis D.L., Patel H.V., Freeman K.B., Garlid K.D. Identification by site-directed mutagenesis of three arginines in uncoupling protein that are essential for nucleotide binding and inhibition. Journal of Biological Chemistry. 1997;272(40):24759–24762. doi: 10.1074/jbc.272.40.24759. [DOI] [PubMed] [Google Scholar]
- 40.Echtay K.S., Esteves T.C., Pakay J.L., Jekabsons M.B., Lambert A.J., Portero-Otín M. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. The EMBO Journal. 2003;22(16):4103–4110. doi: 10.1093/emboj/cdg412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cole M.A., Murray A.J., Cochlin L.E., Heather L.C., McAleese S., Knight N.S. A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Research in Cardiology. 2011;106(3):447–457. doi: 10.1007/s00395-011-0156-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Srivastava S., Baxa U., Niu G., Chen X.L., Veech R. A ketogenic diet increases brown adipose tissue mitochondrial proteins and UCP1 levels in mice. IUBMB Life. 2013;65(1):58–66. doi: 10.1002/iub.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sullivan P.G., Rippy N.A., Dorenbos K., Concepcion R.C., Agarwal A.K., Rho J.M. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Annals of Neurology. 2004;55(4):576–580. doi: 10.1002/ana.20062. [DOI] [PubMed] [Google Scholar]
- 44.Millet L., Vidal H., Andreelli F., Larrouy D., Riou J.-P., Ricquier D. Increased uncoupling protein-2 and-3 mRNA expression during fasting in obese and lean humans. Journal of Clinical Investigation. 1997;100(11):2665–2670. doi: 10.1172/JCI119811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weigle D.S., Selfridge L.E., Schwartz M.W., Seeley R.J., Cummings D.E., Havel P.J. Elevated free fatty acids induce uncoupling protein 3 expression in muscle: a potential explanation for the effect of fasting. Diabetes. 1998;47(2):298–302. doi: 10.2337/diab.47.2.298. [DOI] [PubMed] [Google Scholar]
- 46.Enoch S.J., Schultz T.W., Popova I.G., Vasilev K.G., Mekenyan O.G. Development of a decision tree for mitochondrial dysfunction: uncoupling of oxidative phosphorylation. Chemical Research in Toxicology. 2018;31(8):814–820. doi: 10.1021/acs.chemrestox.8b00132. [DOI] [PubMed] [Google Scholar]
- 47.Terada H. The interaction of highly active uncouplers with mitochondria. Biochimica et Biophysica Acta (BBA) - Reviews on Bioenergetics. 1981;639(3–4):225–242. doi: 10.1016/0304-4173(81)90011-2. [DOI] [PubMed] [Google Scholar]
- 48.Nie T., Zhao S., Mao L., Yang Y., Sun W., Lin X. The natural compound, formononetin, extracted from Astragalus membranaceus increases adipocyte thermogenesis by modulating PPARγ activity. British Journal of Pharmacology. 2018;175(9):1439–1450. doi: 10.1111/bph.14139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tao J., Tao L., Huang W. Formononetin stimulates thermogenesis of Brown adipocytes by promoting the expression of uncoupling protein 1. Xi bao yu fen zi mian yi xue za zhi= Chinese Journal of Cellular and Molecular Immunology. 2020;36(3):242–247. [PubMed] [Google Scholar]
- 50.Antonenko Y.N., Denisov S.S., Khailova L.S., Nazarov P.A., Rokitskaya T., Tashlitsky V.N. Alkyl-substituted phenylamino derivatives of 7-nitrobenz-2-oxa-1, 3-diazole as uncouplers of oxidative phosphorylation and antibacterial agents: involvement of membrane proteins in the uncoupling action. Biochimica et Biophysica Acta (BBA) – Biomembranes. 2017;1859(3):377–387. doi: 10.1016/j.bbamem.2016.12.014. [DOI] [PubMed] [Google Scholar]
- 51.Kigoulet M., Devin A., Avéret N., Vandais B., Guérin B. Mechanisms of inhibition and uncoupling of respiration in isolated rat liver mitochondria by the general anesthetic 2, 6-diisopropylphenol. European Journal of Biochemistry. 1996;241(1):280–285. doi: 10.1111/j.1432-1033.1996.0280t.x. [DOI] [PubMed] [Google Scholar]
- 52.Tainter M., Stockton A., Cutting W. Use of dinitrophenol in obesity and related conditions: a progress report. Journal of the American Medical Association. 1933;101(19):1472–1475. [Google Scholar]
- 53.Chalmers S., Caldwell S.T., Quin C., Prime T.A., James A.M., Cairns A.G. Selective uncoupling of individual mitochondria within a cell using a mitochondria-targeted photoactivated protonophore. Journal of the American Chemical Society. 2012;134(2):758–761. doi: 10.1021/ja2077922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McQuaker S.J., Quinlan C.L., Caldwell S.T., Brand M.D., Hartley R.C. A prototypical small-molecule modulator uncouples mitochondria in response to endogenous hydrogen peroxide production. ChemBioChem. 2013;14(8):993. doi: 10.1002/cbic.201300115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perry R.J., Kim T., Zhang X.-M., Lee H.-Y., Pesta D., Popov V.B. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metabolism. 2013;18(5):740–748. doi: 10.1016/j.cmet.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khan R.S., Dine K., Geisler J.G., Shindler K.S. Mitochondrial uncoupler prodrug of 2, 4-dinitrophenol, MP201, prevents neuronal damage and preserves vision in experimental optic neuritis. Oxidative Medicine and Cellular Longevity. 2017;2017 doi: 10.1155/2017/7180632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goedeke L., Peng L., Montalvo-Romeral V., Butrico G.M., Dufour S., Zhang X.-M. Controlled-release mitochondrial protonophore (CRMP) reverses dyslipidemia and hepatic steatosis in dysmetabolic nonhuman primates. Science Translational Medicine. 2019;11(512) doi: 10.1126/scitranslmed.aay0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kenwood B.M., Weaver J.L., Bajwa A., Poon I.K., Byrne F.L., Murrow B.A. Identification of a novel mitochondrial uncoupler that does not depolarize the plasma membrane. Molecular Metabolism. 2014;3(2):114–123. doi: 10.1016/j.molmet.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rokitskaya T.I., Khailova L.S., Makarenkov A.V., Shunaev A.V., Tatarskiy V.V., Shtil A.A. Carborane derivatives of 1, 2, 3-triazole depolarize mitochondria by transferring protons through the lipid part of membranes. Biochimica et Biophysica Acta (BBA) – Biomembranes. 2019;1861(3):573–583. doi: 10.1016/j.bbamem.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 60.Kanemoto N., Okamoto T., Tanabe K., Shimada T., Minoshima H., Hidoh Y. Antidiabetic and cardiovascular beneficial effects of a liver-localized mitochondrial uncoupler. Nature Communications. 2019;10(1):1–20. doi: 10.1038/s41467-019-09911-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Childress E.S., Salamoun J.M., Hargett S.R., Alexopoulos S.J., Chen S.-Y., Shah D.P. [1, 2, 5] Oxadiazolo [3, 4-b] pyrazine-5, 6-diamine derivatives as mitochondrial uncouplers for the potential treatment of nonalcoholic steatohepatitis. Journal of Medicinal Chemistry. 2020;63(5):2511–2526. doi: 10.1021/acs.jmedchem.9b01440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perry R.J., Zhang D., Zhang X.-M., Boyer J.L., Shulman G.I. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science. 2015;347(6227):1253–1256. doi: 10.1126/science.aaa0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Childress E.S., Alexopoulos S.J., Hoehn K.L., Santos W.L. Small molecule mitochondrial uncouplers and their therapeutic potential. Journal of Medicinal Chemistry. 2018;61(11):4641–4655. doi: 10.1021/acs.jmedchem.7b01182. [DOI] [PubMed] [Google Scholar]
- 64.Tao H., Zhang Y., Zeng X., Shulman G.I., Jin S. Niclosamide ethanolamine improves blood glycemic control and reduces hepatic steatosis in mice. Nature Medicine. 2014;20(11):1263. doi: 10.1038/nm.3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pardo-Andreu G.L., Nunez-Figueredo Y., Tudella V.G., Cuesta-Rubio O., Rodrigues F.P., Pestana C.R. The anti-cancer agent nemorosone is a new potent protonophoric mitochondrial uncoupler. Mitochondrion. 2011;11(2):255–263. doi: 10.1016/j.mito.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 66.Demine S., Renard P., Arnould T. Mitochondrial uncoupling: a key controller of biological processes in physiology and diseases. Cells. 2019;8(8) doi: 10.3390/cells8080795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang H., Jin J., Duan Y., Xie Z., Li Y., Gao A. Mitochondrial uncoupling coordinated with PDH activation safely ameliorates hyperglycemia via promoting glucose oxidation. Diabetes. 2019;68(12):2197–2209. doi: 10.2337/db19-0589. [DOI] [PubMed] [Google Scholar]
- 68.Marrache S., Dhar S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proceedings of the National Academy of Sciences. 2012;109(40):16288–16293. doi: 10.1073/pnas.1210096109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hubbard W.B., Harwood C.L., Geisler J.G., Vekaria H.J., Sullivan P.G. Mitochondrial uncoupling prodrug improves tissue sparing, cognitive outcome, and mitochondrial bioenergetics after traumatic brain injury in male mice. Journal of Neuroscience Research. 2018;96(10):1677–1688. doi: 10.1002/jnr.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Linsinger G., Wilhelm S., Wagner H., Häcker G. Uncouplers of oxidative phosphorylation can enhance a Fas death signal. Molecular and Cellular Biology. 1999;19(5):3299–3311. doi: 10.1128/mcb.19.5.3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vier J., Gerhard M., Wagner H., Häcker G. Enhancement of death-receptor induced caspase-8-activation in the death-inducing signalling complex by uncoupling of oxidative phosphorylation. Molecular Immunology. 2004;40(10):661–670. doi: 10.1016/j.molimm.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 72.Han Y.H., Kim S.W., Kim S.H., Kim S.Z., Park W.H. 2, 4-Dinitrophenol induces G1 phase arrest and apoptosis in human pulmonary adenocarcinoma Calu-6 cells. Toxicology in Vitro. 2008;22(3):659–670. doi: 10.1016/j.tiv.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 73.Han Y.H., Kim S.Z., Kim S.H., Park W.H. 2, 4-Dinitrophenol induces apoptosis in As4. 1 juxtaglomerular cells through rapid depletion of GSH. Cell Biology International. 2008;32(12):1536–1545. doi: 10.1016/j.cellbi.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 74.Han Y.H., Yang Y.M., Park W.H. Carbonyl cyanide p-(trifluoromethoxy) phenylhydroazone induces caspase-independent apoptosis in As4. 1 juxtaglomerular cells. Anticancer Research. 2010;30(7):2863–2868. [PubMed] [Google Scholar]
- 75.Han Y.H., Park W.H. Intracellular glutathione levels are involved in carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone-induced apoptosis in As4. 1 juxtaglomerular cells. International Journal of Molecular Medicine. 2011;27(4):575–581. doi: 10.3892/ijmm.2011.604. [DOI] [PubMed] [Google Scholar]
- 76.Han Y.H., Kim S.H., Kim S.Z., Park W.H. Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) as an O2(∗-) generator induces apoptosis via the depletion of intracellular GSH contents in Calu-6 cells. Lung Cancer. 2009;63(2):201–209. doi: 10.1016/j.lungcan.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 77.Han Y.H., Moon H.J., You B.R., Kim S.Z., Kim S.H., Park W.H. Effects of carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone on the growth inhibition in human pulmonary adenocarcinoma Calu-6 cells. Toxicology. 2009;265(3):101–107. doi: 10.1016/j.tox.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 78.Lim M.L., Minamikawa T., Nagley P. The protonophore CCCP induces mitochondrial permeability transition without cytochrome c release in human osteosarcoma cells. FEBS Letters. 2001;503(1):69–74. doi: 10.1016/s0014-5793(01)02693-x. [DOI] [PubMed] [Google Scholar]
- 79.Chaudhari A.A., Seol J.-W., Kim S.-J., Lee Y.-J., Kang H.-s., Kim I.-s. Reactive oxygen species regulate Bax translocation and mitochondrial transmembrane potential, a possible mechanism for enhanced TRAIL-induced apoptosis by CCCP. Oncology Reports. 2007;18(1):71–76. [PubMed] [Google Scholar]
- 80.Park Y.S., Choi S.E., Koh H.C. PGAM5 regulates PINK1/Parkin-mediated mitophagy via DRP1 in CCCP-induced mitochondrial dysfunction. Toxicology Letters. 2018;284:120–128. doi: 10.1016/j.toxlet.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 81.Andrews P., Thyssen J., Lorke D. The biology and toxicology of molluscicides, Bayluscide. Pharmacology & Therapeutics. 1982;19(2):245–295. doi: 10.1016/0163-7258(82)90064-x. [DOI] [PubMed] [Google Scholar]
- 82.Li Y., Li P.-K., Roberts M.J., Arend R.C., Samant R.S., Buchsbaum D.J. Multi-targeted therapy of cancer by niclosamide: a new application for an old drug. Cancer Letters. 2014;349(1):8–14. doi: 10.1016/j.canlet.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen M., Wang J., Lu J., Bond M.C., Ren X.-R., Lyerly H.K. The anti-helminthic niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry. 2009;48(43):10267–10274. doi: 10.1021/bi9009677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Osada T., Chen M., Yang X.Y., Spasojevic I., Vandeusen J.B., Hsu D. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Research. 2011;71(12):4172–4182. doi: 10.1158/0008-5472.CAN-10-3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wieland A., Trageser D., Gogolok S., Reinartz R., Höfer H., Keller M. Anticancer effects of niclosamide in human glioblastoma. Clinical Cancer Research. 2013;19(15):4124–4136. doi: 10.1158/1078-0432.CCR-12-2895. [DOI] [PubMed] [Google Scholar]
- 86.Suliman M.A., Zhang Z., Na H., Ribeiro A.L., Zhang Y., Niang B. Niclosamide inhibits colon cancer progression through downregulation of the Notch pathway and upregulation of the tumor suppressor miR-200 family. International Journal of Molecular Medicine. 2016;38(3):776–784. doi: 10.3892/ijmm.2016.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Balgi A.D., Fonseca B.D., Donohue E., Tsang T.C., Lajoie P., Proud C.G. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PloS One. 2009;4(9) doi: 10.1371/journal.pone.0007124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fonseca B.D., Diering G.H., Bidinosti M.A., Dalal K., Alain T., Balgi A.D. Structure-activity analysis of niclosamide reveals potential role for cytoplasmic pH in control of mammalian target of rapamycin complex 1 (mTORC1) signaling. Journal of Biological Chemistry. 2012;287(21):17530–17545. doi: 10.1074/jbc.M112.359638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Arend R.C., Londoño-Joshi A.I., Gangrade A., Katre A.A., Kurpad C., Li Y. Niclosamide and its analogs are potent inhibitors of Wnt/β-catenin, mTOR and STAT3 signaling in ovarian cancer. Oncotarget. 2016;7(52):86803. doi: 10.18632/oncotarget.13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Satoh K., Zhang L., Zhang Y., Chelluri R., Boufraqech M., Nilubol N. Identification of niclosamide as a novel anticancer agent for adrenocortical carcinoma. Clinical Cancer Research. 2016;22(14):3458–3466. doi: 10.1158/1078-0432.CCR-15-2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.de Sá Junior P.L., Câmara D.A.D., Porcacchia A.S., Fonseca P.M.M., Jorge S.D., Araldi R.P. The roles of ROS in cancer heterogeneity and therapy. Oxidative Medicine and Cellular Longevity. 2017;2017 doi: 10.1155/2017/2467940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Alasadi A., Chen M., Swapna G., Tao H., Guo J., Collantes J. Effect of mitochondrial uncouplers niclosamide ethanolamine (NEN) and oxyclozanide on hepatic metastasis of colon cancer. Cell Death & Disease. 2018;9(2):1–14. doi: 10.1038/s41419-017-0092-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chai W.-H., Li Y.-R., Lin S.-H., Chao Y.-H., Chen C.-H., Chan P.-C. Antihelminthic niclosamide induces autophagy and delayed apoptosis in human non-small lung cancer cells in vitro and in vivo. Anticancer Research. 2020;40(3):1405–1417. doi: 10.21873/anticanres.14082. [DOI] [PubMed] [Google Scholar]
- 94.Wei W., Liu H., Yuan J., Yao Y. Targeting Wnt/β-catenin by anthelmintic drug niclosamide overcomes paclitaxel resistance in esophageal cancer. Fundamental & Clinical Pharmacology. 2021;35(1):165–173. doi: 10.1111/fcp.12583. [DOI] [PubMed] [Google Scholar]
- 95.Pindiprolu S.K.S., Krishnamurthy P.T., Ghanta V.R., Chintamaneni P.K. Phenyl boronic acid-modified lipid nanocarriers of niclosamide for targeting triple-negative breast cancer. Nanomedicine. 2020;15(16):1551–1565. doi: 10.2217/nnm-2020-0003. [DOI] [PubMed] [Google Scholar]
- 96.Hatamipour M., Jaafari M.R., Momtazi-Borojeni A.A., Ramezani M., Sahebkar A. Nanoliposomal encapsulation enhances in vivo anti-tumor activity of niclosamide against melanoma. Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents) 2019;19(13):1618–1626. doi: 10.2174/1871520619666190705120011. [DOI] [PubMed] [Google Scholar]
- 97.Al-Hadiya B.M. Niclosamide: comprehensive profile. Profiles of Drug Substances, Excipients and Related Methodology. 2005;32:67–96. doi: 10.1016/S0099-5428(05)32002-8. [DOI] [PubMed] [Google Scholar]
- 98.Ray E., Vaghasiya K., Sharma A., Shukla R., Khan R., Kumar A. Autophagy-inducing inhalable Co-crystal formulation of niclosamide-nicotinamide for lung cancer therapy. AAPS PharmSciTech. 2020;21(7):1–14. doi: 10.1208/s12249-020-01803-z. [DOI] [PubMed] [Google Scholar]
- 99.Schweizer M.T., Haugk K., McKiernan J.S., Gulati R., Cheng H.H., Maes J.L. A phase I study of niclosamide in combination with enzalutamide in men with castration-resistant prostate cancer. PloS One. 2018;13(6) doi: 10.1371/journal.pone.0198389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Adagu I.S., Nolder D., Warhurst D.C., Rossignol J.-F. In vitro activity of nitazoxanide and related compounds against isolates of Giardia intestinalis, Entamoeba histolytica and Trichomonas vaginalis. Journal of Antimicrobial Chemotherapy. 2002;49(1):103–111. doi: 10.1093/jac/49.1.103. [DOI] [PubMed] [Google Scholar]
- 101.Theodos C.M., Griffiths J.K., D’onfro J., Fairfield A., Tzipori S. Efficacy of nitazoxanide againstcryptosporidium parvum in cell culture and in animal models. Antimicrobial Agents and Chemotherapy. 1998;42(8):1959–1965. doi: 10.1128/aac.42.8.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McVay C.S., Rolfe R.D. In vitro and in vivo activities of nitazoxanide against Clostridium difficile. Antimicrobial Agents and Chemotherapy. 2000;44(9):2254–2258. doi: 10.1128/aac.44.9.2254-2258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mégraud F., Occhialini A., Rossignol J.F. Nitazoxanide, a potential drug for eradication of Helicobacter pylori with No cross-resistance to metronidazole. Antimicrobial Agents and Chemotherapy. 1998;42(11):2836–2840. doi: 10.1128/aac.42.11.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Korba B.E., Montero A.B., Farrar K., Gaye K., Mukerjee S., Ayers M.S. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antiviral Research. 2008;77(1):56–63. doi: 10.1016/j.antiviral.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 105.de Carvalho L.P.S., Darby C.M., Rhee K.Y., Nathan C. Nitazoxanide disrupts membrane potential and intrabacterial pH homeostasis of Mycobacterium tuberculosis. ACS Medicinal Chemistry Letters. 2011;2(11):849–854. doi: 10.1021/ml200157f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Senkowski W., Zhang X., Olofsson M.H., Isacson R., Höglund U., Gustafsson M. Three-dimensional cell culture-based screening identifies the anthelmintic drug nitazoxanide as a candidate for treatment of colorectal cancer. Molecular Cancer Therapeutics. 2015;14(6):1504–1516. doi: 10.1158/1535-7163.MCT-14-0792. [DOI] [PubMed] [Google Scholar]
- 107.Fan-Minogue H., Bodapati S., Solow-Cordero D., Fan A., Paulmurugan R., Massoud T.F. A c-Myc activation sensor-based high-throughput drug screening identifies an antineoplastic effect of nitazoxanide. Molecular Cancer Therapeutics. 2013;12(9):1896–1905. doi: 10.1158/1535-7163.MCT-12-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang X., Shen C., Liu Z., Peng F., Chen X., Yang G. Nitazoxanide, an antiprotozoal drug, inhibits late-stage autophagy and promotes ING1-induced cell cycle arrest in glioblastoma. Cell Death & Disease. 2018;9(10):1–15. doi: 10.1038/s41419-018-1058-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ripani P., Delp J., Bode K., Delgado M.E., Dietrich L., Betzler V.M. Thiazolides promote G1 cell cycle arrest in colorectal cancer cells by targeting the mitochondrial respiratory chain. Oncogene. 2020;39(11):2345–2357. doi: 10.1038/s41388-019-1142-6. [DOI] [PubMed] [Google Scholar]
- 110.Stockis A., Allemon A.-M., De Bruyn S., Gengler C. Nitazoxanide pharmacokinetics and tolerability in man using single ascending oral doses. International Journal of Clinical Pharmacology and Therapeutics. 2002;40(5):213–220. doi: 10.5414/cpp40213. [DOI] [PubMed] [Google Scholar]
- 111.Corbett J., Goose J. A possible biochemical mode of action of the fasciolicides nitroxynil, hexachlorophene and oxyclozanide. Pesticide Science. 1971;2(3):119–121. doi: 10.1042/bj1210041pa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Axelrod C.L., King W.T., Davuluri G., Noland R.C., Hall J., Hull M. BAM15-mediated mitochondrial uncoupling protects against obesity and improves glycemic control. EMBO Molecular Medicine. 2020 doi: 10.15252/emmm.202012088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schömel N., Gruber L., Alexopoulos S.J., Trautmann S., Olzomer E.M., Byrne F.L. UGcG overexpression leads to increased glycolysis and increased oxidative phosphorylation of breast cancer cells. Scientific Reports. 2020;10(1):1–13. doi: 10.1038/s41598-020-65182-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Serasinghe M.N., Gelles J.D., Li K., Zhao L., Abbate F., Syku M. Dual suppression of inner and outer mitochondrial membrane functions augments apoptotic responses to oncogenic MAPK inhibition. Cell Death & Disease. 2018;9(2):1–13. doi: 10.1038/s41419-017-0044-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Alexopoulos S.J., Chen S.-Y., Brandon A.E., Salamoun J.M., Byrne F.L., Garcia C.J. Mitochondrial uncoupler BAM15 reverses diet-induced obesity and insulin resistance in mice. Nature Communications. 2020;11(1):1–13. doi: 10.1038/s41467-020-16298-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tai Y., Li L., Peng X., Zhu J., Mao X., Qin N. Mitochondrial uncoupler BAM15 inhibits artery constriction and potently activates AMPK in vascular smooth muscle cells. Acta Pharmaceutica Sinica B. 2018;8(6):909–918. doi: 10.1016/j.apsb.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rahbar S., Figarola J.L. Novel inhibitors of advanced glycation endproducts. Archives of Biochemistry and Biophysics. 2003;419(1):63–79. doi: 10.1016/j.abb.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 118.Figarola J.L., Weng Y., Lincoln C., Horne D., Rahbar S. Novel dichlorophenyl urea compounds inhibit proliferation of human leukemia HL-60 cells by inducing cell cycle arrest, differentiation and apoptosis. Investigational New Drugs. 2012;30(4):1413–1425. doi: 10.1007/s10637-011-9711-8. [DOI] [PubMed] [Google Scholar]
- 119.Figarola J.L., Singhal J., Singhal S., Kusari J., Riggs A. Bioenergetic modulation with the mitochondria uncouplers SR4 and niclosamide prevents proliferation and growth of treatment-naïve and vemurafenib-resistant melanomas. Oncotarget. 2018;9(97):36945. doi: 10.18632/oncotarget.26421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Singhal S.S., Figarola J., Singhal J., Leake K., Nagaprashantha L., Lincoln C. 1, 3-Bis (3, 5-dichlorophenyl) urea compound ‘COH-SR4’inhibits proliferation and activates apoptosis in melanoma. Biochemical Pharmacology. 2012;84(11):1419–1427. doi: 10.1016/j.bcp.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Figarola J.L., Rahbar S. Small-molecule COH-SR4 inhibits adipocyte differentiation via AMPK activation. International Journal of Molecular Medicine. 2013;31(5):1166–1176. doi: 10.3892/ijmm.2013.1313. [DOI] [PubMed] [Google Scholar]
- 122.Figarola J.L., Singhal J., Tompkins J.D., Rogers G.W., Warden C., Horne D. SR4 uncouples mitochondrial oxidative phosphorylation, modulates AMP-dependent kinase (AMPK)-Mammalian target of rapamycin (mTOR) signaling, and inhibits proliferation of HepG2 hepatocarcinoma cells. Journal of Biological Chemistry. 2015;290(51):30321–30341. doi: 10.1074/jbc.M115.686352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Singhal S.S., Figarola J., Singhal J., Nagaprashantha L., Berz D., Rahbar S. Novel compound 1, 3-bis (3, 5-dichlorophenyl) urea inhibits lung cancer progression. Biochemical Pharmacology. 2013;86(12):1664–1672. doi: 10.1016/j.bcp.2013.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Urra F.A., Muñoz F., Córdova-Delgado M., Ramírez M.P., Peña-Ahumada B., Rios M. FR58P1a; a new uncoupler of OXPHOS that inhibits migration in triple-negative breast cancer cells via Sirt1/AMPK/β1-integrin pathway. Scientific Reports. 2018;8(1):1–16. doi: 10.1038/s41598-018-31367-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Handeli S., Simon J.A. A small-molecule inhibitor of Tcf/β-catenin signaling down-regulates PPARγ and PPARδ activities. Molecular Cancer Therapeutics. 2008;7(3):521–529. doi: 10.1158/1535-7163.MCT-07-2063. [DOI] [PubMed] [Google Scholar]
- 126.Liu L., Zhi Q., Shen M., Gong F.-R., Zhou B.P., Lian L. FH535, a β-catenin pathway inhibitor, represses pancreatic cancer xenograft growth and angiogenesis. Oncotarget. 2016;7(30):47145. doi: 10.18632/oncotarget.9975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wu M.-Y., Liang R.-R., Chen K., Shen M., Tian Y.-L., Li D.-M. FH535 inhibited metastasis and growth of pancreatic cancer cells. OncoTargets and Therapy. 2015;8:1651. doi: 10.2147/OTT.S82718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Iida J., Dorchak J., Lehman J.R., Clancy R., Luo C., Chen Y. FH535 inhibited migration and growth of breast cancer cells. PloS One. 2012;7(9) doi: 10.1371/journal.pone.0044418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gedaly R., Galuppo R., Daily M.F., Shah M., Maynard E., Chen C. Targeting the Wnt/β-catenin signaling pathway in liver cancer stem cells and hepatocellular carcinoma cell lines with FH535. PloS One. 2014;9(6) doi: 10.1371/journal.pone.0099272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chen Y., Rao X., Huang K., Jiang X., Wang H., Teng L. FH535 inhibits proliferation and motility of colon cancer cells by targeting Wnt/β-catenin signaling pathway. Journal of Cancer. 2017;8(16):3142. doi: 10.7150/jca.19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gustafson C.T., Mamo T., Shogren K.L., Maran A., Yaszemski M.J. FH535 suppresses osteosarcoma growth in vitro and inhibits Wnt signaling through tankyrases. Frontiers in Pharmacology. 2017;8:285. doi: 10.3389/fphar.2017.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang W., Sviripa V.M., Kril L.M., Yu T., Xie Y., Hubbard W.B. An underlying mechanism of dual Wnt inhibition and AMPK activation: mitochondrial uncouplers masquerading as Wnt inhibitors. Journal of Medicinal Chemistry. 2019;62(24):11348–11358. doi: 10.1021/acs.jmedchem.9b01685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Turcios L., Vilchez V., Acosta L.F., Poyil P., Butterfield D.A., Mitov M. Sorafenib and FH535 in combination act synergistically on hepatocellular carcinoma by targeting cell bioenergetics and mitochondrial function. Digestive and Liver Disease. 2017;49(6):697–704. doi: 10.1016/j.dld.2017.01.146. [DOI] [PubMed] [Google Scholar]
- 134.Turcios L., Chacon E., Garcia C., Eman P., Cornea V., Jiang J. Autophagic flux modulation by Wnt/β-catenin pathway inhibition in hepatocellular carcinoma. PloS One. 2019;14(2) doi: 10.1371/journal.pone.0212538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fantin V.R., Berardi M.J., Scorrano L., Korsmeyer S.J., Leder P. A novel mitochondriotoxic small molecule that selectively inhibits tumor cell growth. Cancer Cell. 2002;2(1):29–42. doi: 10.1016/s1535-6108(02)00082-x. [DOI] [PubMed] [Google Scholar]
- 136.Chen H., Wang J., Feng X., Zhu M., Hoffmann S., Hsu A. Mitochondria-targeting fluorescent molecules for high efficiency cancer growth inhibition and imaging. Chemical Science. 2019;10(34):7946–7951. doi: 10.1039/c9sc01410a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang J., He H., Xiang C., Fan X.Y., Yang L.Y., Yuan L. Uncoupling effect of F16 is responsible for its mitochondrial toxicity and anticancer activity. Toxicological Sciences. 2018;161(2):431–442. doi: 10.1093/toxsci/kfx218. [DOI] [PubMed] [Google Scholar]
- 138.Wang J., Fan X.-Y., Yang L.-Y., He H., Huang R., Jiang F.-L. Conjugated 5-fluorouracil with mitochondria-targeting lipophilic cation: design, synthesis and biological evaluation. MedChemComm. 2016;7(10) [Google Scholar]
- 139.He H., Li D.-W., Yang L.-Y., Fu L., Zhu X.-J., Wong W.-K. A novel bifunctional mitochondria-targeted anticancer agent with high selectivity for cancer cells. Scientific Reports. 2015;5:13543. doi: 10.1038/srep13543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Qian K., Chen H., Qu C., Qi J., Du B., Ko T. Mitochondria-targeted delocalized lipophilic cation complexed with human serum albumin for tumor cell imaging and treatment. Nanomedicine: Nanotechnology, Biology and Medicine. 2020;23:102087. doi: 10.1016/j.nano.2019.102087. [DOI] [PubMed] [Google Scholar]
- 141.Holtrup F., Bauer A., Fellenberg K., Hilger R.A., Wink M., Hoheisel J.D. Microarray analysis of nemorosone-induced cytotoxic effects on pancreatic cancer cells reveals activation of the unfolded protein response (UPR) British Journal of Pharmacology. 2011;162(5):1045–1059. doi: 10.1111/j.1476-5381.2010.01125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Díaz-Carballo D., Malak S., Bardenheuer W., Freistuehler M., Reusch H. Cytotoxic activity of nemorosone in neuroblastoma cells. Journal of Cellular and Molecular Medicine. 2008;12(6b):2598–2608. doi: 10.1111/j.1582-4934.2008.00232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Popolo A., Piccinelli A.L., Morello S., Sorrentino R., Osmany C.R., Rastrelli L. Cytotoxic activity of nemorosone in human MCF-7 breast cancer cells. Canadian Journal of Physiology and Pharmacology. 2011;89(1):50–57. doi: 10.1139/y10-100. [DOI] [PubMed] [Google Scholar]
- 144.Cuesta-Rubio O., Frontana-Uribe B.A., Ramírez-Apan T., Cárdenas J. Polyisoprenylated benzophenones in Cuban propolis; biological activity of nemorosone. Zeitschrift für Naturforschung C. 2002;57(3–4):372–378. doi: 10.1515/znc-2002-3-429. [DOI] [PubMed] [Google Scholar]
- 145.Diaz-Carballo D., Malak S., Freistühler M., Elmaagacli A., Bardenheuer W., Reusch H. Nemorosone blocks proliferation and induces apoptosis in leukemia cells. International Journal of Clinical Pharmacology and Therapeutics. 2008;46(8):428–439. doi: 10.5414/cpp46428. [DOI] [PubMed] [Google Scholar]
- 146.Frión-Herrera Y., Gabbia D., Díaz-García A., Cuesta-Rubio O., Carrara M. Chemosensitizing activity of Cuban propolis and nemorosone in doxorubicin resistant human colon carcinoma cells. Fitoterapia. 2019;136:104173. doi: 10.1016/j.fitote.2019.104173. [DOI] [PubMed] [Google Scholar]
- 147.Pardo-Andreu G.L., Nuñez-Figueredo Y., Tudella V.G., Cuesta-Rubio O., Rodrigues F.P., Pestana C.R. The anti-cancer agent guttiferone-A permeabilizes mitochondrial membrane: ensuing energetic and oxidative stress implications. Toxicology and Applied Pharmacology. 2011;253(3):282–289. doi: 10.1016/j.taap.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 148.Wu H.-m., Li Y.-m. In vitro antitumor activity of guttiferone-A in human breast cancer cells is mediated via apoptosis, mitochondrial mediated oxidative stress and reactive oxygen species production. Journal of BUON. 2017;22(6):1500–1504. [PubMed] [Google Scholar]
- 149.Monzote L., Cuesta-Rubio O., Matheeussen A., Van Assche T., Maes L., Cos P. Antimicrobial evaluation of the polyisoprenylated benzophenones nemorosone and guttiferone A. Phytotherapy Research. 2011;25(3):458–462. doi: 10.1002/ptr.3401. [DOI] [PubMed] [Google Scholar]
- 150.Ngouela S., Lenta B.N., Noungoue D.T., Ngoupayo J., Boyom F.F., Tsamo E. Anti-plasmodial and antioxidant activities of constituents of the seed shells of Symphonia globulifera Linn f. Phytochemistry. 2006;67(3):302–306. doi: 10.1016/j.phytochem.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 151.Fromentin Y., Gaboriaud-Kolar N., Lenta B.N., Wansi J.D., Buisson D., Mouray E. Synthesis of novel guttiferone A derivatives: in-vitro evaluation toward Plasmodium falciparum, Trypanosoma brucei and Leishmania donovani. European Journal of Medicinal Chemistry. 2013;65:284–294. doi: 10.1016/j.ejmech.2013.04.066. [DOI] [PubMed] [Google Scholar]
- 152.Monzote L., Lackova A., Staniek K., Cuesta-Rubio O., Gille L. Role of mitochondria in the leishmanicidal effects and toxicity of acyl phloroglucinol derivatives: nemorosone and guttiferone A. Parasitology. 2015;142(9):1239–1248. doi: 10.1017/S0031182015000608. [DOI] [PubMed] [Google Scholar]
- 153.Jara J.A., Castro-Castillo V., Saavedra-Olavarría J., Peredo L., Pavanni M., Jana F. Antiproliferative and uncoupling effects of delocalized, lipophilic, cationic gallic acid derivatives on cancer cell lines. Validation in vivo in singenic mice. Journal of Medicinal Chemistry. 2014;57(6):2440–2454. doi: 10.1021/jm500174v. [DOI] [PubMed] [Google Scholar]
- 154.Fuentes-Retamal S., Sandoval-Acuña C., Peredo-Silva L., Guzmán-Rivera D., Pavani M., Torrealba N. Complex mitochondrial dysfunction induced by TPP+-Gentisic acid and mitochondrial translation inhibition by doxycycline evokes synergistic lethality in breast cancer cells. Cells. 2020;9(2):407. doi: 10.3390/cells9020407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Peredo-Silva L., Fuentes-Retamal S., Sandoval-Acuña C., Pavani M., Maya J.D., Castro-Castillo V. Derivatives of alkyl gallate triphenylphosphonium exhibit antitumor activity in a syngeneic murine model of mammary adenocarcinoma. Toxicology and Applied Pharmacology. 2017;329:334–346. doi: 10.1016/j.taap.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 156.Sandoval-Acuna C., Fuentes-Retamal S., Guzman-Rivera D., Peredo-Silva L., Madrid-Rojas M., Rebolledo S. Destabilization of mitochondrial functions as a target against breast cancer progression: role of TPP(+)-linked-polyhydroxybenzoates. Toxicology and Applied Pharmacology. 2016;309:2–14. doi: 10.1016/j.taap.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 157.Jara J.A., Rojas D., Castro-Castillo V., Fuentes-Retamal S., Sandoval-Acuña C., Parra E. Novel benzoate-lipophilic cations selectively induce cell death in human colorectal cancer cell lines. Toxicology In Vitro. 2020:104814. doi: 10.1016/j.tiv.2020.104814. [DOI] [PubMed] [Google Scholar]
- 158.Rawling T., MacDermott-Opeskin H., Roseblade A., Pazderka C., Clarke C., Bourget K. Aryl urea substituted fatty acids: a new class of protonophoric mitochondrial uncoupler that utilises a synthetic anion transporter. Chemical Science. 2020 doi: 10.1039/d0sc02777d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dyari H.R.E., Rawling T., Chen Y., Sudarmana W., Bourget K., Dwyer J.M. A novel synthetic analogue of ω-3 17,18-epoxyeicosatetraenoic acid activates TNF receptor-1/ASK1/JNK signaling to promote apoptosis in human breast cancer cells. The FASEB Journal. 2017;31(12):5246–5257. doi: 10.1096/fj.201700033R. [DOI] [PubMed] [Google Scholar]
- 160.Al-Zubaidi Y., Pazderka C., Koolaji N., Rahman M.K., Choucair H., Umashankar B. Aryl-urea fatty acids that activate the p38 MAP kinase and down-regulate multiple cyclins decrease the viability of MDA-MB-231 breast cancer cells. European Journal of Pharmaceutical Sciences. 2019;129:87–98. doi: 10.1016/j.ejps.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 161.Rawling T., Choucair H., Koolaji N., Bourget K., Allison S.E., Chen Y.-J. A novel Arylurea fatty acid that targets the mitochondrion and depletes Cardiolipin to promote killing of breast cancer cells. Journal of Medicinal Chemistry. 2017;60(20):8661–8666. doi: 10.1021/acs.jmedchem.7b00701. [DOI] [PubMed] [Google Scholar]
- 162.Triggle D.J. 1, 4-Dihydropyridines as calcium channel ligands and privileged structures. Cellular and Molecular Neurobiology. 2003;23(3):293–303. doi: 10.1023/A:1023632419813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kawase M., Shah A., Gaveriya H., Motohashi N., Sakagami H., Varga A. 3, 5-Dibenzoyl-1, 4-dihydropyridines: synthesis and MDR reversal in tumor cells. Bioorganic & Medicinal Chemistry. 2002;10(4):1051–1055. doi: 10.1016/s0968-0896(01)00363-7. [DOI] [PubMed] [Google Scholar]
- 164.Goto R.N., Sobral L.M., Sousa L.O., Garcia C.B., Lopes N.P., Marín-Prida J. Anti-cancer activity of a new dihydropyridine derivative, VdiE-2N, in head and neck squamous cell carcinoma. European Journal of Pharmacology. 2018;819:198–206. doi: 10.1016/j.ejphar.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 165.Marín-Prida J., Andreu G.L.P., Rossignoli C.P., Durruthy M.G., Rodríguez E.O., Reyes Y.V. The cytotoxic effects of VE-3N, a novel 1, 4-dihydropyridine derivative, involve the mitochondrial bioenergetic disruption via uncoupling mechanisms. Toxicology in Vitro. 2017;42:21–30. doi: 10.1016/j.tiv.2017.03.011. [DOI] [PubMed] [Google Scholar]
- 166.Muscarella D.E., O’brien K.A., Lemley A.T., Bloom S.E. Reversal of Bcl-2–mediated resistance of the EW36 human B-cell lymphoma cell line to arsenite-and pesticide-induced apoptosis by PK11195, a ligand of the mitochondrial benzodiazepine receptor. Toxicological Sciences. 2003;74(1):66–73. doi: 10.1093/toxsci/kfg052. [DOI] [PubMed] [Google Scholar]
- 167.Mlejnek P. Caspase-3 activity and carbonyl cyanide m-chlorophenylhydrazone-induced apoptosis in HL-60 cells. Alternatives to Laboratory Animals. 2001;29(3):243–249. doi: 10.1177/026119290102900313. [DOI] [PubMed] [Google Scholar]
- 168.Niu H., Zhang Y., Tang J., Zhu X., Ye Y., Zhao Y. A bifunctional fluorescent sensor for CCCP-induced cancer cell apoptosis imaging. Chemical Communications. 2020;56(82):12423–12426. doi: 10.1039/d0cc04200e. [DOI] [PubMed] [Google Scholar]
- 169.de Graaf A.O., van den Heuvel L.P., Dijkman H.B.P.M., De Abreu R.A., Birkenkamp K.U., de Witte T. Bcl-2 prevents loss of mitochondria in CCCP-induced apoptosis. Experimental Cell Research. 2004;299(2):533–540. doi: 10.1016/j.yexcr.2004.06.024. [DOI] [PubMed] [Google Scholar]
- 170.Shi L., Zheng H., Hu W., Zhou B., Dai X., Zhang Y. Niclosamide inhibition of STAT3 synergizes with erlotinib in human colon cancer. OncoTargets and Therapy. 2017;10:1767. doi: 10.2147/OTT.S129449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sack U., Walther W., Scudiero D., Selby M., Kobelt D., Lemm M. Novel effect of antihelminthic Niclosamide on S100A4-mediated metastatic progression in colon cancer. Journal of the National Cancer Institute. 2011;103(13):1018–1036. doi: 10.1093/jnci/djr190. [DOI] [PubMed] [Google Scholar]
- 172.Gyamfi J., Lee Y.-H., Min B.S., Choi J. Niclosamide reverses adipocyte induced epithelial-mesenchymal transition in breast cancer cells via suppression of the interleukin-6/STAT3 signalling axis. Scientific Reports. 2019;9(1):1–14. doi: 10.1038/s41598-019-47707-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Han Z., Li Q., Wang Y., Wang L., Li X., Ge N. Niclosamide induces cell cycle arrest in G1 phase in head and neck squamous cell carcinoma through Let-7d/CDC34 axis. Frontiers in Pharmacology. 2019;9:1544. doi: 10.3389/fphar.2018.01544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Weng S., Zhou L., Deng Q., Wang J., Yu Y., Zhu J. Niclosamide induced cell apoptosis via upregulation of ATF3 and activation of PERK in Hepatocellular carcinoma cells. BMC Gastroenterology. 2016;16(1):1–10. doi: 10.1186/s12876-016-0442-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wang C., Zhou X., Xu H., Shi X., Zhao J., Yang M. Niclosamide inhibits cell growth and enhances drug sensitivity of hepatocellular carcinoma cells via STAT3 signaling pathway. Journal of Cancer. 2018;9(22):4150. doi: 10.7150/jca.26948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wei W., Liu H., Yuan J., Yao Y. Targeting Wnt/β-catenin by anthelmintic drug niclosamide overcomes paclitaxel resistance in esophageal cancer. Fundamental & Clinical Pharmacology. 2020 doi: 10.1111/fcp.12583. [DOI] [PubMed] [Google Scholar]
- 177.Lin C.-K., Bai M.-Y., Hu T.-M., Wang Y.-C., Chao T.-K., Weng S.-J. Preclinical evaluation of a nanoformulated antihelminthic, niclosamide, in ovarian cancer. Oncotarget. 2016;7(8):8993. doi: 10.18632/oncotarget.7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yu J., Yang K., Zheng J., Zhao W., Sun X. Synergistic tumor inhibition of colon cancer cells by nitazoxanide and obeticholic acid, a farnesoid X receptor ligand. Cancer Gene Therapy. 2020:1–12. doi: 10.1038/s41417-020-00239-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Panigrahi D.P., Praharaj P.P., Bhol C.S., Mahapatra K.K., Patra S., Behera B.P. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Seminars in Cancer Biology. 2020;66:45–58. doi: 10.1016/j.semcancer.2019.07.015. [DOI] [PubMed] [Google Scholar]
- 180.Hirpara J., Eu J.Q., Tan J.K.M., Wong A.L., Clement M.-V., Kong L.R. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Biology. 2019;25:101076. doi: 10.1016/j.redox.2018.101076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Guerra F., Arbini A.A., Moro L. Mitochondria and cancer chemoresistance. Biochimica et Biophysica Acta (BBA) – Bioenergetics. 2017;1858(8):686–699. doi: 10.1016/j.bbabio.2017.01.012. [DOI] [PubMed] [Google Scholar]
- 182.Sancho P., Barneda D., Heeschen C. Hallmarks of cancer stem cell metabolism. British Journal of Cancer. 2016;114(12):1305–1312. doi: 10.1038/bjc.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Forrest M.D. Why cancer cells have a more hyperpolarised mitochondrial membrane potential and emergent prospects for therapy. bioRxiv. 2015 [Google Scholar]
- 184.Hara K.Y., Kondo A. ATP regulation in bioproduction. Microbial Cell Factories. 2015;14(1):198. doi: 10.1186/s12934-015-0390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Shackelford D.B., Shaw R.J. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nature Reviews Cancer. 2009;9(8):563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Herzig S., Shaw R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nature Reviews Molecular Cell Biology. 2018;19(2):121–135. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hsu C.-C., Wang C.-H., Wu L.-C., Hsia C.-Y., Chi C.-W., Yin P.-H. Mitochondrial dysfunction represses HIF-1α protein synthesis through AMPK activation in human hepatoma HepG2 cells. Biochimica et Biophysica Acta (BBA) – General Subjects. 2013;1830(10):4743–4751. doi: 10.1016/j.bbagen.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 188.Kwon K.-Y., Viollet B., Yoo O.J. CCCP induces autophagy in an AMPK-independent manner. Biochemical and Biophysical Research Communications. 2011;416(3–4):343–348. doi: 10.1016/j.bbrc.2011.11.038. [DOI] [PubMed] [Google Scholar]
- 189.Lee D.-H., Lee T.H., Jung C.H., Kim Y.-H. Wogonin induces apoptosis by activating the AMPK and p53 signaling pathways in human glioblastoma cells. Cellular Signalling. 2012;24(11):2216–2225. doi: 10.1016/j.cellsig.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 190.Costa R., Peruzzo R., Bachmann M., Dalla Monta G., Vicario M., Santinon G. Impaired mitochondrial ATP production downregulates Wnt signaling via ER stress induction. Cell Reports. 2019;28(8):1949–1960. doi: 10.1016/j.celrep.2019.07.050. e1946. [DOI] [PubMed] [Google Scholar]
- 191.Anastas J.N., Moon R.T. WNT signalling pathways as therapeutic targets in cancer. Nature Reviews Cancer. 2013;13(1):11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 192.Sullivan L.B., Chandel N.S. Mitochondrial reactive oxygen species and cancer. Cancer & Metabolism. 2014;2:17. doi: 10.1186/2049-3002-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Sabharwal S.S., Schumacker P.T. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nature Reviews Cancer. 2014;14(11):709–721. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Li N., Oquendo E., Capaldi R.A., Robinson J.P., He Y.D., Hamadeh H.K. A systematic assessment of mitochondrial function identified novel signatures for drug-induced mitochondrial disruption in cells. Toxicological Sciences. 2014;142(1):261–273. doi: 10.1093/toxsci/kfu176. [DOI] [PubMed] [Google Scholar]
- 195.Dlasková A., Hlavatá L., Ježek P. Oxidative stress caused by blocking of mitochondrial Complex I H+ pumping as a link in aging/disease vicious cycle. The International Journal of Biochemistry & Cell Biology. 2008;40(9):1792–1805. doi: 10.1016/j.biocel.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 196.Chatterjee S., Browning E.A., Hong N., DeBolt K., Sorokina E.M., Liu W. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. American Journal of Physiology – Heart and Circulatory Physiology. 2012;302(1):H105–H114. doi: 10.1152/ajpheart.00298.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kinnally K.W., Peixoto P.M., Ryu S.-Y., Dejean L.M. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochimica et Biophysica Acta (BBA) – Molecular Cell Research. 2011;1813(4):616–622. doi: 10.1016/j.bbamcr.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Santulli G., Xie W., Reiken S.R., Marks A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proceedings of the National Academy of Sciences. 2015;112(36):11389–11394. doi: 10.1073/pnas.1513047112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Rao V.K., Carlson E.A., Yan S.S. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2014;1842(8):1267–1272. doi: 10.1016/j.bbadis.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Rasheed M.Z., Tabassum H., Parvez S. Mitochondrial permeability transition pore: a promising target for the treatment of Parkinson's disease. Protoplasma. 2017;254(1):33–42. doi: 10.1007/s00709-015-0930-2. [DOI] [PubMed] [Google Scholar]
- 201.Geisler J.G., Marosi K., Halpern J., Mattson M.P. DNP, mitochondrial uncoupling, and neuroprotection: a little dab'll do ya. Alzheimer's and Dementia. 2017;13(5):582–591. doi: 10.1016/j.jalz.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.De Marchi E., Bonora M., Giorgi C., Pinton P. The mitochondrial permeability transition pore is a dispensable element for mitochondrial calcium efflux. Cell Calcium. 2014;56(1):1–13. doi: 10.1016/j.ceca.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lim H.W., Lim H.Y., Wong K.P. Uncoupling of oxidative phosphorylation by curcumin: implication of its cellular mechanism of action. Biochemical and Biophysical Research Communications. 2009;389(1):187–192. doi: 10.1016/j.bbrc.2009.08.121. [DOI] [PubMed] [Google Scholar]
- 204.Ligeret H., Barthélémy S., Doulakas G.B., Carrupt P.-A., Tillement J.-P., Labidalle S. Fluoride curcumin derivatives: new mitochondrial uncoupling agents. FEBS Letters. 2004;569(1–3):37–42. doi: 10.1016/j.febslet.2004.05.032. [DOI] [PubMed] [Google Scholar]
- 205.Ward M.S., Flemming N.B., Gallo L.A., Fotheringham A.K., McCarthy D.A., Zhuang A. Targeted mitochondrial therapy using MitoQ shows equivalent renoprotection to angiotensin converting enzyme inhibition but no combined synergy in diabetes. Scientific Reports. 2017;7(1):1–14. doi: 10.1038/s41598-017-15589-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Jian C., Fu J., Cheng X., Shen L.-J., Ji Y.-X., Wang X. Low-dose sorafenib acts as a mitochondrial uncoupler and ameliorates nonalcoholic steatohepatitis. Cell Metabolism. 2020;31(5):892–908. doi: 10.1016/j.cmet.2020.04.011. e811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Santandreu F.M., Roca P., Oliver J. Uncoupling protein-2 knockdown mediates the cytotoxic effects of cisplatin. Free Radical Biology and Medicine. 2010;49(4):658–666. doi: 10.1016/j.freeradbiomed.2010.05.031. [DOI] [PubMed] [Google Scholar]
- 208.Rai Y., Anita, Kumari N., Singh S., Kalra N., Soni R. Mild mitochondrial uncoupling protects from ionizing radiation induced cell death by attenuating oxidative stress and mitochondrial damage. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2021;1862(1):148325. doi: 10.1016/j.bbabio.2020.148325. [DOI] [PubMed] [Google Scholar]
- 209.Momcilovic M., Jones A., Bailey S.T., Waldmann C.M., Li R., Lee J.T. In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature. 2019;575(7782):380–384. doi: 10.1038/s41586-019-1715-0. [DOI] [PMC free article] [PubMed] [Google Scholar]