

Abstract
N4-acetylcytidine (ac4C) is an ancient and highly conserved RNA modification that is present on tRNA and rRNA and has recently been investigated in eukaryotic mRNA1–3. However, the distribution, dynamics and functions of cytidine acetylation have yet to be fully elucidated. Here we report ac4C-seq, a chemical genomic method for the transcriptome-wide quantitative mapping of ac4C at single-nucleotide resolution. In human and yeast mRNAs, ac4C sites are not detected but can be induced—at a conserved sequence motif—via the ectopic overexpression of eukaryotic acetyltransferase complexes. By contrast, cross-evolutionary profiling revealed unprecedented levels of ac4C across hundreds of residues in rRNA, tRNA, non-coding RNA and mRNA from hyperthermophilic archaea. Ac4C is markedly induced in response to increases in temperature, and acetyltransferase-deficient archaeal strains exhibit temperature-dependent growth defects. Visualization of wild-type and acetyltransferase-deficient archaeal ribosomes by cryo-electron microscopy provided structural insights into the temperature-dependent distribution of ac4C and its potential thermoadaptive role. Our studies quantitatively define the ac4C landscape, providing a technical and conceptual foundation for elucidating the role of this modification in biology and disease4–6.
Acetylation is an ancient mechanism that regulates biomolecular function. Perhaps the most well conserved of these mechanisms is the enzymatic modification of RNA to form the acetylated nucleobase ac4C. Ac4C occurs in all domains of life, and its formation is catalysed by the acetyltransferases NAT10 in humans and Kre33 in yeast1–3. NAT10 and Kre33 are essential in humans and yeast, respectively, and the four target sites of these enzymes in rRNA and tRNA are also conserved between these two distant eukaryotes1–3. The deposition of ac4C at its two tRNA targets (tRNA-Ser and tRNA-Leu) requires an additional adaptor protein—THUMPD1 in humans and Tan1 in yeast1—and has been implicated in tRNA stability7,8. Conversely, NAT10 is guided towards its two target sites in rRNA by specialized small nucleolar RNAs9. Recently, antibody-based mapping suggested the existence of additional NAT10-regulated ac4C sites in human mRNAs10; however, the lack of base-resolution quantification of any single ac4C site precluded orthogonal validation and functional prioritization on the basis of modification stoichiometries. Thus, the quantitative distribution of ac4C among rRNA, tRNA and mRNA remains to be comparatively defined in any organism.
Nucleotide-resolution ac4C sequencing
To quantitatively study cytidine acetylation in the transcriptome, we developed a chemical method to enable the sensitive detection of ac4C at single-nucleotide resolution. Building on previous work11, we found that the reaction of ac4C with sodium cyanoborohydride (NaCNBH3) under acidic conditions forms the reduced nucleobase N4-acetyltetrahydrocytidine. The altered structure of this reduced nucleobase compared with ac4C causes the incorporation of non-cognate deoxynucleotide triphosphates (dNTPs) upon reverse transcription11, which can be detected via cDNA sequencing. Compared with previous chemistries, this reaction shows faster kinetics and causes increased misincorporation at known ac4C sites in rRNA (Extended Data Fig. 1, Supplementary Note 1). Critically, ac4C-dependent mutations are not observed when the modification is hydrolysed (chemically deacetylated) using mild alkali before analysis12 (Fig. 1a). Integrating these chemistries with next-generation sequencing led to the development of ac4C-seq, a method that enables the transcriptome-wide, quantitative analysis of ac4C at single-nucleotide resolution (Fig. 1b, Methods). Inspection of sequencing data revealed that NaCNBH3 treatment caused C>T misincorporation at acetylated sites, which were reduced upon alkali-induced deacetylation (Fig. 1c). This guided the development of an analytical pipeline for ac4C detection, based on the following observations: C>T misincorporation upon treatment with acid and NaCNBH3; the reduction in C>T misincorporation upon pre-treatment with alkali; and the absence of C>T misincorporation in mock-treated RNA. These three requirements were formalized as two statistical tests, comparing misincorporations in NaCNBH3-treated samples with those in alkali- or mock-treated controls. In practice, excellent signal-to noise ratios could be obtained on the basis of the latter comparison, enabling the former to be used as an optional filter to increase confidence in identified sites (Fig. 1d). To evaluate our ability to quantitatively measure acetylation levels, we applied ac4C-seq to four synthetic RNAs, each harbouring a single ac4C site. In these synthetic RNAs, ac4C was embedded within several sequence contexts, and spiked into complex RNA samples at varying stoichiometries (Supplementary Table 1a). We observed excellent absolute agreement between the synthesized ac4C stoichiometries and the experimentally measured misincorporation levels (Pearson’s R = 0.99) across the entire range of stoichiometries (Fig. 1e). Thus, given sufficient read-depth, ac4C-seq is able to detect and quantify even low-stoichiometry (4%) modifications with excellent accuracy and precision.
Fig. 1 |. Development and application of ac4C-seq in human and yeast.
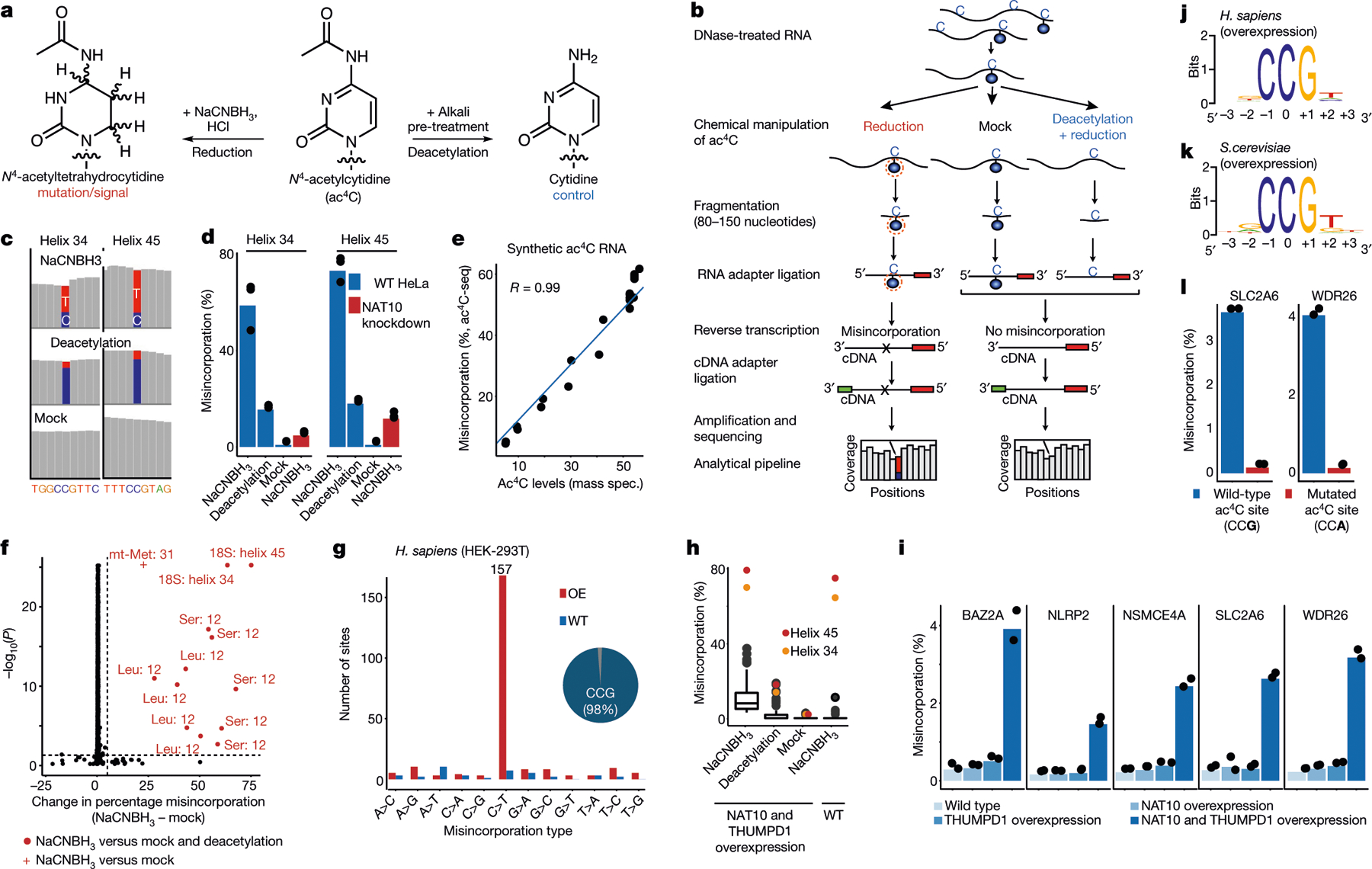
a, Reaction schemes showing the reduction and the deacetylation of ac4C. b, Schematic showing the ac4C-seq procedure: RNA is deacetylated in the pre-treatment step (or mock-pretreated), followed by treatment with NaCNBH3 (or mock treatment). After library preparation as illustrated, ac4C is detected by the analysis of C>T misincorporation. c, Misincorporation rates in total RNA from HeLa cells are shown for known sites in 18S (blue, cytidine; red, thymidine). d, Misincorporation rates in 18S sites in wild-type and NAT10-depleted cells (bars, mean of 3 biological samples; dots, individual measurements). e, Misincorporation rates of 4 synthetic spikes measured by ac4C-seq (y axis) plotted against ac4C stoichiometry as measured by mass spectrometry (x axis). Pearson’s R, n = 1 experiment. f, Statistical significance plotted against the difference in misincorporation rates between NaCNBH3 and mock-treated total RNA from HeLa cells. Vertical dashed line, 5%; horizontal dashed line, P = 0.05 (χ2 test). n = 3 biological samples. g, Frequency of the 12 possible misincorporation patterns (y axis) for sites found in poly(A)-enriched RNA from wild-type (WT) HEK-293T cells and from HEK-293T cells overexpressing NAT10 and THUMPD1 (OE). The pie chart shows the proportion of sites harbouring C>T misincorporations within a CCG motif. h, Misincorporation rate at ac4C sites within CCG motifs identified in g in wild-type cells and in cells overexpressing NAT10 and THUMPD1, shown for RNA treated with NaCNBH3 and indicated controls (n = 2 biological samples for overexpression NaCNBH3- or mock-treated and 1 sample for the rest). For the box plots, the centre line indicates the median, the box boundaries mark the 25th and 75th percentiles, the whiskers indicate ±1.5× the interquartile range (IQR) and outliers are shown as individual dots. i, Misincorporation level (obtained from ac4C-seq) at amplicons spanning ac4C sites in HEK-293T cells, depicted as in d. n = 2 biological samples. j, k, Sequence motif surrounding the ac4C sites identified in indicated organisms. l, Misincorporation rate at two wild-type and mutated ac4C sites in HEK-293T cells overexpressing NAT10/THUMPD1, quantified via targeted ac4C-sequencing, depicted as in d. n = 2 biological samples.
Ac4C in eukaryotic RNA
We next explored the properties of ac4C in eukaryotic RNA. To strengthen this study, we used a cross-evolutionary approach, analysing two human cell lines and the budding yeast, Saccharomyces cerevisiae. Applying ac4C-seq to total RNA from these organisms recapitulated both known sites of ac4C modification in 18S rRNA, as well as the two known sites of ac4C on tRNA: tRNA-Ser and tRNA-Leu1–3 (Fig. 1f, Extended Data Fig. 2a–c, Supplementary Table 2). No additional rRNA or tRNA sites met detection thresholds for ac4C. Acetylation of rRNA and tRNA sites were reduced after the disruption of human NAT10, and eliminated after the mutation of yeast Kre33 (Fig. 1d, Extended Data Fig. 2d). These results suggest that eukaryotic rRNA and tRNA ac4C is well annotated, and that in these abundant RNAs, ac4C-seq demonstrates very good sensitivity and specificity.
Next we explored the properties of ac4C in eukaryotic mRNA10,13. Applying ac4C-seq to poly(A)-enriched mRNA from HEK-293T cells readily identified the known sites on rRNA (Extended Data Fig. 2e). However, only four additional C>T misincorporations passed detection thresholds (Fig. 1g, Supplementary Table 2)—a number consistent with the anticipated false discovery rate (Fig. 1g). To address the possibility that the absence of detectable ac4C in mRNA is unique to HEK-293T cells, we applied ac4C-seq to poly(A)-RNA isolated from HeLa cells and from S. cerevisiae, in which ac4C has been previously suggested to be present using other approaches1,10,13. In both models we detected the known rRNA ac4C sites (Extended Data Fig. 2a–c, Supplementary Table 2). However, no additional sites passed detection thresholds in HeLa cells, and in yeast, three additional sites were identified in mRNA, but they were not eliminated after the mutation of yeast Kre33, and no enrichment was observed for C>T misincorporations—suggesting that they do not represent ac4C sites (Supplementary Table 2). Although these observations do not rule out the existence of rare or low-stoichiometry acetylation sites (Extended Data Fig. 2f–h), we find no confirmatory evidence for the presence of ac4C in eukaryotic mRNA.
To understand the potential for eukaryotic cytidine acetyltransferases to modify mRNA, we co-overexpressed NAT10 and THUMPD1 in HEK-293T cells, and their orthologues Kre33 and Tan1 in yeast (Extended Data Fig. 3a–d). Notably, overexpression of these complexes led to the identification of 146 and 66 putative novel ac4C sites in human and yeast mRNA, respectively (Fig. 1g, Extended Data Fig. 3e, Supplementary Table 2). Misincorporation levels within mRNA remained modest (median 7.7% and 4.9% in human and yeast, respectively) even when NAT10 and THUMPD1 were co-overexpressed at very high levels (Fig. 1h, Extended Data Fig. 3a, d). Targeted deep sequencing of five of these sites (median 120,000 reads per site) recapitulated acetylation upon dual overexpression of NAT10 and THUMPD1 (approximately 3–4% misincorporation), whereas misincorporation rates in RNA from cells in which only one protein was overexpressed were on the order of 0.2%, identical to the wild type (Fig. 1i, Extended Data Fig. 3e, Supplementary Note 2). To characterize substrates of the NAT10–THUMPD1 complex and explore factors that direct its specificity, we performed additional analysis of induced eukaryotic ac4C sites. We found that 154 out of 157 (98%) sites in human mRNA and 73 out of 74 (98.6%) sites in yeast mRNA occurred at a CCG motif, with the central cytidine being acetylated (Fig. 1g, j, k, Extended Data Fig. 3e). It is noteworthy that all four ac4C sites that were previously identified in eukaryotic rRNA and tRNA occur within precisely this motif (Extended Data Fig. 3f). Induced ac4C sites were randomly distributed across genes and displayed no preference for a particular position in a codon (Extended Data Fig. 3g, h). The obligate nature of the CCG motif was validated by plasmid-based reconstitution of an inducible ac4C site, the acetylation of which—dependent on NAT10–THUMPD1—was abolished by mutation of the guanosine immediately downstream of the acetylated site (Fig. 1l). Systematic mutagenesis experiments further indicate that base-paired structural elements may have a role in ac4C deposition; this suggests that ‘CCG’ is required, but is not sufficient, for induced acetylation (Extended Data Fig. 4). Overall, our studies define rRNA and tRNA as the predominant sites of ac4C in eukaryotes, suggest that ac4C is absent or present at very low levels in endogenous eukaryotic mRNA, and demonstrate that RNA acetylation can be induced at hundreds of sites via dual overexpression of NAT10–THUMPD1, invariably within a CCG motif.
Unprecedented ac4C levels in archaeal RNA
A cross-evolutionary analysis of total RNA by liquid chromatography coupled to mass spectrometry (LC–MS) revealed high concentrations of ac4C in the archaeal hyperthermophile Thermococcus kodakarensis14 (Fig. 2a). Motivated by this, we applied ac4C-seq to quantitatively map cytidine acetylation in T. kodakarensis cultured at its optimal growth temperature of 85 °C. We found an unprecedented number (404) of ac4C sites spread across rRNA, tRNA, non-coding (nc)RNAs and mRNA (Extended Data Fig. 5a). Of these sites, 99% occurred within CCG motifs and were highly enriched for C>T misincorporation signatures (Fig. 2b). To validate these identifications, we performed quantitative tandem LC–MS analysis of purified and partially digested T. kodakarensis rRNA15. This revealed 25 uniquely mapped ac4C sites, which fully overlapped with positions identified using ac4C-seq (Fig. 2c, Supplementary Data 1, Supplementary Table 3). Estimates of modification stoichiometry based on LC–MS analysis agreed very well (Pearson’s R = 0.97) with those from ac4C-seq (Fig. 2c, Supplementary Table 4). Deletion of the NAT10 homologue TK0754 (hereafter ‘TkNAT10’; recently reported to acetylate T. kodakarensis tRNA16), but not of the THUMPD1 homologue TK2097 (‘TkTHUMPD1’), caused complete loss of ac4C in all RNA substrates (Fig. 2d); this result was confirmed by ac4C-specific northern blotting and mass spectrometric analysis (Extended Data Fig. 5b–g, Supplementary Table 5a). To understand whether pervasive RNA acetylation is a common feature of archaeal extremophiles, we used ac4C-seq to profile Pyrococcus furiosus and Thermococcus sp. AM4—close euryarchaeal relatives of T. kodakarensis within the order Thermococcales—and the more phylogenetically distant species Methanocaldococcus jannaschii (a Methanococcale from the Euryarchaeota phylum) and Saccharolobus solfataricus (a Sulfolobale from the Crenarchaeota phylum), for evolutionary breadth. This revealed that ac4C is widespread within each of the Thermococcales species, occurring at hundreds of sites across diverse RNA types (Fig. 2e, Supplementary Table 2), almost exclusively within CCG consensus motifs. In T. sp. AM4 and P. furiosus, ac4C was not only widely present, but the precise sites and stoichiometry of ac4C were also highly conserved (Fig. 2f, g, Extended Data Fig. 5h). By contrast, ac4C detected in S. solfataricus was confined to 41 CCG sites mostly in tRNAs (Fig. 2e, Supplementary Table 2), whereas M. jannaschii lacked ac4C entirely, consistent with the absence of an apparent NAT10 homologue in this organism17. These studies establish the existence and regulation of prevalent RNA acetylation in the archaeal order Thermococcales.
Fig. 2 |. Ac4C is present at unprecedented levels across diverse RNA species in archaea.
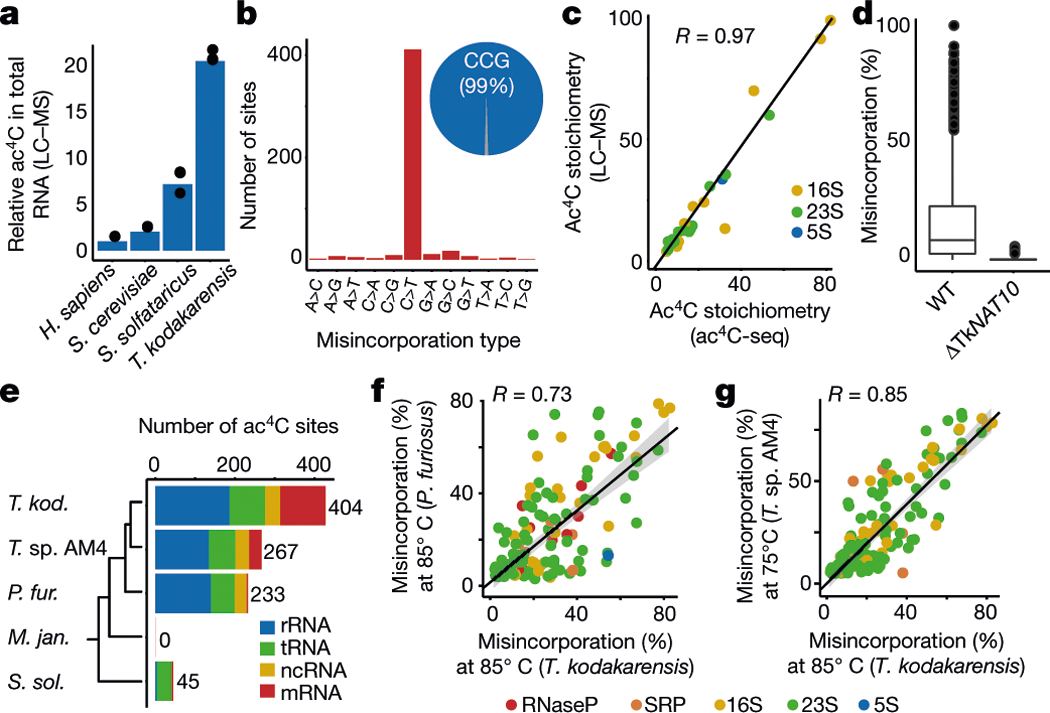
a, Relative quantification of ac4C in total RNA isolated from H. sapiens, S. cerevisiae, S. solfataricus and T. kodakarensis. Mean of n = 3 technical replicates. H. sapiens total RNA was isolated from HeLa cells. b, Distribution of misincorporations (as in Fig. 1g) across all identified sites in T. kodakarensis. Of the C>T misincorporation sites, 99% are embedded within a CCG motif. c, Correlation (Pearson’s R) between ac4C levels as measured by ac4C-seq and those measured by LC–MS, shown for 25 sites that were quantified by both methodologies. n = 2 and 1 independent samples for LC–MS and ac4C-seq experiments, respectively. d, Ac4C-seq quantification of sites identified in wild-type and ΔTkNAT10 strains. Box plot parameters are as in Fig. 1h. n = 4 and 2 independent biological samples for wild-type and ΔTk NAT10, respectively. e, The number of identified ac4C sites in the different RNA types as found in total RNA of different archaeal species. Note that for T. kodakarensis—but not for the others—ac4C-seq was applied also to rRNA-depleted RNA. Non-coding RNAs (ncRNAs) reflect sites in RNaseP RNA, signal-recognition-particle (SRP) RNA and small nucleolar RNA (snRNA), the latter being present only in P. furiosus. The phylogenetic tree represents evolutionary distance between the species. f, g, Correlation between misincorporation levels in ncRNA of T. kodakarensis and P. furiosus (f) and T. sp. AM4 (g), identified by ac4C-seq. Pearson’s R, n = 4 and 1 independent biological samples for T. kodakarensis and other archaea, respectively. Shading indicates 95% confidence interval for predictions from a linear model.
Dynamic acetylation of archaeal RNA
To investigate how ac4C responds to environmental cues, we applied ac4C-seq to RNA from T. kodakarensis cultures grown at 55–95 °C, spanning the range of temperatures at which this organism can be cultivated. These experiments revealed that ac4C across all classes of RNA increases markedly with temperature (Fig. 3a), a finding that was validated by northern blotting and LC–MS analysis (Fig. 3b, Extended Data Fig. 6a). Proteomic analysis of the subsequent gene products indicates that the expression of TkNAT10 is increased at high temperatures (Extended Data Fig. 6b, c, Supplementary Table 5b), which is consistent with increased ac4C. These temperature-dependent patterns of ac4C modification in rRNA, tRNA, ncRNA and mRNA are described in further detail in Fig. 3c, Extended Data Fig. 6d–h and Supplementary Note 3. Notably, the TkNAT10-knockout strain (denoted ΔTkNAT10) showed a temperature-dependent growth lag in comparison to the wild-type strain, beginning at 75 °C and reaching a maximum at 95 °C (Fig. 3d). The reduced fitness of ΔTkNAT10 strains at higher temperatures parallels the increased prevalence of ac4C in wild-type strains under these conditions, suggesting that ac4C is required in particular for growth at high temperatures. If cytidine acetylation is a response to thermal stress, we might expect closely related organisms to also use this mechanism. Indeed, induced acetylations at higher temperatures were also conserved in P. furiosus and T. sp. AM4—two species closely related to T. kodakarensis (Fig. 3e, Extended Data Fig. 6i). Moreover, the precise sites and stoichiometries at which ac4C was induced were also highly conserved in these organisms (Extended Data Fig. 6j). These studies suggest that temperature-dependent cytidine acetylation is a unique adaptive survival strategy and is used by the archaeal order Thermococcales.
Fig. 3 |. Ac4C accumulates in a temperature-dependent manner across all RNA species in archaea and is required for growth at higher temperatures.
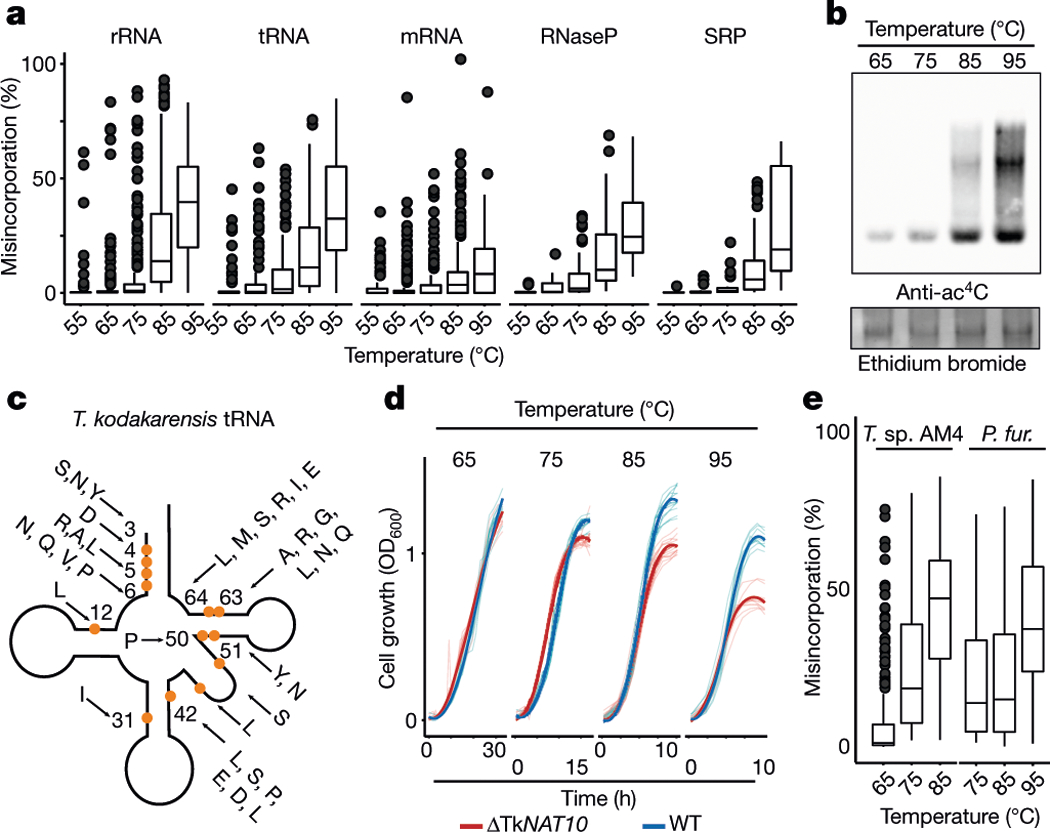
a, Distributions of misincorporation level at ac4C sites across temperatures ranging from 55 to 95 °C. Box plot parameters are as in Fig. 1h. n = 4 biologically independent samples for 85 °C, n = 2 for 65 °C and 75 °C and n = 1 for 55 and 95 °C. b, Immuno-northern blot for the analysis of ac4C in T. kodakarensis total RNA as a function of temperature. Ethidium bromide staining is used to visualize total RNA. Results are representative of two biological replicates. For gel source data, see Supplementary Data 3. c, Schematic representation of a tRNA molecule. A total of 77 ac4C sites found within 19 tRNA species (indicated by the one-letter code of the relevant amino acid) were distributed across 13 distinct positions within the tRNA molecule. Each modified position is indicated by an orange circle. Numbers indicate position within the tRNA. Note that positions in the variable region are not numbered. d, Wild-type T. kodakarensis and ΔTkNAT10 cells were grown at diverse temperatures (65–95 °C), and the optical density at 600 nm (OD600) was measured hourly. The average curve of each replicate is shown by the thick line (n = 11 for 95 °C and n = 12 for each of 65–85 °C), and individual replicates are shown by thin lines. e, Quantification by ac4C-seq of total RNA collected from cells grown at a range of temperatures. Shown are misincorporation levels for ac4C sites identified in P. furiosus and T. sp. AM4. Box plot visualization parameters are as in Fig. 1h. n = 1 biological sample per condition.
Profiling ac4C in an archaeal ribosome
The dynamics of ac4C on the T. kodakarensis ribosome are to our knowledge unprecedented, with both the number of sites and their stoichiometry of modification increasing substantially with temperature (Fig. 3a). In comparison, characterized eukaryotic ribosomes have at most two ac4C sites18 whereas their bacterial counterparts have none18–20. To visualize the distribution of ac4C in T. kodakarensis rRNA, we obtained cryo-electron microscopy (cryo-EM) structures of ribosomes derived from wild-type and ΔTkNAT10 strains with nominal resolutions of 2.95 Å and 2.65 Å, respectively (Extended Data Figs. 7, 8, Supplementary Table 6). This resolution enabled full delineation of the architecture of the T. kodakarensis 70S ribosome—including assignment of the three RNA constituents, associated core proteins, and visualization of modified nucleotides (Fig. 4a, b, Extended Data Fig. 8b, Supplementary Tables 7, 8). Comparing the structures of ribosomes from the wild-type and ΔTkNAT10 strains, we found that the density associated with ac4C was exclusively observed in wild-type ribosomes (Fig. 4b, c, Extended Data Fig. 8b). Cryo-EM maps directly supported the presence of 69 ac4C sites in the T. kodakarensis ribosome grown at 85 °C (Fig. 4a, Supplementary Table 4). The ability to visualize these residues using cryo-EM was consistent with the high stoichiometry estimated at these sites on the basis of the ac4C-seq measurements (Extended Data Fig. 9a, Supplementary Table 4). The cryo-EM analysis enhanced the information available from ac4C-seq by also identifying six locations for the doubly modified nucleoside ac4Cm (Extended Data Fig. 9b–e), which is both acetylated at N4 and methylated at the 2′O sugar and has been previously suggested to have a role in thermostability21,22. To explore the dynamics of ac4C using cryo-EM, we also determined the structure of ribosomes derived from wild-type T. kodakarensis grown at 65 °C (2.55 Å resolution) (Extended Data Figs. 7,8, Supplementary Tables 6,7). Consistent with the results of ac4C-seq, the strain grown at 65 °C exhibited substantially lower ac4C levels than that grown at 85 °C, with only five cytidine residues showing a clear density for acetylation (Extended Data Fig. 8b, Supplementary Table 4).
Fig. 4 |. Cryo-EM structure of wild-type and ac4C-deficient T. kodakarensis ribosomes.
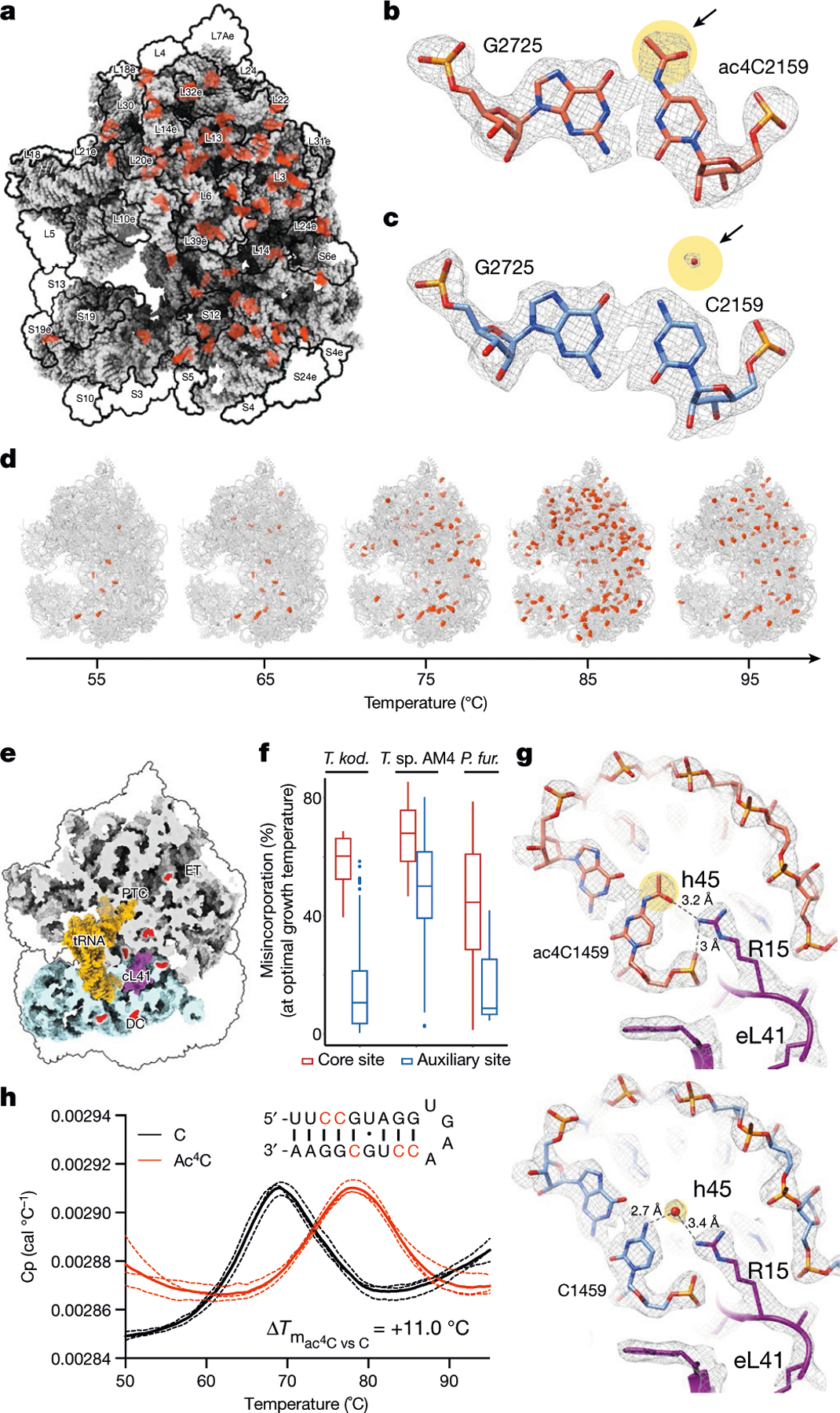
a, Ac4C distribution as observed by a cryo-EM image of wild-type T. kodakarensis grown at 85 °C. Modified residues are highlighted in orange, rRNA in grey and r-proteins are contoured in black. b, c, Ac4Cs participate in Watson–Crick pairing with guanine residues. b, An example of ac4C density shown in mesh. Residues correspond to ac4C2159 and G2725 of LSU. Acetate is highlighted yellow and is indicated by an arrow. c, The same position in the ΔTkNAT10 strain indicates that, in the mutant, the acetyl moiety is replaced by a structured solvent molecule. d, Ac4C in T. kodakarensis ribosomes derived from archaea grown at different temperatures, identified by ac4C-seq and LC–MS. e, ‘Core’ ac4Cs (shown in red) present at high stoichiometries across temperatures are enriched in the intersubunit interface and are in proximity to eL41 and to the ribosomal substrates. The functional ribosome regions indicated are the decoding centre (DC), the peptidyl-transferase centre (PTC) and the protein exit tunnel (ET). tRNA and mRNA are highlighted yellow, eL41 is shown in purple. The tRNA and mRNA coordinates are from PDB structure 4V5D. f, Misincorporation at core and auxiliary sites from T. kodakarensis and their conserved counterparts in P. furiosus and T. sp. AM4, grown at optimal growth temperatures (85 °C for T. kodakarensis and T. sp. AM4 and 95 °C for P. furiosus). n = 4 and 1 independent biological samples for T. kodakarensis and other archaea, respectively. Box plot visualization parameters are as in Fig. 1h. g, A representative example of the electrostatic interaction between ac4C and ribosomal proteins is shown between O(7) of ac4C1459 at h45 of small-subunit (SSU) and R15 of eL41 (top). The same position in the ΔTkNAT10 strain (bottom) implicates a solvent molecule that serves to mediate the same interaction network in the absence of an acetyl group. h, Thermal melting curves of synthetic RNA hairpin containing C (black) or ac4C (red) obtained by differential scanning calorimetry (DSC). Cp, heat capacity; Tm, melting temperature. Data are mean ± s.d. of n = 3 independent experiments.
A notable feature of ac4C in the T. kodakarensis ribosome is that acetylation seems to be spread across core and surface residues in both subunits (Fig. 4a). This contrasts starkly with rRNA base modifications in eukaryotes and bacteria, which are enriched at functional regions near the ribosome core (Extended Data Fig. 9f). Nonetheless, inspection of modification level as a function of temperature revealed a clear pattern of ac4C in archaeal rRNA (Fig. 4d). The seven ac4C residues detected at low temperatures (herein termed ‘core’ sites) were found to concentrate at the interface between the two ribosomal subunits, making direct interactions with the ribosomal substrates (Fig. 4e). Six of these sites envelop an inter-subunit bridge comprising the large-subunit (LSU) ribosomal protein eL41, whereas an additional site is localized at the ribosome exit tunnel (Fig. 4e). Of note, the eukaryotic homologue of eL41 (RPL41) also localizes in an environment that is enriched in modified nucleosides18. Core sites were acetylated at very high levels across all temperatures (median of 77% misincorporation at 85 °C; Extended Data Fig. 9g, h), and were also modified at high levels in T. sp. AM4 and P. furiosus (Fig. 4f), emphasizing a potential role in ribosome function. By contrast, ac4C sites detected only at higher temperatures were modified at lower levels (median 18% at 85 °C) and distributed widely across the ribosome, suggestive of a non-catalytic ‘auxiliary’ role (Extended Data Fig. 9g, h). Considering physical mechanisms that are affected by ac4C, we noted that in the vast majority of sites visualized by cryo-EM (64/70, 91%), the N4-acetyl group present in wild-type ribosomes is replaced by an ordered solvent molecule in the deletion strain ΔTkNAT10 (Fig. 4b, c, Extended Data Fig. 8b, Supplementary Table 4). Similar replacement was observed in unmodified positions from the strain grown at 65 °C (Extended Data Fig. 8b). Ordered solvent molecules are often visualized in near-atomic-resolution structures and can contribute to the structural integrity of protein and RNA architecture; it is tempting to speculate that ac4C may have evolved as a covalent installation to replace tightly bound solvent molecules that might otherwise undergo displacement at high temperatures. Concomitantly, we identified a small subset of positions in which cytidine acetylation created the potential for unique RNA–protein interactions. Representative examples include the interaction of O(7) of ac4C1459—a core site located in helix 45 of the T. kodakarensis small subunit—with Arg15 of eL41 (Fig. 4g) and ac4C1434 of LSU with OP2 of A1786 (Extended Data Fig. 9i). In these examples, the ordered solvent molecule bridges the interactions that are otherwise mediated by the acetyl group (Fig. 4g). Examining the potential influence of ac4C on RNA–RNA interactions, we found that the vast majority (68 out of 69; 99%) of modified residues lie in duplexed rRNA and engage in canonical C–G base pairing. Consistent with the potential for acetylation to strengthen these interactions, biophysical analyses of a synthetic ribosomal RNA hairpin found that its thermal stability is enhanced by the replacement of cytidine with ac4C23,24 (Fig. 4h, Extended Data Fig. 9j). Overall, our structural survey highlights several ways by which dynamic cytidine acetylation at higher temperatures may alter the catalytic properties and physical robustness of the archaeal ribosome.
Conclusion
Here we describe ac4C-seq, a method for the quantitative, nucleotide-resolution profiling of RNA cytidine acetylation. This method leverages acid-catalysed reactivity enhancement to achieve an efficient chemical reduction of ac4C, which was integrated with next-generation sequencing to enable transcriptome-wide detection of ac4C in diverse organisms and RNA species. Applied to eukaryotes, our studies define rRNA and tRNA as the major physiological repositories of ac4C, and suggest that cytidine acetylation is absent or is present at very low levels in endogenous eukaryotic mRNA. This diverges substantially from the findings of previous experiments using antibody-based enrichment10. It remains to be established whether this discrepancy originates from technical differences in the methods (Supplementary Note 2a) or as a result of artefacts caused by antibody promiscuity, the latter of which has substantial precedent in the field25–28 (Supplementary Note 2b).
The application of ac4C-seq in archaea revealed pervasive programs of RNA acetylation. In the context of rRNA base modifications, ac4C in Thermococcales is unprecedented in its prevalence and responsiveness to environmental cues. The dynamic and widespread distribution of ac4C in the T. kodakarensis ribosome challenges our orthodox view of rRNA modifications, in which target sites of rRNA-modifying enzymes are classically conceptualized as being deterministic—that is, each RNA-modifying enzyme catalyses the modification of one or more highly specific sites. The high number and partial modification of ‘auxiliary’ sites in the T. kodakarensis ribosome instead raises the possibility that ac4C catalysis at these positions may be statistical—that is, each site harbours a predefined probability of being targeted by the acetyltransferase, and contributes in an additive manner to overall rRNA function. It remains to be addressed whether such deposition is primarily required for the function of mature ribosomes or to facilitate rRNA folding and processing under increased temperatures. Our results further suggest that such ‘statistical’ deposition of ac4C is not limited to rRNA, but is also widespread in other highly structured RNAs. Collectively, our studies define the ac4C landscape across archaeal and eukaryotic lineages, providing a technical and conceptual foundation for elucidating the role of this modification in biology and disease4–6.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and no allocation to groups was made in this study. Results obtained by ac4C-seq and LC–MS were conducted in different laboratories and were compared only after the data were analysed, making them blind to each other.
Human cell culture
Wild-type HeLa (ATCC) and NAT10-depleted HeLa cells10 were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Quality Biological, 112–013-101) supplemented with 10% fetal bovine serum (FBS, VWR, 89510–194), 25 mM d-glucose, 2 mM l-glutamine, and 1 mM sodium pyruvate. HEK-293T cells (ATCC) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Quality Biological, 112–013-101) supplemented with 10% fetal bovine serum (FBS), 25 mM d-glucose, and 2 mM l-glutamine. All cells were maintained at 37 °C in the presence of 5% CO2, and all cell culture reagents were purchased from Invitrogen unless otherwise noted. Cells were found to be free of mycoplasma contamination and did not undergo authentication.
Microbial growth and media conditions
T. kodakarensis strains—TS559 and their derivatives thereof—were grown as previously described29–31 in artificial seawater (ASW) medium supplemented with vitamins and trace minerals. ASW contains (per litre) 20 g NaCl, 3 g MgCl2·6H2O, 6 g MgSO4·7H2O, 1 g (NH4)2SO4, 200 mg NaHCO3, 300 mg CaCl2·2H2O, 0.5 g KCl, 420 mg KH2PO4, 50 mg NaBr, 20 mg SrCl2·6H2O and 10 mg Fe(NH4)2(SO4)2·6H2O. The trace mineral solution (1,000× per litre) contains 0.5 g MnSO4·H2O, 0.1 g CoCl2·6H2O, 0.1 g ZnSO4·7H2O, 0.01 g CuSO4·5H2O, 0.01 g AlK(SO4)2·12H2O, 0.01 g H3BO3 and 0.01 g Na2MoO4·2H2O. The vitamin mixture (200× per litre) contains 0.2 g niacin, 0.08 g biotin, 0.2 g pantothenate, 0.2 g lipoic acid, 0.08 g folic acid, 0.2 g p-aminobenzoic acid, 0.2 g thiamine, 0.2 g riboflavin, 0.2 g pyridoxine and 0.2 g cobalamin. 5 g/l yeast extract (Y), 5 g/l tryptone (T), 5 g/l pyruvate (Pyr) and 2 g/l elemental sulfur (S˚) were added to rich medium (ASW-YT-Pyr-S˚). ASW-S˚ mixture supplemented with a combination of 20 amino acids formed minimal medium (ASW-aa-S˚). The amino acid mixture contains (20× per litre) 5 g cysteine, 5 g glutamic acid, 5 g glycine, 2.5 g arginine, 2.5 g proline, 2 g asparagine, 2 g histidine, 2 g isoleucine, 2 g leucine, 2 g lysine, 2 g threonine, 2 g tyrosine, 1.5 g alanine, 1.5 g methionine, 1.5 g phenylalanine, 1.5 g serine, 1.5 g tryptophan, 1 g aspartic acid, 1 g glutamine and 1 g valine.
All T. kodakarensis cultures were grown at 55–95 °C under strict anaerobic conditions in sealed vessels with a headspace gas composition of 95% N2/5% H2 at 1 atmosphere at 22 °C; 1 mM agmatine was provided when necessary. Solid medium was prepared by the addition of 1% gelzan, with polysulfides substituting for S˚ (refs. 31,32). Polysulfides were prepared (500×, per 15 ml) by dissolving 10 g Na2S·9H2O and 3 g S˚ with heat to a deep red mixture. Colonies formed on solid medium were observed by lifting cells to polyvinylidene difluoride membranes that were then flash-frozen in liquid N2 before being thawed and stained with Coomassie Brilliant Blue.
P. furiosus strain COM1 was cultured at 75–95 °C in an artificial-seawater-based medium supplemented with cellobiose, maltose, yeast extract, S˚, trace minerals, cysteine and sodium tungstate as previously described33. Thermococcus sp. AM434 was cultured under identical conditions to those for T. kodakarensis.
Yeast growth and media conditions
S. cerevisiae strains were grown at 30 °C in standard YEP medium (1% yeast extract, 2% Bacto Peptone) supplemented with 2% dextrose (YPD). For induction of Tan1 by galactose, cells were washed twice with water, resuspended in YEP medium (1% yeast extract, 2% Bacto Peptone) supplemented with 2% galactose (YPG) and grown at 30 °C for 21 h before collection.
Construction of T. kodakarensis strains markerlessly-deleted for TK0754 or TK2097
Plasmids used to direct the markerless deletion of genomic sequences from the parental strain TS559 were each individually constructed from the parental plasmid pTS70030 and contain approximately 700 bp sequences complementary to both upstream and downstream regions of the respective locus under study29. Each vector also encodes expression cassettes for TK0149 (provides agmatine autotrophy) and TK0664 (provides sensitivity to 6-methylpurine). Strains were constructed as previously described29,30,35. In brief, plasmids incapable of autonomous replication in T. kodakarensis were individually transformed into T. kodakarensis TS559 (ΔTK0149; ΔTK0664; ΔTK0254::TK2276; ΔTK2276)29,30,32,35. Plasmid integration at the desired locus was confirmed by several diagnostic PCR amplicons generated from genomic DNA purified from intermediate strains. Overnight growth in the presence of 1 mM agmatine permitted spontaneous plasmid excision, and colonies were selected on solid media containing 20 amino acids, 6-methylpurine and agmatine. DNA was extracted from 1 ml ASW-YT-Pyr-S˚-agmatine cultures grown from individual 6-MP resistant colonies for use in diagnostic PCRs to confirm the deletion of the desired locus. Final confirmation of each strain included whole-genome sequencing29 to confirm deletion endpoints and to ensure no unanticipated modifications were introduced into the genome at remote locations.
Plasmids for NAT10, Tan1 and THUMPD1 overexpression
Tan1 was synthesized and cloned into pD1201 and pD1231 by ATUM. The remaining plasmids were constructed using Gateway recombination cloning (Thermo Fisher) as follows: NAT10 was amplified from a cDNA plasmid (Dharmacon, accession number BC035558) by PCR and cloned into pDonr-255 with BP Clonase. The insert was sequence-verified and subcloned with LR Clonase into a neomycin-resistant mammalian transfection backbone with CMV promoter and N-terminal 3×Flag–eGFP fusion. The same strategy, NAT10 entry clone, and expression vector were used to generate 3×Flag–NAT10. THUMPD1 was amplified from a cDNA plasmid (Dharmacon, accession number BC000448) by PCR and the entry clone was generated and verified in a similar fashion. This entry clone was then subcloned with LR Clonase into a neomycin-resistant mammalian transfection backbone with CMV promoter, and N-terminal myc tag. Transfection-quality plasmid DNAs were prepared using ZymoPURE II Plasmid Maxiprep Kit (Zymo Research)
Overexpression of eGFP–NAT10 in HEK-293T cells
HEK-293T cells were plated in a 10 cm dish (2.5 × 106 cells per dish in 10 ml DMEM medium) and allowed to adhere and grow for 24 h. eGFP-tagged NAT10 was overexpressed using FuGENE 6 transfection reagent (Promega, E2691). Before transfection, 600 μl of OPTI-MEM (Gibco, 31985062) was incubated with 18 μl FuGENE 6 for 5 min at room temperature before adding 6 μg of eGFP–NAT10 plasmid and incubating for an additional 30 min. Transfection mixture was carefully added to the cell monolayer without changing the medium. Overexpression was carried out by incubating the cells for 24 h at 37 °C under a 5% CO2 atmosphere, after which cells were imaged using an EVOS FL fluorescence microscope at 10× and 40× magnifications.
Co-overexpression of NAT10 and THUMPD1 in HEK-293T cells
HEK-293T cells were seeded into twenty 10 cm dishes (2.5 × 106 cells per dish in 10 ml DMEM medium) and allowed to adhere and grow for 24 h. 3×Flag-tagged NAT10 and myc-tagged THUMPD1 were overexpressed using FuGENE 6 transfection reagent (Promega, E2691). For each 10 cm dish, 600 μl of Opti-MEM I Reduced Serum Medium (Gibco, 31985062) was incubated with 18 μl FuGENE 6 for 5 min at room temperature before adding 3 μg each of NAT10 and THUMPD1 plasmid and incubating for an additional 30 min. Transfection mixtures were carefully added to the cell monolayer without changing the medium. Overexpression was carried out by incubating the cells for 24 h at 37 °C under 5% CO2 atmosphere, after which 19 plates were collected by trypsinization and snap-frozen for total RNA extraction. The remaining plate was collected using ice-cold PBS and pelleted for western blot analysis of overexpression. The cell pellet was resuspended in 500 μl of ice-cold PBS containing protease inhibitor cocktail (1X, EDTA-free, Cell Signaling Technology, 5871S). Samples were then lysed by sonication using a 100 W QSonica XL2000 sonicator (3 × 1 s pulse, amplitude 1, 60 s resting on ice between pulses). The lysate was pelleted by centrifugation (20,817 rcf × 30 min, 4 °C) and quantified using the Qubit 4.0 Fluorometer and Qubit Protein Assay Kit. Protein was run on SDS–PAGE alongside non-transfected control and immunoblotted with anti-Flag-tag (Cell Signaling, 2044), anti-NAT10 (Bethyl Laboratories, A304–385A), and anti-myc-tag (Cell Signaling, 5605) antibodies. For immunoblotting, SDS–PAGE gels were transferred to nitrocellulose membranes (Novex, Life Technologies, LC2001) by electroblotting at 30 V for 1 h using a XCell II Blot Module (Novex). Membranes were blocked using StartingBlock (PBS) Blocking Buffer (Thermo Scientific) for 30 min and incubated overnight at 4 °C in primary antibody. The membranes were washed with TBST buffer and incubated with secondary HRP-conjugated antibody (Cell Signaling, 7074) for 1 h at room temperature. The membranes were again washed with TBST and treated with chemiluminescence reagents (Western Blot Detection System, Cell Signaling) for 1 min, and imaged for chemiluminescent signal using an ImageQuant Las4010 Digital Imaging System (GE Healthcare).
For targeted ac4C-sequencing in cells overexpressing either NAT10, THUMPD1, neither, or both, HEK-293T cells were seeded in replicates in wells of a 6-well plate (0.5 × 106 cells per well in 2 ml DMEM media) and allowed to adhere and grow for 24 h. Cells were transfected using PolyJet (SignaGen Laboratories) according to the manufacturer’s protocol, either with 0.5 μg NAT10, or with 0.5 μg THUMPD1, neither or both. In all samples a total of 50 ng GFP plasmid was used to monitor transfection efficiency. Cells were grown for 24 h before collecting for RNA purification.
Growth analysis of T. kodakarensis
Parental strain TS559 and TkNAT10-deleted T. kodakarensis cells were grown as described above at 65–95 °C (11–12 replicates from each temperature). Growth of liquid cultures was monitored by measurements of optical density at 600 nm at hourly intervals for a total of 33 h. Measurements were used to model cell growth using the ‘locally estimated scatterplot smoothing’ (loess) method36.
Total RNA isolation from yeast, human and archaea
Total RNA from human cells was extracted using TRIzol according to the manufacturer’s protocol. 1 ml TRIzol was used per 1 × 107 cells. The RNA pellet was resuspended by briefly heating at 50 °C in 1.0 ml 1X TE buffer pH 8.0. Samples were quantified by UV absorbance and stored at −80 °C. Typical extractions were carried out with 4 × 107 cells and yielded 400 μg of total RNA.
For targeted ac4C-sequencing, RNA was extracted using Nucleozol (Macherey Nagel) according to the manufacturer’s instructions.
Total RNA was isolated from yeast using hot acidic phenol. In brief, a frozen yeast (S. cerevisiae) pellet was suspended in 1.0 ml AES buffer (50 mM sodium acetate, 10 mM EDTA pH 8.0, 1% SDS) per 0.5 ml pellet volume. To the suspended pellet, 1.0 ml acid-buffered phenol per ml of AES buffer used was added. The sample was mixed by vortexing and incubated in a 65 °C water bath for 30 min, vortexing every 2 min to mix. Samples were put on ice for 10 min and 1.0 ml chloroform:isoamyl alcohol (24:1) was added for each 1.0 ml phenol used. The sample was vortexed to mix and centrifuged at 5,000 rcf for 15 min. The aqueous layer (top) was transferred to a clean tube and extracted three times with an equal volume of acid-buffered phenol:chloroform:isoamyl alcohol (24:23:1). After each extraction the sample was centrifuged at 5,000 rcf for 10 min and the aqueous layer was transferred to a new tube. A final extraction with chloroform:isoamyl alcohol was carried out to remove residual phenol. The aqueous layer was transferred to a clean tube and RNA was precipitated by the addition of an equal volume of 100% isopropanol and 1/9th volume of 3 M sodium acetate. Samples were incubated at −20 °C for 30 min and centrifuged at 12,000 rcf at 4 °C for 15 min. The supernatant was decanted and the pellet was washed with 4 ml ice-cold 70% ethanol. The RNA pellet was resuspended by briefly heating at 50 °C in 1.0 ml 1X TE buffer at pH 8.0. Samples were quantified by UV absorbance and stored at −80 °C. Typical extractions were carried out with cell pellets of 1.0 ml volume and yielded 20 mg of total RNA. Total RNA was isolated from archaeal samples using TRIzol according to the manufacturer’s protocol.
Poly(A) RNA isolation from yeast and human cells
Poly(A) RNA from yeast and human total RNA was isolated by two rounds of purification using the GenElute mRNA miniprep kit (Sigma) according to the manufacturer’s protocol. 500 μg total RNA was used per purification column. A typical yield after two rounds of isolation was 1.2%.
For targeted ac4C-sequencing, poly(A) RNA was isolated from total RNA of HEK-293T cells by two rounds of purification using Dynabeads mRNA DIRECT Kit (Invitrogen), according to the manufacturer’s protocol. 75 μg total RNA was taken from each sample, using 150 μl oligo dT beads. Typical yield after two rounds of isolation was 1.6%.
Ribosome purification
Purification of T. kodakarensis ribosomes of the wild-type and the TkNAT10 deletion strains were conducted similarly to previously documented procedures37. In brief, cell lysis was obtained through sonication in buffer A (20 mM HEPES, pH 7.5, 10.5 mM magnesium acetate, 100 mM ammonium acetate, 0.5 mM EDTA and 6 mM β-mercaptoethanol). Cell debris was discarded by centrifugation at 30,000g for 20 min at 4 °C, and the cytoplasmic fraction was loaded onto a 1.1 M sucrose cushion in buffer B (20 mM HEPES, pH 7.5, 10 mM magnesium acetate, 150 mM potassium acetate 6 mM β-mercaptoethanol). The ribosome-enriched pellet was obtained by overnight centrifugation at 220,000g at 4 °C. The pellet was resuspended in buffer B and ribosome particles were purified on a 10–40% sucrose gradient using a SW-28 rotor, at 43,000g for 17 h at 4 °C. Fractions containing 70S ribosomes were collected, combined and centrifuged at 230,000g overnight at 4 °C. The pellet was resuspended in buffer B and an additional centrifugation step at 200,000g for 1.5 h at 4 °C was designed to remove sucrose traces. The ribosomal pellet was resuspended in buffer C (20 mM HEPES pH 7.5, 10 mM magnesium acetate, 100 mM potassium acetate, 100 mM ammonium acetate and 1 mM DTT), diluted to a concentration of 1 mg/ml aliquoted and stored at −80 °C until further use.
rRNA depletion from total RNA of T. kodakarensis
To deplete abundant T. kodakarensis rRNAs before RNA-seq, we adapted a method originally reported previously38 using reagents provided in the NEBNext rRNA Depletion Kit (NEB, E6310). The protocol in the manual for the kit was followed with the following changes. The NEBNext rRNA Depletion Solution provided in the kit was substituted for an equimolar mixture of 85 oligonucleotides complementary to T. kodakarensis rRNA sequences (Supplementary Table 1b). The concentration of the oligo mix was 85 μM, such that each individual oligo was at 1 μM in the mix. All volumes for the probe hybridization, RNase H treatment and DNase I treatment sections of the protocol were scaled up twofold and 24 μl of 62.5 ng/μl T. kodakarensis RNA was used as the starting material. Instead of bead purification as indicated in the manual, samples were purified using the Monarch RNA Cleanup Kit (NEB, T2030) using the standard protocol. Sixteen depletion reactions were performed as described above for each T. kodakarensis total RNA sample and these were then concentrated into a single depleted RNA sample by pooling them and performing a second round of purification with the Monarch RNA Cleanup Kit. The yield of RNA after depletion was measured using the Qubit RNA BR Assay Kit (Thermo Fisher).
UV spectroscopic analysis of ac4C reduction rates
Model reactions to assess the rate of reduction of ac4C by NaBH4 and NaCNBH3 were performed using free N4-acetylcytidine nucleoside. For NaBH4 reductions, stock solutions of NaBH4 (100 mM) and N4-acetylcytidine (2 mM) were prepared fresh daily in water. Reactions (25 μl) consisted of N4-acetylcytidine (100 μM), NaBH4 (20 mM) and reaction buffer (water, 100 mM sodium acetate (pH 4.5), or 100 mM potassium phosphate (pH 7.5)). At the indicated time point, reactions were adjusted to 50 μl using 100 mM HCl. To normalize pH, a further aliquot of 50 μl 100 mM sodium phosphate (pH 7.2) was added and reactions were transferred to Greiner-UV Star 96-well half-area microplates (655801) for analysis. For NaCNBH3 reductions, stock solutions of NaCNBH3 (1 M) and N4-acetylcytidine (2.5 mM) were prepared fresh daily in water. Reactions (100 μl) consisted of N4-acetylcytidine (100 μM), NaCNBH3 (100 mM) and HCl (100 mM). At the indicated time point, reactions were quenched with 30 μl of 1 M Tris-HCl (pH 8.0), and added to Greiner-UV Star 96-well microplates for analysis. Reduction of N4-acetylcytidine was analysed on a Biotek Synergy plate reader by monitoring the absorbance of N4-acetylcytidine (λmax = 300 nm) and cytidine (λmax = 270 nm). For N4-acetylcytidine reactions, the percentage decrease in N4-acetylcytidine was calculated from absorbance (A) values at 300 nm using the formula: Percentage decrease = (Aac4C(start) − Aac4C(end))/(Aac4C(untreated) – Awater(blank)) × 100.
UV spectroscopic analysis of ac4C deacetylation
Model reactions to assess the rate of acid- and base-induced deacetylation of ac4C were performed using free N4-acetylcytidine nucleoside. Stock solutions of N4-acetylcytidine (2.5 mM) were prepared fresh daily in water. Reactions (50 μl) consisted of N4-acetylcytidine (250 μM) and reaction buffer (KCl/HCl buffer (pH 1) or NaHCO3 buffer (pH 10)) added to a Greiner-UV Star 96-well half-area microplate. Control reactions were set up similarly with cytidine (250 μM). Deacetylation of N4-acetylcytidine was analysed on a Biotek Synergy plate reader by monitoring the absorbance of N4-acetylcytidine (pH 1 λmax = 310 nm; pH 10 λmax = 300 nm) over 18 h. For N4-acetylcytidine reactions, the percentage decrease in N4-acetylcytidine was calculated from λmax absorbance values using the formula: percentage decrease = (Aac4C(start) – Aac4C (end))/(Aac4C(untreated)) × 100.
In vitro transcription of synthetic ac4C-containing RNAs as spike-in controls
In vitro transcription was performed with the HiScribe T7 Kit (New England Biolabs), according to the manufacturer’s instructions using DNA templates containing a T7 promoter upstream of a template sequence harbouring a single cytidine within an ACA, GCA, ACG or GCG sequence context (Supplementary Table 1a). For ac4C-containing transcripts, CTP was replaced in the reaction mixture with ac4CTP (10 mM) as described previously12. In vitro transcription reactions were analysed by denaturing polyacrylamide gel electrophoresis on 10% TBE-urea gels and visualized using SYBR Gold staining. Synthetic RNA products were used in ac4C-seq, LC–MS quantification, and reverse transcription stop experiments, the latter of which were performed as previously described11.
Mass spectrometry analysis of ac4C in synthetic spike-in controls
Mass spectrometry analysis of ac4C reduction in RNA probes was assessed after nuclease digest as described previously12. In brief, in vitro transcribed ac4C RNA was treated with nuclease P1 (2U/10 μg RNA, N8630, Sigma) in 50 μl of buffer containing 100 mM ammonium acetate (pH 5.5), 2.5 mM NaCl and 0.25 mM ZnCl2 for 2 h at 37 °C. Sample volumes were adjusted to 60 μl by adding 3.5 μl of H2O, 6 μl of 10× Antarctic Phosphatase buffer (B0289S, NEB) and 0.5 μl of Antarctic Phosphatase (1 U/10 μg RNA, M0289S, NEB). Samples were further incubated at 37 °C for 2 h, adjusted to 150 μl with RNase-free water and filtered via centrifugation to remove enzymatic constituents (Amicon Ultra 3K, UFC500396). After lyophilization, samples were reconstituted in 10 μl RNase-free water and analysed via LC–MS/MS using reverse phase chromatography (Shimadzu LC-20AD) coupled to a triple-quadrupole mass spectrometer (Thermo TSQ-ultra) operated in positive electrospray ionization mode. Quantification was accomplished by monitoring nucleoside-to-base ion transitions and generating standard curves for each nucleoside using the stable isotope dilution internal standardization method.
Primer extension and reverse transcription stop analysis of ac4C RNAs
Primer extension assays were performed using PAGE-purified model RNAs containing a single site of either ac4C or cytidine produced by in vitro transcription (sequence provided above). For each reaction, RNA (2 μg) was treated in a final reaction volume of 100 μl. For NaBH4-treated samples: 1 M NaBH4 was added to 2 μg RNA in nuclease-free H2O to a final concentration of 100 mM and samples were incubated for 60 min at 37 °C, NaBH4 was quenched with 1 M HCl (15 ml), and neutralized by the addition of 1 M Tris-Cl (pH 8.0) buffer (15 ml). For NaCNBH3 treated samples: 1 M NaCNBH3 was added to 2 μg RNA in nuclease free H2O to a final concentration of 100 mM. Reactions were initiated by the addition of 1 M HCl to a final concentration of 100 mM and samples were incubated 20 min at room temperature (20 °C). The reaction was stopped by neutralizing the pH by the addition of 30 μl 1 M Tris-HCl pH 8.0. For untreated control samples: 1 M HCl was added to 2 μg RNA in nuclease-free water to a final concentration of 100 mM and samples were incubated for 20 min at room temperature (20 °C). Reactions were stopped by neutralizing the pH by the addition of 30 μl 1 M Tris-HCl pH 8.0. Reactions were adjusted to 200 ml with H2O, precipitated with ethanol, desalted with 70% ice-cold ethanol, briefly dried on Speedvac, resuspended in H2O, and quantified by absorbance using a Nanodrop 2000 spectrophotometer. RNA from individual reactions (5 pmol) was incubated with 5′-Cy5 IVT primer (5′-/Cy5/ACTCATCACTTTTCTCCCTCTACACAATC-3′; 3.5 pmol) in a final volume of 50 μl. Individual reactions were heated to 65 °C for 5 min and cooled at a rate of 5 °C per min to a final temperature of 4 °C to facilitate annealing, with the following buffer conditions used for specific RTs: AMV: 1X AMV reaction buffer (NEB), 1.0 mM dNTPs; Superscript III: 500 mM dNTPs; TGIRT: 1X TGIRT reaction buffer (Ingex), 5 mM MgCl2. After annealing, reverse transcriptions were performed as follows: 1) AMV reactions: 100 units RNaseOUT (Invitrogen), 25 U AMV RT, incubate 60 min, 48 °C; 2) Superscript III: 1x SSIII reaction buffer (from 10x stock; Thermo Fisher), 5 mM MgCl2, 10 mM DTT, 100 U RNaseOUT, 500 U Superscript III, incubate 60 min, 48 °C; 3) TGIRT reactions: first add 5 mM DTT, 500 U TGIRT RT, incubate 20 min room temperature, then add 500 mM dNTPs, incubate 1 h, 57 °C. After the indicated incubation time, reactions were adjusted to 200 μl with H2O, extracted with phenol:chloroform, precipitated with ethanol, desalted with 70% ice-cold ethanol, briefly dried on Speedvac, and resuspended in 20 ml of 1X RNA denaturing RNA loading buffer. Samples were heated at 95 °C for 4 min, cooled on ice, loaded onto a 10% denaturing polyacrylamide gel and run at 400 V (20 V/cm) for 5 h. Gels were fluorescently visualized using an ImageQuant Las4010 (GE Healthcare) with red LED excitation (λmax = 630 nm) and a R670 filter, with band intensities quantified by densitometry using Imagequant software. To calculate the product/stop ratio, the fluorescence intensity of the bands observed at the ac4C site (−1, 0 or +1) were divided the total fluorescence intensity of all other primer extension products observed in each gel lane.
Reverse transcription and misincorporation analysis of RNAs by Sanger sequencing
For each reaction, RNA (1 μg) was incubated with either NaCNBH3 (100 mM in H2O + 100 mM HCl) or untreated ‘mock’ control (H2O + 100 mM HCl) in a final reaction volume of 100 ul. Samples were incubated for 20 min at 20 °C. Reactions were stopped by neutralization of pH by the addition of 30 μl 1 M Tris-HCl pH 8.0. Reactions were adjusted to 200 μl with H2O, precipitated with ethanol, desalted with 70% ice-cold ethanol, briefly dried on Speedvac, resuspended in H2O, and quantified by absorbance using a Nanodrop 2000 spectrophotometer. RNA from individual reactions (200 pg) was incubated with 4.0 pmol RT primer in a final volume of 20 μl. Individual reactions were heated to 65 °C for 5 min and transferred to ice for 3 min to facilitate annealing in 1× TGIRT reaction buffer (Ingex), 5 mM MgCl2. After annealing, reverse transcriptions were performed as follows using TGIRT-III; DTT was added to 5 mM along with 100 U TGIRT RT and 25 U RNasin Plus (Promega). The reaction was incubated for 20 min at room temperature. The reverse transcription reaction was initiated by addition of dATP, dTTP and dCTP to 500 mM and dGTP to 250 mM. Reactions were incubated for 1 h at 57 °C. cDNA (2 μl) was used as template in 50 ml PCR reaction with Phusion Hot start flex (NEB). Reaction conditions: 1X supplied HF buffer, 2.5 pmol each forward and reverse primer, 200 mM each dNTPs, 2 U Phusion hot start enzyme, 2 μl template and the following specific conditions:
In vitro transcribed ‘single ac4C’: Primers: IVT rev (PCR primer), IVT forward (PCR primer). Thermocycling conditions: 71 °C annealing, 34 cycles.
Human 18S rRNA, helix 45 ac4C site: Primers: human 18S helix 45 fwd, human 18S helix 45 rev. Thermocycling conditions: 67.4 °C annealing, 34 cycles.
PCR products were run on a 2% agarose gel, stained with SYBR safe and visualized on UV transilluminator at 302 nm. Bands of the desired size were excised from the gel and DNA extracted using QIA-quick gel extraction kit from Qiagen and submitted for Sanger sequencing (GeneWiz) using the forward PCR primer for 18S sites and reverse PCR primer for IVT ‘single ac4C’. Processed sequencing traces were viewed using 4Peaks software. The peak height for each base was measured and the percentage misincorporation was determined using the equation: Percentage misincorporation = (Sum of non-cognate base peaks intensities)/(sum of total base peaks) × 100%.
Ac4C-seq library preparation
Strand-specific ac4C-seq libraries were generated on the basis of previously described protocols39,40. In brief, RNA was first subjected to FastAP Thermosensitive Alkaline Phosphatase (Thermo Scientific), followed by a 3′ ligation of an RNA adaptor using T4 ligase (NEB). Ligated RNA was reverse transcribed using TGIRT-III (InGex), and the cDNA was subjected to a 3′ ligation with a second adaptor using T4 ligase. The single-stranded cDNA product was then amplified for 9–12 cycles in a PCR reaction. Libraries were sequenced on Illumina NextSeq 500 or NovaSeq 6000 platforms generating short paired-end reads, ranging from 25 to 55 bp from each end.
Samples used in ac4C-seq analysis
Human: Three experiments were conducted. In the first experiment, total RNA from wild-type HeLa cells or cells with reduced expression of NAT1010 were treated with NaCNBH3 (with and without alkali pre-treatment) or mock-treated in three biological replicates. In the second, a set of 5 poly(A)-enriched HeLa samples (3 and 2 biological replicates for wild-type and NAT10 knock-down, respectively) were treated with NaCNBH3 or mock-treated. For the third experiment, poly(A)-enriched HEK-293T cells co-overexpressing NAT10 and THUMPD1 (2 biological replicates) and a sample of wild-type cells were treated with NaCNBH3 (with and without alkali pre-treatment) or mock-treated.
Yeast:
Two experiments were conducted. In the first, biological duplicates of wild-type S. cerevisiae cells and cells expressing a catalytic mutant of Kre331 were treated with NaCNBH3 (with and without alkali pre-treatment) or mock-treated. In the second, cells co-overexpressing Kre33 and Tan1 in a Kre33-catalytic mutant strain were analysed in comparison to wild-type S. cerevisiae cells. One replicate of the co-overexpression cells expressed Tan1 under a constitutive GPD promoter, the other under a GAL1-inducible promoter. These cells were grown in YPD and YPG, respectively, along with a matching wild-type sample grown under the same conditions. These four samples were treated with NaCNBH3 or were mock-treated. All libraries of yeast were prepared from poly(A)-enriched RNA.
T. kodakarensis:
A total of 17 samples were analysed, representing 25 treatment conditions. For all samples total RNA was analysed from a single biological sample, unless stated otherwise. TS559 cells grown at 55, 65, 75, 85 and 95 °C were treated with NaCNBH3 or mock-treated. For the 85 °C condition, four biological replicates were assessed, and one of them also underwent alkali pre-treatment. For 65 and 75 °C two biological replicates were assessed. Biological duplicates of cells in which TkNAT10 (TK0754) or TkTHUMPD1 (TK2097) were deleted were treated with NaCNBH3. ΔTkNAT10 samples were also mock-treated. rRNA-depleted RNA from TS559 cells grown at 85 and 95 °C were treated with NaCNBH3 or were treated with NaCNBH3 and mock-treated, respectively. Purified ribosomes from TS559 cells grown at 85 °C were treated with NaCNBH3.
T. sp. AM4:
total RNA from cells grown at 65, 75 and 85 °C was treated with NaCNBH3 or mock-treated.
P. furiosus:
total RNA from cells grown at 75, 85 and 95 °C was treated with NaCNBH3 or mock-treated.
S. solfataricus:
total RNA from cells grown at 85 °C was treated with NaCNBH3 (with and without alkali pre-treatment) or mock-treated. A total of three samples were used, representing a single biological sample.
M. jannaschii:
a single sample was treated with NaCNBH3 (with and without alkali pre-treatment) or mock-treated.
Identification of putative ac4C sites
Reference genomes were generated on the basis of the following genome assemblies: ASM996v1 for T. kodakarensis, ASM27560v1 for P. furiosus, ASM15120v2 was used for T. sp. AM4, ASM700v1 for S. solfataricus and ASM9166v1 for M. jannaschii. For human poly(A)-enriched samples we used the GRCh37/hg19 with UCSC Genes annotations, supplemented with tRNA, rRNA and snRNAs sequences, obtained from the Modomics database41. Samples from total RNA of human cells were aligned to a subset of the full reference containing only the tRNA, rRNA and snRNA sequences. For S. cerevisiae samples the sacCer3 assembly was used in experiments designed to detect modification in mRNA, whereas a limited reference containing only rRNAs and tRNAs (filtered to only retain non-redundant sequences) was used in experiments designed to detect only sites in these non-coding transcripts.
Samples were aligned to the genome using STAR aligner42. For archaeal and S. cerevisiae samples intron size was limited to 500 bases (‘alignIntronMax = 500’). For poly(A)-enriched samples (applicable to some of the human and yeast samples, as indicated in the main text) duplicated reads and chimeric pairs were filtered out by the dedup function of UMI-tools43 (using ‘–chimaeric-pairs = discard’) followed by removal of overlapping reads by the clipOverlap function of bamUtil44. For human and yeast samples aligned to a limited reference containing only the ncRNA sequences mentioned above, multiple mapping was allowed (‘multiMapping = 200’).
Single nucleotide variants were detected using the JACUSA software in pileup mode45, which outputs a tabular format summarizing the abundance of each nucleotide (with minimal coverage of 5 reads) at each position. A custom script was used to extract the misincorporation rate at each position as well as to identify the most abundant nucleotide appearing instead of the wild-type nucleotide (the ‘predominant base conversion’).
For a position to be considered as putatively modified, it had to meet two sets of requirements. First, at the level of an individual NaCNBH3-treated sample compared to a suitable control (whereby the control is in most cases a mock-treated sample, but in some cases is a chemically deacetylated sample or a NAT10-deficient genetic control) the fundamental requirement it had to meet was that the P value obtained from the χ2 test comparing the misincorporation rates in the treated versus control samples was lower than 0.05. In experiments with multiple replicates, the χ2 test was conducted on ‘pooled samples’ combining misincorporation information from all replicates. Second, to reduce the computational load, we applied this statistical framework only to sites matching the minimal criteria below: (1) At least three reads with misincorporations in the NaCNBH3-treated sample (or wild-type sample, when comparing to NAT10-deficient). (2) A misincorporation rate > MIN_RATE in the NaCNBH3-treated sample (for archaea we used a MIN_RATE_TREAT = 2%, for human and yeast with larger genomes and consequently slightly reduced signal:noise ratios we used 3%). (3) A misincorporation rate lower than MAX_RATE_CONT in the control sample (MAX_RATE_CONT = 5% in archaea, 1% in human and S. cerevisiae). (4) Misincorporation rates in the NaCNBH3-treated sample were at least 2% higher than in their control counterparts. (5) The predominant base conversion at the site in the NaCNBH3-treated sample was from cytidine to thymidine (C>T). To eliminate redundancies, positions harbouring identical sequences in a 21-bp window (10 bp upstream + 10 bp downstream) surrounding the putative site were filtered to retain only one. Furthermore, when possible on the basis of the experimental design, we demanded that such a site be reproducibly identified across at least two distinct comparisons. The distinct experimental design for the different organisms (in some cases we monitored distinct temperatures, in others distinct genetic backgrounds, in others we obtained static snapshots under one condition) was taken into consideration, and the precise set of comparisons performed for each organism is detailed in Supplementary Table 2. This set of comparison was used to create a final ‘catalogue of ac4C sites’ for each organism, which was used in downstream analyses. All catalogues, segregated by organism, appear in Supplementary Table 2.
Motif analysis
For each species, we extracted the 20 nt flanking the ac4C positions in its catalogue of ‘significantly modified’ sites. These 21-nt long sequences were used to generate sequence logos using the WebLogo software (available at https://weblogo.berkeley.edu/logo.cgi)46, in which the height of each stack indicates the information content at that position (measured in bits), whereas the height of letters within the stack reflects the relative frequency of the corresponding nucleic acid at that position.
Targeted ac4C-sequencing
mRNA samples treated with NaCNBH3 were incubated with Turbo DNase (Invitrogen) for 30 min at 37 °C. 400 ng of the DNase-treated mRNA was reverse transcribed using TGIRT-III (InGex), with random primers (Applied Biosystems). After cleanup of cDNA using Dynabeads MyOne SILANE beads (Life Technologies), 10 cycles of PCR were carried out using Kapa HiFi HotStart Readymix PCR kit (Kapa Biosystems), and pairs of primers described in Supplementary Table 1a. 1 μl of the PCR reaction was used as template for a second PCR reaction (Kapa HiFi, 25 μl reaction volume, 20 cycles), in which barcoded Illumina adaptors were added. Amplicons were analysed on 2% E-gel EX agarose gels (Invitrogen), and cleaned using two rounds of AMPure XP beads (Beckman Coulter). For targeted ac4C-sequencing of overexpressed sequences, total RNA samples were treated with NaCNBH3 and incubated with Turbo DNase (Invitrogen) for 30 min at 37 °C. 600 ng of the DNase-treated total RNA was reverse transcribed using TGIRT-III (InGex), with random primers (Applied Biosystems). After cleanup of cDNA using Dynabeads MyOne SILANE beads (Life Technologies), 20 cycles of PCR were carried out using Kapa HiFi HotStart Readymix PCR kit (Kapa Biosystems), adding the barcoded Illumina adaptors.
Construction of plasmids for overexpression of wild-type (CCG) and mutated (CCA) ac4C sites
The sequences described in Supplementary Table 1a were cloned using FastDigest SgsI (AscI) and BcuI (SpeI) restriction enzymes (Thermo Scientific) into pZDonor FC plasmid, as a 3′ UTR of a reporter gene47.
Targeted ac4C -sequencing of a pool of sequence variants of BAZ2A mRNA
Pool design.
A 91-base-long sequence surrounding the ac4C site identified in BAZ2A mRNA was used as a wild-type control fragment. Variants of the wild-type BAZ2A fragment were made by introducing a single point mutation at each base of the wild-type sequence, by replacing it with all possible bases. BAZ2A fragments were preceded by an 8-base barcode, allowing each variant to be uniquely mapped, and flanked by SpeI and AscI restriction sites to facilitate cloning, Illumina adaptor sequences to allow sequencing, and primer sequences to allow amplification of the entire construct in the cloning stage.
Cloning of the oligonucleotide pool.
The pool of sequences was cloned as 3′ UTR downstream of a reporter gene in the pZDonor FC plasmid, essentially as described previously48. Specifically, the library was amplified in 5 different PCR reactions, each using 50 pg as a template and 14 cycles. The reactions were combined, cleaned using an QIAquick PCR purification kit (Qiagen), and a total of 540 ng was cut by SgsI (AscI) and BcuI (SpeI) restriction enzymes (FastDigest, Thermo Scientific). After electro-elution from a gel using Midi GeBAflex tubes (GeBA, Kfar Hanagid, Israel), the library was ligated (in 1:1 ratio) to pZDonor FC plasmid digested by SgsI and BcuI, using CloneDirect Rapid Ligation kit (Lucigen Corporation) and transformed into E. cloni 10G electrocompetent cells (Lucigen) in a single cuvette. The bacteria were grown on four 14-cm plates, reaching on average about 1,500 colonies per each sequence variant. Plasmids were purified directly from collected bacterial colonies.
Transfection, treatment and library preparation.
The plasmids pool was transfected to 10-cm plates of HEK-293T cells in replicates using PolyJet reagent (SignaGen Laboratories), either by itself (2 μg) or together with both NAT10 and THUMPD1 (1.5 μg each). For targeted ac4C-sequencing of the library variants, total RNA samples were treated with NaCNBH3 and incubated with Turbo DNase (Invitrogen) for 30 min at 37 °C. 1 μg of the DNase-treated total RNA was reverse transcribed using TGIRT-III (InGex), with random primers (Applied Biosystems). After cleanup of cDNA using Dynabeads MyOne SILANE beads (Life Technologies), half of the cleaned cDNA was used in a 25-cycle PCR reaction, using Kapa HiFi HotStart Readymix PCR kit (Kapa Biosystems), and Illumina adaptors as primers.
Analysis.
SAMtools mpileup was used to assess misincorporation rates at the ac4C site of BAZ2A variants.
mRNA expression analysis
To estimate expression levels, reads were aligned against the human, yeast or T. kodakarensis genome using RSEM (version 1.2.31) in paired-end and strand-specific mode with default parameters49. For robust comparison between different samples, we used trimmed mean of M values (TMM) normalization50 of the RSEM read counts as implemented by the NOISeq package51 in R.
Analysis of codon enrichment and distribution across transcript body
Our analysis identified 146 and 119 putative ac4C sites in mRNA of human and T. kodakarensis, respectively. For each site its relative position within the codon was identified on the basis of the genome annotation. As a control, the distribution of all remaining cytidines embedded in CCG sequences in the examined mRNAs was calculated. For T. kodakarensis, we further calculated the distribution of the putative ac4C sites and the control cytidines between specific codons encoding the different amino acids. For human sites we mapped the location of each ac4C site and control cytidines (as described above) within the transcript body (that is, 5′ UTR, CDS or 3′ UTR) and calculated the distribution across transcript regions.
Multiple alignment of tRNAs
All T. kodakarensis tRNA sequences were multiply aligned against each other using MAFFT v7.402 with default parameters52. Manual inspection of aligned sequences facilitated assignment of ac4C sites into distinct regions within the tRNA structure and into specific positions within a canonical model of a tRNA.
Conservation analysis between archaea
Sequences of 16S, 23S, 5S, RNaseP RNA and SRP RNA were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/) from genome references NC_006624.1, NC_018092.1 and NC_016051.1 for T. kodakarensis, P. furiosus and T. sp. AM4, respectively. Multiple sequence alignment was conducted across all three archaea for each gene separately using the Clustal Omega software with default parameters (https://www.ebi.ac.uk/Tools/msa/clustalo/)53. A custom script was used to detect ac4C at positions conserved between at least two species and assign it with the relevant misincorporation rates as calculated using ac4C-seq across all samples. This dataset was used for archaea conservation-related analysis presented in the main text.
Phylogenetic tree
A phylogenetic tree for the archaea analysed by ac4C-seq was generated using the default parameters of phyloT tree generator (https://phylot.biobyte.de) based on the following NCBI taxonomy IDs: T. kodakarensis, 69014; T. sp. AM4, 246969; P. furiosus, 1185654; S. solfataricus, 555311 and M. jannaschii, 2190.
Comparison between ac4C sites in T. kodakarensis rRNAs as measured by ac4C-seq and LC–MS
A total of 172 ac4C sites at CCG motifs were identified in T. kodakarensis rRNA under the full set of comparisons detailed in Supplementary Table 2. Although LC–MS identified a total of 146 potential ac4C sites, only 25 of these could be uniquely assigned to specific positions within the ribosome, owing to redundancies in the oligo sequences identified in the LC–MS. All comparisons of ac4C between the methods were therefore conducted on a subset of these 25 sites.
Northern blot analysis of ac4C in archaeal total RNA
Immuno-northern blots were performed using Ambion NorthernMax reagents (Thermo Fisher Scientific). The amount of RNA used was dependent upon sample type, with 15 μg used for analysis of human and yeast total RNA, and 3 μg used for hyperthermophilic archaea. Equal amounts of RNA were mixed together with 1 vol of NorthernMax-Gly Sample Loading Dye (Thermo Fisher Scientific), incubated at 65 °C for 30 min, and separated on a 1% agarose-1X Glyoxal Gel prepared using 10X NorthernMax-Gly Gel Prep/Running Buffer (Thermo Fisher Scientific). Gels were run at 75 V for approximately 70 min, or until the dye front had migrated about 3 inches (7.3 cm). Loading controls were analysed by UV-imaging of ethidium bromide before transfer. RNA was transferred onto Amersham Hybond-N+ membranes (GE Healthcare) using a downward capillary method. After transfer, membranes were crosslinked three times at 150 mJ/cm2 in a UV254nm Stratalinker 2400 (Stratagene). Membranes were then blocked in a solution of blocking buffer (5% non-fat milk in 0.1% TBST) for 1 h at room temperature and washed 3 times at 5 min each in 0.1% TBST. Membranes were then incubated overnight at 4 °C with the anti-ac4C antibody (1:10,000 dilution, Abcam) in blocking buffer. Membranes were washed 3 × 5 min in 0.1% TBST and then incubated with HRP-conjugated secondary anti-rabbit IgG in 5% non-fat milk in 0.1% TBST at room temperature for 2 h. Membranes were washed 3 times at 10 min each in 0.1% TBST. SuperSignal ELISA Femto Maximum Sensitivity Substrate reagent (Thermo Fisher Scientific) was added directly to the membrane and signal was detected via chemiluminescent imaging. Typical exposure times ranged from 2 to 20 min depending on the concentration of individual RNA samples. We found that for hyperthermophilic archaea a 2-min exposure time was optimal, but yeast and human RNA required a 15- to 20-min exposure time to yield optimal results.
LC–MS analysis of ac4C in total RNA
For assessment of cellular ac4C levels by LC–MS, total RNA was analysed using a similar method as previously described54. In brief, before UHPLC–MS analysis, 2,000 ng of each oligonucleotide was treated with 0.5 pg/μl of internal standard (IS), isotopically labelled guanosine, [13C] [15N]G (Cambridge Isotope Laboratories). The enzymatic digestion was carried out using Nucleoside Digestion Mix (NEB) according to the manufacturer’s instructions. Finally, the digested samples were lyophilized and reconstituted in 100 μl of RNase-free water, 0.01% formic acid before UHPLC–MS/MS analysis. The UHPLC–MS analysis was accomplished on a Waters XEVO TQ-STM (Waters Corporation) triple quadruple mass spectrometer equipped with an electrospray source (ESI) source maintained at 150 °C and a capillary voltage of 1 kV. Nitrogen was used as the nebulizer gas, which was maintained at a pressure of 7 bar, a flow rate of 500 l/h and a temperature of 500 °C. UHPLC–MS/MS analysis was performed in ESI positive-ion mode using multiple-reaction monitoring (MRM) from ion transitions (m/z 286.16 > 154.07 and m/z 286.16 > 112.06) previously determined for ac4C54. A Waters ACQUITY UPLCTM HSS T3 guard column, 2.1 × 5 mm, 1.8 μm, attached to a HSS T3 column, 2.1 × 50 mm, 1.7 μm were used for the separation. Mobile phases included RNase-free water (18 MΩ/cm) containing 0.01% formic acid (Buffer A) and 50:50 acetonitrile in Buffer A (Buffer B). The digested nucleotides were eluted at a flow rate of 0.5 ml/min with a gradient as follows: 0–2 min, 0–10%B; 2–3 min, 10–15% B; 3–4 min, 15–100% B; 4–4.5 min, 100% B. The total run time was 7 min. The column oven temperature was kept at 35 °C and the sample injection volume was 10 μl. Three injections were performed for each sample. Data acquisition and analysis were performed with Waters software MassLynx V4.1 and TargetLynx. Calibration curves were plotted using linear regression with a weight factor of 1/x.
Preparation of RNase digests and direct nanoflow LC–MS and tandem MS of rRNA fragments
rRNAs were extracted from purified T. kodakarensis 70S ribosomes. An aliquot of the sample (100 μl, 1 mg/ml) was mixed with 800 μl of ISOGEN reagent (Nippon Gene) and passed 100 times through a 23–gauge needle. The sheared sample was mixed with 200 μl of chloroform and centrifuged at 10,000g for 15 min at 4 °C. The resulting upper phase (around 500 μl) was mixed with a glycogen solution (0.5 μl, 20 mg/ml) and isopropanol (500 μl) and centrifuged to yield rRNAs as a precipitate. The precipitate was dissolved in RNase free water and stored at −80 °C until further use. The three rRNA classes (5S, 16S and 23S) were separated by reversed-phase LC through a PLRP-S 4,000 Å column (4.6 × 150 mm, 10 μm, Agilent Technologies). After applying around 10 μg total RNA to the column, the rRNAs were eluted with a 60-min linear gradient of 12–14% (v/v) acetonitrile in 100 mM TEAA, pH 7.0, 0.1 mM diammonium phosphate at a flow rate of 200 μl/min at 60 °C while monitoring the absorbance of the eluate at 260 nm55.
RNA (around 50 ng) was digested with RNase T1 (20 ng) in 100 mM triethylammonium acetate buffer (pH 7.0) at 37 °C for 1 h. The RNA fragments were separated using a direct nanoflow LC–MS system as described56,57. In brief, the digests were injected onto a reversed-phase Develosil C30-UG tip column (150 μm i.d. × 120 mm, 3-μm particle size; Nomura Chemical Co.) equilibrated with solvent A (10 mM TEAA, pH 7, in water:methanol, 9:1). Samples were eluted at 100 nl/min with a 60-min 0–24.5% linear gradient of solvent B (10 mM TEAA, pH 7:acetonitrile, 60:40). The column was subsequently washed with 70% B for 10 min and re-equilibrated with A.
Each LC eluate was sprayed online at −1.4 kV with the aid of a spray-assisting device57 into a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific) operating in the negative ion mode and in the data-dependent mode to automatically switch between MS and tandem MS acquisition. Full-scan mass spectra (m/z = 480–1,980) were acquired at a mass resolution of 350,000. At most, the five most intense peaks, (>100,000 counts per second with a 60-ms maximum injection time), were isolated within a 3-m/z window for fragmentation. Precursors were fragmented by switching to a higher energy collision-induced dissociation mode with a normalized collision energy of 20 or 50%. To retain mass resolution and to increase spectral quality, three tandem mass spectral micro-scans were acquired for each sample. A fixed starting value of m/z = 100 was set for each tandem mass spectrum.
Interpretation of the tandem mass spectra and quantification of modifications
Ariadne58 (http://ariadne.riken.jp/) was used for assignment of the tandem mass spectral peaks in conjunction with the sequence of rRNAs of T. kodakarensis (Gene ID: 3253116, 3253120 and 3253121). The Ariadne search parameters were: the maximum number of missed cleavages was one; two methylations per RNA fragment at any residue position were allowed; an RNA mass tolerance of ±20 ppm and a tandem spectral tolerance of ±50 ppm were allowed.
The quantification of post-transcriptional modification (PTM) was performed by the peak-area-based method. The target oligonucleotide peaks were obtained from extracted-ion chromatograms with their theoretical mass values (±5 ppm). Each peak area was measured using the Xcalibur software (Thermo Fisher Scientific) including a manual determination of the start-end of the peak. The stoichiometry of PTM was calculated from the peak areas obtained by MS with the following equation, in which P and N refer to peak areas of the oligonucleotide with or without PTM, respectively. Stoichiometry (%) = 100 × P/(P + N).
Proteomic analysis of T. kodakarensis
Proteins isolated from cultures of T. kodakarensis were precipitated by trichloroacetic acid (TCA) and washed twice with cold acetone before digestion for MS analysis. In brief, TCA-precipitated proteins were resuspended in 100 mM Tris pH 8.5 containing 8 M urea. Cysteine residues were reduced by 5 mM TCEP for 30 min at room temperature and further modified by 2-chloroacetamide for 30 min in the dark at room temperature. Proteins were first digested by recombinant Lys-C (Promega) overnight at 37 °C with shaking. The urea was diluted to 2 M before additional digestion overnight at 37 °C by the addition of trypsin at a ratio of 1:100 enzyme to substrate (Promega). The digestion reaction was quenched with the addition of formic acid to 5% final concentration. Peptides were quantified by the Pierce Colorimetric Peptide Assay (Thermo Scientific) and diluted in buffer A (5% acetonitrile (ACN), 0.1% formic acid (FA)) such that 1 μg was analysed per technical replicate. Each sample was trapped on an Acclaim PepMap 100 C18 column (5 μm particles 0.3 mm × 5 mm, Thermo Scientific) using the Ultimate 3000 autosampler (Dionex). Using chromatography conditions previously optimized59, peptides were separated on an in-house-packed reverse phase chromatography column (1.9 μm particles (ReproSil, Dr. Maish), 75 μm × 20 cm), directly interfaced to a QExactive Plus (QE+) mass spectrometer (Thermo Scientific). Peptides were eluted over a quick gradient from 2–7% buffer B (80% ACN, 0.1% formic acid) in 10 min before the gradient was gradually increased to 40% buffer B over 6 h before ramping to 95% B in 15 min. The flow was kept at 95% B for 15 min before 20 min of re-equilibration at 2% B before the next injection. Flow rate was 180 nl/min. The application of a 2.5 kV distal voltage electrosprayed the eluting peptides directly into the QE+ mass spectrometer equipped with the Nanospray Flex source (Thermo Scientific). Full MS spectra were recorded on the eluting peptides at a resolving power of 70,000 over a 400 to 1,600 m/z range, followed by higher energy dissociation fragmentation at 30% normalized collision energy on the 15 most intense ions selected from the full MS spectrum. MS2 spectra were collected in the Orbitrap at a resolving power of 17,500. Dynamic exclusion was enabled for 30 seconds60. Mass spectrometer scan functions and HPLC solvent gradients were controlled by the XCalibur data system (Thermo Scientific).
RAW files were extracted into .ms2 file format61,62 using RawDistiller v. 1.0, in-house developed software61. RawDistiller D(g, 6) settings were used to abstract MS1 scan profiles by Gaussian fitting and to implement dynamic offline lock mass using six background polydimethylcyclosiloxane ions as internal calibrants61. MS/MS spectra were first searched using ProLuCID63 with a 10 ppm mass tolerance for peptide and 25 ppm tolerance for fragment ions. Trypsin specificity was imposed on both ends of candidate peptides during the search against a protein database containing 2,301 T. kodakarensis proteins (NCBI 2018–11-09 release), as well as 386 usual contaminants such as human keratins, IgGs and proteolytic enzymes. To estimate false discovery rates (FDR), each protein sequence was randomized (keeping the same amino acid composition and length) and the resulting ‘shuffled’ sequences were added to the database, for a total search space of 5,440 amino acid sequences. A mass of 15.9949 Da was differentially added to methionine residues.
DTASelect v.1.964 was used to select and sort peptide/spectrum matches (PSMs) passing the following criteria set: PSMs were only retained if they had a DeltCn of at least 0.08; minimum XCorr values of 1.0 for singly-, 1.4 for doubly-, and 2.1 for triply-charged spectra; peptides had to be at least 7 amino acids long. Results from each sample were merged and compared using CONTRAST64. Combining all replicate injections, proteins had to be detected by at least 2 peptides and/or 2 spectral counts. Proteins that were subsets of others were removed using the parsimony option in DTASelect on the proteins detected after merging all runs. Proteins that were identified by the same set of peptides (including at least one peptide unique to such protein group to distinguish between isoforms) were grouped together, and one accession number was arbitrarily considered as representative of each protein group.
NSAF765 was used to create the final reports on all detected peptides and non-redundant proteins identified across the different runs. Spectral and protein level FDRs were, on average, 0.17 ± 0.05% and 0.18 ± 0.05%, respectively.
Cryo-EM data acquisition and analysis
3.5 μl of 70S ribosome sample (0.25 mg/ml for the wild-type strains grown at 85 °C and 65 °C, and 0.4 mg/ml for the ac4C-deficient strains) was applied on glow-discharged holey carbon grids (Quantifoil R2/2) coated with a thin layer of continuous carbon film. Grids were blotted (3 s) and plunge-frozen using a Vitrobot Mark IV (FEI, Thermo Fisher Scientific). Micrographs were recorded at liquid nitrogen temperature on a Titan Krios electron microscope (FEI, Thermo Fisher Scientific) operating at 300 kV and equipped with a Falcon 3 direct electron detector (FEI, Thermo Fisher Scientific). Nominal magnification used was 96K and corresponded to a calibrated pixel size of 0.85 Å per pixel, with a dose rate of approximately 1.16 e−/Å2/s and defocus values ranging from −0.5 to −1.5 μm. Automatic data acquisition was performed using EPU (FEI, Thermo Fisher Scientific) and yielded a total of 2,509 micrographs for the WT85 (wild-type grown at 85 °C), 3,115 for the WT65 and 4,211 for the mutant. Micrographs were processed using MotionCor266 to correct for patched frame motion and dose-weighting and contrast transfer function parameters were estimated by CTFFIND 4.167,68. Particle picking, extraction and classifications were performed using Relion 3.069. The 60-Å low-pass-filtered cryo-EM map of the P. furiosus ribosome (EMD-2009) was used as an initial reference and has been used for further particle classification in 3D. Final maps reconstructed from 53,737, 283,424 and 116,586 particles for the WT85, WT65 and mutant strains, respectively, were obtained through multibody refinement with the LSU, the SSU body and SSU head masked individually as demonstrated in Extended Data Fig. 7a69. Density maps were corrected for the modulation transfer function of the detector, and then sharpened by applying a negative B-factor that was estimated using automated procedures in Relion370. Averaged map resolutions were 2.95 Å, 2.55 Å and 2.65 Å for the WT85, WT65 and TkNAT10, respectively and were determined using the gold-standard FSC = 0.143 criterion as implemented in Relion3 and M-triage as implemented in Phenix71 (Extended Data Fig. 7b–d). Local resolutions were estimated using Resmap72 (Extended Data Fig. 8a).
Model building and refinement
The initial template of T. kodakarensis ribosome was derived from the cryo-EM model of P. furiosus ribosome (PDB 4V6U). The model was docked into the electron microscopy density maps using UCSF Chimera73, followed by iterative manual building in Coot74. Coordinates and library files for the modified residues were generated through phenix.elbow75 and were manually docked into the relevant positions using Coot followed by real-space refinement. The final model was subjected to global refinement and minimization in real space using phenix.real_space_refine in Phenix71. MolProbity76 was used to evaluate model geometry. The final refinement parameters are provided in Supplementary Table 6, and map versus model diagrams are in Extended Data Fig. 7e–g. Examples of the ac4C model in density view are shown in Extended Data Fig. 8b.
Biophysical characterization of ac4C containing hairpins
In vitro transcription was performed using the NEB Highscribe T7 highyield RNA synthesis kit according to the manufacturer’s instructions using DNA templates containing a T7 promoter upstream of a template sequence (Supplementary Table 1a). For ac4C-containing transcripts, CTP was replaced in the reaction mixture with ac4CTP (50 mM). Crude in vitro transcription reactions were purified by denaturing polyacrylamide gel electrophoresis (PAGE). Full length product bands were visualized by UV shadowing and excised with a razor blade. RNA was extracted by crushing the gel slices and shaking in 500 mM ammonium acetate with 0.2 mM EDTA pH 8.0. RNA was desalted by four sequential rounds of dilution and concentration in a 1K MWCO centrifugal ultrafiltration device. Before use in DSC and circular dichroism experiments, purified RNAs were analysed for purity by denaturing PAGE and visualized using SYBR Gold Nucleic Acid Gel Stain from Invitrogen. DSC experiments were carried out on a VP-DSC instrument (Microcal). Desalted PAGE purified helix-45 oligos were diluted to 18 μM in 1X Oligo DSC buffer (10 mM phosphate buffer, 50 mM NaCl) and folded by heating to 95 °C for 10 min and rapidly cooled by placing on ice for 10 min. Samples were vacuum degassed with stirring for 8 min at 35 °C. DSC was equilibrated with 550 μl freshly degassed 1x Oligo DSC buffer in sample and reference cells through multiple scan cycles until a stable and flat differential heat flow curve was established. During downscanning, the sample cell was emptied, and 550 μl freshly degassed helix-45 hairpins were loaded between 40 °C and 35 °C. Samples were equilibrated at 35 °C for 15 min and calorimetric data was collected from 35 °C to 120 °C at a scan rate of 1 °C/min. Raw DSC data from each scan was processed by linear baseline subtraction and the absolute value of each baseline was adjusted to allow curves to be observed on a single plot. Melting temperatures were calculated as the mean value of the local maxima of the major transition on each scan (n = 3) and errors were calculated as the standard deviation. CD analyses were performed on a JASCO J-1500 CD Spectrometer using a 1 mm pathlength quartz cuvette. In brief, desalted helix-45 oligos were diluted to 5 μM in 1X melting buffer (100 mM NaCl, 1.97 mM KCl, 0.1 mM EDTA and 8.7 mM sodium phosphate (pH 7.4)) and folding by fast cooling. Denaturation curves were recorded by monitoring the change in ellipticity at 260 nm while the temperature was increased from 30 °C to 95 °C at a rate of 2 °C/min. The minimum points in the first-derivative curves of CD melting spectra were recorded (n = 3) and errors were calculated as standard deviation.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Ac4C-seq datasets generated in this manuscript have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE135826. The cryo-EM density maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD10223 and EMD-10503 for the wild-type strain grown at 85 °C and 65 °C, respectively, and EMD-10224 for the TkNAT10-deletion strain. Model coordinates have been deposited in the Protein Data Bank (PDB) under accession numbers 6SKF, 6TH6 and 6SKG. Mass spectrometry proteomics data has been deposited to the ProteomeXchange Consortium via Pride77,78 partner repository with the dataset identifier PXD014814.
Code availability
Code for the analyses described in this paper is available from the corresponding author upon request.
Extended Data
Extended Data Fig. 1 |. An optimized reaction for sequencing of N4-acetylcytidine in RNA.
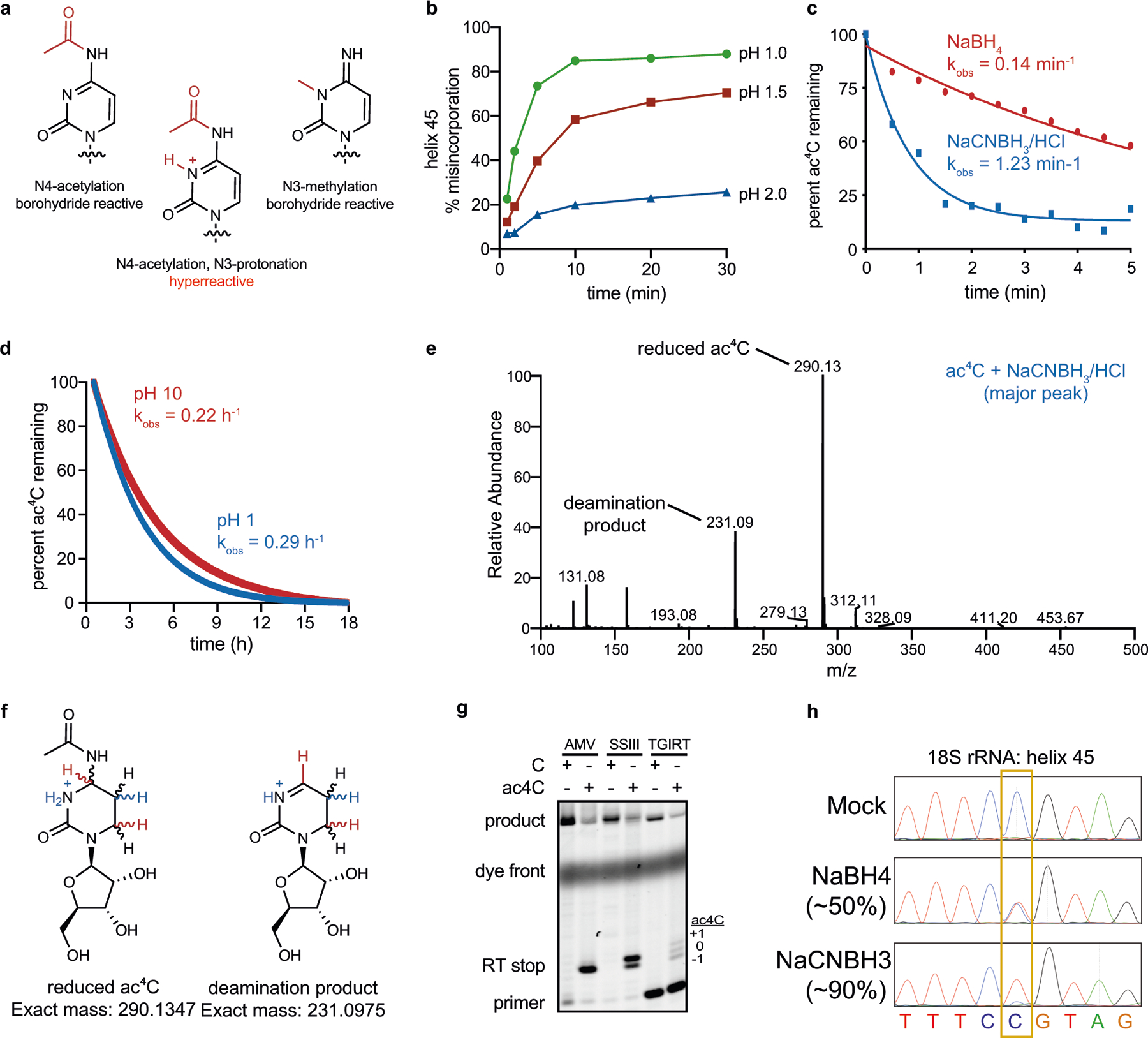
a, Protonation under acidic conditions hyperactivates ac4C, increasing its reactivity with NaCNBH3. Efficient reduction manifests as quantitative misincorporation of deoxynucleotide triphosphates at ac4C upon reverse transcription. b, NaCNBH3-dependent misincorporation at the known ac4C site in human helix 45 is increased at more acidic pH. The percentage misincorporation at ac4C sites after chemical reduction, reverse transcription and PCR was quantified from Sanger sequencing data. One independent experiment. c, Kinetic analysis of ac4C reduction. Reaction progress was assessed by monitoring the disappearance of ac4C absorbance at 300 nm in the presence of first and second-generation hydride donors. Reaction conditions: ac4C (0.1 mM, free nucleoside), reductant (20 mM), H2O. NaBH4 reactions were carried out at pH 10, whereas NaCNBH3 reactions were adjusted to pH 1 using HCl before initiation. Representative of 3 independent experiments. d, Kinetic analysis of the hydrolysis of ac4C at pH values used in NaBH4 (pH 10) and NaCNBH3 (pH 1) reduction reactions. Reaction progress was assessed by monitoring the disappearance of ac4C absorbance at 300 nm. Acid- and base-catalysed hydrolysis occurred at similar rates, and were slow compared to ac4C reduction by NaBH4 and NaCNBH3. Representative of 3 independent experiments. e, LC–MS/MS analysis confirms reduction of ac4C to reduced ac4C in the presence of NaCNBH3. Reaction conditions: ac4C (0.1 mM, free nucleoside), NaCNBH3 (20 mM), HCl pH 1. Representative of 2 independent experiments. f, Exact mass of reduced ac4C and deamination product observed in LC–MS/MS experiments. g, Primer extension analysis of ac4C-containing RNAs after NaCNBH3 treatment (100 mM, pH 1, 37 °C, 1 h). h, Sanger sequence traces of a known ac4C site in helix 45 of human HAP1 cells. C>T misincorporation is exclusively observed at the ac4C site in reduced (NaBH4 and NaCNBH3) but not in mock-treated samples. ac4C sites are highlighted in yellow.
Extended Data Fig. 2 |. Ac4C in eukaryotic cells with wild-type NAT10 expression.
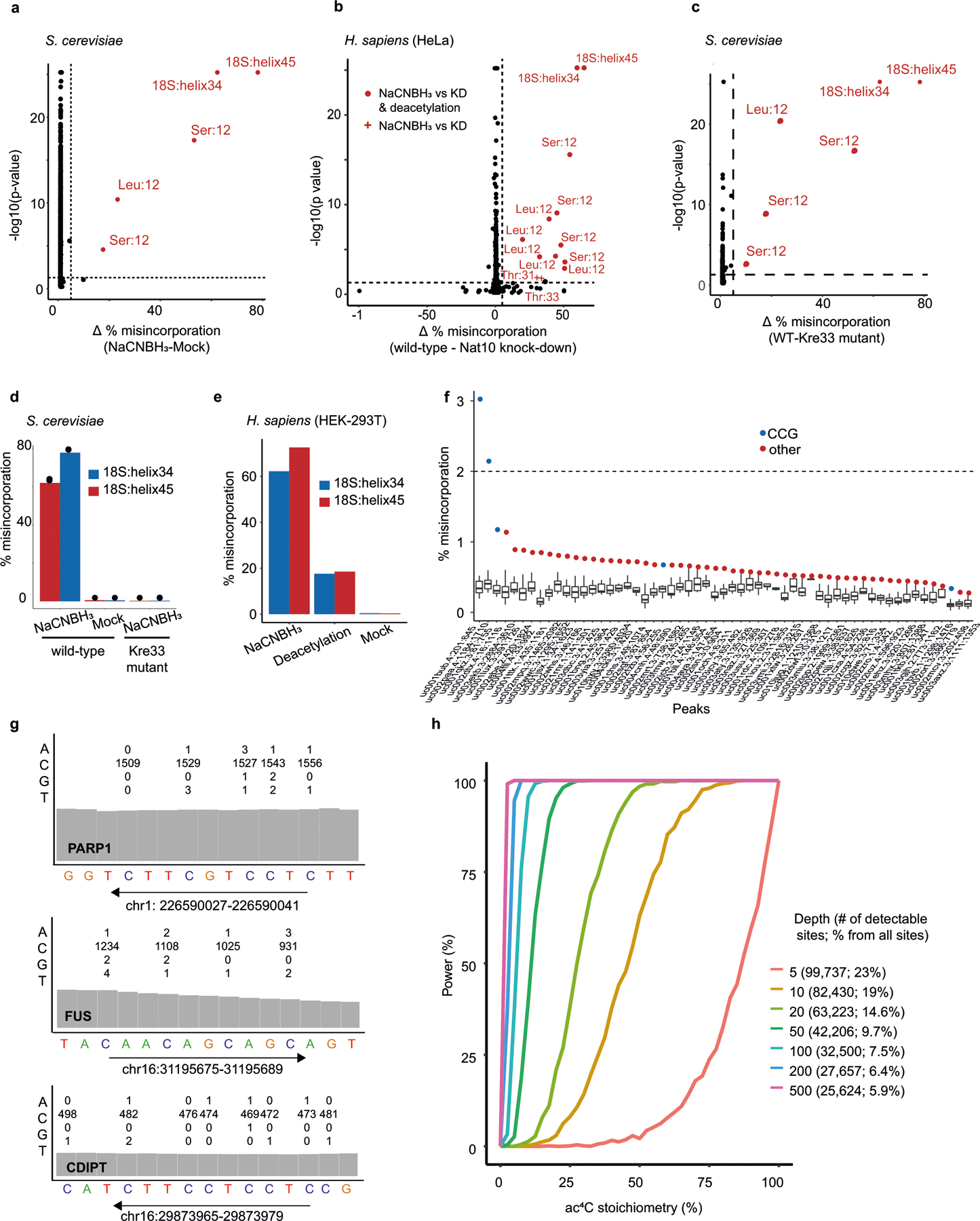
a–c, ac4C-seq was conducted on RNA from S. cerevisiae (a, c) and HeLa cells (b). Statistical significance from the χ2 test is plotted against the difference in misincorporation rates (corresponding to ac4C levels) between NaCNBH3-treated and mock-treated RNA from S. cerevisiae (a), RNA from wild-type and NAT10-depleted cells (b) or from wild-type S. cerevisiae cells and a strain expressing a catalytic mutant of Kre33 (c), treated with NaCNBH3. Sites with a differential misincorporation level >5% and a P value <0.05 are labelled and marked in red. For HeLa cells (b) an additional comparison between NaCNBH3 and deacetylation pre-treatment was conducted. Sites that do not pass significance under these conditions are marked with a plus sign (shown only for sites found significant between NaCNBH3 and mock treatment). Significant sites are labelled with the identity of the molecule and the relative position (or helix) of ac4C. n = 3 biologically independent samples for all but NAT10-depleted HeLa cells, in which case n = 2 biologically independent samples. d, e, Misincorporation level in the two known sites in 18S (helix 34 and helix 45), compared with controls in poly(A)-enriched RNA from wild-type S. cerevisiae cells and S. cerevisiae cells expressing a catalytic mutant of Kre33 (d) and from wild-type HEK-293T cells and HEK-293T cells overexpressing NAT10 and THUMPD1 (e). f, g, ac4C-seq data from poly(A)-enriched RNA from HEK-293T cells overexpressing NAT10 and THUMPD1 on ‘ac4C peaks’ that have been identified previously10 as harbouring ac4C. f, Distribution of misincorporation across each of 57 ‘ac4C peaks’ that had a coverage of more than 400 reads in more than 80% of the cytosines in the peak. For each peak the cytosine harbouring the highest misincorporation rate is indicated in colour, presented in blue if it harbours a CCG motif and red otherwise. g, Traces from the Integrative Genomic Viewer (IGV) browser of three such genes, with highest coverage in the ac4C-seq data. For each gene the 15 bases motif identified in ref. 10 is presented. The numbers above each cytidine indicate the number of bases (A, C, G and T) observed in our data at that position. h, Power analysis for ac4C detection, as a function of sequencing depths and stoichiometries. Each data point in each curve is based on 1,000 simulations. For each sampled depth, numbers in the legend indicate the sequencing depth, which was kept identical for treatment and control samples. In addition, the legend indicates the number of CCG sites found in wild-type HEK-293T samples that have such a minimal depth and the percentage of these detectable CCG sites from all CCG sites in the transcriptome.
Extended Data Fig. 3 |. Ac4C in eukaryotic cells with manipulated NAT10 expression.
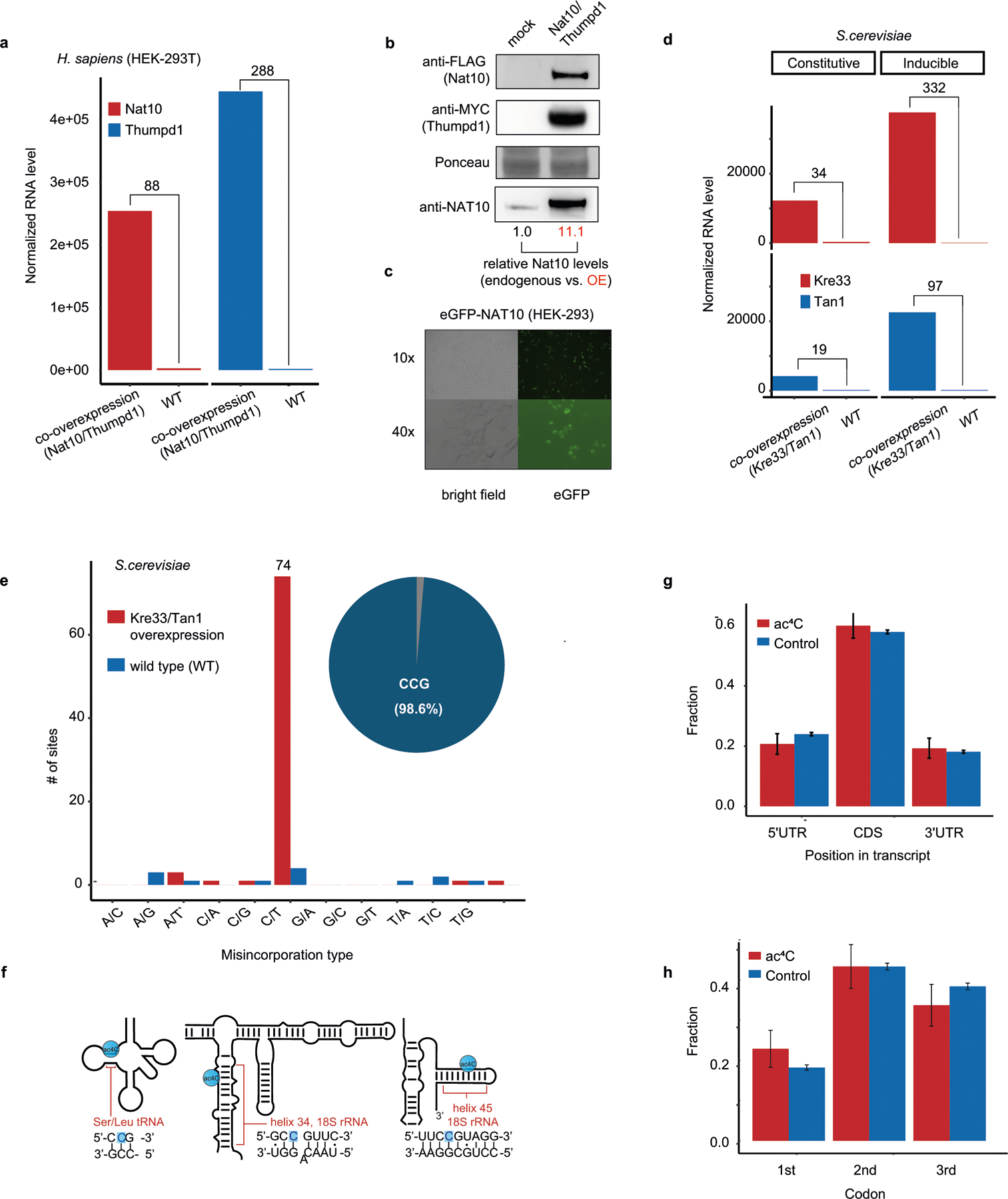
a, RNA expression of NAT10 and THUMPD1 in HEK-293T cells overexpressing both genes compared to wild-type cells. Shown are TMM-normalized read counts. The numbers above the bars indicate fold increase compared with the wild type. b, Immunoblotting analysis of NAT10 and THUMPD1 overexpression in HEK-293T cells. Representative of 3 independent experiments with similar results. For gel source data, see Supplementary Data 3. c, Microscopy images of the eGFP–NAT10 construct, confirming nuclear and nucleolar localization of ectopically expressed N-terminally tagged protein. Representative of 3 independent experiments with similar results. d, RNA expression of Kre33 and Tan1 in wild-type yeast cells and in cells stably overexpressing Kre33 and either stably or inducibly overexpressing Tan1. The numbers above the bars indicate fold increase from the wild type. e, The number of sites displaying each of the 12 possible misincorporation patterns are displayed (bar plot, y axis) for sites found in poly(A)-enriched RNA from both wild-type S. cerevisiae cells and S. cerevisiae cells overexpressing both Kre33 and Tan1. The pie chart displays the proportion of sites harbouring C>T misincorporations that were embedded within a CCG motif (73 out of 74, 98.6%). f, Schematic of the known ac4C sites in human tRNAs (Leu and Ser) and in helix 34 (C1337) and helix 45 (C1842) of human 18S rRNA. The acetylated cytidine residue (highlighted in blue) is embedded within a CCG motif in all known sites. g, Fraction of ac4C sites found within the 5′ UTR, CDS and 3′ UTR (CDS, coding sequence; UTR, untranslated region). Results are shown for the set of ac4C sites in mRNA of HEK-293T cells overexpressing NAT10 and THUMPD1 (red bars, n = 139), and—as controls—for all CCG motifs present within all genes within which any ac4C was found (blue bars, n = 6,129). Error bars representing standard distribution of the binomial distribution. Data are based on 2 biologically independent samples. h, Fraction of ac4C sites at the first, second and third position of each codon, shown for ac4C sites and controls as in g. Data are mean ±s.d. of the binomial distribution and are based on two biologically independent samples.
Extended Data Fig. 4 |. Sequence and structure requirements for deposition of ac4C.
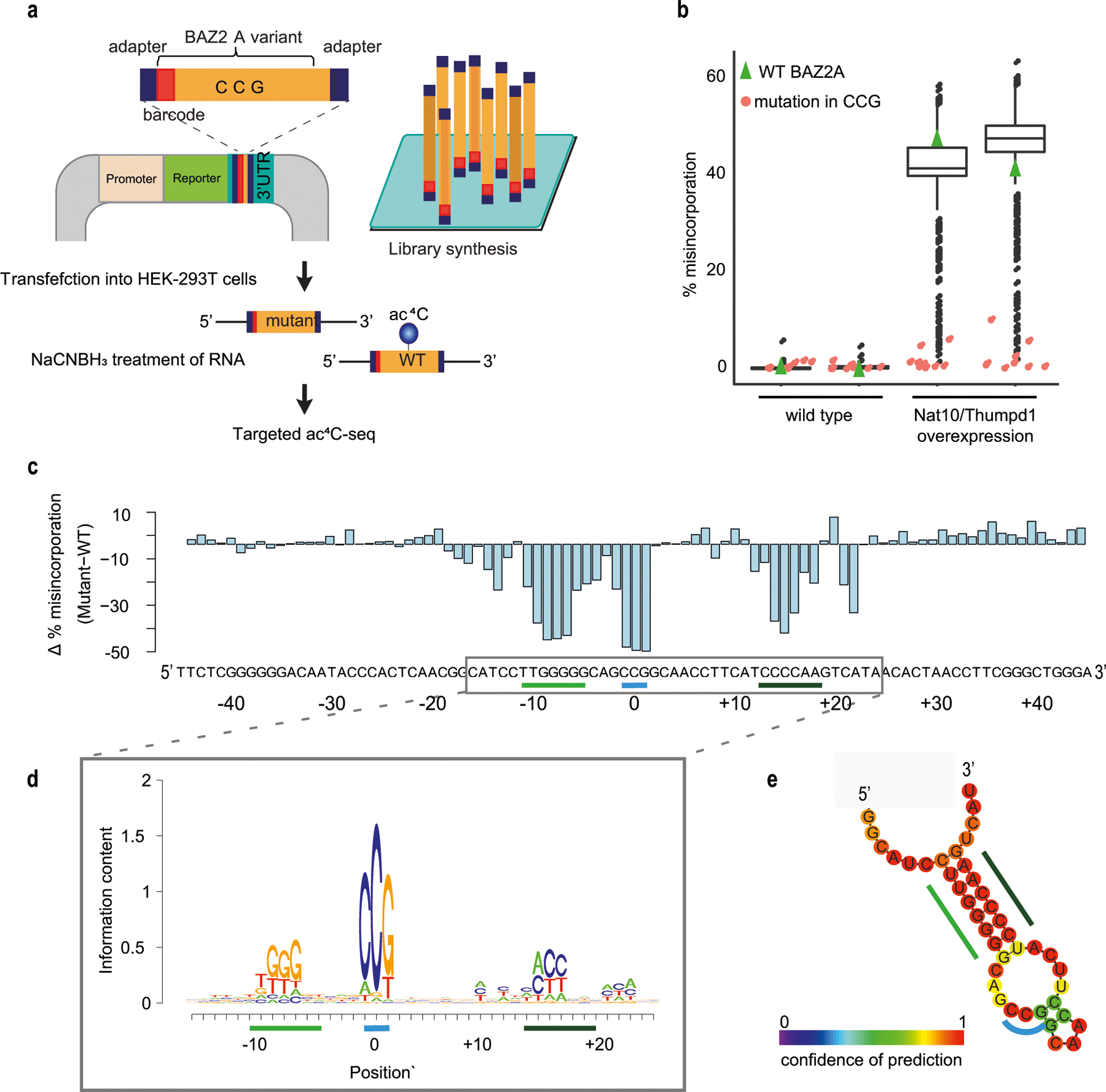
a, Oligonucleotides representing the wild-type sequence surrounding the acetylated site in BAZ2A mRNA, or variants with single mutations across the wild-type sequence, were synthesized as a pool and cloned into the 3′UTR of a reporter gene. The pool of plasmids was transfected into wild-type HEK-293T cells or cells transiently overexpressing NAT10 and THUMPD1. RNA extracted from cells was subjected to targeted ac4C-seq and ac4C levels were estimated on the basis of misincorporation rates. b, Misincorporation rate of oligonucleotides described in a, harbouring the wild-type sequence of BAZ2A (green triangles) or a sequence mutated at the CCG motif and at its surrounding bases (red and black, respectively). Box plot visualization parameters are as in Fig. 1h. n = 2 biologically independent samples. c, The difference in misincorporation rate of oligonucleotides with a single base mutation compared with the wild-type oligonucleotide is shown across all positions of the construct. d, De novo construction of the motifs surrounding the modified cytidine were built on the basis of the contribution of single-base mutations in the BAZ2A sequence to the reduction in misincorporation rate compared to wild-type BAZ2A sequence. e, Secondary structure of the BAZ2A mRNA fragment as predicted by RNAfold. Bases are colour-coded according to confidence level of the prediction. Regions highlighted by a blue and green line in c–e represent the CCG motif and a stem structure surrounding the modified cytidine, respectively.
Extended Data Fig. 5 |. Deletion of TkNAT10 and TkTHUMPD1 in T. kodakarensis.
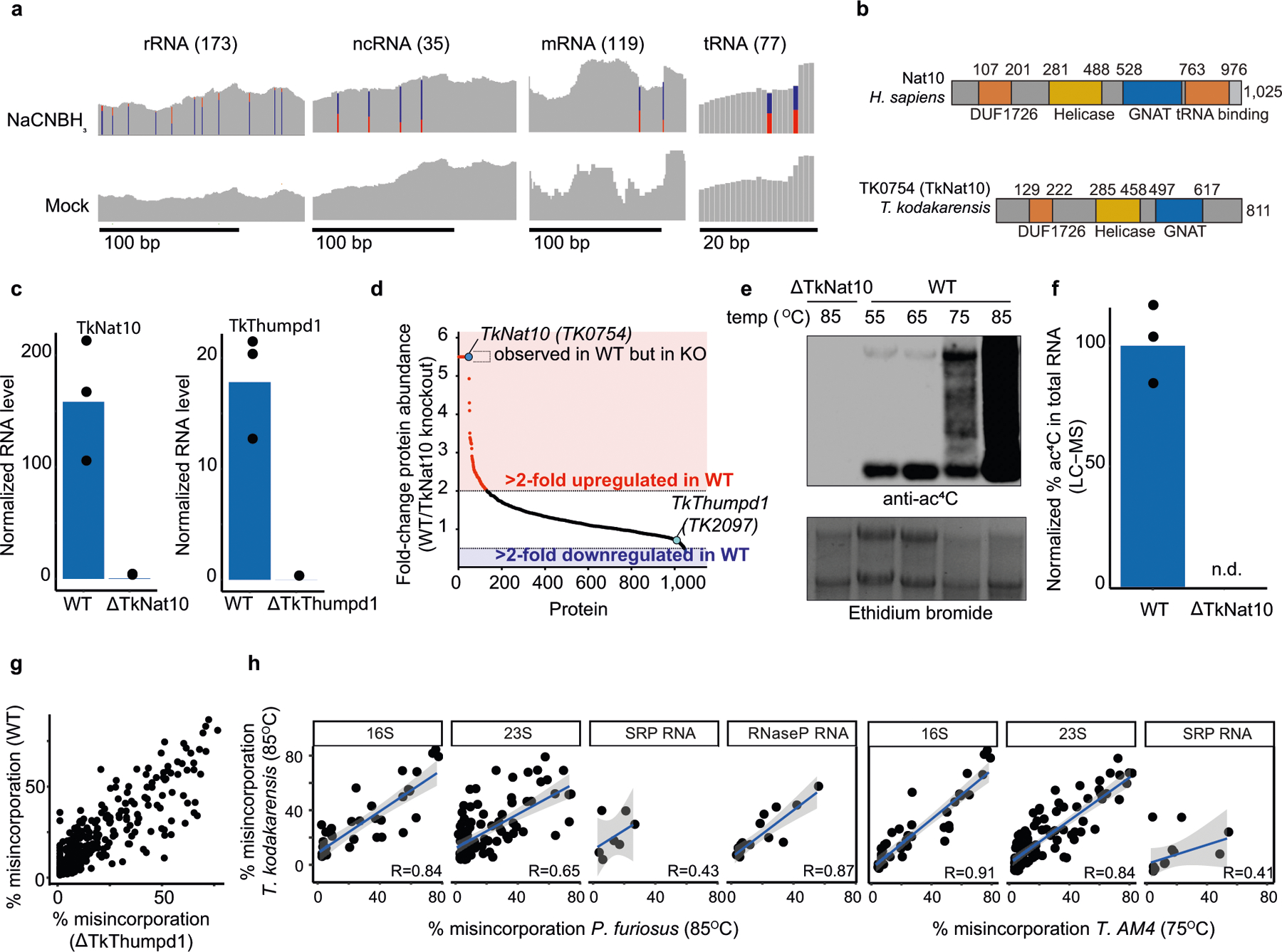
a, Total RNA from T. kodakarensis was analysed via ac4C-seq. IGV browser traces display representative ac4C sites in rRNA, ncRNA, mRNA and tRNA of T. kodakarensis, visualized as in Fig. 1c. The number in parentheses indicates the number of sites identified for each class of molecules. b, Conserved domain architecture of human NAT10 and its homologue in T. kodakarensis, TK0754 (referred to as TkNAT10 in the text). c, Expression of TkNAT10 and TkTHUMPD1 (TK2097) in wild-type T. kodakarensis and the indicated deletion strains was quantified from ac4C-seq data. Shown are mean TMM-normalized values (n = 3 and 2 biological replicates in wild-type and deletion strains, respectively). d, Quantitative LC–MS/MS proteomics analysis of wild-type and ΔTkNAT10 T. kodakarensis. Fold-change in protein abundance was based on comparison of distributed normalized spectral abundance factor for individual proteins. Fold-change for proteins detectable exclusively in the wild-type or the knockout (KO) condition (fold-change = ∞) are graphed at 5.5 and 0.1, respectively, which represents the maximum and minimum of measured values. n = 3 LC–MS/MS runs for each condition. e, Anti-ac4C immuno-northern blot in T. kodakarensis total RNA. Ethidium bromide staining is used to visualize total RNA. Results are representative of two biological replicates. For gel source data, see Supplementary Data 3. f, Relative quantification of ac4C in total RNA isolated from wild-type and ΔTkNAT10 T. kodakarensis strains as measured by LC–MS. Mean of n = 3 technical replicates. n.d., not detectable. g, Scatter-plot depicting misincorporation rate of ac4C sites in wild-type T. kodakarensis is compared with the TkTHUMPD1-deletion strain, showing no effect of the deletion of the gene on the ac4C status. h, Correlation between misincorporation rates in T. kodakarensis compared to P. furiosus and T. sp. AM4 for the different types of ncRNAs identified by ac4C-seq. The Pearson’s correlation coefficient is indicated at the bottom of each plot. n = 4 and 1 independent biological samples for T. kodakarensis and other archaea, respectively. Shading indicates 95% confidence intervals for predictions from a linear model.
Extended Data Fig. 6 |. Ac4C accumulates in a temperature-dependent manner across all RNA species in archaea.
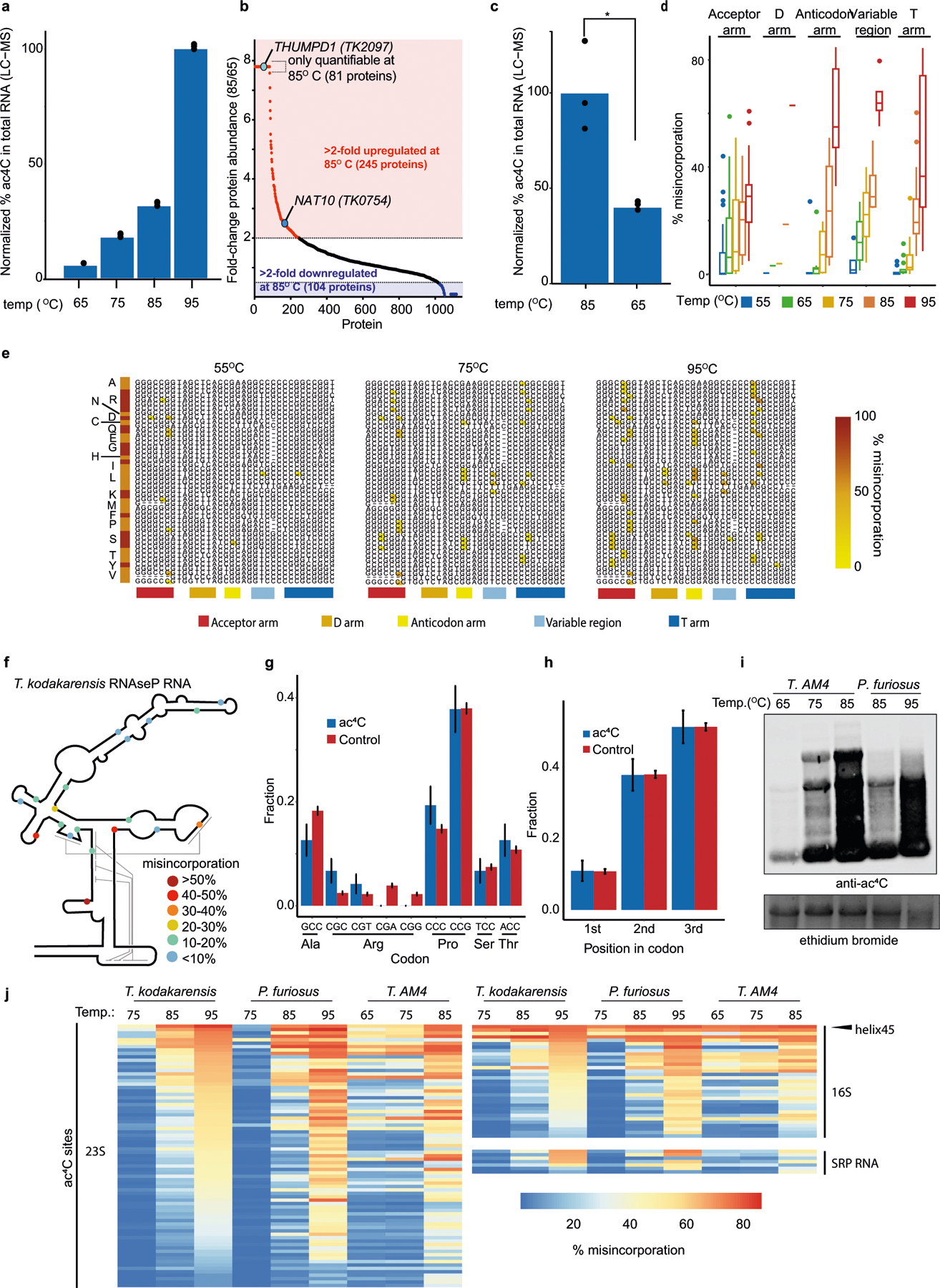
a, Relative quantification of ac4C in total RNA isolated from wild-type T. kodakarensis as a function of temperature as measured by LC–MS. Mean is shown along with individual data points. n = 3 technical replicates. For 65, 75, 85 °C: representative of 2 independent experiments, for 95 °C: 1 experiment. b, c, Quantitative LC–MS/MS proteomics analysis of T. kodakarensis temperature-dependent protein expression. Fold-change in T. kodakarensis protein abundance between growth conditions at 85 °C and 65 °C was based on comparison of distributed normalized spectral abundance factor for individual proteins. Fold-change for proteins detectable exclusively in the 85 °C or 65 °C condition (fold-change = ∞) was set at 7.8 and 0.1, respectively, which represents the maximum and minimum of measured values. n = 3 LC–MS/MS runs for each condition. Student’s t-test, paired, two tailed P = 0.012. d, Misincorporation rates of ac4C sites at distinct regions of T. kodakarensis tRNAs as a function of growth temperature (55–95 °C), segregated into distinct regions within the tRNA molecule. Only sites with a minimal stoichiometry of 5% in any sample are shown. Box plot visualization parameters are as in Fig. 1h. n = 4 biologically independent samples for 85 °C, n = 2 for 65 °C and 75 °C and n = 1 for 55 °C and 95 °C. e, Multiple alignment of 37 tRNA molecules, representing 19 distinct tRNAs in T. kodakarensis, plotted across three distinct temperatures. ac4C sites are coloured on the basis of misincorporation rate (see colour bar). The red–orange bar on the left segregates the aligned sequences into distinct tRNA molecules, identified by the single-letter abbreviation of their amino acid. Selected regions from the multiple alignment, where ac4C is particularly abundant, are shown and colour-coded according to the bottom colour bar. f, Schematic representation of RNaseP RNA in T. kodakarensis. ac4C sites (all in CCG) are marked with circles colour-coded by misincorporation rate measured in cells grown at 85 °C. Fine grey lines indicate regions that base pair in the folded structure of the molecule, according to the model in ref. 79. g, h, Distribution of 119 acetylated cytidine residues (in 86 mRNAs) in T. kodakarensis across different codons (g) and at specific position within codons (h) are shown, and compared to that of 2,245 control non-acetylated cytidines, found at CCG motifs of the same mRNAs. The y axis presents the fraction of cytidines in each position. n = 1 set of sites (comprising 119 ac4Cs and 2,245 Cs) with error bars representing standard deviation of the binomial distribution. i, Anti-ac4C immuno-northern blot in P. furiosus and T. sp. AM4 total RNA as a function of temperature. Ethidium bromide staining was used to visualize total RNA. Results are representative of two biological replicates. For gel source data, see Supplementary Data 3. j, A heat map showing misincorporation rates at conserved ac4C sites in 5S, 16S, 23S, RNase P RNA and SRP RNA of T. kodakarensis, P. furiosus and T. sp. AM4 grown at various temperatures. Rows are ordered according to misincorporation rates quantified in T. kodakarensis grown at 95 °C. The arrowhead indicates the conserved ac4C site at helix 45 (top site in the heat map).
Extended Data Fig. 7 |. Cryo-EM data processing and map reconstruction.
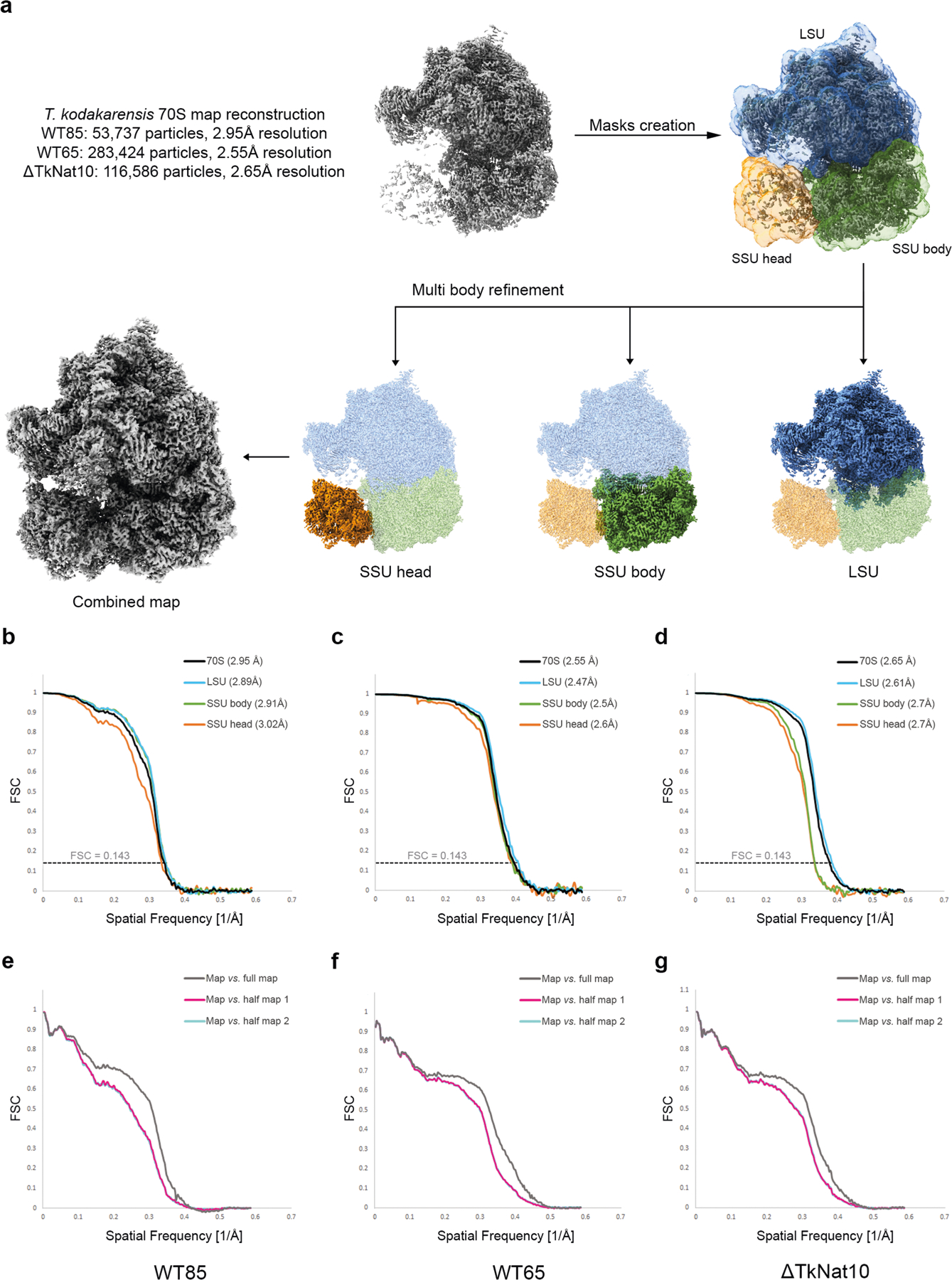
a, Schematic representation of electron microscopy data processing for the T. kodakarensis ribosomes. Data processing was performed in Relion 3 and included motion correction, contrast transfer function correction, particle picking and classification. Initial map reconstruction and post processing was performed by the 3D refinement algorithm implemented in Relion on the complete 70S particle, indicating high residual mobility of the SSU head domain (top left, grey). Further implementation of multibody refinement with individual masks prepared for the LSU (blue), SSU body (green) and SSU head (orange) resulted in the complete reconstruction of the 70S particle. The final map consisting of all three ribosomal domains for the wild-type ribosome derived from cells grown at 85 °C is presented at the bottom left. Fourier shell correlation curves indicating overall (black) and per domain (colour coded according to the relevant masks) resolutions are presented in b for the wild-type strain grown at 85 °C (WT85), c for the wild-type strain grown at 65 °C (WT65) and d for the ΔTkNAT10 ribosomes (mutant). FSC comparisons of full (grey) and half-maps (pink/cyan) to the final refined model are presented in e, f and g for WT85, WT65 and mutant strains, respectively. The excellent agreement between the cyan and pink curves indicates the lack of overfitting.
Extended Data Fig. 8 |. Cryo-EM data quality and ac4C visualization.
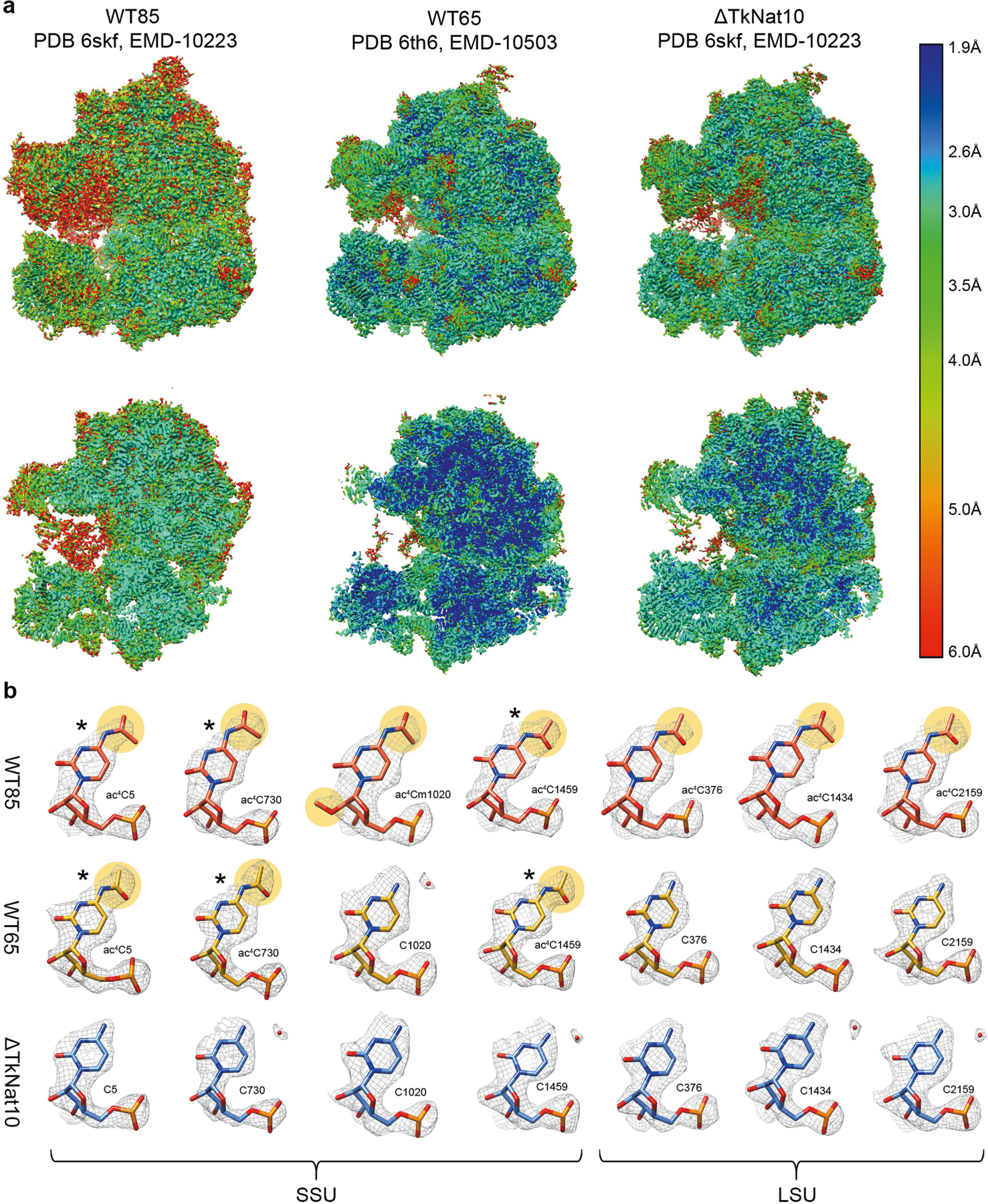
a, Surface (top) and cross-section (bottom) representations of the cryo-EM density maps coloured according to local resolution distribution. Growth conditions and T. kodakarensis strains used in the study along with PDB and EMDB accession codes are indicated. Resolution values are colour-coded according to the right index and are presented in Å. b, Model in density for multiple ac4C positions in wild-type T. kodakaresis grown at 85 °C (orange) and 65 °C (yellow) compared to an ac4C ΔTkNAT10 strain (mutant, blue) indicating the absence of acetate density (highlighted in light orange) in the mutant and in multiple positions of the strain grown at 65 °C. Positions highlighted with an asterisk are also acetylated in the 65 °C strain whereas positions that are unmarked are acetylated only in the archaea grown at 85 °C. These data are in good agreement with both the genomic sequencing and MS approaches described in this manuscript, that similarly indicate that ac4C distribution is highly dependent on growth temperature. A detailed list of ac4C distribution and comparison with other methods is provided in Supplementary Table 4. For a 2D map with ac4C distribution, see Extended Data Fig. 9d.
Extended Data Fig. 9 |. RNA modifications of T. kodakarensis ribosome and thermostability.
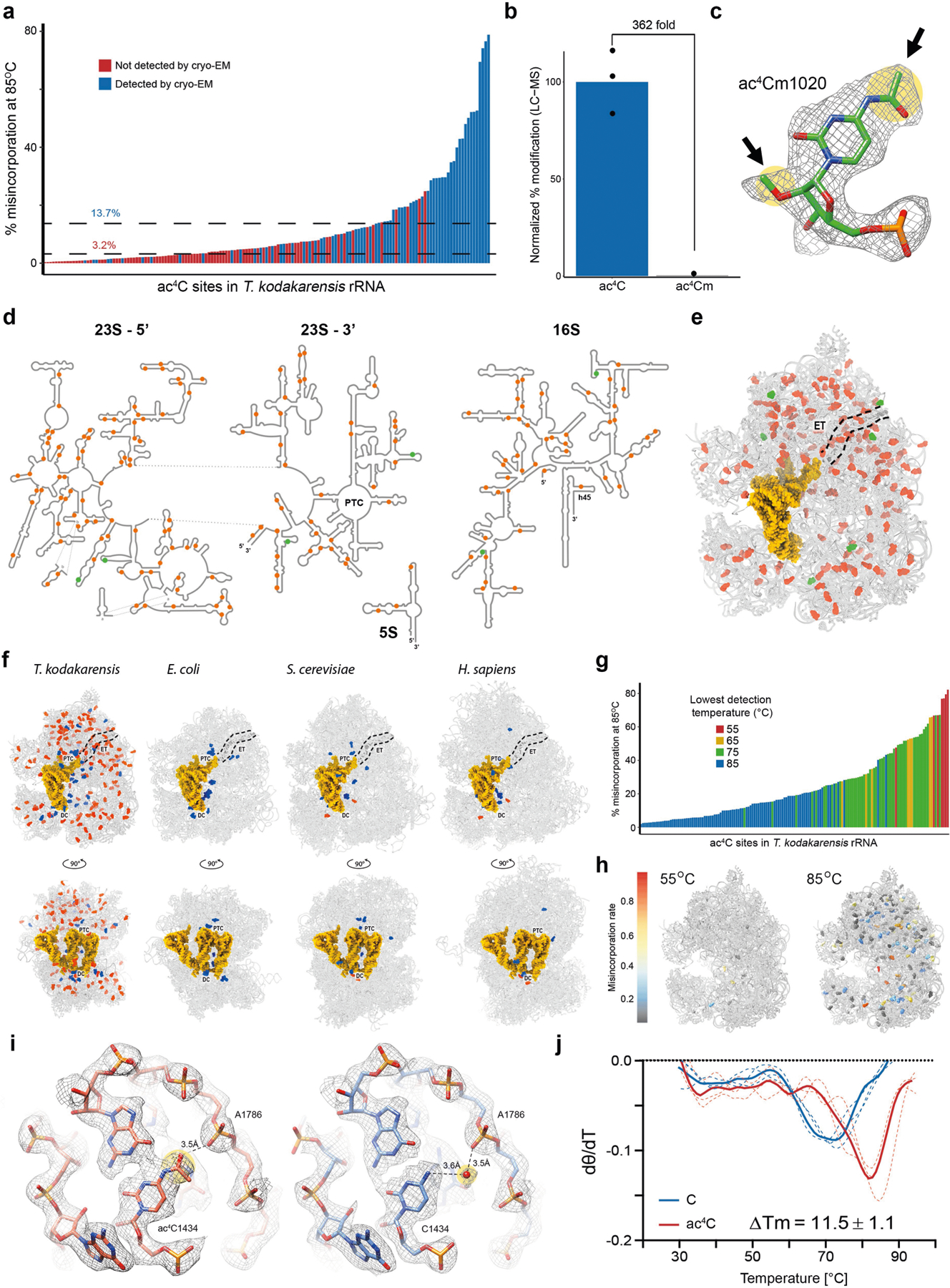
a, Misincorporation level as quantified by ac4C-seq across all ac4C sites identified in ribosomes of T. kodakarensis at 85 °C. Blue and red bars indicate sites that were and were not detected by cryo-EM, respectively. Dashed lines indicate median misincorporation of cryo-EM detected (upper, 13.7%) and not-detected (lower, 3.2%) sites. Acetylation detected by ac4C-seq and also observed in the cryo-EM were generally of medium to high stoichiometry whereas the majority of acetylation sites detected by ac4C-seq but not observed in the cryo-EM map density were of relatively low stoichiometry, rendering them invisible in the ensemble cryo-EM structure, which averages thousands of individual particles for map reconstruction. b–e, Combined cryo-EM and mass spectrometric analysis indicated six ac4C residues that are also methylated at their 2′-O position. Relative quantification of ac4C and ac4Cm detection in T. kodakarensis RNA via LC–MS is presented in b. Mean and individual data points are shown. n = 3 technical replicates. An example of ac4Cm in density is shown in c with acetate and methyl installations indicated by black arrows. A list of ac4Cms is indicated in Supplementary Table 4. 2D (d) and 3D (e) visualization of ac4C and ac4Cm distribution in the T. kodakarensis ribosome with ac4C highlighted orange and ac4Cm green. Data are presented for the T. kodakarensis grown at optimal growth temperature (85 °C). Ac4C positions highlighted in orange include genomic, mass spectrometry and electron microscopy data. Ac4Cm positions are a combination between cryo-EM and mass spectrometry data. In e, RNA and proteins are presented as grey ribbons, modified residues are highlighted as spheres. The protein exit tunnel (ET) is highlighted with a dashed black line, and tRNA is in yellow. The tRNA and mRNA coordinates are from PDB 4V5D. f, A comparative view of RNA modification distribution in E. coli, yeast (S. cerevisiae), human (H. sapiens) and T. kodakarensis. Base modifications are coloured blue, ac4Cs in red, tRNA and mRNAs in yellow. Ribosome functional regions are designated in black with decoding centre (DC), the peptidyl transferase centre (PTC) and the protein exit tunnel (ET) highlighted by a dashed black line. PDB codes for the structures used for comparison are 5AFI, 4V88 and 4UGO, for the E. coli, S. cerevisiae and human ribosome, respectively. g, Misincorporation rate as quantified by ac4C-seq for all ac4C sites in the T. kodakarensis ribosome. The bar colour indicates the lowest growth temperature at which the site was detected. h, 3D representation of the T. kodakarensis ribosome with ac4C sites detected at 55 °C and 85 °C shown and colour-coded according to misincorporation rate in each temperature. i, Ac4Cs were shown to stabilize the T. kodakarensis ribosome via direct interactions with protein and RNA residues. An example of stabilization through RNA– protein interactions is presented in Fig. 4g. RNA–RNA interactions correspond to interactions of ac4C1434 with OP2 of A1786 of LSU. j, Temperature-dependent circular dichroism spectra of synthetic RNAs containing cytidine (blue) or ac4C (red). Solid and dashed lines represent mean and individual measurements, respectively. n = 3 independent experiments. θ, ellipticity at 260 nm.
Supplementary Material
Acknowledgements
We thank S. Fox (Laboratory of Proteomics and Analytical Technology) for LC–MS analyses, C. Grose (Protein Expression Laboratory) for assisting with cloning and preparation of plasmid DNA, P. Blum (University of Nebraska) and B. Mukhopadhyay (Virginia Tech) for archaeal RNA samples, and N. Elad (Electron Microscopy unit, Weizmann Institute of Science) for assistance in setting up the cryo-EM data collection. We thank the Biophysics Resource in the Structural Biophysics Laboratory, Center for Cancer Research, National Cancer Institute for assistance with DSC and circular dichroism spectroscopy studies. We thank E. Westhof for insights into RNA structure. S. Schwartz is funded by the Israel Science Foundation (543165), the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 714023), and the Estate of Emile Mimran. S. Schwartz is the incumbent of the Robert Edward and Roselyn Rich Manson Career Development Chair in Perpetuity. M.S.B. is supported by the Zuckerman STEM Leadership Program, by Ilse Katz Institute for Material Sciences and Magnetic Resonance Research and by the Helen & Milton A. Kimmelman Center for Biomolecular Structure & Assembly. S. Schwartz and M.S.B. are jointly supported by the Weizmann-Krenter-Katz Interdisciplinary Research grant. J.L.M. is supported by the Intramural Research Program of the National Institutes of Health (NIH), the National Cancer Institute, The Center for Cancer Research (ZIA BC011488–06). M.P.W. is supported by the Stowers Institute for Medical Research and the National Institute of General Medical Sciences of the NIH (R01GM112639). T.J.S. is supported by the National Institute of General Medical Sciences of the NIH (R01GM100329) and the Department of Energy, Basic Energy Sciences Division (DE-SC0014597).
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-020-2418-2.
Competing interests The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41586-0202418-2.
Peer review information Nature thanks Danica Fujimori and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Sharma S et al. Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res 43, 2242–2258 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ito S et al. A single acetylation of 18S rRNA is essential for biogenesis of the small ribosomal subunit in Saccharomyces cerevisiae. J. Biol. Chem 289, 26201–26212 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ito S et al. Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). J. Biol. Chem 289, 35724–35730 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larrieu D, Britton S, Demir M, Rodriguez R & Jackson SP Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science 344, 527–532 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tschida BR et al. Sleeping Beauty insertional mutagenesis in mice identifies drivers of steatosis-associated hepatic tumors. Cancer Res 77, 6576–6588 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang H et al. GSK-3β-regulated N-acetyltransferase 10 is involved in colorectal cancer invasion. Clin. Cancer Res 20, 4717–4729 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Kotelawala L, Grayhack EJ & Phizicky EM Identification of yeast tRNA Um44 2′-O-methyltransferase (Trm44) and demonstration of a Trm44 role in sustaining levels of specific tRNASer species. RNA 14, 158–169 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dewe JM, Whipple JM, Chernyakov I, Jaramillo LN & Phizicky EM The yeast rapid tRNA decay pathway competes with elongation factor 1A for substrate tRNAs and acts on tRNAs lacking one or more of several modifications. RNA 18, 1886–1896 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma S et al. Specialized box C/D snoRNPs act as antisense guides to target RNA base acetylation. PLoS Genet 13, e1006804 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arango D et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell 175, 1872–1886.e24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas JM et al. A chemical signature for cytidine acetylation in RNA. J. Am. Chem. Soc 140, 12667–12670 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinclair WR et al. Profiling cytidine acetylation with specific affinity and reactivity. ACS Chem. Biol 12, 2922–2926 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tardu M, Jones JD, Kennedy RT, Lin Q & Koutmou KS Identification and quantification of modified nucleosides in Saccharomyces cerevisiae mRNAs. ACS Chem. Biol 14, 1403–1409 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kowalak JA, Dalluge JJ, McCloskey JA & Stetter KO The role of posttranscriptional modification in stabilization of transfer RNA from hyperthermophiles. Biochemistry 33, 7869–7876 (1994). [DOI] [PubMed] [Google Scholar]
- 15.Taoka M et al. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res 46, 9289–9298 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orita I et al. Random mutagenesis of a hyperthermophilic archaeon identified tRNA modifications associated with cellular hyperthermotolerance. Nucleic Acids Res 47, 1964–1976 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu N et al. tRNA modification profiles and codon-decoding strategies in Methanocaldococcus jannaschii. J. Bacteriol 201, e00690–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma S & Lafontaine DLJ ‘View from a bridge’: a new perspective on eukaryotic rRNA base modification. Trends Biochem. Sci 40, 560–575 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Fischer N et al. Structure of the E. coli ribosome–EF-Tu complex at <3 Å resolution by Cs-corrected cryo-EM. Nature 520, 567–570 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Polikanov YS, Melnikov SV, Söll D & Steitz TA Structural insights into the role of rRNA modifications in protein synthesis and ribosome assembly. Nat. Struct. Mol. Biol 22, 342–344 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawai G et al. Conformational rigidity of N4-acetyl-2′-O-methylcytidine found in tRNA of extremely thermophilic Archaebacteria (Archaea). Nucleosides Nucleotides 11, 759–771 (1992). [Google Scholar]
- 22.Bruenger E et al. 5S rRNA modification in the hyperthermophilic archaea Sulfolobus solfataricus and Pyrodictium occultum. FASEB J 7, 196–200 (1993). [DOI] [PubMed] [Google Scholar]
- 23.Kumbhar BV, Kamble AD & Sonawane KD Conformational preferences of modified nucleoside N4-acetylcytidine, ac4C occur at “wobble” 34th position in the anticodon loop of tRNA. Cell Biochem. Biophys 66, 797–816 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Parthasarathy R, Ginell SL, De NC & Chheda GB Conformation of N4-acetylcytidine, a modified nucleoside of tRNA, and stereochemistry of codon–anticodon interaction. Biochem. Biophys. Res. Commun 83, 657–663 (1978). [DOI] [PubMed] [Google Scholar]
- 25.Safra M et al. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 551, 251–255 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Li X et al. Base-resolution mapping reveals distinct m1a methylome in nuclear- and mitochondrial-encoded transcripts. Mol. Cell 68, 993–1005.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grozhik AV et al. Antibody cross-reactivity accounts for widespread appearance of m1A in 5′ UTRs. Nat. Commun 10, 5126 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Helm M, Lyko F & Motorin Y Limited antibody specificity compromises epitranscriptomic analyses. Nat. Commun 10, 5669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gehring AM, Sanders TJ & Santangelo TJ Markerless gene editing in the hyperthermophilic archaeon Thermococcus kodakarensis. Bio Protoc 7, e2604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hileman TH & Santangelo TJ Genetics techniques for Thermococcus kodakarensis. Front. Microbiol 3, 195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santangelo TJ, Cubonov L, James CL & Reeve JN TFB1 or TFB2 is sufficient for Thermococcus kodakaraensis viability and for basal transcription in vitro. J. Mol. Biol 367, 344–357 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santangelo TJ & Reeve JN Deletion of switch 3 results in an archaeal RNA polymerase that is defective in transcript elongation. J. Biol. Chem 285, 23908–23915 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lipscomb GL et al. Natural competence in the hyperthermophilic archaeon Pyrococcus furiosus facilitates genetic manipulation: construction of markerless deletions of genes encoding the two cytoplasmic hydrogenases. Appl. Environ. Microbiol 77, 2232–2238 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oger P et al. Complete genome sequence of the hyperthermophilic archaeon Thermococcus sp. strain AM4, capable of organotrophic growth and growth at the expense of hydrogenogenic or sulfidogenic oxidation of carbon monoxide. J. Bacteriol 193, 7019–7020 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farkas JA, Picking JW & Santangelo TJ Genetic techniques for the archaea. Annu. Rev. Genet 47, 539–561 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Wickham H ggplot2: Elegant Graphics for Data Analysis (Springer, 2016). [Google Scholar]
- 37.Matzov D et al. The cryo-EM structure of hibernating 100S ribosome dimer from pathogenic Staphylococcus aureus. Nat. Commun 8, 723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morlan JD, Qu K & Sinicropi DV Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS ONE 7, e42882 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shishkin AA et al. Simultaneous generation of many RNA-seq libraries in a single reaction. Nat. Methods 12, 323–325 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Engreitz JM et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 341, 1237973 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Machnicka MA et al. MODOMICS: a database of RNA modification pathways-2013 update. Nucleic Acids Res 41, D262–D267 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith T, Heger A & Sudbery I UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res 27, 491–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jun G, Wing MK, Abecasis GR & Kang HM An efficient and scalable analysis framework for variant extraction and refinement from population-scale DNA sequence data. Genome Res 25, 918–925 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Piechotta M, Wyler E, Ohler U, Landthaler M & Dieterich C JACUSA: site-specific identification of RNA editing events from replicate sequencing data. BMC Bioinformatics 18, 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crooks GE, Hon G, Chandonia J-M & Brenner SE WebLogo: a sequence logo generator. Genome Res 14, 1188–1190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vainberg Slutskin I, Weingarten-Gabbay S, Nir R, Weinberger A & Segal E Unraveling the determinants of microRNA mediated regulation using a massively parallel reporter assay. Nat. Commun 9, 529 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weingarten-Gabbay S et al. Systematic discovery of cap-independent translation sequences in human and viral genomes. Science 351, aad4939 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson MD & Oshlack A A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tarazona S, Garca F, Ferrer A, Dopazo J & Conesa A NOIseq: a RNA-seq differential expression method robust for sequencing depth biases. EMBnet.journal 17, 18–19 (2012). [Google Scholar]
- 52.Katoh K, Misawa K, Kuma K & Miyata T MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30, 3059–3066 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sievers F et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Basanta-Sanchez M, Temple S, Ansari SA, D’Amico A & Agris PF Attomole quantification and global profile of RNA modifications: Epitranscriptome of human neural stem cells. Nucleic Acids Res 44, e26 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamauchi Y et al. Denaturing reversed phase liquid chromatographic separation of non-coding ribonucleic acids on macro-porous polystyrene-divinylbenzene resins. J. Chromatogr. A 1312, 87–92 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Taoka M et al. An analytical platform for mass spectrometry-based identification and chemical analysis of RNA in ribonucleoprotein complexes. Nucleic Acids Res 37, e140 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakayama H, Yamauchi Y, Taoka M & Isobe T Direct identification of human cellular microRNAs by nanoflow liquid chromatography-high-resolution tandem mass spectrometry and database searching. Anal. Chem 87, 2884–2891 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Nakayama H et al. Ariadne: a database search engine for identification and chemical analysis of RNA using tandem mass spectrometry data. Nucleic Acids Res 37, e47 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, Wen Z, Washburn MP & Florens L Evaluating chromatographic approaches for the quantitative analysis of a human proteome on Orbitrap-based mass spectrometry systems. J. Proteome Res 18, 1857–1869 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y, Wen Z, Washburn MP & Florens L Effect of dynamic exclusion duration on spectral count based quantitative proteomics. Anal. Chem 81, 6317–6326 (2009). [DOI] [PubMed] [Google Scholar]
- 61.Zhang Y, Wen Z, Washburn MP & Florens L Improving proteomics mass accuracy by dynamic offline lock mass. Anal. Chem 83, 9344–9351 (2011). [DOI] [PubMed] [Google Scholar]
- 62.McDonald WH et al. MS1, MS2, and SQT-three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Commun. Mass Spectrom 18, 2162–2168 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Xu T et al. ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J. Proteomics 129, 16–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tabb DL, McDonald WH & Yates JR III. DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res 1, 21–26 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y, Wen Z, Washburn MP & Florens L Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Anal. Chem 82, 2272–2281 (2010). [DOI] [PubMed] [Google Scholar]
- 66.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rohou A & Grigorieff N CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mindell JA & Grigorieff N Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol 142, 334–347 (2003). [DOI] [PubMed] [Google Scholar]
- 69.Zivanov J et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rosenthal PB & Henderson R Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol 333, 721–745 (2003). [DOI] [PubMed] [Google Scholar]
- 71.Afonine PV et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kucukelbir A, Sigworth FJ & Tagare HD Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pettersen EF, Goddard TD & Huang CC UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 74.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moriarty NW, Grosse-Kunstleve RW & Adams PD electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D 65, 1074–1080 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Williams CJ et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci 27, 293–315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deutsch EW et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res 45, D1100–D1106 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Perez-Riverol Y et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442–D450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ueda T et al. Mutation of the gene encoding the ribonuclease P RNA in the hyperthermophilic archaeon Thermococcus kodakarensis causes decreased growth rate and impaired processing of tRNA precursors. Biochem. Biophys. Res. Commun 468, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Ac4C-seq datasets generated in this manuscript have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE135826. The cryo-EM density maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD10223 and EMD-10503 for the wild-type strain grown at 85 °C and 65 °C, respectively, and EMD-10224 for the TkNAT10-deletion strain. Model coordinates have been deposited in the Protein Data Bank (PDB) under accession numbers 6SKF, 6TH6 and 6SKG. Mass spectrometry proteomics data has been deposited to the ProteomeXchange Consortium via Pride77,78 partner repository with the dataset identifier PXD014814.