

Abstract
Hexokinase 1 and 2 have been shown to inhibit Bak- and Bax-mediated apoptosis, leading us to combine the histone deacetylase inhibitor romidepsin with clotrimazole or bifonazole, two compounds that reportedly decrease mitochondrial localization of hexokinases. Cancer cell lines derived from breast, kidney, lung, colon or ovarian cancers were treated with a short-term exposure to 25 ng/ml romidepsin combined with either clotrimazole or bifonazole. The combination of romidepsin with 25 μM clotrimazole or bifonazole resulted in increased annexin staining compared to cells treated with any of the drugs alone. Cell death was caspase-mediated, as the pan-caspase inhibitor Q-VD-OPh was found to inhibit apoptosis induced by the combination. A549 lung cancer cells or HCT-116 cells deficient in Bak and Bax were also resistant to apoptosis with the combination implicating the intrinsic apoptotic pathway. We found that a 24 h treatment with clotrimazole or bifonazole decreased total hexokinase 2 expression, resulting in a 76% or 60% decrease, respectively, of mitochondrial expression of hexokinase 2. Mitochondrial hexokinase 1 levels increased 2-fold or less. Our work suggests that the combination of a short-term romidepsin treatment with bifonazole or clotrimazole leads to increased apoptosis, most likely due to decreased mitochondrial expression of hexokinase 2.
Keywords: Histone deacetylase inhibitor, Romidepsin, Hexokinase, Apoptosis
1. Introduction
As a compound class, histone deacetylase inhibitors (HDIs) have been remarkably successful in the treatment of T-cell lymphoma [1]. Vorinostat (suberanilohydroxamic acid or SAHA) was approved by the FDA in 2006 for the treatment of cutaneous T-cell lymphoma [2,3]; romidepsin (RD) was approved for this indication as well in 2009 [4,5] and later for peripheral T-cell lymphoma in 2011 [6,7]; belinostat was approved for this latter indication in 2014 [8]. Despite their success in treating T-cell lymphoma, the HDIs have not proven to be effetive for the treatment of solid tumors [9]. Nearly all clinical trials for solid tumors in which HDIs were used as single agents were unsuccessful; clinical trials with HDIs combined with other chemotherapeutic agents to treat solid tumors have also been largely disappointing [10]. This points to the need for a better understanding of the mechanisms of HDI-mediated cell death to facilitate more effective combinations with HDIs.
Given the emergence of three HDIs approved for the treatment of T-cell lymphoma, it is important to define mechanisms of resistance to them. One recent study found increased levels of tissue transglutaminase, commonly induced by HDI treatment, in MCF7 cells selected for resistance to vorinostat and co-treatment of the resistant cells with transglutaminase inhibitors restored sensitivity to vorinostat [11]. Another group found that the transcription factor GLI1 was overexpressed in HCT-116 colon carcinoma cells that were selected with vorinostat [12]. In the case of romidepsin, it was reported early on that romidepsin was a substrate for the ATP-binding cassette transporter P-glycoprotein (P-gp) [13] and romidepsin-resistant cell lines were found to have increased levels of P-gp expression [13–16]. However, increased expression of P-gp does not appear to explain the resistance of solid tumors to romidepsin and clinical tumor samples taken from patients with resistant disease do not appear to have increased levels of P-gp [17].
To identify alternative mechanisms of resistance to romidepsin, we selected the HuT78 T-cell lymphoma cell line with romidepsin in the presence of P-gp inhibitors to exclude P-gp as a mechanism of resistance. The resulting sublines were found to have increased activation of the mitogen activated protein kinase (MAPK) pathway and increased sensitivity to MEK inhibitors [18]. Activation of the MAPK pathway led to increased phosphorylation and degradation of the proapoptotic protein Bim and MEK inhibitor treatment led to rapid increases in Bim and increased cell death [18]. Thus, the activity of romidepsin is highly dependent on mitochondrial engagement and activation of the intrinsic apoptotic pathway.
The effector proteins Bak and Bax are crucial components of the intrinsic apoptotic pathway; these proteins are members of the Bcl-2 family and effect release of cytochrome c, subsequently leading to formation of the apoptosome [19]. Proapoptotic Bcl-2 family members include Bim, Puma, and Noxa, and push the balance towards cell death while the antiapoptotic members Bcl-2, Mcl-1 and Bcl-XL prevent Bak-or Bax-mediated release of cytochrome c [19]. However, other proteins that are not members of the Bcl-2 family have been shown to affect the ability of cells to undergo apoptosis. Hexokinase 1 and hexokinase 2 (HK1 and HK2), proteins are primarily located in mitochondria, and have been shown to prevent apoptosis mediated by either Bak or Bax [20]. In light of the fact that apoptosis induced by romidepsin and other HDIs is dependent on Bak and/or Bax expression [21,22], we explored whether targeting mitochondrial hexokinases might improve the efficacy of HDIs.
To investigate whether targeting mitochondrial hexokinases was an effective approach, we treated solid tumor cancer cell lines with the HDIs romidepsin and belinostat in combination with two compounds known to disrupt the activity of mitochondrial hexokinases, the azole inhibitors clotrimazole and bifonazole. Our results show that decreasing mitochondrial expression of HK2 may be a potential way to increase the efficacy of HDIs in solid tumors.
2. Materials and methods
2.1. Cell lines
The human cancer cell lines HCT-116 (colon), A549 (lung), 786–0 (renal), IGROV1 (ovarian) and MDA-MB-231 (breast) were obtained from the Division of Cancer Treatment and Diagnosis Tumor Repository, National Cancer Institute (Frederick, MD). HCT-116 cells deficient in Bax were a kind gift of Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD)[23]. HCT-116 cells deficient in Bak or both Bak and Bax were kindly provided by Dr. Richard Youle (NIH, Bethesda, MD)[24]. Cells were periodically checked for mycoplasma with the MycoAlert Mycoplasma Detection Kit (Lonza, Walkersville, MD). A549 cells deficient in Bak, Bax or both (double knock-out, DKO) were generated by our lab using CRISPR technology. Specifically, cells lacking single genes were generated by co-transfecting cells with a CRISPR vector containing Cas9 and guide RNAs to target the gene of interest (BAK1 or BAX) and a homology directed repair (HDR) vector containing a puromycin resistance gene flanked by LoxP sites (obtained from Santa Cruz Biotechnology, Dallas, TX). Cells were subsequently treated with puromycin 48 h later to select for transfected cells. Cells were isolated and protein loss was confirmed by immunoblot, after which cells were transfected with a Cre vector (Santa Cruz) to cause loss of puromycin resistance. To generate the double knockouts, the single knockout cells were subsequently transfected with CRISPR and HDR vectors targeting the second gene.
2.2. Chemicals
Clotrimazole was from Tocris/R&D Systems (Minneapolis, MN); bifonazole was obtained from Sigma-Aldrich (St. Louis, MO). Romidepsin was purchased from Selleck Chemicals (Houston, TX); belinostat was obtained from ChemieTek (Indianapolis, IN), and Q-Vd-OPh was from Apex BioTechnology (Houston, TX).
2.3. Flow cytometry
After treating cells with romidepsin or belinostat alone or in combination with bifonazole or clotrimazole as indicated, floating and adherent cells were combined and incubated with allophycocyanin-labeled annexin V (eBioscience/Thermo Fisher Scientific, Waltham, MA) in combination with SYTOX green DNA dye (Thermo Fisher) according to the manufacturer’s instructions. The percentages of apoptotic cells were determined using FlowJo (v 10.4.2) software (FlowJo, Ashland Oregon).
2.4. Isolation of mitochondria-enriched fractions
Mitochondria-enriched fractions were isolated as previously described [25]. Brie fly, cells were resuspended in isolation bu?er (250 mM sucrose; 20 mM HEPES, pH 7.4; 10 mM KCl; 1.5 mM MgCl2; 1 mM EDTA; 1 mM dithiothreitol) for 3 min after which cells were subjected to 40 strokes in a glass dounce homogenizer. Disrupted cells were then centrifuged twice for 10 min at 1500 × g after which the mitochondrial fraction was isolated by centrifugation at 12,000 × g. Whole cell and mitochondria-enriched fractions were subsequently lysed in RIPA bu?er and fractionation was verified by immunoblot analysis.
2.5. Immunoblot analysis
Cells were harvested and resuspended in RIPA buffer (50 mM Tris -HCl [pH 7.4], 1% Triton X-100, 10% glycerol, 0.1% SDS, 2 mM EDTA, and 0.5% deoxycholate, 50 mM NaCl) containing protease inhibitors (from Bimake, Houston, TX) and subsequently sonicated for 15 min in a bath sonicator. Lysates were then centrifuged at 14,000 RPM for 15 min at 4 °C and after determining protein concentration and approximately 25 μg of protein was subjected to electrophoresis and transferred onto nitrocellulose membranes. Membranes were probed with antibodies against the following proteins: Bax, Bak, Caspase 3, cleaved Caspase 3, Hexokinase 1, Hexokinase 2, Cox IV, VDAC1, or cleaved PARP (all from Cell Signaling Technology, Beverly, MA); total and acetylated histone H3 (both from MilliporeSigma, Burlington, MA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (American Research Products, Belmont, MA) served as a loading control. Proteins were visualized with the Odyssey System (LI-COR, Lincoln, NE) using a 1:8000 dilution of the IRDye 800CW goat anti-mouse secondary antibody or 1:8000 dilution of IRDye 700CW goat anti-rabbit secondary antibody (LI-COR).
2.6. Statistical analysis
Where noted, a studenťs t-test was performed between various treatment regimens using Prism 7 for Mac (GraphPad Software, La Jolla, CA). Differences were considered statistically significant where p < 0.05.
3. Results
3.1. Sub-lethal doses of clotrimazole or bifonazole increase sensitivity to short-term romidepsin treatment
Clotrimazole and bifonazole were previously reported to detach hexokinases from the mitochondria of B16 melanoma cells, with an IC50 of 10–15 μM [26]. While this dose was largely non-toxic, higher doses of 50 μM were found to decrease cell viability. We thus examined whether smaller, non-lethal doses of the compounds might increase sensitivity to romidepsin. The romidepsin combinations were performed as previously described [27,28]. Romidepsin is approved as a 4h infusion and has a short half-life [6,7]. Clinical studies exploring longer romidepsin administration times have resulted in increased toxicities that required dose reductions [29], meaning that long-term administration of romidepsin such as are usually used in 3- or 4-day cytotoxicity assays would not be possible. By treating cells with a 6 h treatment of 25 ng/ml romidepsin and examining apoptosis via annexin staining, we more closely approximate clinical administration of romidepsin, and the efficacy of this schedule in cancer cell lines more closely approximates the clinical profile of romidepsin, with hematologic cancer cell lines showing greater sensitivity [28].
A dose response was performed on HCT-116, A549, IGROV1, MDA-MB-231 and 786–0 cells with either clotrimazole (CTZ) or bifonazole (BIF) at 1, 5 and 25 μM in the presence or absence of 25 ng/ml romidepsin. Briefly, CTZ and BIF are combined with romidepsin for 6 h after which the media is removed and replaced with fresh medium to which CTZ and BIF are again added and incubated with the cells for an additional 42 h. Cells are then harvested and incubated with recombinant human annexin V tagged with allophycocyanin and SYTOX green DNA dye for 20 mins. Results from one experiment with the HCT-116 line treated with the highest concentrations of CTZ or BIF are shown in Fig. 1A. As we previously reported, short-term romidepsin treatment elicited apoptosis of approximately 30%. Treatment with 25 μM of either CTZ or BIF alone did not result in high levels of apoptosis. However, the combination yielded almost complete apoptosis, with annexin positive cells shown in the red box. Results with all cells and all concentrations of CTZ and BIF are shown in Fig. 1B. Addition of even 1 μM CTZ or BIF to romidepsin treatment in the HCT-116 cell line resulted in much higher levels of apoptosis compared to romidepsin treatment alone. Higher concentrations of CTZ and BIF were needed to increase sensitivity of the other cell lines to romidepsin, but all cells responded to the combination of romidepsin with the highest concentrations of clotrimazole or bifonazole.
Fig. 1. Combining clotrimazole or bifonazole with romidepsin leads to increased apoptosis.
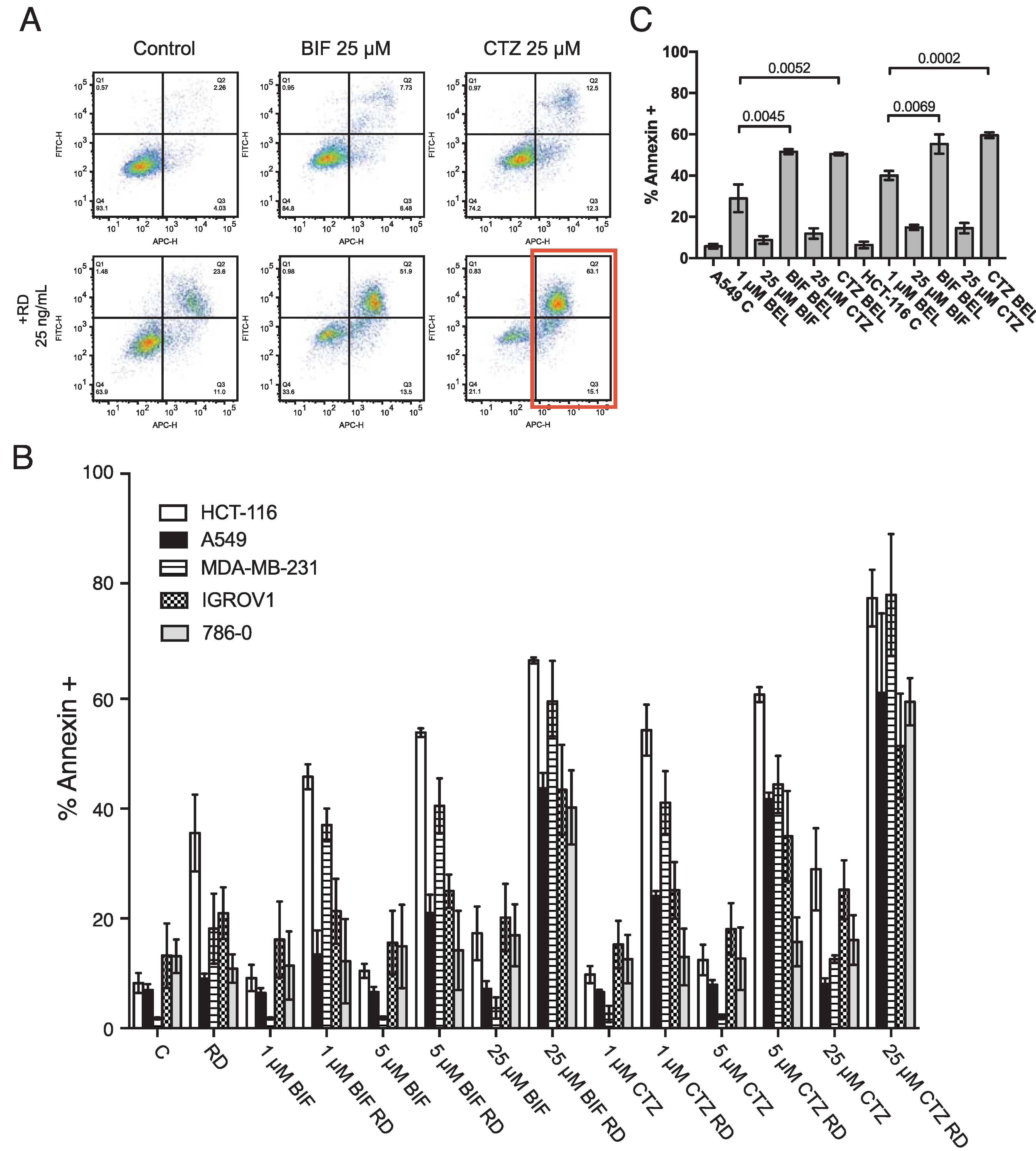
A) HCT-116 cells were left untreated (Control) or treated for 6 h with 25 ng/ml romidepsin alone (RD) or in combination with 25 μM BIF or CTZ (RD+BIF; RD+CTZ). The medium was subsequently removed and cells were incubated in romidepsin-free medium in the absence or presence of CTZ for an additional 42 h, after which cells were stained with APC-labeled recombinant annexin and the DNA dye SYTOX green and assayed by flow cytometry. Cells were also incubated with 25 μM CTZ alone for 48 h (CTZ). The red box denotes annexin-positive cells that were quantitated in subsequent experiments. B) HCT-116, A549, MDA-MB-231, IGROV1 and 786–0 cells were treated as outlined in (A), using 1, 5, or 25 μM CTZ or BIF. The percentages of annexin positive cells were determined and summarized in the graph. At least 3 separate experiments were performed for each condition. C) A549 and HCT116 cells were treated for 48 h with 500 nM or 250 nM belinostat, respectively, alone or in combination with 25 μM CTZ or BIF continuously for 48 h. Cells were then stained with annexin V/SYTOX green and assayed by flow cytometry. Significance (p values) is shown for the bracketed conditions.
Belinostat, currently in development to be administered as a 48 h continuous infusion, was also used in combination with CTZ or BIF. A549 and HCT-116 cells were treated with a concentration of belinostat that was largely non-toxic over a 48 h period and combined with 25 μM of CTZ or BIF. Similar effects were observed when CTZ or BIF were added to belinostat treatment, suggesting that targeting hexokinases might improve solid tumor response to HDIs (Fig. 1C).
3.2. The combination of short-term romidepsin treatment with CTZ or BIF results in caspase-mediated apoptosis
We next sought to confirm results obtained by flow cytometry analysis by examining downstream events in the apoptotic cascade mediated by the combination of romidepsin with CTZ or BIF. A549 cells and HCT-116 cells were treated with romidepsin, CTZ or BIF alone or in combination after 48 h and immunoblotting for cleaved caspase 3 and cleaved PARP was performed. While some caspase and PARP cleavage was observed with romidepsin alone and none was observed with CTZ or BIF alone, the highest amount of caspase and PARP cleavage was noted when the compounds were used in combination (Fig. 2). Apoptosis was subsequently measured in the presence or absence of the pan-caspase inhibitor Q-VD-OPh. As shown in Fig. 3, when A549 or HCT-116 cells were treated with 25 ng/ml romidepsin in combination with 25 μM CTZ or BIF, increased annexin V staining was observed. When cells were treated with the combination in the presence of 25 μM Q-VD-OPh, apoptosis was significantly decreased, suggesting that caspase cleavage is required for the observed apoptosis. Q-VD-OPh also significantly decreased the amount of apoptosis caused by romidepsin alone in the HCT-116 cell line.
Fig. 2. Romidepsin in combination with CTZ or BIF induces PARP and caspase cleavage.
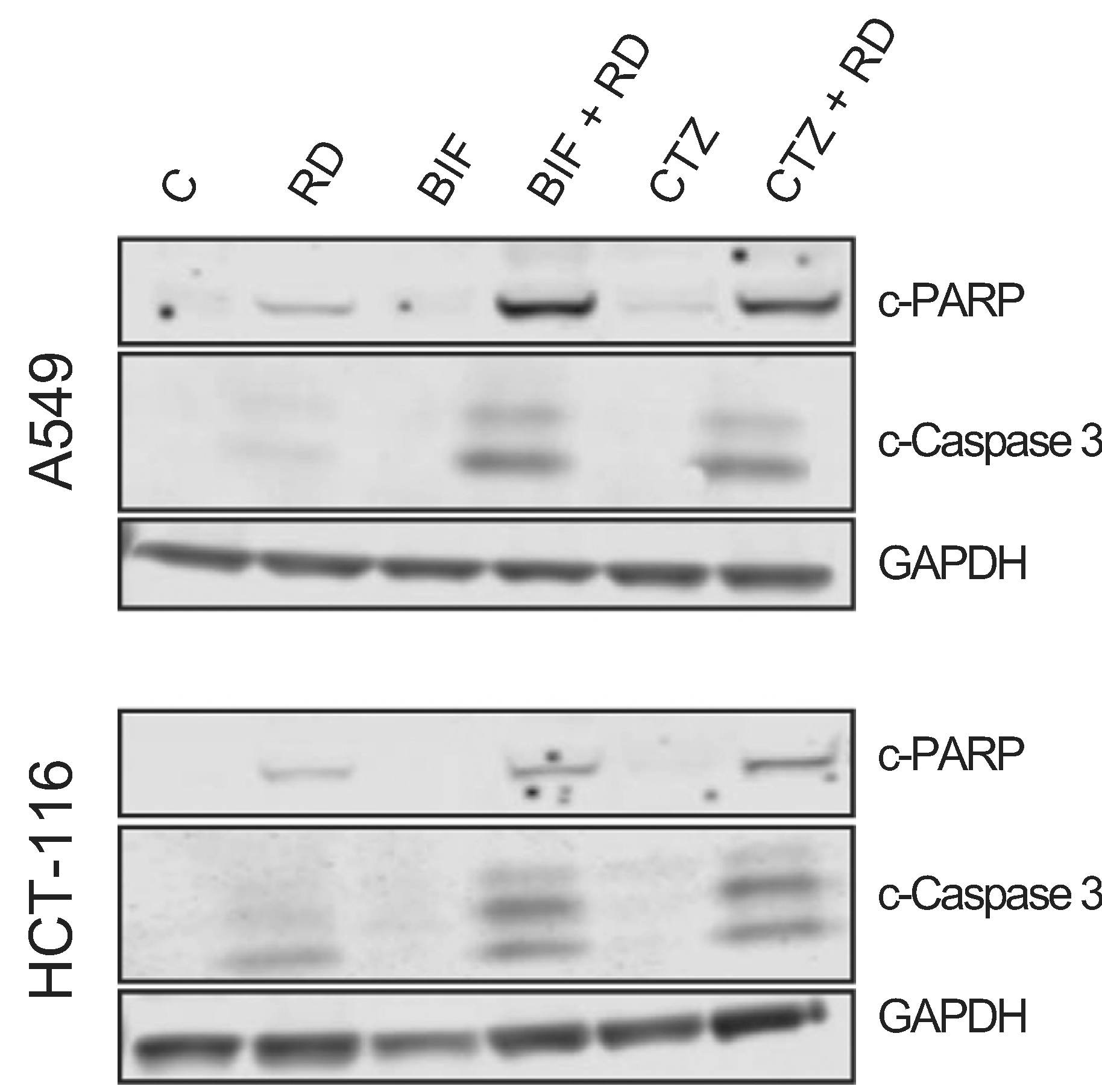
A549 or HCT-116 cells were treated for 6 h with 25 ng/ml romidepsin (RD) alone or in combination with 25 μM clotrimazole (CTZ) or bifonazole (BIF). Subsequently the medium was removed and cells were incubated in romidepsin-free medium in the absence or presence of CTZ or BIF for an additional 42 h. Cells were also treated with 25 μM CTZ or BIF continuously for 48 h. Cleared lysates were then subjected to PAGE and transferred to nitrocellulose. The blot was probed with antibodies to cleaved caspase 3 and cleaved PARP. GAPDH served as a loading control. Results are from one of 3 independent experiments.
Fig. 3. The caspase inhibitor Q-VD-OPh prevents apoptosis mediated by the combination of romidepsin with CTZ or BIF.
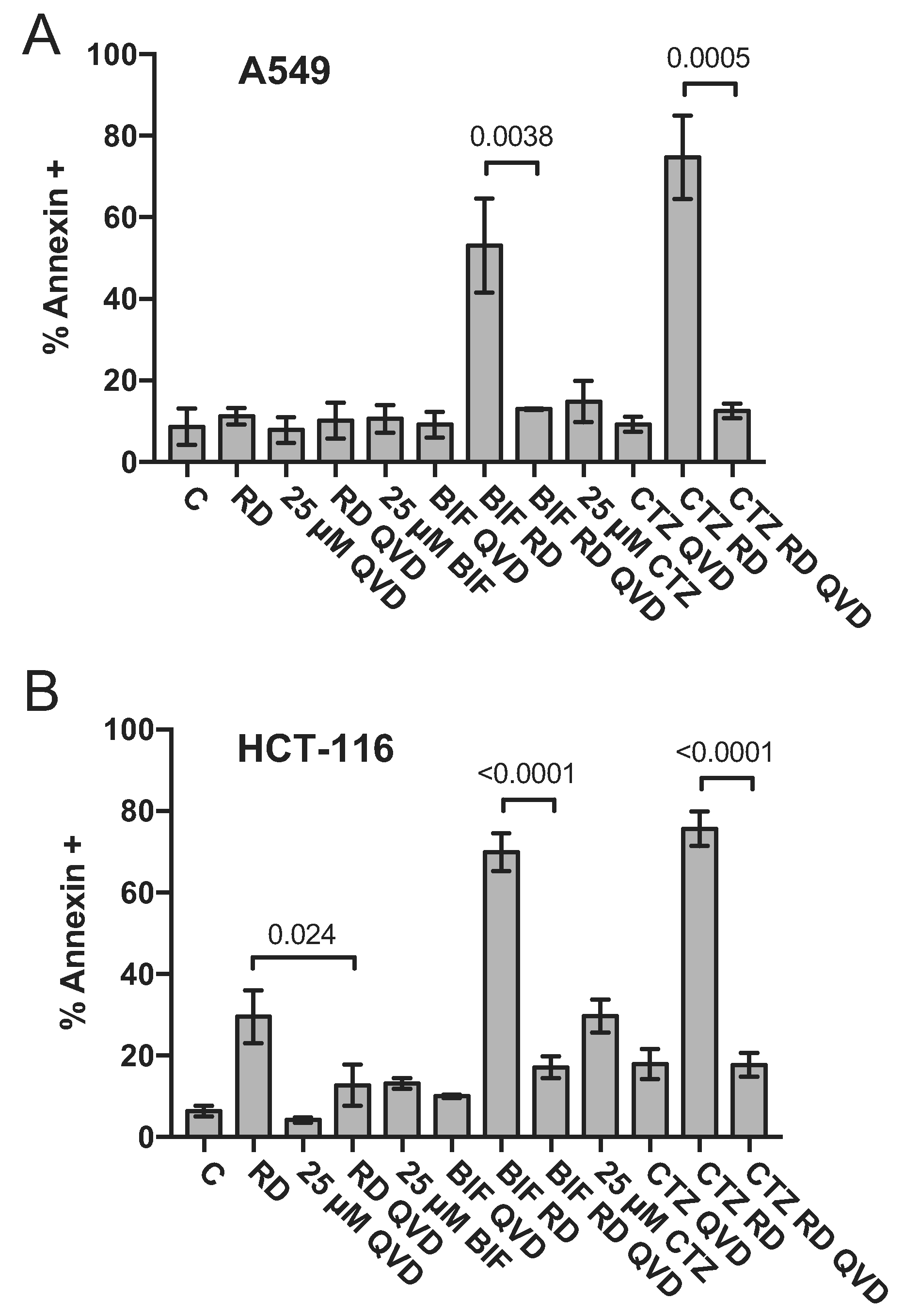
HCT-116 and A549 cells were treated with romidepsin (RD) alone or in combination with 25 μM clotrimazole (CTZ) or bifonazole (BIF) as outlined in Fig. 1A in the presence of absence of 25 μM of the pan-caspase inhibitor Q-VD-OPh. Cells were harvested and stained with annexin V/SYTOX green and assayed by flow cytometry as outlined in Materials and Methods. Percent annexin positive cells determined from at least 3 independent experiments are summarized in the graph. Significance (p values) is shown for the bracketed conditions.
3.3. The apoptosis observed with romidepsin combined with CTZ or BIF requires Bak and Bax expression
As activation of both the intrinsic and extrinsic apoptotic pathways involves caspases, apoptosis generated by the combination of romidepsin and BIF or CTZ was examined in HCT-116 and A549 cells deficient in Bak, Bax or both (double knock-out, DKO). Bak and Bax act as effector proteins to release cytochrome c from the mitochondria, which sets the apoptotic cascade into motion. We have previously shown that Bak and Bax are required for apoptosis to occur in Ras-mutant cell lines treated with romidepsin in combination with a MEK and PI3K inhibitor [27]. Knockout of Bak, Bax or both in the A549 and HCT-116 cell line sets was verified by immunoblot (Fig. 4A). When wild-type cells were treated with romidepsin in combination with 25 μM CTZ or BIF, apoptosis was readily observed, while the effects were blunted in HCT-116 cells lacking Bax. In both A549 and HCT116 DKO cells, apoptosis was almost completely abrogated (Fig. 4B), confirming that the observed apoptosis proceeds via the intrinsic pathway.
Fig. 4. Apoptosis induced by the combination of romidepsin with CTZ or BIF is Bak- and Bax-dependent.
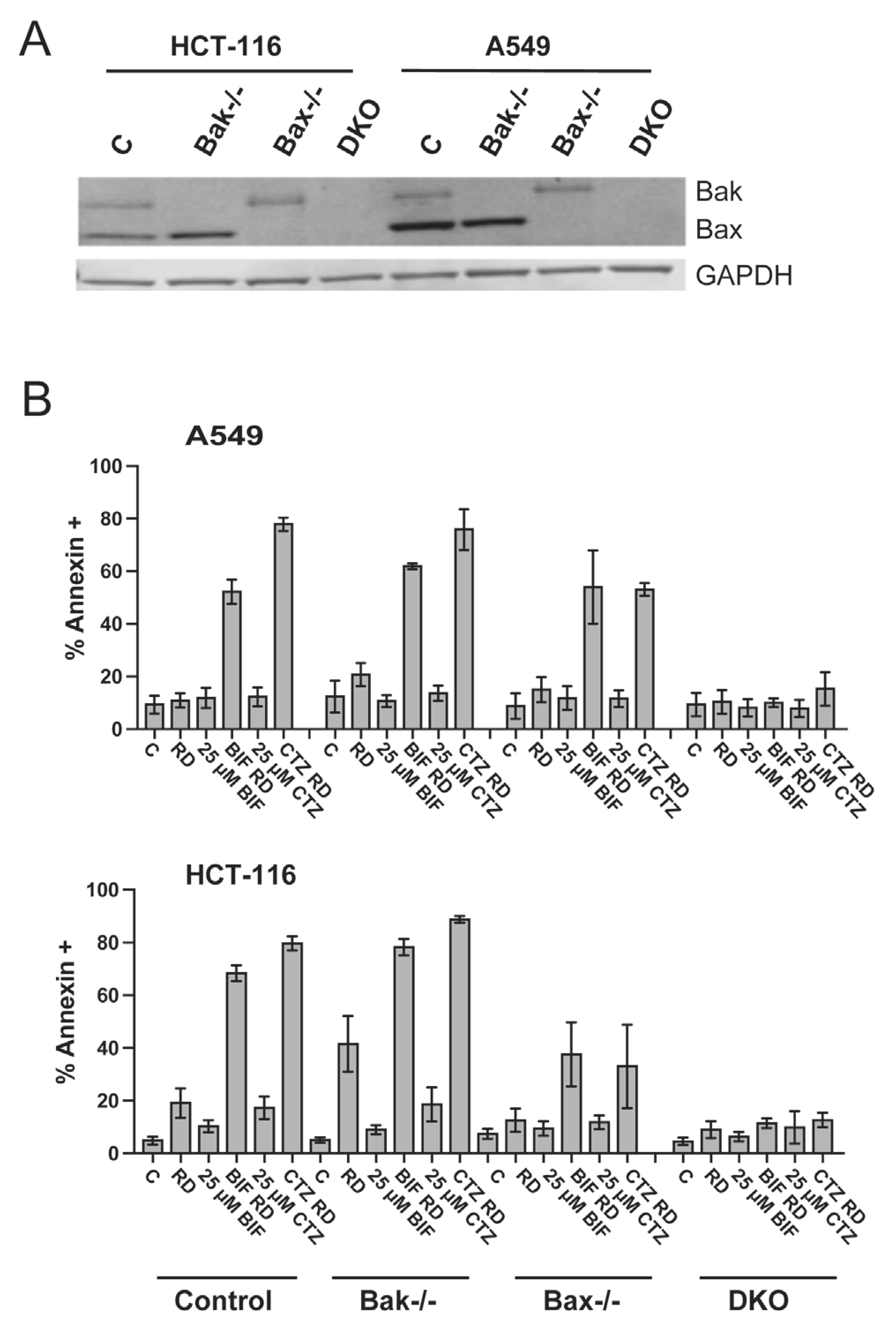
A) Whole cell lysates were obtained from parental HCT-116 and A549 cells or cells lacking Bak, Bax or both (DKO), subjected to PAGE and transferred to nitrocellulose. The blot was subsequently probed with antibodies to Bak and Bax; GAPDH served as a loading control. B) Parental HCT-116 and A549 cells or cells lacking Bak, Bax or both (DKO) were treated with romidepsin alone or in combination with 25 μM CTZ or BIF, as outlined in Fig. 1A. Subsequently, cells were harvested, stained with annexin V/ SYTOX green and assayed by flow cytometry. The percentages of cells that were annexin positive were summarized from at least 3 independent experiments.
3.4. Clotrimazole and bifonazole decrease mitochondrial levels of HK2
We then sought to confirm that treatment with CTZ or BIF decreased mitochondrial localization of hexokinases. HCT-116 cells were treated for 24 h with 25 μM CTZ or BIF after which mitochondria-enriched fractions were obtained as described in the Materials and Methods. We compared expression of HK1 and HK2 in mitochondria-enriched fractions to whole-cell lysates in untreated versus BIF or CTZ treated cells. We found that the mitochondrial fraction had higher levels of the mitochondria markers VDAC1 and COX IV, while expression of the nuclear marker histone H3 and the cytoplasmic marker caspase 3 was predominantly in the whole cell lysates (Fig. 5). Mitochondrial levels of HK1 and HK2 were quantitated and normalized to VDAC expression and are shown in Fig. 5. Levels in treated cells were compared to untreated cells, where levels were given a value of 1. Treatment of HCT-116 cells with CTZ or BIF for 24 h decreased total HK2 levels while total HK1 levels were unchanged; mitochondrial levels of HK1 were slightly increased (Fig. 5). In addition to lower total levels of HK2, treatment with BIF or CTZ treatment resulted in a 52% or 73% decrease in mitochondrial HK2 levels, respectively (Fig. 5). A second biological replicate of the experiment (Fig. S1) demonstrated a 68% or 80% decrease in mitochondrial levels of HK2 after treatment with BIF or CTZ, respectively. This suggests that the target of CTZ and BIF is predominantly HK2. Treatment with CTZ or BIF leads to both a decrease in total levels of HK2 as well as decreased HK2 expression on the mitochondria.
Fig. 5. CTZ and BIF decrease mitochondrial localization of HK2.
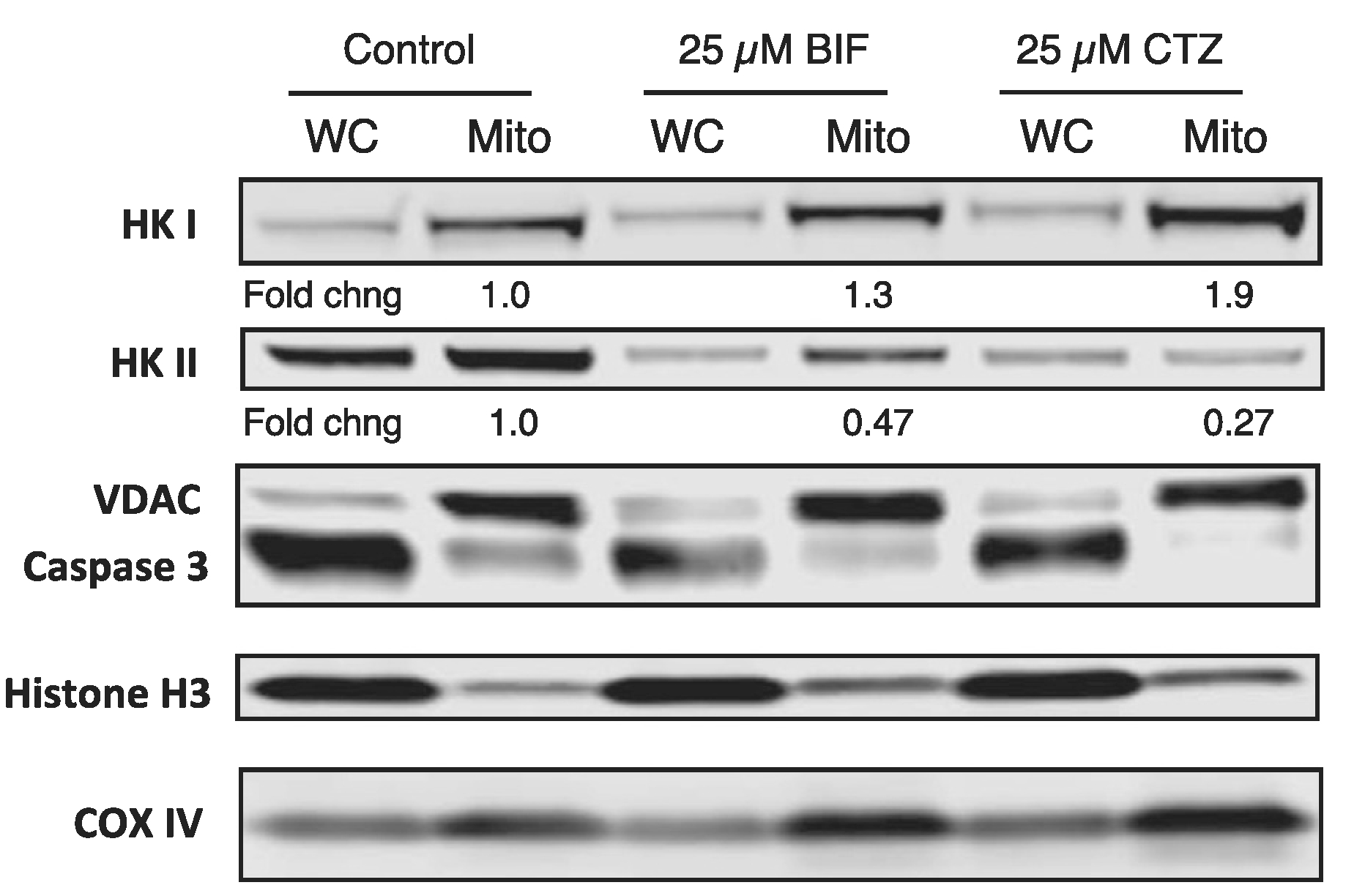
HCT-116 cells were plated and allowed to attach overnight after which cells were left untreated or treated with 25 μM CTZ or BIF for 24 h. Cells were then harvested and whole-cell lysates or mitochondria-enriched fractions were obtained, as outlined in Materials and Methods. Protein was isolated and subjected to PAGE after which the proteins were transferred to a nitrocellulose membrane. The membrane was subsequently probed with antibodies for VDAC1, caspase 3, Cox IV, HK1, HK2 and histone H3. Band intensity was measured on a Li-COR Odyssey scanner. Mitochondrial levels of HK1 and HK2 were normalized to VDAC expression. Fold changes in mitochondrial expression of HK1 and HK2 are compared to levels in untreated cells, which were assigned a value of 1. Results shown are from one of two independent experiments.
4. Discussion
Although HDIs have shown promise in the treatment of T-cell lymphomas, their efficacy in solid tumors has been disappointing and they will clearly need to be combined with other drugs to be more effective. Here we demonstrate that the combination of a short-term, clinically-relevant dose of romidepsin with sublethal doses of either clotrimazole or bifonazole, which led to decreased HK2 expression on the mitochondria, yields high levels of apoptosis in cell line models of colon, lung, renal, ovarian and breast cancer. This suggests that targeting mitochondrial hexokinases in combination with HDI treatment could be an effective treatment strategy.
Hexokinases convert glucose to glucose-6-phosphate—the first step in glycolysis—and HK2 has been linked to the “Warburg effect”, the observation that cancer cells mainly generate ATP by glycolysis, as opposed to oxidative phosphorylation [30,31]. HK2 expression is higher in cancer cells compared to normal cells [31] and it has been linked to cancer metastasis, progression and tumor maintenance. In primary pancreatic ductal carcinoma tumor samples, HK2 was most highly expressed in metastases and stable knockdown of HK2 in cell line xenografts was associated with decreased metastatic potential in mice [32]. Knockdown of HK2 in neuroblastoma cell line models also led to decreased tumor establishment and lung metastases in mice injected with knockdown cells versus control cells [33]. HK2 was shown to be required for tumor establishment and maintenance in mouse models of KRAS-mutant lung cancer and ErbB2-driven breast cancer [34]. Knockdown of HK2 or inhibiton of HK2 by 2-deoxyglucose was found to inhibit growth of KRAS mutant human and murine lung cancer cell line models [35]. Thus, HK2 is an attractive target in cancer.
The exact mechanism by which mitochondrial hexokinases such as HK1 and HK2 prevent apoptosis is unclear. Mitochondrial hexokinases have been shown to bind with the voltage-dependent anion channel 1 (VDAC1), giving them direct access to ATP for use as an energy source [36]. Akt has been shown to promote the interaction between hexokinases and VDAC and therefore prevent apoptosis [37]. Some groups have suggested that HK1 acts to prevent the formation of Bax channels at the outer mitochondrial membrane [38]. HK2 is believed to interact with mitochondrial VDAC1 and prevent mitochondrial permeabilization by Bak and Bax through an as yet undefined mechanism [39]. While the interaction between hexokinases and VDAC proteins has been implicated in prevention of apoptosis, deletion of all VDAC isoforms has not been found to have an effect on apoptosis [40], casting doubt on this theory.
Although our report appears to be the first to combine an HDI with compounds that target mitochondrial hexokinases, previous studies have suggested the possibility that HDIs exert their toxic effects via interference with glucose metabolism by hexokinases. Wardell et al. demonstrated that treating the multiple myeloma cell line OPM2 with valproic acid or SAHA resulted in decreased expression of GLUT1 protein as well as expression of its encoding SLC2A1 mRNA [41]. This was accompanied by an increase in HK1 expression. Despite this, hexokinase activity was apparently inhibited by HDI treatment [41]. They conclude that apoptosis induced by HDIs is due to their ability to affect metabolism. Amoedo et al. saw similar effects when they treated H460 lung cancer cells with sodium butyrate, noting decreased levels of SLC2A1 expression and increased mitochondrial HK1 expression [42]. However, they found that HDI treatment increased hexokinase activity. It is not clear if the observed differences between the two reports are due to model selection or the specific HDI used. However, e?ects of HDIs on cellular metabolic function do seem to contribute to their toxicity.
It is interesting to note that half of the cell lines that showed sensitivity to the romidepsin and clotrimazole/bifonazole combination bear KRAS mutations, specifically the A549, HCT-116, and MDA-MB-231 lines. We previously demonstrated the selective activity of romidepsin in cells bearing KRAS mutation when downstream KRAS signaling pathways were inhibited [27]. This work targeting a hexokinase in combination with romidepsin offers a different strategy by which to exploit the KRAS-directed activity of HDIs, and perhaps a more direct one.
The question is invariably raised as to whether targeting hexokinases will be too toxic for clinical translation. This can be answered in part by the in vivo studies noted above, in which knockout of hexokinases did not lead to excess toxicity. Preclinical studies with clotrimazole to date have been limited. Some reports have suggested that it might be useful alone or in combination with other drugs for the treatment of cancer [43]. Clotrimazole is effective as a single agent in breast, melanoma, lung and colon cancer models in vitro [44–46]. The combination of clotrimazole with cisplatin synergistically increased cisplatin efficacy in a glioblastoma cell line model [47] and the combination of clotrimazole with imatinib led to synergistic toxicity in the T47D breast cancer cell line [48]. Clotrimazole as a single agent was found to increase the survival of rats with intracranial gliomas [49] and decrease tumor growth in an oral squamous cell carcinoma xenograft model [50]. Despite these promising results, the clinical use of clotrimazole as a systemic therapy is limited by its low water solubility and poor bioavailability [43].
A number of interesting combinations of HDIs with targeted therapies have recently been suggested. The combination of the polo-like kinase inhibitor volasertib and the HDI belinostat has been shown to synergistically induce apoptosis in lymphoma cell line models [51]. Belinostat in combination with the CDK inhibitor seliciclib resulted in high levels of apoptosis in non-small cell lung cancer models and did not depend on p53 activity [52]. We recently demonstrated that either romidepsin or belinostat in combination with inhibitors of the MAPK and/or PI3K pathways induced apoptosis selectively in cancers harboring mutant Ras [27]. Those results mirrored those of Ischenko et al., who demonstrated increased efficacy of trichostatin A when combined with a MEK and a PI3K inhibitor [53]. Our data with clotrimazole point to an entirely new therapeutic target not yet exploited in cancer cells and suggest that HDIs sensitize cells to this novel target. We are pursuing medicinal chemistry strategies to develop a compound more suited for preclinical and clinical testing.
Supplementary Material
Acknowledgements
We appreciate the editorial assistance of George Leiman. This research was supported in part by the National Cancer Institute and the Eunice Kennedy Shriver National Institute of Child Health and Human Development. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations:
- HDI
histone deacetylase inhibitor
- RD
romidepsin
- P-gp
P-glycoprotein
- MAPK
mitogen activated protein kinase
- HK
hexokinase
- CTZ
clotrimazole
- BIF
bifonazole
- DKO
double knock-out
Footnotes
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.yexcr.2018.12.012.
References
- [1].Moskowitz AJ, Horwitz SM, Targeting histone deacetylases in T-cell lymphoma, Leuk. Lymphoma (2016) 1–14. [DOI] [PubMed] [Google Scholar]
- [2].Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao J, Reilly J, Ricker J, Richon V, Frankel S, Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL), Blood 109 (2007) 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Olsen E, Kim Y, Kuzel T, Pacheco T, Foss F, Parker S, Frankel S, Chen C, Ricker J, Arduino J, Duvic M, Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma, J. Clin. Oncol. 25 (2007) 3109–3115. [DOI] [PubMed] [Google Scholar]
- [4].Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, Scarisbrick J, Reddy S, Robak T, Becker JC, Samtsov A, McCulloch W, Kim YH, Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma, J. Clin. Oncol. 28 (2010) 4485–4491. [DOI] [PubMed] [Google Scholar]
- [5].Piekarz R, Frye R, Turner M, Wright J, Allen S, Kirschbaum M, Zain J, Prince H, Leonard J, Geskin L, Reeder C, Joske D, Figg W, Gardner E, Steinberg S, Jaffe E, Stetler-Stevenson M, Lade S, Fojo A, Bates S, Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma, J. Clin. Oncol. 27 (2009) 5410–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Piekarz RL, Frye R, Prince HM, Kirschbaum MH, Zain J, Allen SL, Jaffe ES, Ling A, Turner M, Peer CJ, Figg WD, Steinberg SM, Smith S, Joske D, Lewis I, Hutchins L, Craig M, Fojo AT, Wright JJ, Bates SE, Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma, Blood 117 (2011) 5827–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Coiffier B, Pro B, Prince HM, Foss F, Sokol L, Greenwood M, Caballero D, Borchmann P, Morschhauser F, Wilhelm M, Pinter-Brown L, Padmanabhan S, Shustov A, Nichols J, Carroll S, Balser J, Balser B, Horwitz S, Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy, J. Clin. Oncol 30 (2012) 631–636. [DOI] [PubMed] [Google Scholar]
- [8].O’Connor OA, Horwitz S, Masszi T, Van Hoof A, Brown P, Doorduijn J, Hess G, Jurczak W, Knoblauch P, Chawla S, Bhat G, Choi MR, Walewski J, Savage K, Foss F, Allen LF, Shustov A, Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: results of the Pivotal Phase II BELIEF (CLN-19) Study, J. Clin. Oncol. 33 (2015) 2492–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Grant C, Rahman F, Piekarz R, Peer C, Frye R, Robey RW, Gardner ER, Figg WD, Bates SE, Romidepsin: a new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors, Expert Rev. Anticancer Ther. 10 (2010) 997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Grant S, Dai Y, Histone deacetylase inhibitors and rational combination therapies, Adv. Cancer Res 116 (2012) 199–237. [DOI] [PubMed] [Google Scholar]
- [11].Carbone C, Di Gennaro E, Piro G, Milone MR, Pucci B, Caraglia M, Budillon A, Tissue transglutaminase (TG2) is involved in the resistance of cancer cells to the histone deacetylase (HDAC) inhibitor vorinostat, Amino Acids (2016). [DOI] [PubMed] [Google Scholar]
- [12].Falkenberg KJ, Newbold A, Gould CM, Luu J, Trapani JA, Matthews GM, Simpson KJ, Johnstone RW, A genome scale RNAi screen identifies GLI1 as a novel gene regulating vorinostat sensitivity, Cell Death Differ. 23 (2016) 1209–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lee JS, Paull K, Alvarez M, Hose C, Monks A, Grever M, Fojo AT, Bates SE, Rhodamine efflux patterns predict P-glycoprotein substrates in the National cancer Institute drug screen, Mol. Pharmacol. 46 (1994) 627–638. [PubMed] [Google Scholar]
- [14].Xiao JJ, Foraker AB, Swaan PW, Liu S, Huang Y, Dai Z, Chen J, Sadee W, Byrd J, Marcucci G, Chan KK, E?ux of depsipeptide FK228 (FR901228, NSC-630176) is mediated by P-glycoprotein and multidrug resistance-associated protein 1, J. Pharmacol. Exp. Ther. 313 (2005) 268–276. [DOI] [PubMed] [Google Scholar]
- [15].Piekarz RL, Robey RW, Zhan Z, Kayastha G, Sayah A, Abdeldaim AH, Torrico S, Bates SE, T-cell lymphoma as a model for the use of histone deacetylase inhibitors in cancer therapy: impact of depsipeptide on molecular markers, therapeutic targets, and mechanisms of resistance, Blood 103 (2004) 4636–4643. [DOI] [PubMed] [Google Scholar]
- [16].Robey RW, Zhan Z, Piekarz RL, Kayastha GL, Fojo T, Bates SE, Increased MDR1 expression in normal and malignant peripheral blood mononuclear cells obtained from patients receiving depsipeptide (FR901228, FK228, NSC630176), Clin. Cancer Res 12 (2006) 1547–1555. [DOI] [PubMed] [Google Scholar]
- [17].Bates S, Zhan Z, Steadman K, Obrzut T, Luchenko V, Frye R, Robey R, Turner M, Gardner E, Figg W, Steinberg S, Ling A, Fojo T, To K, Piekarz R, Laboratory correlates for a phase II trial of romidepsin in cutaneous and peripheral T-cell lymphoma, Br. J. Haematol. 148 (2010) 256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chakraborty AR, Robey RW, Luchenko VL, Zhan Z, Piekarz RL, Gillet JP, Kossenkov AV, Wilkerson J, Showe LC, Gottesman MM, Collie NL, Bates SE, MAPK pathway activation leads to Bim loss and histone deacetylase inhibitor resistance: rationale to combine romidepsin with an MEK inhibitor, Blood 121 (2013) 4115–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bhola PD, Letai A, Mitochondria-judges and executioners of cell death sentences, Mol. Cell 61 (2016) 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB, Hay N, Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak, Mol. Cell 16 (2004) 819–830. [DOI] [PubMed] [Google Scholar]
- [21].Ierano C, Chakraborty AR, Nicolae A, Bahr JC, Zhan Z, Pittaluga S, Bates SE, Robey RW, Loss of the proteins Bak and Bax prevents apoptosis mediated by histone deacetylase inhibitors, Cell Cycle 12 (2013) 2829–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shao W, Growney JD, Feng Y, O’Connor G, Pu M, Zhu W, Yao YM, Kwon P, Fawell S, Atadja P, Activity of deacetylase inhibitor panobinostat (LBH589) in cutaneous T-cell lymphoma models: defining molecular mechanisms of resistance, Int J. Cancer 127 (2010) 2199–2208. [DOI] [PubMed] [Google Scholar]
- [23].Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B, Role of BAX in the apoptotic response to anticancer agents, Science 290 (2000) 989–992. [DOI] [PubMed] [Google Scholar]
- [24].Wang C, Youle RJ, Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1’s inhibitory effect on Bak, Oncogene 31 (2011) 3177–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pastorino JG, Shulga N, Hoek JB, Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis, J. Biol. Chem. 277 (2002) 7610–7618. [DOI] [PubMed] [Google Scholar]
- [26].Penso J, Beitner R, Clotrimazole and bifonazole detach hexokinase from mitochondria of melanoma cells, Eur. J. Pharmacol. 342 (1998) 113–117. [DOI] [PubMed] [Google Scholar]
- [27].Bahr JC, Robey RW, Luchenko V, Basseville A, Chakraborty AR, Kozlowski H, Pauly GT, Patel P, Schneider JP, Gottesman MM, Bates SE, Blocking downstream signaling pathways in the context of HDAC inhibition promotes apoptosis preferentially in cells harboring mutant Ras, Oncotarget 7 (2016) 69804–69815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Luchenko VL, Litman T, Chakraborty AR, Heffner A, Devor C, Wilkerson J, Stein W, Robey RW, Bangiolo L, Levens D, Bates SE, Histone deacetylase inhibitor-mediated cell death is distinct from its global effect on chromatin, Mol. Oncol 8 (2014) 1379–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Amiri-Kordestani L, Luchenko V, Peer CJ, Ghafourian K, Reynolds J, Draper D, Frye R, Woo S, Venzon D, Wright J, Skarulis M, Figg WD, Fojo T, Bates SE, Piekarz RL, Phase I trial of a new schedule of romidepsin in patients with advanced cancers, Clin. Cancer Res 19 (2013) 4499–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Warburg O, On the origin of cancer cells, Science 123 (1956) 309–314. [DOI] [PubMed] [Google Scholar]
- [31].Mathupala SP, Ko YH, Pedersen PL, Hexokinase-2 bound to mitochondria: cancer’s stygian link to the "Warburg Effect" and a pivotal target for effective therapy, Semin Cancer Biol. 19 (2009) 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Anderson M, Marayati R, Moffitt R, Yeh JJ, Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer, Oncotarget 8 (2017) 56081–56094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Botzer LE, Maman S, Sagi-Assif O, Meshel T, Nevo I, Yron I, Witz IP, Hexokinase 2 is a determinant of neuroblastoma metastasis, Br. J. Cancer 114 (2016) 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, Jha AK, Smolen GA, Clasquin MF, Robey B, Hay N, Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer, Cancer Cell 24 (2013) 213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang H, Wang L, Zhang Y, Wang J, Deng Y, Lin D, Inhibition of glycolytic enzyme hexokinase II (HK2) suppresses lung tumor growth, Cancer Cell Int 16 (2016) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shoshan-Barmatz V, Krelin Y, Shteinfer-Kuzmine A, Arif T, Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics, Front Oncol 7 (2017) 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Stiles BL, PI-3-K and AKT: onto the mitochondria, Adv. Drug Deliv. Rev. 61 (2009) 1276–1282. [DOI] [PubMed] [Google Scholar]
- [38].Schindler A, Foley E, Hexokinase 1 blocks apoptotic signals at the mitochondria, Cell Signal 25 (2013) 2685–2692. [DOI] [PubMed] [Google Scholar]
- [39].Krasnov GS, Dmitriev AA, Lakunina VA, Kirpiy AA, Kudryavtseva AV, Targeting VDAC-bound hexokinase II: a promising approach for concomitant anticancer therapy, Expert Opin. Ther. Targets 17 (2013) 1221–1233. [DOI] [PubMed] [Google Scholar]
- [40].Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD, Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death, Nat. Cell Biol. 9 (2007) 550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wardell S, Ilkayeva O, Wieman H, Frigo D, Rathmell J, Newgard C, McDonnell D, Glucose metabolism as a target of histone deacetylase inhibitors, Mol. Endocrinol. 23 (2009) 388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Amoêdo ND, Rodrigues MF, Pezzuto P, Galina A, da Costa RM, de Almeida FC, El-Bacha T, Rumjanek FD, Energy metabolism in H460 lung cancer cells: effects of histone deacetylase inhibitors, PLoS One 6 (2011) e22264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kadavakollu S, Stailey C, Kunapareddy CS, White S, Clotrimazole as a cancer drug: a short review, Med Chem. (Los Angel.) 4 (2014) 722–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Adinolfi B, Carpi S, Romanini A, Da Pozzo E, Castagna M, Costa B, Martini C, Olesen SP, Schmitt N, Breschi MC, Nieri P, Fogli S, Analysis of the antitumor activity of clotrimazole on A375 human melanoma cells, Anticancer Res 35 (2015) 3781–3786. [PubMed] [Google Scholar]
- [45].Furtado CM, Marcondes MC, Sola-Penna M, de Souza ML, Zancan P, Clotrimazole preferentially inhibits human breast cancer cell proliferation, viability and glycolysis, PLoS One 7 (2012) e30462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Penso J, Beitner R, Clotrimazole decreases glycolysis and the viability of lung carcinoma and colon adenocarcinoma cells, Eur. J. Pharmacol. 451 (2002) 227–235. [DOI] [PubMed] [Google Scholar]
- [47].Khalid MH, Shibata S, Hiura T, Effects of clotrimazole on the growth, morphological characteristics, and cisplatin sensitivity of human glioblastoma cells in vitro, J. Neurosurg. 90 (1999) 918–927. [DOI] [PubMed] [Google Scholar]
- [48].Motawi TM, Sadik NA, Fahim SA, Shouman SA, Combination of imatinib and clotrimazole enhances cell growth inhibition in T47D breast cancer cells, Chem. Biol. Interact. 233 (2015) 147–156. [DOI] [PubMed] [Google Scholar]
- [49].Khalid MH, Tokunaga Y, Caputy AJ, Walters E, Inhibition of tumor growth and prolonged survival of rats with intracranial gliomas following administration of clotrimazole, J. Neurosurg. 103 (2005) 79–86. [DOI] [PubMed] [Google Scholar]
- [50].Wang J, Jia L, Kuang Z, Wu T, Hong Y, Chen X, Leung WK, Xia J, Cheng B, The in vitro and in vivo antitumor effects of clotrimazole on oral squamous cell carcinoma, PLoS One 9 (2014) e98885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Nguyen T, Parker R, Hawkins E, Holkova B, Yazbeck V, Kolluri A, Kmieciak M, Rahmani M, Grant S, Synergistic interactions between PLK1 and HDAC inhibitors in non-Hodgkin’s lymphoma cells occur in vitro and in vivo and proceed through multiple mechanisms, Oncotarget 8 (2017) 31478–31493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ong PS, Wang L, Chia DM, Seah JY, Kong LR, Thuya WL, Chinnathambi A, Lau JY, Wong AL, Yong WP, Yang D, Ho PC, Sethi G, Goh BC, A novel combinatorial strategy using Seliciclib(®) and Belinostat(®) for eradication of non-small cell lung cancer via apoptosis induction and BID activation, Cancer Lett. 381 (2016) 49–57. [DOI] [PubMed] [Google Scholar]
- [53].Ischenko I, Petrenko O, Hayman MJ, A MEK/PI3K/HDAC inhibitor combination therapy for KRAS mutant pancreatic cancer cells, Oncotarget 6 (2015) 15814–15827. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.