

Abstract
Background
Epigenetic mechanisms regulate chromatin accessibility patterns that govern interaction of transcription machinery with genes and their cis-regulatory elements. Mutations that affect epigenetic mechanisms are common in cancer. Because epigenetic modifications are reversible many anticancer strategies targeting these mechanisms are currently under development and in clinical trials.
Main body
Here we review evidence suggesting that epigenetic therapeutics can deactivate immunosuppressive gene expression or reprogram tumor cells to activate antigen presentation mechanisms. In addition, the dysregulation of epigenetic mechanisms commonly observed in cancer may alter the immunogenicity of tumor cells and effectiveness of immunotherapies.
Conclusions
Therapeutics targeting epigenetic mechanisms may be helpful to counter immune evasion and improve the effectiveness of immunotherapies.
Keywords: Epigenetic therapy, Immune evasion, Immunotherapy, DNA methylation, Histone methyltransferase
Background
Epigenetic mechanisms in cancer
Heritable patterns of gene expression not due to DNA sequence variation are maintained and regulated by epigenetic mechanisms. These mechanisms allow genetically identical cells to have distinct gene expression patterns that govern specialization of cellular identity. Patterns of DNA methylation, histone post-translational modifications or nucleosome positioning are epigenetic marks that reversibly store and transmit heritable information (Fig. 1). Writers of epigenetic marks catalyze DNA methylation or post-translational modifications of histones, such as methylation or acetylation of N-terminal histone “tails” extending from the nucleosome structure. These epigenetic marks may be recognized by protein complexes that either alter the chromatin architecture further or regulate enzymatic processes such as transcription factor binding and RNA polymerase processivity. Epigenetic marks may be removed by enzymes such as histone deacetylases or demethylases. In addition, chromatin remodelers can mobilize or exchange histones. Together these mechanisms maintain chromatin accessibility patterns that govern interaction of transcription machinery with genes and cis-regulatory regions. Mutations in genes that regulate the cellular epigenetic state are among the commonest class of mutations found in cancer [1]. These mutations result in reprogrammed gene expression that can directly contribute to cancer and cooperate with other genetic events such as mutations of oncogenes or tumor suppressors, affecting signal transduction and cell life/death pathways. Because epigenetic marks are reversible, many anticancer strategies targeting these mechanisms to rebalance the epigenome back to a more normal state are currently under development and in clinical trials. In addition, the widespread adoption of targeted immunotherapies for cancer has led to the need for a deeper understanding for how epigenetic mechanisms may allow tumor cells to escape immune surveillance.
Fig. 1.
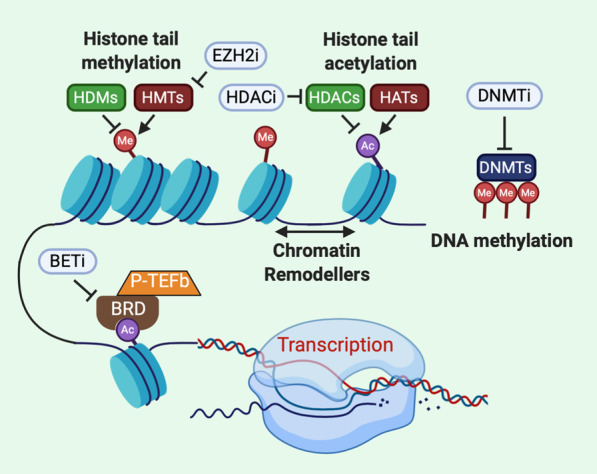
Epigenetic regulation of chromatin accessibility and gene expression. Nucleosomes (blue cylinders) are formed by DNA wrapped around a histone octamer which allows DNA to be condensed into chromatin and finally chromosomes. Epigenetic mechanisms dynamically tune chromatin accessibility especially at cis regulatory elements of gene expression. Post-translational modification of N- and C-terminal histone “tails” regulate nucleosome stability, chromatin compaction and serve as docking sites for proteins that recognize epigenetic marks such as bromodomain proteins (BRD). Histone methyltransferases (HMTs) catalyze the transfer of a methyl group (Me) onto histone tails from donor S-adenyl methionine while this mark is removed by histone demethylases (HDM). Similarly, histone acetyltransferases (HATs) transfer an acetyl group (Ac) from acetyl coenzyme A to histone lysine residues which weakens histone interaction with DNA to increase chromatin accessibility. Histone deacetylases (HDACs) remove the acetyl mark from histones, decreasing chromatin accessibility and subduing gene expression. Chromatin remodelers such as SWI/SNF mobilize and reposition nucleosomes. DNA methyltransferases (DNMT) methylate CpG islands near transcription start sites that inhibit gene expression by impeding transcription factor binding to DNA. Epigenetic inhibitors (white ovals) have been developed to potentially restore a normal cellular epigenetic state to tumor cells. EZH2i, such as tazemetostat, specifically inhibit the gene suppressive methylation of histone H3 by PRC2. HDACi such as entinostat or vorinostat inhibit histone deacetylation to reactivate gene expression. Inhibitors of bromodomain and extra terminal domain proteins (BETi) such as JQ1 or BMS-986158 suppress aberrant gene expression driven by increased BRD activity in cancer cells. DNMT inhibitors (DNMTi) such as azacytidine promote DNA hypomethylation and reactivate expression of tumor suppressor genes
Patterns of DNA methylation regulate gene expression
Methylation of DNA in somatic cells occurs primarily on cytosines that precede a guanine (CpG) and is important for regulation of gene expression. Palindromic methylation patterns are maintained in the genome and transmitted through the germline. CpG dinucleotides are often concentrated within CpG-rich DNA “islands” located around transcription start sites (TSSs) [2]. Hypermethylation of CpG islands may repress expression of nearby genes by inhibiting transcription factor binding and recruiting methyl-CpG binding proteins which in turn affect repressive histone modifying enzymes [3]. Global and local gene methylation pattern changes are frequently found in cancer and atypical methylation patterns have been used to differentiate tumor subtypes [4]. Loss of repeat region methylation is often observed in tumors, and hypomethylation of DNA regions may promote the expression of proto-oncogenes such as observed for ERBB2 and RAS [5]. In addition, hypermethylation of specific CpG-rich regions may silence expression of tumor suppressors such as observed for Rb and p16 [6, 7].
The DNA methyltransferases (DNMT) family of enzymes is responsible for establishing and maintaining patterns of DNA methylation. DNMT3A and DNMT3B establish de novo DNA methylation patterns that are maintained by DNMT1, which recognizes DNA methylation and directs daughter strand methylation after replication [8]. DNA demethylation is expedited by the TET enzymes which convert methyl cytosine into hydroxymethylcytosine, a base modification not recognized by DNMT1 [9]. Mutations of DNMT3A or TET enzymes that cause loss of function are often observed in cancer, dysregulating patterns of DNA methylation.
Histone acetylation regulates chromatin accessibility
Control of lysine acetylation in the histone tail region is important for regulation of chromatin structure, transcription and DNA repair. This highly dynamic modification is regulated by histone lysine acetyltransferases (HATs) and histone deacetylases (HDACs). HATs transfer the acetyl group from acetyl-coenzyme A to the amino group of a lysine in the histone, thereby neutralizing the lysine’s positive charge and weakening the interaction of histone with DNA. In general, histone acetylation causes a more relaxed and accessible chromatin structure that favors binding of proteins such as transcription factors. Thus, acetylation of chromatin is generally associated with transcriptional activation while deacetylation is associated with gene repression [10]. HDACs play a key role in gene expression by removal of the activating histone acetylation and may also have other roles in the cell by controlling acetylation of non-histone and non-nuclear proteins. HDACs are often found overexpressed in cancer where they may silence tumor suppressor genes. In addition, HDACs may be aberrantly recruited to target genes by overexpressed transcription factors or chimeric transcription factors created by chromosomal fusions [10].
Bromodomain-containing proteins recognize acetylated histone
The bromodomain and extra-terminal (BET) protein family that includes BRD2, BRD3 and BRD4 is one of the best characterized families of epigenetic reader proteins in cancer [11, 12]. BET proteins share a conserved structural element consisting of two bromodomains that recognize acetylated lysine on the N-terminal tails of histones H3 and H4. In addition, BRD4 associates with the positive transcription elongation factor (P-TEFb) protein that promotes transcription elongation at paused sites by activating RNA polymerase II [13]. BRD4 is frequently located at transcription start sites, enhancer and super-enhancer regions. Significantly, BRD4 has been reported to promote expression of many transcription factors with roles in cancer development and progression such as Myc. In addition, BRD4 has been reported to recruit the histone methyltransferase NSD2 to the estrogen receptor alpha (ERα) gene to increase ER expression in breast cancer cells [14]. Increased BRD4 expression is associated with poor prognosis in melanoma and hepatocellular carcinoma. In addition, the majority of patients that develop midline carcinoma have chromosomal rearrangements that create a fusion protein between BRD3 or BRD4 and the NUT protein [13]. The BRD-Nut fusion maintains cancer cells in an undifferentiated state of self-renewal by recruiting HATs and sequestering cofactors normally associated with activated, acetylated chromatin away from normal gene targets, leading to reduced expression of differentiation-associated genes. Treatment of BRD4-NUT expressing cell lines with a small molecular inhibitor of BET domain acetyl-lysine binding led to a reactivation of gene expression and induction of differentiation [13]. This finding stimulated the ongoing development of pharmacological approaches to inhibit BET domain proteins.
Histone methylation states regulate transcription
N-terminal and C-terminal tails of histones that extend beyond the nucleosome core are subject to post-translational modifications that influence downstream biological processes such as transcription, replication and chromosomal stability. The basic amino acids lysine, arginine and histidine present in histone tails can serve as a target of methylation. Reversible methylation of histones is orchestrated by histone methyltransferases (HMTs), while histone demethylases (HDMs) remove these marks (Fig. 1). The enzymatic activity of almost all lysine methyltransferases resides in the Su(var)3–9, enhancer-of-zeste and trithorax (SET) catalytic domain. The prime exception being the lysine methyltransferase DOT1L1 which has a unique enzymatic domain. Arginine methyl transferases contain a conserved S-adenosyl methionine (SAM)-binding catalytic core of about 350 amino acids [15]. These enzymatic domains have pockets that bind SAM to be used as a donor co-factor for the transfer of methyl groups to substrates. Histone lysine residues can be modified to mono-, di- or tri-methylated forms (me1, me2 or me3) and histidine can be mono-methylated, but this modification is rare. Arginine may be mono- or di-methylated, and di-methylation may be either symmetrical, meaning that methyl groups are added to both nitrogen atoms in the side chain or asymmetrical in which two methyl groups are added to only one of the side chain nitrogen atoms. Demethylation of histones is accomplished by two main classes of HDMs: the flavin adenine dinucleotide (FAD)-dependent amine oxidases and the Fe(II)/2-oxoglutarate (2-OG)-dependent Jumonji C domain family [15]. Patterns of histone methylation state can change based on cell type, tissue type or cell cycle phase. The reversible nature of histone methylation is important for the response to factors such as DNA damage, mitogen signaling and environmental stress because the balance between the methylated and demethylated states of histones at specific lysine residues can regulate transcriptional activity. For instance, lysine methylation of histone H3 at amino acid residue 4 (H3K4), 36 (H3K36) and 79 (H3K79) are associated with a gene activation state while methylation at lysine 9 (H3K9), 20 (H3K20) and 27 (H3K27) is associated with suppression of gene expression [16]. Loss or gain of HMT or HDM activity can result from missense mutation, deletion, amplification or chromosomal rearrangement affecting the genes encoding these enzymes. Dysfunctional histone methylation and the resultant aberrant gene expression have often been linked to a range of malignancies and clinical outcome.
Enhancer-of-zeste homolog 2 (EZH2) is the catalytic component of the polycomb repressive complex 2 (PRC2) that trimethylates histone H3 at lysine 27 (H3K27) which is associated with chromatin compaction and transcriptional repression [17]. EZH2-mediated H3K27 methylation is an important regulator in several cellular pathways including cell cycle regulation, X-chromosome inactivation and metastasis. EZH2 expression is generally found to be increased in metastatic tumors compared to normal tissues or primary tumor specimens. Increased EZH2 promotes cancer cell growth and an epithelial-mesenchymal transition. Heterozygous activating mutations of EZH2 are found in germinal center-type diffuse large B cell lymphoma and more rarely in thyroid cancer and malignant melanoma [18]. In lymphoma, gain of function EZH2 mutations repress expression of tumor suppressor and late B cell genes, thus locking the B cell in a state of continuous proliferation at the germinal center stage of differentiation. In addition, loss of function and deletion of EZH2 is found in MDS and AML and is associated with global decreases in the repressive H3K27me3 mark which activates expression of oncogenes [19]. Thus, EZH2 can have either an oncogenic or tumor suppressor function, depending on the cellular context.
Main text
Epigenetic therapies may synergize with immunotherapy
A recurring phenotype of cancer cells is aberrant epigenetic mechanisms causing the downregulation of genes involved in the processing or presentation of tumor antigens, leading to immune evasion [20–22]. Many pharmacological agents have been developed that inhibit epigenetic mechanisms and reprogram tumor cell-specific patterns of DNA methylation or post-translational histone modifications. Furthermore, the complex interplay between immune, cancer and stromal cells is important for antitumor immunity. The therapeutic potential of combining epigenetic therapies with immunotherapy was first indicated by reports demonstrating that immune- or inflammatory-related gene signatures were increased upon inhibition of epigenetic mechanisms [23–26]. Furthermore, early clinical studies of combination therapies found that non-small cell lung cancer (NSCLC) patients treated with either DNMT or HDAC inhibitors achieved durable treatment responses when subsequently administered PD1/PD-L1 immune checkpoint inhibitors [27, 28]. These studies and others suggest that epigenetic inhibitors may increase the efficacy of immunotherapy by: (1) enhancing antigenicity and presentation of tumor-associated antigens, (2) reprogramming the tumor microenvironment to counteract immunosuppressive mechanisms and (3) reversing cytotoxic T cell exhaustion (Fig. 2). As our knowledge of how epigenetic mechanisms govern tumor antigen presentation and immune cell function improves, strategies that take advantage of these mechanisms will be important to devise rational combinatorial approaches that bolster response to immunotherapies.
Fig. 2.
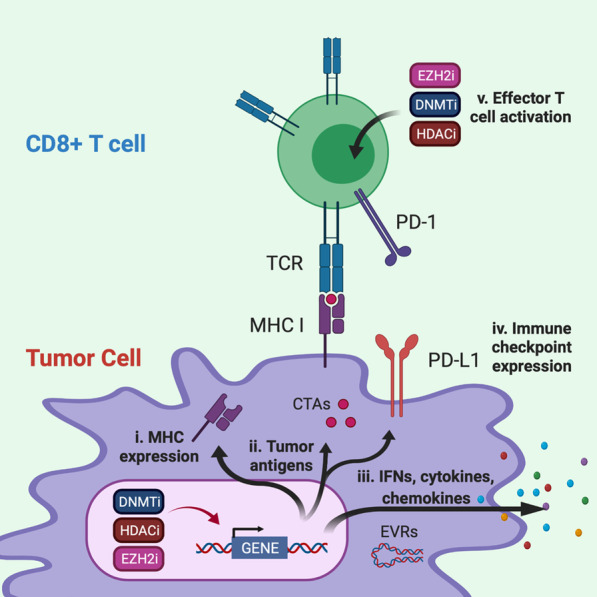
Leveraging inhibition of epigenetic mechanisms to improve immunotherapy. DNA methyltransferase inhibitors (DNMTi), histone deacetylase inhibitors (HDACi) and an inhibitor of histone methylation on histone H3 at lysine 27 (EZH2i) activate immunomodulatory mechanisms that may improve immunotherapy by: (i) increasing gene expression and activation of antigen presentation mechanisms; (ii) increasing gene expression of tumor-associated antigens such as cancer testis antigens (CTAs) MAGE and NY-ESO-1; (iii) upregulating inflammatory genes and pathways that rebalance the secretion of interferons (IFNs), cytokine and chemokines from tumor cells including the expression of normally silent endogenous retroviruses RNAs (EVRs) that activate the interferon response; (iv) upregulating targets of immune checkpoint blockade such as PD-1/PD-1L on both tumors and lymphocytes; and (v) activating the effector T cell population by promoting differentiation of naïve T cells to cytotoxic T cells and inhibiting T cell exhaustion mechanisms
DNA hypomethylation agents synergize with immunotherapy
Inhibitors of DNA methyltransferases (DNMTi) were developed in the 1970s to target aberrant methylation patterns in cancer cells. DNMTi such as 5-azacytidine, decitabine and guadecitabine cause global hypomethylation, and their use as anti-cancer agents has been approved for use in patients with myelodysplastic syndrome or certain leukemias to reactivate tumor suppressor genes. These azanucleosides substitute nitrogen for carbon at the C-5 position of the pyrimidine ring and when incorporated into DNA irreversibly bind DNMT1 resulting in DNMT1 degradation and decreased DNA methylation [29]. The resulting loss of DNA methylation favors the re-expression of aberrantly silenced proteins, including tumor suppressor genes, cancer-associated antigens and components of the antigen presentation machinery.
Emerging evidence suggests that DNMTi hypomethylating agents have potential to improve immunogenicity and immune recognition of cancer cells. Numerous preclinical studies demonstrate that hypomethylating agents significantly increase expression of immunomodulatory pathway genes and tumor antigen presentation mechanisms in a variety of human epithelial cancer cell lines [23, 30]. Antigen presentation mechanisms found upregulated include the type I interferon response pathway upstream of antigen presentation, immune proteasome subunits and endoplasmic reticulum transporters involved in antigen processing prior to presentation and MHC class I genes required for antigen presentation to cytotoxic T cells [23, 24, 31]. For instance, in NSCLC cell lines, 5-azacytidine treatment upregulates the JAK/STAT pathway stimulating the expression of genes involved in antigen presentation and increasing expression of PD-L1, a key ligand-mediator of immune tolerance [27, 32]. DNMTi were also reported to activate the expression of endogenous retroviral double-stranded RNAs (EVRs) that are normally hypermethylated and transcriptionally silent, leading to the induction of a type I interferon response and the activation of MHC I expression [26, 33, 34]. Recent studies indicate that expression of EVRs in renal cell carcinoma, breast, colon and head and neck squamous cell tumors correlated with increased immune infiltration, checkpoint pathway upregulation and higher CD8 + T cell infiltration [35, 36]. Significantly, high EVR expression in tumors correlated with improved response to anti-CTLA4 in melanoma patients and PD-1/PD-L1 blockade in renal cell carcinoma patients [26, 35]. Additionally, DNMTi can upregulate expression of cancer testis antigens (CTAs) which are promising immunotherapy targets expressed in early embryonic and germ cells but normally silenced in mature somatic cells by promoter CpG island DNA methylation [37, 38]. CTAs, such as NY-ESO-1 or MAGE-a, are expressed in a variety of tumor cell types and increased expression is associated with advanced or metastatic disease stage and poor prognosis [39–41].
DNMTi such as decitabine not only affect tumor cells but may also have direct effects on antigen-specific CD8 + T cells. Genome-wide de novo DNA-methylation programs persist in CD8 + T cells after PD-1 immune checkpoint blockade therapy and this restricts the durability of therapy by promoting terminal differentiation of exhausted T cells. Studies in tumor-bearing mice indicate that prior treatment with decitabine followed by administration of anti-PD-L1 prevents the acquisition of exhaustion-associated methylation programs, and T cells retain a greater potential for expansion after immune checkpoint blockade [42]. Furthermore, in a murine ovarian cancer model the efficacy of anti-CTLA-4 was potentiated by combination with decitabine which increased differentiation of naive T cells into effector T cells. This in turn prolonged cytotoxic lymphocyte responses as well as mouse survival [43].
Early-stage clinical trials testing the second-generation DNMTi guadecitabine with ipilimumab (anti-CTLA-4) in metastatic melanoma patients showed promising tumor immunomodulatory and clinical activities [44]. Results indicate that combination therapies utilizing hypomethylating agents promoted upregulation of HLA class I molecules and IFN gamma signaling pathways as well as increased tumor infiltration by CD8 + T cells, hallmarks for sensitivity to immunotherapy [44]. In a phase I clinical trial of relapsed epithelial ovarian cancer, administration of decitabine significantly increased NY-ESO-1 expression, and T cell responses were increased after treatment with the NY-ESO-1 epitope, leading to a favorable response in 60% of patients [45]. Taken together these studies have guided the design and initiation of additional clinical trials that test DNMTi hypomethylating agents in combination with immunotherapies (Table 1).
Table 1.
Clinical trials evaluating combination of epigenetic inhibitors and immunotherapies
| Epigenetic therapy | Immunotherapy | Cancer type | Phase, Trial ID |
|---|---|---|---|
| Histone deacetylase inhibitors (target) | |||
| CXD101 (Pan HDAC) | Nivolumab (PD-1) | Colorectal cancer | I/II, NCT03993626 |
| Domatinostat (HDAC1,2,3) | Avelumab (PD-L1) | GI cancer | II, NCT03812796 |
| Entinostat (HDAC1,2,3) | Pembrolizumab (PD-1) | Bladder cancer | II, NCT03978624 |
| Melanoma | II, NCT03765229 | ||
| MDS | I, NCT02936752 | ||
| Metastatic uveal melanoma | II, NCT02697630 | ||
| Atezolizumab (PD-L1) | Breast cancer | I/II, NCT03280563 | |
| Nivolumab (PD-1) | Cholangiocarcinoma, pancreatic adenocarcinoma | II, NCT03250273 | |
| Aldesleukin (IL-2) | Renal cell carcinoma | I/II, NCT01038778 | |
| Nivolumab (PD-1), Ipilimumab (CTLA-4) | Breast cancer | I, NCT02453620 | |
| Mocetinostat (Pan HDAC) | Durvalumab (PD-L1) | NSCLC | I/II, NCT02805660 |
| Tinostamustine (Pan HDAC) | Nivolumab (PD-1) | Melanoma | I, NCT03903458 |
| Vorinostat (Pan HDAC) | Pembrolizumab (PD-1) | Lymphomas | I, NCT03150329 |
| Renal cell carcinoma | I, NCT02619253 | ||
| NSCLC | I/II, NCT02638090 | ||
| Head and neck | I/II, NCT02538510 | ||
| DNA methyltransferase inhibitors | |||
| Azacytidine | Avelumab (PD-L1) | DLBCL | III, NCT02951156 |
| Alemtuzumab (CD52) | Myeloid malignancies | II, NCT02497404 | |
| Pembrolizumab (PD-1) | AML | II, NCT02845297 | |
| AML | II, NCT03769532 | ||
| Pancreatic cancer | II, NCT03264404 | ||
| MDS | II, NCT03094637 | ||
| Oral azacytidine (CC-486) | Pembrolizumab (PD-1) | Ovarian cancer | II, NCT02900560 |
| NSCLC | II, NCT02546986 | ||
| Melanoma | II, NCT02816021 | ||
| Decitabine | Pembrolizumab (PD-1) | T cell lymphomas | II, NCT03240211 |
| Lymphomas | I, NCT03445858 | ||
| AML | I, NCT03969446 | ||
| Breast cancer | II, NCT02957968 | ||
| Anti-PD-1 antibody | Solid tumors | I/II, NCT02961101 | |
| Dendritic cell vaccine (NY-ESO-1, MAGE-A1 MAGE-A3) | Pediatric brain tumors | I/II, NCT02332889 | |
| Guadecitabine | Atezolizumab (PD-L1) | Urothelial carcinoma | II, NCT03179943 |
| Durvalumab (PD-L1) | Liver, pancreatic, bile duct, gallbladder | I, NCT03257761 | |
| GVAX (Cell vaccine) | Colon cancer | I, NCT01966289 | |
| Ipilimumab (CTLA-4) | Melanoma | I, NCT02608437 | |
| Pembrolizumab (PD-1) | Ovarian | II, NCT02901899 | |
| Prostate, NSCLC | I, NCT02998567 | ||
| Histone modifications (target) | |||
| Tazemetostat (EZH2) | Pembrolizumab (PD-1) | Bladder cancer | I/II, NCT03854474 |
| CPI-1205 (EZH2) | Ipilimumab (CTLA-4) | Solid tumors | I/II, NCT03525795 |
| BMS-986158 (BRD2/3/4, BRDT) | Nivolumab (PD-1) | Advanced tumors | I/II, NCT02419417 |
| Multiple combinations | |||
| Azacytidine, entinostat | Nivolumab | NSCLC | II, NCT01928576 |
| Azacytidine, venetoclax (Bcl-2) | Pembrolizumab | AML | II, NCT04284787 |
| Azacytidine, epacadostat (IDO-1) | Pembrolizumab | Metastatic solid tumors | I/II, NCT02959437 |
| Mocetinostat, guadecitabine | Pembrolizumab | Lung cancer | I, NCT03220477 |
| Vorinostat, temozolomide | Pembrolizumab | Glioblastoma | I, NCT03426891 |
| Vorinostat, tamoxifen | Pembrolizumab | Breast cancer |
II, NCT04190056 II, NCT02395627 |
| Multiple agents | Multiple agents | Breast, prostrate, pancreas, AML | I, NCT03878524 |
| Azacytidine, romidepsin (Pan HDAC) | Pembrolizumab | Colorectal cancer | I, NCT02512172 |
| Azacytidine |
Tremelimumab (CTLA-4) Durvalumab (PD-L1) |
Head and neck cancer | I/II, NCT03019003 |
| Decitabine, tetrahydrouridine | Pembrolizumab | NSCLC | I/II, NCT03233724 |
Histone deacetylase inhibitors promote an immunotherapy favorable microenvironment
Acetylation of lysine on histone tails is commonly observed at promoter and enhancer gene regulatory regions of actively transcribed genes. In cancer cells, de-acetylation of histone lysine residues is commonly associated with hypermethylated and silenced genes. Histone deacetylase inhibitors (HDACi) target these regions to reactivate gene expression. Several HDACi, such as belinostat, panobinostat, romidepsin, vorinostat have received FDA approval for treatment of solid tumors and hematological malignancies and these are reviewed elsewhere [4, 46, 47]. HDACi have been demonstrated to influence tumor immunogenicity and the functional activity of specific immune cells. For instance, treatment of carcinoma cells with HDACi increased the expression of antigen processing enzymes and increased MHC class I expression on the surface of tumor cells [48].
Studies also suggest that HDACi may be useful for reprogramming the tumor microenvironment to deactivate immunosuppressive cells and increase cytotoxic T cell trafficking to the tumor. Cytokines and chemokines from tumor cells such as IL-6, IL-10, TGF-beta, VEGF and CCL2 promote exclusion of cytotoxic T cells from the tumor microenvironment and recruit immunosuppressive cells such as regulatory T cells, myeloid-derived suppressor cells, tumor-associated macrophages, and tolerogenic dendritic cells. Recent reports indicate that HDAC11 and HDAC6 interact with each other in the cytoplasm and nuclei of antigen-presenting cells to coordinate regulation of the IL-10 promoter. Interestingly, HDAC11 was shown to repress IL-10 gene transcriptional activity in APCs while HDAC6 promoted the expression of IL-10 as a transcriptional activator [49]. Overexpression of HDAC11 inhibited IL-10 expression and induced inflammatory APCs that were able to prime naive T cells and restore the responsiveness of tolerant CD4 + T cells [50]. In a mouse melanoma model, pretreatment with HDAC6 inhibitor Nexturastat A followed by administration of anti-PD-1 blocking antibodies significantly decreased tumor growth by causing increased infiltration of CD8 + T cells and natural killers cells as well as a reduction of pro-tumoral M2 macrophages in the tumor microenvironment [51]. Furthermore, cotreatment of syngeneic mouse tumor models with the HDACi entinostat and 5-azacytidine markedly improved the response to both anti–PD-1 and anti–CTLA-4 checkpoint inhibitor antibodies, curing more than 80% of the tumor-bearing mice [52]. Functional studies revealed that rather than alter the level of CD8 + T cell infiltration or antigen presentation mechanisms, entinostat decreased the viability of the myeloid-derived suppressor cell population [52]. Overall these studies suggest that selective HDACi could be used as immunological priming agents to sensitize immunologically "cold" tumors and subsequently improve immune checkpoint blockade therapies.
The promise of combination therapy that includes immune checkpoint blockade and HDACi has led to the recent initiation of several clinical trials. In a phase I study of patients with immune checkpoint inhibitor-resistant metastatic NSCLC, the combination of the anti-PD-1 antibody pembrolizumab and HDACi vorinostat resulted in one confirmed partial response and eight stable disease responses among 24 enrolled patients [53]. Moreover, preliminary results from the ENCORE-601 phase I/II trial evaluating the combination of pembrolizumab and HDACi entinostat demonstrate a favorable response in patients with colorectal cancer and immune checkpoint inhibitor-resistant melanoma or NSCLC [54–56].
Inhibition of histone methylation promotes tumor immunogenicity
Numerous reports suggest that polycomb repressive complex 2 (PRC2)-mediated epigenetic silencing is a key mechanism facilitating immune evasion. For instance, PRC2 suppresses tumor antigen presentation mechanisms by transcriptionally repressing MHC class I antigen presentation pathway genes [57]. Treatment of cancer cell lines with a pharmacological EZH2 inhibitor (EZH2i) promoted antigen-specific T cell killing in vitro, and disruption of EZH2 in mouse models leads to re-establishment of a T cell-mediated anti-tumor response [57]. Furthermore, EZH2i can promote tumor immunogenicity by reactivating the expression of normally silent endogenous retroviruses [34]. EZH2 inhibition was also observed to promote IFN signaling and production of proinflammatory cytokines such as CXCL9 and CXCL10. Also, EZH2 has been reported to be crucial for immune cell differentiation. Targeted disruption of EZH2 in regulatory T cells enhances the anti-tumor immune response in mouse models [58, 59]. EZH2 deficiency in regulatory T cells not only reduces the frequency of these immune suppressive cells in tumors but also alters their function converting them from immunosuppressive to pro-inflammatory cytokine producers in tumors [59]. In a mouse model, inhibition of EZH2 in T cells increases the effectiveness of anti-CTLA-4 therapy [58]. Several EZH2i such as tazemetostat and CPI-1205 are currently in clinical trials as single agent or combination therapies for a variety of cancer types. For instance, the combination of tazemetostat and pembrolizumab is currently being tested in clinical trials of patients with urothelial carcinoma while CPI-1205 is being tested in combination with ipilimumab in patients with advanced solid tumors previously treated with PD-1 or PD-L1 inhibitors.
Targeting microRNAs to improve tumor immunogenicity
Among the diverse epigenetic mechanisms that regulate gene expression, microRNAs (miRNAs) representant an attractive target for future strategies to augment immunotherapy. Non-protein-coding RNAs, including long noncoding RNAs (> 200 nucleotides) and small noncoding RNAs such as enhancer RNAs and miRNAs, can regulate transcription directly or at the post-transcription level to provide another layer of epigenetic control for gene expression. MiRNAs are a group of short (~ 22 nt), evolutionally conserved, single-stranded noncoding RNA molecules that prevent expression of specific target genes through sequence-specific RNA–RNA interactions with the 3′-untranslated regions of target mRNA. MiRNA-mediated gene silencing occurs by mRNA cleavage and degradation, or translational repression, depending on the degree of complementarity between the miRNA and the targeted mRNA [60, 61]. In addition, miRNAs may function as ligands of Toll-like receptor (TLR) to trigger activation of downstream signaling pathways [62]. Currently there are about 2500 curated miRNAs, and it has been estimated that almost 2/3rds of protein-encoding genes are regulated by miRNAs [63]. Compelling evidence demonstrates that miRNA expression is dysregulated in human cancer through various mechanisms, including amplification or deletion of miRNA genes, abnormal transcriptional control of miRNAs, dysregulated epigenetic changes and defects in the miRNA biogenesis machinery [64, 65]. The regulatory roles of miRNAs in metabolic and cellular pathways, especially those controlling cell proliferation, differentiation, apoptosis, and survival, are crucial to tumor initiation and progression [66]. Thus, miRNAs may function as either oncogenes or tumor suppressors depending on specific contexts.
MiRNAs are important to regulate immune cell function, differentiation and interaction with the tumor microenvironment, and these functions have been subject of several recent reviews [67, 68]. For instance, miRNAs such as miR-27a have been reported to alter tumor antigen presentation and colorectal cancer tumors with high miR-27a had reduced T cell infiltration. Increased miR-27a in colorectal cancer was associated with distant metastasis and poor prognosis [69]. In the tumor microenvironment, miRNAs, such as miR-101 and miR-222, regulate the interaction of cancer-associated fibroblasts with tumor cells [70, 71]. MiRNAs have also been reported to regulate immune checkpoints. For instance, miR-34 binds directly to the 3' untranslated region of PDL1 in non-small cell lung cancer cell lines to downregulate PDL-1 expression, resulting in enhanced T cell response [72, 73]. Thus, miRNAs may be useful biomarkers in some contexts to predict response to immunotherapies [68].
Strategies to target miRNAs in cancer are beginning to be developed and tested. Aberrant miRNA expression in cancer may be targeted depending on the cellular context by either restoring miRNA expression that has been lost in cells or inhibiting upregulated miRNAs. Multiple groups are pursing strategies to deliver miRNAs to tumors such as lipid encapsulated miRNA mimics or blockers as potential cancer therapeutics. For instance, a liposomal miR-34a mimic (MRX34) was recently evaluated in patients with advanced solid tumors refractory to all standard treatments [74]. MRX34 treatment with dexamethasone premedication demonstrated some clinical activity, but the trial was closed prematurely due to serious immune-mediated toxicities [74]. Furthermore, in a phase 1 clinical trial, “minicells” loaded with miR-16 and targeted to EGFR were assessed as having an acceptable safety profile and signs of clinical activity in mesothelioma patients [75]. Another group has evaluated an oligonucleotide inhibitor of miR-155, cobomarsen, in DLBCL cell lines and xenograft mouse models [76]. Cobomarsen reduced tumor volume, triggered apoptosis and derepressed direct miR-155 target genes [76]. Although studies such as these are early, they provide proof-of-concept for miRNA-based cancer therapy. As our understanding for how miRNAs may regulate the immune system and response to immunotherapy, a future therapeutic approach may be to integrate these strategies.
Conclusions
Recently, an increasing number of pharmacological agents targeting epigenetic mechanisms have shown promise in clinical trials of cancer patients both as single agents and in combination with other therapies. Insights into epigenetic mechanisms, particularly as they relate to immune cell function and tumor antigen presentation, have informed recent rational strategies for targeting tumor cells and reversing acquired immunotherapy resistance. Epigenetic therapeutics can deactivate immunosuppressive gene expression or reprogram tumor cells to activate antigen presentation mechanisms. Importantly, additional work must be done to determine whether the benefit of integrating epigenetic therapy with immunotherapy may depend upon the type of cancer or some other specific context. Many epigenetic inhibitors have been reported to negatively impact the proliferation of T cells which could hinder the durability of immunotherapy that relies on a persistent T cell population. For instance, inhibition of the EZH2 and the PRC2 complex has been reported to undermine T cell function. EZH2 is essential for the development and maintenance of memory T cells that sustain effector T cell production and associated antitumor function [77]. EZH2-deficient CD8 + T cells were incapable of mediating tumor growth inhibition to the same degree as EZH2-sufficient cells when transferred into mice with preestablished B16-melanoma [77]. In addition, EZH2 has been reported to stimulate polyfunctional cytokine expression on T cells and promote T cell survival by BCL2 signaling [78]. Pharmacological inhibition of EZH2 or short hairpin RNA-mediated knockdown of EZH2 in T cells was reported to elicite poor antitumor immunity [78]. EZH2 + CD8 + T cell numbers and the percentage of EZH2 + CD8 + T cells in tissue samples of ovarian cancer patients were significantly associated with improved overall survival [78]. These studies suggest that more information regarding how epigenetic mechanisms regulate immunity is necessary to determine the circumstances a particular epigenetic therapy may be most beneficial for cancer patients and whether there is a therapeutic window to use such agents in combination with immunotherapy. Identification and validation of biomarkers may be required to determine which patients will derive the greatest benefit from combined epigenetic and immunotherapy. As our understanding of how epigenetic mechanisms may govern immunotherapy effectiveness grows, leveraging these discoveries to counter immune evasion will be valuable to shape future anti-cancer strategies and improve patient outcomes.
Acknowledgements
Figures created with BioRender.com.
Abbreviations
- EZH2
Enhancer of zeste homolog 2
- PRC2
Polycomb repressive complex 2
- H3
Histone H3
- DLBCL
Diffuse large B cell lymphoma
- DNMT
DNA methyltransferase
- HDAC
Histone deacetylase
- CTA
Cancer testis antigen
- EVR
Endogenous viral RNA
- miRNA
MicroRNA
- NSCLC
Non-small cell lung cancer
Authors' contributions
RLB conceived of, wrote and revised the manuscript, JDL reviewed and edited the manuscript. All authors read and approved the manuscript.
Funding
This work was funded by a LLS Specialized Center of Research grant, R01 R01CA195732 and R01 CA180475 (J.D.L.).
Availability of data and material
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lawrence MS, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci USA. 2006;103(5):1412–1417. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 4.Bennett RL, Licht JD. Targeting epigenetics in cancer. Annu Rev Pharmacol Toxicol. 2018;58:187–207. doi: 10.1146/annurev-pharmtox-010716-105106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishigaki M, et al. Discovery of aberrant expression of R-RAS by cancer-linked DNA hypomethylation in gastric cancer using microarrays. Cancer Res. 2005;65(6):2115–2124. doi: 10.1158/0008-5472.CAN-04-3340. [DOI] [PubMed] [Google Scholar]
- 6.Herman JG, et al. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55(20):4525–4530. [PubMed] [Google Scholar]
- 7.Greger V, et al. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum Genet. 1989;83(2):155–158. doi: 10.1007/BF00286709. [DOI] [PubMed] [Google Scholar]
- 8.Okano M, et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257. doi: 10.1016/S0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 9.Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016;30(7):733–750. doi: 10.1101/gad.276568.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, et al. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7(4):397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y, Vakoc CR. Targeting cancer cells with BET bromodomain inhibitors. Cold Spring Harb Perspect Med. 2017;7(7):a026674. doi: 10.1101/cshperspect.a026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan J, et al. Perturbation of BRD4 protein function by BRD4-NUT protein abrogates cellular differentiation in NUT midline carcinoma. J Biol Chem. 2011;286(31):27663–27675. doi: 10.1074/jbc.M111.246975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng Q, et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014;24(7):809–819. doi: 10.1038/cr.2014.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8(4):307–318. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- 16.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 17.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22(2):128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morin RD, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ernst T, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42(8):722–726. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 20.Campoli M, Ferrone S. HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene. 2008;27(45):5869–5885. doi: 10.1038/onc.2008.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christopher MJ, et al. Immune escape of relapsed AML cells after allogeneic transplantation. N Engl J Med. 2018;379(24):2330–2341. doi: 10.1056/NEJMoa1808777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Topper MJ, et al. The emerging role of epigenetic therapeutics in immuno-oncology. Nat Rev Clin Oncol. 2020;17(2):75–90. doi: 10.1038/s41571-019-0266-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5(3):587–598. doi: 10.18632/oncotarget.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siebenkas C, et al. Inhibiting DNA methylation activates cancer testis antigens and expression of the antigen processing and presentation machinery in colon and ovarian cancer cells. PLoS ONE. 2017;12(6):e0179501. doi: 10.1371/journal.pone.0179501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai HC, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21(3):430–446. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiappinelli KB, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162(5):974–986. doi: 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wrangle J, et al. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget. 2013;4(11):2067–2079. doi: 10.18632/oncotarget.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Juergens RA, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1(7):598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghoshal K, et al. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol. 2005;25(11):4727–4741. doi: 10.1128/MCB.25.11.4727-4741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Serrano A, et al. Rexpression of HLA class I antigens and restoration of antigen-specific CTL response in melanoma cells following 5-aza-2'-deoxycytidine treatment. Int J Cancer. 2001;94(2):243–251. doi: 10.1002/ijc.1452. [DOI] [PubMed] [Google Scholar]
- 31.Karpf AR, et al. Inhibition of DNA methyltransferase stimulates the expression of signal transducer and activator of transcription 1, 2, and 3 genes in colon tumor cells. Proc Natl Acad Sci USA. 1999;96(24):14007–14012. doi: 10.1073/pnas.96.24.14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matei D, et al. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012;72(9):2197–2205. doi: 10.1158/0008-5472.CAN-11-3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roulois D, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162(5):961–973. doi: 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canadas I, et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med. 2018;24(8):1143–1150. doi: 10.1038/s41591-018-0116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Panda A, et al. Endogenous retrovirus expression is associated with response to immune checkpoint blockade in clear cell renal cell carcinoma. JCI Insight. 2018 doi: 10.1172/jci.insight.121522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith CC, et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J Clin Invest. 2018;128(11):4804–4820. doi: 10.1172/JCI121476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karpf AR, et al. Increased expression of androgen receptor coregulator MAGE-11 in prostate cancer by DNA hypomethylation and cyclic AMP. Mol Cancer Res. 2009;7(4):523–535. doi: 10.1158/1541-7786.MCR-08-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Smet C, Loriot A, Boon T. Promoter-dependent mechanism leading to selective hypomethylation within the 5' region of gene MAGE-A1 in tumor cells. Mol Cell Biol. 2004;24(11):4781–4790. doi: 10.1128/MCB.24.11.4781-4790.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Velazquez EF, et al. Expression of the cancer/testis antigen NY-ESO-1 in primary and metastatic malignant melanoma (MM)—correlation with prognostic factors. Cancer Immun. 2007;7:11. [PMC free article] [PubMed] [Google Scholar]
- 40.Park TS, et al. Expression of MAGE-A and NY-ESO-1 in primary and metastatic cancers. J Immunother. 2016;39(1):1–7. doi: 10.1097/CJI.0000000000000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kerkar SP, et al. MAGE-A is more highly expressed than NY-ESO-1 in a systematic immunohistochemical analysis of 3668 cases. J Immunother. 2016;39(4):181–187. doi: 10.1097/CJI.0000000000000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghoneim HE, et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell. 2017;170(1):142–157. doi: 10.1016/j.cell.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, et al. Decitabine enhances lymphocyte migration and function and synergizes with CTLA-4 blockade in a murine Ovarian cancer model. Cancer Immunol Res. 2015;3(9):1030–1041. doi: 10.1158/2326-6066.CIR-15-0073. [DOI] [PubMed] [Google Scholar]
- 44.Di Giacomo AM, et al. Guadecitabine plus ipilimumab in unresectable melanoma: the NIBIT-M4 clinical trial. Clin Cancer Res. 2019;25(24):7351–7362. doi: 10.1158/1078-0432.CCR-19-1335. [DOI] [PubMed] [Google Scholar]
- 45.Odunsi K, et al. Epigenetic potentiation of NY-ESO-1 vaccine therapy in human ovarian cancer. Cancer Immunol Res. 2014;2(1):37–49. doi: 10.1158/2326-6066.CIR-13-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–641. doi: 10.1038/nrg.2016.93. [DOI] [PubMed] [Google Scholar]
- 47.Morel D, et al. Combining epigenetic drugs with other therapies for solid tumours - past lessons and future promise. Nat Rev Clin Oncol. 2020;17(2):91–107. doi: 10.1038/s41571-019-0267-4. [DOI] [PubMed] [Google Scholar]
- 48.Setiadi AF, et al. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008;68(23):9601–9607. doi: 10.1158/0008-5472.CAN-07-5270. [DOI] [PubMed] [Google Scholar]
- 49.Cheng F, et al. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol Immunol. 2014;60(1):44–53. doi: 10.1016/j.molimm.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Villagra A, et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol. 2009;10(1):92–100. doi: 10.1038/ni.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Knox T, et al. Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells. Sci Rep. 2019;9(1):6136. doi: 10.1038/s41598-019-42237-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim K, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci USA. 2014;111(32):11774–11779. doi: 10.1073/pnas.1410626111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gray JE, et al. Phase I/Ib study of pembrolizumab plus vorinostat in advanced/metastatic non-small cell lung cancer. Clin Cancer Res. 2019;25(22):6623–6632. doi: 10.1158/1078-0432.CCR-19-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hellman MD, et al. Efficacy/safety of entinostat (ENT) and pembrolizumab (PEMBRO) in NSCLC patients previously treated with anti-PD-(L)1 therapy. In: 19th world conference on lung cancer, 2018.
- 55.Azad NS, et al. ENCORE 601: A phase 2 study of entinostat in combination with pembrolizumab in patients with microsatellite stable metastatic colorectal cancer. J Clin Oncol. 2018;36(15_suppl):3557–3557. doi: 10.1200/JCO.2018.36.15_suppl.3557. [DOI] [Google Scholar]
- 56.Sullivan RJ, et al. Efficacy and safety of entinostat (ENT) and pembrolizumab (PEMBRO) in patients with melanoma previously treated with anti-PD1 therapy. Cancer Res 2019. 79(Proceedings of the 110th Annual meeting of the American Association for Cancer Rasearch: 13 supplement).
- 57.Burr ML, et al. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell. 2019;36(4):385–401. doi: 10.1016/j.ccell.2019.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goswami S, et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J Clin Invest. 2018;128(9):3813–3818. doi: 10.1172/JCI99760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang D, et al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep. 2018;23(11):3262–3274. doi: 10.1016/j.celrep.2018.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 61.Farazi TA, Juranek SA, Tuschl T. The growing catalog of small RNAs and their association with distinct Argonaute/Piwi family members. Development. 2008;135(7):1201–1214. doi: 10.1242/dev.005629. [DOI] [PubMed] [Google Scholar]
- 62.Fabbri M, et al. A new role for microRNAs, as ligands of Toll-like receptors. RNA Biol. 2013;10(2):169–174. doi: 10.4161/rna.23144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Friedman RC, et al. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 65.Garzon R, et al. MicroRNA expression and function in cancer. Trends Mol Med. 2006;12(12):580–587. doi: 10.1016/j.molmed.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 66.Munker R, Calin GA. MicroRNA profiling in cancer. Clin Sci (Lond) 2011;121(4):141–158. doi: 10.1042/CS20110005. [DOI] [PubMed] [Google Scholar]
- 67.Di Martino MT, et al. miRNAs and lncRNAs as novel therapeutic targets to improve cancer immunotherapy. Cancers (Basel) 2021;13(7):1587. doi: 10.3390/cancers13071587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huemer F, et al. miRNA-based therapeutics in the era of immune-checkpoint inhibitors. Pharmaceuticals (Basel) 2021;14(2):89. doi: 10.3390/ph14020089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Colangelo T, et al. Proteomic screening identifies calreticulin as a miR-27a direct target repressing MHC class I cell surface exposure in colorectal cancer. Cell Death Dis. 2016;7:e2120. doi: 10.1038/cddis.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang J, et al. miR-101 represses lung cancer by inhibiting interaction of fibroblasts and cancer cells by down-regulating CXCL12. Biomed Pharmacother. 2015;74:215–221. doi: 10.1016/j.biopha.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 71.Chatterjee A, et al. MicroRNA-222 reprogrammed cancer-associated fibroblasts enhance growth and metastasis of breast cancer. Br J Cancer. 2019;121(8):679–689. doi: 10.1038/s41416-019-0566-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cortez MA, et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst. 2016;108(1):djv303. doi: 10.1093/jnci/djv303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anastasiadou E, et al. Epstein-Barr virus-encoded EBNA2 alters immune checkpoint PD-L1 expression by downregulating miR-34a in B-cell lymphomas. Leukemia. 2019;33(1):132–147. doi: 10.1038/s41375-018-0178-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hong DS, et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br J Cancer. 2020;122(11):1630–1637. doi: 10.1038/s41416-020-0802-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van Zandwijk N, et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: a first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017;18(10):1386–1396. doi: 10.1016/S1470-2045(17)30621-6. [DOI] [PubMed] [Google Scholar]
- 76.Anastasiadou E, et al. Cobomarsen, an oligonucleotide inhibitor of miR-155, slows DLBCL tumor cell growth in vitro and in vivo. Clin Cancer Res. 2021;27(4):1139–1149. doi: 10.1158/1078-0432.CCR-20-3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.He S, et al. Ezh2 phosphorylation state determines its capacity to maintain CD8(+) T memory precursors for antitumor immunity. Nat Commun. 2017;8(1):2125. doi: 10.1038/s41467-017-02187-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao E, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol. 2016;17(1):95–103. doi: 10.1038/ni.3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.