

Abstract
Subarachnoid hemorrhage (SAH) due to rupture of an intracranial aneurysm leads to vasospasm resulting in delayed cerebral ischemia. Therapeutic options are currently limited to hemodynamic optimization and nimodipine, which have marginal clinical efficacy. Nitric oxide (NO) modulates cerebral blood flow through activation of the cGMP-Protein Kinase G (PKG) pathway. Our hypothesis is that SAH results in downregulation of signaling components in the NO-PKG pathway which could explain why treatments for vasospasm targeting this pathway lack efficacy and that treatment with a cell permeant phosphopeptide mimetic of downstream effector prevents delayed vasospasm after SAH. Using a rat endovascular perforation model, reduced levels of NO-PKG pathway molecules were confirmed. Additionally, it was determined that expression and phosphorylation of a PKG substrate: Vasodilator-stimulated phosphoprotein (VASP) was downregulated. A family of cell permeant phosphomimetic of VASP (VP) were designed and shown to have vasorelaxing property that is synergistic with nimodipine in intact vascular tissues ex vivo. Hence, treatment targeting the downstream effector of the NO signaling pathway, VASP, may bypass receptors and signaling elements leading to vasorelaxation and that treatment with VP can be explored as a therapeutic strategy for SAH induced vasospasm and ameliorate neurological deficits.
Keywords: SAH, vasospasm, phosphopeptide mimetics
1. Introduction
Subarachnoid hemorrhage (SAH) due to rupture of an intracranial aneurysm leads to delayed vasospasm resulting in neuroischemia (stroke). The overall morbidity (profound neurologic deficit in 10-20% of survivors) and mortality (50%) are high, and the disease affects a relatively young adult population.(Grasso, Alafaci et al. 2017) (van Gijn, Kerr et al. 2007) Vasoactive pharmacological agents effective in the prevention of delayed vasospasm after SAH remain elusive. Nimodipine, a calcium (Ca2+) channel blocker, is the standard of care and only drug commonly used in the US to prevent neuroischemic events after SAH.(Allen, Ahn et al. 1983, Gelmers, Gorter et al. 1988, Connolly, Rabinstein et al. 2012) Yet the clinical response to nimodipine is minimal (Dorhout Mees, Rinkel et al. 2007) and nimodipine can impair cerebral perfusion by causing systemic hypotension. Attempts have also been made to prevent delayed vasospasm by inhibiting the effect of vasoconstrictors, such as clazosentan (an endothelin receptor antagonist; (Vajkoczy, Meyer et al. 2005) and fasudil (a Rho kinase inhibitor; In the pivotal CONSCIOUS clinical trials, clazosentan demonstrated efficacy in reducing macrovascular vasospasm, however there was no significant clinical effect in reducing stroke or death.(Macdonald, Higashida et al. 2012) Thus, treatment of delayed vasospasm after SAH represents an unmet clinical need in an orphan population with severe clinical consequences.
Preventing vasospasm in the cerebral vasculature after SAH has been refractory to therapeutic approaches that target receptors and upstream signaling events in the vasorelaxation signaling pathway (Macomson, Brophy et al. 2002). Attempts to treat SAH-induced vasospasm with existing vasodilators often fail because of systemic hypotension (leading to decreased cerebral perfusion) and a cerebral vasculature that is refractory to activation of nitric oxide (NO)-dependent signaling pathways(Siuta, Zuckerman et al. 2013). Vasorelaxation of cerebral vessels is mediated by nitric oxide (NO) which activates guanylate cyclase (sGC), leading to increases in cGMP and activation of cGMP-dependent protein kinase (PKG; (Toda, Ayajiki et al. 2009). In vascular smooth muscle, PKG phosphorylates and activates downstream substrates, such as vasodilator-stimulated phosphoprotein (VASP), that modulate contractile and cytoskeletal elements leading to vasorelaxation (Lincoln, Dey et al. 2001).
The hypothesis for this study is that the impaired response of cerebral vessels to vasodilators, i.e. impaired vasorelaxation after SAH, is in part due to downregulation of the signaling elements and substrates in the NO pathway after SAH (Dietrich and Dacey 2000, Macomson, Brophy et al. 2002, Wang, Lee et al. 2014). A non-craniotomy, endovascular perforation rat model of SAH was used to determine modulation of NO signaling pathways after SAH. Utilizing cell-permeant peptide (CPP)-based delivery, a family of rationally designed, cell permeant phosphomimetic peptide of VASP (VP) was developed and tested for the ability to induce vasorelaxation.
2. Materials and Methods
2.1. Materials
All chemicals and reagents were purchased from Sigma unless otherwise described. The peptides used in this study were synthesized by f-moc chemistry and purified using high-performance chromatography by EZBiolab (Parsippany, NJ). Animal procedures followed study protocols (M1800006 and M1800194) approved by the Vanderbilt Institutional Animal Care and Use Committee and adhered to National Institute of Health guidelines for care and use of laboratory animals.
2.2. Non-craniotomy rat endovascular perforation model of SAH
Sprague-Dawley rats were housed under standard 12:12 light-dark cycles and allowed free access to water and food. SAH was induced using the endovascular perforation model with slight modifications (Furnish, Brophy et al. 2010). Briefly, rats were anesthetized with 3% isoflurane and maintained with 2.5-3% isoflurane during surgery. The neurovascular bundle was surgically exposed with a linear incision in the lateral neck. The external carotid artery (ECA) was ligated and a small opening was introduced in the common carotid artery (CCA) using a 30G needle approximately 2 mm proximal to the ECA-CCA bifurcation. A 3-0 nylon monofilament is then inserted through the small opening and advanced through the internal carotid artery (ICA) for 2.5-2.6 mm until resistance was felt. The filament was then advanced 2 mm further to perforate the vessel at the bifurcation of the ICA into the anterior cerebral artery (ACA) and middle cerebral artery (MCA). The filament was withdrawn and the arteriotomy was closed using 10-0 suture. Sham operations exposed and manipulated the cervical carotid artery complex, including ligations and occlusions of branches and passage of the nylon suture to the bifurcation of the ICA, but without intracranial vessel perforation. The neck wound was closed, and animals allowed to recover. Forty-eight hours after SAH induction, rats were perfused with PlasmaLyte (Baxter, IL) under anesthesia using 3% isoflurane. Cerebral vasculature on the ventral side of the brain was isolated and snap frozen. Severity of SAH was graded using a hemorrhage grading scale previously described (Furnish, Brophy et al. 2010).
2.3. Procurement of vascular tissues
Aorta (RA) were procured from Sprague Dawley rats and euthanized. The thoracic and abdominal RA was isolated via an incision along the mid-abdomen, and subcutaneous fat and adventitial tissues were removed. RA was cut into rings of approximately 1–2 mm in width and the endothelium was denuded by gently rolling the luminal surface of each ring at the tip of a fine forceps.
Porcine basilar arteries (PBA) were isolated from adult Yorkshire pigs (Oak Hill Genetics, Ewing, Ill) immediately after euthanasia. PBA were dissected free of subcutaneous fat and adventitial tissues and cut into 2-mm wide rings and the endothelium was denuded by gently rolling the luminal surface of each ring on the muscle bath tissue hook.
2.4. Physiologic measurements
Rings were suspended in a muscle bath containing a bicarbonate buffer (120 mM sodium chloride, 4.7 mM potassium chloride, 1.0 mM magnesium sulfate, 1.0 mM monosodium phosphate, 10 mM glucose, 1.5 mM calcium chloride, and 25 mM sodium bicarbonate, pH 7.4) equilibrated with 95% O2/5% CO2 at 37 °C for 1 h at a resting tension of 1 g and 0.3g for RA and PBA, respectively. RA was then stretched to three times the resting tension and maintained at resting tension for an additional 1 h. This produced the maximal force tension relationship as previously described (Cheung-Flynn, Alvis et al. 2019). After equilibration, the rings were primed with 110 mM potassium chloride (with equimolar replacement of sodium chloride in bicarbonate buffer) to determine functional viability. Viable rings were then tested for contractile response to a dose of agonist [phenylephrine (PE), endothelin (ET), or serotonin (5HT)] to yield submaximal contraction (approximately 60–70% of maximum KCl) and relaxed with either escalating doses of sodium nitroprusside (SNP; 10−10 to 10−6M), nimodipine (NIMO;10−8 to 10−6M) or peptides (10−5 to 10−3M). Force measurements were obtained using the Radnoti force transducer (model 159901A, Radnoti LLC, Monrovia, CA) interfaced with a PowerLab data acquisition system and LabChart software (AD Instruments Inc., Colorado Springs, CO). Contractile responses were defined by stress, calculated using force generated by tissues as follows: stress (x105 N/m2) = force (g) × 0.0987/area, where area = wet weight (mg)/ at maximal length (mm)]/1.055. Relaxation was calculated as percent change in stress compared to the maximal phenylephrine-induced contraction (set as 100%). Each data point was averaged from at least two rings from the same specimen. To determine concurrent signaling events, tissues were frozen in liquid nitrogen at relevant timepoints for Western blot analysis.
2.5. Western blot analysis
Frozen tissues were homogenized in Laemmli buffer and protein concentration determined using Pierce Coomassie protein assay kit (ThermoFisher, CA). Protein samples were separated by SDS-PAGE and transferred onto nitrocellulose membrane and probed with antibodies as follows: endothelial nitric oxide synthase (eNOS), VASP, phospho-Ser239 specific VASP (Cell Signaling, CA), PKG, sGC (ENZO Life Sciences, NY), GAPDH (Millipore, MA), and β-actin (Sigma, MA). Protein-antibody complexes were visualized using Odyssey-CLx (LiCor, NE). Protein levels were normalized to GAPDH and VASP phosphorylation at Ser239 was calculated as a ratio of the phosphorylated VASP to the total VASP level.
2.6. Cytoskeletal dynamics
The amount of filamentous (F-) actin versus globular (G-) actin was measured using the G-actin/F-actin In Vivo Assay kit (Cytoskeleton, Denver, CO), per manufacturer’s protocol. Briefly, RA tissues were pre-contracted with phenylephrine or endothelin and then treated with or without VP3. RA samples were homogenized in 1 ml of lysis buffer (50 mM PIPES pH 6.9, 50 mM NaCl, 5 mM MgCl2 5 mM EGTA, 5% (v/v) Glycerol, 0.1 % Nonidet P40, 0.1% Triton X-100, 0.1% Tween 20, 0.1% 2-mercapto-ethanol, 0.001% Antifoam C, 4 μM Tosyl arginine methyl ester, 15 μM Leupeptin, 10 μM Pepstatin A, 10 mM Benzamidine, 1 mM ATP warmed to 37°C) for 1 min with a mortar and pestle that fit into the 1.5 ml microfuge tube. The lysate was centrifuged at 350 x g for 5 min at 37°C to pellet unbroken cells. The supernatants were centrifuged at 100,000 x g for 1 h at 37°C. Supernatants (contains the G-actin) were transferred to pre-cooled tubes and placed on ice. The pellets (contain F-actin) were resuspended in 1 ml of ice-cold 10 μM cytochalasin D in deionized water, and F- actin was depolymerized by incubating for 1 h on ice with mixing every 15 min. Equal volume of supernatants and pellets along with actin standards (2-20 μg) were separated on SDS-PAGE and transferred to nitrocellulose membrane. The membrane was probed with anti-actin antibody and % F-actin was determined [F-actin/(F-actin+G-actin) x100%]. Each data point was averaged from at least 3 rings per treatment group and compared between groups from the same animal.
2.7. Statistical analysis
Data were reported as mean responses ± standard deviation. Outliers, normality, and statistical significance (P value) were determined using GraphPad Prism version 5.0. Differences between two groups were determined by paired t test for experiments with dependent (matched) pairs. One-way with post hoc tests were used to determine differences among multiple, dependent (matched) samples from the same animal. A P-value < 0.05 was considered statistically significant.
3. Results
3.1. Vasorelaxation is associated with phosphorylation of VASP in normal rat aorta
Treatment of precontracted RA with sodium nitroprusside (SNP; a NO donor) and nimodipine (NIMO; a Ca2+ channel blocker) leads to vasorelaxation (Fig. 1A). Phosphorylation of VASP at S239 increased with SNP but not nimodipine treatment (Fig. 1B and C), suggesting that VASP phosphorylation is associated with vasorelaxation.
Fig. 1. Vasorelaxation is associated with phosphorylation of the VASP protein in rat aorta.
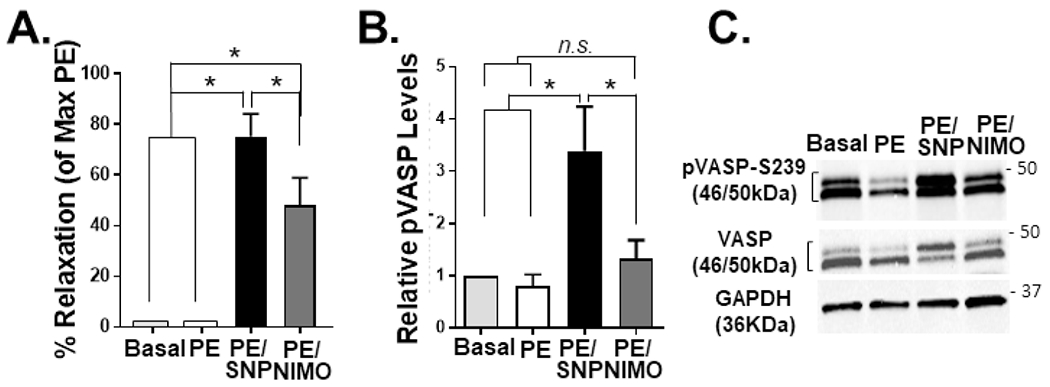
RASM rings were suspended in a muscle bath and left untreated (Basal) or pre-contracted with phenylephrine (PE) followed by sodium nitroprusside (SNP) or nimodipine (NIMO), snap-frozen and protein extracted for immunoblotting experiments (A). % relaxation was determined as a %the maximal PE-induced contraction. Levels of VASP S239 phosphorylation were calculated as a ratio of the phosphorylated protein to total protein and relative levels were calculated by comparing to basal tissues (B). Representative immunoblots shown (C). n=4; *P<0.05; n.s., statistically non-significant.
3.2. Impaired NO signaling in the cerebral vasculature in the non-craniotomy rat endovascular perforation model of SAH
Since SAH leads to cerebral vasospasm, levels of the proteins in the NO/PKG signaling pathway were determined 48 h after endovascular perforation (Fig. 2A and B) in rats (n = 5 SHAM and n=9 SAH). Hemorrhage grades noted were Grade 1, n=5; Grade2, n=2; Grade 3, n=3; and Grade 4; n=1 (Furnish, Brophy et al. 2010). Cerebral vessels were harvested from the ventral aspect of the brain and western blot analysis were performed. SAH led to decreased expression of eNOS, sGC, PKG and the PKG substrate, VASP. Phosphorylation of VASP (S239), the activated isoform, was also decreased. (Fig. 2C and D).
Fig. 2. Concurrent decrease in cerebral perfusion with downregulation of NO signaling pathway elements after SAH in rats.
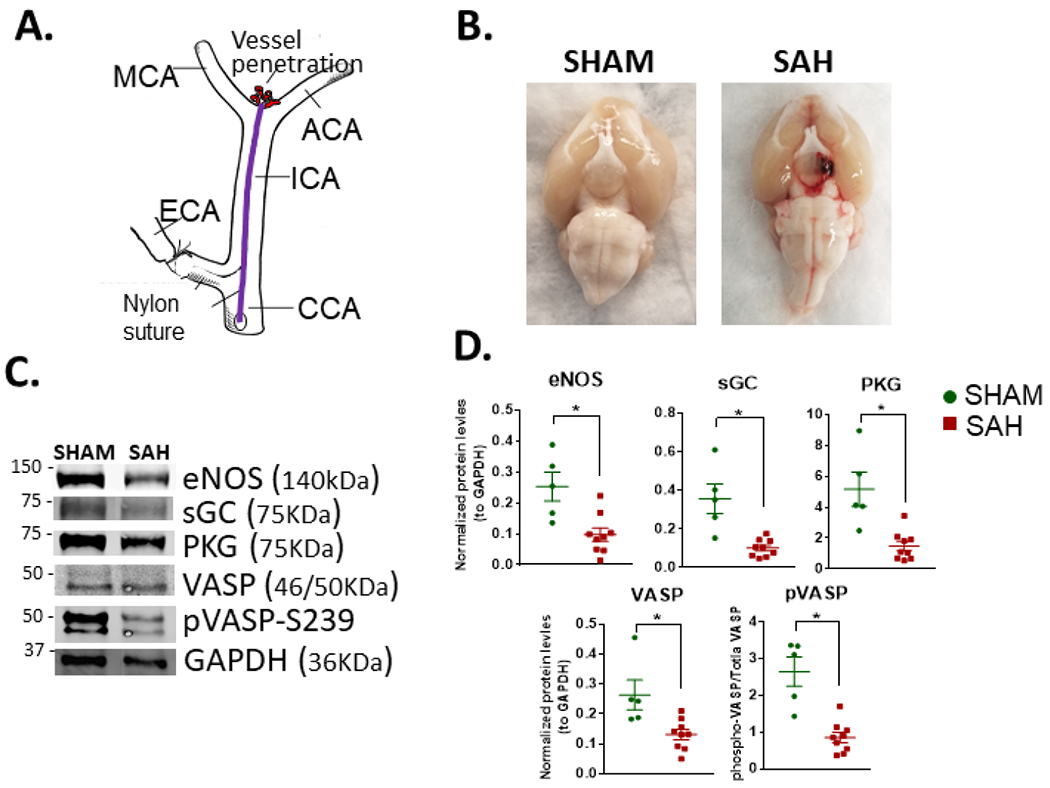
Modified endovascular perforation rat model of SAH (A). Ventral view of brains showing hemorrhage (B). Representative blots (C) and quantitation (D) showing decreased levels of GC. PKG, phospho-VASP S239 (ratio of the phosphorylated protein to total protein levels), and total VASP in cerebral vessels 48 h after hemorrhage. SHAM, n=5; SAH, n=9; *P<0.05 in unpaired t- test.
3.3. Rational design of cell permeant phosphomimetics of VASP
Since the NO-PKG signaling axis is downregulated in cerebral vessels and that VASP phosphorylation was associated with vasorelaxation, it is hypothesized that introduction of a molecular mimetics containing the region surrounding VASP S239 with a phospho-serine may bypass the upstream elements and directly induce vasorelaxation. A family of cell-permeant phosphopeptides of VASP (VP) were designed using a cell permeant domain YARAAARQARA (Ho, Schwarze et al. 2001) attached to cargo of varied length of residues surrounding the S239 (Table 1).
Table 1.
Composition of VASP polypeptide (VP) series. Sequences of polypeptides indicated by single-letter amino acid code. Italics – cell permeant domain; bold – cargo; (pS) denotes phosphoserine S239
| Peptides | Sequences |
|---|---|
| VP1 | YARAAARQARAKLRKV(pS)KQEEASG |
| VP2 | YARAAARQARAKLRKV(pS)KQEEA |
| VP3 | YARAAARQARAKLRKV(pS)K |
| scrVP | YARAAARQARAK(pS)RVLKK |
3.4. VPs relaxes pre-contracted RA
When added directly to the muscle bath, all three VPs relaxed phenylephrine pre-contracted RA in a dose-dependent manner (10−4 to 10−3M; Fig. 3). The peptide (VP3) containing the shortest VASP sequence exhibited the greatest vasorelaxation at the lowest does (highest bioactivity) and was used for further investigation. A peptide containing a scrambled VP3 peptide (scrVP) did not cause relaxation (Fig. 3) demonstrating specificity of the VP3 sequence. A peptide containing only cargo sequence of VP3 did not cause relaxation (Suppl Fig. S1), suggesting that the CPP is required for penetration of VP into tissue to affect contractile responses. Multiple batches of VP3 were synthesized and displayed similar bioactivity (Suppl Fig. S2).
Fig. 3. Vasorelaxation effects of VASP polypeptides (VP) in rat aorta.

RASM rings were suspended in a muscle bath pre-contracted with phenylephrine, followed by treatment with sodium nitroprusside (SNP; 10−9 and 10−8M) or escalating doses of the VASP peptide indicated (x10−3M). VP3 at a dose of 0.25×10−3M generated similar relaxation to that of 10−9M SNP and has the greatest vasorelaxing property among the VP peptides. ScrVP had no vasorelaxing property. % relaxation was determined as a change to the maximal phenylephrine-induced contraction. n=4-7.
3.5. Synergistic vasorelaxation to nimodipine and VP3
Nimodipine acts on Ca2+-dependent signaling pathways which regulate the initiation of contraction and do not affect VASP phosphorylation (Fig. 1). Thus, it was hypothesized that the VP3 peptide and nimodipine would be synergistic. When added to phenylephrine-precontracted RA, low dose VP3 was synergistic with nimodipine in increasing vasorelaxation responses (Fig. 4A and B). Endothelin (ET) has been implicated as a mediator of SAH-induced vasospasm (Fassbender, Hodapp et al. 2000, Juvela 2000, Assenzio, Martin et al. 2015). Vasorelaxation and synergism with nimodipine of VP3 was also determined in endothelin -precontracted RA. The VP3 peptide also demonstrated dose-dependent vasorelaxing properties and was synergistic with nimodipine in endothelin-precontracted RA (Fig. 4C).
Fig. 4. Synergistic vasorelaxation to nimodipine and VP3.

Agonist-precontracted RASM were treated with calcium channel blocker nimodipine (NIMO; 10−7M); VP3 (0.25 X 10−3M); nimodipine followed by VP3 [NIMO/VP3]; VP3 followed by nimodipine [VP3/NIMO]; or simultaneous addition of nimodipine plus VP3 [NIMO+VP3]. Representative muscle bath tracings of force generated in response to phenylephrine, nimodipine and VP3. Dashed lines indicate addition of drugs to RASM (A). % relaxation was determined as a change to the maximal phenylephrine-induced contraction (n = 3-4, *P < 0.05) (B). Endothelin (ET)-precontracted RASM were treated with nimodipine (NIMO, 10−8M); VP3 (0.25×10−3M); nimodipine and VP3 (NIMO/VP3 and VP3/NIMO) (C).
3.6. Mechanism of VP3-induced relaxation in rat aorta
Phosphorylation at S239 induces smooth muscle relaxation by impairing filamentous (F)-actin assembly, suggesting that the mechanism of action (MOA) of VASP is modulation of actin filament dynamics. To demonstrate that the VP3 peptide regulates thin filaments, an actin assay was performed in RA precontracted with either phenylephrine or endothelin, and the proportion of F- vs. G-actin was determined. Treatment with VP3 peptide led to reduced F-actin concentration in phenylephrine- (Fig. 5A; 60.5±12.4 vs. 40.9±16.3%) or endothelin (Fig. 5B; 74.8±8.9 vs. 63.6±10.3%)- precontracted tissues, indicating that the MOA of VP is through prevention of F-actin formation elicited by different contractile agonists.
Fig. 5. Effects of VP3 on actin cytoskeleton dynamics in intact RASM.

PE(A)- or ET (B)-precontracted RASM was treated with VP3 (1 mM) and levels of globular (G-) and F-actin were measured. Top: Representative immunoblots. Bottom, quantitative analysis of % F-actin showed reduction in VP3-treated PE- or ET-precontracted RASM compared to untreated tissue, indicating depolymerization of F-actin by VP3 treatment. n=4; *P<0.05 in paired t-test.
3.7. VP3 relaxes precontracted porcine basilar artery
Different vascular beds show variable responses to contractile agonists. Hence, vasorelaxing properties of VP3 peptide were determined in porcine basilar arteries (PBA). The VP3 peptide directly relaxed PBA pre-contracted with serotonin (5-HT; Fig. 6A) and endothelin (Fig. 6B), vasoconstrictors relevant to vasospasm after SAH (Voldby, Engbaek et al. 1982, Zimmermann and Seifert 1998) and scrVP did not relax agonist-contracted PBA (Fig. 6A) suggesting that VP3 was effective in inducing vasorelaxation of cerebral vessels.
Fig. 6. VASP polypeptide (VP3) relaxed serotonin- and endothelin-precontracted porcine basilar artery.

Basilar artery segments (4mm) were suspended in a muscle bath pre-contracted with serotonin (5-HT; A) or endothelin (ET; B) followed by treatment with SNP (10−8 M), nimodipine (NIMO, 10−8M), or escalating doses of VP3 or scrVP indicated (x10−3 M). % relaxation was determined as a change to the maximal agonist-induced contraction. n=2-6.
4. Discussion
Mechanisms underlying cerebral vasospasm after SAH remain poorly understood. In both cisterna magna blood injection and endovascular perforation rat models of SAH, upstream receptors and enzymes of the NO-PKG signaling pathway are downregulated (Macomson, Brophy et al. 2002, Wang, Lee et al. 2014, Wang, Wang et al. 2020). Here, we confirmed that downregulation of the elements of the NO-PKG signaling pathway and phosphorylation of the PKG substrate VASP, decreased after endovascular perforation in the non-craniotomy rat model. Another substrate protein downstream in the vasorelaxation signaling pathway, small heat shock related protein HSP20, has also been shown to be downregulated after SAH in the non-craniotomy model and this downregulation was associated with impaired relaxation of cerebral vessels ex vivo(Macomson, Brophy et al. 2002). Taken together, these findings suggest the downregulated NO-PKG signaling pathway is an underlying mechanism to pathological vasoconstriction after SAH and implicate that VASP may be contributive in the pathophysiology of the cerebral vessels after SAH (Fig. 7).
Fig. 7. Model of mechanism of SAH-induced vasospasm.

A. In the setting of SAH, NO, guanylate cyclase (GC), and cGMP-dependent protein kinase (PKG) are downregulated in the cerebral vasculature (A). Phosphorylation of PKG substrate VASP is decreased and hence actin-myosin interactions maintain a contractile state. Nimodipine and VASP peptide restore normal vasorelaxation by decreasing intracellular calcium channels (inhibiting thick filament myosin) and bypassing the NO-cGMP-PKG signaling pathway (leading to actin depolymerization; B).
VASP is central to the dynamic reorganization of the cytoskeleton. Phosphorylation of VASP is associated relaxation of smooth muscle from various tissues (Lincoln, Dey et al. 2001, Schafer, Burkhardt et al. 2003, Wu and Gunst 2015). Alternation of VASP expression and phosphorylation contribute to clinical pathology (Pula and Krause 2008). Downregulation of phosphorylation of VASP at Ser239 is associated with impaired vasorelaxation (Benz, Blume et al. 2009) and vascular injury (Cheung-Flynn, unpublished data). Decreased cGMP signaling and PKG expression and PKG-dependent phosphorylation of VASP is associated with neointimal proliferation and vascular dysfunction in atherosclerotic rabbit aorta (Melichar, Behr-Roussel et al. 2004) and hypertension in the Dahl salt sensitive rat (Carrillo-Sepulveda, Panackal et al. 2019). Moreover, overexpression of constitutively-active PKG and the consequent hyperphosphorylation of VASP in rat aorta resulted in the reduction of neointimal proliferation after balloon injury (Sinnaeve, Chiche et al. 2001). In the lung, VASP is repressed and phosphorylation at S239 decreased in pulmonary hypertension and acute lung injury by LPS and hypoxia (Rosenberger, Khoury et al. 2007, Henes, Schmit et al. 2009, Tang, Tian et al. 2014). Decreased pVASP level is associated with hypoxia-induced hypercontractility of pulmonary arteries which is proposed to be a result of reduced PKG activity (Chettimada, Rawat et al. 2012).
In light of the roles VASP play in homeostasis and diseases, it is logical to regard the manipulation of VASP phosphorylation as therapeutic intervention for vascular diseases. CPPs deliver bioactive molecular cargos into cells noninvasively and have provided immense potential for the therapeutic applications (Guidotti, Brambilla et al. 2017). In the present study, we designed cell permeant molecular mimetics of VASP, the VP peptide family, based on the biological roles/activity of VASP and the observed downregulation after SAH. Treatment with VP peptides led to vasorelaxation in ex vivo vascular tissues including rat aorta and porcine basilar artery (Fig. 3, 4 and 6). The shortest sequence that had the highest bioactivity suggested that the ‘KLRKV(pS)K’ region of VASP constitutes the minimum region for vasorelaxing activity.
The cellular cytoskeleton contains a balance of filamentous (F-) and globular (G-) actin. Phosphorylation of VASP has been suggested to play a role in regulating actin cytoskeleton dynamics. VP3 treatment led to a decrease in F-actin of intact RA tissues precontracted by phenylephrine and this property was confirmed in endothelin precontracted tissues (Fig. 5), suggesting that VP3 will likely modulate actin cytoskeleton dynamics in response to multiple contractile agonists. Contraction of vascular smooth muscle requires filamentous actin for the myosin motor to treadmill on. Hence, a reduction in F-actin would lead to a loss of this ability to treadmill and hence vasorelaxation. A cell-permeant peptide of Hsp20 has also been shown to lead to smooth muscle relaxation by disruption of the actin cytoskeleton (Dreiza, Brophy et al. 2005) and prevented delayed vasospasm in the non-craniotomy SAH model (Furnish, Brophy et al. 2010). Thus, targeting downstream elements in the NO-PKG pathway with cell permeant molecular mimetics of downstream phosphoproteins represents a unique and direct approach to the treatment of delayed vasospasm after SAH.
Development of therapeutic intervention to prevent and treat cerebral vasospasm after SAH has been challenging in part due to a cerebral vasculature that is uniquely refractory to vasodilators. It has been proposed that imbalance between vasodilators (e.g. nitric oxide) and vasoconstrictors (e.g. endothelin) occur after SAH, leading to abnormal contraction of cerebral vascular smooth muscle. Attempts to use NO donors or the endothelin receptor antagonist (Clazosentan) to prevent delayed vasospasm after SAH have not been successful (Vajkoczy, Meyer et al. 2005, Fathi, Bakhtian et al. 2011, Macdonald, Higashida et al. 2012). Our data suggest that these strategies failed at least in part because the downregulated NO-PKG signaling pathway which impairs cerebrovascular smooth muscle relaxation machinery. Hence to treat impaired relaxation, strategies that bypass receptors and enzymes of the NO-PKG pathway and PKG substrates are logical targets.
Other CPP-derived peptide therapeutics have been developed to the level of clinical trials (Guidotti, Brambilla et al. 2017), for the treatment of hearing loss and inflammation with a JNK inhibitor peptides (Touchard, Omri et al. 2010, Suckfuell, Lisowska et al. 2014, El Zaoui, Touchard et al. 2015), and myocardial infarction with PKC inhibitor peptides (Bates, Bode et al. 2008). The observations that VP relax cerebral vessels pre-contracted by vasoconstrictors that are upregulated after SAH and that it is synergistic with nimodipine demonstrate the potential relevance of developing VP as a SAH therapeutic strategy. Administration of VP could essentially ‘restore’ NO-PKG signaling by bypassing upstream activators in vasospastic cerebral vessel. Since VASP expression and phosphorylation is lower after SAH, the vasorelaxation effects of VP will likely be greater in cerebral vessels than normal vasculature and hence minimize potential systemic hypotension. In a previous study, restoration of cerebral vascular function was enabled by introducing cell permeant phosphopeptide analogues of HSP20 without leading to hypotension in rat after SAH (Furnish, Brophy et al. 2010).
5. Limitations
The VP peptides were tested on normal large arteries in an ex vivo experimental setup. Both microvascular and macrovascular complications contribute to SAH. It is not known if the VP peptides have an effect on microvessels. Future study should determine in vivo efficacy of VP to alleviate radiographic narrowing, improve cerebral perfusion and neurobehavioral outcomes in animal models of SAH. Neuroischemia after SAH may have other contributory factors such as microthrombi formation, inflammation, apoptosis which are not all addressed by VP, suggesting that an effective therapeutic strategy to alleviate debilitating outcomes of SAH may require multi-prong approaches.
6. Conclusion
The downregulated NO-PKG signaling pathway contributes to SAH-induced vasospasm and explains, at least in part, the refractory responses of cerebral vasculature to vasodilators. Peptide analogues of phosphorylated VASP were rationally designed and developed that directly relax intact vascular smooth muscles. By bypassing the receptors and signaling pathways that are dysregulated in SAH to directlyrelax cerebral vessels and acts synergistically with the current standard of care, nimodipine, VP represents a potential therapy for the unmet need to prevent delayed vasospasm after SAH.
Supplementary Material
Suppl Fig. S1. CPP is required for VP uptake by rat aorta. RASM rings were suspended in a muscle bath pre-contracted with phenylephrine, followed by treatment with sodium nitroprusside (SNP; 10−9 and 10−8M) or escalating doses of the VASP petpides indicated (x10−3M). While VP3 relaxed PE-precontracted tissue dose-dependently, noCPP-VP3 (a peptide containing the cargo sequence of VP3; K(pS)RVLKK) had no vasorelaxing property. % relaxation were determined as a change to the maximal phenylephrine-induced contraction. n=3.
Suppl Fig. S2. Batch performances of VP3. Multiple batches (n=4) of VPS were synthesized independently. Each batch was tested for vasorelaxing properties on PE precontracted rat aorta.
Funding
This work was supported by the National Institutes of Health [R01HL70715-09]; and the Brain Aneurysm Foundation, Hanover, MA [Steph and Holly Chair of Research].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- Allen GS, Ahn HS, Preziosi TJ, Battye R, Boone SC, Boone SC, Chou SN, Kelly DL, Weir BK, Crabbe RA, Lavik PJ, Rosenbloom SB, Dorsey FC, Ingram CR, Mellits DE, Bertsch LA, Boisvert DP, Hundley MB, Johnson RK, Strom JA and Transou CR (1983). “Cerebral arterial spasm--a controlled trial of nimodipine in patients with subarachnoid hemorrhage.” N Engl J Med 308(11): 619–624. [DOI] [PubMed] [Google Scholar]
- Assenzio B, Martin EL, Stankevicius E, Civiletti F, Fontanella M, Boccaletti R, Berardino M, Mazzeo A, Ducati A, Simonsen U and Mascia L (2015). “Cerebrospinal fluid from patients with subarachnoid haemorrhage and vasospasm enhances endothelin contraction in rat cerebral arteries.” PLoS One 10(1): e0116456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates E, Bode C, Costa M, Gibson CM, Granger C, Green C, Grimes K, Harrington R, Huber K, Kleiman N, Mochly-Rosen D, Roe M, Sadowski Z, Solomon S and Widimsky P (2008). “Intracoronary KAI-9803 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction.” Circulation 117(7): 886–896. [DOI] [PubMed] [Google Scholar]
- Benz PM, Blume C, Seifert S, Wilhelm S, Waschke J, Schuh K, Gertler F, Munzel T and Renne T (2009). “Differential VASP phosphorylation controls remodeling of the actin cytoskeleton.” Journal of cell science 122(Pt 21): 3954–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Sepulveda MA, Panackal A, Maracheril R, Maddie N, Patel MN, Ojamaa K, Savinova OV and Gerdes AM (2019). “Triiodothyronine Reduces Vascular Dysfunction Associated with Hypertension by Attenuating Protein Kinase G/Vasodilator-Stimulated Phosphoprotein Signaling.” J Pharmacol Exp Ther 371(1): 88–94. [DOI] [PubMed] [Google Scholar]
- Chettimada S, Rawat DK, Dey N, Kobelja R, Simms Z, Wolin MS, Lincoln TM and Gupte SA (2012). “Glc-6-PD and PKG contribute to hypoxia-induced decrease in smooth muscle cell contractile phenotype proteins in pulmonary artery.” Am J Physiol Lung Cell Mol Physiol 303(1): L64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung-Flynn J, Alvis BD, Hocking KM, Guth CM, Luo W, McCallister R, Chadalavada K, Polcz M, Komalavilas P and Brophy CM (2019). “Normal Saline solutions cause endothelial dysfunction through loss of membrane integrity, ATP release, and inflammatory responses mediated by P2X7R/p38 MAPK/MK2 signaling pathways.” PLoS One 14(8): e0220893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly ES Jr., Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, Hoh BL, Kirkness CJ, Naidech AM, Ogilvy CS, Patel AB, Thompson BG, Vespa P, C. American Heart Association Stroke, R. Council on Cardiovascular, Intervention, N. Council on Cardiovascular, S. Council on Cardiovascular, Anesthesia and C. Council on Clinical (2012). “Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/american Stroke Association.” Stroke 43(6): 1711–1737. [DOI] [PubMed] [Google Scholar]
- Dietrich HH and Dacey RG Jr. (2000). “Molecular keys to the problems of cerebral vasospasm.” Neurosurgery 46(3): 517–530. [DOI] [PubMed] [Google Scholar]
- Dorhout Mees SM, Rinkel GJ, Feigin VL, Algra A, van den Bergh WM, Vermeulen M and van Gijn J (2007). “Calcium antagonists for aneurysmal subarachnoid haemorrhage.” Cochrane Database Syst Rev(3): CD000277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreiza CM, Brophy CM, Komalavilas P, Furnish EJ, Joshi L, Pallero MA, Murphy-Ullrich JE, von Rechenberg M, Ho YS, Richardson B, Xu N, Zhen Y, Peltier JM and Panitch A (2005). “Transducible heat shock protein 20 (HSP20) phosphopeptide alters cytoskeletal dynamics.” Faseb J 19(2): 261–263. [DOI] [PubMed] [Google Scholar]
- El Zaoui I, Touchard E, Berdugo M, Abadie C, Kowalczuk L, Deloche C, Zhao M, Naud MC, Combette JM and Behar-Cohen F (2015). “Subconjunctival injection of XG-102, a c-Jun N-terminal kinase inhibitor peptide, in the treatment of endotoxin- induced uveitis in rats.” J Ocul Pharmacol Ther 31(1): 17–24. [DOI] [PubMed] [Google Scholar]
- Fassbender K, Hodapp B, Rossol S, Bertsch T, Schmeck J, Schutt S, Fritzinger M, Horn P, Vajkoczy P, Wendel-Wellner M, Ragoschke A, Kuehl S, Brunner J, Schurer L, Schmiedeck P and Hennerici M (2000). “Endothelin-1 in subarachnoid hemorrhage: An acute-phase reactant produced by cerebrospinal fluid leukocytes.” Stroke 31(12): 2971–2975. [DOI] [PubMed] [Google Scholar]
- Fathi AR, Bakhtian KD and Pluta RM (2011). “The role of nitric oxide donors in treating cerebral vasospasm after subarachnoid hemorrhage.” Acta Neurochir Suppl 110(Pt 1): 93–97. [DOI] [PubMed] [Google Scholar]
- Furnish EJ, Brophy CM, Harris VA, Macomson S, Winger J, Head GA and Shaver EG (2010). “Treatment with transducible phosphopeptide analogues of the small heat shock-related protein, HSP20, after experimental subarachnoid hemorrhage: prevention and reversal of delayed decreases in cerebral perfusion.” Journal of neurosurgery 112(3): 631–639. [DOI] [PubMed] [Google Scholar]
- Gelmers HJ, Gorter K, de Weerdt CJ and Wiezer HJ (1988). “A controlled trial of nimodipine in acute ischemic stroke.” N Engl J Med 318(4): 203–207. [DOI] [PubMed] [Google Scholar]
- Grasso G, Alafaci C and Macdonald RL (2017). “Management of aneurysmal subarachnoid hemorrhage: State of the art and future perspectives.” Surg Neurol Int 8: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti G, Brambilla L and Rossi D (2017). “Cell-Penetrating Peptides: From Basic Research to Clinics.” Trends Pharmacol Sci 38(4): 406–424. [DOI] [PubMed] [Google Scholar]
- Henes J, Schmit MA, Morote-Garcia JC, Mirakaj V, Kohler D, Glover L, Eldh T, Walter U, Karhausen J, Colgan SP and Rosenberger P (2009). “Inflammation-associated repression of vasodilator-stimulated phosphoprotein (VASP) reduces alveolar-capillary barrier function during acute lung injury.” FASEB J 23(12): 4244–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho A, Schwarze SR, Mermelstein SJ, Waksman G and Dowdy SF (2001). “Synthetic protein transduction domains: enhanced transduction potential in vitro and in vivo.” Cancer Res 61(2): 474–477. [PubMed] [Google Scholar]
- Juvela S (2000). “Plasma endothelin concentrations after aneurysmal subarachnoid hemorrhage.” J Neurosurg 92(3): 390–400. [DOI] [PubMed] [Google Scholar]
- Lincoln TM, Dey N and Sellak H (2001). “Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression.” J Appl Physiol 91(3): 1421–1430. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, Vajkoczy P, Wanke I, Bach D, Frey A, Nowbakht P, Roux S and Kassell N (2012). “Randomized trial of clazosentan in patients with aneurysmal subarachnoid hemorrhage undergoing endovascular coiling.” Stroke 43(6): 1463–1469. [DOI] [PubMed] [Google Scholar]
- Macomson SD, Brophy CM, Miller W, Harris VA and Shaver EG (2002). “Heat shock protein expression in cerebral vessels after subarachnoid hemorrhage.” Neurosurgery 51(1): 204–210; discussion 210–201. [DOI] [PubMed] [Google Scholar]
- Melichar VO, Behr-Roussel D, Zabel U, Uttenthal LO, Rodrigo J, Rupin A, Verbeuren TJ, Kumar HSA and Schmidt HH (2004). “Reduced cGMP signaling associated with neointimal proliferation and vascular dysfunction in late-stage atherosclerosis.” Proc Natl Acad Sci U S A 101(47): 16671–16676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pula G and Krause M (2008). “Role of Ena/VASP proteins in homeostasis and disease.” Handb Exp Pharmacol(186): 39–65. [DOI] [PubMed] [Google Scholar]
- Rosenberger P, Khoury J, Kong T, Weissmuller T, Robinson AM and Colgan SP (2007). “Identification of vasodilator-stimulated phosphoprotein (VASP) as an HIF- regulated tissue permeability factor during hypoxia.” FASEB J 21(10): 2613–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer A, Burkhardt M, Vollkommer T, Bauersachs J, Munzel T, Walter U and Smolenski A (2003). “Endothelium-dependent and -independent relaxation and VASP serines 157/239 phosphorylation by cyclic nucleotide-elevating vasodilators in rat aorta.” Biochem Pharmacol 65(3): 397–405. [DOI] [PubMed] [Google Scholar]
- Sinnaeve P, Chiche JD, Nong Z, Varenne O, Van Pelt N, Gillijns H, Collen D, Bloch KD and Janssens S (2001). “Soluble guanylate cyclase alpha(1) and beta(1) gene transfer increases NO responsiveness and reduces neointima formation after balloon injury in rats via antiproliferative and antimigratory effects.” Circ Res 88(1): 103–109. [DOI] [PubMed] [Google Scholar]
- Siuta M, Zuckerman SL and Mocco J (2013). “Nitric oxide in cerebral vasospasm: theories, measurement, and treatment.” Neurol Res Int 2013: 972417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suckfuell M, Lisowska G, Domka W, Kabacinska A, Morawski K, Bodlaj R, Klimak P, Kostrica R and Meyer T (2014). “Efficacy and safety of AM-111 in the treatment of acute sensorineural hearing loss: a double-blind, randomized, placebo-controlled phase II study.” Otol Neurotol 35(8): 1317–1326. [DOI] [PubMed] [Google Scholar]
- Tang M, Tian Y, Li D, Lv J, Li Q, Kuang C, Hu P, Wang Y, Wang J, Su K and Wei L (2014). “TNF-alpha mediated increase of HIF-1alpha inhibits VASP expression, which reduces alveolar-capillary barrier function during acute lung injury (ALI).” PLoS One 9(7): e102967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda N, Ayajiki K and Okamura T (2009). “Cerebral blood flow regulation by nitric oxide: recent advances.” Pharmacol Rev 61(1): 62–97. [DOI] [PubMed] [Google Scholar]
- Touchard E, Omri S, Naud MC, Berdugo M, Deloche C, Abadie C, Jonet L, Jeanny JC, Crisanti P, de Kozak Y, Combette JM and Behar-Cohen F (2010). “A peptide inhibitor of c-Jun N-terminal kinase for the treatment of endotoxin-induced uveitis.” Invest Ophthalmol Vis Sci 51(9): 4683–4693. [DOI] [PubMed] [Google Scholar]
- Vajkoczy P, Meyer B, Weidauer S, Raabe A, Thome C, Ringel F, Breu V and Schmiedek P (2005). “Clazosentan (AXV-034343), a selective endothelin A receptor antagonist, in the prevention of cerebral vasospasm following severe aneurysmal subarachnoid hemorrhage: results of a randomized, double-blind, placebo-controlled, multicenter phase IIa study.” J Neurosurg 103(1): 9–17. [DOI] [PubMed] [Google Scholar]
- van Gijn J, Kerr RS and Rinkel GJ (2007). “Subarachnoid haemorrhage.” Lancet 369(9558): 306–318. [DOI] [PubMed] [Google Scholar]
- Voldby B, Engbaek F and Enevoldsen EM (1982). “CSF serotonin concentrations and cerebral arterial spasm in patients with ruptured intracranial aneurysm.” Stroke 13(2): 184–189. [DOI] [PubMed] [Google Scholar]
- Wang CJ, Lee PY, Wu BN, Wu SC, Loh JK, Tsai HP, Chung CL, Kassell NF and Kwan AL (2014). “Alteration of basilar artery rho-kinase and soluble guanylyl cyclase protein expression in a rat model of cerebral vasospasm following subarachnoid hemorrhage.” Biomed Res Int 2014: 531508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HB, Wang WQ, Wu QJ, Hou YJ, Li HX, Yang HJ, Yang MF, Sun BL and Zhang ZY (2020). “Negative Allosteric Modulator of mGluR1 Improves Long-Term Neurologic Deficits after Experimental Subarachnoid Hemorrhage.” ACS Chem Neurosci 11(18): 2869–2880. [DOI] [PubMed] [Google Scholar]
- Wu Y and Gunst SJ (2015). “Vasodilator-stimulated phosphoprotein (VASP) regulates actin polymerization and contraction in airway smooth muscle by a vinculin-dependent mechanism.” J Biol Chem 290(18): 11403–11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M and Seifert V (1998). “Endothelin and subarachnoid hemorrhage: an overview.” Neurosurgery 43(4): 863–875; discussion 875-866. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl Fig. S1. CPP is required for VP uptake by rat aorta. RASM rings were suspended in a muscle bath pre-contracted with phenylephrine, followed by treatment with sodium nitroprusside (SNP; 10−9 and 10−8M) or escalating doses of the VASP petpides indicated (x10−3M). While VP3 relaxed PE-precontracted tissue dose-dependently, noCPP-VP3 (a peptide containing the cargo sequence of VP3; K(pS)RVLKK) had no vasorelaxing property. % relaxation were determined as a change to the maximal phenylephrine-induced contraction. n=3.
Suppl Fig. S2. Batch performances of VP3. Multiple batches (n=4) of VPS were synthesized independently. Each batch was tested for vasorelaxing properties on PE precontracted rat aorta.