

Abstract
Field asymmetric ion mobility spectrometry (FAIMS), when used in proteomics studies, provides superior selectivity and enables more proteins to be identified by providing additional gas-phase separation. Here, we tested the performance of cylindrical FAIMS for the identification and characterization of proteoforms by top-down mass spectrometry of heterogeneous protein mixtures. Combining FAIMS with chromatographic separation resulted in a 62% increase in protein identifications, an 8% increase in proteoform identifications, and an improvement in proteoform identification compared to samples analyzed without FAIMS. In addition, utilization of FAIMS resulted in the identification of proteins encoded by lower-abundance mRNA transcripts. These improvements were attributable, in part, to improved signal-to-noise for proteoforms with similar retention times. Additionally, our results show that the optimal compensation voltage of any given proteoform was correlated with the molecular weight of the analyte. Collectively these results suggest that the addition of FAIMS can enhance top-down proteomics in both discovery and targeted applications.
Graphical Abstract
Mass spectrometry (MS)-based proteomics relies on separation of peptides and proteins, as extracts from cells, tissues, and fluids create highly complex mixtures. Because of proteome heterogeneity, multidimensional separations are critical to reduce sample complexity prior to tandem MS.1–3 For molecules of analytes to be sampled for fragmentation in a standard, “data-dependent” mode, its precursor intensity must be above the chemical/instrumental background noise and be one of the more abundant targets being measured.1 Moreover, separation of molecular species occurring at similar mass-to-charge ratios is essential for the acquisition of nonchimeric fragmentation spectra to achieve unambiguous, efficient protein identification by database retrieval. Frequently, the separation technique used online with MS for proteomics is liquid chromatography (LC), because of its versatility and high peak capacity.4
Multidimensional chromatographic separation of digested peptide species helped usher in the technology underlying bottom-up proteomics (BUP).5,6 By combining more than one chromatographic separation method, techniques such as MudPIT increased the number of protein identifications by reducing spectral heterogeneity.5,6 While this technique has been promulgated internationally, similar platforms for in-line top-down proteomics (TDP) analysis of intact proteins have remained in a less-developed state. Off-line separation of proteins prior to LC dramatically increases the number of protein identifications in top-down experiments.3 One technique that could enable multidimensional online protein separation for TDP analysis is field asymmetric ion mobility spectrometry (FAIMS)7 after LC separation and prior to MS analysis, circumventing the limitations faced by multiple hyphenated online or off-line fractionation steps.3
Cylindrical FAIMS is a gas-phase fractionation approach that transmits ions based on their differential mobility in high and low electric fields.7–11 In the context of a typical TDP experiment, intact proteins are ionized by an electrospray source and enter the FAIMS device, composed of an inner and outer electrode.7 A dispersion voltage provides an asymmetric waveform that propels ions toward one of the electrodes. Collision with either of the electrodes halts unstable ions, preventing their introduction to the mass spectrometer. Application of different compensatory DC voltages (compensation voltage, or CV) to the inner electrode of the FAIMS corrects the trajectory of ions that have been displaced due to differences in their mobility at high and low electric fields, allowing them to pass through the FAIMS for detection.7 Hence, FAIMS can function as a gas-phase separation device for intact protein ions.
Prior utilization of FAIMS has improved peptide identification for BUP applications12,13 and functions primarily through improving analyte signal-to-noise.14–16 Its use in TDP analysis has been limited, with most investigators studying intact protein standards. Investigators employing ion-mobility–time-of-flight mass spectrometry systems have shown that IMS influences the arrival time distribution of different peptides, denatured protein charge states, and multimeric native protein structures17–20 Our previous study employed static electrospray ionization (ESI) coupled with a commercial implementation of FAIMS to separate and analyze a simple mixture of heavy and light chains from a monoclonal antibody.21 Without gas-phase fractionation, the light chain dominates the precursor ion signal in MS experiments, resulting in low signal-to-noise and poor detection of the heavy chain. Utilization of FAIMS prior to MS resolved the two antibody chains by favoring the FAIMS stability of the heavy or light chain of the antibody while suppressing the other, all as a function of the applied compensation voltage.21 Additionally, combining liquid extraction surface analysis (LESA) with FAIMS from complex tissue samples provided a clear improvement in the signal-to-noise of intact precursor ion signal, compared to the same LESA samples analyzed without FAIMS.22–26 Building from these initial observations of complex protein samples, we sought to characterize denatured proteoforms from cell extracts using LC-FAIMS-MS/MS. The goal of the present study was to test and benchmark the performance of cylindrical FAIMS during TDP analysis of heterogeneous protein mixtures on an Orbitrap Fusion Lumos mass spectrometer. Our results show that combining FAIMS with reversed-phase LC separation enhances the number of both identified proteins and characterized proteoforms.
RESULTS AND DISCUSSION
Given past success using the FAIMS Pro interface to differentially fractionate reduced antibody chains in the gas phase, we expanded the complexity of our analysis by testing the capacity of LC coupled to FAIMS to separate the four proteins present in the top-down standard (TDS) mixture. We performed replicate injections of TDS over 80 min reversed-phase LC gradients with and without FAIMS prior to MS analysis. In samples that utilized FAIMS, we monitored the precursor ion signal in stepwise 10 V compensation voltage (CV) increments from −30 V to +20 V, using no supplementary carrier gas flow. As expected, the injection without FAIMS showed all four proteins at their expected retention times consistent with previous studies27 (Figure 1, top panel). The most intense protein in the total ion chromatogram (TIC) was carbonic anhydrase (29 kDa), followed by myoglobin (16.7 kDa) and ubiquitin (8.6 kDa) (Figure 1). Trypsinogen (24 kDa) was the least intense peak observed and eluted between ubiquitin and myoglobin. In the no-FAIMS analysis, the intensity of trypsinogen is typically lower, relative to the more-abundant TDS protein peaks in the chromatogram. Analysis of TDS with FAIMS and employing a single, constant CV of −30 V during the entire run resulted in a waveform that favored the stable trajectory of only ubiquitin, as shown on the second panel of Figure 1. As the CV was increased by 10 V in subsequent LC injections, the remaining protein standards were observed with larger protein species generally preferring more positive CVs, consistent with previous observations21 (Figure 1). The exception among the standards was trypsinogen, which contains disulfide bonds and presents lower charge states. Intriguingly, the folded structure of trypsinogen reduces the collisional cross section and affects the protein’s trajectory during differential ion mobility in the gas phase, compared to the other unfolded proteins; we observed trypsinogen to be stable at CV values of −20 V and −10 V, similar to that observed for the much smaller protein, myoglobin. Overall, the total ion intensity of protein standard intensities analyzed with FAIMS versus those without FAIMS was lower; as an example, the NL intensity for trypsinogen was 7.95 × 105 and for myoglobin was 2.8 × 107 (without FAIMS) and decreased to 1 × 105 and 6.85 × 105, respectively, at CV (−20 V) with FAIMS.
Figure 1.
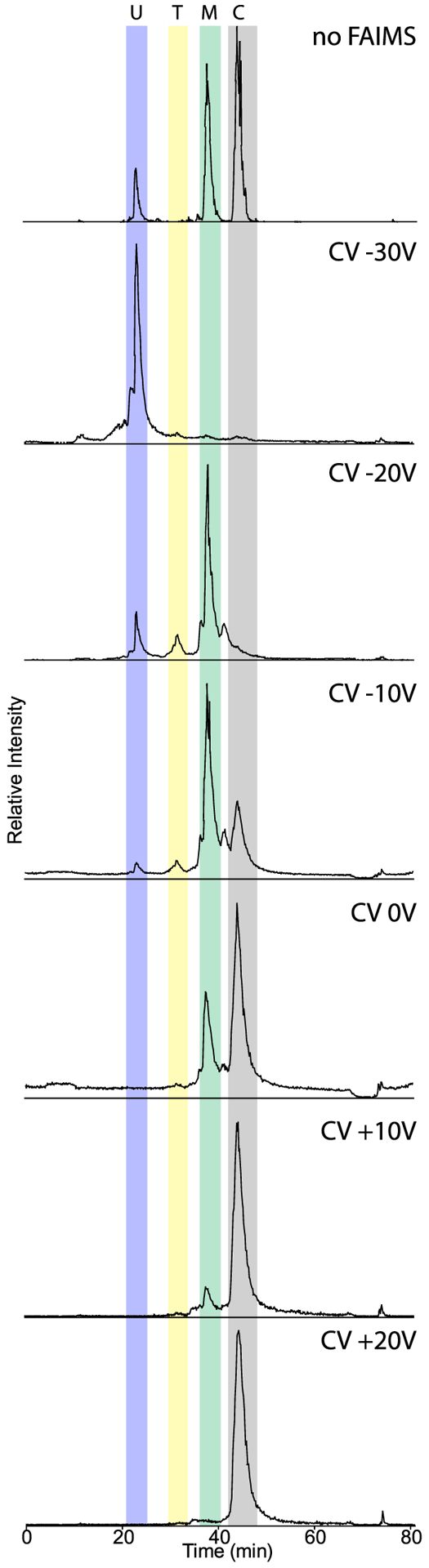
Total ion chromatograms (TICs) of top-down standards using either FAIMS at different CVs or no FAIMS. Top-down standards run without FAIMS (top panel) and with FAIMS using compensation voltages (CV) of −30 V, −20 V, −10 V, 0 V, + 10 V, and +20 V (lower panels). Top-down standard proteins: ubiquitin, 8.6 kDa (U); trypsinogen, 24 kDa (T); myoglobin, 16.7 kDa (M); and carbonic anhydrase, 29 kDa (C).
During TDP analysis of complex protein mixtures, abundant proteins and proteoforms are detected at the expense of lower-abundance forms. The selective stability of TDS proteins at different CVs suggested that FAIMS may be useful as an orthogonal separation technique for proteoforms with retention times similar to more abundant coeluting ions. As a model system for testing the behavior of heterogeneous protein/proteoform populations in the LC-FAIMS-MS/MS system, we employed the 0–30 kDa protein fraction of whole cell extracts from a population of 20 million primary human CD3+ T cells afforded by employing an 8% GELFrEE cartridge.28 This is the optimal molecular weight range for high-resolution TDP analysis and the recommended setting for top-down discovery mode.27 CVs were kept constant during each acquisition and without supplementary carrier gas flow in these experiments. We sought to identify the CVs that would result in the most proteoforms identified from a single injection ranging from −40 V to +30 V. Figure 2A demonstrates that the majority of the proteoforms were detected using a CV of −20 V with a Gaussian-like distribution.
Figure 2.

Protein and proteoform identifications with or without FAIMS. (A) Histogram of CD3+ T cell proteoforms identified at each compensation voltage (CV). (B, C) Venn diagrams of proteins (panel (B)) and proteoforms (panel (C)) identified from CD3+ T cells subjected to top-down proteomics analysis with FAIMS or without FAIMS.
In BUP, the use of the FAIMS Pro interface has been shown to increase the number of protein identifications when used as an orthogonal fractionation method.12,13 Similarly, we tested if FAIMS separation would result in gains in protein or proteoform identifications. We compared eight replicate injections of <30 kDa proteins from CD3+ T cells without FAIMS to eight injections with FAIMS applying CVs between −40 V and +30 V. Analysis of all 16 runs (with and without FAIMS) resulted in 367 protein identifications (Figure 2B) 1,537 proteoform identifications (Figure 2C). The application of FAIMS resulted in an 8% increase in proteoform identifications (Figure 2C) and a 62% increase of protein identifications, with 337 proteins identified with FAIMS, compared to 208 proteins identified without FAIMS (Figure 2B). The increase in identifications occurred over the same LC separation conditions and instrument method acquisition time.
To characterize the diversity of proteins identified from each replicate with and without FAIMS, we generated a heatmap using protein spectral counts (Figure 3A). From eight identical injections without FAIMS, we observed a conserved and stable population of proteins that were detected (Figure 3A). In contrast, changing the CV by 10 V for each injection resulted in the identification of a fraction of proteins that were unique to each CV (Figure 3A). The CV values with the largest number of new unique IDs were −40 V, −30 V, and −20 V. These results demonstrate that, in TDP experiments, FAIMS can (1) boost the identification of proteins/proteoforms that would otherwise be not fragmented and identified with only the LC separation alone, and (2) function as a selection filter for the identification of specific proteins/proteoforms from complex samples.
Figure 3.
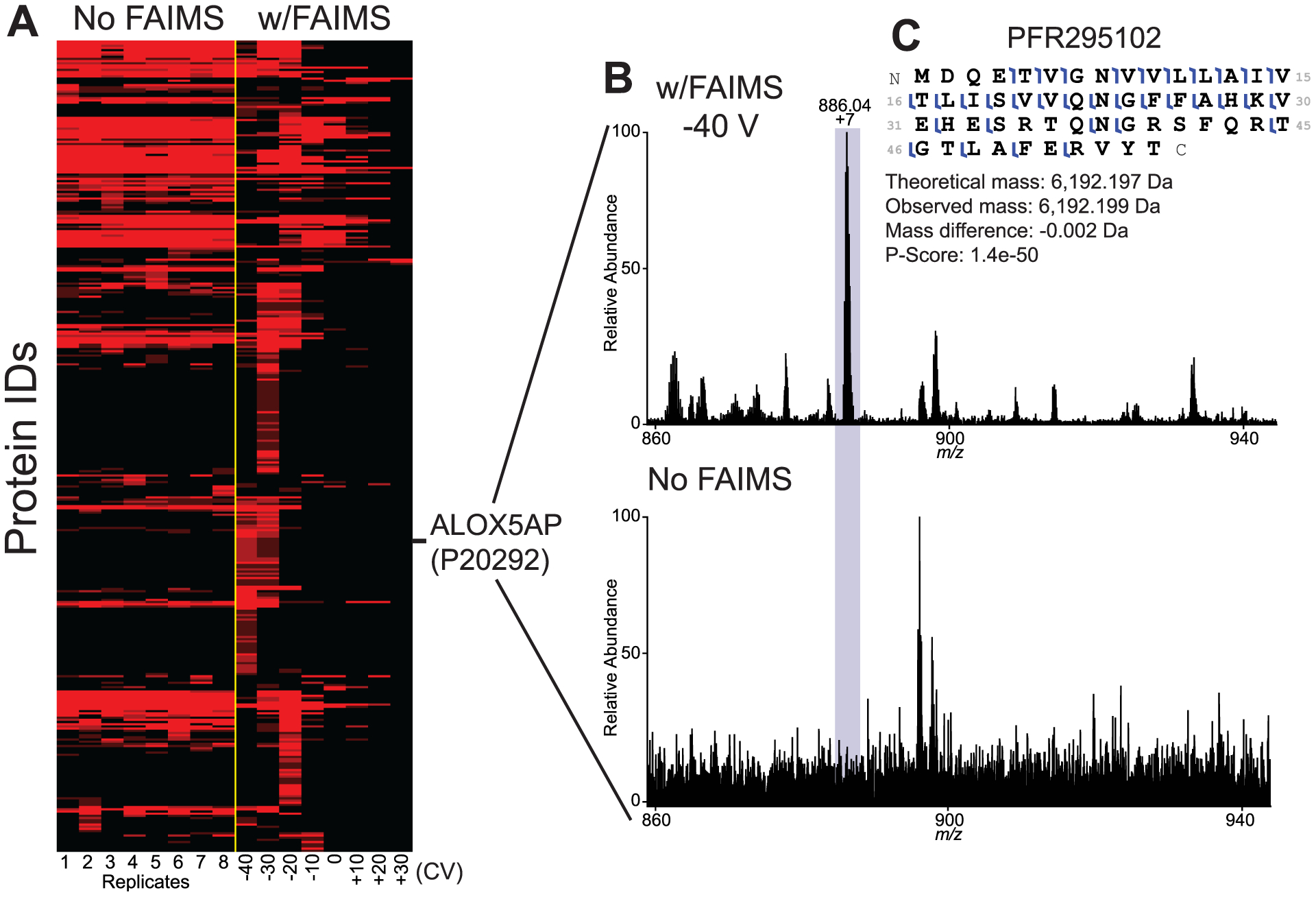
Changing the compensation voltage (CV) results in detection of a hidden population of proteins through improved signal-to-noise values. (A) Protein-level heatmap of CD3+ T cell proteins identified without FAIMS (8 technical replicates) or with FAIMS applying different CVs from −40 V to +30 V. CVs for injections analyzed with FAIMS are indicated at the bottom of the heatmap. (B) Mass spectrum of proteoform PFR295102 from UniProt accession P20292 (Arachidonate 5-lipoxygenase-activating protein) analyzed with FAIMS (top panel) and without FAIMS (lower panel) at same retention time. (C) Fragmentation map of PFR295102 showing 52% sequence coverage in the sample using FAIMS.
FAIMS improves the signal-to-noise ratio by enhancing for the analyte of interest, relative to background. Stable ions pass through the FAIMS unit, while other ions are deposited onto the FAIMS electrodes. Past experiments with FAIMS would suggest that the identification of new proteins at specific compensation voltages during orthogonal separation might be due to an improvement of signal-to-noise of those proteins.21,22,24 To test this possibility, we compared precursor spectra (with or without FAIMS) at retention times where a given protein was only identified by FAIMS. While there were several positive examples throughout the experiment, we highlighted 20 proteoforms (Figure S1 in the Supporting Information) and focused our analysis on PFR295102 from Arachidonate 5-lipoxygenase-activating protein (UniProt accession P20292; see Figure 3B). At a retention time of 41.57 min and a CV value of −40 V, the dominant 7+ precursor ion was observed with FAIMS. At the identical retention time (without FAIMS), the precursor ion was not observable above the background signal. Fragmentation spectra of the sample that utilized FAIMS showed high-quality protein characterization with fragment ions mapping to 52% of backbone positions (Figure 3C). Given the importance of Arachidonate 5-lipoxygenase-activating protein in leukotriene synthesis and inflammation,29–32 the capacity of FAIMS to facilitate characterization of this proteoform and others demonstrates an example of the potential impact of this technique in the analysis of complex biological samples. Additionally, all 20 additional proteoforms presented in Figure S1 showed clear differences in the signal-to-noise ratio in the precursor scans and have roles either directly implicated in specific T cell function or immunity.
We tested to see if proteins and proteoforms identified with FAIMS might be less abundant in the cell lysate than those identified without FAIMS. To evaluate this premise, we correlated the proteins identified in our experiments to mRNA transcriptome data from CD3-positive cells obtained from the Expression Atlas (see https://www.ebi.ac.uk/gxa/home). Comparing the expression level of all proteins identified either with FAIMS (mean = 208.9 ± 29.8) or without FAIMS (mean = 408.9 ± 46.2), using box-and-whisker plots, results were statistically significant (Student’s t-test, p < 0.016) indicating a lower number of transcripts per million (TPM) for proteins identified with FAIMS (see Figure S2A in the Supporting Information). The significance is even higher (p = 7.6 × 10−11) if we compare the TPM of all proteins identified without FAIMS (mean = 408.9 ± 46.2) with those that were uniquely identified with FAIMS (mean = 88.4 ± 8.8) (Figure S2B in the Supporting Information). Therefore, FAIMS improved proteome coverage by increasing the dynamic range of identified proteins.
Based on the ion selectivity noticed with the use of FAIMS, we expected precursor ion coselection and cofragmentation events to decrease, leading to improved fragmentation spectra and proteoform identification. To investigate this assumption, we compared sequence coverage, P-score,33 and C-score34 from proteoforms identified in the FAIMS and no FAIMS analysis using box and whisker plots (see Figure S3 in the Supporting Information). For all three parameters, the use of FAIMS improved the values and their difference is supported by strong statistical significance (Student’s t-test) with p < 4.1 × 10−75 for sequence coverage, p < 6.7 × 10−49 for P-score, and p < 2.2 × 10−28 for C-score (see Figure S3). These results suggest that FAIMS improves the identification and characterization of novel proteins and proteoforms from heterogeneous samples derived from specific cell types in part through improvement of signal-to-noise ratios of both precursor ions and their fragmentation products.
Aiming to understand the effect of supplementary gas flow on protein and proteoform fractionation in the FAIMS Pro device, we replicated the 8 injections with FAIMS using CVs from −40 V to +30 V and applied a supplementary carrier gas flow of 1 L/min. Increasing the gas amount inside the FAIMS device, we expected a higher residence time and more collisions, leading to better ion selectivity. Using these conditions, we identified 212 proteins and 704 proteoforms, representing a decrease of 37% and 32%, respectively, in the number of identifications, compared to the analysis performed without supplemental gas. The use of 1 L/min of gas resulted in reduced overall ion signal, suggesting a lower sensitivity under these conditions due to ion collision. Plotting the molecular weight of the identified proteoforms at each CV for both gas conditions, we observed an inter-relationship between them (see Figures S4A and S4B in the Supporting Information). The Pearson coefficient of correlation (r) between CV and proteoform molecular weight was 0.72 at 1L/min gas levels and 0.69 without supplementary carrier gas flow with an overlap in the 95% confidence intervals (see Figures S4C and S4D in the Supporting Information). These results show that when the cylindrical FAIMS is operated at higher gas levels, overall sensitivity is reduced and selectivity of proteoforms by mass is not improved. These results highlight the efficacy of cylindrical FAIMS as a mass selection filter in TDP acquisitions and opens a new range of possibilities for the field.
CONCLUSION
The FAIMS Pro interface coupled to high-resolution MS after reversed-phase LC increased the number of protein and proteoform identifications in TDP experiments performed on 0–30 kDa protein mixtures derived from human CD3+ T cells. The multidimensional LC-FAIMS separation occurred over the same instrument acquisition time as samples employing LC-MS alone, but resulted in 62% more protein identifications. Additionally, FAIMS can function as an effective molecular weight filter for proteoforms, providing a mechanism for predictive selectivity of targets from complex samples. Ultimately, FAIMS improvements are attributable to improved signal-to-noise ratios, resulting in the identification of proteoforms in samples that have coeluting species and highly complex matrices. The use of FAIMS in the TDP field will help to achieve deeper proteome characterizations in both discovery and targeted modes of operation.
Supplementary Material
ACKNOWLEDGMENTS
This research was conducted as part of the National Resource for Translational and Developmental Proteomics under Grant No. P41 GM108569 from the National Institute of General Medical Sciences and the National Institutes of Health, under Grant No. UH3 CA246635-02 (N.L.K.).
Footnotes
The authors declare the following competing financial interest(s): N.L.K. consults for Thermo Fisher Scientific. M.B. and R.H. are employees of Thermo Fisher Scientific.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.1c00402.
Details regarding the experimental methods; Figures S1–S4; associated references (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.1c00402
Contributor Information
Vincent R. Gerbasi, Northwestern University, National Resource for Translational and Developmental Proteomics, Evanston, Illinois 60208, United States; Pacific Northwest National Laboratories, Richland, Washington 99352, United States
Rafael D. Melani, Northwestern University, National Resource for Translational and Developmental Proteomics, Evanston, Illinois 60208, United States.
Susan E. Abbatiello, Northeastern University, Boston, Massachusetts 02115, United States; Thermo Fisher Scientific, San Jose, California 98665, United States
REFERENCES
- (1).Zhang Y; Fonslow BR; Shan B; Baek MC; Yates JR 3rd Chem. Rev 2013, 113 (4), 2343–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Cox J; Mann M Annu. Rev. Biochem 2011, 80, 273–99. [DOI] [PubMed] [Google Scholar]
- (3).Tran JC; Zamdborg L; Ahlf DR; Lee JE; Catherman AD; Durbin KR; Tipton JD; Vellaichamy A; Kellie JF; Li M; Wu C; Sweet SM; Early BP; Siuti N; LeDuc RD; Compton PD; Thomas PM; Kelleher NL Nature 2011, 480 (7376), 254– 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zhang X; Fang A; Riley CP; Wang M; Regnier FE; Buck C Anal. Chim. Acta 2010, 664 (2), 101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Link AJ; Eng J; Schieltz DM; Carmack E; Mize GJ; Morris DR; Garvik BM; Yates JR 3rd Nat. Biotechnol 1999, 17 (7), 676–82. [DOI] [PubMed] [Google Scholar]
- (6).Washburn MP; Wolters D; Yates JR 3rd Nat. Biotechnol 2001, 19 (3), 242–7. [DOI] [PubMed] [Google Scholar]
- (7).Cooper HJ J. Am. Soc. Mass Spectrom 2016, 27 (4), 566–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Guevremont RJ Chromatogr A 2004, 1058 (1–2), 3–19. [PubMed] [Google Scholar]
- (9).Shvartsburg AA; Tang K; Smith RD J. Am. Soc. Mass Spectrom 2005, 16 (1), 2–12. [DOI] [PubMed] [Google Scholar]
- (10).Swearingen KE; Moritz RL Expert Rev. Proteomics 2012, 9 (5), 505–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Purves RW; Prasad S; Belford M; Vandenberg A; Dunyach JJ J. Am. Soc. Mass Spectrom 2017, 28 (3), 525–538. [DOI] [PubMed] [Google Scholar]
- (12).Hebert AS; Prasad S; Belford MW; Bailey DJ; McAlister GC; Abbatiello SE; Huguet R; Wouters ER; Dunyach JJ; Brademan DR; Westphall MS; Coon JJ Anal. Chem 2018, 90 (15), 9529–9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Meyer JG; Niemi NM; Pagliarini DJ; Coon JJ Nat. Methods 2020, 17 (12), 1222–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bonneil E; Pfammatter S; Thibault PJ Mass Spectrom. 2015, 50 (11), 1181–95. [DOI] [PubMed] [Google Scholar]
- (15).Saba J; Bonneil E; Pomies C; Eng K; Thibault PJ Proteome Res. 2009, 8 (7), 3355–66. [DOI] [PubMed] [Google Scholar]
- (16).Venne K; Bonneil E; Eng K; Thibault P Anal. Chem 2005, 77 (7), 2176–86. [DOI] [PubMed] [Google Scholar]
- (17).Koeniger SL; Merenbloom SI; Clemmer DE J. Phys. Chem. B 2006, 110 (13), 7017–21. [DOI] [PubMed] [Google Scholar]
- (18).Laos V; Bishop D; Lang CA; Marsh NM; Cantrell KL; Buratto SK; Singh AK; Bowers MT Biochemistry 2020, 59 (4), 499–508. [DOI] [PubMed] [Google Scholar]
- (19).Laos V; Do TD; Bishop D; Jin Y; Marsh NM; Quon B; Fetters M; Cantrell KL; Buratto SK; Bowers MT ACS Chem. Neurosci 2019, 10 (9), 4112–4123. [DOI] [PubMed] [Google Scholar]
- (20).Srebalus CA; Li J; Marshall WS; Clemmer DE Anal. Chem 1999, 71 (18), 3918–27. [DOI] [PubMed] [Google Scholar]
- (21).Melani RD; Srzentic K; Gerbasi VR; McGee JP; Huguet R; Fornelli L; Kelleher NL MAbs 2019, 11 (8), 1351– 1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Griffiths RL; Cooper HJ Anal. Chem 2016, 88 (1), 606–9. [DOI] [PubMed] [Google Scholar]
- (23).Griffiths RL; Creese AJ; Race AM; Bunch J; Cooper HJ Anal. Chem 2016, 88 (13), 6758–66. [DOI] [PubMed] [Google Scholar]
- (24).Griffiths RL; Hughes JW; Abbatiello SE; Belford MW; Styles IB; Cooper HJ Anal. Chem 2020, 92 (4), 2885–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Griffiths RL; Kocurek KI; Cooper HJ Methods Mol. Biol 2020, 2084, 191–201. [DOI] [PubMed] [Google Scholar]
- (26).Sarsby J; Griffiths RL; Race AM; Bunch J; Randall EC; Creese AJ; Cooper HJ Anal. Chem 2015, 87 (13), 6794–800. [DOI] [PubMed] [Google Scholar]
- (27).Donnelly DP; Rawlins CM; DeHart CJ; Fornelli L; Schachner LF; Lin Z; Lippens JL; Aluri KC; Sarin R; Chen B; Lantz C; Jung W; Johnson KR; Koller A; Wolff JJ; Campuzano IDG; Auclair JR; Ivanov AR; Whitelegge JP; Pasa-Tolic L; Chamot-Rooke J; Danis PO; Smith LM; Tsybin YO; Loo JA; Ge Y; Kelleher NL; Agar JN Nat. Methods 2019, 16 (7), 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Toby TK; Fornelli L; Srzentic K; DeHart CJ; Levitsky J; Friedewald J; Kelleher NL Nat. Protoc 2019, 14 (1), 119–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Goodarzi K; Goodarzi M; Tager AM; Luster AD; von Andrian UH Nat. Immunol 2003, 4 (10), 965–73. [DOI] [PubMed] [Google Scholar]
- (30).Matsumoto T; Funk CD; Radmark O; Hoog JO; Jornvall H; Samuelsson B Proc. Natl. Acad. Sci. U. S. A 1988, 85 (1), 26–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ott VL; Cambier JC; Kappler J; Marrack P; Swanson BJ Nat. Immunol 2003, 4 (10), 974–81. [DOI] [PubMed] [Google Scholar]
- (32).Tager AM; Bromley SK; Medoff BD; Islam SA; Bercury SD; Friedrich EB; Carafone AD; Gerszten RE; Luster AD Nat. Immunol 2003, 4 (10), 982–90. [DOI] [PubMed] [Google Scholar]
- (33).Meng F; Cargile BJ; Miller LM; Forbes AJ; Johnson JR; Kelleher NL Nat. Biotechnol 2001, 19 (10), 952–7. [DOI] [PubMed] [Google Scholar]
- (34).LeDuc RD; Fellers RT; Early BP; Greer JB; Thomas PM; Kelleher NL J. Proteome Res 2014, 13 (7), 3231–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.