

Abstract
This Review describes the increasing demand for organic synthesis to facilitate fragment-based drug discovery (FBDD), focusing on polar, unprotected fragments. In FBDD, X-ray crystal structures are used to design target molecules for synthesis with new groups added onto a fragment via specific growth vectors. This requires challenging synthesis which slows down drug discovery, and some fragments are not progressed into optimisation due to synthetic intractability. We have evaluated the output from Astex's fragment screenings for a number of programs, including urokinase-type plasminogen activator, hematopoietic prostaglandin D2 synthase, and hepatitis C virus NS3 protease-helicase, and identified fragments that were not elaborated due, in part, to a lack of commercially available analogues and/or suitable synthetic methodology. This represents an opportunity for the development of new synthetic research to enable rapid access to novel chemical space and fragment optimisation.
Herein is described the concept of fragment sociability and the opportunities for organic chemistry to address the challenges of fragment elaboration.
Introduction
In view of the increasing interest in and success of fragment-based drug discovery (FBDD), this Review describes the current chemistry challenges within the field: the design of new fragments and the elaboration of weakly binding fragments into nM leads guided by X-ray crystal structures.1 Ideally, synthetic elaboration of fragment hits in three-dimensions from many different growth vectors is experimentally worked out prior to fragment screening.2,3 This will increase the chance of success during fragment-to-lead optimisation.4–6
As the field of small molecule drug discovery has advanced, the demand for molecular complexity has increased in line with ambitions to modulate the functions of increasingly complex human protein systems.7,8 This evolution has resulted in calls to the synthetic organic chemistry community for advances in synthesis methodology to keep pace with the demands of modern drug design in an attempt to avoid situations where desired molecules cannot be synthesised or, more commonly, are avoided in favour of designs that are more accessible.9 Such calls are being met by recent advances in broadly impactful organic synthesis methodology including transition metal catalysed couplings,10–14 electrocatalysis,15–18 photocatalysis,19–23 and C–H activation24–27 together with new technologies such as high-throughput experimentation (HTE),28–30 flow chemistry,31,32 and artificial intelligence.33–35
Fragment-based drug discovery
Over the past 20 years fragment-based drug discovery (FBDD) has become widely used in pharma, biotech and academic institutes to identify over 40 compounds in clinical trials and 4 launched drugs pexidartinib,36 vemurafenib,37 erdafitinib,38 and venetoclax.6,39 In FBDD, the binding of low molecular weight fragments to their target protein is typically characterised by high resolution X-ray crystallography.40–42 This is critical not only in understanding and optimising the interactions underlying fragment binding, but in providing insights for the progression into high affinity leads.43
The fragment-to-lead process involves the bespoke design of small molecules with 3D shape and electrostatics complementary to the target protein.44–46 In order to engage in additional interactions with the protein, substituents need to be added to the starting fragment at precise positions referred to as growth vectors (Fig. 1). Clearly, the direction and synthetic tractability of growth vector elaboration are critical in defining the suitability of a fragment for further development, together with other properties relevant to drug discovery.1,47 Furthermore, fragment elaboration may also reveal cryptic subpockets that result from residue movement to accommodate binding of the ligand.48,49 In addition to these factors, commercial availability of closely related analogues50 with exemplified growth vectors, heterocycle core modifications, or well-established scaffold modifications are important to consider when selecting which fragment to consider as a suitable starting point for a fragment-to-lead program.
Fig. 1. Example of a protein–fragment crystal structure that is used to identify specific growth vectors (arrows) to guide fragment-to-lead elaboration.

For fragment optimisation, access to close analogues of the fragment hit determines the speed with which fragments can be evaluated and prioritised. This involves the analysis of protein fragment X-ray crystal structures to identify suitable growth vectors for the introduction of common functional groups onto the fragment and should be limited to a HAC (heavy atom count) ≤ 6 (Fig. 2). Frequently, closely related analogues can be obtained from commercial suppliers to determine structure activity relationships (SAR).51 However, this process can be a limiting factor for unexemplified fragments given the timelines of many commercial drug discovery projects. Recently, several groups have explored synthetic methods to address this issue, such as the concept of ‘poised fragments’ by Brennan and colleagues which utilise pre-functionalised fragments and the Spring group with derivatization of DOS-derived fragments.52–54
Fig. 2. Representative examples of common functional groups added during fragment elaboration. Note – not an exhaustive list of functional groups. HAC = number of non-hydrogen atoms (heavy atom count).

In this Review, we describe a retrospective comparison of fragments from screening campaigns at Astex Pharmaceuticals. Through this retrospective analysis we have developed a working definition of fragment sociability: an unsociable fragment is one that has limited (if any) synthetic methodology to enable growth vector elaboration and few commercially available close analogues. In contrast, a sociable fragment is one supported with robust synthetic methodology that enables every growth vector to be elaborated and a significant number of commercially available close analogues. To illustrate the concept of sociability we identified two X-ray hits from each of the selected programs (uPA, H-PGDS, and HCV NS3 protease-helicase); an unsociable fragment that was down prioritized due to synthetic intractability and a sociable fragment that progressed to a lead compound (Table 1).
Fragments that have been identified binding to three protein targets and classified by sociability as determined by published methods and close analogues available on eMolecules®.
| Protein target | Unsociable fragments | Sociable fragments | ||||
|---|---|---|---|---|---|---|
| Structure | Published method | Commercially available close analogues | Structure | Published method | Commercially available close analogues | |
| Urokinase-like plasminogen activator (uPA) |
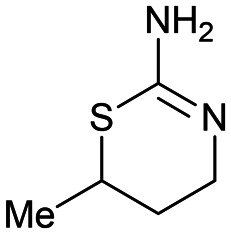 1 1 |
No55 | <5 |
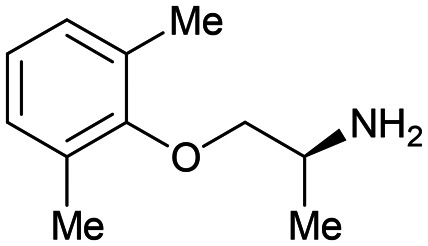 2 2 |
Yes56 | >100 |
| Hematopoietic prostaglandin D2 synthase (H-PGDS) |
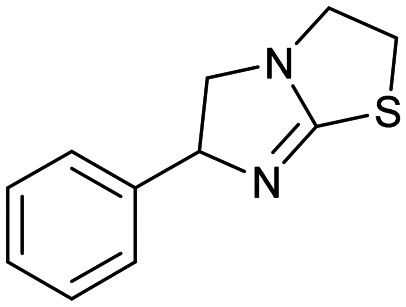 3 3 |
Yes57 | <10 |
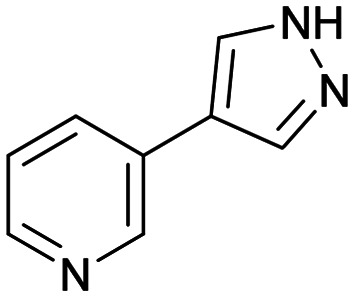 4 4 |
Yes58 | >250 |
| Hepatitis C virus NS3 protease-helicase |
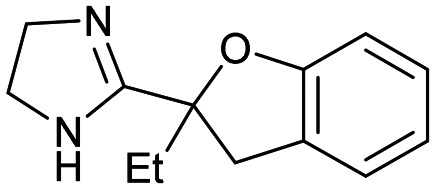 5 5 |
Yes59 | <5 |
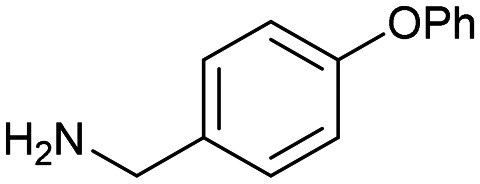 6 6 |
Yes60 | >1000 |
Results and discussion
Urokinase-type plasminogen activator (uPA)
Urokinase-type plasminogen activator (uPA) is a trypsin-like serine protease that catalyses the conversion of plasminogen to plasmin through amide bond hydrolysis.61 Plasmin is responsible for a number of proteolytic processes that degrade components of the extracellular matrix enabling cellular migration.62 As such, uPA is linked to metastasis in cancer and therefore a target for therapeutic intervention.63
We screened our fragment library against uPA and detected 105 X-ray hits bound in the catalytic site of the enzyme.64 This enabled the team to identify the essential binding pharmacophore as the charged ammonium or amidine acting as a formal hydrogen bond donor (Fig. 3A). The pharmacophore interacts with the backbone carbonyls of Gly219 and Ser190 as well as forming a salt bridge with the side chain of Asp189. Among the fragments binding to the active site of the enzyme we identified the fragments 1 and 2 (Fig. 3A and C). Additionally, both fragments make beneficial hydrophobic interactions with the protein.
Fig. 3. Urokinase-type plasminogen activator (uPA)-fragment co-complexes A) overlay of unsociable fragment 1 (orange) and sociable fragment 2 (green) bound to the active site of uPA. B) Overlay of sociable fragment 2 (green) and lead compound 7 (pink). C) Properties and biochemical potencies of unsociable fragment 1, sociable fragment 2, and lead compound 7. Red circle – binding pharmacophore, blue circle and arrow – growth vector.

Fragment 1 was a ligand efficient starting point (LE > 0.51) for fragment-to-lead program. The X-ray structure of fragment 1 bound to uPA identified several potential growth vectors from the saturated ring. Fortuitously, during the initial screening campaign Abbott Laboratories (now Abbvie) reported on the development of napthamidine inhibitors of uPA which identified that growth towards the catalytic triad (Ser195, His57, and Asp102) resulted in a substantial gain in affinity.65 Guided by the crystal structure and literature information the optimal growth vector to access the catalytic machinery was identified as the 5-position from 1 (Fig. 3A and C). Although commercially available from several suppliers, the exact synthetic route of 1 is not reported55 and close analogues that are elaborated at the 5-position were not commercially available, therefore early SAR could not be easily obtained. This situation would require a substantial investment in chemistry resource at an early stage in the program to develop a bespoke diastereo- and enantioselective synthetic route to close analogues of 1.66 Due to this intractability, fragment 1 was not selected for optimisation against uPA.
In contrast to fragment 1, the orally active drug mexiletine 2 was identified as a more attractive fragment hit for follow-up.67 While the clog P of 2 was quite high, this fragment has clear developability and substantial chemistry reported in the literature, there was inherent confidence in this fragment as a suitable starting point. Thorough exploration of the active site pocket from the 4-position of the fragment was enabled by chemistries such as Suzuki–Miyaura cross-coupling and amide bond formation. This facilitated rapid SAR gathering and led to the lead compound 7, a potent inhibitor of uPA with an IC50 = 0.07 μM (LE = 0.31) (Fig. 3B and C).64
Hematopoietic prostaglandin D2 synthase (H-PGDS)
H-PGDS is an enzyme responsible for the isomerisation of prostaglandin H2 to prostaglandin D2 (PGD2).68 The biological effects of PGD2 include vasodilation, bronchoconstriction, inhibition of platelet aggregation among others. As such, H-PGDS has been indicated as a target for allergic rhinitis and other inflammatory disorders.69
Fragment screening against H-PGDS identified 76 fragments binding to the catalytic site of the protein.70 These fragments contain a similar chemical architecture that compliments the binding pocket with a polar heterocycle linked via a carbon–carbon bond to an aromatic ring. This is illustrated in both fragments 3 and 4 (Fig. 4A and B). Additionally, the pyrazole moiety of 4 binds to the protein through a donor–acceptor interaction with Asp96 and Tyr152. This was an unexpected result as H-PGDS is known to bind lipophilic aryl pharmacophores, whereas our fragment screening identified a strong polar interaction within the lipophilic pocket.58
Fig. 4. Hematopoietic prostaglandin D2-synthase (H-PGDS)-fragment co-complexes A) overlay of unsociable fragment 3 (orange) and sociable fragment 4 (green). B) Overlay of sociable fragment 3 (green) and lead compound 8 (pink). C) Properties and biochemical potencies of fragments 3, 4 and the lead compound 8. Red circle – binding pharmacophore, blue circle and arrow – growth vector.

Similar to compound 2, fragment 3 is an approved oral drug ((+/−)-tetramisole, (+)-dexamisole, and (−)-levamisole),57 and therefore represents an attractive starting point for hit-to-lead. This fragment does not undergo a formal hydrogen bond donor or acceptor interaction but has a beneficial π–π-stacking interaction with Trp104 and excellent shape complementarity with the protein (Fig. 4A). However, the route to 3 is linear and does not lend itself to the rapid generation of analogues, in particular from the sp3-growth vectors of the core bicycle. Thus, limited access to close analogues and a linear synthetic route rendered compound 3 an unsociable fragment. Coupled with the sub-optimal ligand efficiency (LE ∼ 0.29) this hit was down prioritised in favour of 4.
Fragment 4 is a weak but ligand efficient inhibitor of H-PGDS and the aromatic substituent of 4 forms a π–π-stacking interaction with Trp104 (Fig. 4B). As with all sociable fragments, there were a substantial number of commercially available analogues that facilitated rapid follow up of fragment 4 from the 3-position of the pyrazole and from the 4-position of the other aromatic ring. Once the commercially available compounds were exhausted, reliable synthetic chemistry enabled rapid progress to the lead compound 8 which transitioned into lead optimization (Fig. 4C).58
Hepatitis C virus NS3 protease-helicase (HCV NS3 protease-helicase)
The hepatitis C virus genome encodes ten viral proteins that ensure the propagation of the viral particle.71,72 Of these proteins, the NS3 protein is a bifunctional enzyme that contains an N-terminal serine protease domain and a C-terminal helicase domain that are closely associated in the full length protein.73 While there had been extensive studies on the isolated protease domain74 and early reports on the helicase domain,75 we performed the first fragment screen on the full-length protein.
The fragment screen was performed against the HCV NS3 full-length genotype 1b holozyme.76 The output from the screen identified a novel binding site at the interface of the protease-helicase domains.77 It is a relatively lipophilic binding site with acidic amino acid residues (Asp527, Asp79, Glu628) lining the entrance to the tunnel. This was reflected in the prevalence of lipophilic fragment hits, such as compounds 5 and 6 (Fig. 5A and C).
Fig. 5. Hepatitis C virus NS3 protease-helicase (HCV NS3 protease-helicase) – fragment co-complexes A) overlay of unsociable fragment 5 (orange) and sociable fragment 6 (green). B) Overlay of sociable fragment 6 (green) and lead compound 9 (pink). C) Properties and biochemical potencies of fragments 5, and 6 and the lead compound 9. Red circle – binding pharmacophore, blue circle and arrow – growth vector.

Compound 5, the commercially available efaroxan,59 contains a semi-saturated bicycle with a quaternary stereocentre that interacts with the entrance to the tunnel of the protein (Fig. 5A) This was an essential growth vector to interact with the acidic residues around the entrance of the pocket. Another key growth vector that needs to be synthetically enabled originates from the 5-position of the aromatic ring. While 5 itself is commercially available and there is literature precedent for this compound, there is a limited number of commercially available analogues and the synthetic route is a lengthy linear process.78,79 This necessitates the installation of the desired substitution or a synthetic handle at an early stage of the synthesis which limits throughput and lengthens the design-synthesise-test paradigm of medicinal chemistry, making this an unsociable fragment.80
The HCV NS3 fragment co-complexes of 6 enabled the effective deployment of structure-based drug design (SBDD) to identify viable growth vectors to target specific residues and sub-pockets within the protein to rapidly improve affinity. Examining this structural data, a systematic exploration of the SAR in the tunnel site was carried out whilst maintaining the benzylamine as the binding pharmacophore. This process was facilitated by the large number of commercially available analogues with this motif. Additionally, the facile introduction of the benzylic stereocentre and ease of amine substitution enabled the efficient growth of the ligand around the tunnel entrance. Finally, exploration of the entrance to the tunnel from the benzylic amine growth vector culminated in the identification of the lead molecule 9, a potent allosteric inhibitor (IC50 = 0.1 μM, LE = 0.38) of the HCV NS3 protease-helicase (Fig. 5B and C).77 When the aforementioned factors are considered, it is clear that compound 6 can be classified as a sociable fragment.
Method for the identification of unsocial fragments
The fragment network, recently described by Hall and co-workers, is based on a graph database, which is a data structure that is common in social media applications. In social media a node in the network represents a person and each edge in the network represents a friendship between two people. A person with many friendships can be thought of as sociable. In the fragment network a node in the network represents a fragment molecule and an edge represents a relationship between two fragments (based on their similarity). By analogy to social media we denote a fragment with many edges to be a sociable fragment.81
To identify unsocial fragments, we utilized the fragment network on all fragments in the current version of our core screening library (1651 compounds). For each fragment, we interrogated the corresponding node in the fragment network and focused on edges between this node and neighbouring nodes that have a higher heavy atom count, indicating commercially available analogues that are growth vector enabled. By grouping the connecting edges by positional growth vector and comparing the ratio of observed positions at which a fragment is grown to the maximum theoretical number of growth points, we could estimate how many growth points are synthetically accessible. We then ranked the compounds in our fragment library according to this ratio of observed growth points. The number of protein targets against which each fragment had been observed as an X-ray hit was also used to rank the least sociable fragments. For the purposes of this work, only commercially available fragments available in eMolecules® were considered in the analysis.82 This analysis resulted in the identification of 30 putative unsociable fragments.
The fragment network is our preferred methodology for the rapid assessment of sociability for a large number of fragments, however, sociability can also be assessed by a collection of substructure searches and analysis of individual fragments in standard searching tools such as SciFinder®. To further confirm the initial analysis performed by the fragment network, we performed a manual assessment of the 30 compounds for commercial availability of close analogues on SciFinder®. We subsequently went further and manually assessed the literature for synthetic methods to access the target molecule and analogues thereof within 4-synthetic steps. Robustness of the chemistry with a focus on commonly utilised medicinal chemistry reactions was also considered to target suitable quantities of material.83 This manual process led to the identification of 12 compounds within our fragment library that we consider as unsociable fragments (Scheme 1).
Scheme 1. 12 fragments contained within our fragment library are considered unsociable fragments. These are examples of fragments that require organic methodology development to become sociable.

Synthetic routes could be envisioned for these unsocial fragments, but they may not be consistent with the time constraints of typical drug discovery projects with the pressure of pressing unmet medical needs. Some of these fragments could become more synthetically tractable if minor changes were made to the structure (e.g. introduction of a carbonyl at C-4 of 1 or removal of the –Et from 5) or selecting different growth vectors. However, in general these changes disrupt the protein binding interactions and therefore are not suitable from a SBDD perspective. If there was methodology to selectively functionalize these unsociable fragments at each and every carbon growth vector with the common functional groups added during fragment elaboration (Fig. 2) while maintaining the binding pharmacophore, we would consider this a valuable development in FBDD and organic chemistry as a whole.
The Astex fragment library has been subjected to constant analysis to improve performance. These analyses have resulted in evolution of the library over several generations taking into account these learnings.1 Two of these key factors – synthetic tractability and synthetic vectors – have influenced the design and chemical space occupied by the library and are important aspects influencing which fragments that are included in the library. This correlates to the low rate of unsociability for the fragments contained within the Astex library. However, if more esoteric fragments were socialized then they would warrant inclusion into our library and provide an opportunity to identify novel starting points for drug discovery.
Identification of false positives – the eye of an organic chemist
Computational analysis (such as our fragment network) can result in sociable fragments being identified as unsociable (apparent unsociable fragment), so it is important to engage a skilled organic chemist prior to assessing if a compound is or is not sociable. As the implications of an apparently unsociable fragment can down-prioritise an otherwise valuable fragment in favour of other chemical matter. For example, this is observed for simple bond disconnections or single-step functional group transformations and will be discussed in more detail below.
One class of false positive is a double scaffold in which two ring systems are joined by a simple linker or single bond (Scheme 2). Such molecules do not score well in the fragment network analysis because the growth points on each ring system are not well represented in commercial databases; yet analogues would be facile to synthesise as the apparent unsociable fragment yields two highly social compounds upon disconnection. These functionalised reagents represent good starting points for diversification through robust chemical transformations.52
Scheme 2. Examples of apparently unsociable fragments and the single bond transformation that yields functionalised sociable reagents that enable rapid analogue synthesis via robust organic methods.

A second type of false positive manifests when a simple functional group modification can result in an otherwise social fragment being misidentified. As a representative example, the dihydrobenzothiazine-dioxide (10) is poorly socialised when analysed by the fragment network.84 However, ‘simplification’ to the reduced dihydrobenzothiazine (11) results in identification of several commercially available analogues.85 It is important to note that synthetic elaboration of every carbon position of 11 is exemplified thus enabling access to every growth vector of 10 through a single-synthetic transformation step. As such, complex molecules should be simplified to the core scaffold by a single bond-forming or bond breaking chemical transformation to identify near neighbours (Scheme 3). We expect that the recent advances in AI to make a significant impact in this area of fragment sociability and growth vector elaboration in the near future.86,87
Scheme 3. Example of false positive ‘unsociable fragment’ based on functional group manipulation. Simplification of fragment 10 results in a more sociable compound 11 that is growth vector enabled at each carbon atom (selected commercially available examples identified by the fragment network).

Conclusions
FBDD is a key hit-finding technology for drug discovery and has enabled the discovery of several approved drugs. However, as the field of FBDD has developed over the past 20 years it has revealed the need for further development in the field of organic synthesis to successfully functionalise specific growth vectors of polar, unprotected small molecules using medicinal chemistry relevant transformations.7,88 This Review is intended to inspire the development of organic methodology targeted at unsociable fragments in an effort to socialise them and thereby facilitate the development of novel medicines.1,89
Conflicts of interest
The authors are employees of Astex Pharmaceuticals.
Supplementary Material
Acknowledgments
We would like to thank our Astex colleagues, in particular Gianni Chessari, Rachel Grainger, Christopher Johnson, David Twigg, and Andrew Woodhead for their insightful comments during the preparation of this manuscript.
Biographies
Biography
Jeffrey D. St. Denis.

Jeffrey D. St. Denis received his BSc (hons) in Biology with a minor in chemistry from Niagara University (2008). He then moved to McGill University to pursue his MSc (2010) in organic chemistry under the supervision of Prof. James. L. Gleason. He then went on to obtain his PhD in organic chemistry from the University of Toronto (2014) under the supervision of Prof. Andrei K. Yudin. Currently, he is a Senior Research Associate – Medicinal Chemistry at Astex Pharmaceuticals in Cambridge, UK a drug discovery company in Cambridge, UK that has pioneered fragment-based drug design.
Biography
Richard Hall.

Richard Hall received his BSc in chemistry from UMIST (1992) and his PhD in theoretical chemistry from the University of Manchester (1995) under the supervision of Prof. Ian Hillier. He is currently Director of Computational Chemistry and Informatics at Astex Pharmaceuticals in Cambridge, UK.
Biography
Christopher Murray.

Christopher Murray received his BA (1986) in chemistry and PhD in theoretical chemistry (1989) from the University of Cambridge. Currently, he is Senior VP of Discovery Technology at Astex where he has contributed to the design and exploitation of fragment libraries. He has extensive experience of structure-based drug-design and has helped discover several compounds that have entered into human clinical trials, including the approved FGFR-inhibitor erdafitinib.
Biography
Tom Heightman.

Tom Heightman received his MA in Chemistry from the University of Oxford (1993) and his PhD from the ETH in Zurich (1998). Since then he has led medicinal chemistry teams in big pharma, biotech and academia, contributing to the discovery of multiple INDs across a range of therapeutic areas and protein families. He is currently VP & Global Head of Oncology Chemistry at AstraZeneca, based in Cambridge, UK and Boston, US.
Biography
David Rees.

David Rees received his PhD in 1982 (Cambridge, UK, A. J. Pearson) and worked as a post-doc with E. J. Corey (Harvard). Currently, he is Chief Scientific Officer at Astex. Previously, with colleagues at Organon, he discovered Sugammadex, an anaesthetic reversal agent used in over 40 countries. He served as President of the RSC Organic Division and as Trustee of the RSC. In 2020 he was elected to the Fellowship of the Academy of Medical Sciences.
Notes and references
- (a) Murray C. W. Rees D. C. Angew. Chem., Int. Ed. 2016;55:488–492. doi: 10.1002/anie.201506783. [DOI] [PubMed] [Google Scholar]; (b) Keserű G. M. Erlanson D. A. Ferenczy G. G. Hann M. M. Murray C. W. Pickett S. D. J. Med. Chem. 2016;59:8189–8206. doi: 10.1021/acs.jmedchem.6b00197. [DOI] [PubMed] [Google Scholar]
- Ray P. C. Kiczun M. Huggett M. Lim A. Prati F. Gilbert I. H. Wyatt P. G. Drug Discovery Today. 2017;22:43–56. doi: 10.1016/j.drudis.2016.10.005. [DOI] [PubMed] [Google Scholar]
- O'Brien P. Downes T. D. Jones S. P. Klein H. F. Wheldon M. C. Atobe M. Bond P. S. Firth J. D. Chan N. S. Waddelove L. Hubbard R. E. Blakemore D. C. De Fusco C. Roughley S. D. Vidler L. R. Whatton M. A. Woolford A. J.-A. Wrigley G. L. Chem. – Eur. J. 2020;26:8969–8975. doi: 10.1002/chem.202001123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troelsen N. S. Shanina E. Gonzalez-Romero D. Danková D. Jensen I. S. A. Śniady K. J. Nami F. Zhang H. Rademacher C. Cuenda A. Gotfredsen C. H. Clausen M. H. Angew. Chem. 2020;59:2204–2210. doi: 10.1002/anie.201913125. [DOI] [PubMed] [Google Scholar]
- (a) Johnson C. N. Erlanson D. A. Jahnke W. Mortenson P. N. Rees D. C. J. Med. Chem. 2018;61:1774–1784. doi: 10.1021/acs.jmedchem.7b01298. [DOI] [PubMed] [Google Scholar]; (b) Johnson C. N. Erlanson D. A. Murray C. W. Rees D. C. J. Med. Chem. 2017;60:89–99. doi: 10.1021/acs.jmedchem.6b01123. [DOI] [PubMed] [Google Scholar]; (c) Mortenson P. N. Erlanson D. A. de Esch I. J. P. Jahnke W. Johnson C. N. J. Med. Chem. 2019;62:3857–3872. doi: 10.1021/acs.jmedchem.8b01472. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A. de Esch I. J. P. Jahnke W. Johnson C. N. Mortenson P. M. J. Med. Chem. 2020;63:4430–4444. doi: 10.1021/acs.jmedchem.9b01581. [DOI] [PubMed] [Google Scholar]
- Grygorenko O. O. Volochnyuk D. M. Ryabukhin S. V. Judd D. B. Chem. – Eur. J. 2020;26:1196–1237. doi: 10.1002/chem.201903232. [DOI] [PubMed] [Google Scholar]
- Eastgate M. D. Schmidt M. A. Fandrick K. R. Nat. Rev. Chem. 2017;1:0016. doi: 10.1038/s41570-017-0016. [DOI] [Google Scholar]
- Blakemore D. C. Castro L. Churcher I. Rees D. C. Thomas A. W. Wilson D. M. Wood A. Nat. Chem. 2018;10:383–394. doi: 10.1038/s41557-018-0021-z. [DOI] [PubMed] [Google Scholar]
- Feng K. Quevedo R. E. Kohrt J. T. Oderinde M. S. Reilly U. White M. C. Nature. 2020;580:621–627. doi: 10.1038/s41586-020-2137-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana R. Pathak T. P. Sigman M. S. Chem. Rev. 2011;111:1417–1492. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Lennox A. J. Lloyd-Jones G. C. Chem. Soc. Rev. 2014;43:412–443. doi: 10.1039/C3CS60197H. [DOI] [PubMed] [Google Scholar]; (b) St. Denis J. D. Lee C. F. Yudin A. K. ACS Catal. 2015;5:5373–5379. doi: 10.1021/acscatal.5b00790. [DOI] [Google Scholar]
- Dorel R. Grugel C. P. Haydl A. M. Angew. Chem., Int. Ed. 2019;58:17118–17129. doi: 10.1002/anie.201904795. [DOI] [PubMed] [Google Scholar]
- (a) Everson D. A. Weix D. J. J. Org. Chem. 2014;79:4793–4798. doi: 10.1021/jo500507s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee C. F. Holownia A. Bennett J. M. Elkins J. M. St. Denis J. D. Adachi S. Yudin A. K. Angew. Chem., Int. Ed. 2017;56:6264–6267. doi: 10.1002/anie.201611006. [DOI] [PubMed] [Google Scholar]
- Kingston C. Palkowitz M. D. Takahira Y. Vantourout J. C. Peters B. K. Kawamata Y. Baran P. S. Acc. Chem. Res. 2020;53:72–83. doi: 10.1021/acs.accounts.9b00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M. Kawamata Y. Baran P. S. Chem. Rev. 2017;117:13230–13319. doi: 10.1021/acs.chemrev.7b00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francke R. Little R. D. Chem. Soc. Rev. 2014;43:2492–2521. doi: 10.1039/C3CS60464K. [DOI] [PubMed] [Google Scholar]
- Horn E. J. Rosen B. R. Baran P. S. ACS Cent. Sci. 2016;2:302–308. doi: 10.1021/acscentsci.6b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twilton J. Le C. Zhang P. Shaw M. H. Evans R. W. MacMillan D. W. C. Nat. Rev. Chem. 2017;1:0052. doi: 10.1038/s41570-017-0052. [DOI] [Google Scholar]
- Shaw M. H. Twilton J. MacMillan D. W. C. J. Org. Chem. 2016;81:6898–6926. doi: 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prier C. K. Rankic D. A. MacMillan D. W. C. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero N. A. Nicewicz D. A. Chem. Rev. 2016;116:10075–10166. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- McAtee R. C. McClain E. J. Stephenson C. R. J. Trends Chem. 2019;1:111–125. doi: 10.1016/j.trechm.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K. Yamada S. Kaneda T. Itami K. Chem. Rev. 2017;117:9302–9332. doi: 10.1021/acs.chemrev.7b00021. [DOI] [PubMed] [Google Scholar]
- (a) Chen M. S. White M. C. Science. 2007;318:783–787. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]; (b) St. Denis J. D. Lee C. F. Yudin A. K. Org. Lett. 2015;17:5764–5767. doi: 10.1021/acs.orglett.5b02861. [DOI] [PubMed] [Google Scholar]
- Cernak T. Dykstra K. D. Tyagarajan S. Vachal P. Krska S. W. Chem. Soc. Rev. 2016;45:546–576. doi: 10.1039/C5CS00628G. [DOI] [PubMed] [Google Scholar]
- Lyons T. W. Sanford M. S. Chem. Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger R. Heightman T. D. Ley S. V. Lima F. Johnson C. N. Chem. Sci. 2019;10:2264–2271. doi: 10.1039/C8SC04789H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buitrago Santanilla A. Regalado E. L. Pereira T. Shevlin M. Bateman K. Campeau L.-C. Schneeweis J. Berritt S. Shi Z.-C. Nantermet P. Lui Y. Hemly R. Welch C. J. Vachal P. Davies I. W. Cernak T. Dreher S. D. Science. 2015;347:49–53. doi: 10.1126/science.1259203. [DOI] [PubMed] [Google Scholar]
- Shevlin M. ACS Med. Chem. Lett. 2017;8:601–607. doi: 10.1021/acsmedchemlett.7b00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo W.-J. Ishitani H. Saito Y. Laroche B. Kobayashi S. J. Org. Chem. 2020;85:5132–5145. doi: 10.1021/acs.joc.9b03416. [DOI] [PubMed] [Google Scholar]
- Plutschack M. B. Pieber B. Gilmore K. Seeberger P. H. Chem. Rev. 2017;117:11796–11893. doi: 10.1021/acs.chemrev.7b00183. [DOI] [PubMed] [Google Scholar]
- Struble T. J. Alvarez J. C. Brown S. P. Chytil M. Cisar J. DesJarlais R. L. Engkvist O. Frank S. A. Greve D. R. Griffin D. J. Hou X. Johannes J. W. Kreatsoulas C. Lahue B. Mathea M. Mogk G. Nicolaou C. A. Palmer A. D. Price D. J. Robinson R. I. Salentin S. Xing L. Jaakkola T. Green W. H. Barzilay R. Coley C. W. Jensen K. F. J. Med. Chem. 2020;63:8667–8682. doi: 10.1021/acs.jmedchem.9b02120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida A. F. Moreira R. Rodrigues T. Nat. Rev. Chem. 2019;3:589–604. doi: 10.1038/s41570-019-0124-0. [DOI] [Google Scholar]
- Segler M. H. S. Preuss M. Waller M. P. Nature. 2018;555:604–610. doi: 10.1038/nature25978. [DOI] [PubMed] [Google Scholar]
- Lamb Y. N. Drugs. 2019;79:1805–1812. doi: 10.1007/s40265-019-01210-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag G. Tsai J. Zhang J. Zhang C. Ibrahim P. Nolop K. Hirth P. Nat. Rev. Drug Discovery. 2012;11:873–886. doi: 10.1038/nrd3847. [DOI] [PubMed] [Google Scholar]
- Murray C. W. Newell D. R. Angibaud P. MedChemComm. 2019;10:1509–1511. doi: 10.1039/C9MD90044F. [DOI] [Google Scholar]
- Baker M. Nat. Rev. Drug Discovery. 2013;12:5–7. doi: 10.1038/nrd3926. [DOI] [PubMed] [Google Scholar]
- Hall R. J. Mortenson P. N. Murray C. W. Prog. Biophys. Mol. Biol. 2014;116:82–91. doi: 10.1016/j.pbiomolbio.2014.09.007. [DOI] [PubMed] [Google Scholar]
- Patel D. Bauman J. D. Arnold E. Prog. Biophys. Mol. Biol. 2014;116:92–100. doi: 10.1016/j.pbiomolbio.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I.-J. Hubbard R. E. J. Comput.-Aided Mol. Des. 2009;23:603–620. doi: 10.1007/s10822-009-9280-5. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A. Fesik S. W. Hubbard R. E. Jahnke W. Jhoti H. Nat. Rev. Drug Discovery. 2016;15:605–619. doi: 10.1038/nrd.2016.109. [DOI] [PubMed] [Google Scholar]
- Johnson J. A. Nicolaou C. A. Kirberger S. E. Pandey A. K. Hu H. Pomerantz W. C. K. ACS Med. Chem. Lett. 2019;10:1648–1654. doi: 10.1021/acsmedchemlett.9b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach A. R. Hann M. M. Curr. Opin. Chem. Biol. 2011;15:489–496. doi: 10.1016/j.cbpa.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Ogata K. Isomura T. Kawata S. Yamashita H. Kubodera H. Wodak S. J. Molecules. 2010;15:4382–4400. doi: 10.3390/molecules15064382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray C. W. Verdonk M. L. Rees D. C. Trends Pharmacol. Sci. 2012;33:224–232. doi: 10.1016/j.tips.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Murray C. W. Carr M. G. Callaghan O. Chessari G. Congreve M. Cowan S. Coyle J. E. Downham R. Figueroa E. Frederickson M. Graham B. McMenamin R. O'Brien M. A. Patel S. Phillips T. R. Williams G. Woodhead A. J. Woolford A. J. J. Med. Chem. 2010;53:5942–5955. doi: 10.1021/jm100059d. [DOI] [PubMed] [Google Scholar]
- Woodhead A. J. Angove H. Carr M. G. Chessari G. Congreve M. Coyle J. E. Cosme J. Graham B. Day P. J. Downham R. Fazal L. Feltell R. Figueroa E. Frederickson M. Lewis J. McMenamin R. Murray C. W. O'Brien M. A. Parra L. Patel S. Phillips T. Rees D. C. Rich S. Smith D. M. Trewartha G. Vinkovic M. Williams B. Woolford A. J. J. Med. Chem. 2010;53:5956–5969. doi: 10.1021/jm100060b. [DOI] [PubMed] [Google Scholar]
- Helal C. J. Bundesmann M. Hammond S. Holmstrom M. Klug-McLeod J. Lefker B. A. McLeod D. Subramanyam C. Zakaryants O. Sakata S. ACS Med. Chem. Lett. 2019;10:1104–1109. doi: 10.1021/acsmedchemlett.9b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg F. W. Kettle J. G. Kogej T. Perry M. W. D. Tomkinson N. P. Drug Discovery Today. 2015;20:11–17. doi: 10.1016/j.drudis.2014.09.023. [DOI] [PubMed] [Google Scholar]
- Cox O. B. Krojer T. Collins P. Monteiro O. Talon R. Bradley A. Fedorov O. Amin J. Marsden B. D. Spencer J. von Delft F. Brennan P. E. Chem. Sci. 2016;7:2322–2330. doi: 10.1039/C5SC03115J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg D. G. Kondo N. Mitchell S. L. Galloway W. R. Sore H. F. Madin A. Spring D. R. Angew. Chem., Int. Ed. 2016;55:12479–12483. doi: 10.1002/anie.201606496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd S. L. Fowler E. Reinhardt T. Compton T. Mateu N. Newman H. Bellini D. Talon R. McLoughlin J. Krojer T. Aimon A. Bradley A. Fairhead M. Brear P. Díaz-Sáez L. McAuley K. Sore H. F. Madin A. O'Donovan D. H. Huber K. V. M. Hyvönen M. von Delft F. Dowson C. G. Spring D. R. Chem. Sci. 2020;11:10792–10801. doi: 10.1039/D0SC01232G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöberl A. Magosch K.-H. Justus Liebigs Ann. Chem. 1970;742:74–84. doi: 10.1002/jlac.19707420110. [DOI] [PubMed] [Google Scholar]
- Roselli M. Carocci A. Budriesi R. Micucci M. Toma M. Di Cesare Mannelli L. Lovece A. Catalano A. Cavalluzzi M. M. Bruno C. De Palma A. Contino M. Perrone M. G. Colabufo N. A. Chiarini A. Franchini C. Ghelardini C. Habtemariam S. Lentini G. Eur. J. Med. Chem. 2016;121:300–307. doi: 10.1016/j.ejmech.2016.05.046. [DOI] [PubMed] [Google Scholar]
- Raeymaekers A. H. M. Allewijn F. T. N. Vandenberk J. Demoen P. J. A. Van Offenwert T. T. T. Janssen P. A. J. J. Med. Chem. 1966;9:545–551. doi: 10.1021/jm00322a023. [DOI] [PubMed] [Google Scholar]
- Saxty G. Norton D. Affleck K. Clapham D. Cleasby A. Coyle J. Day P. Frederickson M. Hancock A. Hobbs H. Hutchinson J. Le J. Leveridge M. McMenamin R. Mortenson P. Page L. Richardson C. Russell L. Sherriff E. Teague S. Uddin S. Hodgson S. Med. Chem. Commun. 2014;5:134–141. doi: 10.1039/C3MD00280B. [DOI] [Google Scholar]
- Chan S. L. F. Brown C. A. Morgan N. G. Eur. J. Pharmacol. 1993;230:375–378. doi: 10.1016/0014-2999(93)90577-5. [DOI] [PubMed] [Google Scholar]
- Fotsch C. Sonnenberg J. D. Chen N. Hale C. Karbon W. Norman M. H. J. Med. Chem. 2001;44:2344–2356. doi: 10.1021/jm0004547. [DOI] [PubMed] [Google Scholar]
- Michael J. D. Curr. Pharm. Des. 2004;10:39–49. doi: 10.2174/1381612043453559. [DOI] [PubMed] [Google Scholar]
- Tarui T. Majumdar M. Miles L. A. Ruf W. Takada Y. J. Biol. Chem. 2002;277:33564–33570. doi: 10.1074/jbc.M205514200. [DOI] [PubMed] [Google Scholar]
- Schweinitz A. Steinmetzer T. Banke I. J. Arlt M. J. Sturzebecher A. Schuster O. Geissler A. Giersiefen H. Zeslawska E. Jacob U. Kruger A. Sturzebecher J. J. Biol. Chem. 2004;279:33613–33622. doi: 10.1074/jbc.M314151200. [DOI] [PubMed] [Google Scholar]
- Frederickson M. Callaghan O. Chessari G. Congreve M. Cowan S. R. Matthews J. E. McMenamin R. Smith D.-M. Vinković M. Wallis N. G. J. Med. Chem. 2008;51:183–186. doi: 10.1021/jm701359z. [DOI] [PubMed] [Google Scholar]
- Wendt M. D. Rockway T. W. Geyer A. McClellan W. Weitzberg M. Zhao X. Mantei R. Nienaber V. L. Stewart K. Klinghofer V. Giranda V. L. J. Med. Chem. 2004;47:303–324. doi: 10.1021/jm0300072. [DOI] [PubMed] [Google Scholar]
- Latli B. D'Amou K. Casida J. E. J. Med. Chem. 1999;42:2227–2234. doi: 10.1021/jm980721x. [DOI] [PubMed] [Google Scholar]
- Monk J. P. Brogden R. N. Drugs. 1990;40:374–411. doi: 10.2165/00003495-199040030-00005. [DOI] [PubMed] [Google Scholar]
- Kanaoka Y. Urade Y. Prostaglandins, Leukotrienes Essent. Fatty Acids. 2003;69:163–167. doi: 10.1016/S0952-3278(03)00077-2. [DOI] [PubMed] [Google Scholar]
- Thurairatnam S., in Progress in Medicinal Chemistry, ed. G. Lawton and D. R. Witty, Elsevier, 2012, vol. 51, pp. 97–133 [Google Scholar]
- Deaton D. N. Do Y. Holt J. Jeune M. R. Kramer H. F. Larkin A. L. Orband-Miller L. A. Peckham G. E. Poole C. Price D. J. Schaller L. T. Shen Y. Shewchuk L. M. Stewart E. L. Stuart J. D. Thomson S. A. Ward P. Wilson J. W. Xu T. Guss J. H. Musetti C. Rendina A. R. Affleck K. Anders D. Hancock A. P. Hobbs H. Hodgson S. T. Hutchinson J. Leveridge M. V. Nicholls H. Smith I. E. D. Somers D. O. Sneddon H. F. Uddin S. Cleasby A. Mortenson P. N. Richardson C. Saxty G. Bioorg. Med. Chem. 2019;27:1456–1478. doi: 10.1016/j.bmc.2019.02.017. [DOI] [PubMed] [Google Scholar]
- Bartenschlager R. Lohmann V. J. Gen. Virol. 2000;81:1631–1648. doi: 10.1099/0022-1317-81-7-1631. [DOI] [PubMed] [Google Scholar]
- Reed K. E. and Rice C. M., in The Hepatitis C Viruses, ed. C. H. Hagedorn and C. M. Rice, Springer, Berlin Heidelberg, 2000, pp. 55–84, 10.1007/978-3-642-59605-6_4 [DOI] [Google Scholar]
- Beran R. K. Pyle A. M. J. Biol. Chem. 2008;283:29929–29937. doi: 10.1074/jbc.M804065200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashfaq U. A. Javed T. Rehman S. Nawaz Z. Riazuddin S. Virol. J. 2011;8:1–10. doi: 10.1186/1743-422X-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.-S. Wang C.-C. Wu H.-N. FEBS Lett. 2010;584:2356–2362. doi: 10.1016/j.febslet.2010.04.020. [DOI] [PubMed] [Google Scholar]
- Yao N. Reichert P. Taremi S. S. Prosise W. W. Weber P. C. Structure. 1999;7:1353–1363. doi: 10.1016/S0969-2126(00)80025-8. [DOI] [PubMed] [Google Scholar]
- Saalau-Bethell S. M. Woodhead A. J. Chessari G. Carr M. G. Coyle J. Graham B. Hiscock S. D. Murray C. W. Pathuri P. Rich S. J. Richardson C. J. Williams P. A. Jhoti H. Nat. Chem. Biol. 2012;8:920–925. doi: 10.1038/nchembio.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer P. Brunel P. Imbert T. Bioorg. Med. Chem. Lett. 1999;9:3021–3022. doi: 10.1016/S0960-894X(99)00531-4. [DOI] [PubMed] [Google Scholar]
- Couture K. Gouverneur V. Mioskowski C. Bioorg. Med. Chem. Lett. 1999;9:3023–3026. doi: 10.1016/S0960-894X(99)00530-2. [DOI] [PubMed] [Google Scholar]
- Chapleo C. B. Myers P. L. Butler R. C. M. Davis J. A. Doxey J. C. Higgins S. D. Myers M. Roach A. G. Smith C. F. C. J. Med. Chem. 1984;27:570–576. doi: 10.1021/jm00371a003. [DOI] [PubMed] [Google Scholar]
- Hall R. J. Murray C. W. Verdonk M. L. J. Med. Chem. 2017;60:6440–6450. doi: 10.1021/acs.jmedchem.7b00809. [DOI] [PubMed] [Google Scholar]
- eMolecules® website: https://www.emolecules.com/
- Roughley S. D. Jordan A. M. J. Med. Chem. 2011;54:3451–3479. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- Xing L. Jing T. Zhang J. Guo M. Miao X. Jiang F. Zhai X. Bioorg. Chem. 2018;81:689–699. doi: 10.1016/j.bioorg.2018.09.019. [DOI] [PubMed] [Google Scholar]
- Monika M. Anju G. Asian J. Pharm. Clin. Res. 2015;8:41–46. [Google Scholar]
- Empel C. Koenigs R. M. Angew. Chem., Int. Ed. 2019;58:17114–17116. doi: 10.1002/anie.201911062. [DOI] [PubMed] [Google Scholar]
- Johansson S. Thakkar A. Kogej T. Bjerrum E. Genheden S. Bastys T. Kannas C. Schliep A. Chen H. Engkvist O. Drug Discovery Today: Technol. 2020 doi: 10.1016/j.ddtec.2020.06.002. [DOI] [PubMed] [Google Scholar]
- Boström J. Brown D. G. Young R. J. Keserü G. M. Nat. Rev. Drug Discovery. 2018;17:709–727. doi: 10.1038/nrd.2018.116. [DOI] [PubMed] [Google Scholar]
- Palmer N. Peakman T. M. Norton D. Rees D. C. Org. Biomol. Chem. 2016;14:1599–1610. doi: 10.1039/C5OB02461G. [DOI] [PubMed] [Google Scholar]