

Abstract
Dioxin was historically one of the most common industrial contaminants with several major industry accidents, as well as governmental actions involving military service, having exposed large numbers of the worldwide population over the past century. Previous rat studies have demonstrated the ability of dioxin (2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)) exposure to promote the epigenetic transgenerational inheritance of disease susceptibility in subsequent generations. The types of disease previously observed include puberty abnormalities, testis, ovary, kidney, prostate and obesity pathologies. The current study was designed to use an epigenome-wide association study (EWAS) to identify potential sperm DNA methylation biomarkers for specific transgenerational diseases. Therefore, the transgenerational F3 generation dioxin lineage male rats with and without a specific disease were compared to identify differential DNA methylation regions (DMRs) as biomarkers for disease. The genomic features of the disease-specific DMRs were characterized. Observations demonstrate that disease-specific epimutation DMRs exist for the transgenerational dioxin lineage rats that can potentially be used as epigenetic biomarkers for testis, kidney, prostate and obesity diseases. These disease-specific DMRs were associated with genes that have previously been shown to be linked with the specific diseases. This EWAS for transgenerational disease identified potential epigenetic biomarkers and provides the proof of concept of the potential to develop similar biomarkers for humans to diagnose disease susceptibilities and facilitate preventative medicine.
Keywords: EWAS, Epigenetics, Transgenerational, DNA Methylation, Sperm, Dioxin, TCDD, Prostate, Kidney, Obesity, Testis, Pathology
Introduction
Agent Orange was an herbicide/defoliant sprayed across Vietnam and Southeast Asia by the US Air Force during the Vietnam War. The most toxic byproduct contaminant present in Agent Orange was dioxin – 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) [1, 2]. The soldiers handling this chemical were heavily exposed and thus at risk for exposure to TCDD. In addition, dioxin (TCDD) is a common pollutant from industry in most urban areas. Several major industrial accidents have occurred where the general populations were exposed [3, 4]. Dioxins are extremely lipophilic and persist in both the exposed individuals and the environment. The half-life of TCDD in the human body has been estimated to range from 7 to 11 years [1, 2]. In rodents, TCDD has a half-life of weeks to years and can cause liver disease, weight loss, thymic atrophy and immune suppression [5, 6]. In humans, direct exposure to TCDD can influence chronic diseases such as lymphomas and leukemias [7]. Vietnam officials reported around 400,000 people killed or maimed, and 500,000 children born with birth defects after being exposed to Agent Orange [8]. Prostate cancer, multiple myeloma, type II diabetes, and spina bifida in children were reported to be associated with Agent Orange exposure [9]. TCDD exposure happened not only in Vietnam. In the 1970s, Italy, China and Taiwan had industrial accidents that exposed populations to TCDD [3, 4, 10]. Various human exposures to TCDD have been documented and associated with a large number of different diseases [11–13]. The Developmental Origins of Health and Disease (DOHaD) paradigm is an expanding field of research focusing on the effects of chemical exposures such as dioxin on early-life development and the propagation of non-communicable disease into adulthood [14]. However, the majority of these epidemiology studies have focused on direct adult and fetal exposures [15]. In the Seveso Italy population, health effects in the grandchildren have been shown, even three decades after the dioxin exposure [16]. In addition to health effects of direct parental exposures on the offspring, a number of studies have been conducted on the health consequences of ancestral exposures in future generations.
The biological mechanism underlying these phenomena are epigenetic transgenerational inheritance processes, a form of non-genetic inheritance [17]. Epigenetics is defined as molecular factors or processes around DNA that regulate genome activity, independent of DNA sequence, and are mitotically stable [18]. Epigenetic transgenerational inheritance involves the transmission of an altered epigenome and phenotypes through the germline across generations in the absence of continued direct environmental exposures [17, 19]. During fetal development, the primordial germ cells (PGCs) undergo DNA demethylation and then upon gonadal sex determination a remethylation in a sex specific manner in order to generate the sperm or egg [20]. Environmental exposures during this period of development can alter the reprogramming of germline epigenetics, and sometimes the altered DNA methylation appears to become permanently programmed, similar to the DNA methylation of an imprinted gene [18, 21]. The epigenetic changes are propagated from the male and female germline to the zygote, and subsequently to the embryo stem cells and subsequently all somatic cells, which then will result in an altered epigenome and transcriptome in the subsequent generations [18]. Various environmental exposures such as nutrition, stress and chemical insults have been shown to promote the epigenetic transgenerational inheritance of adult onset disease in a wide variety of organisms from plants to humans [18]. These epigenetic changes could be used as potential biomarkers of exposure and disease [22]. Epigenetic molecular processes involve DNA methylation, histone modifications, non-coding RNA, chromatin structure, and RNA methylation [18]. A variety of environmental exposures and toxicants have been shown to promote the epigenetic transgenerational inheritance of disease [17, 21, 23–26]. Recent studies have demonstrated that ancestral environmental exposures promote the concurrent alterations of three different epimutations in sperm involving differential DNA methylated regions (DMRs), differential histone retention sites (DHRs) and ncRNA [27, 28]. The agricultural fungicide vinclozolin [29, 30], pesticide DDT (dichloro-diphenyl-trichloroethane) [31, 32], herbicide atrazine [23], and herbicide glyphosate [33] have all been shown to promote the epigenetic transgenerational inheritance of disease. In addition, the pathologies observed appear to be associated with unique epigenetic signatures of DMRs [23, 29, 31]. The current study investigates DMRs in association with specific transgenerational diseases.
In previous studies, we have shown that dioxin (i.e., TCDD) was able to promote epigenetic transgenerational inheritance of disease and DNA methylation epimutations in sperm [34, 35]. When the F1 generation offspring directly exposed in utero were studied at one year of age, they were found to have a higher incidence of prostate disease in the males, primordial follicle loss in the females and polycystic ovarian disease compared to the control lineage [34]. The subsequent F3 generation great-grand offspring not directly exposed also appeared to have a significant increase in the frequency of male kidney disease, primordial follicle loss, polycystic ovarian disease, and female multiple disease compared to the control lineage [34]. As previously described, dioxin was found to promote major pathology through both direct exposure and ancestral exposure. These results are relevant to the human population since more data are accumulating on the health consequences of ancestral exposures to dioxin in future generations [34–37]. Understanding the biological mechanisms underlying this toxicant exposure induced transgenerational epigenetic inheritance is further investigated in the current study. The potent and persistent environmental contaminants such as dioxins should be major environmental concerns today for human health [38]. Dioxin-induced alterations have been shown to be transmitted to the subsequent generations through the male germline to influence primordial germ cells reprogramming, which is a significant developmental window for disease susceptibility [22, 34, 36, 39, 40].
Individual animals were studied, and the specific pathologies in these transgenerational model systems were associated with specific epigenetic signatures (i.e., DMRs) for each toxicant exposure. Disease specific DMRs were identified for a number of these transgenerational pathologies, which shows that the establishment of an epigenetic biomarker for a specific disease and exposure is possible [23, 31, 33]. Although our previous study identified the ability of dioxin to promote the epigenetic transgenerational inheritance of pathologies and sperm epigenetic alterations in the transgenerational F3 generation [34], the potential that disease-specific epigenetic biomarkers exist has not been investigated. The current study provides evidence that these DMRs can be used as epigenetic disease specific biomarkers after an ancestral exposure to dioxin.
Results
Animal Breeding
As previously described [34], F0 generation outbred Sprague Dawley female rats were administered daily intraperitoneal injections of dioxin (TCDD 100 ng/kg BW/day) or dimethyl sulfoxide (vehicle control) during embryonic days E8-E14 of gestation [34]. The lowest observable adverse effects level (LOAEL) is 160 ng/kg/BW [41]. Therefore, the exposure used in the current study is a low exposure level. The intraperitoneal exposure was used to better control the exposure dose as compared to oral administration. The F3 generation is the first not directly exposed, thus called the transgenerational generation. All the animals were aged to 1 year and then euthanized by CO2 inhalation and cervical dislocation for pathology and sperm epigenetic analyses [34]. No sibling or cousin breeding was used to prevent any inbreeding artifacts in the control or dioxin lineages. All protocols and studies were approved by the Washington State University Animal Care and Use Committee (protocol IACUC # 6252).
Pathology Analysis
The archived pathology slides from the previous study [34] were used to reanalyze the pathology with more advanced digital pathology procedures. Images of the different pathology histologies have been previously reported [34]. Pathology analysis was performed by analyzing digitally captured images of histology sections of testis, kidney, and prostate. Two individuals blinded to exposure evaluated each tissue image for abnormalities. If there was disagreement about disease status, then a third individual blinded to exposure evaluated the tissue, as described in the Supplemental Methods. The disease parameters were identified and quantified, as previously described [23, 29, 31, 42]. The various tissue histological parameters used to identify pathology are outlined in the Methods, as well as other pathology conditions. For the F3 generation dioxin lineage male pathology, the individual animals are listed with a (+) that indicates presence of disease / pathology and a (−) that indicates the absence of disease (Table 1). To assess a statistical alteration in the dioxin lineage pathology, a comparison with the control lineage involving a vehicle exposure was analyzed for pathology as presented in Supplemental Table S1. The control animal lineage had minimal disease. For a specific pathology, individuals were only selected for epigenetic analysis if they had that single pathology. Animals with multiple diseases (≥2) were identified, but only one animal had multiple disease, so no further analysis of this was performed. This strategy allows for a more accurate association with epimutations and eliminates the confounding presence of other diseases. The dioxin induced transgenerational diseases / pathologies that had sufficient numbers of animals was prostate disease (3 males), kidney disease (6 males), obesity (4 males), and testis disease (8 males), Table 1. These individuals were used to investigate the sperm disease epigenetic biomarkers.
Table 1. Dioxin transgenerational pathology.
F3 generation dioxin lineage male rat pathology. The individual animals for the dioxin lineage males are listed and a (+) indicates presence of disease and (−) absence of disease. The animals with shaded (+) or (0) were used for the epigenetic analysis due to the presence of only one disease, except for the multiple (≥2) disease or no disease (0). The n/a indicates not analyzed and the totals provide the ratio of diseased / total animals, and % disease.
| Molecular ID | Late Puberty | Testis Disease | Prostate Disease | Kidney Disease | Obesity | Tumor | Multiple Disease | Total Disease |
|---|---|---|---|---|---|---|---|---|
| DX14 | − | − | + | + | − | − | + | 2 |
| DX9 | − | − | − | − | − | − | − | 0 |
| DX10 | − | − | − | − | − | − | − | 0 |
| DX11 | − | + | − | − | − | − | − | 1 |
| DX12 | − | − | − | − | − | − | − | 0 |
| DX13 | − | − | − | + | − | − | − | 1 |
| DX1 | − | − | − | + | − | − | − | 1 |
| DX2 | + | − | − | − | − | − | − | 1 |
| DX4 | − | − | − | − | + | − | − | 1 |
| DX5 | − | − | − | − | − | − | − | 0 |
| DX6 | − | − | − | − | − | − | − | 0 |
| DX7 | − | − | − | − | + | − | − | 1 |
| DX8 | − | − | − | − | − | − | − | 0 |
| DX18 | − | − | − | − | + | − | − | 1 |
| DX15 | − | + | − | − | − | − | − | 1 |
| DX16 | − | + | − | − | − | − | − | 1 |
| DX25 | − | − | − | − | + | − | − | 1 |
| DX26 | − | − | − | − | − | − | − | 0 |
| DX27 | − | − | − | − | − | − | − | 0 |
| DX28 | − | − | − | − | − | − | − | 0 |
| DX29 | − | − | − | − | − | − | − | 0 |
| DX34 | − | − | − | + | − | − | − | 1 |
| DX35 | − | − | − | − | − | − | − | 0 |
| DX30 | − | − | − | − | − | − | − | 0 |
| DX31 | − | − | − | − | − | − | − | 0 |
| DX32 | − | − | n/a | − | − | − | − | n/a |
| DX33 | − | − | − | + | − | − | − | 1 |
| DX21 | − | − | + | − | − | − | − | 1 |
| DX19 | − | − | + | − | − | − | − | 1 |
| DX20 | − | − | − | − | − | − | − | 0 |
| DX22 | − | + | − | − | − | − | − | 1 |
| DX23 | − | − | + | − | − | − | − | 1 |
| DX24 | − | + | − | − | − | − | − | 1 |
| DX41 | − | + | − | − | − | − | − | 1 |
| DX42 | − | + | − | − | − | − | − | 1 |
| DX43 | − | − | − | − | − | − | − | 0 |
| DX40 | − | − | − | + | − | − | − | 1 |
| DX36 | − | − | − | − | − | − | − | 0 |
| DX37 | − | − | − | − | − | − | − | 0 |
| DX38 | − | − | − | − | − | − | − | 0 |
| DX39 | − | − | − | − | − | − | − | 0 |
| DX53 | − | − | − | + | − | − | − | 1 |
| DX50 | − | + | − | − | − | − | − | 1 |
| DX51 | − | − | − | + | − | − | − | 1 |
| DX52 | − | − | − | − | − | − | − | 0 |
| DX44 | − | − | − | − | − | − | − | 0 |
| DX45 | − | − | − | − | − | − | − | 0 |
| DX46 | + | − | − | − | − | − | − | 1 |
| DX47 | − | − | − | − | − | − | − | 0 |
| DX48 | − | − | − | − | − | − | − | 0 |
| DX49 | − | − | + | − | − | − | − | 1 |
| Totals | 2/51 = 4% | 8/51 = 16% | 5/50 = 10% | 8/51 = 16% | 4/51 = 8% | 0/51 = 0% | 1/51 = 2% |
Sperm DNA methylation analysis
The archived sperm samples maintained at −80 °C from the previous study [34] were used to reanalyze the epigenetics with more advanced MeDIP-Seq technology than the tiling arrays previously used [34] on individual animals with specific disease. Sperm samples were collected from the dioxin lineage F3 generation individual males for epigenetic analysis. Within the dioxin lineage, individual males with no disease were compared to individuals with a single specific disease (testis, prostate, kidney, or obesity) in order to determine the disease specific differential DNA methylation regions (DMRs) (Figure 1A–D). This eliminates the confounding effects of multiple disease and allows disease specific biomarkers to be identified.
Figure 1.
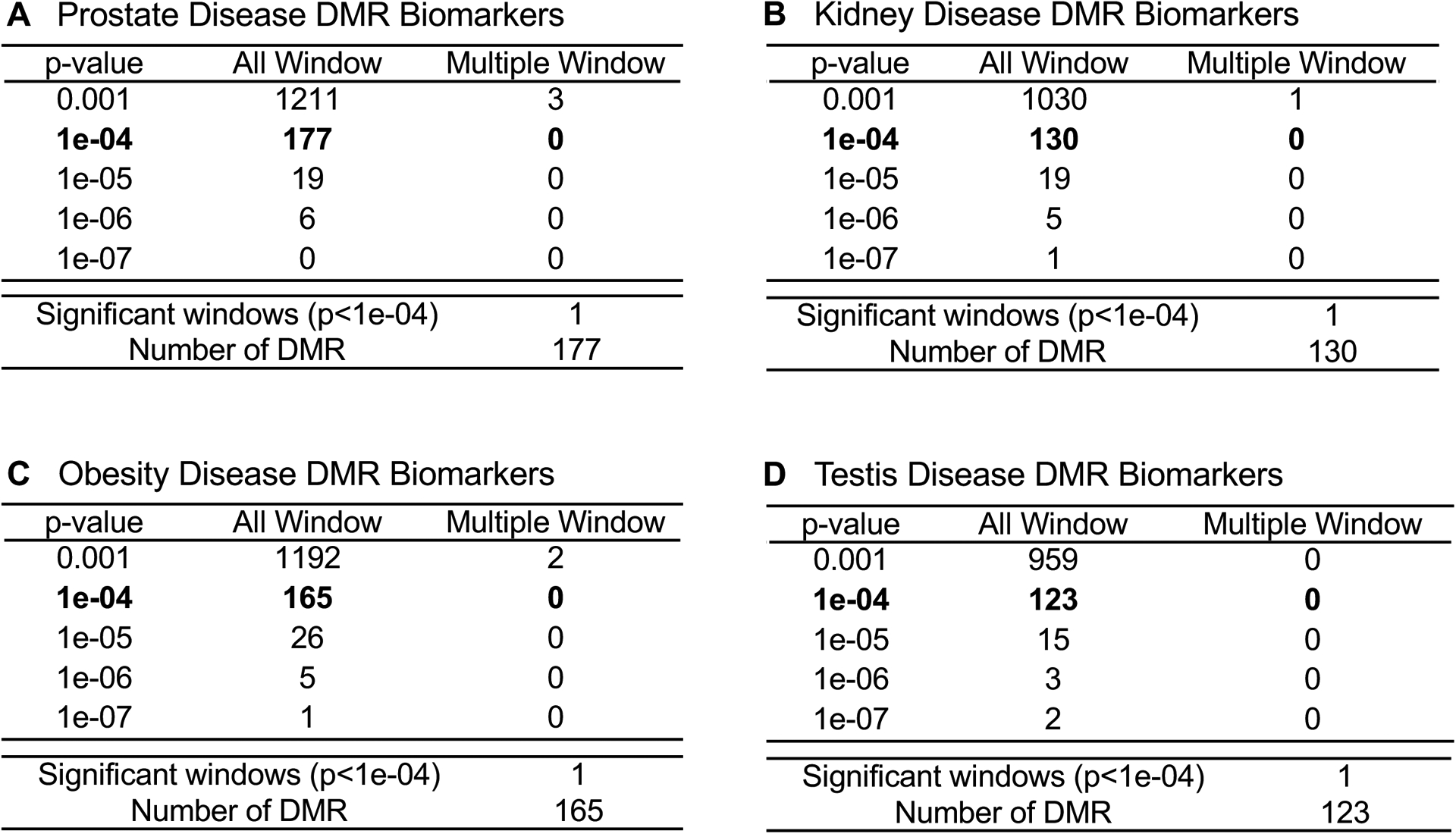
DMR identification and numbers. The number of DMRs found using different p-value cutoff thresholds. The All Window column shows all DMRs. The Multiple Window column shows the number of DMRs containing at least two significant windows (1 kb each). The number of DMRs with the number of significant windows (1 kb per window) at a p-value threshold p<1e-04 for DMR. (A) Prostate disease DMRs; (B) Kidney disease DMRs; (C) Obesity disease DMRs; and (D) Testis disease DMRs.
The sperm samples were collected, then the DNA extracted, fragmented and the methylated DNA immunoprecipitated (MeDIP) using a methyl-cytosine antibody [27, 28]. The methylated DNA fragments were sequenced for an MeDIP-Seq analysis, as described in the Supplemental Methods section [21, 27, 28]. The DMR numbers are listed in Figure 1 for different edgeR statistical p-value cutoff thresholds, and p<1e-04 (diseased versus non-diseased) were selected as the threshold for all subsequent analyses. The total number of DMRs (All Windows) if present for each disease and multiple neighboring 1000 bp windows (Multiple Window) are shown (Figure 1).
In our previous study, the reported transgenerational F3 generation sperm dioxin versus control lineage DMRs used three pools of different animals to determine the dioxin induced sperm DNA epimutations with tiling arrays [34]. In the current study, individual animals were used to identify the transgenerational F3 generation dioxin induced disease sperm DMR epimutations with MeDIP-Seq. With an edgeR p<1e-04, 177 DMRs were identified for the animals with prostate disease (Figure 1A). The animals with kidney disease had 130 DMRs (Figure 1B). The obesity disease group was found to have 165 DMRs (Figure 1C). The animals showing testis disease had 123 DMRs (Figure 1D). None of these different groups displayed any DMRs with multiple neighboring 1000 bp windows (Figure 1A–D). In conclusion, the different diseases had altered DNA methylation in the F3 generation sperm at a p<1e-04. The disease specific DMRs with an edgeR p<1e-04 threshold are presented in Supplemental Tables S2–S5. The log-fold change in DNA methylation is presented and an increase in methylation was associated with 40% of the testis disease DMRs, 47% of the prostate disease DMRs, 42% of the kidney disease DMRs, and 35% of the obesity DMRs. The others all had a decrease in DNA methylation.
The disease specific DMR chromosomal locations are presented in Figure 2 where the DMR locations are represented by red arrowheads, and DMR clusters by black boxes. The prostate, kidney, obesity and testis disease biomarkers did not have any DMRs on the Y chromosome or the mitochondrial DNA (MT). Therefore, the DMRs were identified on nearly all chromosomes. DMR length and CpG density are shown in Figure 3. The CpG density of the DMRs for all comparisons was 1–4 CpG per 100 bp being predominant, which is characteristic of a low-density CpG desert. These observations were similar to our past studies with other ancestral exposures [34]. The DMR lengths for each disease biomarker were 1–4 kb with 1 kb being predominant, Figure 3. In general, the DMRs are 1 kb in size with around 10 CpGs, as previously reported [43]. The DMR genomic features and chromosomal locations are presented in Supplemental Tables S2–S5. For the different disease DMR biomarker comparisons with non-disease (prostate disease DMRs, kidney disease DMRs, obesity disease DMRs, and testis disease DMRs), a principal component analysis (PCA) demonstrates a clustered separation of the prostate, kidney, obesity, and testis disease samples compared to the non-disease based on read depth at DMR sites (Supplemental Figure S1). The PCA on DMR sites was performed to assess the potential presence of any outlier samples, of which none were observed. Therefore, the disease samples were distinct from the non-disease samples for each of the pathologies when considering read depth at DMR sites.
Figure 2.
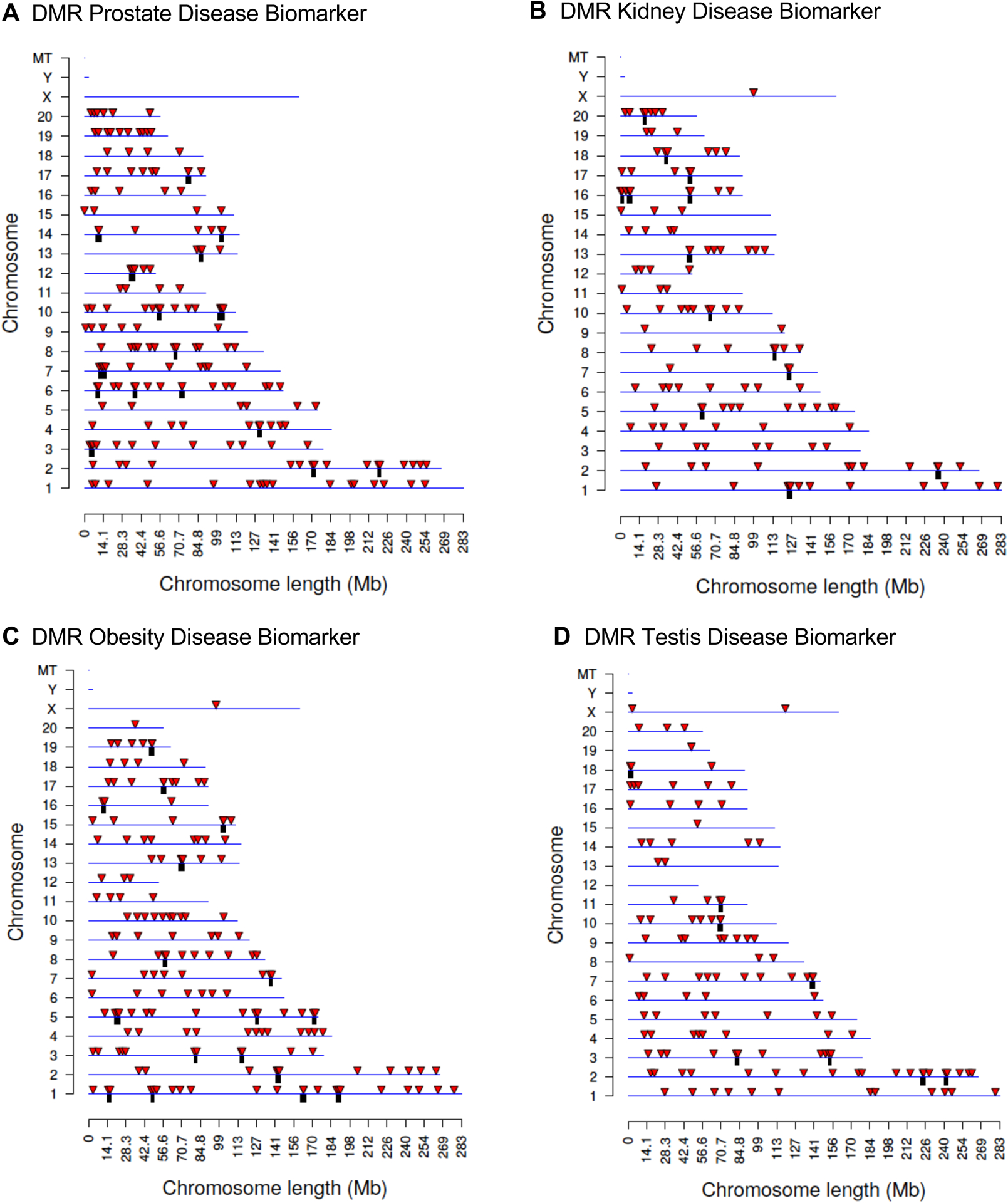
DMR chromosomal locations. The DMR locations on the individual chromosomes is represented with an arrowhead and a cluster of DMRs with a black box. All DMRs containing at least one significant window at the selected p-value p<1e-04 threshold are shown. The chromosome number and size of the chromosome (megabase) are presented. (A) Prostate disease DMRs; (B) Kidney disease DMRs; (C) Obesity disease DMRs; and (D) Testis disease DMRs.
Figure 3.
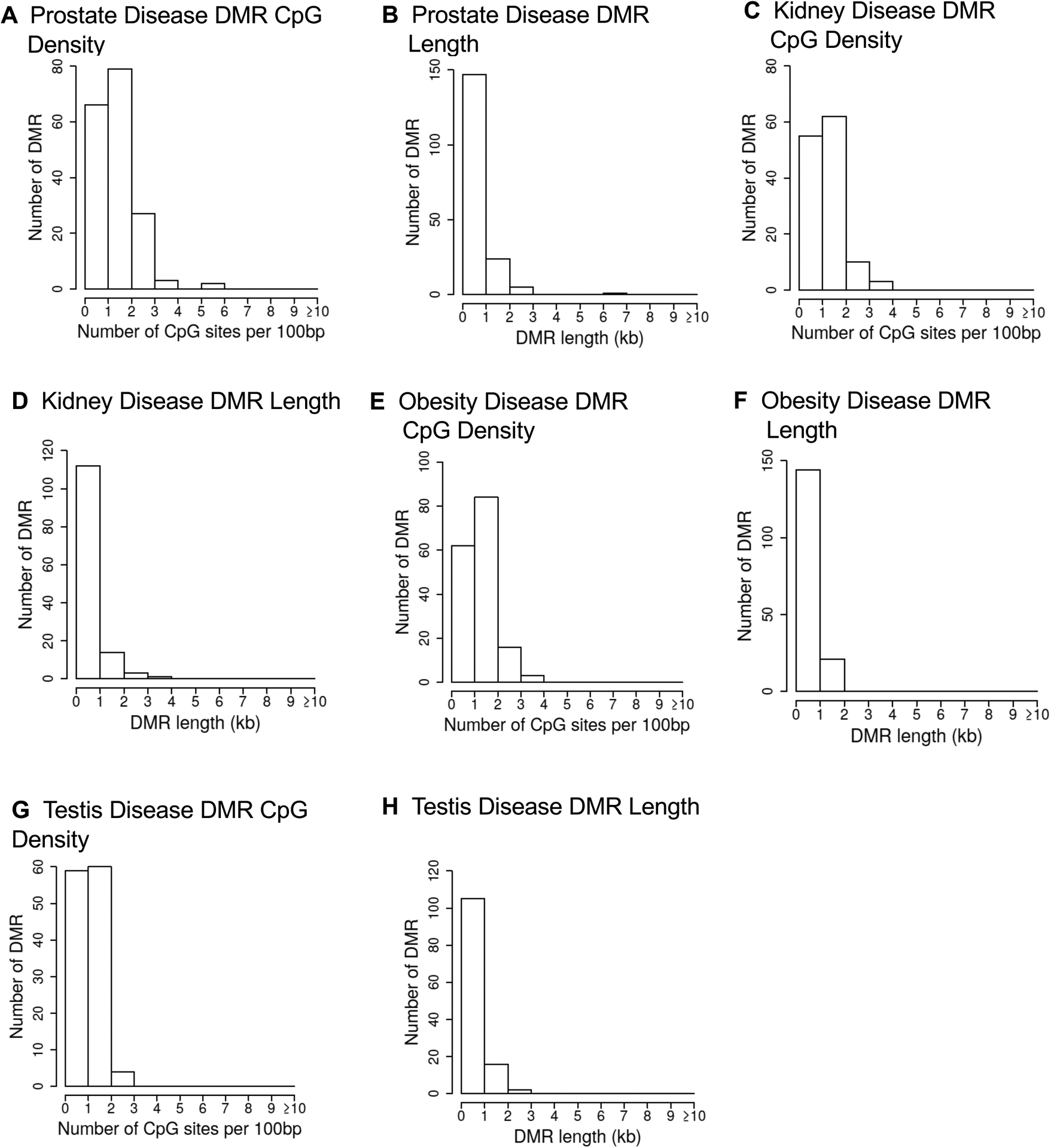
DMR genomic features. The number of DMRs at different CpG densities. All DMRs at a p-value threshold of p<1e-04 are shown. (A) Prostate disease DMR CpG density; (B) Prostate disease DMR length; (C) Kidney disease DMR CpG density; (D) Kidney disease DMR length; (E) Obesity disease DMR CpG density; (F) Obesity disease DMR length; (G) Testis disease DMR CpG density; (H) Testis disease DMR length.
To compare and identify overlapping DMRs for each disease (testis, prostate, kidney and obesity), a Venn diagram of chromosomal location overlaps at the p<1e-04 threshold was used (Figure 4A). Negligible overlap is observed at the p-value (p<1e-04) threshold between the differential transgenerational DMR disease sites. This overlap analysis was further investigated with an extended overlap of the p<1e-04 DMRs. The DMRs with p<1e-04 were compared to DMRs with p<0.05 statistical threshold to allow for an increased potential to identify overlaps when a less stringent p-value was used. In all the comparisons, between 8 to 17% overlaps were observed for each comparison, Figure 4B. An overlap of the disease-specific DMRs with a p<0.05 is shown in Figure 4C. The total number of DMRs is dramatically increased at this lower statistical threshold. Only 63 DMRs had an overlap with all diseases at p<0.05, Figure 4C. These 63 overlapping p<0.05 DMRs were compared to the p<1e-04 disease specific DMRs and few overlapping DMRs were identified, Figure 4D. Therefore, no overlapping group of DMRs for all diseases was identified, but some overlap is observed between comparisons of two diseases. Observations indicate that the DMRs identified are primarily specific to one disease / pathology, but approximately 15–25 DMR overlapped at the reduced statistical threshold for specific disease comparisons.
Figure 4.
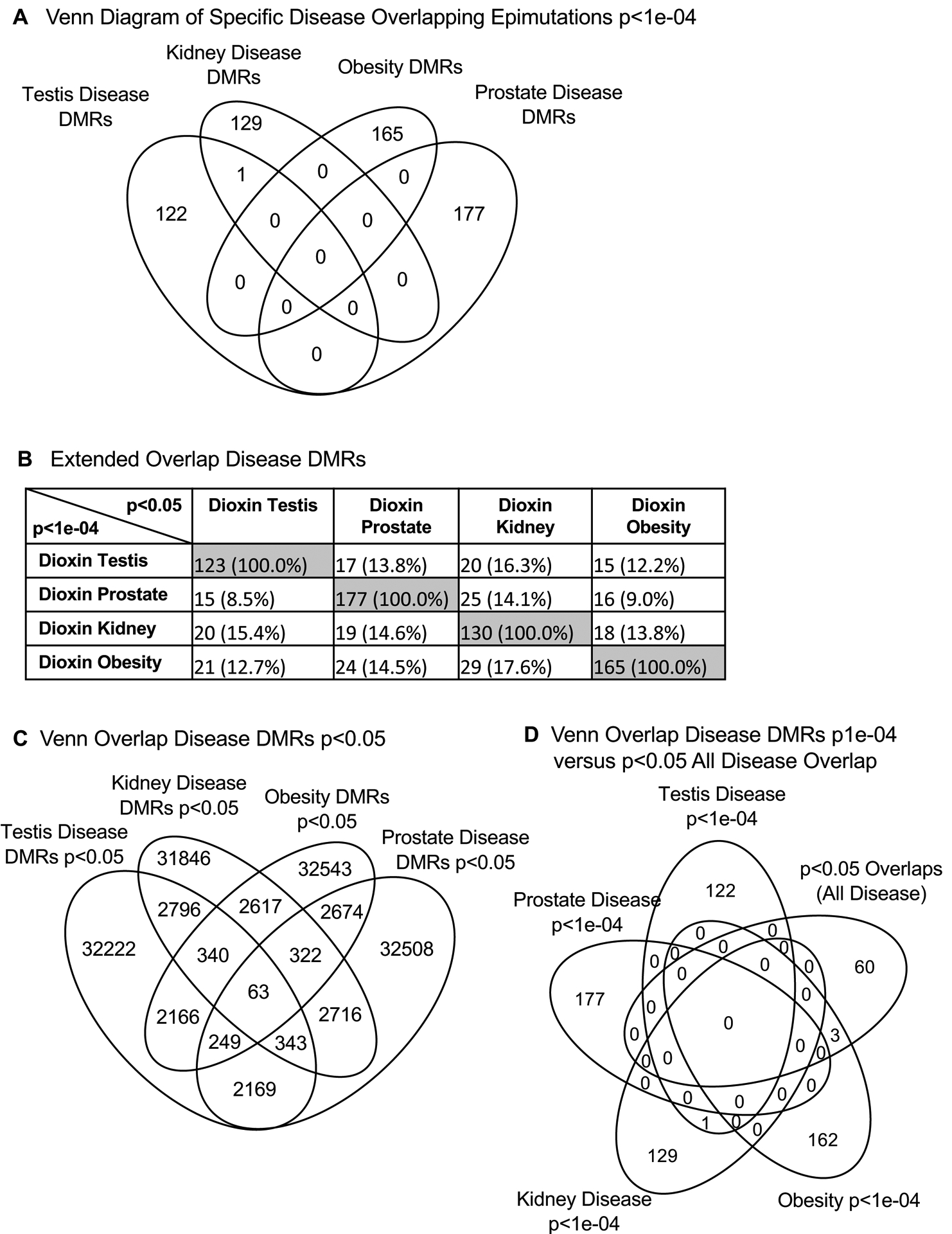
Overlap of disease DMRs. (A) Overlap of specific disease overlap epimutations p<1e-04. Venn diagram overlap analysis for specific disease states. (B) An extended overlap of disease DMRs. The p-value data set at p<1e-04 is compared to the p<0.05 data to identify potential overlap between the different pathologies with DMR number and percentage of the total presented. The gray highlight is the expected 100% overlap. (C) Overlap of the different disease DMRs at p<0.05. The Venn diagram identified 63 DMRs at p<0.05 in common between the different diseases. (D) Venn diagram overlap of the different diseases DMRs at p<1e-04 with the 63 common overlapping p<0.05 DMRs.
DMR Gene Associations
Genes associated with the DMRs were identified for each disease specific DMR data set. The DMRs with a gene within 10 kb distance, in order to include promoters, were determined as well as the associated genes and gene functional categories (Supplemental Tables S2–S5). The DMRs with a p<1e-04 were used for this analysis on the different diseases. For all the different disease specific DMRs approximately 50% had gene associations. The DMR associated gene categories demonstrated several relevant gene categories such as signaling, metabolism, transcription, receptor and cytoskeleton for all the different disease DMR signatures, Figure 5A. A cellular KEGG pathway analysis was conducted to determine the associated genes for each DMR data set, as described in the Supplemental Methods. The top ten pathways with associated genes are presented, Figure 5B. The cellular pathways identified also had signaling and critical cellular processes involved.
Figure 5.
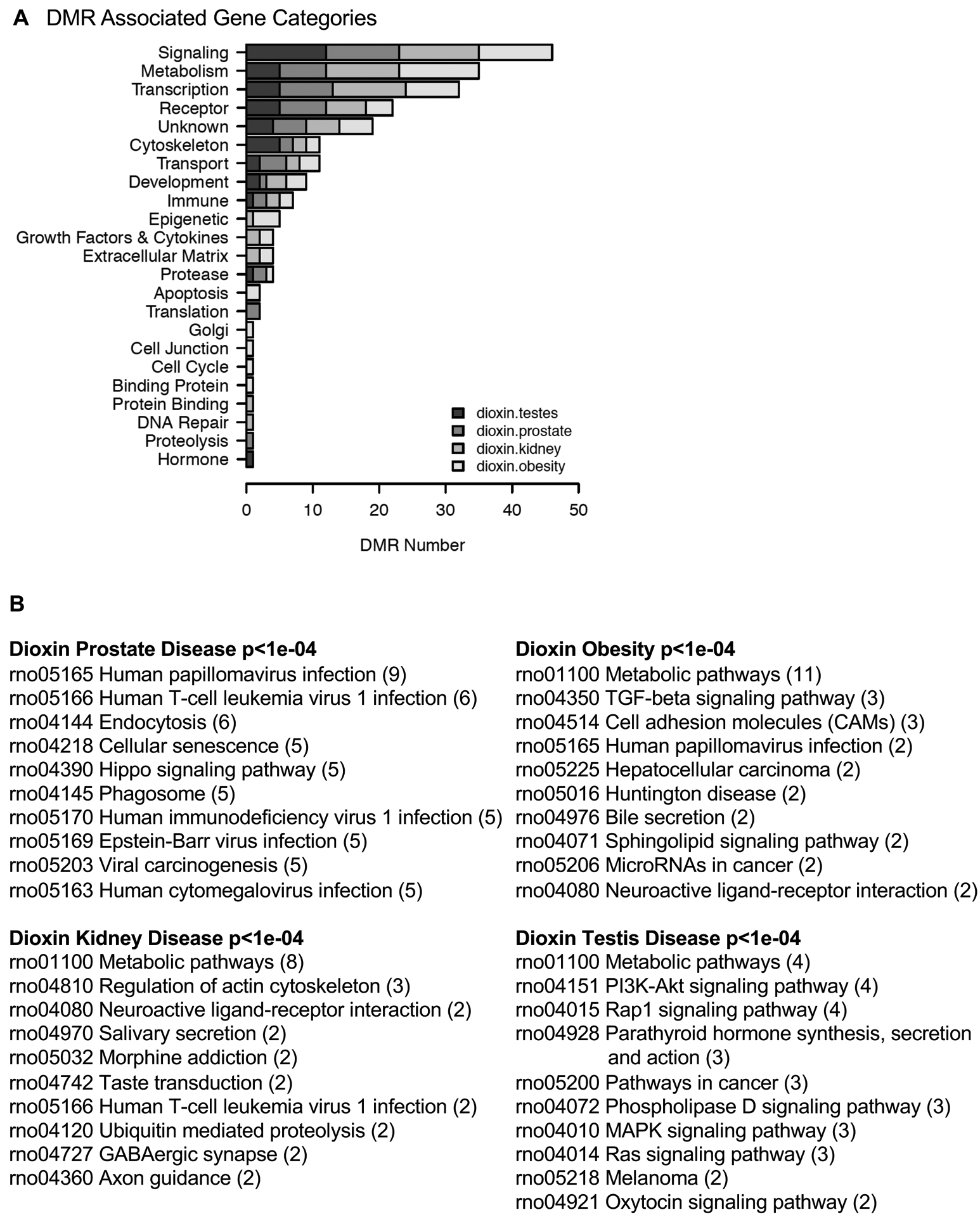
Disease DMR associated gene categories. (A) DMR associated gene categories. The different gene categories and number of DMRs in each category is presented with a legend indicating the different disease DMR sets. (B) KEGG pathways for the different disease DMR associated genes. The top ten pathways with the number of associated genes in brackets are presented.
Potential dioxin transgenerational disease specific DMR associated genes were analyzed for genes previously shown to associate with the specific diseases, Figures 6 & 7. The prostate disease DMR associated genes had a number of genes linked to prostate physiology and some linked to subfertility, Figure 6A. The kidney disease DMR associated genes had a large number of genes linked to kidney physiology and disease and some linked to kidney hypocalciuria, Figure 6B. The obesity DMR associated genes had a number of genes linked to obesity and some associated with diabetes and insulin resistance, Figure 7A. The testis disease DMR associated genes had a number of genes linked to testis physiology and male infertility, Figure 7B.
Figure 6.
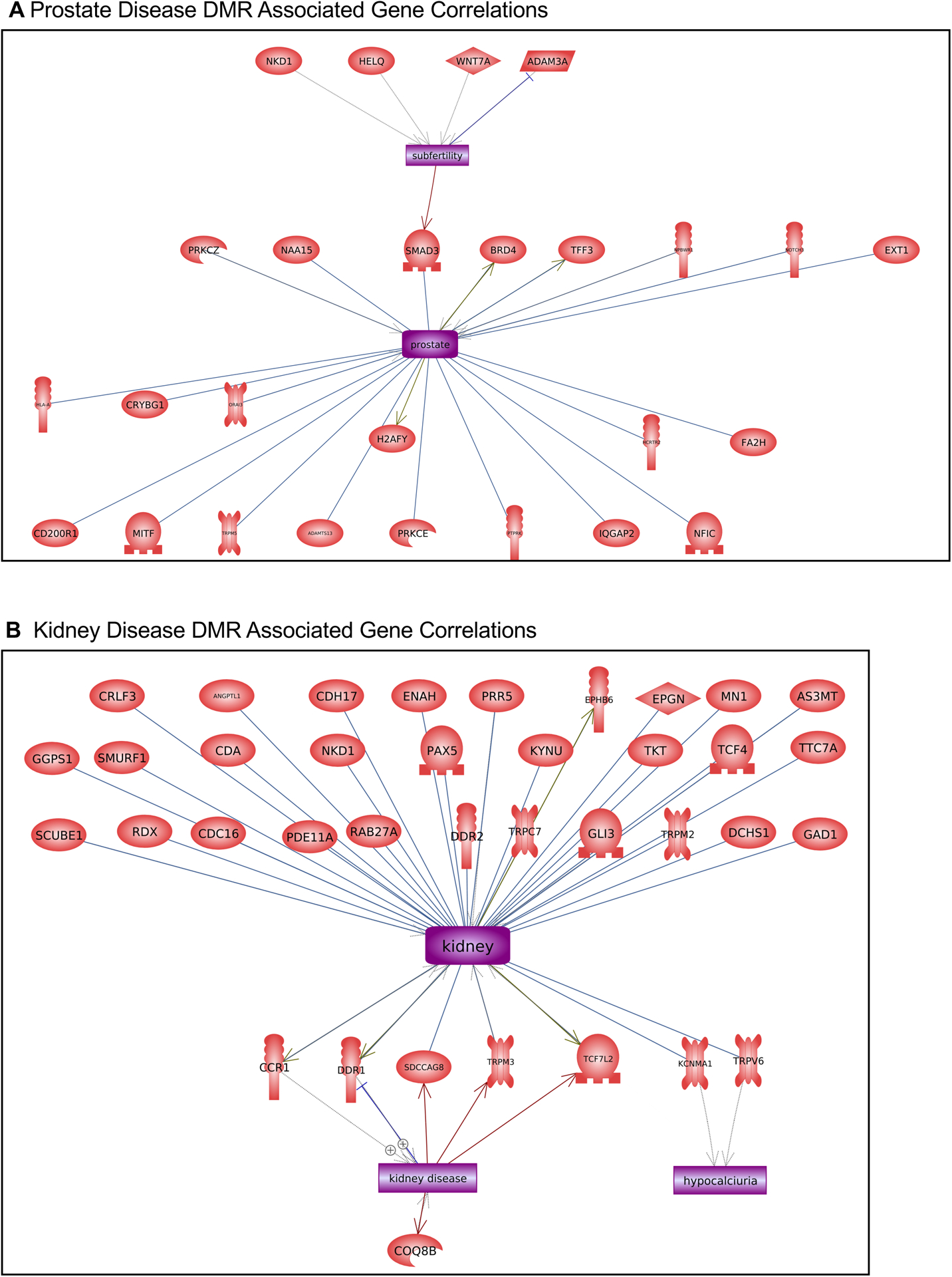
Disease DMR associated gene correlations. The disease DMR associated genes that correlate with the specific disease tissue functions and pathologies for each individual pathology are presented. Direct gene links to pathologies and physiologic processes are shown. (A) Prostate disease and (B) kidney disease.
Figure 7.
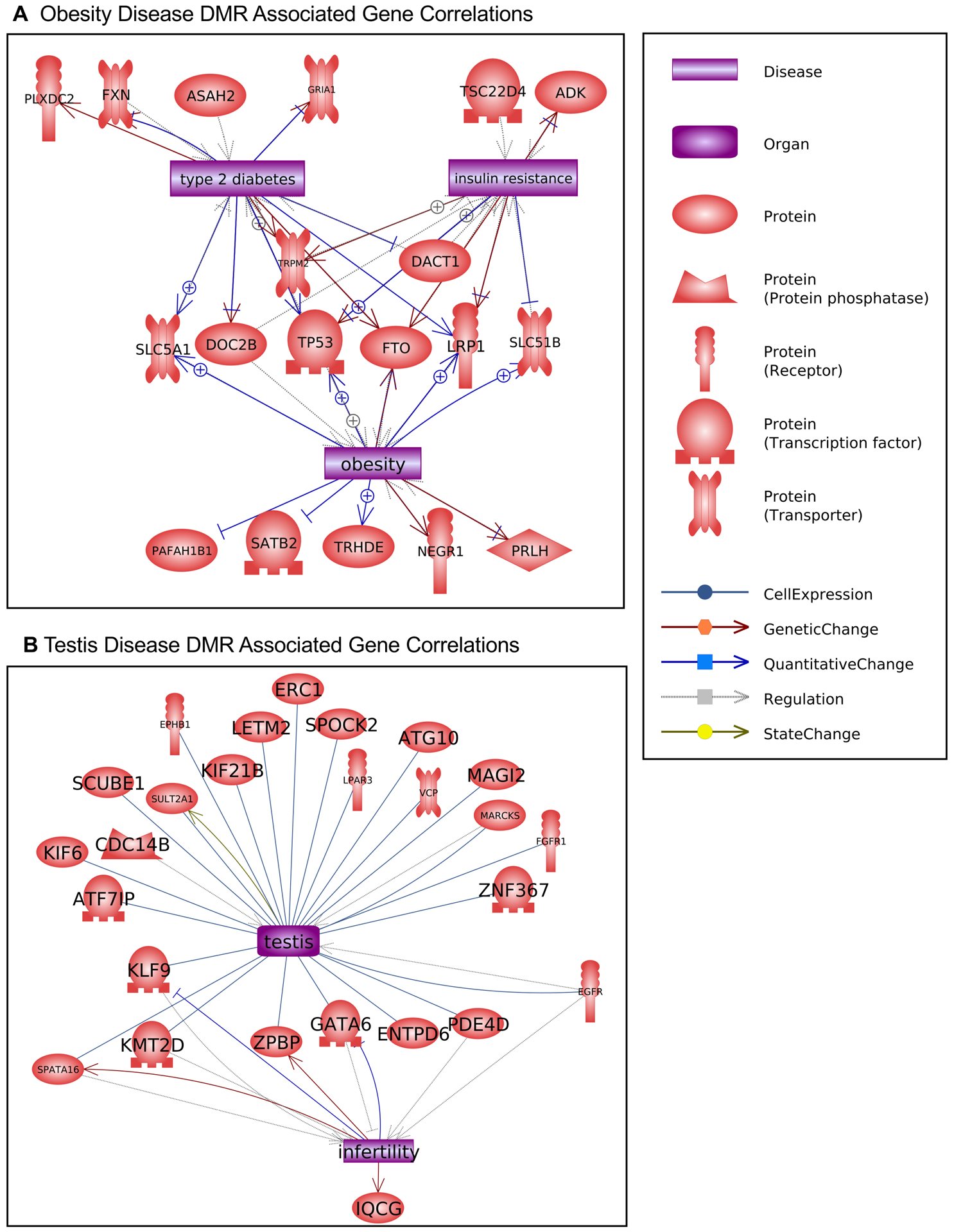
Disease DMR associated gene correlations. The disease DMR associated genes that correlate with the specific disease tissue functions and pathologies for each individual pathology are presented. Direct gene links to pathologies and physiologic processes are shown. (A) obesity and (B) testis disease.
Discussion
Humans and other animals are exposed to a wide array of man-made toxicants. Many of them act as environmental toxicants or endocrine disruptors that can exhibit differential effects across the lifespan [40]. Various human exposures to dioxin (TCDD) have been documented and associated with a large number of different diseases and pathologies. However, the majority of these epidemiology studies have focused on direct adult and fetal exposures [15]. Despite having thousands of man-made substances released into our environment each year, prospective risk assessment studies of these potentially harmful chemicals on human health are often not required by current regulations [44].
Human toxicant exposures have also been associated with military service. During the Vietnam War (1964–1975), thousands of pounds of Agent Orange, a highly toxic herbicide and defoliant, were sprayed by the U.S. military over large areas of dense jungles in south and central Vietnam to destroy the ground cover it provided to enemy troops [45]. An estimated 1.5 million American servicemen are believed to have been exposed to dioxin as a consequence of Operation Ranch Hand. Some of these veterans reported skin rashes (chloracne), cancer, psychological symptoms, birth defects in their children, and other health issues [1]. Agriculture crops and inhabited villages were also sprayed, resulting in the exposure of Vietnamese residents. Many of them continue to experience a wide range of health issues including high incidence of early pregnancy loss, congenital birth defects and serious health problems, such as cancers, in surviving children [46–50].
Since transgenerational inheritance of these detrimental health effects or diseases to subsequent generations can occur, the populations present today that were ancestrally exposed to dioxin (TCDD) is of critical relevance. Dioxin exposure has been linked in the past to epigenetic modifications in rats [22, 34, 35, 37, 40, 51, 52] and in humans [53–56]. The present study shows DNA methylation alterations associated with ancestral exposure to dioxins and disease-specific epimutation biomarkers for different pathologies identified. The pathologies studied include kidney disease, obesity, testis disease, and prostate disease. Kidney and prostate diseases are especially relevant to human populations since both are a major cause of disease and mortality among male humans [57, 58]. According to the Centers of Disease Control (CDC), the prevalence of obesity in the USA was 42.4 million in 2017–2018 [59]. The association of epigenetic biomarkers with these diseases could become particularly valuable indicators of disease susceptibility in the human population. Genome-wide association studies (GWAS) have found specific genetic mutations associated with these human pathologies, however these genetic mutations typically appear in less than 1% of the diseased population.
In contrast, epigenetic alterations called epimutations seem to have a higher frequency and appear in most individuals with the disease [23, 29, 31]. The current study supports this observation where the number of differential DNA methylation regions (DMRs) in the transgenerational males is between 100 and 200 at an edgeR p<1e-04 threshold for each individual pathology (Figure 1). A subpopulation of DMRs overlapping between the different individual disease pathologies was not identified (Figure 4B). An overlap analysis of the disease-specific DMRs demonstrates some of the DMRs overlap between two different diseases, but none overlapped between all four different diseases, Figure 4. Therefore, an overlapping set of DMR associated with all general disease susceptibility was not observed. The DMR associated genes show that the most affected gene categories were signaling, metabolism, and transcription. In addition, a large number of previously identified disease-associated genes were present in the DMR associated gene list, Figures 6 & 7.
A limitation of the current study was the low numbers of animals with a specific individual disease. Although an edgeR p-value was used to identify and analyze the disease biomarker DMRs, analysis for multiple testing error for false discovery rate (FDR) provided p-values for the disease biomarkers of >0.1. Previous studies have demonstrated limitations in FDR analysis due to the presumptions in the multiple testing parameters [60–65]. Therefore, the low sample number is a limitation in the current analysis. Potential higher variability in the data needs to be considered even though higher edgeR values were used, but this does not address multiple testing corrections. Future studies will need to use higher n-values to reduce this analysis limitation [60–65]. The current disease specific epimutation (i.e., DMRs) needs to be validated in future studies.
Observations suggest dioxin induced transgenerational DMRs present in sperm appear associated with specific diseases. This indicates the existence of potential disease specific biomarkers could be used to assess transgenerational transmission of various pathology susceptibilities in the offspring. Such epigenetic biomarkers would also allow potential preventative therapeutics to be used or developed. Although more extensive studies in humans are required, the current study supports the concept that associated pathology DMRs could be utilized as epigenetic biomarkers. Further analysis is needed to determine if the use of these biomarkers is feasible for early detection of disease susceptibility, prior to the actual onset of diseases. Our first study examining the transgenerational endocrine disruptor atrazine showed epigenetic inheritance of disease and sperm epimutations, and also supports the concept that epigenetic biomarkers for disease can be identified and potentially employed for diagnosis [31]. Subsequently, DDT and vinclozolin induced transgenerational DMRs were identified [29, 31]. The current study showed dioxin induction of transgenerational disease and also suggests such environmental biomarkers can be identified and potentially become a diagnostic tool for disease susceptibility in the future. Epigenetic biomarkers have a high frequency of association with pathologies, and their incorporation into medical diagnostics will facilitate preventative medicine and disease management.
Methods
Animal studies and breeding
As previously described [34], female and male rats of an outbred strain Hsd:Sprague Dawley SD (Harlan) at 70 to 100 days of age were fed ad lib with a standard rat diet and ad lib tap water. Timed-pregnant females on days 8 through 14 of gestation [66] were administered daily intraperitoneal injections of dioxin (TCDD 100 ng/kg BW/day) or dimethyl sulfoxide (DMSO), as previously described [34].
As previously described [34], the gestating female rats treated were designated as the F0 generation. F1-F3 generation control and dioxin lineages were housed in the same room and racks with lighting, food and water as previously described [19, 22, 67]. All experimental protocols for the procedures with rats were pre-approved by the Washington State University Animal Care and Use Committee (protocol IACUC # 6252). All methods were performed in accordance with the relevant guidelines and regulations. The animal tissues and sperm samples from the previous study [34] have been archived and were used for the current study.
Tissue harvest and histology processing
As previously described [34], at 12 months of age, rats were euthanized by CO2 inhalation and cervical dislocation for tissue harvest. Testis, prostate, and kidney were fixed in Bouin’s solution (Sigma) followed by 70% ethanol, then processed for paraffin embedding and hematoxylin and eosin (H & E) staining by standard procedures for histopathological examination. Paraffin five micron sections were processed, stained, and processed by Nationwide Histology, Spokane WA, USA.
Histopathology examination and disease classification
Archived histology slides or paraffin blocks from the previous study were stored in standard archive containers and organized files in the dark at room temperature 20–25°C [34] were used for a new histology analysis for the current study. The images of the various pathologies are presented in a previous study [34]. The tissue sections were reimaged and reanalyzed with a digital pathology procedure where an image of the tissue section is captured electronically and corrected for area for histopathology analysis. The oversight of the pathology analysis involved the co-author, Dr. Eric Nilsson, DVM/PhD, with over 20 years of pathology analysis experience in rats [23, 24]. The Washington Animal Disease Diagnostic Laboratory (WADDL) at the Washington State University College of Veterinary Medicine has board certified veterinary pathologists and assisted in initially establishing the criteria for the pathology analyses and identifying parameters to assess [67]. The tissues evaluated histologically were selected from previous literature showing them to have pathology in transgenerational models [17, 21, 23, 25, 26, 32, 34, 67–69], with an emphasis on reproductive organs. Stained testis, prostate, and kidney slides were imaged through a microscope using 4x objective lenses (testis and prostate) or 10x objective lenses (kidney). Tiled images were captured using a digital camera. Tiled images for each tissue were photo-merged into a single image using Adobe Photoshop (ver. 21.1.2, Adobe, Inc.). The image area was captured in pixels and then converted into mm2. This allowed for correction of abnormality counts based on the size of the tissue sample. Images were evaluated and pathology features digitally marked using Photoshop software. Raw counts were then divided by the measured area for each sample. Histopathology readers were trained to recognize the specific abnormalities evaluated for this study in rat testis, ventral prostate and kidney. Two different pathology readers were used if they agreed and three different readers if they disagreed for each tissue that were blinded for the readers to the exposure lineage groups. A set of quality control (QC) slides were generated for each tissue and were read by each reader prior to evaluating any set of experimental slides. These QC slide results were monitored for reader accuracy and concordance. Previous studies by the laboratory help confirm and validate the pathology analysis [17, 21, 23, 25, 26, 32, 34, 67–69].
As previously described [18], testis histopathology criteria included the presence of vacuoles in the seminiferous tubules, azoospermic atretic seminiferous tubules, and ‘other’ abnormalities including sloughed spermatogenic cells in the center of the tubule and a lack of a tubule lumen. As previously described [24, 70], prostate histopathology criteria included the presence of vacuoles in the glandular epithelium, atrophic glandular epithelium and hyperplasia of prostatic gland epithelium. Kidney histopathology criteria included reduced size of glomerulus, thickened Bowman’s capsule, and the presence of proteinaceous fluid-filled cysts > 50μm in diameter. A cutoff was established to declare a tissue ‘diseased’ based on the mean number of histopathological abnormalities plus 1.5 standard deviations from the mean of control group tissues, as assessed by each of the individual observers blinded to the treatment groups. This number (i.e., greater than 1.5 standard deviations) was used to classify rats into those with and without testis, prostate, or kidney disease in each lineage. Two individuals blinded to exposure evaluated each tissue image for abnormalities. If there was disagreement about disease status, then a third individual blinded to exposure evaluated the tissue. Obesity was assessed with an increase in body mass and intra-abdominal adiposity and subcutaneous fat at the time of euthanasia, as previously described [34]. The results for pathology, as previously described [34], were expressed as the proportion of affected animals that exceeded a predetermined threshold (testis, prostate, kidney disease frequency, tumor frequency, obese frequency). Groups were analyzed for statistical differences using Fisher’s exact test.
Epididymal sperm collection and DNA isolation
The protocol is described in detail in reference [34]. Briefly, the epididymis was dissected free of fat and connective tissue, then, after cutting open the cauda, placed into 6 ml of phosphate buffer saline (PBS) for 20 minutes at room temperature. Further incubation at 4°C immobilized the sperm. The tissue was then minced, the released sperm pelleted at 4°C 3,000 × g for 10 minutes, then resuspended in NIM buffer and stored at −80°C for further processing. An appropriate amount of rat sperm suspension was used for DNA extraction. Previous studies have shown mammalian sperm heads are resistant to sonication unlike somatic cells [71, 72]. Somatic cells and debris were therefore removed by brief sonication (Fisher Sonic Dismembrator, model 300, power 25), then centrifugation and washing 1–2 times in 1X PBS. The resulting pellet was resuspended in 820 μL DNA extraction buffer and 80 μl 0.1M DTT was added, then incubated at 65°C for 15 minutes. 80 μl proteinase K (20 mg/ml) was added and the sample was incubated at 55°C for 2–3 hours under constant rotation. Proteins were removed by addition of protein precipitation solution (300 μl, Promega A795A), incubation for 15 minutes on ice, then centrifugation at 13,500 rpm for 30 minutes at 4°C. One ml of the supernatant was precipitated with 2 μl of GlycoBlue (Invitrogen, AM9516) and 1 ml of cold 100 % isopropanol. After incubation, the sample was spun at 13,500 × g for 30 minutes at 4°C, then washed with 70% cold ethanol. The pellet was air-dried for about 5 minutes then resuspended in 100 μl of nuclease free water.
Methylated DNA Immunoprecipitation (MeDIP)
The archived sperm samples were prepared from previously collected samples as described [34]. The protocol is described in detail in reference [34]. Genomic DNA was sonicated and run on 1.5% agarose gel for fragment size verification. The sonicated DNA was then diluted with 1X TE buffer to 400 μl, then heat-denatured for 10 minutes at 95°C, and immediately cooled on ice for 10 minutes to create single-stranded DNA fragments. Then 100 μl of 5X IP buffer and 5μg of antibody (monoclonal mouse anti 5-methyl cytidine; Diagenode #C15200006) were added, and the mixture was incubated overnight on a rotator at 4°C. The following day, magnetic beads (Dynabeads M280 Sheep anti-Mouse IgG; Life Technologies 11201D) were pre-washed per manufacturer’s instructions, and 50μl of beads were added to the 500μl of DNA-antibody mixture from the overnight incubation, then incubated for 2 hours on a rotator at 4°C. After this incubation, the samples were washed three times with 1X IP buffer using a magnetic rack. The washed samples were then resuspended in 250μl digestion buffer (5mM Tris PH 8, 10.mM EDTA, 0.5% SDS) with 3.5μl Proteinase K (20mg/ml), and incubated for 2–3 hours on a rotator at 55°C. DNA clean-up was performed using a Phenol-Chloroform-Isoamyl-Alcohol extraction, and the supernatant precipitated with 2μl of GlycoBlue (20mg/ml), 20μl of 5M NaCl and 500μl ethanol in −20°C freezer for one to several hours. The DNA precipitate was pelleted, washed with 70% ethanol, then dried and resuspended in 20μl H2O or 1X TE. DNA concentration was measured in Qubit (Life Technologies) with the ssDNA kit (Molecular Probes Q10212).
MeDIP-Seq Analysis
As previously described [27], MeDIP DNA was used to create libraries for next generation sequencing (NGS) using the NEBNext Ultra RNA Library Prep Kit for Illumina (San Diego, CA) starting at step 1.4 of the manufacturer’s protocol to generate double stranded DNA from the single-stranded DNA resulting from MeDIP. After this step, the manufacturer’s protocol was followed indexing each sample individually with NEBNext Multiplex Oligos for Illumina. The WSU Spokane Genomics Core sequenced the samples on the Illumina HiSeq 2500 at PE50, with a read size of approximately 50 bp and approximately 15–20 million reads per pool.
Statistics and Bioinformatics
The DMR identification and annotation methods follow those presented in previous published papers [23, 27]. Data quality was assessed using the FastQC program (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and reads were cleaned and filtered to remove adapters and low quality bases using Trimmomatic (28). The reads for each MeDIP were mapped to the Rnor 6.0 rat genome using Bowtie2 [73] with default parameter options. The mapped read files were then converted to sorted BAM files using SAMtools [74]. The MEDIPS R package [75] was used to calculate differential coverage between control and exposure sample groups. The reference genome was split into 1000 bp windows. Windows with an average of at least 10 reads per sample were selected for differential analysis. The edgeR p-value [76] was used to determine the relative difference between the two groups for each genomic window. Windows with an edgeR p-value less than an arbitrarily selected threshold were considered DMR. The site edges were extended until no genomic window with an edgeR p-value less than 0.1 remained within 1000 bp of the DMR. The edgeR p-value was used to assess the significance of the DMR identified. Differential epimutation sites were annotated using the biomaRt R package [77] to access the Ensembl database [78]. The DMR associated genes were then automatically sorted into functional groups using information provided by the DAVID [79] and Panther [80] databases incorporated into an internal curated database (www.skinner.wsu.edu under genomic data). A Pathway Studio, Elsevier, database and network tool was used to assess physiological and disease process gene correlations. All molecular data has been deposited into the public database at NCBI (GEO # GSE157539) and R code computational tools available at GitHub (https://github.com/skinnerlab/MeDIP-seq) and www.skinner.wsu.edu.
Supplementary Material
Supplemental Figure S1. DMR principal component analysis. The first two principal components are used. The underlying data is the RPKM read depth for DMR only genomic windows. (A) Prostate disease DMRs PCA; (B) Kidney disease DMRs PCA; (C) Obesity disease DMRs PCA; (D) Testis disease DMRs PCA.
Supplemental Table S1. Dioxin study control transgenerational pathology. F3 generation control lineage male rat pathology. Vehicle dimethyl sulfoxide (DMSO) exposure used in the F0 generation gestating female. The individual animals for the control lineage males are listed with a (+) to indicate the presence of the pathology or (−) for absence of the pathology. The number of disease/total animals is presented and percentage disease.
Supplemental Table S2. DMR Site List Prostate Disease p<1e-04. DMR name, chromosome, start, stop, length, number signature windows, minimum p-value, max log-fold change, CpG number, CpG density, gene annotation, and gene category are presented.
Supplemental Table S3. DMR Site List Kidney Disease p<1e-04. DMR name, chromosome, start, stop, length, number signature windows, minimum p-value, max log-fold change, CpG number, CpG density, gene annotation, and gene category are presented.
Supplemental Table S4. DMR Site List Obesity p<1e-04. DMR name, chromosome, start, stop, length, number signature windows, minimum p-value, max log-fold change, CpG number, CpG density, gene annotation, and gene category are presented.
Supplemental Table S5. DMR Site List Testis Disease p<1e-04. DMR name, chromosome, start, stop, length, number signature windows, minimum p-value, max log-fold change, CpG number, CpG density, gene annotation, and gene category are presented.
Acknowledgments
We acknowledge Dr. Ingrid Sadler-Riggleman, Ms. Michelle Pappalardo, Mr. Ryan Thompson, Ms. Skylar Shea Davidson, Ms. Makena Horne, Ms. Emma Impala, and Ms. Rachel LaRosa for technical assistance. We acknowledge Ms. Amanda Quilty for editing and Ms. Heather Johnson for assistance in preparation of the manuscript. We thank the Genomics Core laboratory at WSU Spokane for sequencing data. This study was supported by John Templeton Foundation (50183 and 61174) (https://templeton.org/) grants to MKS and NIH (ES012974) (https://www.nih.gov/) grant to MKS. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Funding Sources
This study was supported by John Templeton Foundation (50183 and 61174) (https://templeton.org/) grants to MKS and NIH (ES012974) (https://www.nih.gov/) grant to MKS. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Animal Studies
All experimental protocols for the procedures with rats were pre-approved by the Washington State University Animal Care and Use Committee (protocol IACUC # 6252), and all methods were performed in accordance with the relevant guidelines and regulations.
Competing Interests
The authors declare no competing financial or other interest.
References
- 1.IOM, I.o.M.U.C.t.R.t.H.E.i.V.V.o.E.t.H. Health Effects of Herbicides Used in Vietnam. Veterans and Agent Orange; 1994 13 April 2020]; Available from: https://www.ncbi.nlm.nih.gov/books/NBK236356/ [Google Scholar]
- 2.National Academies of Sciences, E., and Medicine. Veterans and Agent Orange: Update 11. Veterans and Agent Orange; 2018; Available from: https://www.nap.edu/catalog/25137/veterans-and-agent-orange-update-11-2018. [PubMed] [Google Scholar]
- 3.Eskenazi B, Mocarelli P, Warner M, Needham L, Patterson DG Jr., Samuels S, Turner W, Gerthoux PM, and Brambilla P, Relationship of serum TCDD concentrations and age at exposure of female residents of Seveso, Italy. Environ Health Perspect, 2004. 112(1): p. 22–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ni HG, Zeng H, Tao S, and Zeng EY, Environmental and human exposure to persistent halogenated compounds derived from e-waste in China. Environ Toxicol Chem, 2010. 29(6): p. 1237–47. [DOI] [PubMed] [Google Scholar]
- 5.Birnbaum LS and Tuomisto J, Non-carcinogenic effects of TCDD in animals. Food Addit Contam, 2000. 17(4): p. 275–88. [DOI] [PubMed] [Google Scholar]
- 6.Grassman JA, Masten SA, Walker NJ, and Lucier GW, Animal models of human response to dioxins. Environ Health Perspect, 1998. 106 Suppl 2: p. 761–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spaulding SW, The possible roles of environmental factors and the aryl hydrocarbon receptor in the prevalence of thyroid diseases in Vietnam era veterans. Curr Opin Endocrinol Diabetes Obes, 2011. 18(5): p. 315–20. [DOI] [PubMed] [Google Scholar]
- 8.York G and Mick H, Last Ghost of the Vietnam War, in The Globe and Mail. 2008, Phillip Crawley: Toronto, ON Canada. [Google Scholar]
- 9.IOM, Veterans and Agent Orange Update 2010. (Institute of Medicine). 2010, Washington D.C.: The National Academies Press. [Google Scholar]
- 10.Guo YL, Hsu PC, Hsu CC, and Lambert GH, Semen quality after prenatal exposure to polychlorinated biphenyls and dibenzofurans. Lancet, 2000. 356(9237): p. 1240–1. [DOI] [PubMed] [Google Scholar]
- 11.Igarashi TM, Bruner-Tran KL, Yeaman GR, Lessey BA, Edwards DP, Eisenberg E, and Osteen KG, Reduced expression of progesterone receptor-B in the endometrium of women with endometriosis and in cocultures of endometrial cells exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fertil Steril, 2005. 84(1): p. 67–74. [DOI] [PubMed] [Google Scholar]
- 12.Resuehr D, Glore DR, Taylor HS, Bruner-Tran KL, and Osteen KG, Progesterone-dependent regulation of endometrial cannabinoid receptor type 1 (CB1-R) expression is disrupted in women with endometriosis and in isolated stromal cells exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Fertil Steril, 2012. 98(4): p. 948–56 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruner-Tran KL and Osteen KG, Dioxin-like PCBs and endometriosis. Syst Biol Reprod Med, 2010. 56(2): p. 132–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haugen AC, Schug TT, Collman G, and Heindel JJ, Evolution of DOHaD: the impact of environmental health sciences. J Dev Orig Health Dis, 2015. 6(2): p. 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carpenter DO, Polychlorinated biphenyls (PCBs): routes of exposure and effects on human health. Rev Environ Health, 2006. 21(1): p. 1–23. [DOI] [PubMed] [Google Scholar]
- 16.Baccarelli A, Giacomini SM, Corbetta C, Landi MT, Bonzini M, Consonni D, Grillo P, Patterson DG, Pesatori AC, and Bertazzi PA, Neonatal thyroid function in Seveso 25 years after maternal exposure to dioxin. PLoS Med, 2008. 5(7): p. e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anway MD, Cupp AS, Uzumcu M, and Skinner MK, Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science, 2005. 308(5727): p. 1466–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nilsson E, Sadler-Riggleman I, and Skinner MK, Environmentally Induced Epigenetic Transgenerational Inheritance of Disease. Environmental Epigenetics, 2018. 4(2): p. 1–13, dvy016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skinner MK, Manikkam M, and Guerrero-Bosagna C, Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol Metab, 2010. 21(4): p. 214–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reik W, Dean W, and Walter J, Epigenetic reprogramming in mammalian development. Science, 2001. 293(5532): p. 1089–93. [DOI] [PubMed] [Google Scholar]
- 21.Guerrero-Bosagna C, Settles M, Lucker B, and Skinner M, Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. Plos One, 2010. 5(9): p. 1–17, e13100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manikkam M, Guerrero-Bosagna C, Tracey R, Haque MM, and Skinner MK, Transgenerational actions of environmental compounds on reproductive disease and identification of epigenetic biomarkers of ancestral exposures. PLoS ONE 2012. 7(2): p. 1–12, e31901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McBirney M, King SE, Pappalardo M, Houser E, Unkefer M, Nilsson E, Sadler-Riggleman I, Beck D, Winchester P, and Skinner MK, Atrazine Induced Epigenetic Transgenerational Inheritance of Disease, Lean Phenotype and Sperm Epimutation Pathology Biomarkers. PLoS One, 2017. 12(9): p. 1–37, e0184306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anway MD and Skinner MK, Transgenerational effects of the endocrine disruptor vinclozolin on the prostate transcriptome and adult onset disease. Prostate, 2008. 68(5): p. 517–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manikkam M, Tracey R, Guerrero-Bosagna C, and Skinner M, Pesticide and Insect Repellent Mixture (Permethrin and DEET) Induces Epigenetic Transgenerational Inheritance of Disease and Sperm Epimutations. Reproductive Toxicology 2012. 34(4): p. 708–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manikkam M, Tracey R, Guerrero-Bosagna C, and Skinner M, Plastics Derived Endocrine Disruptors (BPA, DEHP and DBP) Induce Epigenetic Transgenerational Inheritance of Obesity, Reproductive Disease and Sperm Epimutations. PLoS ONE, 2013. 8(1): p. 1–18, e55387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ben Maamar M, Sadler-Riggleman I, Beck D, McBirney M, Nilsson E, Klukovich R, Xie Y, Tang C, Yan W, and Skinner MK, Alterations in sperm DNA methylation, non-coding RNA expression, and histone retention mediate vinclozolin-induced epigenetic transgenerational inheritance of disease. Environmental Epigenetics, 2018. 4(2): p. 1–19, dvy010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skinner MK, Ben Maamar M, Sadler-Riggleman I, Beck D, Nilsson E, McBirney M, Klukovich R, Xie Y, Tang C, and Yan W, Alterations in sperm DNA methylation, non-coding RNA and histone retention associate with DDT-induced epigenetic transgenerational inheritance of disease. Epigenetics & Chromatin 2018. 11(1): p. 8, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nilsson E, King SE, McBirney M, Kubsad D, Pappalardo M, Beck D, Sadler-Riggleman I, and Skinner MK, Vinclozolin induced epigenetic transgenerational inheritance of pathologies and sperm epimutation biomarkers for specific diseases. PLoS One, 2018. 13(8): p. 1–29, e0202662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stouder C and Paoloni-Giacobino A, Transgenerational effects of the endocrine disruptor vinclozolin on the methylation pattern of imprinted genes in the mouse sperm. Reproduction, 2010. 139(2): p. 373–9. [DOI] [PubMed] [Google Scholar]
- 31.King SE, McBirney M, Beck D, Sadler-Riggleman I, Nilsson E, and Skinner MK, Sperm Epimutation Biomarkers of Obesity and Pathologies following DDT Induced Epigenetic Transgenerational Inheritance of Disease. Environ Epigenet, 2019. 5(2): p. 1–15, dvz008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skinner MK, Manikkam M, Tracey R, Guerrero-Bosagna C, Haque MM, and Nilsson E, Ancestral dichlorodiphenyltrichloroethane (DDT) exposure promotes epigenetic transgenerational inheritance of obesity. BMC Medicine, 2013. 11: p. 228, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ben Maamar M, Beck D, Nilsson E, Kubsad D, and Skinner MK, Glyphosate Induced Transgenerational DNA Methylation and Histone Retention Sperm Epigenetic Biomarkers for Disease. Epigenetcs, 2020(In press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manikkam M, Tracey R, Guerrero-Bosagna C, and Skinner MK, Dioxin (TCDD) induces epigenetic transgenerational inheritance of adult onset disease and sperm epimutations. PLoS ONE, 2012. 7(9): p. 1–15, e46249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nilsson E, Larsen G, Manikkam M, Guerrero-Bosagna C, Savenkova M, and Skinner M, Environmentally Induced Epigenetic Transgenerational Inheritance of Ovarian Disease. PLoS ONE, 2012. 7(5): p. 1–18, e36129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruner-Tran KL, Ding T, Yeoman KB, Archibong A, Arosh JA, and Osteen KG, Developmental exposure of mice to dioxin promotes transgenerational testicular inflammation and an increased risk of preterm birth in unexposed mating partners. PLoS One, 2014. 9(8): p. e105084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding T, Mokshagundam S, Rinaudo PF, Osteen KG, and Bruner-Tran KL, Paternal developmental toxicant exposure is associated with epigenetic modulation of sperm and placental Pgr and Igf2 in a mouse model. Biol Reprod, 2018. 99(4): p. 864–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.(CONTAM), E.P.o.C.i.t.F.C., Knutsen HK, Alexander J, Barregård L, M. B, Brüschweiler B, Ceccatelli S, Cottrill B, Dinovi M, Edler L, Grasl-Kraupp B, Hogstrand C, Nebbia CS, Oswald IP, Petersen A, Rose M, Roudot A-C, Schwerdtle T, Vleminckx C, Vollmer G, Wallace H, Fürst P, Håkansson H, Halldorsson T, Lundebye A-K, Pohjanvirta R, Rylander L, Smith A, van Loveren H, Waalkens‐Berendsen I, Zeilmaker M, Binaglia M, Gómez Ruiz JA, Horváth Z, Christoph E, Ciccolallo L, Bordajandi LR, Steinkellner H, and Hoogenboom L, Risk for animal and human health related to the presence of dioxins and dioxin-like PCBs in feed and food. EFSA Journal, 2018. 16(11): p. e05333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pilsner JR, Parker M, Sergeyev O, and Suvorov A, Spermatogenesis disruption by dioxins: Epigenetic reprograming and windows of susceptibility. Reprod Toxicol, 2017. 69: p. 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bruner-Tran KL, Gnecco J, Ding T, Glore DR, Pensabene V, and Osteen KG, Exposure to the environmental endocrine disruptor TCDD and human reproductive dysfunction: Translating lessons from murine models. Reprod Toxicol, 2017. 68: p. 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greene JF, Hays S, and Paustenbach D, Basis for a proposed reference dose (RfD) for dioxin of 1–10 pg/kg-day: a weight of evidence evaluation of the human and animal studies. J Toxicol Environ Health B Crit Rev, 2003. 6(2): p. 115–59. [DOI] [PubMed] [Google Scholar]
- 42.Kubsad D, Nilsson EE, King SE, Sadler-Riggleman I, Beck D, and Skinner MK, Assessment of Glyphosate Induced Epigenetic Transgenerational Inheritance of Pathologies and Sperm Epimutations: Generational Toxicology. Scientific Reports, 2019. 9(1): p. 6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skinner MK and Guerrero-Bosagna C, Role of CpG Deserts in the Epigenetic Transgenerational Inheritance of Differential DNA Methylation Regions. BMC Genomics 2014. 15(1): p. 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Melnick R, Lucier G, Wolfe M, Hall R, Stancel G, Prins G, Gallo M, Reuhl K, Ho SM, Brown T, Moore J, Leakey J, Haseman J, and Kohn M, Summary of the National Toxicology Program’s report of the endocrine disruptors low-dose peer review. Environ Health Perspect, 2002. 110(4): p. 427–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lewis J, Smokey Bear in Vietnam. Environmental History, 2006. 11(3): p. 598–603. [Google Scholar]
- 46.Schecter A, Pavuk M, Amirova DA, Grosheva EI, Papke, Ryan JJ, Adibi J, and Piskac AL, Characterization of dioxin exposure in firefighters, residents, and chemical workers in the Irkutsk Region of Russian Siberia. Chemosphere, 2002. 47(2): p. 147–56. [DOI] [PubMed] [Google Scholar]
- 47.Anh NT, Nishijo M, Tai PT, Maruzeni S, Morikawa Y, Anh TH, Van Luong H, Dam PM, Nakagawa H, Son le K, and Nishijo H, Maternal risk factors associated with increased dioxin concentrations in breast milk in a hot spot of dioxin contamination in Vietnam. J Expo Sci Environ Epidemiol, 2014. 24(5): p. 489–96. [DOI] [PubMed] [Google Scholar]
- 48.Nghi TN, Nishijo M, Manh HD, Tai PT, Van Luong H, Anh TH, Thao PN, Trung NV, Waseda T, Nakagawa H, Kido T, and Nishijo H, Dioxins and Nonortho PCBs in Breast Milk of Vietnamese Mothers Living in the Largest Hot Spot of Dioxin Contamination. Environ Sci Technol, 2015. 49(9): p. 5732–42. [DOI] [PubMed] [Google Scholar]
- 49.Schecter A, Pavuk M, Constable JD, Dai le C, and Papke O, A follow-up: high level of dioxin contamination in Vietnamese from agent orange, three decades after the end of spraying. J Occup Environ Med, 2002. 44(3): p. 218–20. [DOI] [PubMed] [Google Scholar]
- 50.Tai PT, Nishijo M, Anh NT, Maruzeni S, Nakagawa H, Van Luong H, Anh TH, Honda R, Kido T, and Nishijo H, Dioxin exposure in breast milk and infant neurodevelopment in Vietnam. Occup Environ Med, 2013. 70(9): p. 656–62. [DOI] [PubMed] [Google Scholar]
- 51.Rowlands JC, Budinsky RA, Aylward LL, Faqi AS, and Carney EW, Sex ratio of the offspring of Sprague-Dawley rats exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in utero and lactationally in a three-generation study. Toxicol Appl Pharmacol, 2006. 216(1): p. 29–33. [DOI] [PubMed] [Google Scholar]
- 52.Sanabria M, Cucielo MS, Guerra MT, Dos Santos Borges C, Banzato TP, Perobelli JE, Leite GA, Anselmo-Franci JA, and De Grava Kempinas W, Sperm quality and fertility in rats after prenatal exposure to low doses of TCDD: A three-generation study. Reprod Toxicol, 2016. 65: p. 29–38. [DOI] [PubMed] [Google Scholar]
- 53.Chang ET, Boffetta P, Adami HO, Cole P, and Mandel JS, A critical review of the epidemiology of Agent Orange/TCDD and prostate cancer. Eur J Epidemiol, 2014. 29(10): p. 667–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chevrier J, Warner M, Gunier RB, Brambilla P, Eskenazi B, and Mocarelli P, Serum dioxin concentrations and thyroid hormone levels in the Seveso Women’s Health Study. Am J Epidemiol, 2014. 180(5): p. 490–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye M, Warner M, Mocarelli P, Brambilla P, and Eskenazi B, Prenatal exposure to TCDD and atopic conditions in the Seveso second generation: a prospective cohort study. Environ Health, 2018. 17(1): p. 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ames J, Warner M, Siracusa C, Signorini S, Brambilla P, Mocarelli P, and Eskenazi B, Prenatal dioxin exposure and neuropsychological functioning in the Seveso Second Generation Health Study. Int J Hyg Environ Health, 2019. 222(3): p. 425–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pernar CH, Ebot EM, Wilson KM, and Mucci LA, The Epidemiology of Prostate Cancer. Cold Spring Harb Perspect Med, 2018. 8(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hill NR, Fatoba ST, Oke JL, Hirst JA, O’Callaghan CA, Lasserson DS, and Hobbs FD, Global Prevalence of Chronic Kidney Disease - A Systematic Review and Meta-Analysis. PLoS One, 2016. 11(7): p. e0158765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hales CM, Carroll MD, Fryar CD, and Ogden CL, Prevalence of Obesity and severe obesity among adults: United States, 2017–2018. NCHS data brief, 2020(360): p. 1–8. [PubMed] [Google Scholar]
- 60.Devlin B, Roeder K, and Wasserman L, Analysis of multilocus models of association. Genet Epidemiol, 2003. 25(1): p. 36–47. [DOI] [PubMed] [Google Scholar]
- 61.Higdon R, van Belle G, and Kolker E, A note on the false discovery rate and inconsistent comparisons between experiments. Bioinformatics, 2008. 24(10): p. 1225–8. [DOI] [PubMed] [Google Scholar]
- 62.Yang H and Churchill G, Estimating p-values in small microarray experiments. Bioinformatics, 2007. 23(1): p. 38–43. [DOI] [PubMed] [Google Scholar]
- 63.Bretz F, Landgrebe J, and Brunner E, Multiplicity issues in microarray experiments. Methods Inf Med, 2005. 44(3): p. 431–7. [PubMed] [Google Scholar]
- 64.Jung SH, Sample size and power calculation for molecular biology studies. Methods Mol Biol, 2010. 620: p. 203–18. [DOI] [PubMed] [Google Scholar]
- 65.Nilsson R, Bjorkegren J, and Tegner J, On reliable discovery of molecular signatures. BMC Bioinformatics, 2009. 10: p. 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nilsson EE, Anway MD, Stanfield J, and Skinner MK, Transgenerational epigenetic effects of the endocrine disruptor vinclozolin on pregnancies and female adult onset disease. Reproduction, 2008. 135(5): p. 713–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anway MD, Leathers C, and Skinner MK, Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology, 2006. 147(12): p. 5515–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Manikkam M, Haque MM, Guerrero-Bosagna C, Nilsson E, and Skinner MK, Pesticide methoxychlor promotes the epigenetic transgenerational inheritance of adult onset disease through the female germline. PLoS ONE, 2014. 9(7): p. 1–19, e102091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tracey R, Manikkam M, Guerrero-Bosagna C, and Skinner M, Hydrocarbons (jet fuel JP-8) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. Reproductive Toxicology, 2013. 36: p. 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taylor JA, Richter CA, Ruhlen RL, and vom Saal FS, Estrogenic environmental chemicals and drugs: mechanisms for effects on the developing male urogenital system. J Steroid Biochem Mol Biol, 2011. 127(1–2): p. 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang TT Jr. and Yanagimachi R, Inner acrosomal membrane of mammalian spermatozoa: its properties and possible functions in fertilization. Am J Anat, 1985. 174(3): p. 249–68. [DOI] [PubMed] [Google Scholar]
- 72.Calvin HI, Isolation of subfractionation of mammalian sperm heads and tails. Methods Cell Biol, 1976. 13: p. 85–104. [DOI] [PubMed] [Google Scholar]
- 73.Langmead B and Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nature Methods, 2012. 9(4): p. 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome S Project Data Processing, The Sequence Alignment/Map format and SAMtools. Bioinformatics, 2009. 25(16): p. 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lienhard M, Grimm C, Morkel M, Herwig R, and Chavez L, MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics, 2014. 30(2): p. 284–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robinson MD, McCarthy DJ, and Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 2010. 26(1): p. 139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Durinck S, Spellman PT, Birney E, and Huber W, Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nature Protocols, 2009. 4(8): p. 1184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, Gil L, Giron CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Johnson N, Juettemann T, Kahari AK, Keenan S, Martin FJ, Maurel T, McLaren W, Murphy DN, Nag R, Overduin B, Parker A, Patricio M, Perry E, Pignatelli M, Riat HS, Sheppard D, Taylor K, Thormann A, Vullo A, Wilder SP, Zadissa A, Aken BL, Birney E, Harrow J, Kinsella R, Muffato M, Ruffier M, Searle SM, Spudich G, Trevanion SJ, Yates A, Zerbino DR, and Flicek P, Ensembl 2015. Nucleic Acids Research, 2015. 43(Database issue): p. D662–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang da W, Sherman BT, and Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols, 2009. 4(1): p. 44–57. [DOI] [PubMed] [Google Scholar]
- 80.Mi H, Muruganujan A, Casagrande JT, and Thomas PD, Large-scale gene function analysis with the PANTHER classification system. Nature Protocols, 2013. 8(8): p. 1551–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. DMR principal component analysis. The first two principal components are used. The underlying data is the RPKM read depth for DMR only genomic windows. (A) Prostate disease DMRs PCA; (B) Kidney disease DMRs PCA; (C) Obesity disease DMRs PCA; (D) Testis disease DMRs PCA.
Supplemental Table S1. Dioxin study control transgenerational pathology. F3 generation control lineage male rat pathology. Vehicle dimethyl sulfoxide (DMSO) exposure used in the F0 generation gestating female. The individual animals for the control lineage males are listed with a (+) to indicate the presence of the pathology or (−) for absence of the pathology. The number of disease/total animals is presented and percentage disease.
Supplemental Table S2. DMR Site List Prostate Disease p<1e-04. DMR name, chromosome, start, stop, length, number signature windows, minimum p-value, max log-fold change, CpG number, CpG density, gene annotation, and gene category are presented.
Supplemental Table S3. DMR Site List Kidney Disease p<1e-04. DMR name, chromosome, start, stop, length, number signature windows, minimum p-value, max log-fold change, CpG number, CpG density, gene annotation, and gene category are presented.
Supplemental Table S4. DMR Site List Obesity p<1e-04. DMR name, chromosome, start, stop, length, number signature windows, minimum p-value, max log-fold change, CpG number, CpG density, gene annotation, and gene category are presented.
Supplemental Table S5. DMR Site List Testis Disease p<1e-04. DMR name, chromosome, start, stop, length, number signature windows, minimum p-value, max log-fold change, CpG number, CpG density, gene annotation, and gene category are presented.