

INTRODUCTION
As humans age, many changes take place in the bone marrow, which may have clinical consequences. Differences in the marrow between young and old have been demonstrated in both animal models and in humans, involving alterations in overall cell number, senescence, lineage differentiation, cellular composition, and function of the hematopoietic stem cell (HSC). These changes likely contribute to a higher rate of cytopenias, predominantly anemia and lymphopenia, and a risk of hematological malignancy.
As stem cells reproduce, they naturally acquire mutations that are then passed on to their progeny—producing a clonal hematopoiesis. Acquired mutations found in HSCs are not necessarily pathogenic and they normally and inevitably accumulate over time.1 Clonal hematopoiesis of indeterminate potential (CHIP) is described as the presence of known somatic mutations in genes long associated with myeloid hematological disorders and is common in the normal aging population. It remains unclear whether clonal hematopoiesis is a disease or an accompaniment of normal aging. In retrospective population studies, CHIP has been shown associated with the development of both hematological malignancy (lymphoid and myeloid) and atherosclerosis (cardiovascular and cerebrovascular disease).2
ALTERED BLOOD COUNTS WITH AGING
Anemia in the elderly is prevalent, in 11% of men and 10% of women 65 years and older, and it is multifactorial.3 Common etiologies include anemia of chronic disease associated with inflammation, chronic kidney disease, nutritional deficiencies (such as iron, vitamin B12, and folate deficiencies), and clonal disorders, such as myelodysplastic syndrome (MDS). These topics are covered elsewhere in this issue.
There has been no specific decrease in the erythroid population noted when evaluating marrow function. Levels of erythropoietin (EPO), a hormone that stimulates erythropoiesis, are increased. It is possible that, as humans age, their response to EPO stimulation is decreased and higher levels are required to maintain a normal hemoglobin level. Patients with inflammatory disorders have lower blood concentrations of EPO, suggesting less hormonal stimulation as an explanation for anemia in this circumstance.4
An elevated white blood cell count is an independent marker of all-cause mortality,5 specifically correlating with cancer or cardiovascular-related death,6 in all populations, including the elderly. Leukocytosis may reflect generalized inflammation.
Monocytes increase with age. In murine models, monocytes from aged mice promote intimal hyperplasia, a feature of atherosclerosis, compared with monocytes from young mice. In addition, up-regulation of genes involved in cellular adhesion and inflammation is a feature of aged monocytes.7 In humans, alterations occur in the phenotype and cell surface markers of monocytes from classical CD14+CD16− to nonclassical CD14+CD16+, with an increase in nonclassical monocytes seen with age. Nonclassical monocytes secrete proinflammatory cytokines8 but with unknown consequences.
PHYSIOLOGY AND PATHOPHYSIOLOGY OF STEM CELL AGING
Hematopoiesis is the continuous process by which blood cells are produced, maintained at normal levels, and increased in response to demand. HSCs have 3 critical properties: differentiation into progenitors of all lineages; high proliferative capacity, as a progenitor giving rise to many thousands of mature blood cells; and self-renewal to maintain the HSC pool. The major point where lineages diverge is between 2 common progenitors—myeloid and lymphoid. The primitive myeloid progenitor is responsible for the production of granulocytes, erythroid cells (red cell precursors), and megakaryocytes (platelet precursors) and the lymphoid progenitor for the production of lymphocytes. The marrow stroma, vasculature, and cytokines of the marrow environment play a role in stem cell biology.9
Mouse Models
In young mice, HSC cell turnover is infrequent, approximately 1 division every 30 days. In comparing young and old mice, there is a 2-fold increase of HSCs in the older mouse, measured by competitive repopulation. In the competitive repopulation assay, marrow cells from young and old mice are mixed, and these cell mixtures are transplanted to repopulate irradiated recipients. Skewing, or increased numbers, of myeloid progenitor cells compared with lymphoid progenitor cells also has been observed in mice using transplantation of younger and older mouse bone marrow and ascertaining myeloid and lymphoid chimerism.10 Telomere shortening, which occurs with age, may play a role in myeloid skewing by activation of interferon gamma, thus inducing an inflammatory response.11
Numbers and Senescence
Human stem cells are difficult to study. Immunophenotyping identifies HSCs by specific cell surface markers using antibodies. Standard immunotyping is fluorescence based, and there are limits to the number of cell surface markers that can be identified on a single cell. Additionally, sample quality (especially dilution with peripheral blood) and sampling site may affect results.
In humans, as with mice, the bone marrow seems to have increased numbers of multipotent stem cells with increased age, by immunophenotype, despite its hypocellular morphologic appearance. Aged HSCs have a higher rate of cell division and proliferation (active phases of the cell cycle) than do younger stem cells, which tend to be quiescent (inactive phase of the cell cycle).12
Lineage
As cells age, they show up-regulation in genes associated with growth and proliferation as well as those associated with myeloid malignancies.13 By immunophenotyping of lymphoid and myeloid cell markers in humans, there is a preservation of numbers of myeloid cells and a decrease in B-lymphoid cells, skewing the myeloid-to-lymphoid ratio and resulting in myeloid predominance,12,14 an effect also seen in mice. Skewing may explain the greater increased prevalence of myeloid compared with lymphoid malignancies apparent with age.15
The reduction in cells differentiated to the B-lymphoid lineage, combined with an overall reduction in T-cell production,16 may explain the lymphopenia associated with aging and contribute to impaired immunity in the elderly.
Cellular Factors
Many cellular factors have been shown to contribute to HSC aging: molecular signaling pathways, such as those involved in DNA repair; changes in mitochondrial function, causing reduced enzymatic activity and the acquisition of somatic mutations; reduction in cell polarity, resulting in altered cell division; and shortening of telomeres.17–20
Function
Although HSC numbers increase with age, their functionality may be impaired. Oxidative DNA damage may hinder self-replication.12 In HSC transplants, older age of the donor associates with decreased overall survival,21–23 and, with autologous stem cell transplants, older patients have poorer engraftment.24 In addition, stem cells from older donor animals have reduced engraftment compared with those from younger mice.
AGING CLONES
Defining clonality is difficult. Classically, in the field of hematology-oncology, clones are described as a uniform population of malignant cells. Malignant clones evolve, acquiring further subclones, and ultimately may cause disease. Clonality also applies, however, to normal hematopoiesis. As cells divide, they, by definition, produce a clone of themselves. During this process cell alterations may occur, including mutations, which may be either pathogenic or nonpathogenic.
Because HSCs must undergo multiple divisions to sustain hematopoiesis, they are susceptible, over time, to accumulate mutations, which are inherited by their descendant cells. Mutations also occur due to environmental factors, such as chemicals and radiation. The average person acquires 10 to 20 nonpathogenic passenger mutations per stem cell by middle age.1 Mutations likely confer a survival advantage over non-mutated stem cells, resulting in proliferation of a clonal population or clonal hematopoiesis. Skewed X chromosome inactivation, particularly in older patients, was an early if nonspecific indication of this phenomenon.25
Clonal hematopoiesis has been detected using a barcode method—measuring for the presence of somatic mutations without a specific driver gene. In 1 large study looking at an Icelandic population, the presence of clonal hematopoiesis increased from 0.5% in those less than 35 years to more than 50% for subjects older than 85 years.26 Clonal hematopoiesis was associated with a reduction in overall survival, a finding that likely reflects a degree of overall cell damage.
The population size and lifetime dynamics of HSCs were recently investigated by measuring naturally occurring somatic mutations in a single individual (with normal blood counts and no evidence of hematological disease) and imputing an evolutionary tree. The subject had no mutations associated with myeloid malignancy (CHIP). By computation, there seemed to be hundreds of thousands of stem cells at any 1 time, a much higher figure than previously estimated, with cell division at intervals of 2 months to 20 months. There was a close ancestral relationship between differentiated granulocytes and differentiated B lymphocytes but not between granulocytes and T lymphocytes, suggesting divergence in ancestry.
THE DEFINITION AND PATHOPHYSIOLOGY CLONAL HEMATOPOIESIS OF INDETERMINATE POTENTIAL
CHIP is a form of clonal hematopoiesis where a somatic driver mutation associated with hematological malignancy is present without morphologic evidence of bone marrow dysplasia or neoplasia.27 A clone size, or variant allele fraction (VAF), of at least 2% has been used in the literature as a defined cutoff, although smaller clones can be detected using deeper sequencing techniques. Such mutations occur in leukocytes and have been found in a substantial proportion of the healthy aging population by next-generation sequencing. CHIP may represent a preneoplastic phase of hematological malignancy, in particular myeloid disorders, but also is associated with lymphoid malignancy and plasma cell dyscrasias.28–31 Most people with these mutations do not develop a hematological malignancy, with a risk of progression of 0.5% to 1% per year.32
There are several specific myeloid genes associated with CHIP, the most common being DNMT3A, TET2, and ASXL1, and some spliceosome mutations. CHIP mutations preferentially affect the myeloid lineage and natural killer cells based on a higher VAF than seen that in B cells and T cells of the same sample.33
CLONAL HEMATOPOIESIS OF INDETERMINATE POTENTIAL AND HEMATOLOGICAL DISORDERS
Few patients with CHIP actually develop myeloid neoplasms despite the association.27 It is estimated that its presence doubles the risk of developing a hematological malignancy—however, these are not common malignancies, with MDS having an incidence of 0.22 per 100,000 to 13.2 per 100,000 and acute myeloid leukemia (AML) having an incidence of 0.6 to 11 per 100,000, combining all ages, genders, and risk factors.34
CHIP has been associated with all types of hematological malignancy, including MDS, AML, chronic lymphocytic leukemia, acute lymphoblastic leukemia/lymphoma, myeloproliferative neoplasms, lymphoma, and myeloma, although in studies the overall numbers of patients who developed hematological malignancy is low.32 The presence of greater than 1 somatic mutation and increased VAF positively predict for malignant progression. Certain mutations (such as IDH and spliceosome mutations) are correlated more strongly with this progression than are the more common DNMT3A, TET2, and ASXL1. These latter mutations may represent a founder event that renders the environment more susceptible to further leukemogenesis rather than a direct neoplastic trigger.35
In one recent study, the presence of CHIP mutations in AML patients was sought in samples collected years prior to disease development to identify ways of predicting risk of progression to AML. The presence of a CHIP mutation (except DNMT3A and TET2) doubled the risk of developing AML per 5% increase in clone size. DNMT3A and TET2 conferred a lower risk of progression than TP53 and U2AF1 (a spliceosome mutation) which conferred a higher risk. AML patients had significantly higher numbers of mutations and larger clones than a control group without AML. Mutations commonly seen in AML, such as FLT3, NPM1, and CEBPA, were absent in historic samples, suggesting that these occur later in leukemogenesis. Red cell distribution width (RDW) was identified as a routine clinical test that had a significant correlation to risk of progression to AML and may act as a screening tool.30
CHIP mutations have been observed in aplastic anemia at a rate of 47%, with BCOR, BCORL1, and PIGA the most commonly seen mutations, correlating with a better response to therapy and higher progression-free survival. DNMT3A and ASXL1 also were common but were collectively associated with poorer outcomes.36
CLONAL HEMATOPOIESIS OF INDETERMINATE POTENTIAL AND ATHEROSCLEROSIS
CHIP has been associated with an increased atherosclerotic risk,2,37 in particular, cardiovascular and cerebrovascular disease. Cardiovascular disease has been shown 2-fold to 4-fold higher in patients with CHIP, with an increase in both myocardial infarction and coronary artery calcification, using retrospective data. This association was significantly correlated with a higher VAF (>10%). The presence of greater than 1 somatic mutation was linked to an increased prevalence of peripheral vascular disease and diabetes33 compared with those with 1 mutation. Both DNMT3A and TET2 have been associated with worse long-term clinical outcomes in patients with congestive heart failure.38
Mutational loss of TET2 has been associated with increased risk of atherosclerosis in murine models. Explanations include an inflammatory response with macrophage dysfunction causing plaque build-up or leukocytosis effect similar to that seen in myeloproliferative disorders. The loss of TET2 increased monocytes and interleukin (IL)-6 in murine and human bone marrow.39,40 TET2 seems to play a role in the regulation of IL-6, an inflammatory cytokine, and may be associated with a proinflammatory state and increased atherosclerotic risk. With further study, this inflammatory process may become a target of future therapeutic agents.
CLONAL CYTOPENIAS OF UNDETERMINED SIGNIFICANCE
Idiopathic cytopenia of undetermined significance (ICUS) is used to describe an unexplained cytopenia not meeting MDS diagnostic criteria that cannot be explained by any other hematologic or nonhematologic disease process.41 Clinical confusion has long existed as to the optimal follow-up for these patients. Recently, it has been shown that many patients with unexplained cytopenia harbor a myeloid somatic mutation. In one study, this was true for approximately two-thirds of patients who historically had been diagnosed with ICUS.42
Clonal cytopenia of undetermined significance (CCUS) is defined as a cytopenia and associated myeloid somatic mutation in the absence of morphologic evidence of MDS or an MDS defining cytogenetic abnormality43,44 (Fig. 1). In a study looking at patients with unexplained cytopenia (previously classed as ICUS), the presence of a myeloid somatic mutation (CCUS) significantly increased the risk of progression to a myeloid neoplasm (95% risk vs 9% risk at 10 years). Risk of progression to leukemia was similar to that of low-risk MDS. Risk factors for progression identified included the presence of spliceosome mutations, having greater than 1 mutation, and a large clone size. These data suggest that these patients should be clinically monitored as high-risk patients, similarly to patients with MDS, rather than persons with standard CHIP, who have a much lower progression risk and are likely to have a normal life span.42 Additionally, patients with ICUS, in whom CCUS has been excluded, have a lower risk of progression than those with an identified mutation.
Fig. 1.
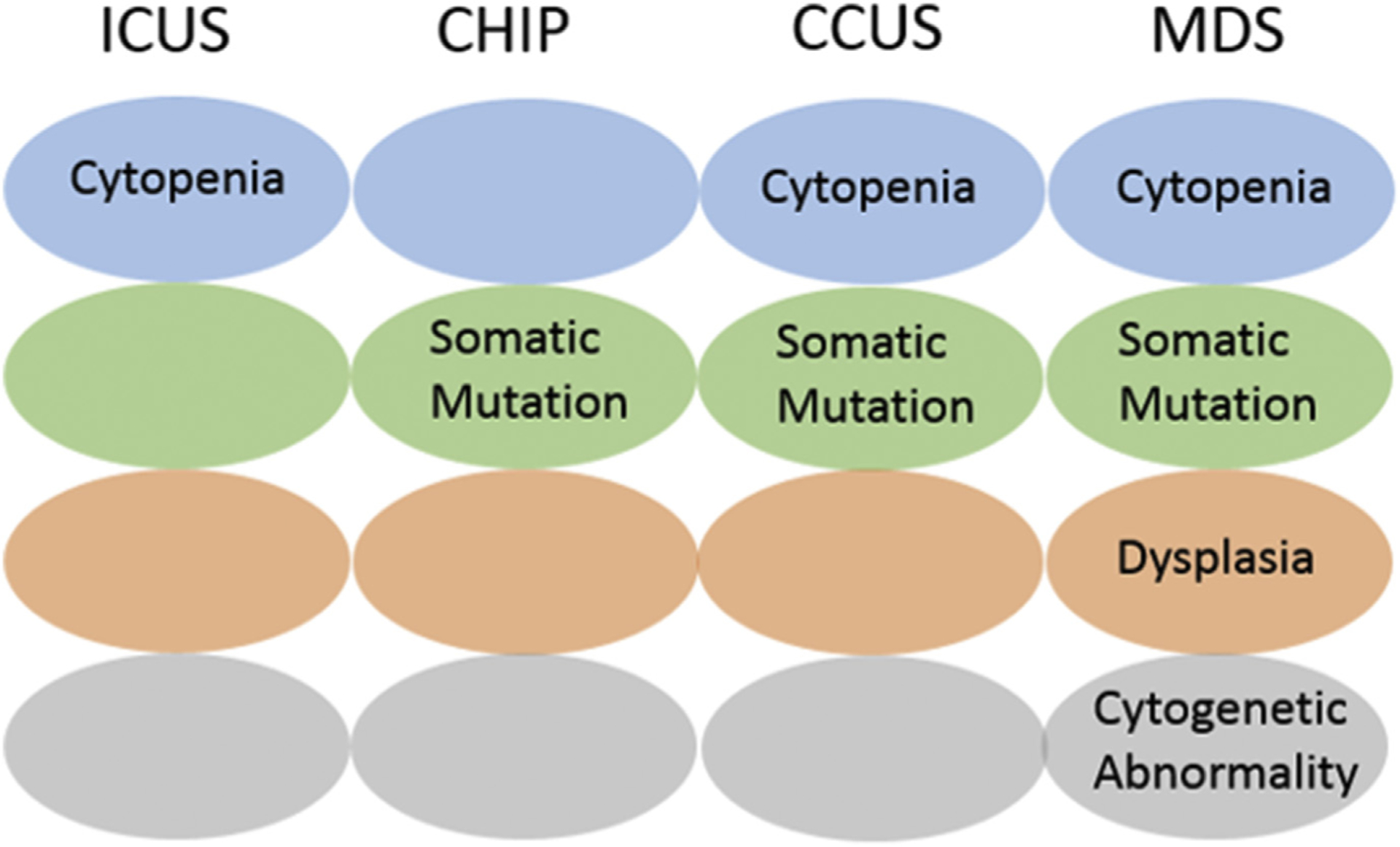
Distinction between ICUS, CHIP, CCUS, and MDS.
SUMMARY
With age, changes take place in bone marrow hematopoiesis that can have an impact on health and longevity. Overall, human stem cells seem to become more poorly functional and, possibly as a result, have an increase in both their number and cell dividing capacity. Myeloid skewing also becomes apparent with age and may contribute to the increased rates of myeloid malignancy in the elderly. This skewing, coupled with a reduction in T-cell production, results in a decrease in lymphocytes, which may contribute to a decrease in immunity seen in the elderly. Although erythroid progenitors do not seem to decrease with age, increased rates of anemia may be explained by changes in their response to EPO.
Clonal hematopoiesis is prevalent with aging and is increasingly identified with the advent of more sequencing. A majority of individuals with CHIP do not develop hematological malignancy and many of those who do develop it acquire a further driver mutation that promotes leukemogenesis. Certain higher-risk populations with CHIP have been identified—in particular those with more than 1 mutation and a higher allele burden. CCUS also is classed as higher risk with a significant rate of progression to hematological malignancy demonstrated in 1 study. The association between CHIP and atherosclerosis warrants further study, given the high incidence and prevalence of atherosclerosis in the general population, particularly in older age groups. Prospective studies further investigating clonal hematopoiesis are needed both to develop a method of risk stratification for the clinical hematologist and to identify any potential future therapeutic targets.
KEY POINTS.
With age, both the overall number of stem cells and the rate of cell division increase, but functionally these hematopoietic stem cells seem inferior.
A decrease in lymphoid progenitors contributes to lymphopenia and likely to decreased immunity in the elderly.
Clonal hematopoiesis of indeterminate potential (CHIP) is defined as the presence of a somatic mutation associated with hematological malignancy in the absence of a cytopenia.
CHIP is common in the elderly and has been associated with an increased risk of both hematological malignancy and atherosclerosis.
Clonal cytopenia of undetermined significance (CCUS) is defined as cytopenias associated with a myeloid somatic mutation but not meeting the criteria for myelodysplastic syndrome. CCUS has a significant rate of progression to myeloid neoplasm.
Footnotes
Disclosure Statement: The authors have nothing to disclose.
REFERENCES
- 1.Cooper JN, Young NS. Clonality in context: hematopoietic clones in their marrow environment. Blood 2017;130(22):2363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 2017;377(2):111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guralnik JM, Eisenstaedt RS, Ferrucci L, et al. Prevalence of anemia in persons 65 years and older in the United States: evidence for a high rate of unexplained anemia. Blood 2004;104(8):2263–8. [DOI] [PubMed] [Google Scholar]
- 4.Ershler WB, Sheng S, McKelvey J, et al. Serum erythropoietin and aging: a longitudinal analysis. J Am Geriatr Soc 2005;53(8):1360–5. [DOI] [PubMed] [Google Scholar]
- 5.Willems JM, Trompet S, Blauw GJ, et al. White blood cell count and C-reactive protein are independent predictors of mortality in the oldest old. J Gerontol A Biol Sci Med Sci 2010;65(7):764–8. [DOI] [PubMed] [Google Scholar]
- 6.Grimm RH, Neaton JD, Ludwig W, et al. Prognostic importance of the white blood cell count for coronary, cancer, and all-cause mortality. JAMA 1985;254(14): 1932–7. [PubMed] [Google Scholar]
- 7.Martinez L, Gomez C, Vazquez-Padron R. Age-related changes in monocytes exacerbate neointimal hyperplasia after vascular injury. Oncotarget 2015;6(19): 17054–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seidler S, Zimmermann HW, Bartneck M, et al. Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol 2010;11(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sallman DA, Cluzeau T, Basiorka AA, et al. Unraveling the pathogenesis of MDS: the NLRP3 inflammasome and pyroptosis drive the MDS phenotype. Front Oncol 2016;6:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sudo K, Ema H, Morita Y, et al. Age-associated characteristics of murine hematopoietic stem cells. J Exp Med 2000;192(9):1273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Bryant MA, Dent JJ, et al. Hematopoietic lineage skewing and intestinal epithelia degeneration in aged mice with telomerase RNA component deletion. Exp Gerontol 2015;72:251–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pang WW, Price EA, Sahoo D, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A 2011;108(50):20012–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akunuru SG. Hartmut, aging, clonality, and Rejuvenation of hematopoietic stem cells. Trends Mol Med 2016;22(8):701–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuranda K, Vargaftig J, de la Rochere P, et al. Age-related changes in human hematopoietic stem/progenitor cells. Aging Cell 2011;10(3):542–6. [DOI] [PubMed] [Google Scholar]
- 15.Signer RAJ, Montecino-Rodriguez E, Witte ON, et al. Age-related defects in B lymphopoiesis underlie the myeloid dominance of adult leukemia. Blood 2007; 110(6):1831–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorshkind K, Montecino-Rodriguez E, Signer RA. The ageing immune system: is it ever too old to become young again? Nat Rev Immunol 2009;9(1):57–62. [DOI] [PubMed] [Google Scholar]
- 17.Yao YG, Ellison FM, McCoy JP, et al. Age-dependent accumulation of mtDNA mutations in murine hematopoietic stem cells is modulated by the nuclear genetic background. Hum Mol Genet 2007;16(3):286–94. [DOI] [PubMed] [Google Scholar]
- 18.Van Zant G, Liang Y. Concise review: hematopoietic stem cell aging, life span, and transplantation. Stem Cells Transl Med 2012;1(9):651–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calado RT, Dumitriu B. Telomere dynamics in mice and humans. Semin Hematol 2013;50(2):165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Florian MC, Geiger H. Concise review: polarity in stem cells, disease, and aging. Stem Cells 2010;28(9):1623–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kollman C, Howe CW, Anasetti C, et al. Donor characteristics as risk factors in recipients after transplantation of bone marrow from unrelated donors: the effect of donor age. Blood 2001;98(7):2043–51. [DOI] [PubMed] [Google Scholar]
- 22.Kollman C, Spellman SR, Zhang MJ, et al. The effect of donor characteristics on survival after unrelated donor transplantation for hematologic malignancy. Blood 2016;127(2):260–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finke J, Schmoor C, Bethge WA. Prognostic factors affecting outcome after allogeneic transplantation for hematological malignancies from unrelated donors: results from a randomized trial. Biol Blood Marrow Transplant 2012;18(11):1716–26. [DOI] [PubMed] [Google Scholar]
- 24.Woolthuis CM, Mariani N, Verkaik-Schakel RN, et al. Aging impairs long-term hematopoietic regeneration after autologous stem cell transplantation. Biol Blood Marrow Transplant 2014;20(6):865–71. [DOI] [PubMed] [Google Scholar]
- 25.Anger B, Janssen JW, Schrezenmeier H, et al. Clonal analysis of chronic myeloproliferative disorders using X-linked DNA polymorphisms. Leukemia 1990;4(4): 258–61. [PubMed] [Google Scholar]
- 26.Zink F, Stacey SN, Norddahl GL, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017;130(6):742–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126(1): 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371(26):2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012;150(2):264–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abelson S, Collord G, Ng SWK, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018;559(7714):400–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Desai P, Mencia-Trinchant N, Savenkov O, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med 2018;24(7):1015–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371(26): 2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arends CM, Galan-Sousa J, Hoyer K, et al. Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia 2018;32(9): 1908–19. [DOI] [PubMed] [Google Scholar]
- 34.Lubeck DP, et al. Systematic literature review of the global incidence and prevalence of myelodysplastic syndrome and acute myeloid leukemia. Blood 2016; 128(22):5930. [Google Scholar]
- 35.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014;20(12):1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshizato T, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med 2015;373(1):35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuster JJ, MacLauchlan S, Zuriaga MA, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017;355(6327):842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dorsheimer L, Assmus B, Rasper T, et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol 2019;4(1):25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cull AH, Snetsinger B, Buckstein R, et al. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol 2017;55:56–70.e13. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Q, Zhao K, Shen Q, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015;525(7569):389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valent P, Horny HP, Bennett JM, et al. Definitions and standards in the diagnosis and treatment of the myelodysplastic syndromes: consensus statements and report from a working conference. Leuk Res 2007;31(6):727–36. [DOI] [PubMed] [Google Scholar]
- 42.Malcovati L, Gallì A, Travaglino E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017;129(25):3371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cargo CA, Rowbotham N, Evans PA, et al. Targeted sequencing identifies patients with preclinical MDS at high risk of disease progression. Blood 2015; 126(21):2362–5. [DOI] [PubMed] [Google Scholar]
- 44.Kwok B, Hall JM, Witte JS, et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood 2015;126(21):2355–61. [DOI] [PMC free article] [PubMed] [Google Scholar]