

Abstract
Objective:
The underlying cause of amyotrophic lateral sclerosis (ALS), a progressive neurodegenerative disorder, remains unknown. However, there is strong evidence that one pathophysiological mechanism, toxic protein misfolding and/or aggregation, may trigger motor neuron dysfunction and loss. Since the clinical and pathological features of sporadic and familial ALS are indistinguishable, all forms of the disease may be better understood and ultimately treated by studying pathogenesis and therapy in models expressing mutant forms of SOD1.
Methods:
We developed a cellular model in which cell death depended on the expression of G93A SOD1, a mutant form of superoxide dismutase found in familial ALS patients that produces toxic protein aggregates. This cellular model was optimized for high throughput screening to identify protective compounds from a >50,000-member chemical library.
Results:
Three novel chemical scaffolds were selected for further study following screen implementation, counter-screening and secondary testing, including studies with purchased analogs. All three scaffolds blocked SOD1 aggregation in high content screening assays and data on the optimization and further characterization of these compounds will be reported separately.
Conclusions:
These data suggest that optimization of these chemicals scaffolds may produce therapeutic candidates for ALS patients.
Keywords: ALS therapy, protein aggregation, high throughput screening
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a rapid, fatal neurodegenerative disease with a worldwide prevalence of ~87,000 patients [1]. Disability is significant and patient care costs during the late stages of the disease can exceed $200,000 per year. Riluzole, the only clinically approved therapeutic for ALS, extends median survival by only 2–3 months [2–4]. While there is clearly a great need for new ALS therapies, the identification of treatments has been impeded because the mechanism(s) directly underlying the most common sporadic form of ALS remain(s) unknown. There are, however, a number of candidate hypotheses for the molecular etiology of ALS including glutamate excitotoxicity, impaired mitochondrial function, oxidative stress, aberrant protein aggregation, defective RNA processing/transport, inflammation, and apoptotic cell death [5–9].
Considerable progress in the understanding of ALS has been made through the study of a familial form of the disease, caused by mutations in the gene encoding copper/zinc superoxide dismutase type 1 (SOD1) [10, 11]. Missense mutations in SOD1 cause about 20% of familial ALS cases, whose clinical and pathological features are indistinguishable from those in sporadic ALS. ALS promoting alleles do not in all cases inhibit enzyme activity. However, all of these mutations produce gain of function effects associated with an increased propensity of the protein to misfold and aggregate. Studies of sporadic ALS patients also support a role for aberrant protein aggregation. A neuropathological hallmark of sporadic ALS is the presence of ubiquitinated cytoplasmic inclusions in the lower motor neurons of these patients. These inclusions are composed, at least in part, of the TAR DNA binding protein (TDP-43) and most recently familial ALS kindreds have been identified that carry mutations in the TDP-43 gene, providing evidence that TDP-43 aggregation may be a cause rather than a result of the disease [12–16]. In addition to providing additional support for protein aggregation toxicity in ALS, mutant forms of SOD1 have been important in the development of cellular and animal models of the disease. The similarities in the clinical and pathological features of familial and sporadic ALS have led investigators to use the FALS phenotype as a strategy for elucidating disease pathogenesis and defining novel treatments in both forms of the disease. Mice expressing human mutant G93A SOD1 recapitulate many of the clinical and neuropathological features of ALS and are the most commonly used animal model to test compounds for efficacy in treating the disease [8, 17, 18]. Some of the compounds that are efficacious in transgenic mice have gone on to clinical testing. We have been involved in these preclinical studies and the subsequent clinical trials and have helped develop standards for ALS mouse studies being performed as a basis of early phase clinical trials [19, 20].
We initiated our studies by using a PC12 cell model of mutant SOD1 aggregation [21, 22]. These cells express wild-type or mutant SOD1 as a fusion with yellow fluorescent protein, making it possible to easily follow changes in the cellular distribution of SOD1 protein via epifluorescent microscopy as misfolding and aggregation commence. We employed these cells to develop a high throughput assay with which we screened a chemical library of >50,000 compounds. The screen identified compounds that blocked mutant SOD1 toxicity, and, in the vast majority of cases, also blocked mutant SOD1 aggregation. The ultimate goal is to develop a disease-modifying therapy to delay symptoms, slow progression, and permit recovery in those who have clinical illness.
MATERIALS AND METHODS
Cell culture
PC12 cells were maintained in DMEM supplemented with 10% heat-inactivated horse serum and 1% penicillin/streptomycin. PC12 Tet-Off SOD1-YFP stable lines were maintained in the same media containing an additional 100 μg/ml G418, 200 μg/ml hygromycin and 0.1 μg/ml doxycycline. Cells were grown at 37 °C in a 5% CO2-humidified atmosphere. The expression of SOD1-YFP fusion protein was induced by the withdrawal of doxycycline from the media 5 days prior to screening.
Mutant SOD1-induced cytotoxicity protection assay
PC12 cells expressing G93A SOD1-YFP were seeded at 15,000 cells/well in 96-well plates. Cells were incubated 24 h prior to compound addition. Compounds were screened as singletons at a final concentration of 5 μM. Two types of control wells were included on every plate: negative control wells containing DMSO, and positive control wells containing 700 nM radicicol or 5 μM CMB-003299. After 24 h incubation with the compounds, MG132 was added at a final concentration of 100 nM. MG132 is a well characterized proteasome inhibitor compound [23], which would be expected to enhance the appearance of protein aggregation by blocking the proteosomal clearance of aggregated proteins (see also the ‘mutant SOD1 aggregation assay: automated high content screening protocol’ below). Cell viability was measured 48 h later using the fluorescent viability probe, Calcein-AM (Molecular Probes). Briefly, cells were washed twice with PBS, Calcein-AM was added at a final concentration of 1 μM for 20 min at room temperature, and fluorescence intensity was read in a POLARstar fluorescence plate reader (BMG). Fluorescence data were coupled with compound structural data, then stored and analyzed using the CambridgeSoft Chemoffice Enterprise Ultra software package.
Follow up on hits
Compounds that restored cell viability to >60% were considered potential primary screening hits. All primary hits were re-tested in duplicate using the original screening format. Confirmed hits, as well as purchased analogs, were then assayed in six point dose response experiments. The highest compound concentration tested was 100 μM, which was decreased by one-third with each subsequent dose. To refine assay conditions, a twelve point dose response was used to determine the potency and efficacy of synthesized compounds. The highest compound concentration tested was 32 μM, which was decreased by one-half with each subsequent dose. Cell viability was assayed as described previously and dose response curves were fitted and EC50 values were determined using the CambridgeSoft BioAssay Enterprise software.
Mutant SOD1 aggregation assay: automated high content screening protocol
Actives from the protection screen were tested in the high content aggregation assay to determine their ability to block mutant SOD1 aggregation. G85R SOD1-YFP cells were plated at 5,000 cells/well in 96-well plates. Test compounds were added at 5 μM, 10 μM and 30 μM in duplicate. MG132 was then added at 1 μM final concentration and cells were grown for an additional 24 h. To assay protein aggregation, cell culture medium was removed and cells washed once with HBSS, then incubated in HBSS containing 5 μg/ml of the Image-iT WGA plasma membrane dye (Molecular Probes) for 15 min at 37 °C. Cells were then fixed in 4% paraformaldehyde in PBS, and imaged using a Cellomics Arrayscan® high throughput microscopy system. Arrayscan images were acquired using a 20x objective and the XF93 TRITC/FITC filter set (TRITC in channel 1 for the Image-iT WGA plasma membrane dye, and FITC in channel 2 for SOD1-YFP). Images were analyzed using Cellomics Spot Detector software. Software parameters were set to maximally differentiate cells from background and maximize recognition of SOD1 aggregates without recognizing background cytoplasmic staining as a spot. Data were recorded as spot count (aggregate)/object (cell).
RESULTS
SOD1 aggregate formation in PC12 cells and reversal by pharmacological treatment
Studies employed PC12 cells that express wild-type or mutant SOD1 (G93A or G85R) as a YFP fusion protein from a doxycycline-inducible promoter [21, 22]. The properties of these SOD1 mutant proteins in PC12 cells was extensively characterized in the prior studies, providing a strong base of information on the properties of the cell lines for their use as the basis of the development of screening assays. SOD1 is diffusely localized in PC12 cells that express wild-type SOD1 [22 and data not shown] (Figure 1). In contrast, G85R SOD1 shows heterogeneous patterns of localization; in most cells, G85R is diffusely localized throughout the cell. However in ~5% of the cells, G85R SOD1 is localized in large juxta-nuclear aggregates. Following inhibition of the proteasome by treatment with MG132, up to 75% of cells expressing G85R SOD1 contain such protein aggregates (Figure 1) while no aggregation is observed in MG132 treated cells expressing wild-type SOD1. Cells expressing G93A mutant SOD1, likewise, show an intermediate level of protein aggregation: none of the cells developed protein aggregates in the absence of MG132, and ~75% of the cells have protein aggregates following treatment with MG132 (Figure 1). Similar effects are observed in cells treated with bortezomib (Velcade®, data not shown), indicating that these effects are likely to be due to MG132-induced proteasome inhibition and not due to an off-target effect of MG132.
Figure 1. Mutant but not wild type SOD1 forms protein aggregates in cells treated with the proteasome inhibitor MG132.
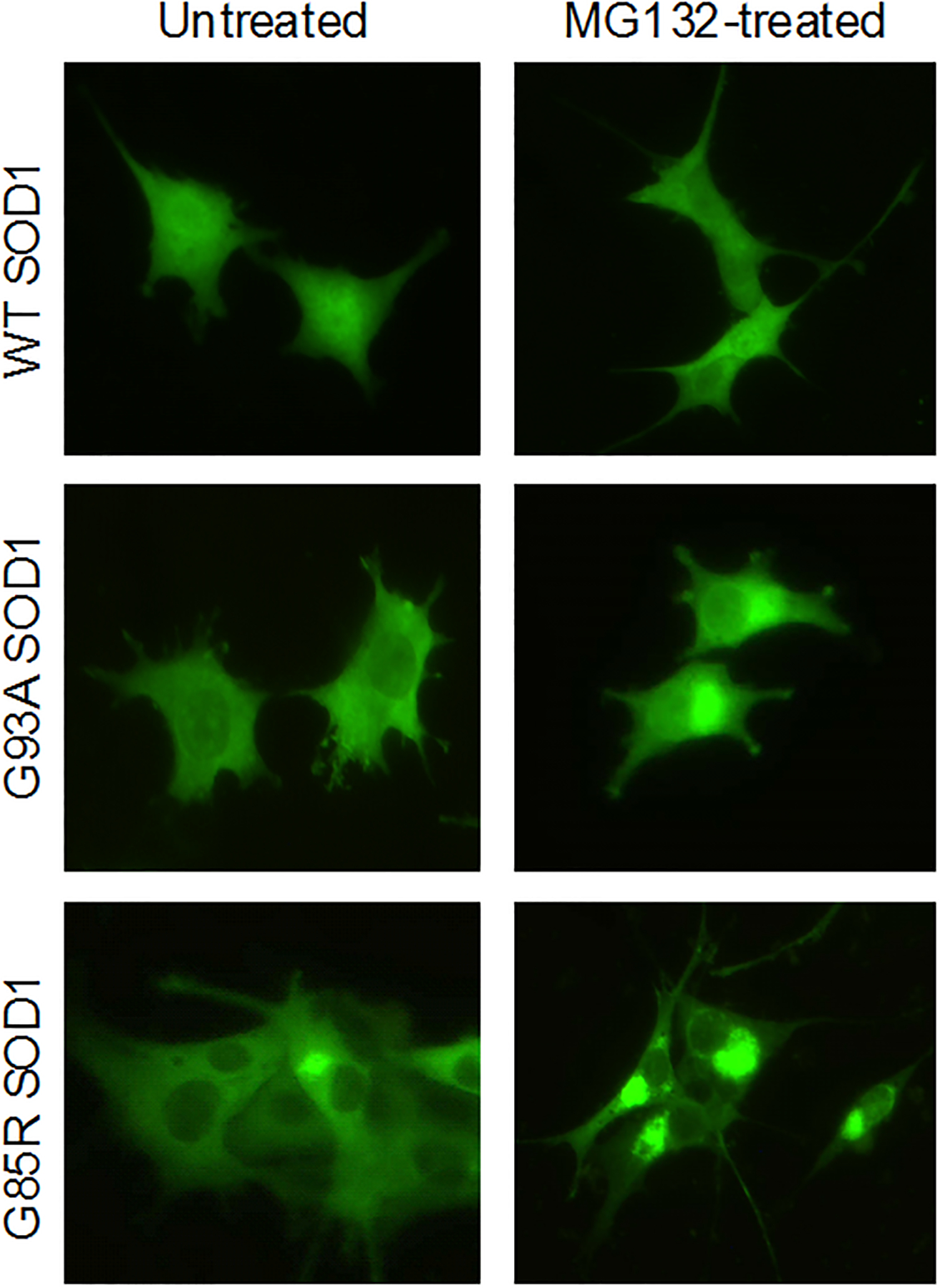
Fluorescence micrographs of PC12 cells expressing YFP tagged wild type (WT), G93A mutant (G93A) and G85R mutant (G85R) SOD1 proteins. The micrographs show the effect of treating cells with 200 nM MG132 for 24 h. The wild type SOD1 cells are unaffected while cells expressing mutant SOD1 show an elevated fraction of cells with large peri-nuclear aggregates.
To determine whether SOD1 aggregation in this model could be pharmacologically manipulated, G93A or G85R SOD1 cells were co-treated with MG132 and the HSP90 inhibitors geldanamycin or radicicol. These compounds induce heat shock transcription factor HSF-1, which in turn induces the heat shock response leading to the synthesis of chaperone proteins [24]. Therefore, this treatment is expected to reduce aggregation by protein refolding and/or degradation of misfolded proteins via the ubiquitin/proteasome pathway. As shown in Figure 2, treatment with radicicol reduced the proportion of protein aggregates by over 50%. Geldanamycin produced virtually identical effects (data not shown).
Figure 2. Radicicol decreases mutant SOD1 aggregation induced by the proteasome inhibitor MG132.

Fluorescence micrographs of PC12 cells expressing YFP tagged G93A SOD1 or G85R SOD1 proteins. The cells were untreated, treated for 24 h with 200 nM MG132 to induce protein aggregation, or co-treated with MG132 and radicicol. Without radicicol treatment, cells show large peri-nuclear aggregates. The aggregates are reduced in radicicol-treated cells. While the behavior of the two cell lines is generally similar, G85R SOD1 cells show ‘brighter’ aggregates and more contrast between the aggregates and the cytoplasm.
To automate this assay for use as a screening assay, the protocol was adapted for use with a Cellomics Arrayscan® high content microscopy system. Because the most robust high content assays measure events on a per cell basis, it was first necessary to identify a fluorescent stain to mark whole cells in a fashion that was compatible with YFP SOD1 fluorescence. An Image-iT conjugated wheat germ agglutinin (WGA) dye proved to be a useful marker (Figure 3–A). A computer algorithm using Cellomics Spot Detector software was developed, which allowed detection of changes in SOD1 aggregation produced by radicicol in a statistically significant fashion (Figure 3–B). Protein aggregates were identified in about 75% of cells treated with MG132 and in about 35% of cells treated with MG132 plus radicicol. Reliable high throughput screening requires a Z’ value > 0 [25]. The Z’ value in this assay was 0.12, indicating that the assay was capable of identifying compounds that perform as well or better than the positive control.
Figure 3. Automated detection of mutant SOD1 aggregates.

A. Compound microscope fluorescence micrographs of the same G85R SOD1 cells using GFP filter set to image the SOD1-YFP aggregates and the TRITC filter set to image the Image-iT WGA plasma membrane stain. B. Aggregate detection with the Cellomics Arrayscan 3.5 using spot detector software to image cells with Image-iT in channel 1 and SOD1-YFP aggregates in channel 2. Data are expressed as spot count (aggregates) per object (cell).
Assay development - G93A SOD1 aggregation is cytotoxic and inhibition of aggregation is cytoprotective
A series of protocol optimization experiments was conducted following the establishment of the above high content assay for SOD1 aggregation. In one such set of experiments, incubation times were varied to determine the effect of incubation time on protein aggregation. As shown in Figure 4, cells expressing G93A SOD1 were highly sensitive to MG132 toxicity after 48 hours of exposure to the compound. In quantitative terms, cells expressing no SOD1, wild-type SOD1, and G85R SOD1 were sensitive to MG132, with an IC50 of approximately 400 nM while cells expressing G93A SOD1 were approximately 5-fold more sensitive to MG132 (IC50 ~75 nM). We hypothesized that cell death resulted from the formation of a toxic aggregated form of G93A SOD1 and tested this idea in two experiments. In the first study, MG132 was removed at 24 hours, a time when the cells showed 100% viability. If the G93A SOD1 cells were simply MG132 sensitive, washing out the compound at this time should be protective. However, if MG132 toxicity in these cells resulted from the formation of a toxic entity within cells, cell viability should decrease despite the drug washout, which was observed as shown in Figure 4–C.
Figure 4. Proteasome inhibition is selectively toxic to PC12 cells expressing mutant G93A SOD1.

A. Treatment with 100 nM MG132 for 24 h shows no toxicity against any of the cell lines, although large numbers of aggregates are seen in the G85R SOD1 and G93A SOD1 cell lines. B. Treatment with 100 nM MG132 is selectively toxic to the G93A SOD1 cell line after 48 h. C. Washing of the cells at 24 h to remove compound does not reverse toxicity to the G93A SOD1 cell line suggesting that an irreversible toxic event, potentially related to the aggregation of G93A SOD1, has been triggered prior to wash out.
Secondly, if MG132 toxicity was due to protein aggregation, compounds that reduce aggregation should be cytoprotective. To test this idea, viability was measured in cells co-treated with MG132 and the HSP90 inhibitor radicicol. As shown in Figure 5, radicicol had a significant and dose related protective effect, consistent with the idea that toxicity was due to protein aggregation. We, therefore, decided to exploit this observation to develop an optimized cell viability protocol for high throughput screening and to screen a library of structurally diverse compounds for protective chemicals. Statistical analysis of the resulting protocol using radicicol as a positive control produced a Z’ value of 0.55, which predicted positive performance during assay implementation [25].
Figure 5. Radicicol protects PC12 cells expressing mutant G93A SOD1 from the toxic effects of the proteasome inhibitor MG132.

Qualitatively similar results were obtained with geldanamycin (not shown).
Lastly, this leaves the question that while both G85R and G93A SOD1 aggregate, only the G93A mutant produces cell toxicity under our experimental conditions. We hypothesize that the G93A cells might show selective sensitivity because of the production of toxic aggregated intermediate forms, which would not be produced by wild-type SOD1 and that they were less likely to be produced by the G85R mutation, which appears to localize much more exclusively in the juxta-nuclear aggregate (compare G93A and G85R in Figure 1). Consistent with this idea, it has recently been reported that disease duration in familial SOD1 ALS patients correlates with the aggregation propensity of the underlying mutation [26]. Thus, since different aggregation promoting alleles vary in their ability to accelerate disease, it might be reasonable to expect that alleles could show different toxicities in cell models.
Screening strategy, implementation, and recovery of actives
A library of approximately 50,000 small molecules, described in Table 1, was assembled for screening. The library was constructed to address two goals: maximization of chemical structural diversity and inclusion of biologically well-characterized compounds, such as FDA-approved and clinically-tested compounds, on the expectation that these latter compounds, if active, could be more rapidly moved into clinical testing.
Table 1.
Compound collections used for screening
| Name | Rationale | Collection and source | Number | Composition and Comments |
|---|---|---|---|---|
| Validation Set | Neuroactive compounds | CB145 – various chemical suppliers (145) | 145 | FDA approved & investigational compounds for use in screen characterization and validation |
| Test Set | Biological safety and tolerance in humans, otherwise well-studied compounds |
CB8000 – Spectrum Library (MSDS) LOPAC Library (Sigma-Aldrich) (3280)SPECS, MayBridge Hits Kit (4720) | 8,000: 1636 FDA approved, 1644 ‘biological reagents’, 4720 diverse chemistry | FDA-approved drugs plus biochemical reagents. The Spectrum Library is the commercial version of a set assembled for an NINDS drug screening consortium [35]. The approved drugs are supplemented with compounds with known biochemical, clinical, and pharmacological activity, many of which are known to be neuroactive. Structurally diverse compounds were selected for drug-like properties. |
| Production Set | Drug-like structurally diverse compounds |
CB42000 – Maybridge HitsKit ®, SPECS (17,000); Proprietary compounds in-licensed from large chemical companies (25,000) | 42,000 | Significant numbers of analogs available many have already been screened for therapeutic applications by others. Proprietary compounds selected in silico to have drug-like properties [36]. Will require chemical optimization. |
Implementation was performed using the cytotoxicity protection assay as the primary screen and active compounds were then tested in the protein aggregation assay. Our rationale for carrying out screening operations in this way was based on the strategy that 1) identifying compounds that protect cells from an established disease relevant insult would arguably be more pertinent to clinical therapy development than simply screening for compounds on the basis of altering a cellular phenotype, 2) it could be possible to find compounds that increased viability in cells carrying juxta-nuclear aggregates, as these large aggregates have been argued in the literature to potentially be protective rather than toxic and 3) this screening approach would be less duplicative of prior studies in which compound effects on aggregation were scored directly [27]. Operationally, the primary screening ran well producing an average Z’ factor of 0.5 and an average Z factor of 0.6. Using 60% viability as the cutoff for activity, 68 confirmed actives were recovered (0.13% confirmed hit rate). These actives were then tested for auto-fluorescence and inhibition of protein synthesis to eliminate artifactual positives (see below). Following testing in the high content protein aggregation screen, confirmed primary actives were grouped into compounds that were positive or negative in affecting protein aggregation. The vast majority of compounds blocked or reversed protein aggregation. This compound collection included riluzole, which tested negative, indicating that our actives differed in mechanism from the sole clinically approved drug for ALS.
Counterscreens to eliminate fluorescence artifacts, protein synthesis inhibitors and highly cytotoxic compounds
Assays for autofluorescence identified only one compound with significant autofluorescence, a low artifact rate that might be due to the use of Calcein-AM to measure cell viability. Calcein-AM is a high quantum yield reagent, and, thus, only highly fluorescent compounds would produce a comparable signal. The ability of compounds to produce protective effects by reducing the cellular concentration of SOD1 was estimated using a modification of the high content assay, in which YFP fluorescence was measured with a plate reader prior to counting the number of cells per well with ArrayScan. This yielded a value for YFP fluorescence per cell, which is approximately equivalent to the SOD1 content per cell. No compounds tested active in this assay. Lastly, all active compounds were tested for nonspecific cytotoxicity in untransfected PC12 cells. The vast majority of the active compounds lacked or had extremely low non-specific cytotoxicity. One possible explanation for the low level of cytotoxicity associated with active compounds is that the primary protection screen selected for cytoprotection and thus potentially against toxic compounds.
Chemistry Analysis
The screening library was analyzed to improve on the selection of active compounds using a ligand-based computational approach including substructure and similarity searching and clustering techniques. Iterative statistical clustering of HTS actives has been shown to improve selection and prioritization of chemical series. These chemoinformatic methodologies were used to aid in the identification and validation of the chemical series. Confirmed active compounds from the screen were used to identify structurally related compounds in our larger screening library.
In addition, structural analogs were selected from an extensive search of over two million unique available commercial compounds based upon a combination of chemical substructure or similarity searching. The structure activity relationship (SAR) was developed by testing 40 to 100 structural analogs per series. Each structurally similar group of compounds, or chemical clusters, was used as a guide for prioritizing hits. Our analysis identified 17 distinct chemical scaffolds or chemotypes containing structurally-related active and inactive compounds plus 15 singleton hits. Compounds or series are de-prioritized or deselected from review when structurally similar compounds were inactive, a low number of active analogs were found, toxicity liabilities were identified, no preliminary SAR could be established, or based upon a medicinal chemistry review. All active compounds were reordered and re-tested from dry powder to confirm their activities. Compounds were determined to be > 90% pure as determined by LC/MS or 1H NMR spectrometry. At the end of this iterative process three distinct and validated chemical series were selected for further work. Figure 6 shows representative structures for each of these chemical scaffolds.
Figure 6.

Representative chemical structures: (1) Arylsulfanyl pyrazolones – CMB-003299, (2) cyclohexane-1,3-diones – CMB-050378, and (3) pyrimidine-2,4,6-triones – CMB-052802.
Arylsulfanyl pyrazolones (ASP)
The most potent arylsulfanyl pyrazolones (ASP) show 100% efficacy as compared with the positive control, radicicol, which showed only 80% efficacy at the optimum dose. The most potent compounds produced ED50 values between 400 nM and 15 μM in the cytotoxicity protection assay. One of the most potent compounds in the series was CMB-003299 (Figure 6–1), which produced an ED50 = 400 nM. Compounds that were active in the protection assay were active in the protein aggregation assay with lower, but similar relative potency. While the aggregation assay is subject to high experimental variability, it is possible to implement this assay in a dose response format in order to estimate compound potency. CMB-003299 produced an ED50 of ~9 μM when tested in this way and compounds from the other chemical scaffolds showed similarly lower aggregation assay potency when compared to their potency in the cytotoxicity protection assay (see below). Despite the potency differential, it is reasonable to assume that these compounds protect, at least in part, by reducing mutant SOD1 aggregation. For example, a partial reduction in protein aggregation may be adequate to restore cell viability, yet not be scored as positive in the high content protein aggregation assay. Alternately, the observed compound potency differences between the two assays may indicate that compound effects on protein aggregation are secondary to another upstream activity. These compounds are only very weakly cytotoxic or not generally considered to be cytotoxic (LD50 ≥ 100 μM).
Cyclohexane-1,3-dione (CHD)
The cyclohexane-1,3-dione (CHD) chemical series produced biological effects similar to the arylsulfanyl pyrazolones: these compounds are efficacious with good correlation between activity in the protection and protein aggregation assays and lack of cytotoxicity. One of the most potent compounds from the CHD series was CMB-050378 (Figure 6–2), which produced an ED50 = 0.8 μM. CMB-050378 was active when tested in the protein aggregation assay and produced an ED50 of ~10 μM. While not generally as potent as the ASP compounds, the predicted potential pharmacological properties appeared promising, thus providing a rationale for further work (data not shown).
Pyrimidine 2,4,6-trione (PYT)
One of the more interesting compounds in the PYT series was CMB-052802 (Figure 6–3), which produced an ED50 = 6.0 μM. Compounds from this series show 100% efficacy in the protection assay with a good correlation in activity between the protection and protein aggregation assays. For example, CMB-052802 produced an ED50 of ~32 μM when tested in the protein aggregation assay. Thus, it is reasonable to assume that these compounds protect PC12 cells against mutant SOD1-induced cytotoxicity, at least in part, by reducing SOD1 protein aggregation. These compounds are not cytotoxic. The chemical optimization of each of these series will be discussed in future reports.
DISCUSSION
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive and fatal neurodegenerative disorder for which there is no effective treatment. Identifying new therapeutic agents depends in large part on understanding the molecular mechanism(s) that produce the disease. Work on therapies for diseases in which disease mechanism is not, or only partially, defined requires proceeding on the best, although inconclusive evidence. Possible targets for ALS therapy include glutamate excitotoxicity, impaired mitochondrial function, oxidative stress, defective RNA processing/transport, and aberrant protein aggregation [20]. We have found aberrant protein aggregation to be one of the more compelling of these targets in part because: 1) aberrant protein aggregation is a feature of virtually all neurodegenerative diseases and 2) recent findings provide a link between the aggregated protein seen in patients with the sporadic form of the disease and disease causality [12–16]. Sporadic ALS patients have aggregates of the protein TDP-43 [12, 13, 15] and the identification of familial ALS TDP-43 mutations strongly suggests that TDP-43 aggregation may be a cause rather than a result of the disease [14, 16, 28]. We therefore developed a screening system to identify compounds that would protect cells against the toxic effects of aggregated mutant SOD1 protein to develop clinical candidate compounds appropriate for entry into GMP and GLP IND enabling studies.
We developed a novel assay protocol in which treating PC12 expressing G93A SOD1 (but not wild type SOD1) with the proteasome inhibitor MG132 leads to cell death within 48 hours. MG132 treatment of these G93A SOD1 cells (and not wild type SOD1 cells) produced prominent juxta-nuclear SOD1 aggregates, consistent with protein aggregation causing, or at least contributing to, cell death. To test this idea, we co-treated the cells with geldanamycin or radicicol, HSP90 inhibitors that significantly induce chaperone protein synthesis, and which would be expected to decrease the formation of protein aggregates. As anticipated, treatment with an HSP90 inhibitor blocked the formation of the SOD1 aggregates, and in addition protected against cell death, consistent with the idea that cell death was a consequence of protein aggregation.
Cell lines expressing mutant SOD1 as fluorescent protein fusions were created to provide the basis for high throughput screens (HTS), and two assays (high content and protection) were ultimately developed using these cell lines [21, 22]. We optimized this protocol for high throughput screening and, using radicicol as a positive control, screened a >50,000 member compound library composed of structurally diverse chemistry and a small group of FDA approved drugs and biochemical reagents. From this screen we recovered 68 actives which were characterized in a series of secondary counter-screens: cytotoxicity (to eliminate toxic compounds), protein synthesis inhibition (to eliminate compounds acting through decreasing G93A SOD1 levels), fluorescence (to eliminate compounds which mimicked the Calcein-AM cell viability reagent used in the assay) and protein aggregation (to identify compounds which blocked protein aggregation and were thus likely to produce cell protection by decreasing protein aggregation). The protection assay ran well as a HTS and yielded a manageable hit rate (0.13%) with good screening statistics. Commercially available analogs of the remaining actives were purchased and these compounds plus the initial hits were tested in the screening assay in a dose response format to 1) identify the most potent and efficacious actives and 2) to determine whether the chemical substitution patterns of the active compounds suggested an early rational structure activity relationship (SAR) for the scaffold. Lastly, the hit compounds were subjected to chemistry review to identify chemical scaffolds with potential for convenient synthetic modification and which had ‘drug like’ structures and physical properties. The fact that the active compounds identified by the screen fell into structurally related classes provided support for the idea that the assay was selective for specific types of compounds. Also supportive of this idea, the vast majority of the actives were also active in the high content aggregation assay, an assay with high selectivity in its own right. This result demonstrates that the protection screen was identifying a rare subset of compounds having biological properties consistent with our initial screening hypothesis.
Corcoran and co-workers [27] had previously developed a screen similar to the above protein aggregation high content assay, used it to screen a 20,000 compound chemical file, and identified cardiac glycosides and histone deacetylase (HDAC) inhibitors as actives. We anticipated that we could potentially identify additional novel actives in our study by 1) implementing a cytotoxicity protection assay as the primary screen to potentially find protective compounds that produce minor effects on protein aggregation and 2) by screening a different and larger chemical file. It also seems reasonable that some compounds might be protective without anti-aggregation activity and thus only be identified as hits via this experimental design.
Chemistry review resulted in the selection of three scaffolds for optimization: arylsulfanyl pyrazolone (ASP), pyrimidine-2,4,6-trione (PYT) and cyclohexane-1,3-dione (CHD). On the basis of these findings, efforts are now ongoing to further optimize all three chemical scaffolds so as to produce candidate compounds for evaluation in ALS mouse models. Analog syntheses in these studies are being guided by the protection screen applied in conjunction with a panel of in vitro assays to guide the synthesis of analogs with good pharmacological properties for rodent studies. A critical intermediate goal is to test analogs in G93A SOD1 mice, as results from studies in ALS animal models have been a highly important factor for the selection of compounds to be progressed into human clinical trials [17, 20, 29, 30].
Two recent publications on mice strains carrying mutant SOD1 transgenes are of particular relevance to the development of compounds affecting protein aggregation [31, 32]. These studies demonstrated that larger SOD1 aggregates only become prominent at later stages of disease in these mice and suggest that soluble forms of SOD1 may initiate disease with the larger aggregates becoming a factor only in later disease stages. If this is reflective of the course of human familial or sporadic ALS, anti-protein aggregation therapeutics might have little efficacy during early disease stages. However, at this point we do not know the exact target of our leads and there is some suggestive evidence that compound effects on protein aggregation could be secondary to another activity. We also do not know whether these compounds affect intermediate aggregate forms of SOD1, as these forms are not scored in our assay. In any event, our planned studies to evaluate optimized analogs of our leads in G93A SOD1 mice should shed light on this issue and provide critical information on the value of pursuing this approach. There is, in addition, a general ongoing controversy in the literature regarding the predictive value of the G93A SOD1 mouse model for compound efficacy in the clinic. Two studies have recently appeared on the development of TDP-43 transgenic mice [33, 34], providing a potential alternate mouse model for drug testing and with potentially increased relevance to sporadic ALS.
The results of our ongoing investigations will be reported separately. The findings reported here provide encouragement in the hit optimization and chemoinformatic paradigms to pursue the optimization of these chemical leads with the goal of accelerating the pace to translate drugs to clinical trials in ALS patients.
ACKNOWLEGEMENTS
This work was supported by grants from the National Institutes of Health [1R43NS057849], the ALS Association (TREAT program), Department of Defense [AL093052] and the Veterans Administration at the Edith Nourse Rogers Memorial Veterans Hospital, Bedford, MA.
Footnotes
Disclosure of Interests: The authors have had no involvements that might raise the question of bias in the work reported or in the conclusions, implications or opinions stated.
REFERENCES
- 1.Brown RWJ, The Motor Neuron Diseases, in Harrison’s Principles of Internal Medicine, Fauchi AS, et al. , Editors. 1998, McGraw-Hill: New York. p. 2368–2371. [Google Scholar]
- 2.Bensimon G, Lacomblez L, and Meininger V, A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med, 1994. 330(9): p. 585–91. [DOI] [PubMed] [Google Scholar]
- 3.Francis K, Bach JR, and DeLisa JA, Evaluation and rehabilitation of patients with adult motor neuron disease. Arch Phys Med Rehabil, 1999. 80(8): p. 951–63. [DOI] [PubMed] [Google Scholar]
- 4.Miller RG, Mitchell JD, Lyon M, and Moore DH, Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Amyotroph Lateral Scler Other Motor Neuron Disord, 2003. 4(3): p. 191–206. [PubMed] [Google Scholar]
- 5.Bruijn LI, Miller TM, and Cleveland DW, Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci, 2004. 27: p. 723–49. [DOI] [PubMed] [Google Scholar]
- 6.Kwiatkowski TJ Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. , Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science, 2009. 323(5918): p. 1205–8. [DOI] [PubMed] [Google Scholar]
- 7.Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. , Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science, 2009. 323(5918): p. 1208–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, et al. , Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem, 2005. 93(5): p. 1087–98. [DOI] [PubMed] [Google Scholar]
- 9.Rothstein JD, Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol, 2009. 65 Suppl 1: p. S3–9. [DOI] [PubMed] [Google Scholar]
- 10.Brown RH Jr. and Robberecht W, Amyotrophic lateral sclerosis: pathogenesis. Semin Neurol, 2001. 21(2): p. 131–9. [DOI] [PubMed] [Google Scholar]
- 11.Gaudette M, Hirano M, and Siddique T, Current status of SOD1 mutations in familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord, 2000. 1(2): p. 83–9. [DOI] [PubMed] [Google Scholar]
- 12.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. , TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun, 2006. 351(3): p. 602–11. [DOI] [PubMed] [Google Scholar]
- 13.Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, et al. , Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol, 2007. 61(5): p. 427–34. [DOI] [PubMed] [Google Scholar]
- 14.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. , TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science, 2008. 319(5870): p. 1668–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strong MJ, Volkening K, Hammond R, Yang W, Strong W, Leystra-Lantz C, et al. , TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci, 2007. 35(2): p. 320–7. [DOI] [PubMed] [Google Scholar]
- 16.Yokoseki A, Shiga A, Tan CF, Tagawa A, Kaneko H, Koyama A, et al. , TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol, 2008. 63(4): p. 538–42. [DOI] [PubMed] [Google Scholar]
- 17.Traynor BJ, Bruijn L, Conwit R, Beal F, O’Neill G, Fagan SC, et al. , Neuroprotective agents for clinical trials in ALS: a systematic assessment. Neurology, 2006. 67(1): p. 20–7. [DOI] [PubMed] [Google Scholar]
- 18.Wong PC, Cai H, Borchelt DR, and Price DL, Genetically engineered mouse models of neurodegenerative diseases. Nat Neurosci, 2002. 5(7): p. 633–9. [DOI] [PubMed] [Google Scholar]
- 19.Cudkowicz ME, Andres PL, Macdonald SA, Bedlack RS, Choudry R, Brown RH Jr., et al. , Phase 2 study of sodium phenylbutyrate in ALS. Amyotroph Lateral Scler, 2009. 10(2): p. 99–106. [DOI] [PubMed] [Google Scholar]
- 20.Ryu H and Ferrante RJ, Translational therapeutic strategies in amyotrophic lateral sclerosis. Mini Rev Med Chem, 2007. 7(2): p. 141–50. [DOI] [PubMed] [Google Scholar]
- 21.Matsumoto G, Kim S, and Morimoto RI, Huntingtin and mutant SOD1 form aggregate structures with distinct molecular properties in human cells. J Biol Chem, 2006. 281(7): p. 4477–85. [DOI] [PubMed] [Google Scholar]
- 22.Matsumoto G, Stojanovic A, Holmberg CI, Kim S, and Morimoto RI, Structural properties and neuronal toxicity of amyotrophic lateral sclerosis-associated Cu/Zn superoxide dismutase 1 aggregates. J Cell Biol, 2005. 171(1): p. 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee DH and Goldberg AL, Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol, 1998. 8(10): p. 397–403. [DOI] [PubMed] [Google Scholar]
- 24.Morimoto RI, Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev, 2008. 22(11): p. 1427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang JH, Chung TD, and Oldenburg KR, A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen, 1999. 4(2): p. 67–73. [DOI] [PubMed] [Google Scholar]
- 26.Prudencio M, Hart PJ, Borchelt DR, and Andersen PM, Variation in aggregation propensities among ALS-associated variants of SOD1: correlation to human disease. Hum Mol Genet, 2009. 18(17): p. 3217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corcoran LJ, Mitchison TJ, and Liu Q, A novel action of histone deacetylase inhibitors in a protein aggresome disease model. Curr Biol, 2004. 14(6): p. 488–92. [DOI] [PubMed] [Google Scholar]
- 28.Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, et al. , TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet, 2008. 40(5): p. 572–4. [DOI] [PubMed] [Google Scholar]
- 29.Benatar M, Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol Dis, 2007. 26(1): p. 1–13. [DOI] [PubMed] [Google Scholar]
- 30.Scott S, Kranz JE, Cole J, Lincecum JM, Thompson K, Kelly N, et al. , Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler, 2008. 9(1): p. 4–15. [DOI] [PubMed] [Google Scholar]
- 31.Karch CM, Prudencio M, Winkler DD, Hart PJ, and Borchelt DR, Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proc Natl Acad Sci U S A, 2009. 106(19): p. 7774–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Farr GW, Zeiss CJ, Rodriguez-Gil DJ, Wilson JH, Furtak K, et al. , Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc Natl Acad Sci U S A, 2009. 106(5): p. 1392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wegorzewska I, Bell S, Cairns NJ, Miller TM, and Baloh RH, TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A, 2009. 106(44): p. 18809–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, et al. , TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 107(8): p. 3858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heemskerk J, Tobin AJ, and Bain LJ, Teaching old drugs new tricks. Meeting of the Neurodegeneration Drug Screening Consortium, 7–8 April 2002, Washington, DC, USA. Trends Neurosci, 2002. 25(10): p. 494–6. [DOI] [PubMed] [Google Scholar]
- 36.Lipinski CA, Lombardo F, Dominy BW, and Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 1997. 23(1): p. 3–25. [DOI] [PubMed] [Google Scholar]