

Abstract
BACKGROUND:
Neonatal mouse cardiomyocytes undergo a metabolic switch from glycolysis to oxidative phosphorylation, which results in a significant increase in reactive oxygen species (ROS) production that induces DNA damage. These cellular changes contribute to cardiomyocyte cell cycle exit and loss of the capacity for cardiac regeneration. The mechanisms that regulate this metabolic switch and the increase in ROS production have been relatively unexplored. Current evidence suggests that elevated ROS production in ischemic tissues occurs due to accumulation of the mitochondrial metabolite succinate during ischemia via succinate dehydrogenase (SDH), and this succinate is rapidly oxidized at reperfusion. Interestingly, mutations in SDH in familial cancer syndromes have been demonstrated to promote a metabolic shift into glycolytic metabolism, suggesting a potential role for SDH in regulating cellular metabolism. Whether succinate and SDH regulate cardiomyocyte cell cycle activity and the cardiac metabolic state remains unclear.
METHODS:
Here, we investigated the role of succinate and succinate dehydrogenase (SDH) inhibition in regulation of postnatal cardiomyocyte cell cycle activity and heart regeneration.
RESULTS:
Our results demonstrate that injection of succinate in neonatal mice results in inhibition of cardiomyocyte proliferation and regeneration. Our evidence also shows that inhibition of SDH by malonate treatment after birth extends the window of cardiomyocyte proliferation and regeneration in juvenile mice. Remarkably, extending malonate treatment to the adult mouse heart following myocardial infarction injury results in a robust regenerative response within 4 weeks following injury via promoting adult cardiomyocyte proliferation and revascularization. Our metabolite analysis following SDH inhibition by malonate induces dynamic changes in adult cardiac metabolism.
CONCLUSIONS:
Inhibition of SDH by malonate promotes adult cardiomyocyte proliferation, revascularization, and heart regeneration via metabolic reprogramming. These findings support a potentially important new therapeutic approach for human heart failure.
Keywords: heart regeneration, myocardial infarction, succinate dehydrogenase, metabolism, cell cycle
INTRODUCTION
Cardiovascular disease remains the leading cause of death in the world1. Both vascular and myocardial damage arise from acute cardiovascular events such as myocardial infarction (MI). The limited capacity of the adult heart to repair itself represents a major barrier in cardiovascular medicine and often leads to heart failure. In contrast, the neonatal mouse heart has the ability to regenerate following MI, with the newly formed cardiac tissue being derived primarily from the proliferation of the pre-existing cardiomyocytes2–4. During postnatal development, exposure to high levels of atmospheric oxygen following birth results in a metabolic switch in energy utilization from glycolysis to oxidative phosphorylation5. This metabolic switch results in increased mitochondrial reactive oxygen species (ROS) production, causing cardiomyocyte DNA damage and contributing to the postnatal cardiomyocyte cell cycle arrest in mice6. Thus, understanding the metabolic state of the mammalian heart following birth can lead to important insights towards restoring adult cardiomyocyte cell cycle activity and subsequent regenerative abilities following injury.
Recent studies have demonstrated that the metabolite succinate accumulates during ischemia, which is a conserved phenomenon across vertebrates7–9. Different models suggest that succinate accumulation occurs either through reverse activity of the enzyme complex succinate dehydrogenase (SDH, also known as complex II), or via canonical tricarboxylic acid (TCA) cycle7, 9. SDH activity plays a central role in succinate accumulation in the proposed models owing to its involvement in both the TCA cycle and the electron transport chain (ETC)10. Subsequently upon reperfusion, the high levels of accumulated succinate are rapidly metabolized into fumarate, which results in a burst of ROS production via reverse activity of mitochondrial complex I7, 11. More importantly, administration of the SDH competitive inhibitor, malonate, prevents the accumulation of succinate and the subsequent metabolization that increases ROS levels during ischemia/reperfusion injury, emphasizing the link between SDH and ROS production7, 11, 12.
SDH plays an important role in metabolism and cell cycle activity, as it is the first mitochondrial protein to be identified as a tumor suppressor13. Mutations in SDH in familial cancer syndromes promote a metabolic shift into glycolysis that drives cell division13–15. Interestingly, metabolic reprogramming to glycolysis is essential during zebrafish heart regeneration, which is concomitant with a significant reduction in SDH activity as well16. However, whether succinate and SDH activity directly contribute to the limited regenerative capability of the heart after injury is unknown. In this study, we aim to determine the role of succinate and SDH in regulation of postnatal cardiomyocyte cell cycle activity and heart regeneration.
METHODS
Detailed methods are available in the Data Supplement. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
CD-1 mice were obtained from Charles River Laboratories. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Wisconsin-Madison. All experiments were performed on age and sex matched mice, with an equal ratio of male to female mice for neonatal experiments and only male mice for adult experiments.
Neonatal Myocardial Infarction
Neonatal mice at postnatal day 1 (P1) or day 7 (P7) were used for myocardial infarction (MI) surgery. Neonatal mice were subjected to MI surgeries as previously described2. Briefly, neonates were anesthetized by hypothermia on ice. Lateral thoracotomy at the fourth intercostal space was performed by blunt dissection of the intercostal muscles after skin incision. A C-1 tapered needle attached to a 6–0 prolene suture (Ethicon Inc., Bridgewater, NJ) was passed through the mid-ventricle below the left anterior descending coronary artery (LAD) and tied off to induce MI. The prolene suture was used to suture the ribs together and seal the chest wall incisions, and the skin was closed using adhesive glue (3M). The mice then were warmed on a heating pad until recovery. Sham-operated mice underwent the same procedure including hypothermic anesthesia, but not LAD ligation.
Adult Myocardial Infarction
Adult male mice (8-week-old) were subjected to MI by ligation of the proximal aspect of the LAD coronary artery. In brief, mice were anaesthetized using 3% isoflurane, then mice were intubated with PE50 tubing and placed on a mouse ventilator at 120–130 breaths per minute with a stroke volume of 150 microliters and maintained on 2% isoflurane. A left lateral incision through the fourth intercostal space was made to expose the heart. After visualizing the left coronary artery, 7–0 suture was placed through the myocardium in the anterolateral wall and secured as previously described17, 18. Coronary artery entrapment was confirmed by observing blanching of the distal circulation (ventricular apex). ECG was used to confirm MI by noting ST segment changes. The lungs were over inflated, and the ribs and muscle layers were closed by absorbable sutures. The skin is closed by additional suturing using 6–0 nylon. The mouse was recovered from the anesthesia and extubated.
Drug Administration
Neonatal mice were weighed and injected daily with either saline, 100 mg/kg dimethyl succinate (Sigma), 100 mg/kg dimethyl malonate (Sigma), or 100 μg/kg Atpenin A5 (Enzo Life Sciences). Dimethyl succinate and dimethyl malonate were dissolved in saline. Stock solution of Atpenin A5 at 0.1mg/ml was initially prepared by dissolving in DMSO (Sigma) and then further diluted with saline before injection. Saline was used as a vehicle control for all experiments. Neonatal mice were given intravenous injections for the first 5 days after birth, followed by intraperitoneal injections until completion of the injection time course. Adult mice were injected intraperitoneally daily with either saline or 100 mg/kg dimethyl malonate post-MI for 2 or 4 weeks. To track cardiomyocyte proliferation, we added 0.25 mg/ml 5-bromodeoxyuridine (BrdU, Sigma) to the drinking water for 2 weeks post-MI. Fresh BrdU-containing water was changed every 2 days.
Histology
Hearts were harvested and fixed in 4% paraformaldehyde (PFA) in PBS solution overnight at 4 °C, processed for paraffin embedding, and sectioned in intervals. Masson’s trichrome staining was performed according to the manufacture’s protocol (Newcomer Supply, Middleton, WI). Scar size measurements were quantified from at least 3 sections of the heart from ligature to apex. ImageJ was used to quantify the fibrotic scar, and the average scar area for each heart was plotted.
Metabolite analysis
Metabolites were extracted with cold liquid chromatography–mass spectrometry (LC–MS) grade 3/1 butanol/methanol (v/v) following previously established method19. Tissue extracts containing polar metabolites were dried under nitrogen flow and subsequently dissolved in LC–MS grade water for analysis. Protein pellets were removed by centrifugation. Samples were analyzed using a Thermo Q-Exactive mass spectrometer coupled to a Vanquish Horizon Ultra-High Performance Liquid Chromatograph (UHPLC). Metabolites were separated on a 2.1 × 100 mm, 1.7 μM Acquity UPLC BEH C18 Column (Waters) at a 0.2 ml per min flow rate and 30 °C column temperature, with a gradient of solvent A (97/3 H2O/methanol, 10 mM TBA, 9 mM acetate, pH 8.2) and solvent B (100% methanol). The gradient was: 0 min, 5% B; 2.5 min, 5% B; 17 min, 95% B; 21 min, 95% B; 21.5 min, 5% B. Data were collected on a full scan negative mode. The identification of metabolites reported was based on exact m/z and retention times, which were determined with chemical standards. Data were analyzed with Maven. Relative metabolite levels were normalized to internal standard Tryptophan (13C11) and expressed relative to levels measured in the control group.
Echocardiography
Transthoracic echocardiography was performed by using a Visual Sonics 770 ultrasonograph with a 25-MHz transducer (Visual Sonics, Toronto) as described previously20. Mice were lightly anesthetized with 1% isoflurane and maintained on a heated platform. Two-dimensionally guided M-mode images from a parasternal long axis (PLAX) of the LV were acquired at the tip of the papillary muscles. Wall thickness and chamber diameters were measured in both diastole and systole. Fractional shortening was calculated as (LVDd-LVDs)/LVDd × 100, where LVDd is LV diameter in diastole and LVDs is LV diameter in systole. Ejection fraction was calculated as [(7.0/(2.4 + LVDd)(LVDd)3 − (7.0/(2.4 + LVDs)(LVDs)3/(7.0/(2.4 + LVDd)(LVDd)3 × 100 and LV mass was calculated by using the formula [1.05 × ((Posterior Wall diastole+Anterior Walldiastole+LVDd)3 – (LVDd)3)]. All parameters were measured over at least three consecutive cycles.
Statistical Analysis
All graphs are presented as means ± SE. Statistical analysis was performed using Prism 9 (GraphPad Software). Two-tailed unpaired Student’s t-test was performed to determine the difference between the treatment group and control group. One-way ANOVA was performed by Tukey’s multiple comparison test to determine the differences of group mean among treatment groups. Two-way ANOVA with Bonferroni’s post-hoc test was used to determine the difference among the treatment groups and different time points. The level of significance was set at P < 0.05.
RESULTS
Succinate Reduces Cardiomyocyte Proliferation and Heart Regeneration in Neonatal Mice
To determine whether an increase in succinate levels impacts cardiomyocyte cell cycle activity and neonatal mouse heart regeneration, neonatal mice were injected with dimethyl succinate (100 mg/kg) daily for 7 days following myocardial infarction at postnatal day1 (P1) (Figure 1A). We tested multiple doses (50, 100, 150 mg/kg) and we found that 100 mg/kg was the minimum effective dose that reduces cardiomyocyte proliferation (data not shown). To determine whether succinate reduces cardiomyocyte proliferation, we performed immunostaining of the mitosis marker pH3 at 7 days post-MI. Our results revealed a reduction in cardiomyocyte mitosis in succinate injected MI hearts compared to controls (Figure 1B & C).
Figure 1. Succinate reduces cardiomyocyte proliferation and blocks heart regeneration in neonatal mice following myocardial infarction (MI).
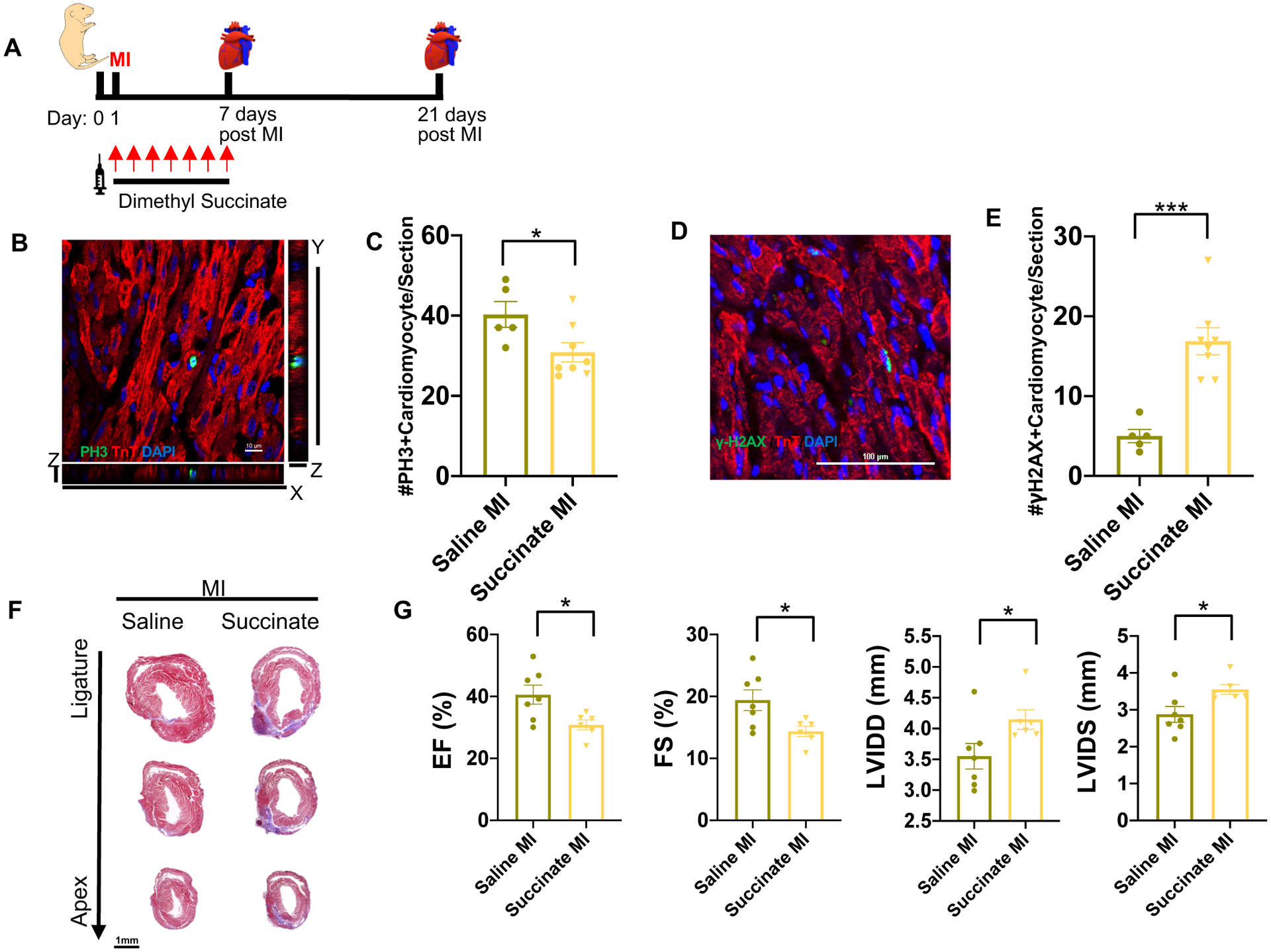
(A) Schematic of injection period and myocardial infarction strategy in neonatal mice. (B) High magnification Z-stack image of a mitotic cardiomyocyte following immunostaining of pH3 and cTnT at 7 days post-MI. Scale bar, 10 μm. (C) Quantification of the number of mitotic cardiomyocytes per section showing a significant decrease in cardiomyocyte mitosis following dimethyl succinate injection. (D) Immunostaining of the DNA double-strand breaks marker γH2AX. Scale bar, 100 μm. (E) Quantification of cardiomyocytes with increased γH2AX foci demonstrating a significant increase in DNA damage in succinate-treated mice compared to controls. (F) Trichrome staining demonstrating persistence of the fibrotic scar following MI in the dimethyl succinate-injected mice compared to saline-injected controls. Scale bar, 1 mm. (G) Echocardiography at 21 days post-MI showing a significant reduction in the cardiac function of dimethyl succinate-injected mice following MI compared to saline-injected controls as measured by ejection fraction (EF), fractional shortening (FS), left ventricle internal diameter diastole (LVIDD) and left ventricle internal diameter systole (LVIDS). (n=5–8 mice per group). *P< 0.05, ***P<0.0001 by two-tailed unpaired Student’s t-test.
Loss of cardiomyocyte cell cycle activity occurs due to increase in cardiomyocyte DNA damage as a consequence of the metabolic switch to oxidative phosphorylation and the subsequent rise in ROS levels. To determine whether succinate induces cardiomyocyte DNA damage, we performed immunostaining of γH2AX, which is a marker for DNA double-strand breaks. We quantified a significant increase in cardiomyocytes with γH2AX foci in succinate-treated mice (Figure 1D & E). Our results demonstrate that in neonatal mice high levels of succinate can induce cardiomyocyte DNA damage and reduce the proliferative potential of pre-existing cardiomyocytes, which is the main source of the newly formed cardiomyocytes during cardiac regeneration3, 21.
To further establish the effects of succinate on neonatal heart regeneration, we performed trichrome staining at 21 days post-MI to assess regeneration and fibrosis in control and dimethyl succinate injected mice. As expected, saline-injected control mice demonstrated complete heart regeneration. In contrast, dimethyl succinate-injected mice showed lack of regeneration with persistence of a fibrotic scar (Figure 1F). This lack of regeneration was also evident in the significant reduction in cardiac function of dimethyl succinate-injected mice compared to saline-injected controls, as measured by ejection fraction (EF), fractional shortening (FS), left ventricle internal diameter diastole (LVIDD), and left ventricle internal diameter systole (LVIDS) (Figure 1G & Table I in the Data Supplement). Together, these results reveal that succinate injection during the first week of life can result in premature cardiomyocyte cell cycle exit, which inhibits the neonatal cardiac regenerative response.
Malonate Extends the Cardiac Regenerative Window in Postnatal Hearts
Although our results demonstrate that exogenous administration of succinate can inhibit cardiomyocyte proliferation and regeneration, it remains unclear whether succinate dehydrogenase (SDH) activity contributes to cardiomyocyte cell cycle exit in the postnatal heart. Thus, we wanted to determine whether the SDH complex competitive inhibitor, malonate, could extend the cardiomyocyte proliferative window and improve the cardiac regeneration capacity of the juvenile, normally non-regenerative 7-day-old mice. We injected dimethyl malonate (100 mg/kg, daily) in neonatal mice directly after birth for 2 weeks with an MI performed once the mice reached P7. We then analyzed the hearts following injury to determine whether malonate results in prolongation of the neonatal regenerative window (Figure 2A). To examine whether malonate stimulates cardiomyocyte proliferation, we performed MI in 7-day-old mice and analyzed their hearts at 7 days post-MI (14-day-old) by immunostaining for markers of proliferation. We measured a significant increase in the number of cardiomyocytes undergoing mitosis as evident by pH3 staining in the dimethyl malonate-injected mice compared to saline injected controls (Figure 2B & C). We also quantified a significant increase in the number of cardiomyocytes undergoing cytokinesis by Aurora B staining, suggesting that a significantly higher number of cardiomyocytes are undergoing complete cell division (Figure 2D & E). Furthermore, there was a significant reduction in cardiomyocyte cell size at 21 days post-MI in the dimethyl malonate injected mice as quantified by wheat germ agglutinin staining (WGA), suggestive of newly formed cardiomyocytes and a reduction in cardiomyocyte hypertrophy (Figure 2F & G). To determine whether this increase in cardiomyocyte proliferation results in improved regeneration in the P7 mouse heart, we performed trichrome staining at 21 days post-MI to quantify structural regeneration and fibrosis. As expected, lack of myocardium regeneration and persistence of fibrotic scarring was detected below the ligature plane of the saline-injected controls. In contrast, mice that were injected with dimethyl malonate demonstrated complete heart regeneration (Figure 2H & Figure I in the Data Supplement). More importantly, cardiac function assessed by EF, FS, LVIDD, and LVIDS in the dimethyl malonate injected hearts was restored to normal levels (Figure 2I & Table II in the Data Supplement). These data indicate that SDH inhibition by malonate can promote cardiomyocyte proliferation and extend the regenerative capacity of the neonatal mouse heart beyond 1 week after birth, resulting in a complete regenerative response following MI in P7 juvenile mice.
Figure 2. Malonate promotes cardiomyocyte proliferation and heart regeneration in the postnatal heart following MI.
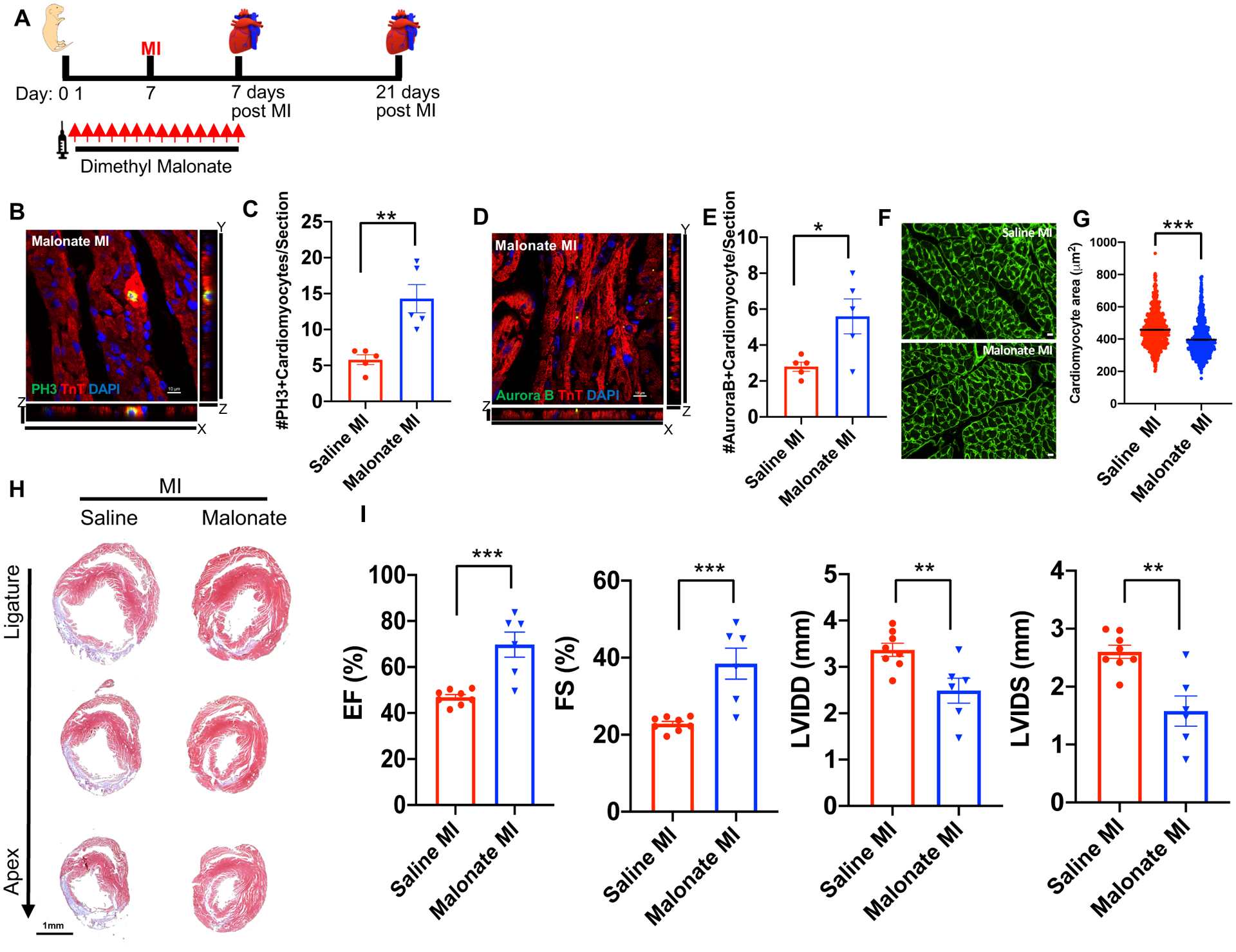
(A) Schematic of dimethyl malonate injection and MI strategy. (B, C) Immunostaining and quantification of pH3 positive cardiomyocytes showing a significant increase in the levels of mitotic myocytes in dimethyl malonate injected hearts at 7 days post-MI compared to controls. Scale bar, 10 μm. (D, E) Immunostaining and quantification of Aurora B positive cardiomyocytes demonstrating a significant increase in myocyte cytokinesis in the dimethyl malonate injected mice. Scale bar, 10 μm. (F, G) Wheat germ agglutinin (WGA) staining and cell size quantification showing decrease in the cardiomyocyte size in malonate injected hearts at 21 days post-MI. Quantitative analyses represent counting of multiple fields from independent samples per group (~ 700 cells per group). Scale bar, 10 μm. (H) Trichrome staining of malonate injected hearts at 21 days post-MI at P7, showing complete regeneration in dimethyl malonate injected mice compared to control. Scale bar, 1 mm. (I) Left ventricular systolic function quantified by EF, FS, LVIDD and LVIDS at 3 weeks post-MI demonstrating functional recovery in dimethyl malonate injected MI hearts compared to the saline injected controls. (n=5–8 mice per group). *P< 0.05, **P<0.005, ***P<0.0001 by two-tailed unpaired Student’s t-test.
SDH Inhibition by Atpenin A5 Recapitulates the Regenerative Effect of Malonate
To establish that malonate promotes cardiomyocyte proliferation and heart regeneration via SDH inhibition, we used a similar treatment strategy using Atpenin A5, which is a potent inhibitor of SDH (Figure 3A)22. To determine whether Atpenin A5 treatment can stimulate cardiomyocyte proliferation in the non-regenerative heart, we performed MI in P7 mice and analyzed their hearts at 7 days post-MI for markers of proliferation. We quantified a significant increase in the percentage of cardiomyocytes undergoing mitosis and cytokinesis as evident by pH3 and Aurora B staining, respectively, in the Atpenin A5 injected mice compared to controls (Figure 3B–E). To determine whether SDH inhibition by Atpenin A5 regenerates the non-regenerating P7 mouse heart similar to malonate, we performed trichrome staining at 21 days post-MI to quantify fibrosis and myocardial regeneration. Atpenin A5-injected mice demonstrated myocardial thickness at the infarct zone and a significant reduction in scar size compared to controls (Figure 3F & G). This was concomitant with restoration of cardiac function in Atpenin A5-injected mice (Figure 3H & Table III in the Data Supplement). Our results demonstrate that Atpenin A5 restores cardiac structure and function similar to malonate, suggesting that SDH inhibition is a central mechanism by which malonate promotes heart regeneration.
Figure 3. SDH inhibition by Atpenin A5 promotes cardiomyocyte mitosis and regeneration in the postnatal heart following MI.
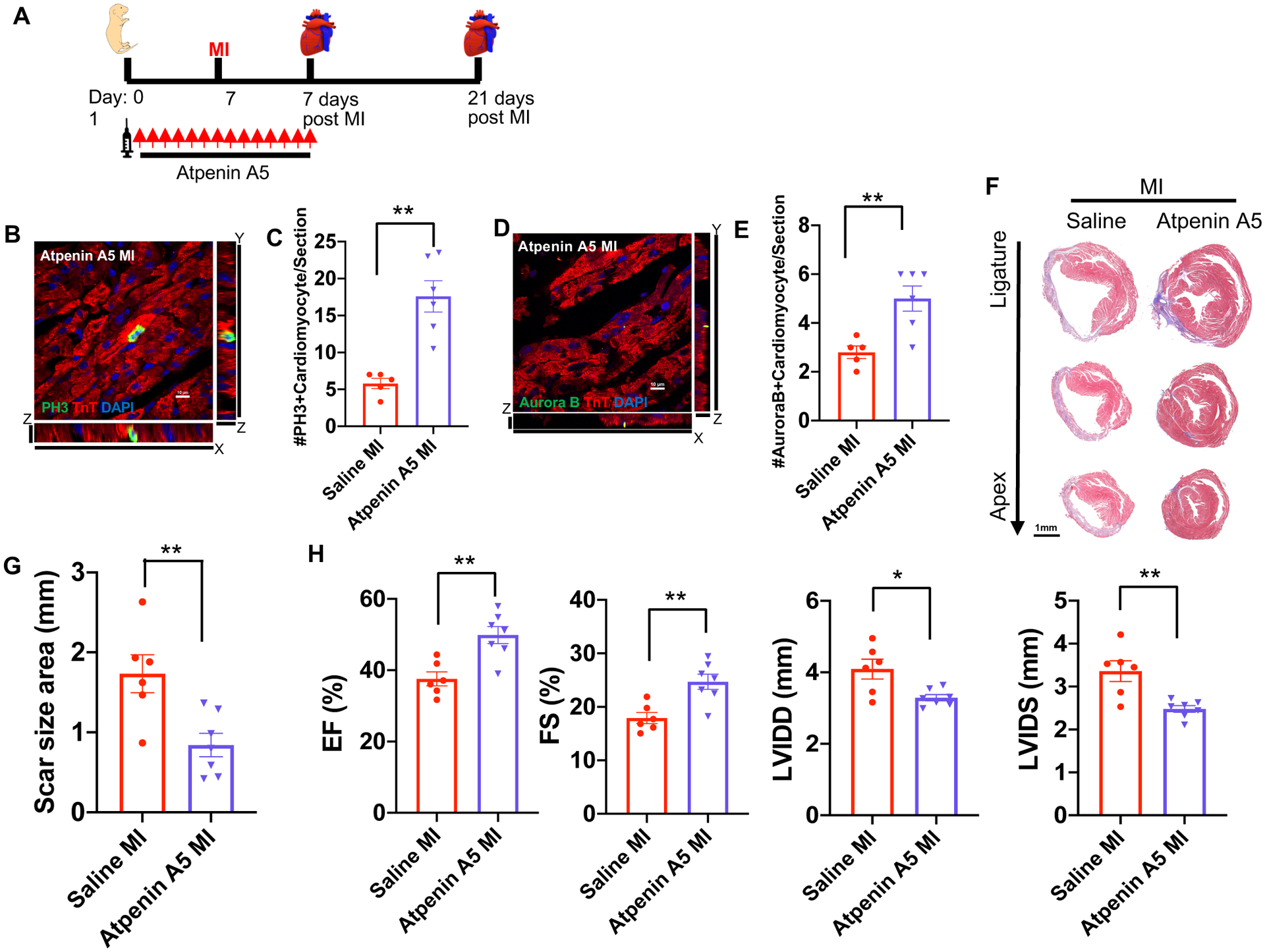
(A) Schematic of Atpenin A5 injection and MI strategy. (B) Z-stack confocal image of a mitotic cardiomyocyte stained with pH3 and cTnT. Scale bar, 10 μm. (C) Quantification of mitotic cardiomyocytes showing a significant increase in the number of mitotic cardiomyocytes in Atpenin A5 injected mice at 7 days post-MI compared to controls. (D) Z-stack confocal image of an Aurora B positive cardiomyocyte. Scale bar, 10 μm. (E) Quantification of cardiomyocytes undergoing cytokinesis showing a significant increase in the number of cardiomyocytes in cytokinesis in Atpenin A5 injected mice at 7 days post-MI compared to controls. (F) Trichrome staining of Atpenin A5 injected mice at 3 weeks post-MI at P7 demonstrating myocardial regeneration and a significant reduction in scar size in Atpenin A5 injected mice compared to controls. Scale bar, 1 mm. (G) Quantification of scar size by ImageJ software from serial sections from ligature to apex. (H) Echocardiography measurements of EF, FS, LVIDD and LVIDS at 3 weeks post-MI showing restoration of cardiac function in Atpenin A5 injected mice compared to controls. (n=6–8 mice per group). *P< 0.05, **P<0.005 by two-tailed unpaired Student’s t-test.
Malonate Promotes Cardiomyocyte Proliferation and Heart Regeneration in Adult Mice Following Myocardial Infarction
The ability of malonate to promote cardiomyocyte proliferation and heart regeneration beyond the 1-week postnatal regenerative window in mice raises the question of whether malonate can metabolically reprogram the adult heart to a regenerative state following injury. To address this question, we performed MI in 8-week-old mice and injected either saline or dimethyl malonate (100 mg/kg) within an hour following MI and continued this treatment daily for two weeks (Figure 4A). Although SDH inhibition by malonate results in cardioprotection from reperfusion injury, whether malonate can protect the myocardium following infarction remains undetermined. To determine whether malonate protects against infarction similar to reperfusion injury, we performed a viability stain using triphenyltetrazolium chloride (TTC) at 3 days post-MI. Quantification of the non-viable myocardium (white) in heart sections below the ligature showed no significant difference in both saline and malonate injected mice, suggesting that malonate did not protect against myocardial necrosis following infarction (Figure 4B & Figure II in the Data Supplement). In addition, there was no difference in the number of apoptotic cardiomyocytes at 3- or 7-days post-MI in saline- and dimethyl malonate-injected hearts as quantified by TdT-mediated dUTP nick-end labelling (TUNEL) staining (Figure 4C & Figure III-A in the Data Supplement). These results demonstrate that malonate did not protect against infarction and cardiomyocyte death following adult MI.
Figure 4. Malonate promotes adult cardiomyocyte proliferation following MI.
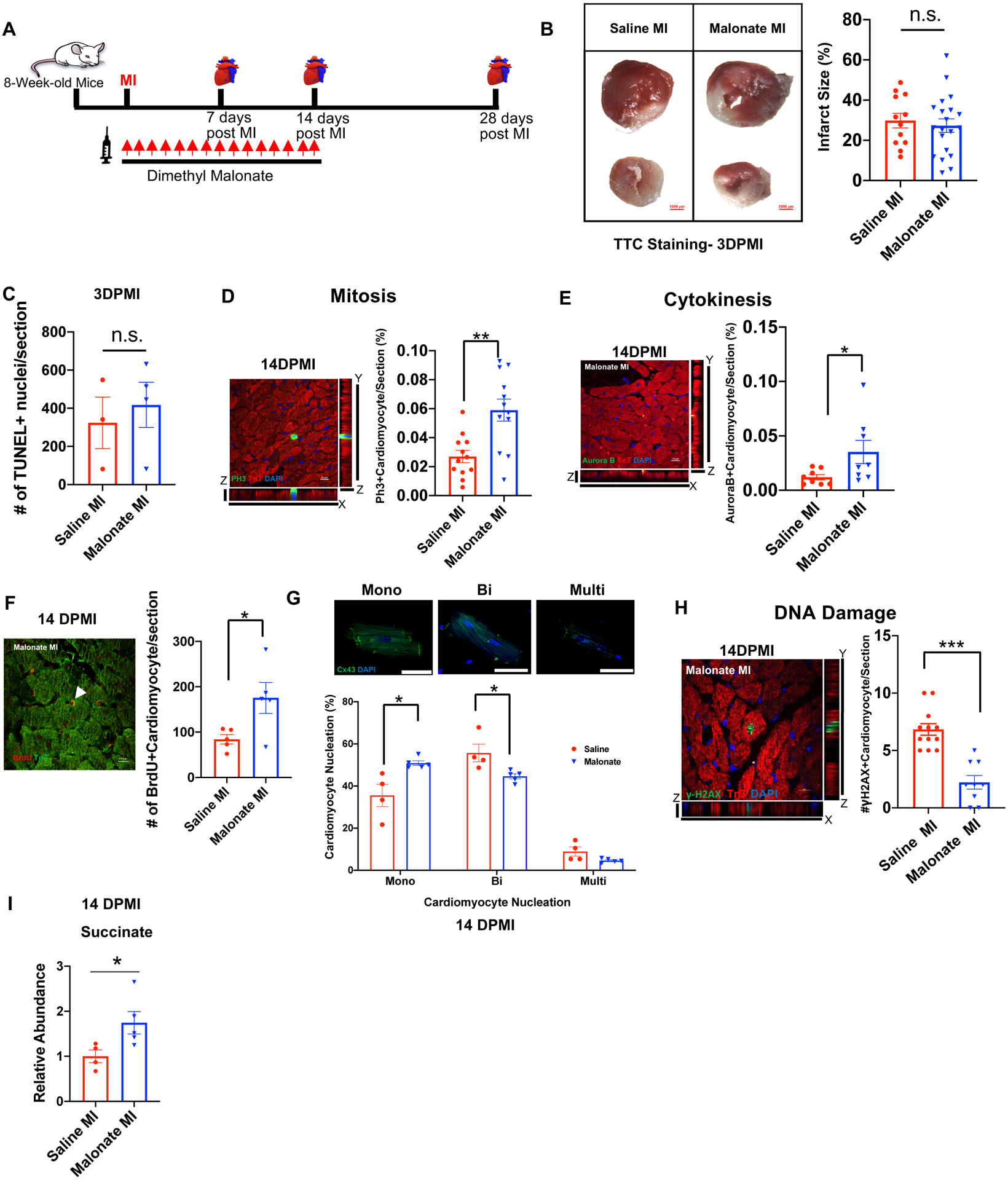
(A) Schematic of dimethyl malonate injection following adult MI. (B) TTC viability stain and quantification showing no significant difference in myocardial necrosis (white) in both saline and dimethyl malonate injected mice at 3 days post-MI (DPMI). Scale bar, 1 mm. (C) Quantification of TUNEL positive cardiomyocytes demonstrating no significant difference between TUNEL positive myocytes in saline and malonate injected mice at 3 days post-MI. (D) Z-stack confocal image of a pH3 positive cardiomyocyte at 14-days post-MI. Scale bar, 10 μm. Quantification of the percentage of pH3 positive cardiomyocytes showing a significant increase in the numbers of mitotic cardiomyocytes in dimethyl malonate injected hearts at 14-days post-MI compared to controls. (E) Z-stack confocal image of an Aurora B positive cardiomyocyte at 14-days post-MI. Scale bar, 10 μm. Quantification of the percentage of Aurora B positive cardiomyocytes demonstrating a significant increase in the numbers of cardiomyocytes undergoing cytokinesis in dimethyl malonate injected hearts at 14-days post-MI compared to controls. (F) Representative image and quantification of BrdU positive cardiomyocytes demonstrating a significant increase in BrdU positive cardiomyocytes at 14 days post-MI in malonate-treated mice. Scale bar, 10 μm. (G) Cardiomyocyte nucleation staining with connexin 43 (Cx43) and DAPI and quantification at 14 days post-MI demonstrating a significant increase in mononucleated cardiomyocytes as well as a significant decrease in binucleated cardiomyocytes following malonate treatment. Scale bar, 50 μm. (H) Immunostaining and quantification of the DNA double-strand breaks marker γH2AX demonstrating a significant decrease in DNA damage in malonate-treated mice compared to controls. Scale bar, 10 μm. (I) Relative intracellular abundance of succinate showing a significant increase in succinate levels in malonate-treated mice at 14 days post-MI (n=3–6 mice per group). *P< 0.05, **P<0.005, ***P<0.0001 by two-tailed unpaired Student’s t-test. n.s. indicates not significant.
To determine whether malonate stimulates adult cardiomyocyte proliferation, we performed immunostaining for the mitosis marker pH3 at 7- and 14 days post-MI. We quantified a significant increase in the number of cardiomyocytes undergoing mitosis in dimethyl malonate-injected hearts compared to saline-injected controls at both 7- and 14-days post-MI (Figure 4D & Figure III-B&D in the Data Supplement). In addition, there was a significant increase in the number of cardiomyocytes undergoing cytokinesis in dimethyl malonate-injected hearts as demonstrated by Aurora B staining at 7- and 14 days post-MI (Figure 4E & Figure III-C&E in the Data Supplement). Furthermore, there was a significant increase in 5-bromodeoxyuridine (BrdU) incorporation in cardiomyocytes in malonate-treated mice at 14 days post-MI (Figure 4F). This was concomitant with a remarkable increase in the number of mononucleated cardiomyocytes and a significant decrease in the number of binucleated cardiomyocytes in the malonate-treated mice (Figure 4G). Additionally, we quantified a significant decrease in the number of cardiomyocytes with γH2AX foci in malonate-treated mice indicating a significant reduction in cardiomyocyte DNA damage (Figure 4H). These results suggest that malonate can promote adult cardiomyocyte cell cycle activity following MI.
To determine whether SDH inhibition regulates cardiomyocyte cell cycle activity by modulating succinate levels, we measured the levels of intracellular succinate by liquid chromatography–mass spectrometry (LC–MS). Interestingly, there was a significant increase in succinate levels in the hearts of malonate-treated mice at 14 days post-MI (Figure 4I). This is in line with multiple studies showing that SDH inhibition and subsequent blockade of oxidative phosphorylation is accompanied by an increase in succinate levels, which promotes metabolic reprogramming to aerobic glycolysis23–25. This is distinct from the cardioprotective role of malonate in reperfusion injury, which confers cardioprotection by inhibiting reverse activity of SDH that prevents succinate accumulation7. This suggests that malonate might promote cardiomyocyte cell cycle activity via metabolic reprogramming of the adult heart to a regenerative state, rather than preventing succinate accumulation.
We then collected the hearts of both saline and dimethyl malonate-treated adult mice at 14- and 28-days post-MI and quantified structural regeneration by trichrome staining. By 14 days, fibrotic scarring in heart sections was evident in both dimethyl malonate-treated mice and saline-treated control (Figure 5A & Figure IV in the Data Supplement). Quantification showed a trend for reduction in fibrosis in the dimethyl malonate-treated samples compared to controls at 14 days post-MI, but the difference was not significant (Figure 5A). By 28 days post-MI, trichrome staining from the dimethyl malonate-injected hearts showed remarkable restoration of the myocardium with minimal fibrosis compared to the saline-injected controls that showed ventricle dilation and significant fibrotic scarring, as expected from an adult MI (Figure 5A & Figure V in the Data Supplement). Quantification of fibrosis demonstrated a significant reduction of scar size in the dimethyl malonate-treated mice at 28 days post-MI. Notably, there was incomplete regeneration in one mouse heart that was subjected to a large infarct (Figure V in the Data Supplement), indicating that the size of infarction can impact the regenerative response, similar to neonatal mouse heart regeneration26. Whether longer duration of malonate treatment can regenerate larger infarcts is yet to be determined.
Figure 5. Malonate restores cardiac structure and function following adult MI.
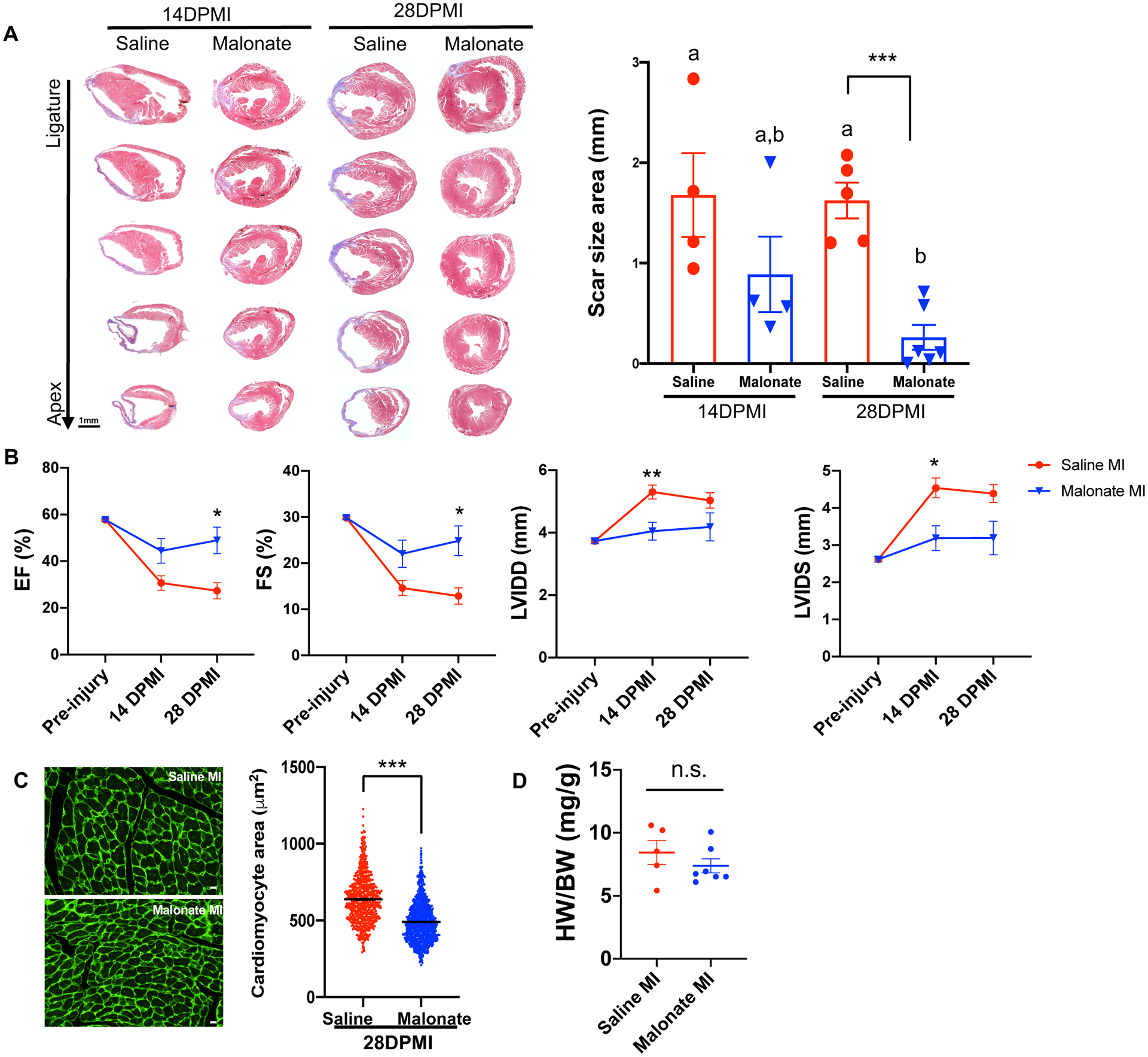
(A) Trichrome staining of heart sections from saline and dimethyl malonate-injected mice at 14 and 28 days following adult MI, showing restoration of cardiac structure and no fibrotic scarring by 28 days post-MI in dimethyl malonate injected mice. Quantification of scar size demonstrating a significant reduction in fibrosis in dimethyl malonate treated mice at 28 days post-MI. (B) Echocardiography of cardiac function measured by EF, FS, LVIDD and LVIDS at 14- and 28-days post-MI showing a significant functional recovery in dimethyl malonate injected hearts compared to saline injected controls at 28 days post-MI. (C) Wheat germ agglutinin (WGA) staining and cell size quantification showing decrease in cardiomyocyte size in dimethyl malonate injected hearts at 4 weeks post-MI. Quantitative analyses represent counting of multiple fields from 5–6 independent samples per group (750 ~ 1000 cells per group). Scale bar, 10 μm. (D) Heart weight-to-body weight ratios at 28 days post-MI showing no significant difference between saline and malonate injected mice. (n=5–6 mice per group). *P< 0.05, **P< 0.005, ***P<0.0001 by two-tailed unpaired Student’s t-test. One-way ANOVA was performed by Tukey’s multiple comparison test to determine the differences of group mean among treatment groups. Different letters indicate significant differences among groups. n.s. indicates not significant.
To determine whether this restoration of cardiac structure was accompanied by improvement in cardiac function, we performed echocardiographic measurements of both saline and dimethyl malonate-injected mice at 14- and 28-days post-MI. Our echocardiographic measurements demonstrated a reduction in cardiac function at 14 days post-MI (Figure 5B & Table IV in the Data Supplement). There was a trending improvement in cardiac function of dimethyl malonate injected hearts compared to controls by 14 days post-MI, but the difference was not significant (Figure 5B). Remarkably, there was a significant improvement of cardiac function in dimethyl malonate-injected mice as measured by EF and FS at 28 days post-MI (Figure 5B). Interestingly, there was a significant reduction in cardiomyocyte size in dimethyl malonate-injected mice compared to controls at 28 days post-MI as quantified by WGA staining (Figure 5C). No significant differences in heart weight to body weight ratios were detected (Figure 5D). Our results reveal that malonate is capable of restoring cardiac structure and function following adult MI. Collectively, these results demonstrate that malonate promotes adult cardiomyocyte proliferation and heart regeneration following adult MI.
Malonate Induces a Metabolic Shift in the Adult Heart and Promotes Revascularization Following Infarction
The metabolic switch from glycolysis to oxidative phosphorylation results in loss of the endogenous cardiac regenerative ability in mammals6. Furthermore, a metabolic switch to glycolysis promotes cardiomyocyte proliferation during zebrafish heart regeneration16. Interestingly, mutations in SDH have been demonstrated to induce a metabolic shift into glycolysis in familial cancer syndromes that promotes cell division and angiogenesis13–15. These studies together with our comprehensive results strongly suggest that SDH inhibition may promote a cardiac regenerative response by modulating the cardiac metabolic state. To determine whether inhibition of SDH promotes metabolic reprogramming in the adult heart, we treated adult mice for 2 weeks with dimethyl malonate and performed metabolomics using LC-MS (Figure 6A). Several metabolites in the TCA cycle, such as α-ketoglutarate, decreased following SDH inhibition by malonate, except for succinate, which increased as expected (Figure 6B). This is consistent with previous studies showing inhibition of aerobic respiration in response to SDH inhibition, since SDH participates in both the TCA cycle and the electron transport chain (ETC)10. More importantly, this was accompanied by a shift in metabolites in other pathways of glucose metabolism (Figure 6C). These results are consistent with previous studies demonstrating that SDH inhibition stimulates a metabolic shift to glycolysis, which is a metabolic state that can promote heart regeneration.
Figure 6. Malonate induces a dynamic metabolic shift in the adult heart and promotes revascularization following MI.
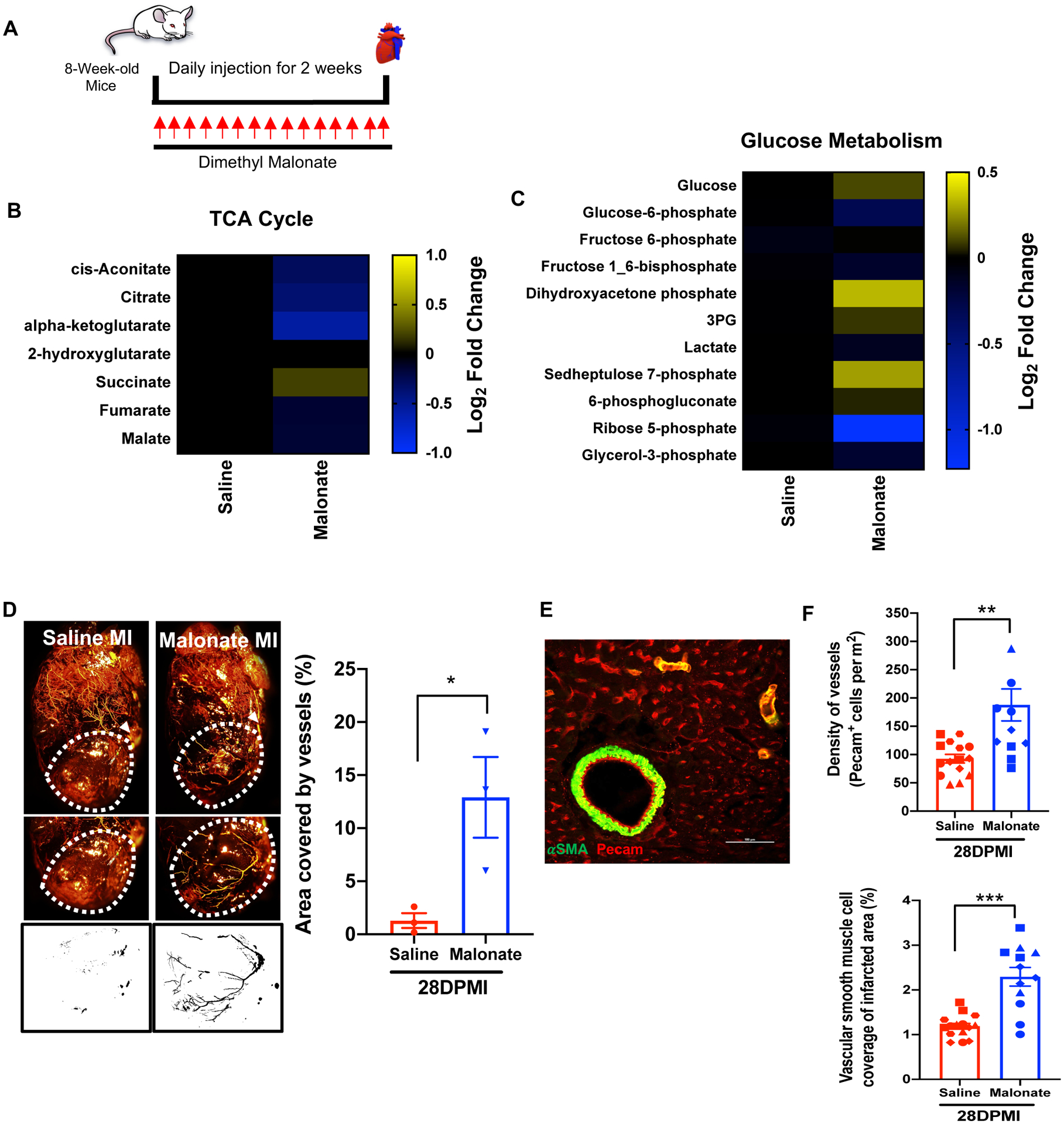
(A) Schematic of malonate administration for metabolomics. (B & C) Metabolomic changes of tricarboxylic acid (TCA) cycle and glucose metabolism in saline and malonate treated mice at 14 days following treatment. Relative abundance of metabolites in malonate-treated mice is compared to saline-treated mice and presented as a heatmap on a log2 scale demonstrating a dynamic change in TCA cycle and glucose metabolism in malonate-treated mice. (D) Coronary vessel casting by MICROFIL injection at 28 days post-MI showing a significant increase in revascularization of the infarct zone in malonate-treated mice compared to controls. Quantification of vasculature in region of interest (ROI) by analyzing binarized images for grey level intensity by ImageJ demonstrating a significant increase in vascular density in the infarct zone. (E) Immunostaining with the endothelial marker PECAM and vascular smooth muscle cell marker α-SMA. Scale bar, 100 μm. (F) Quantification of vascular lineages demonstrating a significant increase in endothelial capillary density and vascular smooth muscle cells in the infarct zone at 28 days post-MI. (n=3–4 mice per group). *P< 0.05, **P<0.005, ***P<0.0001 by two-tailed unpaired Student’s t-test.
Cardiomyocyte proliferation is key to replenishing the lost myocardium following injury; however, complete regeneration following infarction requires coronary artery formation and revascularization to supply the newly formed myocardium with oxygenated blood. This response has been demonstrated to be activated during neonatal mouse regeneration and is a hallmark for complete regeneration following infarction27. Interestingly, glycolysis plays in important role in angiogenesis28, 29. Since malonate treatment induces a cardiac metabolic shift in the adult heart, we wanted to determine whether malonate promotes heart regeneration by inducing coronary artery formation and revascularization following injury. To address this question, we performed coronary casting of saline and dimethyl malonate injected hearts at 28 days post-MI. There was a remarkable formation of coronary arteries below the ligature site in the dimethyl malonate injected mice compared to controls (Figure 6D). In addition, there was a significant increase in capillary density and vascular smooth muscle cells in the infarct zone, suggesting an increase in newly synthesized vessels ranging in size from small capillaries to large-diameter vessels (Figure 6E & F). These results reveal that malonate can induce myocardial regeneration by stimulating adult cardiomyocyte proliferation as well as revascularization post-MI.
Malonate Treatment at 1-Week Following Infarction Promotes Heart Regeneration
To determine whether malonate can promote a regenerative response following the establishment of infarction, we performed MI in 8-week-old mice and started malonate treatment at 1-week post-MI (Figure 7A). The reduction of cardiac function was confirmed at 1-week post-MI by echocardiography (Figure 7D). Mice were then randomized and treated with saline or malonate for a period of 4 weeks, and hearts were harvested at 6 weeks post-MI (Figure 7A). Trichrome staining at 6 weeks post-MI showed a remarkable increase in myocardial thickness with minimal fibrosis in malonate-treated mice (Figure 7B & Figure VI in the Data Supplement). Fibrosis quantification demonstrated a significant reduction in scar size in the malonate-treated mice at 6 weeks post-MI (Figure 7C). Remarkably, there was a significant increase in cardiac function in malonate-treated mice at 4-weeks and 6-weeks post-MI (Figure 7D & Table V in the Data Supplement). Our results demonstrate that injection of malonate in adult mice at 1-week post-MI promoted myocardial regeneration and functional improvement over time. Together, these results demonstrate that SDH inhibition by malonate can stimulate a cardiac regenerative response following the establishment of infarction in the adult heart.
Figure 7. Malonate treatment starting 1-week post-MI promotes myocardial regeneration.

(A) Schematic of malonate injection and MI strategy. (B) Trichrome staining of heart sections from saline and malonate-injected mice at 6 weeks post-MI. (C) Scar size quantification by ImageJ from serial sections per heart showing a significant reduction in scar size in malonate treated mice at 6 weeks post-MI. (D) Serial echocardiography measurements of EF, FS, LVID, LVIDS showing significant functional recovery in malonate-injected mice over time compared to controls. (n=6 mice per group). *P< 0.05, **P<0.005, ***P<0.0001 by two-tailed unpaired Student’s t-test. Two-way ANOVA with Bonferroni’s post-hoc test was used to determine the difference among the treatment groups and different time points. #P<0.05.
DISCUSSION
Systolic heart failure often occurs as a consequence of the inability of the adult mammalian heart to regenerate following injury such as MI. Models of mammalian endogenous heart regeneration provide an opportunity to identify new approaches to restore adult human heart regeneration30. Lineage tracing studies demonstrated that proliferation of the pre-existing cardiomyocytes is the main source of the newly formed functional myocardium during endogenous regeneration. Thus, stimulating adult cardiomyocyte proliferation represents an important target towards regenerating the adult human heart following injury.
The metabolic switch in energy utilization of the postnatal heart and the subsequent increase in ROS production has emerged as an important factor in loss of this regenerative response6. The mechanisms that regulate this metabolic switch remain unclear. In this study, our results demonstrate a powerful link in succinate metabolism and succinate dehydrogenase (SDH) activity to the regenerative response of the mammalian heart. We demonstrate that high levels of succinate can induce cardiomyocyte DNA damage and inhibit cardiomyocyte proliferation and regeneration. More importantly, we demonstrate that inhibition of SDH activity by malonate can also restore a cardiac regenerative response in the adult heart by stimulating adult cardiomyocyte cell cycle re-entry and revascularization, important hallmarks of endogenous heart regeneration. This regenerative effect is largely due to SDH inhibition, since Atpenin A5, a potent inhibitor of SDH, can recapitulate the regenerative effect of malonate. Furthermore, our analysis demonstrates that SDH inhibition induces metabolite level changes that are consistent with a metabolic shift from oxidative phosphorylation to glycolysis in the adult heart. Although we did not detect an increase in lactate levels, serial metabolite analysis at multiple timepoints can reveal the extent of the dynamic cardiac metabolic shift following SDH inhibition. The metabolic switch from aerobic respiration to glycolysis is implicated by previous studies defining SDH as a tumor suppressor, where SDH mutations result in a metabolic reprogramming to glycolysis that promotes cancer growth13–15.
Malonate has been demonstrated to play a cardioprotective role in reperfusion injury by inhibiting reverse activity of SDH, which prevents succinate accumulation and the subsequent redox insult and cardiac damage7. Interestingly, SDH inhibition by malonate for 2 weeks increased succinate levels, a consequence of inhibition of oxidative phosphorylation23–25. In contrast to reperfusion injury, our results demonstrate that malonate does not exhibit a cardioprotective role following myocardial infarction. The progression in restoration of cardiac structure and function over time strongly suggests a stimulation of a regenerative response by malonate following infarction, rather than protection. Furthermore, we demonstrate that malonate treatment starting 1-week post-MI promotes myocardial regeneration and functional improvement over time. Collectively, these results reveal a novel role for SDH in its ability to metabolically reprogram the adult heart to a regenerative state. This underscores the translational potential of SDH inhibition as a powerful metabolic target for promoting adult heart regeneration.
There is an emerging appreciation for the role of metabolism in controlling cell state. Adult neural stem cell activity changes from a quiescent to a proliferative state via a metabolic shift by a single metabolite31. Similarly, metabolic reprogramming regulates macrophage function in response to different stimuli32, 33. Interestingly, a recent study demonstrated that metabolic reprogramming is required for cardiomyocyte proliferation during zebrafish heart regeneration16. The potential metabolic targeting of multiple cell types by systemic administration of malonate explains the striking regenerative effect following adult myocardial infarction. In this study, we demonstrate that SDH inhibition promotes adult cardiomyocyte proliferation and revascularization following injury. The overall impact of malonate on other cell types will need to be further investigated. For example, SDH inhibition by malonate has been shown to promote an anti-inflammatory state of macrophages following lipopolysaccharide stimulation34. Thus, whether SDH inhibition regulates the inflammatory response following infarction remains unclear. In addition, mutations in SDH have been shown to promote DNA methylation, demonstrating an interplay between epigenetics and metabolism14, 23, 35. Whether SDH inhibition by malonate can modulate the epigenetic landscape and the transcriptional activity of multiple cardiac cell types needs to be determined.
Understanding the effect of malonate on the heart as well as other tissues will be an essential step prior to translating our findings to the clinic for treatment of heart failure. Targeted delivery to the heart would avoid any off-target effects from systemic SDH inhibition. Furthermore, additional safety and pharmacokinetic studies are warranted owing to the role of SDH inhibition in multiple cancers, which will pave the way to harnessing the regenerative effect of malonate due to its simplicity and efficacy. Malonate provides an opportunity for transient SDH inhibition, but the long-term effects need to be determined as well. Promoting adult heart regeneration by SDH inhibition has enormous implications for treatment of systolic heart failure patients.
Supplementary Material
Clinical Perspective.
What Is New?
We found that malonate, a competitive inhibitor of succinate dehydrogenase (SDH), promotes adult cardiomyocyte proliferation, revascularization of the infarct zone, and myocardial regeneration following infarction.
We also found that SDH inhibition by malonate is consistent with a metabolic shift from oxidative phosphorylation to glucose metabolism in the adult heart.
What Are the Clinical Implications?
Transient inhibition of SDH represents an important metabolic target to promote adult heart regeneration following infarction.
The long-term effects of systemic SDH inhibition needs to be closely evaluated owing to its role as a tumor suppressor.
Sources of Funding
Funding for this project was provided by the UW School of Medicine and Public Health from the Wisconsin Partnership Program (A.I.M.), an American Heart Association Career Development Award 19CDA34660169 (A.I.M.), NIH/NCATS through CTSA award UL1TR002373 to the UW Institute for Clinical and Translational Research, a postdoctoral training award from the Stem Cell & Regenerative Medicine Center at UW-Madison (J.B.) and NIH/NHLBI under Ruth L. Kirschstein NRSA T32 HL007936 to the UW Cardiovascular Research Center (J.B.).
Non-Standard Abbreviations and Acronyms
- ANOVA
analysis of variance
- BrdU
5-bromodeoxyuridine
- DNA
deoxyribonucleic acid
- EF
ejection fraction
- ETC
electron transport chain
- FS
fractional shortening
- LVIDD
left ventricle internal diameter diastole
- LVIDS
left ventricle internal diameter systole
- MI
myocardial infarction
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- TCA
tricarboxylic acid
Footnotes
Disclosures
The authors declare no competing interests.
REFERENCES
- 1.Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation. 2020;141:e139–e596. [DOI] [PubMed] [Google Scholar]
- 2.Mahmoud AI, Porrello ER, Kimura W, Olson EN and Sadek HA. Surgical models for cardiac regeneration in neonatal mice. Nat Protoc. 2014;9:305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN and Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN and Sadek HA. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci U S A. 2013;110:187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Webster WS and Abela D. The effect of hypoxia in development. Birth Defects Res C Embryo Today. 2007;81:215–28. [DOI] [PubMed] [Google Scholar]
- 6.Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hochachka PW and Dressendorfer RH. Succinate accumulation in man during exercise. Eur J Appl Physiol Occup Physiol. 1976;35:235–42. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, Wang YT, Miller JH, Day MM, Munger JC and Brookes PS. Accumulation of Succinate in Cardiac Ischemia Primarily Occurs via Canonical Krebs Cycle Activity. Cell Rep. 2018;23:2617–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King A, Selak MA and Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25:4675–82. [DOI] [PubMed] [Google Scholar]
- 11.Kula-Alwar D, Prag HA and Krieg T. Targeting Succinate Metabolism in Ischemia/Reperfusion Injury. Circulation. 2019;140:1968–1970. [DOI] [PubMed] [Google Scholar]
- 12.Valls-Lacalle L, Barba I, Miro-Casas E, Ruiz-Meana M, Rodriguez-Sinovas A and Garcia-Dorado D. Selective Inhibition of Succinate Dehydrogenase in Reperfused Myocardium with Intracoronary Malonate Reduces Infarct Size. Sci Rep. 2018;8:2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gottlieb E and Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer. 2005;5:857–66. [DOI] [PubMed] [Google Scholar]
- 14.Her YF and Maher LJ 3rd. Succinate Dehydrogenase Loss in Familial Paraganglioma: Biochemistry, Genetics, and Epigenetics. Int J Endocrinol. 2015;2015:296167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tseng PL, Wu WH, Hu TH, Chen CW, Cheng HC, Li CF, Tsai WH, Tsai HJ, Hsieh MC, Chuang JH et al. Decreased succinate dehydrogenase B in human hepatocellular carcinoma accelerates tumor malignancy by inducing the Warburg effect. Sci Rep. 2018;8:3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Honkoop H, de Bakker DE, Aharonov A, Kruse F, Shakked A, Nguyen PD, de Heus C, Garric L, Muraro MJ, Shoffner A et al. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. Elife. 2019;8:e50163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar D, Hacker TA, Buck J, Whitesell LF, Kaji EH, Douglas PS and Kamp TJ. Distinct mouse coronary anatomy and myocardial infarction consequent to ligation. Coron Artery Dis. 2005;16:41–4. [DOI] [PubMed] [Google Scholar]
- 18.Singla DK, Hacker TA, Ma L, Douglas PS, Sullivan R, Lyons GE and Kamp TJ. Transplantation of embryonic stem cells into the infarcted mouse heart: formation of multiple cell types. J Mol Cell Cardiol. 2006;40:195–200. [DOI] [PubMed] [Google Scholar]
- 19.Seim GL, John SV and Fan J. Metabolomic and Lipidomic Analysis of Bone Marrow Derived Macrophages. Bio-protocol. 2020;10:e3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, Powers PA and Moss RL. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res. 2002;90:594–601. [DOI] [PubMed] [Google Scholar]
- 21.Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER and Sadek HA. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013;497:249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyadera H, Shiomi K, Ui H, Yamaguchi Y, Masuma R, Tomoda H, Miyoshi H, Osanai A, Kita K and Omura S. Atpenins, potent and specific inhibitors of mitochondrial complex II (succinate-ubiquinone oxidoreductase). Proc Natl Acad Sci U S A. 2003;100:473–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–52. [DOI] [PubMed] [Google Scholar]
- 24.Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231–9. [DOI] [PubMed] [Google Scholar]
- 25.Sciacovelli M, Guzzo G, Morello V, Frezza C, Zheng L, Nannini N, Calabrese F, Laudiero G, Esposito F, Landriscina M et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 2013;17:988–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bryant DM, O’Meara CC, Ho NN, Gannon J, Cai L and Lee RT. A systematic analysis of neonatal mouse heart regeneration after apical resection. J Mol Cell Cardiol. 2015;79:315–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Das S, Goldstone AB, Wang H, Farry J, D’Amato G, Paulsen MJ, Eskandari A, Hironaka CE, Phansalkar R, Sharma B et al. A Unique Collateral Artery Development Program Promotes Neonatal Heart Regeneration. Cell. 2019;176:1128–1142 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–63. [DOI] [PubMed] [Google Scholar]
- 29.Eelen G, de Zeeuw P, Simons M and Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. 2015;116:1231–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sadek H and Olson EN. Toward the Goal of Human Heart Regeneration. Cell Stem Cell. 2020;26:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knobloch M, Pilz GA, Ghesquiere B, Kovacs WJ, Wegleiter T, Moore DL, Hruzova M, Zamboni N, Carmeliet P and Jessberger S. A Fatty Acid Oxidation-Dependent Metabolic Shift Regulates Adult Neural Stem Cell Activity. Cell Rep. 2017;20:2144–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelly B and O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seim GL, Britt EC, John SV, Yeo FJ, Johnson AR, Eisenstein RS, Pagliarini DJ and Fan J. Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-γ stimulation. Nature Metabolism. 2019;1:731–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Dabritz JHM, Gottlieb E, Latorre I et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell. 2016;167:457–470 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaelin WG Jr. and McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153:56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.