

Abstract
Pediatric T cell acute lymphoblastic leukemia (T-ALL) cells frequently contain mutations in the interleukin-7 (IL-7) receptor pathway or respond to IL-7 itself. To target the IL-7 receptor on T-ALL cells, murine monoclonal antibodies (MAbs) were developed against the human IL-7Rα chain and chimerized with human IgG1 constant regions. Crystal structures demonstrate that the two MAbs bound different IL-7Rα epitopes. The MAbs mediated antibody-dependent cell-mediated cytotoxicity (ADCC) against patient-derived xenograft (PDX) T-ALL cells, which was improved by combining two MAbs. In vivo, the MAbs showed therapeutic efficacy via ADCC-dependent and independent mechanisms in minimal residual and established disease. PDX T-ALL cells that relapsed following a course of chemotherapy displayed elevated IL-7Rα, and MAb treatment is effective against relapsing disease, suggesting the use of anti-IL7Rα MAbs in relapsed T-ALL patients or patients that do not respond to chemotherapy.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common malignancy in children. T cell acute lymphoblastic leukemia (T-ALL) is an aggressive, hematological malignancy accounting for 15% of pediatric and 25% of adult ALL cases. Current treatment protocols result in an overall survival rate of 70% for T-ALL patients [1], however relapse occurs in 20–25% of children [2], and in over half of adult patients [3]. Despite intensive chemo-radiotherapy treatment and transplantation, 50–70% of relapsed patients succumb to disease, therefore, novel salvage regimens are urgently needed [4].
While several immunotherapies have been developed for B-ALL, limited options exist for T-ALL patients for whom treatment fails to cure. The alpha chain of interleukin 7 receptor (IL-7Rα, CD127) is one potential target in T-ALL [5]. IL-7Rα together with the γc chain (CD132) comprise the receptor for the stromal-produced cytokine IL-7. The IL-7Rα is expressed on normal T cells during most immature and mature stages and is required for T cell development and survival [6]. The majority (60–70%) of patient T-ALL samples express IL-7Rα and respond to IL-7, although positive samples show a wide range of expression [7–10]. Oncogenic gain-of-function mutations in IL-7Rα have been identified in about 10% of pediatric T-ALL patients [11–13] and many other mutations in T-ALL cells are components of the IL-7 receptor signaling pathway [5, 14]. We therefore evaluated whether targeting IL-7Rα with a monoclonal antibody would have a therapeutic benefit against T-ALL.
We generated two new chimeric monoclonal antibodies (MAbs) against human IL-7Rα, 4A10, and 2B8, that recognize non-overlapping IL-7Rα epitopes. These antibodies were used to demonstrate that patient derived T-ALL cells express IL-7Rα, and that this expression increases after exposure to 4–6 weeks of multi-agent chemotherapy. Furthermore, we demonstrate IL-7Rα MAbs mediate potent antibody-dependent cell mediated cytotoxicity (ADCC) in vitro and effective anti-leukemia responses in vivo using minimal residual, established, and relapsing patient-derived xenografts (PDXs).
Materials and methods
IL-7Rα production, screening of murine MAbs, Fab production for crystal structure determination
The extracellular domains (ECD) of the wild-type (WT) and a T-ALL mutant of the IL-7Rα were expressed from Schneider S2 insect cells and purified as described previously [15]. The T-ALL IL-7Rα mutant consists of the wt IL-7Rα ECD protein sequence with the following C-terminal extension of PILLTCPT. This protein sequence corresponds to patient 2 and is similar in sequence to the T-ALL insertion seen in patient 1 described below as described previously [12]. The purified homodimeric form of this T-ALL mutant and the monomeric WT hIL-7Rα ECD were subsequently conjugated to KLH. These conjugates were used to immunize and boost mice and hybridomas were created under contract with Precision Antibody (Columbia, MD). Supernatants were screened on Baf3 cells transfected with IL-7Rα using anti-mouse IgG1 APC as a secondary (BioLegend). Two positive clones were identified, 4A10 and 2B8, from the T-ALL mutein and were sequenced. The monomeric WT hIL-7Rα ECD was non-immunogenic in the mice used. Human IL-7 was expressed from bacteria and purified as previously reported [15].
Some experiments utilized chimeric antibodies that were generated as human IgG1 molecules under contract with GenScript (Piscataway NJ). In addition, the murine 4A10 IgG1 hybridoma was expressed in high levels using Celline bioreactor flasks grown with serum-free hybridoma media (Gibco, Thermo Scientific). For in-house chimerized antibody protein production, codon optimized genes for HEK293 cells of the variable heavy and light domains were designed and synthesized by DNA 2.0, Inc. (now Atum, Inc). These genes were subcloned into a kappa light and heavy chain expression vectors to be secreted as a human IgG1 molecule. These chimerized MAbs were transiently transfected and expressed from Freestyle HEK293F cells as described by the manufacturer (Thermo Scientific). Full-length murine or human IgG1 proteins were purified to homogeneity employing protein A/G or A chromatography resin (Thermo Scientific). A human IgG1 isotype control, Cetuximab (an anti-EGFR MAb), was purchased from the NCI pharmacy. Fab fragments were generated using standard immobilized papain cleavage reactions and the fragments were purified by protein A and size-exclusion chromatography methods and used for SPR binding studies and crystal structural determinations (Supplementary Methods).
Retroviral infection of D1 cells
Generation of D1-hIL-7Rα WT and D1-hIL-7Rα P1 have been described previously [12]. Briefly, the D1 thymocyte cell line was transduced with a pMIG retroviral vector expressing GFP and either full length WT IL-7Rα, mutant IL-7Rα (P1) (p.Leu242-Leu243 insAsnProCys), or an empty pMIG vector as a control. The mutant IL-7Rα sequence was obtained from patient 1 (P1) [12]. Transfected cells were maintained in culture medium containing 50 ng/mL of recombinant murine IL-7 (Peprotech).
MAbs and flow cytometry
A commercial anti-human IL-7Rα APC (A019D5, cat #351316), anti-human CD4 FITC (OKT4, cat #317408), and anti-human CD45 APC/FIRE (2D1, cat #368517) were purchased from BioLegend. An anti-murine CD45 FITC (30-F11, cat #553079) was purchased from BD Biosciences. A secondary anti-human IgG Fc APC (HP6017, cat #409306) was purchased from BioLegend. Cells were collected using a BD Canto ll with FACS DIVA software. Data analysis was performed using FlowJo (Tree Star, Inc.). Center values are median fluorescence intensity (MFI).
ADCC assay
ADCC activity was determined by measuring lactate dehydrogenase (LDH) release using Cytotoxicity Detection KitPLUS (Roche). Briefly, T-ALL target cells (1 × 104) were incubated in triplicate with 10 μg/mL of the anti-IL-7Rα MAbs or recombinant human IgG1 Fc (Bio X cell) as an isotype control for 30 min. Human NK effector cells isolated from healthy donors were added at the indicated effector to target (E:T) ratios. After a 4 h incubation, the reaction mixture was prepared according to manufacturer’s instructions and added to the samples. LDH release was measured using a Molecular Devices VersaMax ELISA reader. The percentage of cytotoxicity was calculated as: %lysis = (effector + target mix − effector cell alone)-target cell alone/lysed targets-targets alone × 100. Most experiments were repeated two or more times with similar results.
In vivo experiments
For survival studies, D1-hIL-7Rα P1 (mutant IL-7Rα) or D1-pMIG (empty vector) cells (1 × 105) were injected intravenously via tail vein into 26 sub-lethally irradiated RAG1−/− mice (Jackson Laboratory). Mice were prophylactically treated with SMZ antibiotics (0.08 mg/mL in drinking water) for 3 days prior to irradiation. One day after cell injection, mice were randomized into treatment groups and treated with either a single injection of anti-IL-7Rα 4A10 (10 mice, 250 μg intravenously), or PBS (10 mice, 0.2 mL intravenously) as a control. Mice receiving D1-pMIG cells as a control were treated with PBS (6 mice, 0.2 ml intravenously). D1-pMIG cells do not express human IL-7Rα or form leukemia. Mice were euthanized when they became clinically moribund. In another experiment, 3 mice from each group were assessed for disease burden 15 days post cell transfer. Experiments were repeated once with similar results.
PDXs and therapeutic treatments
T-ALL#5 is a pediatric sample obtained from Dr. Curt Civin from the University of Maryland School of Medicine. CBAT44179, CBAT37614, and CBAT27299 are pediatric T-ALL samples obtained from The Public Repository of Xenografts (PRoXe; Dana-Farber Cancer Institute). PDX cells were expanded in NOD.Cg-PrkdcSCIDIl2rgtm1wjl/SzJ (NSG: The Jackson Laboratory), and a cell bank was made from spleen, or a combination of spleen and bone marrow. Therapeutic treatments are described in Supplementary Methods. Samples were MTBM tested prior to engraftment and found to be free of mycoplasma contamination. Most experiments were performed twice with similar results.
Mice were maintained at the National Cancer Institute (NCI)-Frederick, Maryland. Animal care was provided in accordance with the US NIH Animal Use and Care guidelines. Experiments were approved by the NCI-Frederick Animal Care and Use Committee. All mice used were 6–12 weeks old. Groups contained a minimum of 4 mice.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7 (GraphPad Software). Differences were calculated using an unpaired two-tailed Student’s t-test with P values being shown. Kaplan-Meier survival curves were analyzed using the Log-rank test with P values being shown. A P value of less than 0.05 was considered statistically significant. Error bars are standard deviation. Center values are mean. Multiple comparisons were not used.
Results
Anti-IL-7Rα 4A10 and 2B8 bind different epitopes on the IL-7Rα chain
To generate new anti-human IL-7Rα MAbs, the extracellular domain of an oncogenic IL-7Rα mutant P 2 (C-terminal sequence of PILLTCPT with the underlined amino acids containing observed mutations observed in patient 2) which has the capacity to form homodimers was expressed from S2 insect cells [16], the homodimer purified, coupled to KLH, and used to immunize mice. Hybridomas were generated and two positive clone supernatants were identified by screening against murine cells transfected with human IL-7Rα. Specificity of clones anti-IL-7Rα 2B8 and anti-IL-7Rα 4A10 was demonstrated in triplicate by binding to the murine D1 thymocyte cell line [17] transfected with pMIG empty vector (Supplemental Fig. S1A, top panel), versus D1 cells transfected with either WT IL-7Rα (Supplemental Fig. S1,A, middle panel), or mutant IL-7Rα P1 (Supplemental Fig. S1,A, bottom panel) [12]. The mean fluorescence intensity was significantly increased when the MAbs were used together (Supplemental Figs. S1,A and S1, B). Transfected cells expressed identical levels of IL-7Rα (Supplemental Figs. S1,A and S1,B). Both MAbs also bound normal human lymphocytes and the MFI was significantly increased when both MAbs were used together (Supplemental Fig. S2A bottom, right panel). Jurkat cells with (unfilled histograms) and without (filled histograms) transfected IL-7Rα were used as controls (Supplemental Fig. S2B). Neither 2B8 or 4A10 induced rapid internalization of IL-7Rα as measured by disappearance of the receptor from the cell surface after binding and incubation at 37 °C for 1 h (Supplemental Fig. S3). Binding affinities of the Fab fragments to immobilized IL-7Rα ECD were determined using surface plasmon resonance (Supplemental Fig. S4A–S4,D). In this SPR assay, wt IL-7 bound IL-7Rα ECD with a Kd of 67.0 ± 2.4 nM [Kd = k-1k-2/k1(k2 + k-2), k1 = 7.0 × 105 M−1s−1, k-1 = 9.4 × 10−2 s−1, k2 = 9.0 × 10−4 s−1, k-2 = 9.1 × 10−4 s−1] similar to previously published results [16]. The 2B8 Fab bound the IL-7Rα ECD with a Kd of 8.7 ± 0.1 nM (Kd = koff/kon = 1.2 × 10−4 s−1/1.4 × 104 M−1s−1). The 4A10 Fab bound the IL-7Rα ECD ~9-fold stronger than the 2B8 Fab with a Kd of 1.0 ± 0.1 nM (Kd = 2.7 × 10−4 s−1/2.6 × 105 M−1s−1) similar to the binding behavior of the MAbs to full-length IL-7Rα on human lymphocytes. We observed no binding of an unrelated Fab fragment of Cetuximab (anti-EGFR hIgG1) to the IL-7Rα coupled surface (Supplemental Fig. S4,B).
Biophysical, cell biological, and structure determinations revealed that 2B8 and 4A10 have two distinct binding epitopes on the IL-7Rα. Co-injections of the 2B8 and 4A10 MAbs displayed higher SPR response changes to an IL-7Rα sensor chip relative to co-injections of the individual MAbs alone, suggesting non-overlapping IL-7Rα epitopes (Supplemental Fig. S4,E). Similarly, the MAbs recognized WT and mutant IL-7Rα expressing cells equally well and showed additive binding by increased MFI by flow cytometry (Supplemental Fig. S4, F), indicating that they recognized non-overlapping epitopes.
To determine the precise IL-7Rα binding epitopes of the two MAbs, crystal structures were determined of the Fab/IL-7Rα complexes both to 2.9 Å resolution (Fig. 1, Supplemental Fig. S5 and Table S1). In addition, we solved the crystal structure of the uncomplexed 4A10 Fab to 1.8 Å resolution. The 4A10 Fab bound IL-7Rα to an epitope distal to the membrane, interacting with IL-7Rα residues from β-strands C1, C’1, E1, and corresponding loop region of the D1 domain. In contrast, the 2B8 Fab bound IL-7Rα to a membrane proximal epitope, interacting with IL-7Rα residues from β-strands A2, G2, and loop region of the D2 domain (Fig. 1a). Both Fab/IL-7Rα complexes buried similar amount of surface areas in their interfaces (2B8 Fab/IL-7Rα = 1854 Å2 vs. 4A10 Fab/IL-7Rα = 1903 Å2) and the MAbs utilized both heavy and light chain complementary determining region residues to engage IL-7Rα. The 4A10 Fab binds just on the periphery of the IL-7 binding epitope to IL-7Rα [16]. In a similar manner, the 2B8 Fab interacts on the periphery of the γc [16] and TSLPR [18] epitopes to the IL-7Rα (Fig. 1b). The MAb epitopes are close to the cytokine and second receptor epitopes and showed inhibition of IL-7 receptor signaling at low IL-7 concentrations (data not shown).
Fig. 1.
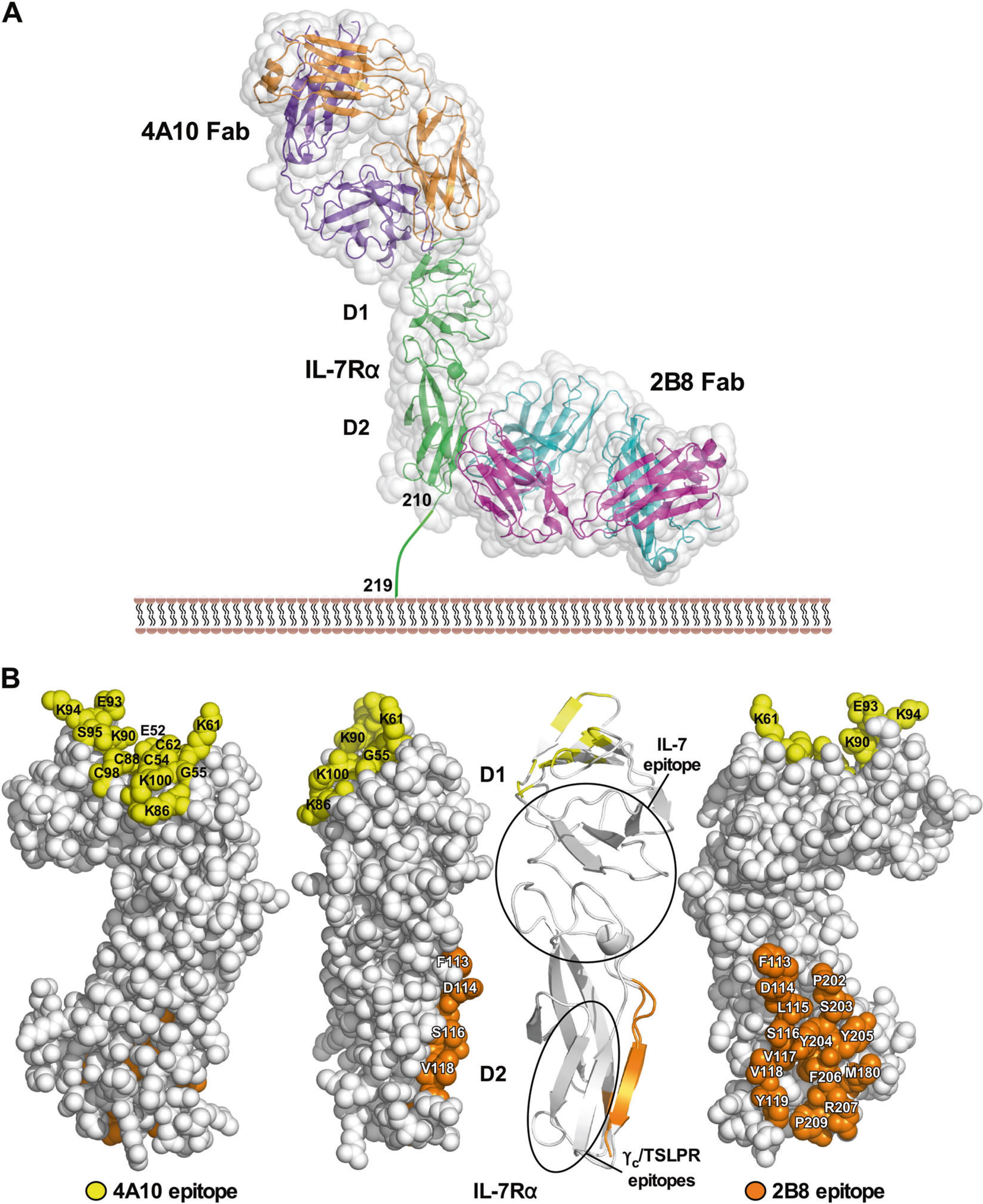
4A10 and 2B8 epitopes on the IL-7Rα ECD. Two crystal structures were determined of the Fab fragments of 4A10 and 2B8 that were bound to the IL-7Rα ECD. a Surface and ribbon diagrams showing where the two MAb Fab fragments bind to the IL-7Rα ECD (green). The 4A10 Fab heavy and light chains are colored orange and purple, respectively, while the 2B8 Fab heavy and light chains are colored in cyan and magenta, respectively. The first and second fibronectin type 3 domains of the IL-7Rα ECD are labeled D1 and D2 accordingly. b Different structural views of the IL-7Rα highlighting the binding epitopes of 4A10 and 2B8. The binding epitopes of IL-7, γc (predicted), and TSLPR are circled on the ribbon diagram
Anti-IL-7Rα MAbs mediate ADCC against patient derived T-ALL cells
The murine MAbs were chimerized with human IgG1 heavy chain sequences to optimize ADCC. Lactate dehydrogenase (LDH) release from target cells was used to quantitate cell-mediated cytotoxicity by fresh human NK cell effectors in vitro. D1-hIL-7Rα P1 cells, which express abundant mutant IL-7Rα, or D1-pMIG (empty vector, no IL-7Rα expression) were used in triplicate as targets (Fig. 2a, left panel). Anti-IL-7Rα 4A10 mediated 60% cytotoxicity at effector to target (E:T) ratios of 30:1 and 90:1, while no killing was observed in D1-hIL-7Rα P1 cells without MAbs (Fig. 2a, right panel). No killing was observed in control D1-pMIG cells. These controls indicate the specificity of anti-IL-7Rα 4A10 in mediating ADCC by targeting cell surface expressed IL-7Rα.
Fig. 2.

IL-7Rα is expressed on T-ALL and anti-IL-7Rα mediates ADCC. a IL-7Rα expression and ADCC of murine D1-hIL-7Rα P1 and D1-pMIG. (Left) D1-hIL-7Rα P1 and D1-pMIG gated on GFP+ cells. Histogram of IL-7Rα using a commercial MAb (blue) with an unstained (red) control. (Right) LDH release assay using D1-hIL-7Rα P1 or D1-pMIG as target cells and freshly isolated human NK cells as effectors. Cells were incubated for 4 h in medium alone, or medium with 10 μg/mL of anti-IL-7Rα 4A10. b IL-7Rα expression and ADCC of 4 human PDX T-ALL. (Left) Patient derived leukemia cells gated on human CD45. Histogram of IL-7Rα (blue) with an unstained (red) control. (Right) LDH release assay using patient derived leukemia cells as targets and freshly isolated human NK cells as effectors. Cells were incubated for 4 h in medium with isotype control, medium with 10 μg/mL anti-IL-7Rα 4A10, 10 μg/mL anti-IL-7Rα 2B8, or the combination of both MAbs
To determine whether IL-7Rα is a relevant target in human T-ALL, a panel of cells from four patient derived xenografts (PDXs) were evaluated for IL-7Rα expression and as ADCC targets. Staining with a commercial anti-IL-7Rα that recognizes a different epitope from our MAbs, a range of IL-7Rα expression was observed, T-ALL#5 > CBAT44179 > CBAT27299 > CBAT37614 (Fig. 2b, left panel). ADCC efficacy against these four PDXs was evaluated in triplicate (Fig. 2b right panel). ADCC was mediated by anti-IL-7Rα 4A10 more effectively than by anti-IL-7Rα 2B8, perhaps relating to higher loading or to a different orientation of the MAbs directed to different epitopes. Combining anti-IL-7Rα 4A10 with anti-IL-7Rα 2B8 showed increased ADCC compared to each Mab alone, particularly against the target CBAT27299 and CBAT37614 which express very low levels of IL-7Rα. Although CBAT27299 and CBAT37614 express low levels of IL-7Rα, the receptors are functional as indicated by STAT-5 phosphorylation following IL-7 stimulation (data not shown). Fratricide of NK cells has been observed in multiple myeloma patients treated with Daratumumab [19]. NK cells have low IL-7Rα expression (Supplemental Fig. 6a) and anti-IL-7Rα-induced NK cell fratricide by ADCC was not observed (Supplemental Fig. 6b).
Anti-IL-7Rα controls the growth of mutant IL-7Rα-driven leukemia in vivo
To evaluate the therapeutic potential of anti-IL-7Rα against leukemia driven by an IL-7Rα mutation, we first evaluated effects against the murine D1 thymocyte line that was transformed to leukemia with the P1 mutant human IL-7Rα. This artificial leukemia, driven by mutant human IL-7Rα, was grown in Rag1−/− mice which lack T and B cells but contain NK cells that can mediate ADCC. Based on previous experience, experiments were planned for 3–20 mice per group. No statistical methods were used to determine sample size. Experiments were performed in an unblinded fashion. Six to 12-week-old male and female mice were injected intravenously with either D1-hIL-7Rα P1 cells (20 mice) or D1-pMIG cells (6 mice) as a control, and randomized by distributing experimental groups across multiple cages and litters. This experimental model with mutant IL-7Rα has been shown to develop aggressive leukemia formation necessitating euthanasia by day 18, while D1-pMIG mice do not develop any signs of leukemia and D1-pMIG cells are not detectable by FACS in the blood [12, 20]. Anti-IL-7Rα 4A10 was initially evaluated rather than 2B8 because of higher ADCC activity (Fig. 2). A single intra venous injection of MAb one day after D1-hIL-7Rα P1 cell transfer significantly extended the survival of treated mice compared to untreated mice (Fig. 3a). In another experiment, D1-hIL-7Rα P1 bearing Rag1−/− mice treated with anti-IL-7Rα 4A10 were euthanized 15 days post leukemia engraftment. Untreated mice (3 mice) developed significant splenomegaly as demonstrated by a 2-fold increase in splenic cellularity, while three mice treated with a single intravenous injection of 250 μg anti-IL-7Rα 4A10 one day after leukemia cell engraftment showed a comparable splenic size and cellularity to a naïve Rag1−/− mouse (Fig. 3b). At day 15, mice treated with a single injection of anti-IL-7Rα 4A10 had undetectable numbers of GFP positive leukemia cells in their spleens, bone marrow, or blood compared to untreated mice, which had a 10–40% leukemia burden in these organs (Fig. 3c). Histologically, mice treated with a single injection of 4A10 had rare small clusters of neoplastic cells in liver and kidney, compared to untreated mice which had high leukemic burden (Supplemental Fig. S7). Thus, in an artificial leukemia model driven by an oncogenic mutant IL-7Rα, MAbs directed against this target showed therapeutic efficacy.
Fig. 3.

Anti-IL-7Rα is effective in controlling the growth of cells with mutated IL-7Rα in RAG1 deficient mice. D1-hIL-7RP1 (IL-7Rα mutant) and D1-pMIG (empty vector) were injected into RAG1−/− mice on day 0 followed by injection of PBS (untreated) or anti-IL-7Rα 4A10 (250 μg, treated) on day 1. a Kaplan Meier curve depicting survival prolongation in D1-hIL-7Rα P1 bearing mice treated with anti-IL-7Rα 4A10. Survival analysis by Log-rank (Mantel-Cox) test. D1-hIL-7Rα P1, treated (n = 10), control mice were either IL-7Rα mutant D1-hIL7Rα P1, untreated (n = 10), or empty vector D1-pMIG, untreated (n = 6) as indicated. b In another experiment, spleens from antibody treated mice (n = 3) were significantly smaller than those of untreated mice (n = 3) at day 15 post cell engraftment. One naïve mouse was included for comparison. c Mice were injected and treated as in a with GFP+ D1-hIL-7Rα P1 cells, and analyzed for leukemia 15 days post cell engraftment in the spleen (left), bone marrow (middle) and peripheral blood (right) compared to untreated control mice (n = 3 for both groups). Leukemia burden was determined by flow cytometry of the green fluorescent protein. Bars represent mean values
Anti-hIL-7Rα monoclonal antibodies control the growth of PDX T-ALL cells in vivo via ADCC-dependent and independent mechanisms
Anti-IL-7Rα MAbs were then evaluated against T-ALL PDX cells in mice. NOD.Cg-PrkdcSCIDIl2rgtm1wjl/SzJ (NSG) mice have been shown to be a superior host for transplanting human leukemia cells to establish xenografts [21]. Leukemia xenografts established in NSG mice closely model human disease, and have been widely used in preclinical treatment studies [22–24]. However, NSG mice lack NK cells, the major mediators of ADCC that our MAbs were engineered to mediate. Nevertheless, anticipating that there could be ADCC-independent inhibitory mechanisms, perhaps by blocking IL-7 receptor signaling, our initial experiments were performed in male and female NSG mice that were 6–12 weeks in age. PDX T-ALL#5 cells (which express WT, not mutant IL-7Rα) were injected intravenously into 28 NSG mice on day 0, and mice were randomized by distributing experimental groups across multiple cages and litters. Intravenous injection of anti-IL-7Rα 4A10 began one day after cell transfer and continued weekly for 5 weeks (Supplemental Fig. S8A). Leukemia engraftment was monitored weekly by accessing the percentage of human CD45 positive cells present in peripheral blood of a subset of mice (n = 3 for each time point). Surprisingly, in the absence of NK cells, leukemia growth was well controlled compared to isotype control treated mice whose blood was 80% engrafted with leukemia 29 days post leukemia cell transfer (Supplemental Fig. S8,B). In another experiment, untreated mice (n = 3) developed a 5-fold increase in splenic cellularity compared to antibody treated mice (n = 3) at 33 days post leukemia engraftment (Supplemental Fig. S8,C). At day 32, treated mice (n = 3) lacked detectable human CD45+ leukemia cells in their spleens compared to controls (n = 3) whose spleens were 80% positive (Supplemental Fig. S8,d, top panel). Bone marrow in treated mice was also significantly improved compared to controls, which were nearly 100% leukemia cells (Supplemental Fig. S8,D, bottom panel). Control treated (n = 13) T-ALL#5 bearing NSG mice developed clinical signs necessitating euthanasia at a median of day 38, compared to treated mice (n = 15) that had a significantly increased median survival of day 48 (Supplemental Fig. S8,E). Two sickly mice were euthanized from the control group for non-leukemia reasons. Thus, anti-IL-7Rα showed significant efficacy in the absence of NK cells mediating ADCC, possibly by inhibiting survival signals from host IL-7, which is active on the human IL-7 receptor. Additional studies (data not shown) using non-chimerized murine MAbs also showed efficacy, and because they were murine IgG1 subclass, which performs poorly at ADCC, it also supports the concept that non-ADCC mechanisms can inhibit leukemia growth in vivo, such as blocking IL-7Rα signaling pathway, complement dependent cytotoxicity, and/or enhanced phagocytosis.
Since the MAbs were effective at mediating ADCC against T-ALL cells in vitro, we explored whether an in vivo model that included NK cells was feasible. This would require that the human leukemia could grow in the presence of mouse NK cells. Fortunately, PDX T-ALL#5 cells were able to expand in 6–8-week-old male and female NOD.SCID mice, which have murine NK cells [25] and have been used as a preclinical model to evaluate antibodies as a monotherapy against xenografts [26, 27]. After PDX injection, mice were randomized by distributing experimental groups across multiple cages and litters. Treatment with 11 weekly intravenous injections of either anti-IL-7Rα or isotype control were begun 1 day after leukemia cell transfer (Fig. 4a). Leukemia burden was monitored weekly in a subset of mice (n = 2–3) by assessing the percentage of human CD45+ cells present in peripheral blood (Fig. 4b). Mice treated with anti-IL-7Rα 4A10 showed delayed leukemia development compared to control mice whose nucleated blood cells were nearly 100% human CD45+ by day 47 post leukemia cell transfer. Treatment significantly increased survival by nearly 4 weeks (median survival of 75 days; n = 7) compared to controls (median survival of 48 days, n = 6) that developed clinical signs, necessitating euthanasia between days 46–53 (Fig. 4c). One mouse in the control group did not develop leukemia and was excluded from analysis. At 33 days post leukemia cell transfer, bone marrow of treated mice had undetectable hCD45+ leukemia cells compared to a 50% leukemia burden in control mice (Fig. 4d and Supplemental Fig. S9). Thus, the presence of NK cells, which are optimal ADCC effectors, increased efficacy of MAb therapy, extending survival by 1 month compared to 10 days in the absence of NK cells.
Fig. 4.
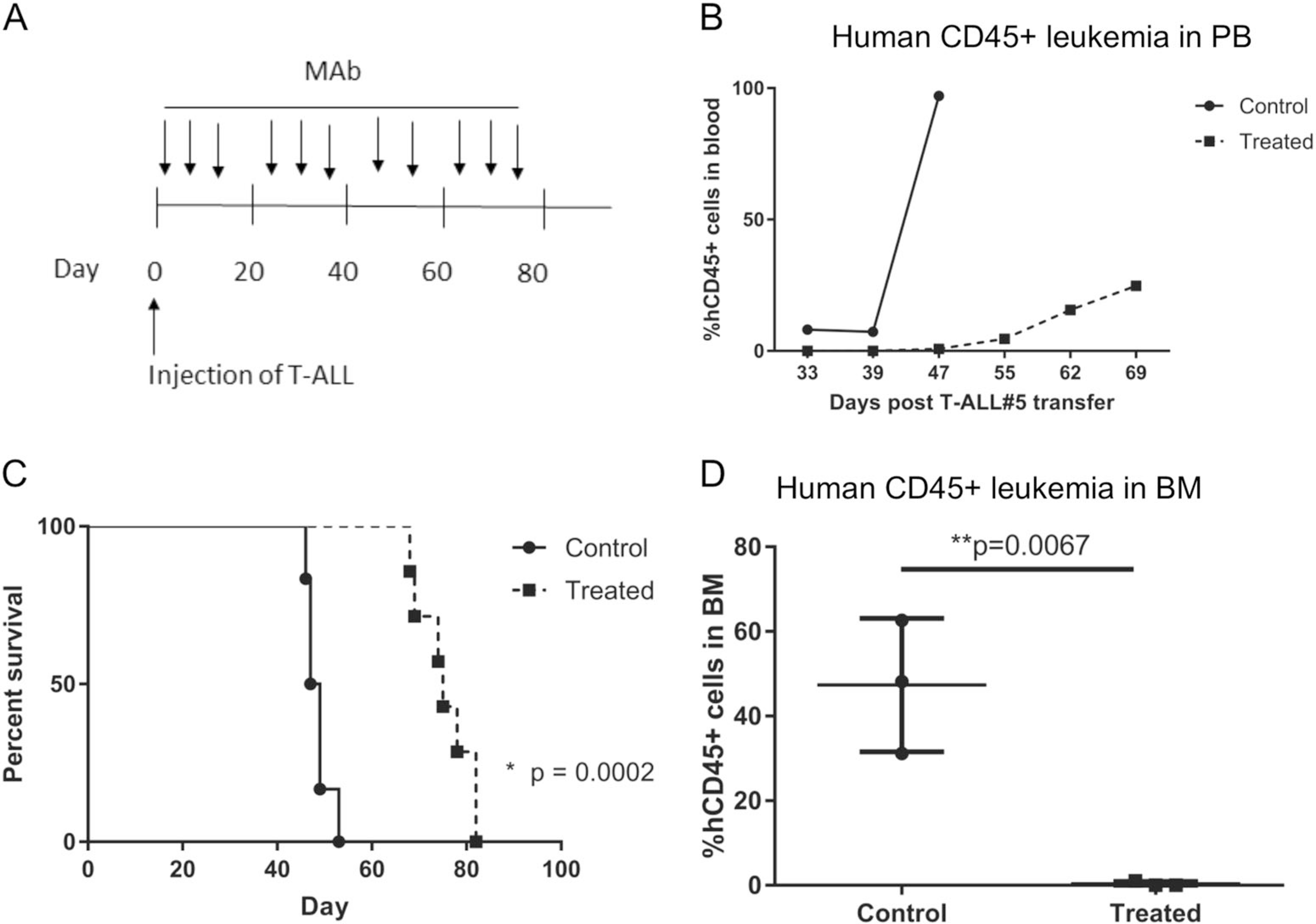
Anti-IL-7Rα controls the growth of patient- derived T-ALL xenografts in NOD. SCID mice. a Experimental protocol. Patient derived T-ALL xenografts were established by intravenous injection in NOD. SCID mice on day 0. Mice were given weekly injections of anti-IL-7Rα or isotype control beginning on day 1 for a total of 11 injections. b Reduction of leukemia burden in the peripheral blood of a subset of treated mice (n = 2–3) compared to isotype treated controls (n = 2–3). c Kaplan Meier curve depicting survival prolongation in xenografts by anti-IL-7Rα 4A10 (treated; n = 7) compared to isotype control treated mice (control; n = 6). One mouse that did not develop leukemia was excluded from the control group. Survival analysis by Log-rank (Mantel-Cox) test. d Leukemia burden in the bone marrow by anti-hIL7Rα 4A10 (treated) at D33, compared to isotype control treated mice. Leukemia burden was determined by flow cytometry using antibodies specific for human CD45. Bars represent mean values and n = 3 for both groups
Anti-hIL-7Rα monoclonal antibodies are effective in controlling established leukemia
Anti-IL-7Rα MAbs were evaluated in a model of advanced disease. PDX T-ALL#5 cells were injected intravenously into 28 NSG mice on day 0, and mice were randomized by distributing experimental groups across multiple cages and litters. Intravenous injection of anti-IL-7Rα 4A10 began when 1% leukemic blasts were detected in the blood of a subset of mice, and continued weekly for 4 weeks (Fig. 5a). Untreated mice (n = 3) showed a 6-fold increase in splenic cellularity and a 4-fold increase in liver cellularity compared to antibody treated mice (n = 3) at 33 days post leukemia engraftment (Fig. 5b). At D33, leukemia burden in the spleen, blood, liver, and lungs of antibody treated mice was significantly reduced compared to untreated mice (Fig. 5c). Thus, MAb therapy was effective in a model of advanced leukemia.
Fig. 5.
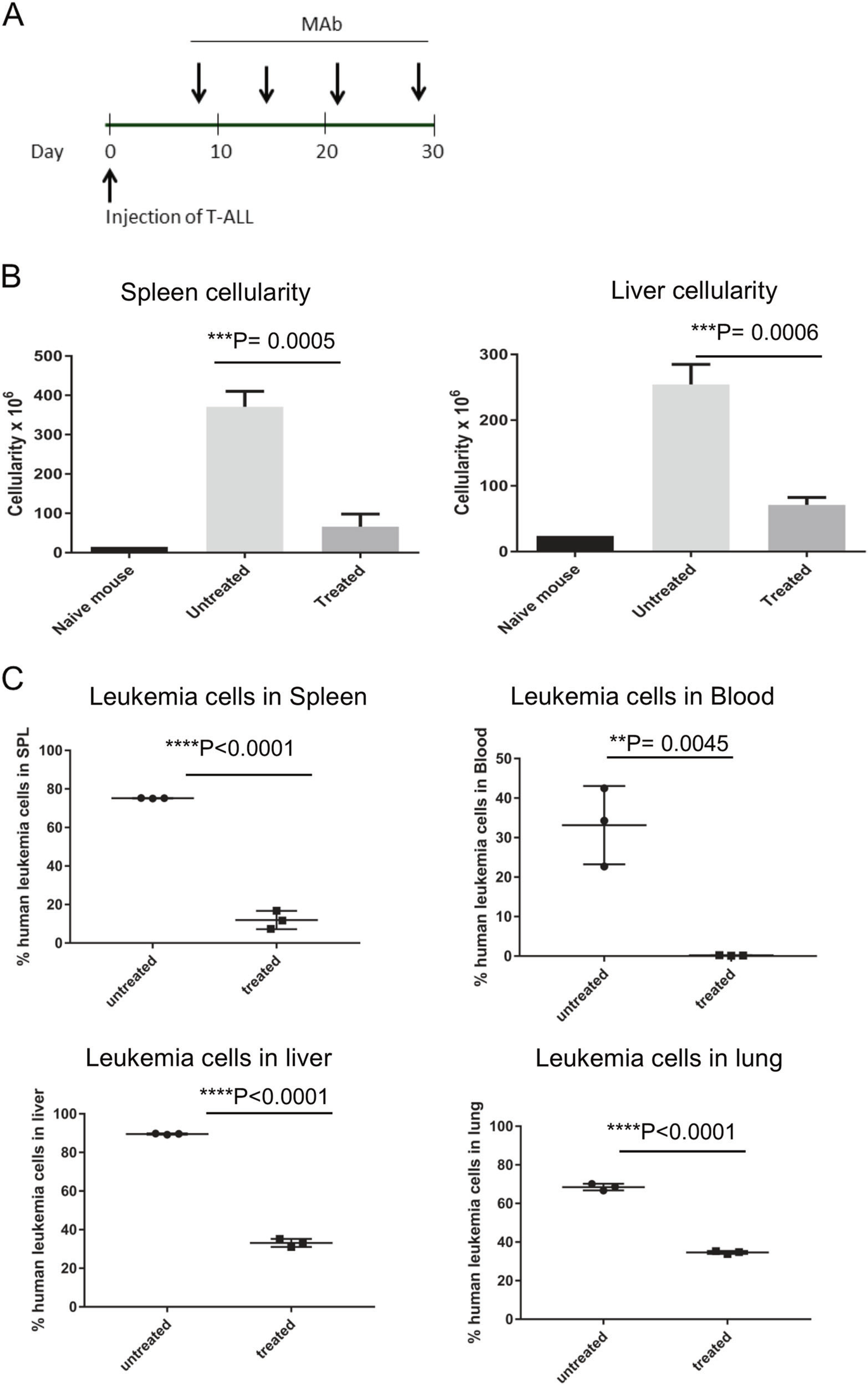
Anti-IL-7Rα controls the growth of established patient derived T-ALL xenografts in NSG mice. a Experimental protocol. Patient derived T-ALL xenografts were established by intravenous injection into NSG mice on day 0. When leukemia burden reached 1% in peripheral blood, mice were given weekly injections of anti-IL-7Rα or isotype control beginning on day 7 for a total of 4 injections. b Spleens and livers from antibody treated mice (n = 3) had significantly decreased cellularity compared to untreated mice (n = 3) at day 33 post cell engraftment. c Leukemia burden in the spleen, blood, liver, lung, and bone marrow was significantly decreased by anti-hIL7Rα 4A10 (treated) at D33, compared to isotype control (untreated) mice. Leukemia burden was determined by flow cytometry using commercial antibodies specific for human CD4 and human IL-7 receptor. Bars represent mean values and n = 3 for both groups
Combining two different anti-hIL-7Rα MAbs may increase efficacy in bone marrow
Because combining two different MAbs loaded more antibody on the target cell (Supplemental Fig. S4D) and increased ADCC efficacy (Fig. 2), the effect of combining the two was evaluated in vivo. T-ALL#5 bearing NOD.SCID mice were treated one day after leukemia cell transfer with either anti-IL-7Rα 4A10, anti-IL-7Rα 2B8, or the combination weekly for 9 weeks, or left untreated. Bone marrow of untreated mice (n = 5) was nearly 100% engrafted with leukemia at day 41. Bone marrow can act as a therapy-resistant reservoir of T-ALL cells. Single MAb therapy was effective in bone marrow of 3 out of 4 mice treated with anti-IL-7Rα 4A10, and 4 of 5 mice treated with anti-IL-7Rα 2B8 at day 55 post leukemia cell transfer, whereas the combination was effective in 5 of 5 (Supplemental Fig. 10a). However, survival was similar in single MAb and combined MAb treatment groups (Supplemental Fig. 10b).
Chemotherapy increased IL-7Rα expression on PDX T-ALL cells and antibody therapy is effective on relapsing leukemia
Multi-agent chemotherapy is the current treatment strategy for ALL [28]. Although the majority of patients are cured, little progress has been made in the treatment of patients who relapse [29]. Analysis of relapsed patients concluded that most relapses occur due to the expansion of clones that were resistant to initial treatment [30]. To determine whether leukemia cells in relapsed patients could be sensitive to anti-IL-7Rα, this chemotherapy regimen was modeled in PDX- bearing NOD.SCID mice. Once sufficient disease burden was detected (>1% human CD45+ cells in the peripheral blood), mice were treated with a combination of daily dexamethasone and weekly vincristine for 4 weeks. One week after the last chemotherapy treatment, relapsing/refractory leukemia hCD45+ leukemia cells were analyzed for human IL-7Rα expression by flow cytometry. Leukemia cells from treated mice (n = 3) had an increased percentage and a 100-fold increase in MFI of IL-7Rα (Fig. 6b) compared to the starting population (Fig. 6a).
Fig. 6.
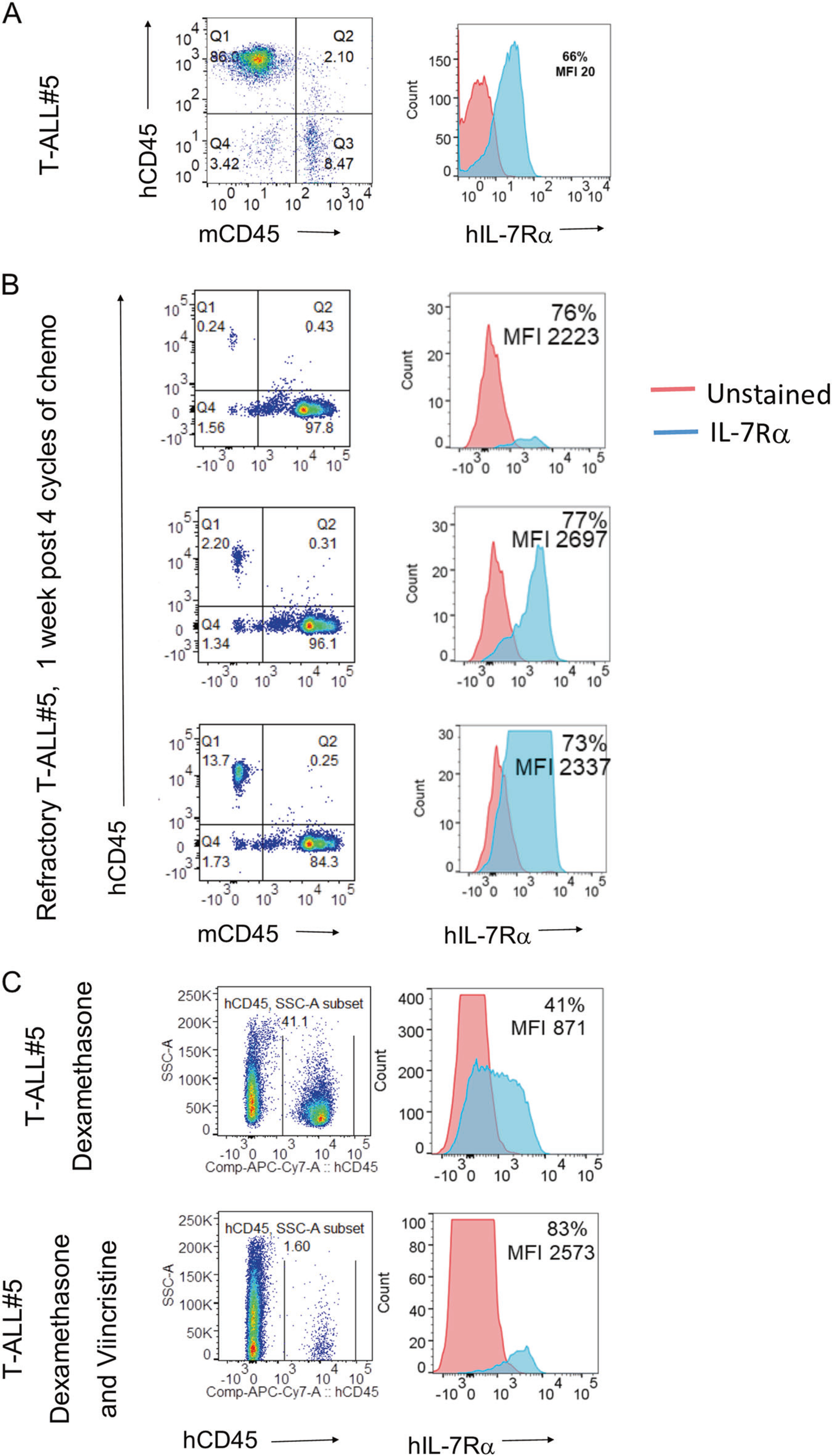
Chemotherapy resistant patient derived leukemia cells express high levels of IL-7Rα. a Percentage of human CD45 positive leukemia cells in the T-ALL PDX cell bank (left). Expression of IL-7Rα on freshly thawed T-ALL PDX cells (right). b Patient derived T-ALL xenografts were injected into NOD.SCID mice on day 0. When leukemia burden reached 1% in peripheral blood, mice were treated with a combination of dexamethasone (5 mg/kg, daily) and vincristine (0.15 mg/kg, weekly) for 4 weeks. One week after the last chemotherapy treatment, relapsing/refractory hCD45+ leukemia cells (left) were analyzed for hIL-7Rα expression by flow cytometry (right) at day 68 post leukemia transfer. Relapsing leukemia cells had increased expression and MFI of IL-7Rα (right side; n = 3). c Patient derived T-ALL xenografts were injected into NOD.SCID mice on day 0. When leukemia burden reached 1% in peripheral blood, mice were randomized into dexamethasone (5 mg/kg, daily), or a combination of dexamethasone and vincristine (0.15 mg/kg, weekly) treatment groups and treated for 6 weeks. Relapsing leukemia cells in surviving mice were analyzed at day 95 (2.5 weeks after last chemotherapy treatment) for hIL-7Rα expression by flow cytometry. Relapsing leukemia cells (left) in mice receiving dexamethasone alone had increased MFI, and mice treated with a combination of dexamethasone and vincristine had increased levels and MFI of IL-7Rα (right; n = 1)
In another experiment, T-ALL#5 engrafted NOD.SCID mice (n = 20) with >1% hCD45+ cells in the peripheral blood were randomized into treatment groups across multiple cages and litters and given either daily dexamethasone, weekly vincristine, or dexamethasone and vincristine in combination (n = 5 mice for each group). Following 6 weeks of chemotherapy, relapsing leukemia cells were analyzed at day 95 for IL-7Rα expression by flow cytometry. Relapsing leukemia cells in mice receiving both dexamethasone and vincristine were nearly all positive for IL-7Rα (Fig. 6c, bottom panel). Only 40% of the relapsing leukemia cells in mice treated with dexamethasone alone displayed significant IL-7Rα expression (Fig. 6c, top panel), which was less than the starting population (Fig. 6a). Both treatment groups had greatly increased MFI compared to the starting population.
Because the IL-7 receptor delivers survival signals to cells, perhaps chemotherapy selected for high IL-7Rα expression. This suggests that anti-IL-7Rα may be a novel agent that could be integrated into existing regimens for relapsed T-ALL. To evaluate this possibility, T-ALL#5 was engrafted into NOD.SCID mice, and the mice were monitored weekly for the presence of human CD45+ leukemia cells. Once the leukemic burden in the blood reached 1%, mice were treated with dexamethasone and vincristine for 4 cycles (Fig. 7a). Leukemia burden was assessed at day 61 post leukemia cell transfer (last day of chemotherapy) in a subset of mice (n = 3) and a small population of chemotherapy resistant leukemia was observed (Fig. 7b). Mice received either a single intravenous injection of anti-IL-7Rα 4A10 combined with 2B8 (250 μg of each MAb) or PBS on day 69. Mice receiving chemotherapy followed by a single injection of MAbs (n = 10) had reduced human CD45 positive leukemia cells at day 75 post leukemia transfer compared to mice that received chemo alone (n = 10) (Fig. 7c). This demonstrates that anti-IL-7Rα can be effective against chemotherapy resistant leukemia in relapsed/refractory T-ALL.
Fig. 7.
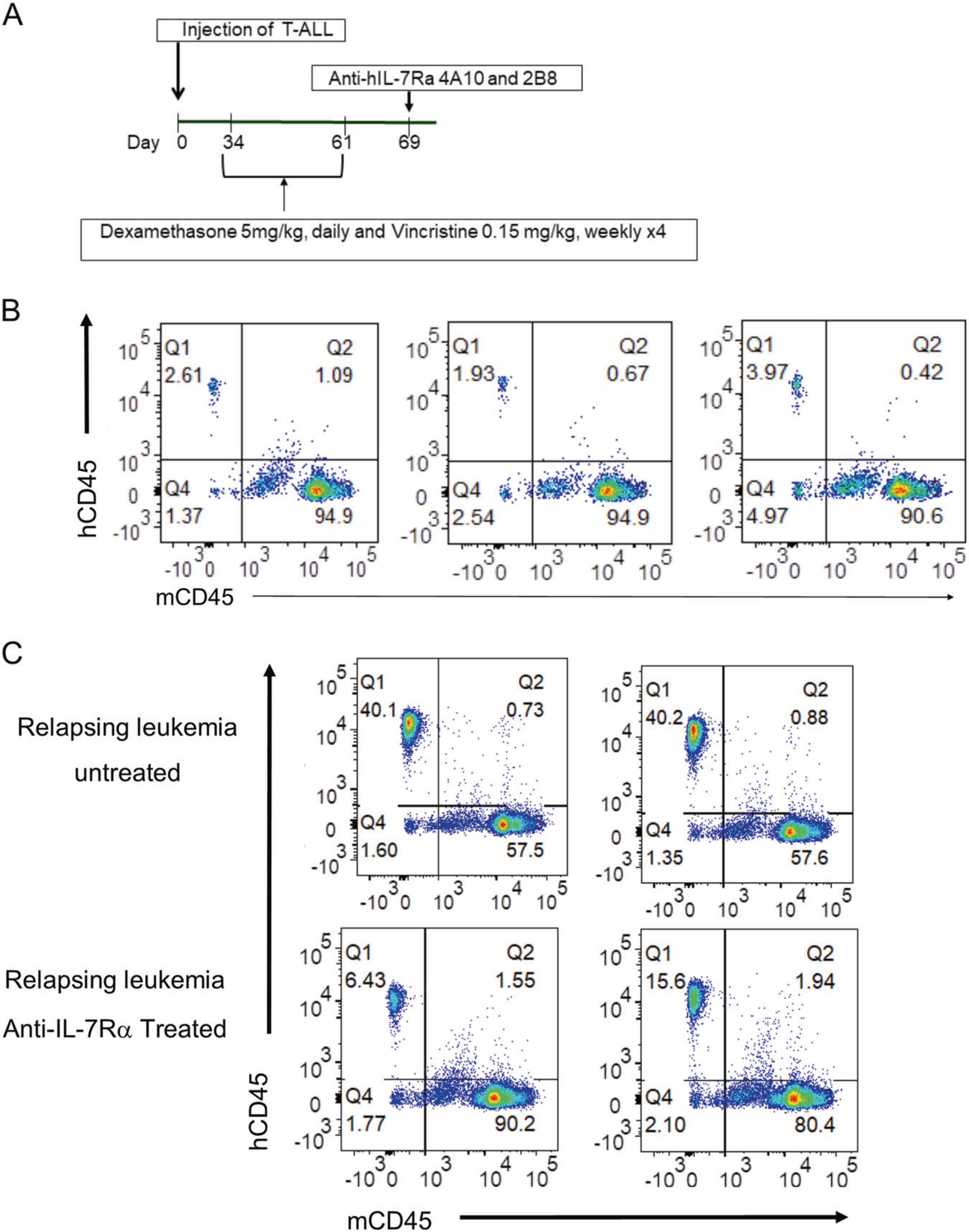
Anti-IL-7Rα is effective in controlling the growth of chemotherapy resistant leukemia in NOD.SCID mice. a Experimental protocol. Patient derived T-ALL xenografts were established by intravenous injection into 20 NOD.SCID mice on day 0. When leukemia burden reached 1% in peripheral blood, mice were treated with a combination of dexamethasone (5 mg/kg, daily) and vincristine (0.15 mg/kg, weekly) for 4 weeks. On day 69, mice were given a single intravenous injection of anti-IL-7Rα 4A10 and 2B8 (250 μg of each MAb) or PBS on day 69. b Chemotherapy resistant leukemia was detected in the blood of mice at day 61 (n = 3). c Growth of chemotherapy resistant leukemia was controlled by a single injection of anti-human IL-7Rα 4A10 combined with 2B8 at day 75 compared to mice that received chemo alone (n = 2)
Discussion
The IL-7 receptor pathway is one of the most vulnerable to mutations in immature T cells that can lead to acute lymphoblastic leukemia. Mutations occur in the IL-7Rα chain, in several signaling components both positive and negative, and IL-7 itself can promote survival of T-ALL cells in the absence of mutations in the pathway [5, 14]. Here, we have shown that newly developed antibodies against IL-7Rα can direct ADCC and other inhibitory mechanisms and have therapeutic benefit against PDX T-ALL cells in mice.
The anti-IL-7Rα MAbs were chimerized to human IgG1 constant regions. This follows the chimeric design of Rituxan which is directed against CD20 and is standard of care for B cell lymphoma. Chimerization is done to reduce immunogenicity of mouse IgG, but of equal importance, murine IgG1 is a poor mediator of ADCC in either mouse or man. The anti-leukemia mechanism of Rituxan is thought to be largely from ADCC [31], but also from other mechanisms. Our MAbs also mediate ADCC. However, perhaps because IL-7 receptor delivers survival signals to lymphocytes, there appears to be an additional ADCC-independent mechanism by which the MAbs inhibit leukemia growth. This was suggested in two ways, first that there is a therapeutic effect, albeit reduced, in mice lacking NK cells. Second, the unchimerized mouse MAbs, which do not mediate ADCC, showed therapeutic benefit.
Our MAbs did not induce rapid internalization, which is a different property from the anti-IL-7Rα MAb described in a related paper (Akkapeddi P et al. Leukemia, 2019 Mar 8. doi:10.1038s41375–019-0438–8. [Epub ahead of print]) which does internalize rapidly. Lack of rapid internalization would be desirable for promoting ADCC as in our model. On the other hand, delivery of a toxin conjugate would be favored by internalization as in the model described in the related paper.
Other therapeutic MAbs are suggested to mediate ADCC that are also augmented by additional inhibitory effects. In treatment of breast cancer, anti-Her2 (Trastuzumab) juxtamembrane region induces both uncoupling from Her3, as well as ADCC [32]. In treatment of squamous cell carcinoma and colon cancer, the mechanism of anti-EGFR (Cetuximab) is based on blocking receptor dimerization, as well as ADCC [33]. It is therefore possible that T-ALL cells in patients will be killed by anti-IL-7Rα MAbs based on several possible mechanisms ranging from blocked signaling to targeting cells for ADCC. We also recognize that, analogous to anti-CD20 killing normal B cells, one can anticipate killing of the patient’s normal T cells.
We note that the MAbs extended life of leukemic mice but were not curative, even in the treatment of an artificial leukemia that was driven by the MAb target, mutant IL-7Rα (Fig. 3). The mechanism of this leukemia escape is under current investigation. In the case of PDX leukemia escape (Fig. 4), we observed downregulation of IL-7Rα on the cell surface on relapsed cells (not shown). Since cells relapsing from chemotherapy showed upregulation of IL-7Rα (Fig. 6), and relapsing leukemia responds to MAb therapy (Fig. 7), it may therefore be optimal to alternate cycles of treatment with chemotherapy followed by treatment with MAbs.
Agents that target the IL-7 receptor pathway could also find clinical use in other malignancies in which this pathway has been implicated. These include B-ALL [34] and non-hematopoietic malignancies such as non-small cell lung cancer [35], breast [36], colorectal [37], renal [38], esophageal [39], and central nervous system cancers [40]. In addition to treatment with unmodified MAbs, they could be coupled to drugs or toxins or used in combination with other chemotherapeutics or biologics. For example, we recently reported combining agents targeting the IL-7 receptor and Ras pathways in a T-ALL model [41]. The V-regions of these MAbs could be used to direct CAR-T cells. A major goal is to reduce the toxicity of therapy for ALL, which although often curative, can lead to life-long side effects.
T-ALL is a heterogenous disease, and other authors have reported variable IL-7 receptor expression in T-ALL samples [8, 42]. This heterogeneity could impact the effectiveness of anti-IL-7Rα and we are actively evaluating T-ALL PDXs with a range of IL-7 receptor expression, including ETPs, which are reported to express higher levels of IL-7 receptor than non-ETP subtypes [42]. One ETP T-ALL sample, CBAT27299 which has low IL-7 receptor expression, was included in this study. We observed anti-IL-7Rα mediated ADCC in vitro, but were unable to evaluate this PDX in vivo due to lack of engraftment in NOD.SCID mice.
The downside of targeting the IL-7 receptor pathway is immunosuppression. T cells can be expected to be depleted because IL-7Rα is expressed on T cells at most immature and mature stages, and we have shown that the MAbs target normal T cells for ADCC (not shown). However, T cell depletion also occurs in conventional chemotherapy and risk of infection is considered manageable. In pediatric ALL patients, the thymus is functional and can restore T cell populations, unlike adults which have poor thymic function.
In conclusion, targeting IL-7Rα with monoclonal antibodies engineered for ADCC offers a promising new therapeutic mechanism for treatment of T-ALL and possibly other malignancies.
Supplementary Material
Acknowledgements
We thank Dr. Curt Civin for T-ALL#5, Roberta Matthai for flow cytometry, and Kelli Czarra for expert animal technical work. X-ray data were collected using the National Institutes of Health GM/CA 23ID beamline at the Advanced Photon Source at Argonne National Laboratory, which is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02–06CH11357. STRW thank Drs. Craig Ogata and Michael Becker of the GM/CA staff for their assistance. This research was supported by extramual support from the National Institute of Allergy and Infectious Diseases and National Cancer Institute of the NIH (STRW) and the Children’s Cancer Foundation (SKD) and intramural support from the NCI-NIH (JPS, STRW, and SKD).
Footnotes
Compliance with ethical standards
Conflict of interest Several of the co-authors (JAH, LK, WL, STRW, and SKD) are listed as co-inventors on the patent application “IL-7R-alpha specific antibodies for treating acute lymphoblastic leukemia” filed by the NIH (DHHS). U.S. Patent Application No. 62/238,612. The remaining authors declare that they have no conflict of interest.
Supplementary information The online version of this article (https://doi.org/10.1038/s41375–019-0531–8) contains supplementary material, which is available to authorized users.
References
- 1.Takahashi H, Kajiwara R, Kato M, Hasegawa D, Tomizawa D, Noguchi Y, et al. Treatment outcome of children with acute lymphoblastic leukemia: the Tokyo Children’s Cancer Study Group (TCCSG) Study L04–16. Int J Hematol. 2018;108:98–108. [DOI] [PubMed] [Google Scholar]
- 2.Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol. 2011;29:551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vitale A, Guarini A, Chiaretti S, Foa R. The changing scene of adult acute lymphoblastic leukemia. Curr Opin Oncol. 2006;18:652–9. [DOI] [PubMed] [Google Scholar]
- 4.Locatelli F, Schrappe M, Bernardo ME, Rutella S. How I treat relapsed childhood acute lymphoblastic leukemia. Blood. 2012; 120:2807–16. [DOI] [PubMed] [Google Scholar]
- 5.Cramer SD, Aplan PD, Durum SK. Therapeutic targeting of IL-7Ralpha signaling pathways in ALL treatment. Blood. 2016; 128:473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazzucchelli R, Durum SK. Interleukin-7 receptor expression: intelligent design. Nat Rev Immunol. 2007;7:144–54. [DOI] [PubMed] [Google Scholar]
- 7.Barata JT, Cardoso AA, Nadler LM, Boussiotis VA. Interleukin-7 promotes survival and cell cycle progression of T-cell acute lymphoblastic leukemia cells by down-regulating the cyclin-dependent kinase inhibitorp27(kip1). Blood. 2001;98:1524–31. [DOI] [PubMed] [Google Scholar]
- 8.Karawajew L, Ruppert V, Wuchter C, Kosser A, Schrappe M, Dorken B, et al. Inhibition of in vitro spontaneous apoptosis by IL-7 correlates with bcl-2 up-regulation, cortical/mature immunophenotype, and better early cytoreduction of childhood T-cell acute lymphoblastic leukemia. Blood. 2000;96:297–306. [PubMed] [Google Scholar]
- 9.Silva A, Laranjeira AB, Martins LR, Cardoso BA, Demengeot J, Yunes JA, et al. IL-7 contributes to the progression of human T-cell acute lymphoblastic leukemias. Cancer Res. 2011;71:4780–9. [DOI] [PubMed] [Google Scholar]
- 10.Touw I, Pouwels K, van Agthoven T, van Gurp R, Budel L, Hoogerbrugge H, et al. Interleukin-7 is a growth factor of precursor B and T acute lymphoblastic leukemia. Blood. 1990;75: 2097–101. [PubMed] [Google Scholar]
- 11.Shochat C, Tal N, Bandapalli OR, Palmi C, Ganmore I, te Kronnie G, et al. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukemias. J Exp Med. 2011;208:901–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zenatti PP, Ribeiro D, Li W, Zuurbier L, Silva MC, Paganin M, et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet. 2011;43:932–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tal N, Shochat C, Geron I, Bercovich D, Izraeli S. Interleukin 7 and thymic stromal lymphopoietin: from immunity to leukemia. Cell Mol Life Sci. 2014;71:365–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wickham J Jr, Walsh ST. Crystallization and preliminary X-ray diffraction of human interleukin-7 bound to unglycosylated and glycosylated forms of its alpha-receptor. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63(Pt 10):865–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McElroy CA, Dohm JA, Walsh ST. Structural and biophysical studies of the human IL-7/IL-7Ralpha complex. Structure. 2009;17:54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim K, Khaled AR, Reynolds D, Young HA, Lee C-K, Durum SK. Characterization of an interleukin-7-dependent thymic cell line derived from ap53−/− mouse. J Immunol Methods. 2003;274: 177–84. [DOI] [PubMed] [Google Scholar]
- 18.Verstraete K, Peelman F, Braun H, Lopez J, Van Rompaey D, Dansercoer A, et al. Structure and antagonism of the receptor complex mediated by human TSLP in allergy and asthma. Nat Commun. 2017;8:14937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Zhang Y, Hughes T, Zhang J, Caligiuri MA, Benson DM, et al. Fratricide of NK cells in daratumumab therapy for multiple myeloma overcome by ex vivo-expanded autologous NK cells. Clin Cancer Res. 2018;24:4006–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Senkevitch E, Li W, Hixon JA, Andrews C, Cramer SD, Pauly GT, et al. Inhibiting Janus Kinase 1 and BCL-2 to treat T cell acute lymphoblastic leukemia with IL7-Ralpha mutations. Oncotarget. 2018;9:22605–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agliano A, Martin-Padura I, Mancuso P, Marighetti P, Rabascio C, Pruneri G, et al. Human acute leukemia cells injected in NOD/LtSz-scid/IL-2Rgamma null mice generate a faster and more efficient disease compared to other NOD/scid-related strains. Int J Cancer. 2008;123:2222–7. [DOI] [PubMed] [Google Scholar]
- 22.Bride KL, Vincent TL, Im SY, Aplenc R, Barrett DM, Carroll WL, et al. Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia. Blood. 2018;131:995–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmfeldt L, Mullighan CG. Generation of human acute lymphoblastic leukemia xenografts for use in oncology drug discovery. Curr Protoc Pharm. 2015;68:14.32.1–14.32.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schewe DM, Alsadeq A, Sattler C, Lenk L, Vogiatzi F, Cario G, et al. An Fc-engineered CD19 antibody eradicates MRD in patient-derived MLL-rearranged acute lymphoblastic leukemia xenografts. Blood. 2017;130:1543–52. [DOI] [PubMed] [Google Scholar]
- 25.Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol. 1995;154:180–91. [PubMed] [Google Scholar]
- 26.Agnusdei V, Minuzzo S, Frasson C, Grassi A, Axelrod F, Satyal S, et al. Therapeutic antibody targeting of Notch1 in T-acute lymphoblastic leukemia xenografts. Leukemia. 2014; 28:278–88. [DOI] [PubMed] [Google Scholar]
- 27.Lee EM, Yee D, Busfield SJ, McManus JF, Cummings N, Vairo G, et al. Efficacy of an Fc-modified anti-CD123 antibody (CSL362) combined with chemotherapy in xenograft models of acute myelogenous leukemia in immunodeficient mice. Haematologica. 2015;100:914–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiaretti S, Jabbour E, Hoelzer D. “Society of Hematologic Oncology (SOHO) state of the art updates and next questions”-treatment of ALL. Clin Lymphoma Myeloma Leuk. 2018;18: 301–10. [DOI] [PubMed] [Google Scholar]
- 29.Nguyen K, Devidas M, Cheng SC, La M, Raetz EA, Carroll WL, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia. 2008;22:2142–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson NS, Yang B, Yang A, Loeser S, Marsters S, Lawrence D, et al. An Fcgamma receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell. 2011;19:101–13. [DOI] [PubMed] [Google Scholar]
- 32.Garrett JT, Arteaga CL. Resistance to HER2-directed antibodies and tyrosine kinase inhibitors: mechanisms and clinical implications. Cancer Biol Ther. 2011;11:793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brand TM, Iida M, Wheeler DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther. 2011;11:777–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ming J, Jiang G, Zhang Q, Qiu X, Wang E. Interleukin-7 up-regulates cyclin D1 via activator protein-1 to promote proliferation of cell in lung cancer. Cancer Immunol Immunother. 2012;61:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Rawi MA, Rmali K, Watkins G, Mansel RE, Jiang WG. Aberrant expression of interleukin-7 (IL-7) and its signalling complex in human breast cancer. Eur J Cancer. 2004;40:494–502. [DOI] [PubMed] [Google Scholar]
- 37.Maeurer MJ, Walter W, Martin D, Zitvogel L, Elder E, Storkus W, et al. Interleukin-7 (IL-7) in colorectal cancer: IL-7 is produced by tissues from colorectal cancer and promotes preferential expansion of tumour infiltrating lymphocytes. Scand J Immunol. 1997;45:182–92. [DOI] [PubMed] [Google Scholar]
- 38.Trinder P, Seitzer U, Gerdes J, Seliger B, Maeurer M. Constitutive and IFN-gamma regulated expression of IL-7 and IL-15 in human renal cell cancer. Int J Oncol. 1999;14:23–31. [DOI] [PubMed] [Google Scholar]
- 39.Kim MJ, Choi SK, Hong SH, Eun JW, Nam SW, Han JW, et al. Oncogenic IL7R is downregulated by histone deacetylase inhibitor in esophageal squamous cell carcinoma via modulation of acetylated FOXO1. Int J Oncol. 2018;53:395–403. [DOI] [PubMed] [Google Scholar]
- 40.Cosenza L, Gorgun G, Urbano A, Foss F. Interleukin-7 receptor expression and activation in nonhaematopoietic neoplastic cell lines. Cell Signal. 2002;14:317–25. [DOI] [PubMed] [Google Scholar]
- 41.Cramer SD, Hixon JA, Andrews C, Porter RJ, Rodrigues GOL, Wu X, et al. Mutant IL-7Ralpha and mutant NRas are sufficient to induce murine T cell acute lymphoblastic leukemia. Leukemia. 2018;32:1795–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maude SL, Dolai S, Delgado-Martin C, Vincent T, Robbins A, Selvanathan A, et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood. 2015;125:1759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.