

Abstract
The occurrence of multidrug resistance (MDR) associated with the overexpression of the ATP-binding cassette (ABC) protein ABCB1 in cancer cells remains a significant obstacle to successful cancer chemotherapy. Therefore, discovering modulators that are capable of inhibiting the drug efflux function or expression of ABCB1 and re-sensitizing multidrug-resistant cancer cells to anticancer agents is of great clinical importance. Regrettably, due to potential adverse events associated with drug-drug interactions and toxicity in patients, researchers have struggled to develop a synthetic inhibitor of ABCB1 that is clinically applicable to improve the effectiveness of chemotherapy. Alternatively, through drug repositioning of approved drugs, we discovered that the FMS-like tyrosine kinase-3 (FLT3) inhibitor midostaurin blocks the drug transport function of ABCB1 and resensitizes ABCB1-overexpressing multidrug-resistant cancer cells to conventional chemotherapeutic drugs. Our findings were further supported by results demonstrating that midostaurin potentiates drug-induced apoptosis in ABCB1-overexpressing cancer cells and inhibits the ATPase activity of ABCB1. Considering that midostaurin is a clinically approved anticancer agent, our findings revealed an additional action of midostaurin and that patients with multidrug-resistant tumors may benefit from a combination therapy of midostaurin with standard chemotherapy, which should be further investigated.
Keywords: Chemoresistance, Combination chemotherapy with PKC412, Drug repositioning, Modulator, P-glycoprotein
1. Introduction
The overexpression of the ATP-Binding Cassette (ABC) drug transporter ABCB1 (P-glycoprotein/MDR1) is a major contributing factor in the development of multidrug resistance (MDR) in cancer cells [1,2]. ABCB1 was the first human ABC protein discovered to be capable of reducing the intracellular concentration and cytotoxicity of ABCB1 substrate drugs by transporting drugs directly out of cancer cells using energy from ATP hydrolysis [1–4]. Consequently, patients with multidrug-resistant tumors become insensitive to a broad spectrum of chemotherapeutic agents, which often leads to cancer recurrence and treatment failure [5,6]. ABCB1 is especially infamous for contributing to the MDR phenotype in metastatic breast cancer [7], multiple myeloma (MM) [8–13], acute myelogenous leukemia (AML) [14], chronic myeloid leukemia (CML) [15] and chronic lymphocytic leukemia (CLL) [16]. Moreover, as an important endogenous protective mechanism against xenobiotics, ABCB1 is highly expressed in cells forming the blood-brain barrier (BBB) and blood-tissue barrier (BTB) sites, such as those in the liver, kidney and gastrointestinal tract, affecting the absorption, distribution, metabolism, and elimination of almost all classes of drugs [17,18]. Therefore, identifying factors or drugs that may alter the function and/or protein expression of ABCB1 is of importance to drug developers and clinicians. It is worth noting that in addition to ABCB1, ABCC1 (multidrug-resistance protein 1, MRP1) and ABCG2 (breast cancer resistance protein, BCRP) are two other members of the ABC protein superfamily that have often been linked to the development of the MDR phenotype in cancer patients [6,19].
Although many innovative strategies have been proposed over the years, a practical approach to reversing MDR mediated by ABCB1 is to directly increase the accumulation and efficacy of anticancer agents by transiently blocking the drug transport function of ABCB1 with a selective, competitive modulator of ABCB1 in multidrug-resistant cancer cells [20,21]. Unfortunately, previous work on developing clinically applicable novel synthetic inhibitors of ABCB1 ended in failure, mostly due to complications associated with adverse drug-drug interactions, high intrinsic toxicity and the lack of selectivity caused by the overlapping substrate specificity of ABCB1, ABCC1 and ABCG2 [5,21,22]. An alternative approach is to apply the drug repositioning (also referred to as repurposing) approach to identify US Food and Drug Administration (FDA)-approved therapeutic agents that are capable of re-sensitizing ABCB1-overexpressing multidrug-resistant cancer cells to standard chemotherapeutic drugs [20,21,23–27].
In the present study, we investigated the effect of midostaurin on the drug transport function of ABCB1 and the chemosensitivity of ABCB1-overexpressing multidrug-resistant cancer cells. Midostaurin (PKC412) is an orally bioavailable multikinase inhibitor that has been approved recently by the FDA and the European Medicines Agency (EMA) for the treatment of adult patients with newly diagnosed fms-like tyrosine kinase-3 (FLT3)-mutated AML and advanced systemic mastocytosis (SM) [28]. Midostaurin was initially found to be an inhibitor of angiogenesis [29] and protein kinase C (PKC) activity [30], with a significant antiproliferative activity in human cancer cell lines [31] and in murine xenograft models [32,33]. It was not until more recently that midostaurin was discovered to have a potent activity against FLT3 tyrosine kinase [34], a mutant form of KIT proto-oncogene receptor tyrosine kinase that drives advanced SM [35]. More importantly, clinical studies have demonstrated the favorable safety profile of midostaurin and the significant benefits of treating patients with newly diagnosed FLT3-mutated AML with midostaurin in combination with standard chemotherapy [34,36]. Our data revealed some additional effects of midostaurin. This drug selectively inhibits the transport function of ABCB1, enhances drug-induced apoptosis and more significantly, reverses MDR in ABCB1-overexpressing cancer cells at nanomolar non-toxic concentrations. In summary, our study indicates that midostaurin could potentially be used in combination with other chemotherapeutic drugs to treat patients with drug-resistant cancer associated with the overexpression of ABCB1, and this needs further investigation.
2. Materials and methods
2.1. Chemicals
Phosphate-buffered saline (PBS), fetal calf serum (FCS), Dulbecco’s Modified Eagle’s medium (DMEM), Iscove’s Modified Dulbecco’s Medium (IMDM), RPMI-1640 medium, trypsin-EDTA, penicillin and streptomycin were purchased from Gibco, Invitrogen (CA, USA). Tools Cell Counting (CCK-8) Kit was purchased from Biotools Co., Ltd (Taipei, Taiwan). Annexin V: FITC Apoptosis Detection Kit was purchased from BD Pharmingen (San Diego, CA, USA). Midostaurin (PKC412) was purchased from Selleckchem (Houston, TX, USA). Verapamil, MK-571, Ko143 and all other chemicals were purchased from Sigma (St. Louis, MO, USA), unless stated otherwise.
2.2. Cell culture conditions
The human KB-3–1 epidermal cancer cells and the ABCB1-over-expressing variant KB-V1 cells, transfected human embryonic kidney (HEK) 293 cells overexpressing human ABCB1 (MDR19-HEK293) or human ABCC1 (MRP1-HEK293) or wild-type human ABCG2 (R482-HEK293), as well as mouse fibroblast NIH3T3 cells and NIH3T3 cells transfected with human ABCB1 (NIH3T3-G185) were cultured in DMEM. The human OVCAR-8 ovarian cancer cells and the ABCB1-overexpressing variant NCI-ADR-RES cells were cultured in RPMI-1640. KB-V1 cells were maintained in 1 mg/mL vinblastine [2], NIH3T3-G185 cells were maintained in 60 ng/mL colchicine [37], and stable transfectants were maintained in media containing 2 mg/mL G418 [38]. All cells were grown in medium supplemented with 10% FCS, 2mM L-glutamine and 100 units of penicillin/streptomycin/mL at 37 °C in 5% CO2 humidified air and maintained in drug-free medium for 7 days prior to assay.
2.3. Fluorescent drug accumulation assay
To monitor the drug efflux function of ABCB1, ABCC1 and ABCG2, we used calcein as a fluorescent substrate of ABCB1 and ABCC1, and pheophorbide A (PhA) as a fluorescent substrate of ABCG2 as described previously [39]. Briefly, calcein-AM (0.5 μM) or PhA (1 μM) was added to 3 × 105 cell suspension in 4 mL of IMDM supplemented with 5% FCS in the presence or absence of midostaurin (10 μM) or verapamil (20 μM), a reference inhibitor of ABCB1, MK-571 (25 μM), a reference inhibitor of ABCC1 or Ko143 (3 μM), a reference inhibitor of ABCG2. Cells were washed and resuspended in ice-cold PBS. The intracellular accumulation of calcein (excitation and emission wavelengths of 485 and 535 nm) or PhA (excitation and emission wavelengths of 395 and 670 nm) was measured using a FACScan flow cytometer (Becton-Dickinson Biosciences, San Jose, CA, USA) and analyzed using CellQuest software and FlowJo software (Tree Star, Inc., Ashland, OR, USA) according to the method described by Gribar et al. [40].
2.4. Cell viability assay
Cell viability assays were performed according to the method described by Ishiyama et al. [41] to evaluate the cytotoxicity of therapeutic drugs in various cell lines. Briefly, 5000 cells were seeded in 96-well plates containing 100 μL of culture medium and maintained at 37 °C for 24 h. An additional 100 μL of the respective regimen was added to each well to make a final volume of 200 μL and incubated for an additional 72 h. CCK-8 reagent was used to determine the toxicity of drugs in HEK293 cells and HEK293 cells stably transfected with human ABCB1, ABCC1 or ABCG2, whereas MTT reagent was used for attached cancer cells. IC50 values were calculated from fitted concentration-response curves obtained from at least three independent experiments. For the drug resistance reversal experiments, a nontoxic concentration of midostaurin or a reference inhibitor of ABCB1, ABCC1 or ABCG2, was added to the cytotoxicity assays. The extent of reversal was determined based on the calculated fold-reversal (FR) values, as described previously [38,42].
2.5. Immunoblotting
Cells were treated with increasing concentrations of midostaurin for 72 h before they were harvested and subjected to SDS-polyacrylamide electrophoresis. For the Western blot immunoassay, each blot was then incubated in blocking buffer containing 5% (w/v) milk powder in 0.1% TBS-Tween (25 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 0.1% Tween 20) for 1 h before the addition of primary antibody 1: 3000 dilution of C219 or 1:100000 dilution of α-tubulin for the detection of ABCB1 and positive loading control tubulin, respectively. 1:100000 dilution of the horseradish peroxidase-conjugated goat anti-mouse IgG was used as the secondary antibody. Signals were detected using an Immobilon enhanced chemiluminescence (ECL) kit from Merck Millipore (Billerica, MA, USA), as described previously [38].
2.6. Apoptosis assay
The percentage of apoptotic cells in the total cell population was determined using the conventional annexin V–FITC and propidium iodide (PI) staining method, as described previously [27]. Briefly, cells were first treated with DMSO, colchicine, midostaurin or a combination of colchicine and midostaurin for 48 h before being harvested, centrifuged and resuspended in FACS buffer containing 1.25 μg/mL annexin V–FITC (PharMingen) and 0.1 mg/mL PI and incubated for 15 min at room temperature. FACScan equipped with CellQuest software (BD Biosciences) was used to analyze labeled cells (10000 per sample) after each regimen. Early apoptotic cells with intact plasma membranes are shown as phosphatidylserine (PS)-positive and PI-negative cells (lower right dot-plot quadrant), whereas necrotic or late apoptotic cells with leaky membranes are shown as PS-positive and PI-positive cells (upper right dot-plot quadrant) [43].
2.7. ABCB1 ATPase assay
The effect of midostaurin on the vanadate (Vi)-sensitive ATPase activity of ABCB1 was determined based on the endpoint Pi assay using ABCB1-overexpressing High-Five insect cell membrane vesicles (Invitrogen, Carlsbad, CA, USA) as described previously [3]. Briefly, High-Five cells were infected with recombinant baculovirus carrying the MDR1 gene with a hexahistidine (His6) tag at the C-terminal end as described previously [44]. Total membrane vesicles were prepared by hypotonic lysis and differential centrifugation as previously reported [45]. Total membranes (100 μg protein/mL) were incubated in the presence or absence of 0.3 mM sodium orthovanadate in a buffer containing 50 mM MES-Tris pH 6.8, 50 mM KCl, 10 mM MgCl2, 1 mM EGTA, 1 mM ouabain and 2 mM DTT. Basal ATPase activity was measured in the presence of 1% v/v DMSO, whereas midostaurin-modulated activity was measured in the presence of increasing concentrations of midostaurin dissolved in DMSO. The reaction was started by the addition of 5 mM ATP at 37 °C and stopped after 20 min by addition of 2.5% SDS, and the amount of inorganic phosphate (Pi) released was quantified using a colorimetric method. The Vi-sensitive activity was determined and the IC50 value was calculated from fitted concentration-response curve obtained from three independent experiments using GraphPad Prism software v.7 as described previously [3].
2.8. Docking of midostaurin in the drug-binding pocket of ABCB1
A homology model of human ABCB1 was generated using the mouse P-glycoprotein crystal structure (PDB:5KPI) [46] as a template and the human ABCB1 sequence (UniprotKB:P08183) using SWISS-MODEL [47]. Ligand and receptor structures were prepared for docking using MGLTools software [48]. AutoDock Vina software was used for extensive docking of midostaurin. A total of 33 residues of ABCB1 were set as flexible side chains: L65, M68, M69, F72, Q195, F303, I306, Y307, Y310, F314, F336, L339, I340, F343, Q347, N721, Q725, F728, F732, F759, F770, F938, F942, M949, Y953, F957, L975, F978, V982, M986, Q990, S993 and F994. The receptor grid was centered at x = 20, y = 60 and z = 5 Å, whereas the inner box dimensions were 50 × 50 × 50 Å and the exhaustiveness was set at 100.
2.9. Quantification and statistical analysis
Experimental data and IC50 values are presented as mean ± standard deviation (SD) calculated from at least three independent experiments, unless stated otherwise. KaleidaGraph (Reading, PA, USA) and GraphPad Prism (La Jolla, CA, USA) software were used for curve plotting and statistical analysis. The difference between mean values of experimental and control or improvement in fit was analyzed by two-sided Studenťs t-test and labeled “statistically significant” if the probability, p, was less than 0.05.
3. Results
3.1. Midostaurin inhibits the drug transport function of ABCB1
To examine the effect and selectivity of midostaurin on drug transport mediated by the major MDR-linked ABCB transporters ABCB1, ABCC1 and ABCG2, we first measured the accumulation of their respective fluorescent substrate drugs in parental HEK293 cells and HEK293 cells transfected with human ABCB1 (MDR19-HEK293), human ABCC1 (MRP1-HEK293) or human ABCG2 (R482-HEK293) cells in the presence of DMSO (solid lines), 10 μM of midostaurin (shaded, solid lines), or a reference inhibitor (dotted lines) of ABCB1 (verapamil at 20 μM), or ABCC1 (MK-571 at 25 μM) or ABCG2 (Ko143 at 1 μM), and analyzed as described in Materials and methods. We discovered that midostaurin markedly increased the accumulation of calcein-AM, a known substrate of ABCB1 [49], in MDR19-HEK293 cells (Fig. 1A). In contrast, midostaurin had little effect on the accumulation of calcein, a known fluorescent substrate of ABCC1 in MRP1-HEK293 cells (Fig. 1B) or pheophorbide A (PhA), a known fluorescent substrate of ABCG2 [50] in R482-HEK293 cells (Fig. 1C). Knowing that midostaurin inhibits ABCB1-mediated drug transport in MDR19-HEK293 cells, we further examined the effect of midostaurin on drug efflux mediated by ABCB1 in paired drug-sensitive and ABCB1-overexpressing MDR cancer cell lines. We found that ABCB1-mediated transport of calcein-AM was fully inhibited by 10 μM of midostaurin (shaded, solid lines) in both ABCB1-overexpressing human KB-V1 epidermal cancer cells (Fig. 1D) and human NCI-ADR-RES ovarian cancer cells (Fig. 1E). Of note, midostaurin had no significant effect on the accumulation of fluorescent drugs in HEK293 cells or in drug-sensitive parental KB-3–1 and OVCAR-8 cancer cell lines (Fig. 1A–E, left panels).
Fig. 1. Midostaurin inhibits ABCB1-mediated drug transport in ABCB1-overexpressing cells.

Drug-sensitive parental pcDNA-HEK293 cells (A - C, left panels), ABCB1-transfected MDR19-HEK293 cells (A, right panel), ABCC1-transfected MRP1-HEK293 cells (B, right panel), ABCG2-transfected R482-HEK293 cells (C, right panel), human KB-3–1 epidermal cancer cells (D, left panel), ABCB1-overexpressing variant KB-V1 cancer cells (D, right panel), human OVCAR-8 ovarian cancer cells (E, left panel) and ABCB1-overexpressing variant NCI-ADR-RES cancer cells (E, right panel), were treated with calcein-AM, a known fluorescent substrate of ABCB1 and ABCC1, or PhA, a known fluorescent substrate of ABCG2, in the presence of DMSO (control, solid lines), or 10 μM midostaurin (shaded, solid lines), or 20 μM of ABCB1 reference inhibitor verapamil (A, D and E, dotted lines), or 25 μM of ABCC1 reference inhibitor MK-571 (B, dotted lines), or 1 μM of ABCG2 reference inhibitor Ko143 (C, dotted lines) as indicated. The intracellular accumulation of the respective fluorescent substrate in each cell line was analyzed immediately by flow cytometry as described previously [38]. Representative histograms of three independent experiments and immunoblots of total cell expression of ABCB1 (A, D and E, inset) or ABCC1 (B, inset) or ABCG2 (C, inset) in total cell lysate protein (10 μg/lane) from pcDNA-HEK293, MDR19-HEK293, MRP1-HEK293, R482- HEK293, KB-3–1, KB-V-1, OVCAR-8 and NCI-ADR-RES cells are shown.
3.2. Midostaurin reverses ABCB1-mediated multidrug resistance
We examined the reversal effect of midostaurin on drug resistance mediated by ABCB1, ABCC1 or ABCG2 in MDR19-HEK293, MRP1-HEK293 and R482-HEK293 cells, respectively. At non-toxic concentrations of 20 nM and 50 nM, midostaurin restored the chemosensitivity of MDR19-HEK293 cells to paclitaxel, a known substrate chemotherapy drug of ABCB1 [51], by approximately 3-fold and 7-fold, respectively (Fig. 2A). In contrast, midostaurin had no significant effect on etoposide resistance mediated by ABCC1 in MRP1-HEK293 cells (Fig. 2B) or topotecan resistance mediated by ABCG2 in R482-HEK293 cells (Fig. 2C), which is consistent with what we observed in the drug accumulation assays (Fig. 1). Of note, verapamil (5 μM), MK-571 (25 μM) and Ko143 (1 μM) are reference inhibitors for the reversal of drug resistance mediated by ABCB1, ABCC1 and ABCG2. Our data indicate that midostaurin selectively reverses ABCB1-mediated multidrug resistance. The fold-reversal (FR) value [42] representing the chemosensitization effect of midostaurin on the extent of drug resistance of the respective multidrug-resistant cell line to a particular substrate drug was calculated as the ratios of IC50 values in the absence and presence of midostaurin (see Table 1). Next, the chemosensitizing effect of midostaurin was further tested in ABCB1-overexpressing human cancer cell lines. At 200 nM, midostaurin significantly resensitized drug-resistant KB-V1 and NCI-ADR-RES cancer cells to paclitaxel by approximately 20-fold and 24-fold, respectively (Fig. 2D and E). We found that in addition to paclitaxel, midostaurin was able to resensitize KB-V1 and NCI-ADR-RES cancer cells to other known substrate drugs of ABCB1, such as vincristine and colchicine [51,52] in a concentration-dependent manner (Table 2). It is worth noting that midostaurin at the tested concentrations had no significant effect on the proliferation of HEK293 cells, drug-sensitive KB-3–1 or OVCAR-8 parental cancer cell lines. In contrast, verapamil at a non-toxic concentration of 5 μM significantly enhanced the cytotoxicity of vincristine in KB-3–1 and OVCAR-8 cells (Table 2), which is consistent with previous studies reporting that verapamil enhances the cytotoxicity of vincristine by 3-fold in drug-sensitive cancer cells at a non-toxic concentration of 6.6 μM [53,54]. Overall, our results revealed that midostaurin is capable of reversing ABCB1-mediated multidrug resistance in cancer cells at nanomolar concentrations.
Fig. 2. Midostaurin re-sensitizes ABCB1-overexpressing cells to substrate chemotherapy drug of ABCB1.

Parental HEK293 cells (A - C, left panels), MDR19-HEK293 cells (A, right panel), MRP1-HEK293 cells (B, right panel) and R482-HEK293 cells (C, right panel) were treated with increasing concentrations of ABCB1 substrate chemotherapy drug paclitaxel, ABCC1 substrate chemotherapy drug etoposide, or ABCG2 substrate chemotherapy drug SN-38, respectively, in the presence of DMSO (open circles), or midostaurin at 20 nM (open squares) or at 50 nM (filled squares), or a reference inhibitor as a positive control (filled circles). (D) Drug-sensitive parental KB-3–1 cancer cells (left panel) and ABCB1-overexpressing variant KB-V1 cancer cells (right panel), (E) drug-sensitive parental OVCAR-8 cancer cells (left panel) and ABCB1-over-expressing variant NCI-ADR-RES cancer cells (right panel) were treated with paclitaxel in the presence of DMSO (open circles), or midostaurin at 50 nM (open squares) or at 100 nM (filled squares) or at 200 nM (open triangles). Points, mean values from at least three independent experiments; bars; S.E.M.
Table 1.
Chemosensitization effect of midostaurin on drug resistance mediated by ABCB1, ABCC1 or ABCG2 in HEK293 cells.
| Treatment | Concentration (nM) | Mean IC50a ± SD and (FRb) |
|
|---|---|---|---|
| pcDNA-HEK293 [nM] | MDR19-HEK293 (ABCB1) [nM] | ||
| Paclitaxel | – | 1.67 ± 0.18 (1.0) | 813.43 ± 100.82 (1.0) |
| + midostaurin | 20 | 1.47 ± 0.15 (1.1) | 295.11 ± 41.97** (2.8) |
| + midostaurin | 50 | 1.34 ± 0.14 (1.2) | 118.47 ± 16.94*** (6.9) |
| + verapamil | 5000 | 1.29 ± 0.15 (1.3) | 6.85 ± 1.20*** (118.7) |
| pcDNA-HEK293 [nM] | MRP1-HEK293 (ABCC1) [μM] | ||
| Etoposide | – | 282.62 ± 52.41 (1.0) | 77.95 ± 12.67 (1.0) |
| + midostaurin | 20 | 234.00 ± 40.53 (1.2) | 80.21 ± 16.67 (1.0) |
| + midostaurin | 50 | 237.94 ± 41.26 (1.2) | 70.31 ± 12.09 (1.1) |
| + MK-571 | 25000 | 172.78 ± 35.09* (1.6) | 14.17 ± 1.47*** (5.5) |
| pcDNA-HEK293 [nM] | R482-HEK293 (ABCG2) [nM] | ||
| SN-38 | – | 4.61 ± 0.89 (1.0) | 125.14 ± 19.03 (1.0) |
| + midostaurin | 20 | 5.17 ± 0.93 (0.9) | 134.28 ± 18.11 (0.9) |
| + midostaurin | 50 | 4.87 ± 0.98 (0.9) | 133.55 ± 19.81 (0.9) |
| + Ko143 | 1000 | 4.60 ± 0.92 (1.0) | 2.74 ± 0.66*** (45.7) |
Abbreviation: FR, fold-reversal.
P < 0.05
P < 0.01
P < 0.001.
IC50 values are mean ± SD calculated from dose-response curves obtained from at least three independent experiments using cytotoxicity assay as described in Materials and methods.
FR values were calculated by dividing IC50 values of cells treated with drug substrate in the absence of midostaurin or a reference inhibitor verapamil, MK-571 or Ko143 by IC50 values of cells treated with the same drug in the presence of midostaurin or a reference inhibitor.
Table 2.
Chemosensitization effect of midostaurin on ABCBl-mediated drug resistance in human cáncer cell lines.
| Treatment | Concentration (nM) | Mean IC50a ± SD and (FRb) |
|
|---|---|---|---|
| KB-3–1 [nM] | KB-V-1 [nM] | ||
| Paclitaxel | – | 2.01 ± 0.65 (1.0) | 2104.90 ± 266.55 (1.0) |
| + midostaurin | 50 | 1.96 ± 0.64 (1.0) | 521.20 ± 53.09*** (4.0) |
| + midostaurin | 100 | 1.74 ± 0.53 (1.1) | 217.19 ± 31.18*** (9.7) |
| + midostaurin | 200 | 1.53 ± 0.40 (1.0) | 104.25 ± 26.72*** (20.2) |
| + verapamil | 5000 | 2.28 ± 0.88 (0.9) | 74.08 ± 11.89*** (28.4) |
| Colchicine | – | 9.25 ± 3.57 (1.0) | 806.40 ± 85.74 (1.0) |
| + midostaurin | 50 | 11.21 ± 4.50 (0.8) | 342.11 ± 31.36*** (2.4) |
| + midostaurin | 100 | 11.75 ± 4.93 (0.8) | 215.85 ± 19.06*** (3.7) |
| + midostaurin | 200 | 12.06 ± 5.05 (0.8) | 76.76 ± 18.00*** (10.5) |
| + verapamil | 5000 | 6.54 ± 2.60 (1.4) | 210.53 ± 30.01*** (3.8) |
| Vincristine | – | 1.76 ± 0.46 (1.0) | 3199.33 ± 473.63 (1.0) |
| + midostaurin | 50 | 2.08 ± 0.57 (0.8) | 1595.80 ± 273.79** (2.0) |
| + midostaurin | 100 | 2.29 ± 0.67 (0.8) | 729.65 ± 63.57*** (4.4) |
| + midostaurin | 200 | 2.38 ± 0.69 (0.7) | 262.26 ± 50.76*** (12.2) |
| + verapamil | 5000 | 0.38 ± 0.12** (4.6) | 56.08 ± 6.30*** (57.1) |
| OVCAR-8 [nM] | NCI-ADR-RES [μM] | ||
| Paclitaxel | – | 7.14 ± 1.58 (1.0) | 7.53 ± 1.41 (1.0) |
| + midostaurin | 50 | 5.77 ± 1.51 (1.2) | 2.07 ± 0.28** (3.6) |
| + midostaurin | 100 | 4.83 ± 1.14 (1.5) | 1.02 ± 0.14** (7.3) |
| + midostaurin | 200 | 4.50 ± 1.02 (1.6) | 0.31 ± 0.03*** (23.6) |
| + verapamil | 5000 | 2.74 ± 0.48** (2.6) | 0.26 ± 0.04*** (29.0) |
| Colchicine | – | 25.09 ± 7.42 (1.0) | 2.98 ± 0.58 (1.0) |
| + midostaurin | 50 | 26.75 ± 8.53 (0.9) | 1.50 ± 0.48* (2.0) |
| + midostaurin | 100 | 27.19 ± 8.74 (0.9) | 0.63 ± 0.13** (4.7) |
| + midostaurin | 200 | 27.16 ± 9.24 (0.9) | 0.32 ± 0.10** (9.3) |
| + verapamil | 5000 | 19.72 ± 7.39 (1.3) | 0.76 ± 0.16** (3.9) |
| Vincristine | – | 14.58 ± 1.72 (1.0) | 6.88 ± 1.32 (1.0) |
| + midostaurin | 50 | 13.90 ± 2.26 (1.0) | 2.68 ± 0.38** (2.6) |
| + midostaurin | 100 | 12.45 ± 1.99 (1.2) | 1.44 ± 0.16** (4.8) |
| + midostaurin | 200 | 11.01 ± 1.86 (1.3) | 0.65 ± 0.09** (10.6) |
| + verapamil | 5000 | 3.52 ± 0.66*** (4.1) | 0.37 ± 0.06** (18.6) |
Abbreviation: FR, fold-reversal.
P < 0.05
P < 0.01
P < 0.001.
IC50 values are mean ± SD calculated from dose-response curves obtained from at least three independent experiments using cytotoxicity assay as described in Materials and methods.
FR values were calculated by dividing IC50 values of cells treated with an ABCB1 drug substrate in the absence of midostaurin or verapamil, a reference inhibitor of ABCB1 by IC50 values of cells treated with the same drug in the presence of midostaurin or verapamil.
3.3. Midostaurin does not downregulate ABCB1 protein expression in cancer cells
In addition to inhibiting ABCB1-mediated drug transport, previous studies have shown that the chemosensitivity of ABCB1-overexpressing cancer cells can be restored by transient down-regulation of ABCB1 in these MDR cancer cells [26,55,56]. To this end, we examined the effect of midostaurin on ABCB1 protein expression by treating KB-V1 and NCI-ADR-RES cancer cells with increasing concentrations of midostaurin (0–200 nM) for 72 h, before being processed by immunoblotting, as described in Materials and methods. We found that midostaurin has no significant effect on the protein expression of ABCB1 in either KB-V1 (Fig. 3A) or NCI-ADR-RES (Fig. 3B) cell lines, indicating that the chemosensitization of ABCB1-overexpressing cancer cells by midostaurin is not due to down regulation of expression of this transporter.
Fig. 3. Midostaurin has no significant e?ect on ABCB1 protein expression in human cancer cells.
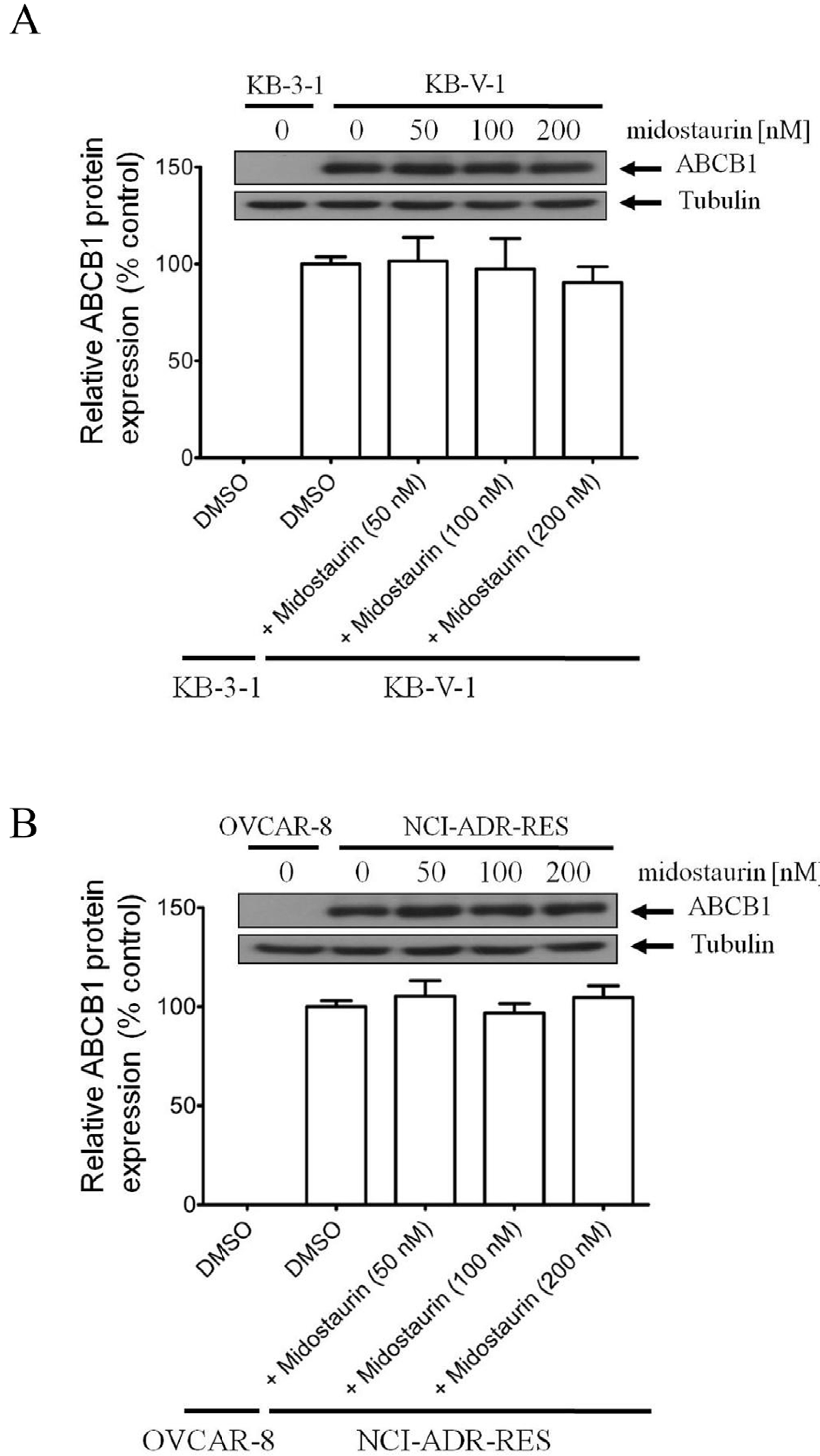
(A) Human KB-3–1 epidermal cancer cells and ABCB1-overexpressing variant KB-V1 cancer cells, as well as (B) human OVCAR-8 ovarian cancer cells and ABCB1-overexpressing variant NCI-ADR-RES cancer cells were treated with DMSO or midostaurin at various concentrations (50 nM, 100 nM and 200 nM) for 72 h before processed for immunoblotting and quantification as described previously [38]. α-Tubulin was used as an internal loading control. Values are presented as mean ± S.D. calculated from three independent experiments.
3.4. Midostaurin enhances drug-induced apoptosis in ABCB1-overexpressing cancer cells
Next, we determined the effect of midostaurin on apoptosis induction by colchicine, a known inducer of apoptosis [52] and a substrate drug of ABCB1, in ABCB1-overexpressing human cancer cells. Drug-sensitive parental KB-3–1 cells and drug-resistant variant KB-V1 cells were treated with DMSO, colchicine or a combination of colchicine and midostaurin for 48 h and processed as described previously [27]. As expected, colchicine induced a high level of apoptosis in KB-3–1 cancer cells (Fig. 4A, upper panels), from 4% basal level to approximately 57% of early and late apoptosis (Fig. 4B). In contrast, colchicine failed to induce a significant level of apoptosis in ABCB1-expressing KB-V1 cancer cells (Fig. 4A, lower panels). Notably, without affecting colchicine-induced apoptosis in KB-3–1 cells, midostaurin significantly increased the level of colchicine-induced apoptosis in KB-V1 cells, from 7% basal level to approximately 34% of early and late apoptosis (Fig. 4B). Our results indicate that midostaurin enhances the apoptotic effect of ABCB1 substrate-drug in ABCB1-overexpressing MDR cancer cells.
Fig. 4. Midostaurin potentiates drug-induced apoptosis in ABCB1-overexpressing cancer cells. (A).
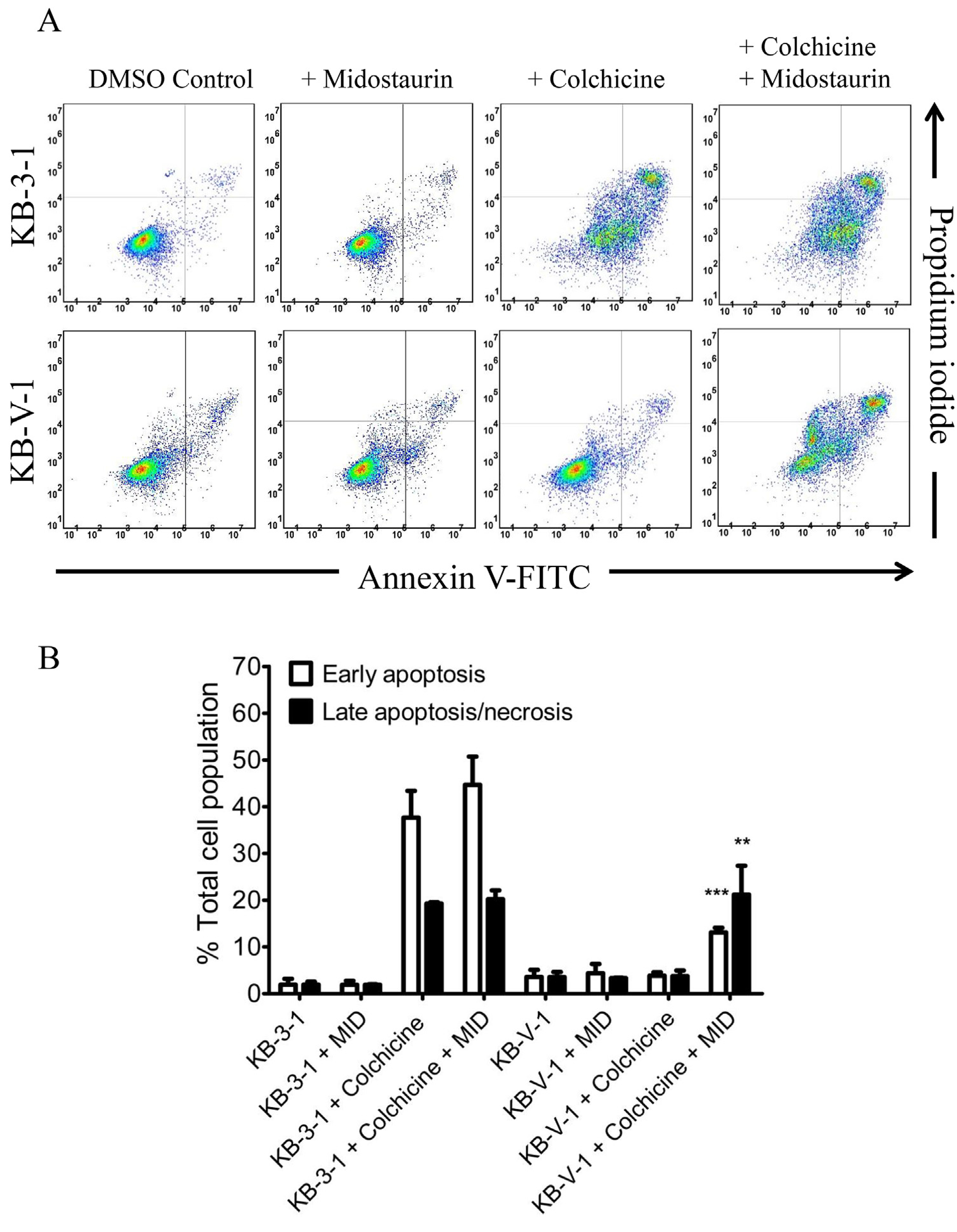
The effect of midostaurin on colchicine-induced apoptosis was determined in drug-sensitive parental human KB-3–1 cancer cells (top panels) and ABCB1-overexpressing KB-V1 cells (lower panels). Cells were treated with either DMSO (control), 500 nM colchicine (+colchicine) or a combination of 500 nM colchicine and 200 nM of midostaurin (+colchi-cine + midostaurin) for 48 h, isolated and analyzed by flow cytometry as described previously [27]. The dot plots of representative experiments are shown. (B) Quantification of colchicine-induced apoptosis in KB 3–1 and KB-V1 cancer cells. The early apoptosis and late apoptosis/necrosis data are given as mean values ± S.D. calculated from at least three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
3.5. Midostaurin inhibits the ATPase activity of ABCB1
We examined the effect of midostaurin on vanadate (Vi)-sensitive ATPase activity of ABCB1 and performed docking analysis of midostaurin with molecular modeling of ABCB1 to gain insight into the interaction between midostaurin and the substrate-binding sites of ABCB1. We discovered that midostaurin partially inhibited the ATPase activity of ABCB1 in a concentration-dependent manner to approximately 50% of the basal activity (basal, 87.8 ± 7.2 nmoles Pi/min/mg protein) and an IC50 value of approximately 10 μM (Fig. 5). Given that substrate transport mediated by ABCB1 is coupled to ATP hydrolysis [57,58], our data indicate that midostaurin attenuates the transport activity of ABCB1.
Fig. 5. Midostaurin inhibits vanadate-sensitive ATPase activity of ABCB1.
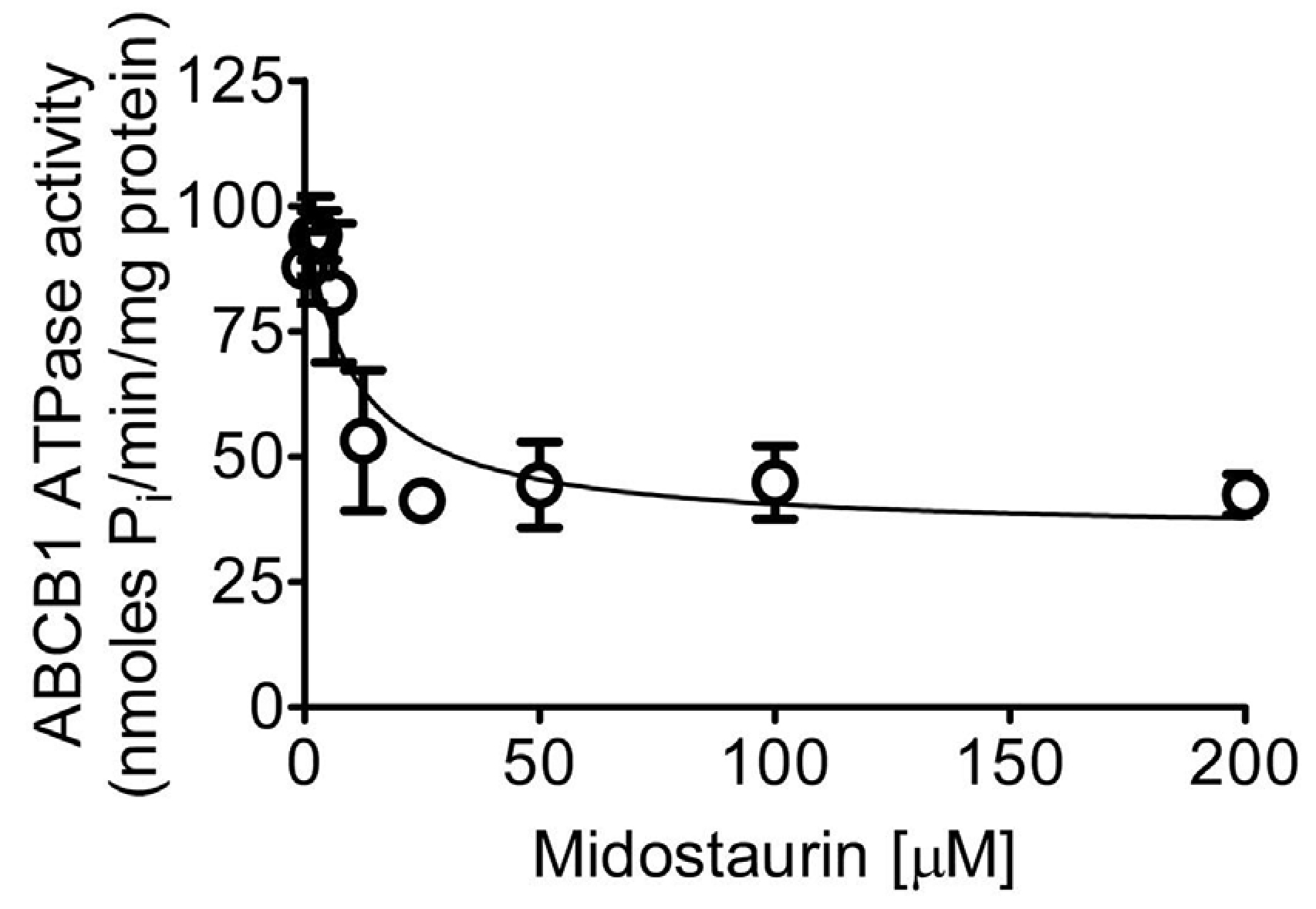
The effect of midostaurin at indicated concentrations ranging from 0 to 200 μM on vanadate-sensitive ABCB1 ATP hydrolysis was determined by endpoint Pi liberation assays as described in the Methods section. Data are presented as mean ± S.D. from three independent experiments.
3.6. Docking of midostaurin in the drug-binding pocket of ABCB1
As the crystal structure of human ABCB1 has yet to be solved in the inward-open conformation, which is the conformation of the transporter when the substrate binds to the transmembrane region of ABCB1, we generated a homology model of human ABCB1 based on the mouse P-gp crystal structure (PDB:5KPI). We used this homology model to dock midostaurin and to further understand the interaction between midostaurin and residues in the drug-binding pocket of ABCB1. Analysis of the lowest energy docking poses (Fig. 6A) identified hydrophobic interactions between midostaurin and several aromatic and hydrophobic residues located at the substrate-binding pocket of ABCB1, which are presented in Fig. 6B. Only residues within 4 Å of the midostaurin molecule are shown in Fig. 6B. Collectively, our results indicate that midostaurin interacts at the drug-binding pocket of ABCB1 and inhibits the activity of ABCB1.
Fig. 6. Docking of midostaurin in the drug-binding pocket of ABCB1.
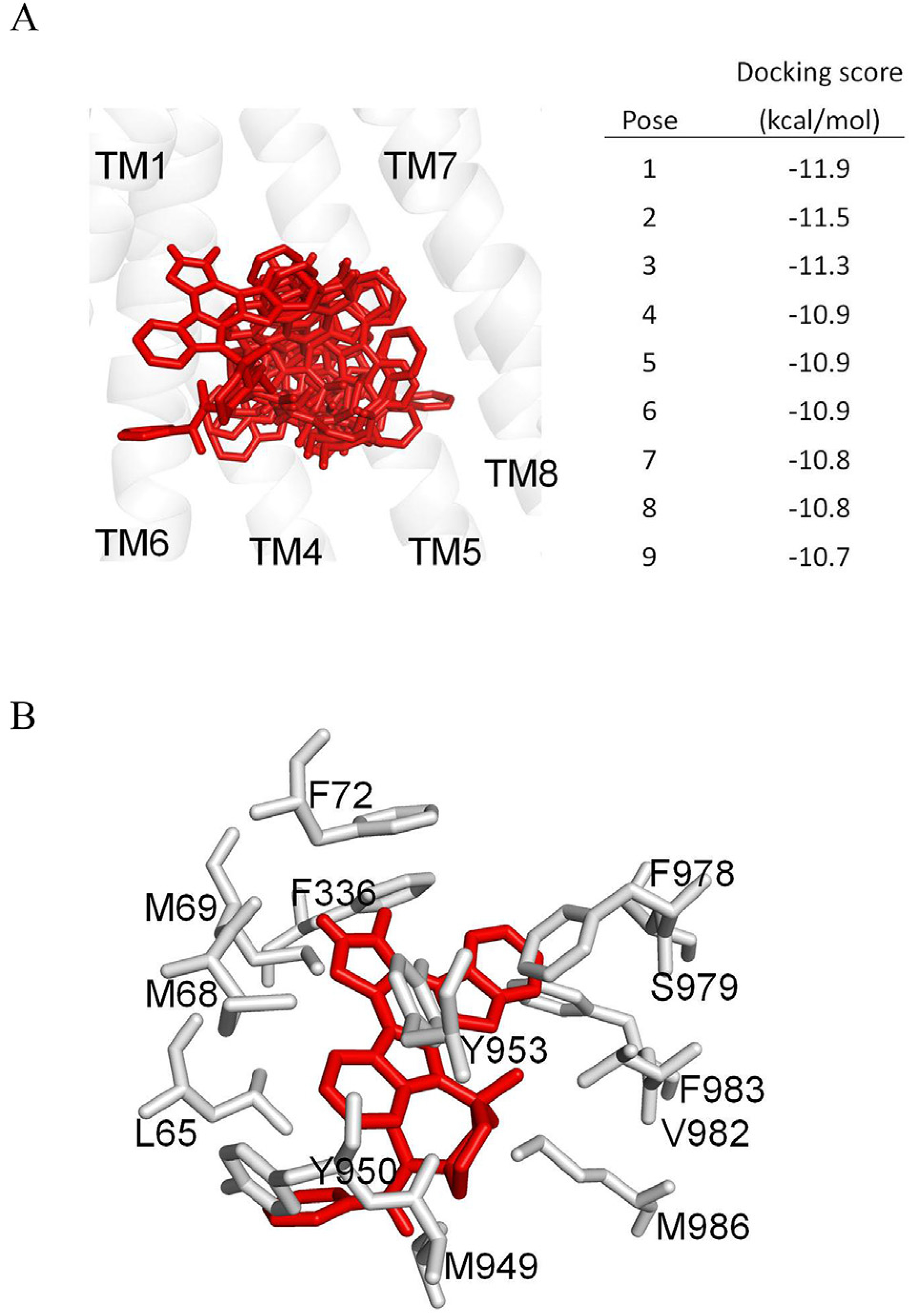
(A) The homology model of ABCB1, based on the PDB:5KPI mouse P-glycoprotein structure, was used for exhaustive docking using AutoDock Vina software. The receptor grid was centered at x = 20, y = 60 and z = 5 Å, and 33 residues in the binding-pocket were set as flexible. The box size was 50 × 50 × 50 Å and the exhaustiveness was set at 100. The first nine poses with the highest docking scores are shown in red sticks. For the purpose of clarity, only transmembrane helices 1 and 4 to 8 are presented in grey as a cartoon representation. The figure was prepared using PyMOL software. (B) The lowest docking score pose binding mode of midostaurin is presented in red sticks. Residues within 4 Å distance of midostaurin are shown in grey. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.7. Overexpression of ABCB1 does not reduce the chemosensitivity of cancer cells to midostaurin
Knowing that the overexpression of ABCB1 can lead to reduced efficacy of numerous protein kinase inhibitors in cancer cells [26,55,56], including the FLT3 inhibitor crenolanib [59], we determined the cytotoxicity of midostaurin in drug-sensitive, ABCB1-negative cells and drug-resistant ABCB1-overxpressing cells. The relative-resistance (RR) value, representing the extent of acquired cellular resistance to midostaurin caused by the overexpression of ABCB1, is calculated by dividing the IC50 value of midostaurin in ABCB1-overexpressing cell lines by the IC50 value of midostaurin in the respective ABCB1-negative parental lines. As shown in Table 3, ABCB1-negative cells and ABCB1-overexpressing cells are equally sensitive to midostaurin treatment, indicating that the overexpression of ABCB1 does not lead to reduced susceptibility of cancer cells to midostaurin.
Table 3.
Sensitivity of ABCBl-expressing cells and their respective drug-sensitive parental cells to midostaurin.
| Cell line | Transporter expressed | IC50 [μM]a | R.Rb |
|---|---|---|---|
| KB-3–1 | – | 0.64 ± 0.11 | 1.0 |
| KB-V-1 | ABCB1 | 0.74 ± 0.23 | 1.2 |
| OVCAR-8 | – | 2.03 ± 0.36 | 1.0 |
| NCI-ADR-RES | ABCB1 | 3.16 ± 0.83 | 1.6 |
| NIH3T3 | – | 0.62 ± 0.11 | 1.0 |
| NIH3T3-G185 | ABCB1 | 0.84 ± 0.17 | 1.4 |
Abbreviation: RR, relative-resistance.
IC50 values are mean ± SD calculated from dose-response curves obtained from at least three independent experiments using cytotoxicity assay as described in Materials and methods.
RR values were calculated by dividing IC50 values of ABCBl-overexpressing multidrug-resistant cells by IC50 values of respective drug-sensitive parental cells treated with midostaurin.
4. Discussion
Multidrug resistance caused by the overexpression of ABCB1 remains a significant challenge in the treatment of many solid tumors and blood cancer [6,19]. More specifically, studies have shown that the overexpression of ABCB1 or ABCG2 with FLT3 internal tandem duplications is associated with poor response to chemotherapy in AML [60,61]. In addition, it was reported that the FLT3 inhibitors ponatinib, quizartinib and crenolanib interact directly with ABCB1 and/or ABCG2. Sen et al. demonstrated that ponatinib inhibits ABCB1- and ABCG2-mediated drug transport and produces synergistic cytotoxicity with substrate drugs of ABCB1 and ABCG2 [62]. Similarly, Bhullar et al., showed that quizartinib enhances uptake of substrates of ABCB1 and ABCG2, and resensitizes ABCG2-overexpressing cancer cells to anticancer agents at concentrations below 1 μM [63]. In contrast, Mathias et al. discovered that crenolanib is a substrate of ABCB1, but does not affect the ABCB1-mediated drug transport or protein expression of ABCB1 [59]. These results prompted us to examine the potential interactions of midostaurin with the MDR-linked ABC proteins ABCB1, ABCC1 and ABCG2.
Mutations that lead to constitutive activation of FLT3 are associated with cancer progression and poor prognosis in approximately 30% of AML patients [64]. Midostaurin is highly effective against FLT3 tyrosine kinase [34] and mutant forms of KIT proto-oncogene receptor tyrosine kinase that drive advanced systemic mastocytosis [35]. Midostaurin directly inhibits FLT3 kinase activity in vitro with an IC50 value of approximately 500 nM and was found to be non-toxic in murine hematopoietic Ba/F3 cells at 100 nM [65]. Clinical studies demonstrated that the steady-state midostaurin plasma levels reached 0.2–0.7 μmol/L in patients with solid tumors, which are 20–70 times the IC50 values in cell line studies [30], and that 100–200 mg can be administered safely as a daily dose in combination with other chemotherapeutic agents [30,66,67]. In the present study, we discovered that midostaurin re-sensitizes ABCB1-overexpressing cells to multiple substrate drugs of ABCB1 at clinically relevant nanomolar concentrations. Although other mechanisms are also possible, our results show that midostaurin enhances drug-induced apoptosis and restores the chemosensitivity of ABCB1-overexpressing cancer cells, most likely by directly inhibiting the drug transport function of ABCB1, and not by altering the protein expression of this transporter in cancer cells. Interaction of midostaurin at the drug-binding pocket of and ABCB1 was supported by the results of in silico docking analysis and concentration-dependent inhibition of the ATPase activity of ABCB1 by midostaurin. Knowing that inhibition of ABCB1 ATPase activity is associated with the presence of a high-affinity substrate or inhibitor at the substrate-binding site of ABCB1 [68], it is likely that midostaurin binds to the substrate-binding site of ABCB1 and inhibits the binding of substrate drugs at the same site, consequently attenuating the transport of these drugs (see Fig. 7). Moreover, our results indicate that midostaurin is selective for ABCB1 relative to ABCC1 and ABCG2, as midostaurin has no significant effect on drug efflux or MDR mediated by ABCC1 or ABCG2. Collectively, these results suggest that midostaurin is a well-tolerated drug that can be safely administered to patients at doses achieving sufficient plasma levels to inhibit cancer proliferation and reverse ABCB1-mediated MDR at pharmacologically relevant concentrations.
Fig. 7. A graphic illustration of midostaurin modulating the drug efflux function of ABCB1 in cancer cells.
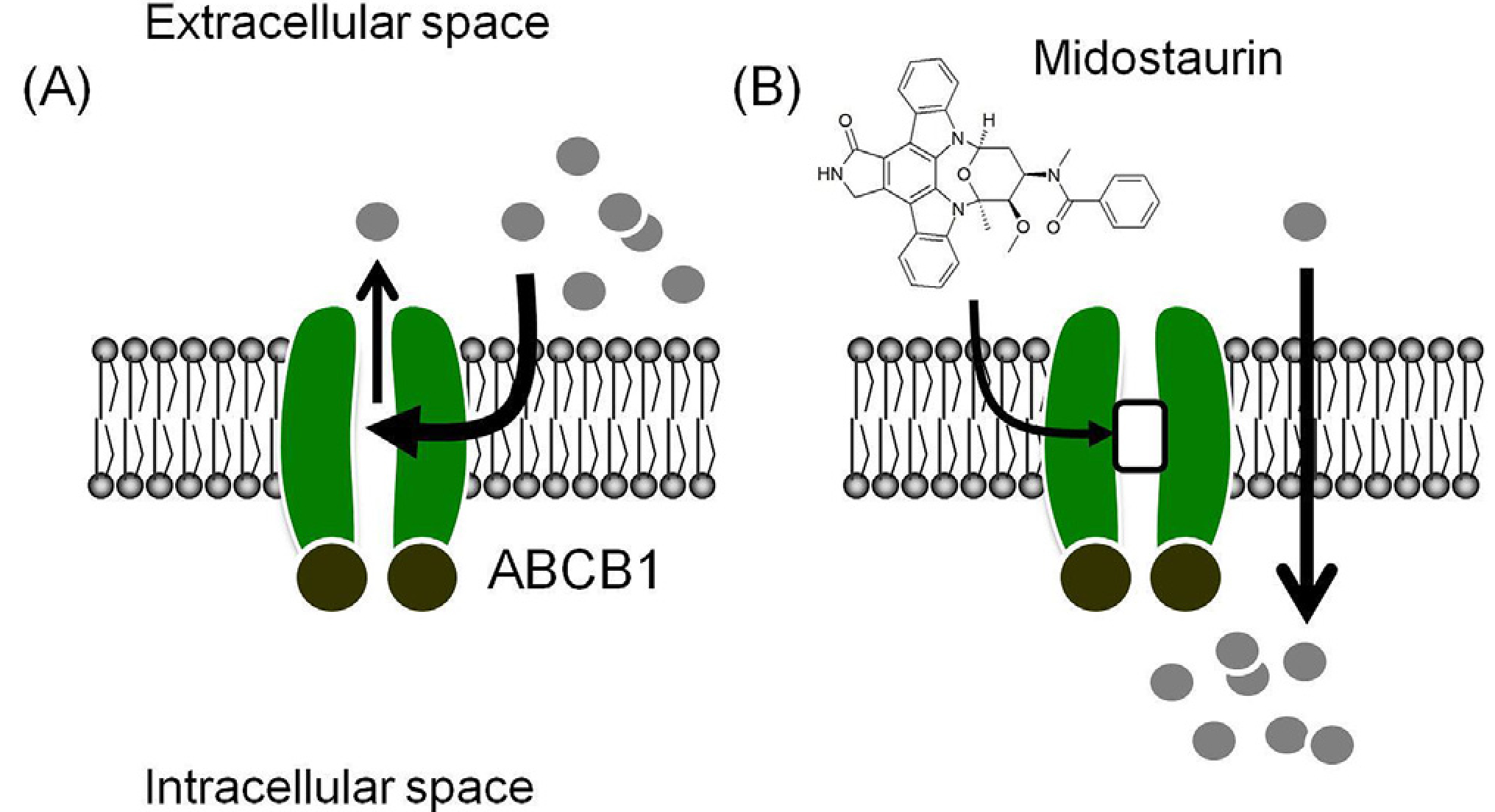
(A) In the absence of midostaurin, anticancer drugs (grey circles) are effluxed out of ABCB1-overepressing cancer cells by ABCB1 (green), preventing the accumulation of therapeutic drugs in cancer cells, which leads to the MDR phenotype. (B) In the presence of midostaurin, the drug effux function of ABCB1 is blocked by midostaurin and the chemosensitivity of ABCB1-overexpressing cancer cells is hence restored. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
It is worth mentioning that a recent phase I clinical trial evaluating the use of nilotinib as a co-adjuvant treatment with doxorubicin in patients with sarcomas concluded that combination therapy of doxorubicin with nilotinib as an inhibitor of ABCB1 and ABCC1 is feasible, and warrants further phase II study [69]. The conclusion from this study supports the results from preclinical studies demonstrating that nilotinib directly inhibits the function of ABCB1 and consequently reverses ABCB1-mediated multidrug resistance in cancer cells and in a tumor xenograft model [25,70–72]. Midostaurin, similar to nilotinib, is an approved tyrosine kinase inhibitor that interacts strongly with ABCB1 and inhibits the drug efflux function of ABCB1 and resensitizes ABCB1-overexpressing cancer cells to conventional chemotherapeutic drugs. Moreover, a reason for giving midostaurin to newly diagnosed AML patients is to circumvent the development of resistance mechanisms, such as the overexpression of ABCB1, in patients with relapsed/ refractory AML [73]. Other studies also indicate that ABCB1 polymorphisms may contribute to poorer outcomes in AML patients treated with standard chemotherapy, such as cytarabine and anthracycline [74–78]. Our results suggest that the overexpression of ABCB1 is unlikely to play a major role in the development of midostaurin resistance in cancer patients, and patients with newly diagnosed FLT3-mutated AML (or at least a subset of relapsed/refractory AML) might benefit from treatment with midostaurin in combination with standard chemotherapy to overcome MDR associated with the overexpression of ABCB1.
In summary, our study revealed that in addition to inhibiting FLT3, midostaurin is also a potent and selective modulator of ABCB1 that can re-sensitize MDR cancer cells to chemotherapeutic agents at non-toxic concentrations. Although unfavourable clinical responses might occur in combination therapy [79,80], and encouraging experimental results do not always translate into clinical outcomes [21], our results suggest that concomitant administration of midostaurin may improve the therapeutic efficacy of anticancer agents for the treatment of multidrug-resistant tumors, and should thus be further evaluated in clinical practice.
Acknowledgments
We thank George Leiman, Laboratory of Cell Biology, Center for Cancer Research, NCI, NIH for editing this manuscript. This work was supported by funds from the Ministry of Science and Technology of Taiwan (MOST-107–2320-B-182–017), Chang Gung Medical Research Program (BMRPC17 and CMRPD1G0112) and the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, USA (SL and SVA).
Abbreviations:
- MDR
multidrug resistance
- ABC
ATP-binding cassette
- FLT3
fms-like tyrosine kinase-3
- AML
acute myeloid leukemia
- CCK-8
Cell Counting Kit-8
- MTT
3-(4,5-dimethylthiazol-yl)-2,5-diphenyllapatinibrazolium bromide
- Vi
sodium orthovanadate
- FR
fold-reversal
- RR
relative-resistance
Footnotes
Conflict of interest The authors declare no conflict of interest.
References
- [1].Dano K, Active outward transport of daunomycin in resistant Ehrlich ascites tumor cells, Biochim. Biophys. Acta 323 (1973) 466–483. [DOI] [PubMed] [Google Scholar]
- [2].Shen DW, Fojo A, Chin JE, Roninson IB, Richert N, Pastan I, Gottesman MM, Human multidrug-resistant cell lines: increased mdr1 expression can precede gene amplification, Science 232 (1986) 643–645. [DOI] [PubMed] [Google Scholar]
- [3].Ambudkar SV, Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells, Methods Enzymol. 292 (1998) 504–514. [DOI] [PubMed] [Google Scholar]
- [4].Gottesman MM, Fojo T, Bates SE, Multidrug resistance in cancer: role of ATP-dependent transporters, Nat. Rev. Cancer 2 (2002) 48–58. [DOI] [PubMed] [Google Scholar]
- [5].Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM, Targeting multidrug resistance in cancer, Nat. Rev. Drug Discov. 5 (2006) 219–234. [DOI] [PubMed] [Google Scholar]
- [6].Gillet JP, Gottesman MM, Mechanisms of multidrug resistance in cancer, Methods Mol. Biol. 596 (2010) 47–76. [DOI] [PubMed] [Google Scholar]
- [7].Kovalev AA, Tsvetaeva DA, Grudinskaja TV, Role of ABC-cassette transporters (MDR1, MRP1, BCRP) in the development of primary and acquired multiple drug resistance in patients with early and metastatic breast cancer, Exp. Oncol. 35 (2013) 287–290. [PubMed] [Google Scholar]
- [8].Pilarski LM, Belch AR, Intrinsic expression of the multidrug transporter, P-glycoprotein 170, in multiple myeloma: implications for treatment, Leuk. Lymphoma 17 (1995) 367–374. [DOI] [PubMed] [Google Scholar]
- [9].Pilarski LM, Szczepek AJ, Belch AR, Deficient drug transporter function of bone marrow-localized and leukemic plasma cells in multiple myeloma, Blood 90 (1997) 3751–3759. [PubMed] [Google Scholar]
- [10].Schwarzenbach H, Expression of MDR1/P-glycoprotein, the multidrug resistance protein MRP, and the lung-resistance protein LRP in multiple myeloma, Med. Oncol. 19 (2002) 87–104. [DOI] [PubMed] [Google Scholar]
- [11].Nakagawa Y, Abe S, Kurata M, Hasegawa M, Yamamoto K, Inoue M, Takemura T, Suzuki K, Kitagawa M, IAP family protein expression correlates with poor outcome of multiple myeloma patients in association with chemotherapy-induced overexpression of multidrug resistance genes, Am. J. Hematol. 81 (2006) 824–831. [DOI] [PubMed] [Google Scholar]
- [12].Tsubaki M, Satou T, Itoh T, Imano M, Komai M, Nishinobo M, Yamashita M, Yanae M, Yamazoe Y, Nishida S, Overexpression of MDR1 and survivin, and decreased Bim expression mediate multidrug-resistance in multiple myeloma cells, Leuk. Res. 36 (2012) 1315–1322. [DOI] [PubMed] [Google Scholar]
- [13].Hofmeister CC, Yang X, Pichiorri F, Chen P, Rozewski DM, Johnson AJ, Lee S, Liu Z, Garr CL, Hade EM, Ji J, Schaaf LJ, Benson DM Jr., Kraut EH, Hicks WJ, Chan KK, Chen CS, Farag SS, Grever MR, Byrd JC, Phelps MA, Phase I trial of lenalidomide and CCI-779 in patients with relapsed multiple myeloma: evidence for lenalidomide-CCI-779 interaction via P-glycoprotein, J. Clin. Oncol. 29 (2011) 3427–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ross DD, Novel mechanisms of drug resistance in leukemia, Leukemia 14 (2000) 467–473. [DOI] [PubMed] [Google Scholar]
- [15].Maia RC, Vasconcelos FC, Souza PS, Rumjanek VM, Towards comprehension of the ABCB1/P-glycoprotein role in chronic myeloid leukemia, Molecules (2018) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Matthews C, Catherwood MA, Larkin AM, Clynes M, Morris TC, Alexander HD, MDR-1, but not MDR-3 gene expression, is associated with unmutated IgVH genes and poor prognosis chromosomal aberrations in chronic lymphocytic leukemia, Leuk. Lymphoma 47 (2006) 2308–2313. [DOI] [PubMed] [Google Scholar]
- [17].Bodo A, Bakos E, Szeri F, Varadi A, Sarkadi B, The role of multidrug transporters in drug availability, metabolism and toxicity, Toxicol. Lett. 140–141 (2003) 133–143. [DOI] [PubMed] [Google Scholar]
- [18].Leslie EM, Deeley RG, Cole SP, Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense, Toxicol. Appl. Pharmacol. 204 (2005) 216–237. [DOI] [PubMed] [Google Scholar]
- [19].Wu CP, Hsieh CH, Wu YS, The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy, Mol. Pharm. 8 (2011) 1996–2011. [DOI] [PubMed] [Google Scholar]
- [20].Wu CP, Calcagno AM, Ambudkar SV, Reversal of ABC drug transporter-mediated multidrug resistance in cancer cells: evaluation of current strategies, Curr. Mol. Pharmacol. 1 (2008) 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shukla S, Wu CP, Ambudkar SV, Development of inhibitors of ATP-binding cassette drug transporters: present status and challenges, Expert Opin. Drug Metabol. Toxicol. 4 (2008) 205–223. [DOI] [PubMed] [Google Scholar]
- [22].Shukla S, Ohnuma S, Ambudkar SV, Improving cancer chemotherapy with modulators of ABC drug transporters, Curr. Drug Targets 12 (2011) 621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shi Z, Tiwari AK, Shukla S, Robey RW, Singh S, Kim IW, Bates SE, Peng X, Abraham I, Ambudkar SV, Talele TT, Fu LW, Chen ZS, Sildenafil reverses ABCB1- and ABCG2-mediated chemotherapeutic drug resistance, Cancer Res. 71 (2011) 3029–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shukla S, Chen ZS, Ambudkar SV, Tyrosine kinase inhibitors as modulators of ABC transporter-mediated drug resistance, Drug Resist. Updates 15 (2012) 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tiwari AK, Sodani K, Dai CL, Abuznait AH, Singh S, Xiao ZJ, Patel A, Talele TT, Fu L, Kaddoumi A, Gallo JM, Chen ZS, Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models, Cancer Lett. 328 (2013) 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang SQ, Liu ST, Zhao BX, Yang FH, Wang YT, Liang QY, Sun YB, Liu Y, Song ZH, Cai Y, Li GF, Afatinib reverses multidrug resistance in ovarian cancer via dually inhibiting ATP binding cassette subfamily B member 1, Oncotarget 6 (2015) 26142–26160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hsiao SH, Lu YJ, Li YQ, Huang YH, Hsieh CH, Wu CP, Osimertinib (AZD9291) attenuates the function of multidrug resistance-linked ATP-binding cassette transporter ABCB1 in vitro, Mol. Pharm. 13 (2016) 2117–2125. [DOI] [PubMed] [Google Scholar]
- [28].Rasko JEJ, Hughes TP, First approved kinase inhibitor for AML, Cell 171 (2017) 981. [DOI] [PubMed] [Google Scholar]
- [29].Fabbro D, Ruetz S, Bodis S, Pruschy M, Csermak K, Man A, Campochiaro P, Wood J, O’Reilly T, Meyer T, PKC412–a protein kinase inhibitor with a broad therapeutic potential, Anti Cancer Drug Des. 15 (2000) 17–28. [PubMed] [Google Scholar]
- [30].Propper DJ, McDonald AC, Man A, Thavasu P, Balkwill F, Braybrooke JP, Caponigro F, Graf P, Dutreix C, Blackie R, Kaye SB, Ganesan TS, Talbot DC, Harris AL, Twelves C, Phase I and pharmacokinetic study of PKC412, an inhibitor of protein kinase C, J. Clin. Oncol. 19 (2001) 1485–1492. [DOI] [PubMed] [Google Scholar]
- [31].Fabbro D, Buchdunger E, Wood J, Mestan J, Hofmann F, Ferrari S, Mett H, O’Reilly T, Meyer T, Inhibitors of protein kinases: CGP 41251, a protein kinase inhibitor with potential as an anticancer agent, Pharmacol. Ther. 82 (1999) 293–301. [DOI] [PubMed] [Google Scholar]
- [32].Ikegami Y, Yano S, Nakao K, Effects of the new selective protein kinase C inhibitor 4’-N-benzoyl staurosporine on cell cycle distribution and growth inhibition in human small cell lung cancer cells, Arzneim. Forsch. 46 (1996) 201–204. [PubMed] [Google Scholar]
- [33].Ikegami Y, Yano S, Nakao K, Antitumor effect of CGP41251, a new selective protein kinase C inhibitor, on human non-small cell lung cancer cells, Jpn. J. Pharmacol. 70 (1996) 65–72. [DOI] [PubMed] [Google Scholar]
- [34].Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, Thiede C, Prior TW, Dohner K, Marcucci G, Lo-Coco F, Klisovic RB, Wei A, Sierra J, Sanz MA, Brandwein JM, de Witte T, Niederwieser D, Appelbaum FR, Medeiros BC, Tallman MS, Krauter J, Schlenk RF, Ganser A, Serve H, Ehninger G, Amadori S, Larson RA, Dohner H, Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation, N. Engl. J. Med. 377 (2017) 454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gotlib J, Kluin-Nelemans HC, George TI, Akin C, Sotlar K, Hermine O, Awan FT, Hexner E, Mauro MJ, Sternberg DW, Villeneuve M, Huntsman Labed A, Stanek EJ, Hartmann K, Horny HP, Valent P, Reiter A, Efficacy and safety of midostaurin in advanced systemic mastocytosis, N. Engl. J. Med. 374 (2016) 2530–2541. [DOI] [PubMed] [Google Scholar]
- [36].DeAngelo DJ, George TI, Linder A, Langford C, Perkins C, Ma J, Westervelt P, Merker JD, Berube C, Coutre S, Liedtke M, Medeiros B, Sternberg D, Dutreix C, Ruffie PA, Corless C, Graubert TJ, Gotlib J, Efficacy and safety of midostaurin in patients with advanced systemic mastocytosis: 10-year median follow-up of a phase II trial, Leukemia 32 (2018) 470–478. [DOI] [PubMed] [Google Scholar]
- [37].Currier SJ, Kane SE, Willingham MC, Cardarelli CO, Pastan I, Gottesman MM, Identification of residues in the first cytoplasmic loop of P-glycoprotein involved in the function of chimeric human MDR1-MDR2 transporters, J. Biol. Chem. 267 (1992) 25153–25159. [PubMed] [Google Scholar]
- [38].Wu CP, Shukla S, Calcagno AM, Hall MD, Gottesman MM, Ambudkar SV, Evidence for dual mode of action of a thiosemicarbazone, NSC73306: a potent substrate of the multidrug resistance linked ABCG2 transporter, Mol. Cancer Therapeut. 6 (2007) 3287–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wu CP, Hsiao SH, Sim HM, Luo SY, Tuo WC, Cheng HW, Li YQ, Huang YH, Ambudkar SV, Human ABCB1 (P-glycoprotein) and ABCG2 mediate resistance to BI 2536, a potent and selective inhibitor of Polo-like kinase 1, Biochem. Pharmacol. 86 (2013) 904–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gribar JJ, Ramachandra M, Hrycyna CA, Dey S, Ambudkar SV, Functional characterization of glycosylation-deficient human P-glycoprotein using a vaccinia virus expression system, J. Membr. Biol. 173 (2000) 203–214. [DOI] [PubMed] [Google Scholar]
- [41].Ishiyama M, Tominaga H, Shiga M, Sasamoto K, Ohkura Y, Ueno K, A combined assay of cell viability and in vitro cytotoxicity with a highly water-soluble tetrazolium salt, neutral red and crystal violet, Biol. Pharm. Bull. 19 (1996) 1518–1520. [DOI] [PubMed] [Google Scholar]
- [42].Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG, Ashby CR Jr., Huang Y, Robey RW, Liang YJ, Chen LM, Shi CJ, Ambudkar SV, Chen ZS, Fu LW, Lapatinib (Tykerb GW572016), Reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2, Cancer Res. 68 (2008) 7905–7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Anderson HA, Maylock CA, Williams JA, Paweletz CP, Shu H, Shacter E, Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells, Nat. Immunol. 4 (2003) 87–91. [DOI] [PubMed] [Google Scholar]
- [44].Ramachandra M, Ambudkar SV, Chen D, Hrycyna CA, Dey S, Gottesman MM, Pastan I, Human P-glycoprotein exhibits reduced a?nity for substrates during a catalytic transition state, Biochemistry 37 (1998) 5010–5019. [DOI] [PubMed] [Google Scholar]
- [45].Kerr KM, Sauna ZE, Ambudkar SV, Correlation between steady-state ATP hydrolysis and vanadate-induced ADP trapping in Human P-glycoprotein. Evidence for ADP release as the rate-limiting step in the catalytic cycle and its modulation by substrates, J. Biol. Chem. 276 (2001) 8657–8664. [DOI] [PubMed] [Google Scholar]
- [46].Esser L, Zhou F, Pluchino KM, Shiloach J, Ma J, Tang WK, Gutierrez C, Zhang A, Shukla S, Madigan JP, Zhou T, Kwong PD, Ambudkar SV, Gottesman MM, Xia D, Structures of the multidrug transporter P-glycoprotein reveal asymmetric ATP binding and the mechanism of polyspecificity, J. Biol. Chem. 292 (2017) 446–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Arnold K, Bordoli L, Kopp J, Schwede T, The SWISS-MODEL workspace: a webbased environment for protein structure homology modelling, Bioinformatics 22 (2006) 195–201. [DOI] [PubMed] [Google Scholar]
- [48].Sanner MF, Python: a programming language for software integration and development, J. Mol. Graph. Model. 17 (1999) 57–61. [PubMed] [Google Scholar]
- [49].Hollo Z, Homolya L, Davis CW, Sarkadi B, Calcein accumulation as a fluorometric functional assay of the multidrug transporter, Biochim. Biophys. Acta 1191 (1994) 384–388. [DOI] [PubMed] [Google Scholar]
- [50].Robey RW, Steadman K, Polgar O, Morisaki K, Blayney M, Mistry P, Bates SE, Pheophorbide a is a specific probe for ABCG2 function and inhibition, Cancer Res. 64 (2004) 1242–1246. [DOI] [PubMed] [Google Scholar]
- [51].Kartner N, Riordan JR, Ling V, Cell surface P-glycoprotein associated with multidrug resistance in mammalian cell lines, Science 221 (1983) 1285–1288. [DOI] [PubMed] [Google Scholar]
- [52].Riordan JR, Ling V, Purification of P-glycoprotein from plasma membrane vesicles of Chinese hamster ovary cell mutants with reduced colchicine permeability, J. Biol. Chem. 254 (1979) 12701–12705. [PubMed] [Google Scholar]
- [53].Tsuruo T, Iida H, Naganuma K, Tsukagoshi S, Sakurai Y, Promotion by verapamil of vincristine responsiveness in tumor cell lines inherently resistant to the drug, Cancer Res. 43 (1983) 808–813. [PubMed] [Google Scholar]
- [54].Tsuruo T, Iida H, Yamashiro M, Tsukagoshi S, Sakurai Y, Enhancement of vincristine-and adriamycin-induced cytotoxicity by verapamil in P388 leukemia and its sublines resistant to vincristine and adriamycin, Biochem. Pharmacol. 31 (1982) 3138–3140. [DOI] [PubMed] [Google Scholar]
- [55].Cuestas ML, Castillo AI, Sosnik A, Mathet VL, Downregulation of mdr1 and abcg2 genes is a mechanism of inhibition of efflux pumps mediated by polymeric amphiphiles, Bioorg. Med. Chem. Lett 22 (2012) 6577–6579. [DOI] [PubMed] [Google Scholar]
- [56].Natarajan K, Bhullar J, Shukla S, Burcu M, Chen ZS, Ambudkar SV, Baer MR, The Pim kinase inhibitor SGI-1776 decreases cell surface expression of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and drug transport by Pim-1-dependent and -independent mechanisms, Biochem. Pharmacol. 85 (2013) 514–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM, Biochemical, cellular, and pharmacological aspects of the multidrug transporter, Annu. Rev. Pharmacol. Toxicol. 39 (1999) 361–398. [DOI] [PubMed] [Google Scholar]
- [58].Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM, P-glycoprotein: from genomics to mechanism, Oncogene 22 (2003) 7468–7485. [DOI] [PubMed] [Google Scholar]
- [59].Mathias TJ, Natarajan K, Shukla S, Doshi KA, Singh ZN, Ambudkar SV, Baer MR, The FLT3 and PDGFR inhibitor crenolanib is a substrate of the multidrug resistance protein ABCB1 but does not inhibit transport function at pharmacologically relevant concentrations, Invest. N. Drugs 33 (2015) 300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hunter HM, Pallis M, Seedhouse CH, Grundy M, Gray C, Russell NH, The expression of P-glycoprotein in AML cells with FLT3 internal tandem duplications is associated with reduced apoptosis in response to FLT3 inhibitors, Br. J. Haematol. 127 (2004) 26–33. [DOI] [PubMed] [Google Scholar]
- [61].Tiribelli M, Geromin A, Michelutti A, Cavallin M, Pianta A, Fabbro D, Russo D, Damante G, Fanin R, Damiani D, Concomitant ABCG2 overexpression and FLT3-ITD mutation identify a subset of acute myeloid leukemia patients at high risk of relapse, Cancer 117 (2011) 2156–2162. [DOI] [PubMed] [Google Scholar]
- [62].Sen R, Natarajan K, Bhullar J, Shukla S, Fang HB, Cai L, Chen ZS, Ambudkar SV, Baer MR, The novel BCR-ABL and FLT3 inhibitor ponatinib is a potent inhibitor of the MDR-associated ATP-binding cassette transporter ABCG2, Mol. Cancer Therapeut. 11 (2012) 2033–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bhullar J, Natarajan K, Shukla S, Mathias TJ, Sadowska M, Ambudkar SV, Baer MR, The FLT3 inhibitor quizartinib inhibits ABCG2 at pharmacologically relevant concentrations, with implications for both chemosensitization and adverse drug interactions, PLoS One 8 (2013) e71266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mattison RJ, Ostler KR, Locke FL, Godley LA, Implications of FLT3 mutations in the therapy of acute myeloid leukemia, Rev. Recent Clin. Trials 2 (2007) 135–141. [DOI] [PubMed] [Google Scholar]
- [65].Weisberg E, Boulton C, Kelly LM, Manley P, Fabbro D, Meyer T, Gilliland DG, Griffin JD, Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412, Cancer Cell 1 (2002) 433–443. [DOI] [PubMed] [Google Scholar]
- [66].Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C, Fox E, Ehninger G, Feldman EJ, Schiller GJ, Klimek VM, Nimer SD, Gilliland DG, Dutreix C, Huntsman-Labed A, Virkus J, Giles FJ, Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multitargeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3, J. Clin. Oncol. 28 (2010) 4339–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, Grandin W, Lebwohl D, Wang Y, Cohen P, Fox EA, Neuberg D, Clark J, Gilliland DG, Griffin JD, Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412, Blood 105 (2005) 54–60. [DOI] [PubMed] [Google Scholar]
- [68].Ambudkar SV, Cardarelli CO, Pashinsky I, Stein WD, Relation between the turnover number for vinblastine transport and for vinblastine-stimulated ATP hydrolysis by human P-glycoprotein, J. Biol. Chem. 272 (1997) 21160–21166. [DOI] [PubMed] [Google Scholar]
- [69].Alemany R, Moura DS, Redondo A, Martinez-Trufero J, Calabuig S, Saus C, Obrador-Hevia A, Ramos RF, Villar VH, Valverde C, Vaz MA, Medina J, Felipe-Abrio I, Hindi N, Taron M, Martin-Broto J, Nilotinib as co-adjuvant treatment with doxorubicin in patients with sarcomas: A phase I trial of the Spanish Group for Research on Sarcoma, Clin. Cancer Res. (2018) 5239–5249. [DOI] [PubMed] [Google Scholar]
- [70].Hegedus C, Ozvegy-Laczka C, Apati A, Magocsi M, Nemet K, Orfi L, Keri G, Katona M, Takats Z, Varadi A, Szakacs G, Sarkadi B, Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: implications for altered anticancer effects and pharmacological properties, Br. J. Pharmacol. 158 (2009) 1153–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Tiwari AK, Sodani K, Wang SR, Kuang YH, Ashby CR Jr., Chen X, Chen ZS, Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters, Biochem. Pharmacol. 78 (2009) 153–161. [DOI] [PubMed] [Google Scholar]
- [72].Hiwase DK, White D, Zrim S, Saunders V, Melo JV, Hughes TP, Nilotinib-mediated inhibition of ABCB1 increases intracellular concentration of dasatinib in CML cells: implications for combination TKI therapy, Leukemia 24 (2010) 658–660. [DOI] [PubMed] [Google Scholar]
- [73].Pratz KW, Levis MJ, Bench to bedside targeting of FLT3 in acute leukemia, Curr. Drug Targets 11 (2010) 781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Green H, Falk IJ, Lotfi K, Paul E, Hermansson M, Rosenquist R, Paul C, Nahi H, Association of ABCB1 polymorphisms with survival and in vitro cytotoxicty in de novo acute myeloid leukemia with normal karyotype, Pharmacogenomics J. 12 (2012) 111–118. [DOI] [PubMed] [Google Scholar]
- [75].Doxani C, Voulgarelis M, Zintzaras E, MDR1 mRNA expression and MDR1 gene variants as predictors of response to chemotherapy in patients with acute myeloid leukaemia: a meta-analysis, Biomarkers 18 (2013) 425–435. [DOI] [PubMed] [Google Scholar]
- [76].Jakobsen Falk I, Fyrberg A, Paul E, Nahi H, Hermanson M, Rosenquist R, Hoglund M, Palmqvist L, Stockelberg D, Wei Y, Green H, Lotfi K, Impact of ABCB1 single nucleotide polymorphisms 1236C > T and 2677G > T on overall survival in FLT3 wild-type de novo AML patients with normal karyotype, Br. J. Haematol. 167 (2014) 671–680. [DOI] [PubMed] [Google Scholar]
- [77].He H, Yin J, Li X, Zhang Y, Xu X, Zhai M, Chen J, Qian C, Zhou H, Liu Z, Association of ABCB1 polymorphisms with prognostic outcomes of anthracycline and cytarabine in Chinese patients with acute myeloid leukemia, Eur. J. Clin. Pharmacol. 71 (2015) 293–302. [DOI] [PubMed] [Google Scholar]
- [78].Megias-Vericat JE, Montesinos P, Herrero MJ, Moscardo F, Boso V, Rojas L, Martinez-Cuadron D, Hervas D, Boluda B, Garcia-Robles A, Rodriguez-Veiga R, Martin-Cerezuela M, Cervera J, Sendra L, Sanz J, Miguel A, Lorenzo I, Poveda JL, Sanz MA, Alino SF, Impact of ABC single nucleotide polymorphisms upon the efficacy and toxicity of induction chemotherapy in acute myeloid leukemia, Leuk. Lymphoma 58 (2017) 1197–1206. [DOI] [PubMed] [Google Scholar]
- [79].Stewart CF, Leggas M, Schuetz JD, Panetta JC, Cheshire PJ, Peterson J, Daw N, Jenkins JJ 3rd, Gilbertson R, Germain GS, Harwood FC, Houghton PJ, Gefitinib enhances the antitumor activity and oral bioavailability of irinotecan in mice, Cancer Res. 64 (2004) 7491–7499. [DOI] [PubMed] [Google Scholar]
- [80].Leggas M, Panetta JC, Zhuang Y, Schuetz JD, Johnston B, Bai F, Sorrentino B, Zhou S, Houghton PJ, Stewart CF, Gefitinib modulates the function of multiple ATP-binding cassette transporters in vivo, Cancer Res. 66 (2006) 4802–4807. [DOI] [PubMed] [Google Scholar]