

Summary
This protocol describes the use of the deterministic barcoding in tissue for spatial omics sequencing platform to construct a multi-omics atlas on fixed frozen tissue samples. This approach uses a microfluidic-based method to introduce combinatorial DNA oligo barcodes directly to the cells in a tissue section fixed on a glass slide. This technique does not directly resolve single cells but can achieve a near-single-cell resolution for spatial transcriptomics and spatial analysis of a targeted panel of proteins.
For complete details on the use and execution of this protocol, please refer to Liu et al. (2020).
Subject areas: Genomics, Sequencing, RNA-seq
Graphical abstract
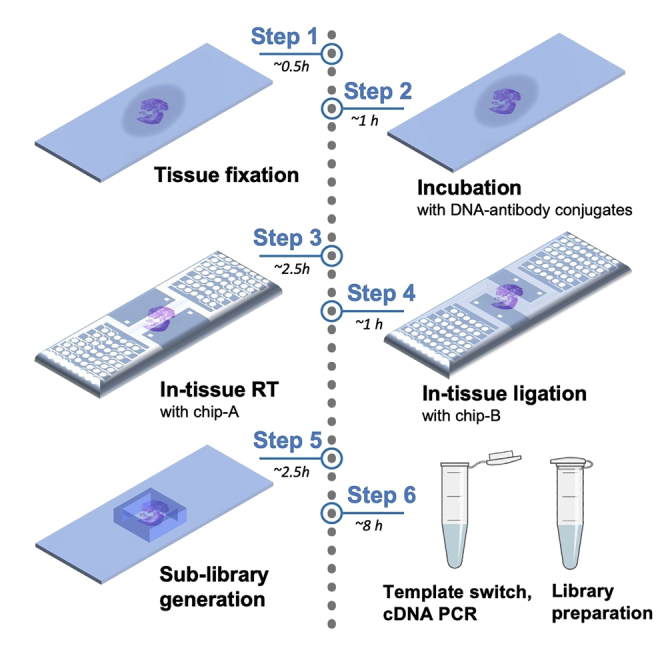
Highlights
-
•
Simultaneous transcriptomic and proteomic analysis on fixed tissue slides
-
•
10- and 25-μm spot size for varying resolution and near-single-cell analysis
-
•
DBiT-seq can be conducted with common laboratory equipment and reagents
-
•
DBiT-seq produces high-quality RNA sequencing data with high spatial resolution (10 μm)
This protocol describes the use of the deterministic barcoding in tissue for spatial omics sequencing platform to construct a multi-omics atlas on fixed frozen tissue samples. This approach uses a microfluidic-based method to introduce combinatorial DNA oligo barcodes directly to the cells in a tissue section fixed on a glass slide. This technique does not directly resolve single cells but can achieve a near-single-cell resolution for spatial transcriptomics and spatial analysis of a targeted panel of proteins.
Before you begin
The protocol below describes the reagents, equipment, and specific experimental steps for using the deterministic barcoding in tissue for spatial omics sequencing (DBiT-seq) platform on frozen fixed tissue slides. The 10 μm, 25 μm, 50 μm microfluidic channel width were designed and validated to provide spatial profiling at different resolutions.
CRITICAL: Work in an RNase-free environment when the microfluidic device is not on the tissue slide. Use RNaseZapTM or other commercially available cleaner solution and filter-tips. Clean surfaces and gloves with RNaseZapTM.
Fabricating the silicon wafer device mold
Timing: ~0.5 days
-
1.
Prepare a high-resolution computer-aided-design (CAD) file with the desired microfluidics chip design. A CAD file is also available in the link provided.
-
2.
Prepare chrome photomasks of the microfluidics chip by printing the high-resolution CAD files onto a glass substrate (Figure 1).
Note: We have outsourced this step (Front Range Photomask, USA).
Using these masks, prepare a replica mold as follows:
-
3.
Start the process by cleaning a 4-inch silicon wafer with 100% acetone (Aldrich) and then 100% isopropanol (Aldrich), then dry with compressed air.
Note: Acetone and isopropanol are mildly toxic. Use proper PPE when handling and discard waste in the appropriate containers.
-
4.
Bake the wafer at 180°C for 10 min on a hot plate to dry it out.
-
5.
Use a spin coater to evenly spread SU-8 2010 photoresist (MicroChem) for 10 μm device or SU-8 2025 (MicroChem) for 25 μm and 50 μm device onto the wafer at 500 rpm for 5 s followed by 1100 rpm (10 μm device), 3500 rpm (25 μm device) or 1750 rpm (50 μm device) for 40 s.
-
6.
Soft bake the wafer for 3 min at 65°C (25 μm and 50 μm device) and 4 min (10 μm device) or 6 min (25 μm and 50 μm device) at 95°C.
-
7.
Expose the SU-8 on the wafer through the photomask using Mask Aligner with a dose of 150 mJ/cm2 UV.
-
8.
For the post exposure bake, bake the wafer for 1 min at 65°C (25 μm and 50 μm device) and 5 min (10 μm and 25 μm device) or 6 min (50 μm device) at 95°C.
-
9.
Develop the SU-8 for 4 min (10 μm and 25 μm device) or 5 min (50 μm device) in a bath of ~50 mL SU-8 developer (MicroChem).
-
10.
Rinse the wafer with 100% isopropanol and dry with compressed air.
-
11.
Perform a hard bake by baking the wafer for 10 min at 180°C.
Figure 1.

Microfluidic photomask design
(Left) AutoCAD design for the 25 micron-width fifty channel Chip-A and Chip-B. (Right) AutoCAD design for 10 micron-width channel Chip-A and Chip-B.
 Pause point: The replica mold can now be stored and reused indefinitely.
Pause point: The replica mold can now be stored and reused indefinitely.
Creating the acrylic clamps
-
12.
Prepare the pattern and dimensions for the barcoding clamp and lysis clamp.
-
13.
Peel covering of acrylic sheet and place it in the laser cutting machine.
-
14.
Select the program dimensions and cut two pieces for the top and bottom of the barcoding clamp.
-
15.
Place the top blank piece into the laser cutter and select the pattern to cut out the four holes in the corner for the screws and nuts.
-
16.
Remove cut-out scraps with a pipet tip or a similar pointed tool.
-
17.
Repeat steps 15 and 16 for the bottom blank piece.
-
18.
Repeat steps 13 to 17 for the lysis clamp using the correct dimensions and pattern with an additional hole in the center of the top piece (Example shown in Figure 2).
Note: The acrylic clamps can be reused indefinitely.
Figure 2.

Key parts for setup
PDMS reservoirs and acrylic clamps used in DBiT-seq.
(A) Inlet Reservoir.
(B) Barcoding Clamp.
(C) Lysis Reservoir.
(D) Lysis Clamp.
Preparing the microfluidic device and reagent reservoirs
-
19.
Thoroughly mix polydimethylsiloxane (PDMS) elastomer base and curing agent (these come together) at a 10:1 ratio (see Figure 3).
-
20.
Pour the mixture into the silicon device mold.
Note: Aim for a chip height of about 5 mm.
-
21.
Place in a vacuum desiccator until all bubbles dissipate from the mixture about 30–60 min.
-
22.
Cure in an oven at 65°C–70°C for a minimum of 2 h and up to overnight.
Note: Make sure to place on an even surface to prevent uneven curing!
-
23.
Cut out the cured device and hole punch each of the 50 inlets and outlets for the channels.
Note: Cut both Chip-A and Chip-B in the approximate size of a glass slide.
Note: Make sure there are no PDMS pieces left in the inlets or outlets.
-
24.
Thoroughly clean the surface of the device with scotch tape.
-
25.
Pour mixture in a container large enough to cut out two roughly 25 × 25 × 5 mm PDMS pieces.
-
26.
For the barcoding reservoir, cut out a 20 × 20 mm piece in the center, barely large enough to surround the inlets of the chips when placed above.
-
27.
For the lysis reservoir, depending on the device size used, cut out the center to barely surround the barcoded region of the tissue (1 × 1 mm for the 10 μm channel device, 2.5 × 2.5 mm for the 25 μm channel device).
Note: The reservoirs are reusable. Thoroughly wash with 70% ethanol after each use.
Figure 3.

Step-by-step visual guide for making Chip-A and Chip-B
Preparing barcode B
-
28.
Thoroughly mix the ligation linker with each barcode B1-50 at a 1:1 ratio.
-
29.
Place the 50 mixes in a thermal cycle and heat to 97°C to anneal.
-
30.
Slowly cool to room temperature at a rate of −0.1°C/s.
-
31.
Store at −20°C for up to 6 months.
Preparing the tissue slide
-
32.
Fix in 4% paraformaldehyde (PFA) in PBS.
Note: PFA is moderately toxic and should be handled in a chemical fume hood with proper PPE. It should be disposed of in the proper waste container. This step is optional.
-
33.
25% sucrose overnight bath.
-
34.
Embed in OCT frozen on dry ice.
-
35.
Section at ~10 μm thickness for the 25 μm channel device and ~5 μm thickness for the 10 μm channel device onto Poly-L-lysine slides.
-
36.
Store slides at −80°C for up to 6 months until use.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TotalSeqTM antibodies | BioLegend | See Table S1 |
| Chemicals, peptides, and recombinant proteins | ||
| Maxima H Minus Reverse Transcriptase (200 U/L) | Thermo Fisher Scientific | EP0751 |
| dNTP Mix | Thermo Fisher Scientific | R0192 |
| RNase Inhibitor | Enzymatics | Y9240L |
| SUPERase•InTM RNase Inhibitor | Thermo Fisher Scientific | AM2694 |
| T4 DNA Ligase | New England Biolabs | M0202L |
| KAPA Pure Beads | Kapa Biosystems | KK8002 |
| DynabeadsTM MyOneTM Streptavidin C1 | Thermo Fisher Scientific | 65001 |
| Proteinase K, recombinant, PCR grade | Thermo Fisher Scientific | EO0491 |
| Kapa HiFi HotStart ReadyMix PCR Kit | Kapa Biosystems | KK2601 |
| Formaldehyde solution | Sigma | F8775-25ML |
| NEBuffer 3.1 | New England Biolabs | B7203S |
| T4 DNA Ligase Reaction Buffer | New England Biolabs | B0202S |
| EvaGreen Dye, 20× in water | Biotium | 31000-T |
| Oligonucleotides | ||
| Primers, ligation linkers, DNA barcodes | IDT | See Table S2 |
| Critical commercial assays | ||
| DNA Clean & Concentrator with Zymo-Spin I Columns | Research Products International | ZD4004 |
| Nextera XT DNA Preparation Kit | Illumina | FC-131-1024 |
| Other | ||
| Multiplate PCR plate, 96-well, clear | Bio-Rad | MLL9601 |
| 50 mL Falcon Tube | Corning | 352070 |
| BioDot Pure 8 – Strip PCR tubes w/ Optically Clear Flat Caps | Dot Scientific Inc. | 403-8PCR |
| SYLGARDTM 184 Silicone Elastomer Base and Curing Agent | Dow Corning | 4019862 |
| SuperChipTM Poly-L-Lysine Slides | Electron Microscopy Sciences | 63478-AS |
| RNaseZapTM RNase Decontamination Solution | Thermo Fisher Scientific | AM9780 |
| Silicon wafer | WaferPro | C04004 |
| Photoresist SU-8 2010 | Microchem Laboratory | SU-8 2010 |
Materials and equipment
-
•
This protocol requires using a microfluidic device fabricated with PDMS using soft lithography.
-
•
A hole punching machine (SCHMIDT® Manual Press) is needed to punch the inlet and outlet holes in the PDMS chip after its fabrication.
-
•
The tissue of interest should be placed on the center of a poly-L-lysine coated glass slide. (CatLog no. 63478-AS, electron microscopy sciences).
-
•
A custom-designed acrylic clamp with screws is needed to hold the PDMS device and the glass slide together firmly.
-
•
The silicon wafer used for fabricating the PDMS mold is purchased from WaferPro (CatLog. No. C04004).
-
•
The photoresist is purchased from MicroChem Laboratory (CatLog. No. SU-8 2010).
-
•
A homemade laboratory vacuum system (Figure 4) is needed to applying suction to move fluid in the microfluidic channels.
-
•
Microscopy – The tissue of interest can be scanned and imaged using EVOS (Thermo Fisher EVOS fl), typically at a magnification of 10×. Any suitable optical microscope can be used.
-
•
A Laser Engraver Cutter is used to cut out the acrylic barcode and lysis clamps.
-
•
A “humidified chamber” (Figure 4) to prevent reagent evaporation during incubation.
Figure 4.

Key equipment used in DBiT-seq
Hole punch machine, the key device for DBiT-seq, and the house vacuum lines. (Left) SCHMIDT® Manual Press used for hole punching 2-mm diameter holes for the inlets and outlets of Chip-A and Chip-B. (Top Right) Example of a “humidified chamber” to prevent reagent evaporation during the incubation steps. (Bottom Right) House vacuum used for pulling reagents through the microchannels.
Recipes
PBS-RI
| Reagent | Volume (μL) |
|---|---|
| 1× PBS | 5,000 |
| RNase Inhibitor (40 U/μL) (Enzymatics) | 7 |
| Total | 5,007 |
80% Ethanol
| Reagent | Volume (mL) |
|---|---|
| 100% Ethanol | 4 |
| RNase-free water | 1 |
| Total | 5 |
RT mixture
| Reagent | Volume (μL) |
|---|---|
| 5× Maxima RT buffer | 50 |
| RNase-free water | 32.8 |
| RNase Inhibitor (Enzymatics) | 1.6 |
| Superase•In RNase Inhibitor (Ambion) | 3.2 |
| dNTPs (10 mM stock) | 12.5 |
| Maxima H Minus Reverse Transcriptase | 25 |
| PBS-RI | 100 |
| Total | 225.1 |
Ligation mix
| Reagent | Volume (μL) |
|---|---|
| RNase-free water | 69.5 |
| 10× T4 Ligase Buffer | 27 |
| T4 DNA Ligase (400 U/μL) | 11 |
| RNase Inhibitor (Enzymatics) | 2.2 |
| Superase•In RNase Inhibitor (Ambion) | 0.7 |
| 5% Triton-X100 | 5.4 |
| 1× NEB buffer 3.1 with 1% RI (Enzymatics) | 115.8 |
| Total | 231.6 |
Template switch mix
| Reagent | Volume (μL) |
|---|---|
| 20% Ficoll PM-400 | 44 |
| 5× Maxima RT buffer | 44 |
| dNTPs (10 mM stock) | 22 |
| RNase Inhibitor (Enzymatics) | 5.5 |
| Maxima H Minus Reverse Transcriptase | 11 |
| Template Switch Primer (100 μM stock) | 5.5 |
| RNase-free water | 88 |
| Total | 220 |
PCR mix
| Reagent | Volume (μL) |
|---|---|
| 2× Kapa HiFi HotStart master mix | 110 |
| Primer1 BC_0062 (10 μM) | 8.8 |
| Primer2 BC_0108 (10 μM) | 8.8 |
| RNase-free water | 92.4 |
| Total | 220 |
Flow Wash Buffer
| Reagent | Volume (mL) |
|---|---|
| 1× PBS | 4 |
| 10 % Triton X-100 | 0.04 |
| Superase•In RNase Inhibitor | 0.01 |
| Total volume | 4.05 |
1× Lysis Solution
| Reagent | Volume (μL) |
|---|---|
| 1× PBS | 50 |
| 2× Lysis Buffer | 50 |
| Proteinase K (20 mg/mL) (Thermo) | 10 |
| Total | 110 |
2× Lysis Buffer
| Reagent | Stock concentration | Final concentration (2×) | Volume (mL) |
|---|---|---|---|
| Tris, pH 8.0 | 1 M | 20 mM | 0.5 |
| NaCl | 5 M | 400 mM | 2 |
| EDTA, pH 8.0 | 0.5 M | 100 mM | 5 |
| SDS | 10% | 4.4 % | 11 |
| RNase-free Water | NA | NA | 6.5 |
| Final Volume | 25 |
1× B&W Buffer with 0.05% Tween-20
| Reagent | Volume |
|---|---|
| 1M Tris-HCl pH 8.0 | 100 μL |
| EDTA, 0.5M | 20 μL |
| 5M NaCl | 4 mL |
| Tween 20 10% | 100 μL |
| RNase-free water | 15.78 mL |
| Total | 20 mL |
2× B&W Buffer
| Reagent | Volume |
|---|---|
| 1M Tris-HCl pH 8.0 | 500 μL |
| EDTA, 0.5M | 100 μL |
| 5M NaCl | 20 mL |
| RNase-free water | 29.4 mL |
| Total | 50 mL |
Step-by-step method details
Tissue preparation
This section describes the steps to do right before introducing the barcodes to the tissue.
-
1.
Remove stored sections from the freezer and allow to warm to room temperature for 10 min.
-
2.
Clean sections by pipetting 2 mL of PBS-RI across the tissue.
-
3.
If sections are not yet fixed, then fix here with 1 mL 4% PFA by applying on top of the tissue and incubate for 20 min at room temperature.
-
4.
Wash the slides 3 times with 1 mL of PBS and then dip the slide in a 50 mL falcon tube with DI water.
-
5.
Dry the section with gentle air flow.
-
6.
Take a full pre-scan image of the section at the desired optical resolution (Figure 10A). Recommended 10× objective.
-
7.
Block the tissue slide with 1% BSA and 1% RNase Inhibitor in 1× PBS for 30 min at 4°C.
-
8.
Remove the blocking buffer and briefly air-dry.
-
9.
Add a cocktail of the desired DNA-antibody conjugates + blocking buffer and incubate for 30 min at 4°C.
-
10.
Wash the slide with 1× PBS three times.
-
11.
Dip in a 50 mL falcon tube with DI water and gently dry with air flow.
-
12.
Attach Chip-A as shown in Figure 5 centered on the tissue and take another scan of the channels laying on the tissue (Figure 10C).
-
13.Permeabilize the tissue section.
-
a.Firmly attach the barcoding clamp across the center of the device & slide.
-
b.Place the inlet reservoir above the inlets.
-
c.Add ~1 mL 0.5% Triton X-100 in PBS to the reservoir.
-
i.Use a 10 μL pipet tip to pipet up and down inside the inlets to remove bubbles.
-
i.
-
d.Attach the vacuum head to the outlets as shown in Figure 6 and pull the solution through the 50 channels.Note: Make sure all channels are filled with solution.
-
e.Stop the vacuum and incubate for 20 min at room temperature.
-
f.Remove remaining permeabilization solution from the reservoir and the inlets/outlets.
-
g.Add ~1 mL 0.5× PBS-RI to the reservoir and flow for about 10 min to wash and clean the channels of Chip-A.
-
a.
Figure 10.

Example scanning of the tissue slide
(A) Initial full scan of the tissue section prior to DBiT-seq (step 6).
(B) Final scan after barcoding and removing the device (step 26).
(C) Scan of Chip-A channels placed on the tissue (step 12).
(D) Scan of Chip-B channels across the tissue (step 20).
Figure 5.

Assembly to create the setup for the PDMS reservoir and acrylic clamps
(A) Chip-A with the inlet reservoir and barcoding clamp.
(B) Side view of Chip-A.
(C) The lysis reservoir with the lysis clamp on top of the barcoded tissue.
(D) Side view of the lysis setup.
Figure 6.

Chip-A with Acrylic clamp and house vacuum attached
In-tissue reverse transcription
This applies the set of barcode A using Chip-A.
-
14.Prepare the RT mixture.
-
a.For one experiment, prepare 50 μL of 5× RT Buffer, 32.8 μL of RNase-free water, 1.6 μL Enzymatics RNase inhibitor, 3.2 μL SUPERase•InTM RNase Inhibitor, 12.5 μL of 10 mM dNTPs each, 25 μL of Maxima H Minus Reverse Transcriptase, and 100 μL 0.5× PBS-RI.
-
a.
-
15.Prepare barcode solution.
-
a.In a 96-well PCR plate, add 4 μL of the RT mixture in each of 50 wells.
-
b.Add 1 μL of each 25 μM Barcode A1-50 to the wells and mix thoroughly.
-
a.
-
16.Introduce final barcode A mixture to the tissue.
-
a.Pipet each barcode A mixture directly into the inlets following the sequence as indicated in Figure 7. This sequence corresponds to the ordering of the channels across the center region of interest (for example, channels from top to bottom of Chip A correspond to 1-50 in the pipetting order).
-
b.Apply the vacuum and pull the solution until all channels are full.Note: May take as short as 10 s or as long as 2 min depending on channel dimensions (smaller channels may take longer to fill).CRITICAL: Do not over vacuum! Make sure the inlets do not run out of barcode A mixture which will cause the channels to lose the mixture and fill with air.
-
c.Incubate in the humidified chamber at room temperature for 30 min followed by 42°C for 90 min for in-cell reverse transcription (Figure 8).
-
a.
-
17.
Aspirate remaining barcode A mixture from inlets/outlets.
-
18.
Clean the channels by flushing 1× NEB buffer 3.1 with 1% Enzymatics RNase Inhibitor for 10 min.
-
19.
Peel off Chip-A and briefly dip the slide in a 50 mL falcon tube with DI water and gently dry with air flow.
Figure 7.

Sequential numbering of the inlet holes and pipetting order for Barcodes A1-50 and B1-50
Figure 8.

Deterministic tissue barcoding chemistry workflow
In-tissue ligation
This applies barcode B using Chip-B.
-
20.Attach Chip-B centered on the tissue.
-
a.Scan the channels over the tissue to ensure proper placement (Figure 10D).
-
b.Firmly clamp as before.
-
a.
-
21.Prepare the ligation mixture.
-
a.For one experiment, prepare 69.5 μL RNase-free water, 27 μL 10× T4 Ligation buffer (NEB), 11 μL T4 DNA Ligase (400 U/μL, NEB), 2.2 μL RNase inhibitor (40 U/μL, Enzymatics), 0.7 μL SUPERase•InTM RNase Inhibitor (20 U/μL, Ambion), and 5.4 μL of 5% Triton X-100. Mix with 115.8 μL 1× NEB buffer 3.1 with 1% RNase Inhibitor (Enzymatics).
-
a.
-
22.Prepare barcode B mixture.
-
a.In a 96-well PCR plate, add 4 μL of the ligation mixture in each of 50 wells.
-
b.Add 1 μL of each 25 μM Barcode B1-50 to the wells and mix thoroughly.
-
a.
-
23.Introduce final barcode B mixture to the tissue.
-
a.Pipet each barcode B mixture directly into the inlets.
-
b.Apply the vacuum and pull the solution until all channels are full. May take as short as 10 s or as long as 2 min.CRITICAL: Do not over vacuum! Make sure the inlets do not run out of barcode B mixture which will cause the channels to lose the mixture and fill with air.
-
c.Incubate in the humidified chamber at 37°C for 30 min for in-cell ligation of barcode B (Figure 8).
-
a.
-
24.Clean the channels with wash buffer for 10 min.
-
a.Prepare wash buffer by mixing 4 mL of 1× PBS, 40 μL of 10% Triton X-100, and 10 μL of SUPERase•InTM RNase Inhibitor.
-
a.
-
25.
Peel off Chip-B and dip the slide into a 50 mL falcon tube with DI water and gently dry with air flow.
-
26.
Take a final post-scan of the tissue section (Figure 10B).
Lysis and sub-library generation
The tissue-derived cDNA is collected in this step.
-
27.
Place the lysis reservoir directly around the barcoded tissue region.
Note: Try to cover as little extra tissue as possible.
-
28.
Tightly apply the lysis clamp.
-
29.Add lysis solution to lysis reservoir depending on channel dimensions (~20 μL for the 25 μm channels, ~10 μL for the 10 μm channels).
-
a.Lysis solution is made with 50 μL 1× PBS, 50 μL of 2× lysis buffer, and 10 μL of proteinase K solution (20 mg/mL).
-
b.2× lysis buffer is made up of 20 mM Tris (pH 8.0), 400 mM NaCl, 100 mM EDTA (pH 8.0), and 4.4% SDS.
-
a.
-
30.
Place in the humidified chamber and tightly wrap the chamber with parafilm and incubate at 55°C for 2 h.
-
31.
Collect the lysate into a 1.5 mL centrifuge tube. Add the same amount of extra lysis solution to wash out the reservoir and retrieve as much lysate as possible.
-
32.
Immediately store at −80°C until next step.
cDNA purification
The cDNA is purified and bound to DynabeadsTM MyOneTM Streptavidin C1 beads in this step.
-
33.Prepare 40 μL DynabeadsTM MyOneTM Streptavidin C1 beads (Thermo Fisher) per sub-library.
-
a.Wash 3 times with 800 μL of 1× B&W buffer with 0.05% Tween-20.
-
b.Resuspend beads in 100 μL 2× B&W buffer + 2 μL SUPERase•InTM RNase Inhibitor.
-
a.
-
34.
Purify lysate following the DNA Clean & Concentrator with Zymo-Spin IC Columns (RPI Research Products) protocol. Use 100 μL water to elute DNA.
-
35.
Add 100 μL of resuspended DynabeadsTM MyOneTM Streptavidin C1 magnetic beads to each lysate.
-
36.
Rotate at 30 RPM and room temperature for 60 min for binding to occur.
-
37.Wash beads.
-
a.Wash twice with 400 μL of 1× B&W buffer with 5 min rotation after resuspending beads.
-
b.Wash once more with 400 μL 10 mM Tris containing 0.1% Tween-20 with 5 min rotation after resuspension.
-
a.
Template switch
This step performs template switching and adds the second PCR handle.
-
38.
Resuspend the streptavidin beads bound with cDNA in solution containing 44 μL of 5× Maxima RT buffer (Thermo Fisher), 44 μL of 20% Ficoll PM-400 solution, 22 μL of 10 mM dNTPs each (Thermo Fisher), 5.5 μL RNase Inhibitor (Enzymatics), 11 μL of Maxima H Minus Reverse Transcriptase (Thermo Fisher), 5.5 μL of 100 μM of template switch primer, and 88 μL of RNase-free water.
-
39.
Rotate beads at 30 RPM and room temperature for 30 min followed by rotation at 42°C for 90 min.
PCR
In this step, cDNA is amplified and separated from the Dynabeads.
-
40.Wash beads and resuspend.
-
a.Wash once with 400 μL of 10 mM Tris and 0.1% Tween-20 solution and once more with 400 μL of RNase-free water.
-
b.Resuspend in the PCR mix solution containing 110 μL of 2× Kapa HiFi HotStart Master Mix (Kapa Biosystems), 8.8 μL each of 10 μM stocks of primers 1 and 2, and 92.4 μL of RNase-free water. Transfer 200 μL to four PCR tubes with 50 μL in each.
-
a.
-
41.
Perform PCR to detach cDNA from beads.
| PCR cycling conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial Denaturation | 95°C | 3 min | 1 |
| Denaturation | 98°C | 20 s | 5 cycles |
| Annealing | 65°C | 45 s | |
| Extension | 72°C | 3 min | |
-
42.
Remove the DynabeadsTM from the PCR solution using a magnetic tube holder.
-
43.
Add Evagreen (Biotium) at a 1× concentration.
Note: This step is optional.
-
44.
Perform PCR again with the following thermocycling conditions. Cycling can also be halted once the qPCR signal begins to plateau.
| PCR cycling conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial Denaturation | 95°C | 3 min | 1 |
| Denaturation | 98°C | 20 s | 15 cycles |
| Annealing | 65°C | 20 s | |
| Extension | 72°C | 3 min | |
| Final Extension | 72°C | 5 min | 1 |
| Hold | 4°C | Forever | |
cDNA purification
This is the final purification step for the cDNA before building a library.
-
45.
Add 80 μL KAPA pure beads for 100 μL of the cDNA sample.
Note: Ensure that beads have been equilibrated to room temperature and are fully resuspended before use.
-
46.
Mix thoroughly by pipetting.
-
47.
Incubate the tubes at room temperature for at least 10 min to bind the cDNA to the beads.
-
48.
Place the tubes on a magnet to capture the beads and incubate until the liquid is clear.
-
49.
Carefully remove and discard the supernatant.
-
50.
Keep the tubes on the magnet and add 200 μL of 80% ethanol.
-
51.
Incubate the tubes at room temperature for at least 30 s.
-
52.
Carefully remove and discard the ethanol.
-
53.
Repeat steps 50–52.
-
54.
Try to remove all residual ethanol and dry the beads at room temperature for 3–5 min or until all the ethanol has evaporated.
-
55.
Remove the tubes from the magnet and resuspend the beads in 15 μL of PCR-grade water or elution buffer depending on downstream application.
-
56.
Incubate the tubes at room temperature for at least 10 min to elute the cDNA off the beads.
-
57.
Place the tubes back on the magnet and incubate until the liquid is fully clear.
-
58.
Transfer the clear supernatant to a new tube.
-
59.
Perform Bioanalyzer QC to obtain cDNA length profile and concentration.
Library preparation
This step describes the process to building a library for sequencing.
-
60.
Dilute 0.75 to 1 ng of purified cDNA in PCR-grade water to a total of 5 μL.
-
61.
Add 10 μL of Tagment DNA buffer and 5 μL of Amplicon Tagment Mix to the cDNA for a total of 20 μL.
-
62.
Mix thoroughly and incubate at 55°C for 5 min.
-
63.
Add and mix 5 μL of NT Buffer to the solution and incubate at room temperature for 5 min.
-
64.
In this order, add 15 μL of the PCR master mix, 8 μL of water, and 1 μL of each 10 μM primer (P5 primer and indexed P7 primer) to the mix for a total of 50 μL.
-
65.
Perform PCR with the following thermocycling conditions.
| PCR cycling conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial Denaturation | 95°C | 30 s | 1 |
| Denaturation | 95°C | 10 s | 12 cycles |
| Annealing | 55°C | 30 s | |
| Extension | 72°C | 30 s | |
| Final Extension | 72°C | 5 min | 1 |
| Hold | 4°C | Forever | |
-
66.
Purify the resulting PCR reaction mix at a 0.7× ratio of KAPA pure beads according to the manufacturer’s manual to generate an Illumina-compatible sequencing library (as in steps 45–59).
-
67.
Perform quality control analysis (Bioanalyzer) to obtain more accurate concentration and yield to prepare for sequencing.
Expected outcomes
When using Qubit or Bioanalyzer, the resulting cDNA concentration should be larger than 2 ng/μL, but generally less than 20 ng/μL. The value can vary substantially with different tissue types. The general cDNA size should be peaked over 1000 bp and the average size will also be larger than 1000 bp. For the ADT library, it should have a dominant sharp peak around 256 bp and the concentration is usually larger than 7 ng/μL. An example of these are shown in Figure 9.
Figure 9.

Example bioanalyzer results for the barcoded cDNA (top), protein library (middle), and cDNA library (bottom)
Limitations
Although DBiT-seq is close to single-cell level spatial mapping, it does not directly resolve single cells. Our approach potentially allows for high resolution immunofluorescence or fluorescence in situ hybridization (FISH) on the same tissue slide to facilitate cell segmentation, identification, and the deconvolution of the data in each pixel to computationally derive single-cell spatial omics.
There is a fundamental limit of pixel size. The validation experiments in our previous study indicate the diffusion distance is ~1 μm. Thus, the theoretical pixel size can be as small as ~2 μm (Liu et al., 2020). However, because the tissue section thickness is 5 μm, the tissue deformation upon the application of the clamp may partially block the microchannel flow if the channels are too shallow. According to our observations, the achievable smallest pixel size is ~5 μm, in which most pixels contain one or a fraction of a cell, making computational convolution more feasible. On the other hand, because the tissue section is typically 5 to 7 μm, it is unlikely to yield the meaningful data with the pixel size << 5 μm due to high aspect ratio.
The number of flow channels in current DBiT-seq device is still limited such that the mappable area with 10 μm pixel size is 1 mm × 1 mm. It can be increased by increasing the number of barcodes to 100 × 100 or 200 × 200, giving rise to 2 mm × 2 mm mappable area. Alternatively, a serpentine microfluidic channel design can dramatically increase the mappable area without the need to increase the number of DNA barcodes, which has been demonstrated in our laboratory with 5 repeats of microfluidic channels to increase the mappable area to 10 mm.
Formaldehyde-fixed tissue slide is used as the starting material in DBiT-seq. Unfortunately, Formalin-fixed paraffin-embedded (FFPE) tissues, the most abundant archivable specimens in clinical tissue banks, are incompatible with most spatial transcriptome mapping techniques at cellular level (spot size ~10 μm) due to the problems in RNA degradation in storage and RNA damage in extraction. In theory, in-tissue barcoding may obviate the problems associated with tissue decrosslinking and mRNA extraction prior to reverse transcription. Our results have demonstrated the feasibility to perform DBiT-seq on FFPE tissue samples although the performance is yet to be further improved (unpublished data).
Following our current DBiT protocol, lysis has to be carried out on the flow-barcoded tissue region to extract and collect cDNAs of interest. To extend the utility of DBiT-seq to further combine with other spatial analysis, how to perform DBiT-seq in a completely non-destructive manner and retain intact tissue sections after the DBiT-seq experiment is critical.
As discussed in trouble shooting, the variability in microfluidic flow is unavoidable experimentally. However, researchers in the field of scRNA-seq have seen the variability of DNA barcode beads and developed computational methods to perform normalization, which can be adopted for DBiT-seq. However, to better correct this channel-to-channel variation, new computational methods can be developed specifically for DBiT-seq. In theory, such normalization can be more reliable and more accurate because the pixels do not vary in a completely random manner like in scRNA-seq but usually co-vary within the same microchannel, which can be considered to develop image-driven computational normalization.
The reliability of the experiment can depend highly on the type of tissue, especially when using the 10-micron channel-width device. DBiT-seq with 25-micron and 50-micron devices are quite robust with most tissue types. The latter two can be easily clamped tightly to ensure that reagents do not leak through the channels.
The highest resolution 10-micron device also has the characteristic of very thin channel walls which collapse more readily if clamped too tightly. This potential issue causes uneven or porous sections of tissue more difficult to process. For example, folding of the tissue during the sectioning process or damage to the tissue or holes cause issues to the microfluidic flow. Furthermore, denser or stiffer tissue such as those that are highly collagenous (sclera, for example) also have difficulties. To test the compatibility of a tissue section with this protocol, fluorescently labeled barcode A and B can be used as substitutes to barcode A1-50 and B1-50 to visualize proper flow and reactions occurring. The 10-micron device requires very thin and flat tissue sections to maximize its success, and porous and folded sections are not so robust at this resolution.
Troubleshooting
Problem 1
Possible leakage across channels.
Potential solution
To prevent this from happening, use an acrylic clamp to firmly hold the PDMS device against the tissue of interest on the poly-L-lysine glass slide. Ensure that the screws are tightened enough to prevent crosstalk across channels. However, tightening the clamp too firmly may obstruct the flow of fluid in the channels. Use a microscope to make sure the channel walls are stable and not collapsed.
Problem 2
Tissue sample is placed off-center on the poly-L-lysine slide.
Potential solution
For optimal results, it is advised that the tissue section be placed at the center or as close to the center of the slide as possible such that the channels can cover it. This can be done by drawing diagonal lines with sharpie on the back of the slide and placing the tissue section on the intersection of the two lines. The sharpie can be erased after with 70% ethanol.
Problem 3
Fluid in channels not flowing as expected.
Potential solution
This problem may be caused by several factors:
Firstly, if the tissue of interest is too thick, it may affect the ability of fluids to flow within the channels. To mitigate this problem and to obtain optimal results, ensure that the thickness of the tissue of interest after sectioning is under 7 μm for the 10 μm channel device and about 10 μm for the 25 μm channel device.
The custom-designed acrylic clamp used to firmly hold the PDMS against the tissue on the glass slide may be tightened too tightly. It is recommended to slightly adjust the tightness of the screws to ensure that fluids in the channel can flow unimpededly.
Trapped air bubbles in the channels or inlets may also impede the flow of fluid in the channels. It is sometimes necessary to gently pipette the fluid into the inlets up and down a few times to remove any trapped air bubbles.
The suctioning power of the house vacuum may be too low. Adjust this by opening and closing the valves as necessary to ensure that the fluid within the channels can flow freely.
Problem 4
Adding the wrong barcode to an inlet during the reverse transcription and ligation step.
Potential solution
To ensure that the right barcode is placed into the right well, a guide for adding the barcodes to the inlets has been included here. However, if the barcodes are swapped for any reason, take note of the inset of interest and account for those barcodes in the data analysis.
Problem 5
Evaporation of fluid from inlets and channels during reverse transcription step and ligation step.
Potential solution
It is recommended to cover the device in a petri dish and keep inside a sealed wet chamber before placing in the oven or incubator.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Rong Fan (rong.fan@yale.edu).
Materials availability
Microfluidic devices and the associated design files generated in this study are available upon request in accordance with the Materials Transfer Agreement (MTA) of Yale University.
Data and code availability
The accession number for the analyzing the data generated by the corresponding Cell paper is submitted to GEO: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137986.
Code for sequencing data analysis is available (https://github.com/rongfan8/DBiT-seq).
Acknowledgments
The molds for microfluidic chips were fabricated at the Yale University School of Engineering and Applied Science (SEAS) Nanofabrication Center. This sequencing service was conducted at Yale Stem Cell Center Genomics Core Facility that was supported by the Connecticut Regenerative Medicine Research Fund and the Li Ka Shing Foundation. It was also conducted using the sequencing facility at the Yale Center for Genomic Analysis (YCGA). This research was supported by the Packard Fellowship for Science and Engineering (to R.F.), Stand-Up-to-Cancer (SU2C) Convergence 2.0 Award (to R.F.), and Yale Stem Cell Center Chen Innovation Award (to R.F.). It was supported in part by grants from the NIH (U54CA209992, R01CA245313, and UG3CA257393 to R.F.). Y.L. was supported by the Society for Immunotherapy of Cancer (SITC) Postdoctoral Fellowship.
Author contributions
Conceptualization, R.F.; methodology, Y.L., Y.D., and G.S.; experimental investigation, Y.L., Y.D., G.S., A.E., and X.Q.; resources, G.S., Y.L, Y.D., and Z.B.; writing – original draft, G.S., X.Q., A.E., and Y.D.; writing – review & editing, R.F., Y.L., G.S., A.E., X.Q., Y.D., and Z.B.
Declaration of interests
R.F., Y.L., and Y.D. are inventors of a patent application related to this work. R.F. is a co-founder of IsoPlexis, Singleron Biotechnologies, and AtlasXomics and a member of their scientific advisory boards with financial interests, which could affect or have the perception of affecting the author's objectivity. The interests of R.F. were reviewed and managed by Yale University Provost's Office in accordance with the university's conflict of interest policies.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100532.
Contributor Information
Graham Su, Email: graham.su@yale.edu.
Rong Fan, Email: rong.fan@yale.edu.
Supplemental information
References
- Liu Y., Yang M., Deng Y., Su G., Enninful A., Guo C.C., Tebaldi T., Zhang D., Kim D., Bai Z. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell. 2020;183:1665–1681.e18. doi: 10.1016/j.cell.2020.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the analyzing the data generated by the corresponding Cell paper is submitted to GEO: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137986.
Code for sequencing data analysis is available (https://github.com/rongfan8/DBiT-seq).