

Abstract
Background:
To mitigate increased risk of premature cardiovascular disease in antiretroviral therapy (ART) suppressed adults living with HIV (PWH), low dose methotrexate (LDMTX) was evaluated in a multicenter randomized placebo controlled clinical trial of 176 PWH taking various ART regimens (ACTG A5314). Given shared methotrexate (MTX) and tenofovir (TFV) pharmacokinetic (PK) pathways, a substudy was carried out to investigate whether LDMTX alters TFV exposure.
Methods:
Adults virally suppressed on ART for >24 weeks were randomized to LDMTX or placebo. The first 66 participants taking a tenofovir disoproxil fumarate-containing regimen underwent intensive PK sampling over 24 h following the second dose of LDMTX 10 mg or placebo. TFV and MTX levels were quantified using validated mass spectrometry methods. TFV PK between LDMTX and placebo groups were compared and MTX PK was characterized.
Results:
Forty-eight participants completed this substudy (n=20 on LDMTX and 28 on placebo). Baseline characteristics were balanced except for PI-use (25% in LDMTX and 43% in placebo groups). For TFV, AUC6 (primary endpoint), and AUC24,imputed, Cmax, and Cmin (secondary endpoints) were on average 22%, and 24%, 27%, and 31% less in the LDMTX versus placebo groups, with reductions in secondary endpoints reaching statistical significance. Additional analyses suggested a greater reduction in the absence of PI although not significant.
Conclusion:
Lower TFV AUC24,imputed and Cmax indicates that LDMTX reduces TFV exposure in PWH. However, this change was modest, not warranting a change in TFV dosing at this time. Further studies of TFV PK with LDMTX, especially without PI co-administration, are warranted.
Keywords: tenofovir, methotrexate, pharmacokinetics, drug interactions, transporters
Introduction
A recent study of the AIDS Clinical Trials Group (ACTG), A5314, investigated the use of low dose methotrexate (LDMTX) to reduce inflammation and also improve endothelial function associated with chronic HIV infection1. This study, a phase II trial, investigated the safety and efficacy of LDMTX in participants with adequately controlled HIV on antiretroviral therapy (ART). In this pharmacokinetic (PK) substudy, we hypothesized that tenofovir (TFV), given as tenofovir disoproxil fumarate (TDF), a common component of ART regimens, may be subject to drug-drug interactions with methotrexate (MTX) given that both drugs are renal organic ion transporter (OAT) 1 and OAT3 substrates.
MTX is primarily eliminated renally unchanged via filtration and active secretion via OAT1 and OAT32,3 but exhibits other complex pharmacological characteristics, including variable absorption (tmax 0.7–4 h) and saturable dose-dependent absorption (28 to 88% bioavailability)5,6. It is also significantly metabolized by intestinal flora and intracellularly to more active polyglutamate derivatives retained in the cells until reverse conversion for elimination5. Coadministration of OAT-transported substrates, such as penicillins7,8 and ciprofloxacin9 have resulted in increased, potentially toxic, MTX levels. MTX has not been identified a perpetrator of renal transport-related drug-drug interactions10, but studies are limited.
Similar to MTX, TFV is also eliminated by renal filtration and active secretion by OAT1 and OAT311,12. TFV, administered as TDF, is poorly absorbed (approximately 25% bioavailability)13. For some known TFV drug interactions, definitive mechanisms are unclear. In the case of higher TFV with protease inhibitors (e.g. atazanavir14), inhibition at the apical membrane resulting in higher TFV accumulation in proximal tubule cells has been proposed. For TFV increasing raltegravir exposure15, OAT1 inhibition has been suggested as a potential mechanism16.
For MTX, little is known regarding the use of low dose oral MTX in the context of ART. One study observed no difference in MTX half-life following high dose intravenous administration to ART treated PWH17, but did not investigate the impact of MTX on TFV PK.
Such interactions may result in increased TFV or MTX levels, which may result in TFV-mediated renal toxicity via accumulation in renal proximal tubule cells or toxic MTX exposure. Unexpected interactions that may decrease exposure of TFV or MTX, could potentiate loss of viral suppression or loss of MTX anti-inflammatory efficacy, respectively.
We therefore sought to evaluate the potential drug-drug interaction between LDMTX and TFV in people living with HIV (PWH). In this A5314 substudy enrolling TFV-treated participants, intensive PK samples were collected and analyzed for TFV and MTX concentrations. Using PK and statistical analyses, we compared TFV exposure between those receiving active MTX to those receiving placebo and characterized low dose MTX exposure in the context of TFV-containing ART.
Methods
A5314 study design
A5314 is a phase II double blind, randomized placebo-controlled trial assessing the safety and efficacy of LDMTX on endothelial function and inflammation in PWH who have been virologically suppressed with continuous ART (NCT01949116)1. The study included men and women at least 40 years of age, with or at risk for atherosclerotic cardiovascular disease with ART-suppressed HIV (CD4+ T-cell count ≥ 400 cells/mm3 and HIV-1 RNA level<40 copies/mL for at least 24 weeks prior to study entry). Participants who met the enrollment criteria were randomized 1:1 to LDMTX or placebo. From entry through week 1 (lead-in period), participants took 5 mg once weekly by mouth of either MTX or placebo. Participants were then titrated to 10mg/week over 12 weeks, at which point they received the maximum dose of 15mg/week until week 24. If a participant did not meet the criteria for dose escalation at the protocol-defined dose escalation time, then the participant remained on his/her current dose until the next study visit at which time the participant was re-evaluated for dose escalation. All participants received 1 mg folic acid daily from study entry until 4 weeks after completion of study treatment, regardless of randomization.
PK Study Design
Participants were required to be on steady state TFV (defined as continuous ART for ≥24 weeks before enrollment without any changes to their basic regimen in the prior 12 weeks) and self-reported adherence for the last 4 doses. Intensive PK sampling was performed after the second dose of MTX (10 mg). LDMTX (or placebo) and TFV were administered at the same time and serial sampling was carried out at 0 (pre-dose), 0.5, 1, 2, 4, and 6 hours post-dosing (protocol version 2.0) for analysis of both TFV and MTX (for those on active drug) levels; under version 1.0 of the protocol some participants were also sampled at 8, 12 and 24 hours post-dosing. The reason for the 6 hour sampling was to enhance enrollment by limiting the time commitment for the study, but to still allow intensive PK sampling during the day while clinics remained open.
A sample size of approximately 21 evaluable participants per treatment group was chosen to provide 95% power to detect a clinically relevant 40% increase in TFV AUC (in LDMTX versus placebo arms) with a relaxed type-one error rate (1-sided 5%). Given the absence of appropriate control within the A5314, analysis plans for MTX exposure involved a simple characterization of the MTX AUC to be compared descriptively against historical controls.
MTX and TFV Assay Development and Sample Quantification
For study participants randomized to placebo, plasma samples were assayed only for TFV and not MTX while those randomized to LDMTX were analyzed for both TFV and MTX using validated liquid chromatography coupled with tandem mass spectrometry methods (LC-MS/MS). MTX was fortified with a deuterated internal standard, and extracted from 50 μL of plasma by protein precipitation with acetonitrile (ACN). The extracted samples were separated on an Agilent® Zorbax XDB-C8 high performance liquid chromatography (HPLC) column (2.1×50mm, 5μm), and detected on a Sciex API5000 mass spectrometer. The method had a lower limit of quantitation (LLOQ) of 5 ng/mL, with a calibration range of 5–500 ng/mL. During sample analysis, the coefficient of variation (CV) for quality control samples (QC) ranged from 3.15% to 4.39%. Similar to MTX, TFV was fortified with a deuterated internal standard, and then 50 μL of sample was extracted by protein precipitation with ACN. The sample extracts were separated on a Phenomenex® Synergi Polar-RP HPLC column (150 × 2.0mm, 4μm), then detected on a Sciex API 5000 mass spectrometer. The LLOQ for TFV was 5 ng/mL, with a calibration range of 5–1000 ng/mL. The CV of the QC during TFV sample analysis ranged from 5.18% to 8.36%.
PK and Statistical Analysis
PK parameter outcomes included the area under the plasma concentration vs. time curve (AUC), and peak concentration (Cmax) for MTX and TFV, and the trough concentration (Cmin) for TFV. Parameters were estimated using non-compartmental analysis in WinNonlin v.6.2.1® (Certara L.P., Princeton, NJ, USA). For AUC calculations, samples below the LLOQ were treated as missing except for the pre-peak TFV or MTX concentrations, which was set to 0 if below the LLOQ. AUC was calculated using the linear up-log down trapezoidal rule from 0 to 6 h (AUC6) and 0 to 24 h (AUC24 and AUC24i, where AUC24 was available only in participants enrolled under Version 1.0 and AUC24i was estimated for all participants using the pre-dose concentration as the imputed 24 h TFV concentration for participants enrolled under Version 2.0). Imputed concentrations assume there is no difference between the concentrations between 0 and 24 h for steady state dosing. For TFV, Cmin was calculated from the raw data at time 0.
AUC6 was the primary endpoint, while all other PK parameters (AUC24, AUC24i, Cmax, Cmin) were secondary endpoints for TFV exposure. The primary endpoint for MTX was AUC6 and the secondary endpoint was Cmin. TFV PK parameters were compared between placebo and LDMTX treatment groups, while MTX PK was characterized only in the context of TFV-containing ART. PK parameters were summarized using geometric means with 90% confidence intervals; imputed zero concentrations were set to 0.1 ng/mL to facilitate log transformation. Distributions were compared between LDMTX and placebo groups using two sample t-tests with unequal variance and were interpreted at the 10% nominal level of significance (two-sided test) without adjustment for multiple comparisons. This change to the analysis plan specified in the protocol (use of a 10% two-sided test versus a 5% one-sided test) was made prior to review of the data. All tests were performed on natural log-transformed PK parameters. Statistical analyses were conducted with SAS Version 9.4 (SAS Institute Inc, Cary, North Carolina, USA).
Results
Participant Demographics
Of the 66 participants enrolled in the substudy, 18 were excluded from PK analysis due to missed doses of TFV or failure to meet protocol criteria for dose-escalation to 10 mg of MTX at the time of the substudy. Forty-eight participants completed PK sampling (n=20 on LDMTX, n=28 on placebo; a subset of participants were sampled through 24 hours: n=7 on LDMTX and n=10 on placebo); all were taking TFV in the form of TDF. Participants were 92% male, 48% white and 46% black; characteristics were balanced across treatment arms with the exception of concomitant protease inhibitor (PI)-use (25% LDMTX, 43% placebo). Complete demographic information for evaluable participants in PK substudy is presented in Table 1.
Table 1.
Demographic Information of the participants in the PK substudy. SD: standard deviation
| Treatment Group | ||||
|---|---|---|---|---|
| Characteristic | Total (N=48) | LDMTX (N=20) | Placebo (N=28) | |
| Sex | M | 44 (92%) | 17 (85%) | 27 (96%) |
| F | 4 (8%) | 3 (15%) | 1 (4%) | |
| Race | Black or African American | 22 (46%) | 9 (45%) | 13 (46%) |
| White | 24 (50%) | 10 (50%) | 14 (50%) | |
| More than One Race | 1 (2%) | 0 (0%) | 1 (4%) | |
| Unknown | 1 (2%) | 1 (5%) | 0 (0%) | |
| Race/Ethnicity | White Non-Hispanic | 23 (48%) | 9 (45%) | 14 (50%) |
| Black Non-Hispanic | 22 (46%) | 9 (45%) | 13 (46%) | |
| Hispanic (Regardless of Race) | 2 (4%) | 2 (10%) | 0 (0%) | |
| More than one race | 1 (2%) | 0 (0%) | 1 (4%) | |
| Age (years) | Median (Q1-Q3) | 54 (49–59) | 56 (52–61) | 52 (49–57) |
| BMI (kg/m2) | # missing | 2 | 0 | 2 |
| Median (Q1-Q3) | 27.5 (24.3–31.1) | 29.1 (24.5–31.2) | 27.4 (24.1–29.8) | |
| Calculated Creatinine Clearance (mL/min) | Median (Q1-Q3) | 98 (84–115) | 110 (86–116) | 96 (81–110) |
| Entry CD4+ Cell Count (cells/mm3) | Median (Q1-Q3) | 731 (625–946) | 687 (539–968) | 756 (649–918) |
| Does the ARV regimen include a concomitant NNRTI | Yes | 24 (50%) | 12 (60%) | 12 (43%) |
| Does the ARV regimen include a concomitant PI | Yes | 17 (35%) | 5 (25%) | 12 (43%) |
| Does the ARV regimen include a concomitant II | Yes | 10 (21%) | 3 (15%) | 7 (25%) |
PK Results
Effect of MTX on TFV PK parameters
Table 2 details the descriptive statistics of TFV PK parameters by treatment group. The geometric mean (GM) (90% CI) for the primary endpoint, TFV AUC6, was 967 (802, 1166) ng·h/mL for the LDMTX group and 1239 (1105, 1390) ng·h/mL for placebo (geometric mean ratio (GMR)= 0.78, 90% CI [0.64, 0.96], p=0.06). Similar results were observed for AUC24 (GMR=0.64, 90% CI [0.45, 0.91], p=0.08) and AUC24i (GMR=0.76, 90% CI [0.61, 0.93], p=0.033). In addition, mean Cmax was lower in the LDMTX group versus the placebo group (GM =231 ng/mL versus 315 ng/mL, GMR=0.73, 90% CI [0.60, 0.90], p= 0.027). Trough TFV concentrations (Cmin) did not differ between arms (GMR=0.69, 90% CI [0.34, 1.40], p=0.39). Figure 1 depicts the average concentration time profile of TFV with and without MTX co-administration.
Table 2.
PK parameters for TFV after co-administration with LDMTX. Data represent Geometric mean (GM) with 90% CIs, except for geometric mean ratio (GMR) of LDMTX/placebo.
| LDMTX (n=20) | Placebo (n=28) | LDMTX/Placebo | ||
|---|---|---|---|---|
| GM (90% CI) | GM (90% CI) | GMR (90% CI) | p-value | |
| AUC6 (ng•h/mL) | 967 (802, 1166) | 1239 (1105, 1390) | 0.78 (0.64, 0.96) | 0.06 |
| PI-based ART | 1275 (874, 1859) | 1343 (1127, 1600) | 0.95 (0.68, 1.32) | 0.80 |
| Non-PI-based ART | 882 (706, 1101) | 1167 (992, 1372) | 0.76 (0.58, 0.98) | 0.08 |
| AUC24 (ng•h/mL)* | 2235 (1511, 3306) | 3481 (2948, 4110) | 0.64 (0.45, 0.91) | 0.08 |
| AUC24i (ng•h/mL) | 2647 (2218, 3160) | 3503 (3087, 3975) | 0.76 (0.61, 0.93) | 0.033 |
| PI-based ART | 3331 (2202,5039) | 3873 (3205, 4679) | 0.86 (0.60, 1.23) | 0.52 |
| Non-PI-based ART | 2453 (1995, 3014) | 3249 (2718, 3883) | 0.75 (0.58, 0.98) | 0.08 |
| Cmax (ng/mL) | 231 (188, 283) | 315 (284, 349) | 0.73 (0.60, 0.90) | 0.027 |
| PI-based ART | 314 (234,420) | 341 (291, 400) | 0.92 (0.69, 1.22) | 0.62 |
| Non-PI-based ART | 209 (162, 268) | 296 (256, 343) | 0.70 (0.53, 0.93) | 0.045 |
| Cmin (ng/mL) | 43 (24, 79) | 63 (41, 97) | 0.69 (0.34, 1.40) | 0.39 |
| PI-based ART | 70 (45, 110) | 51 (18, 146) | 1.37 (0.27, 6.93) | 0.62 |
| Non-PI-based ART | 37 (16, 82) | 73 (58, 92) | 0.50 (0.23, 1.11) | 0.17 |
n=7 for LDMTX and 10 for placebo. AUC, area under concentration-time curve, AUC24i, AUC from 0 to 24 h with imputed 24 h concentration from 0 h, PI, protease inhibitor.
Figure 1.
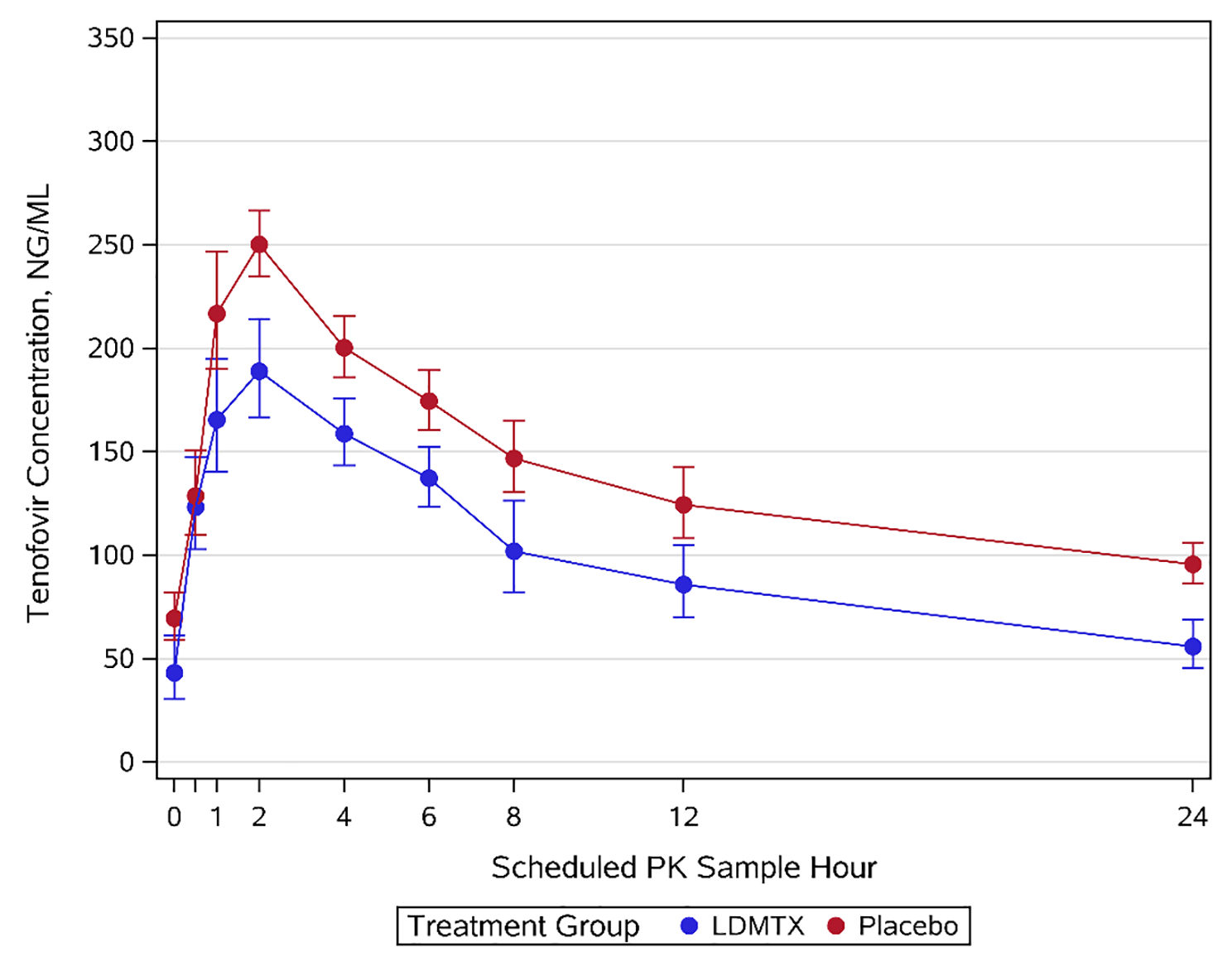
Geometric mean plasma concentration-time profile of TFV. Blue line, TFV with LDMTX; red line, TFV with placebo. Error bars indicate standard error.
Due to the interaction between TFV and PIs and the observed treatment group imbalance, a sub-group analysis by concomitant PI use was performed. This analysissuggested lower TFV concentrations in the presence of LDMTX compared to placebo when taken in conjunction with a non PI-based regimen; this difference was not apparent in the context of co-administration with a PI regimen (Figure 2). Specifically, a greater effect of MTX on TFV exposure was observed for participants who were not on PIs (non PI) for LDMTX compared to placebo (Cmax GMRnonPI=0.7, 90% CI [0.53, 0.93], p=0.045; AUC6 GMRnonPI =0.76, 90% CI [0.58, 0.98], p=0.08; AUC24i GMRnonPI=0.75, 90% CI [0.58, 0.98], p=0.08). While a formal interaction test was not statistically significant (p>0.3), this test is underpowered given the small study sample.
Figure 2.
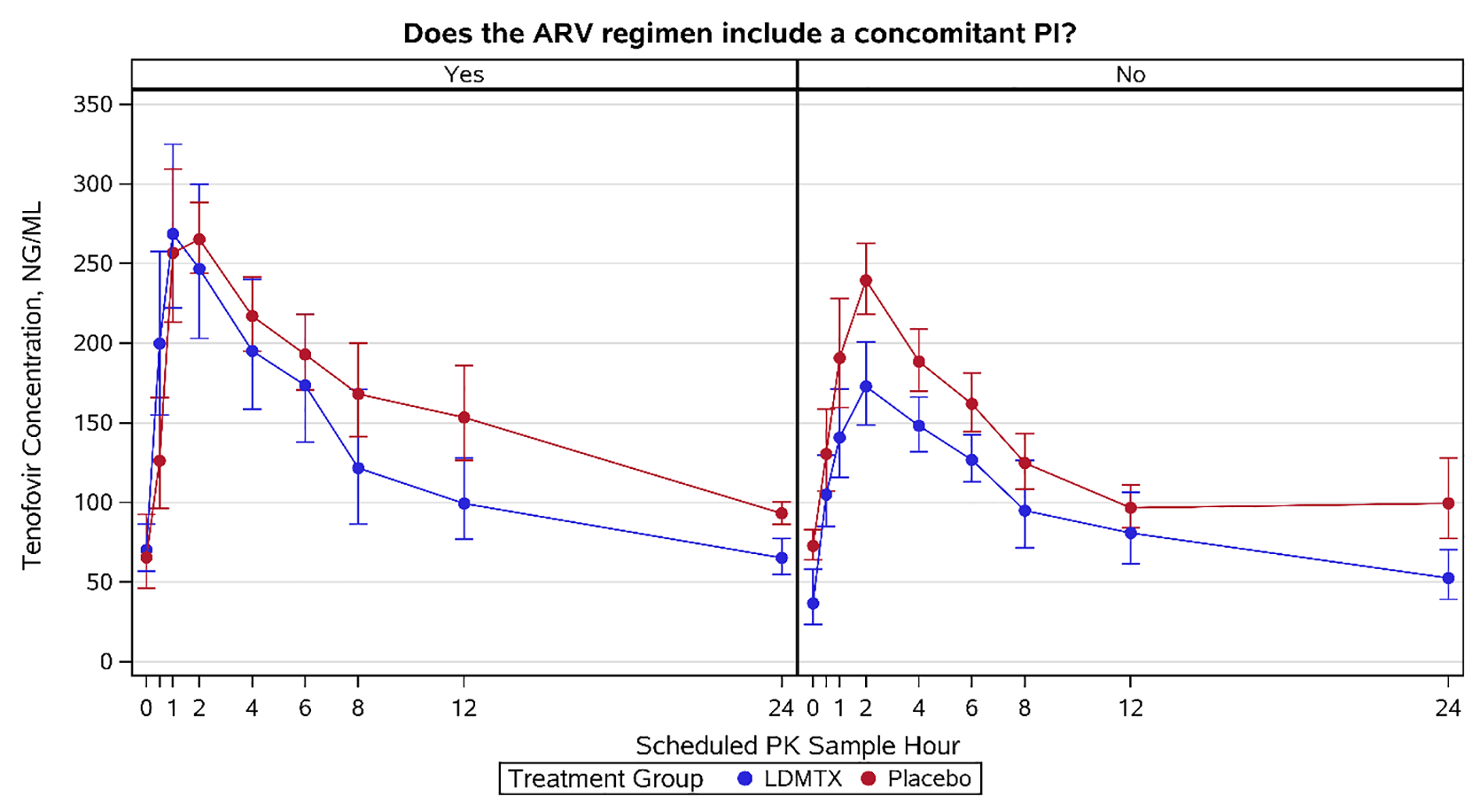
Geometric mean plasma concentration-time profile of TFV in the context of LDMTX. Blue line, TFV with LDMTX; red line, TFV with placebo. Error bars indicate standard error.
MTX PK in the context of TFV- containing ART
MTX was characterized in the context of TFV. MTX AUC6 was estimated to be 492 (434, 558) ng·h/mL, and Cmax was 144 (127, 164) ng/mL. The geometric mean concentration-time profiles for MTX in the presence of TFV are shown in Supplemental Figure 1. MTX exposure did not appear to differ by PI use on visual evaluation.
Discussion
Common renal elimination transporter pathways of TFV and MTX raise concern for potential drug-drug interactions. In this study, we investigated the impact of LDMTX on TFV PK via intensive venous sampling in a subset of participants chronically suppressed with TFV-containing ART who were randomized to either active LDMTX or placebo as part of the A5314 study1. Our results demonstrate decreased TFV exposure in the presence of LDMTX, including a 22% reduction in the geometric mean AUC6. Similar results were seen for AUC24, AUC24i, and Cmax (with 36%, 24%, and 27% reductions, respectively). While the differences in GMR for some PK parameters did not reach statistical significance, these results demonstrate an overall trend towards decreased TFV exposure in the presence of MTX. However this magnitude of decrease in TFV exposure is likely not clinically significant towards providing adequate viral suppression. As reported for the parent trial1, very few participants in the trial as a whole experienced a HIV-1 RNA level above the assay limit of quantification (40 copies/mL), and all were evenly distributed by treatment group. Among participants with measures above the assay limit of quantification, none had confirmed viral load failures (HIV-1 RNA >200 copies/mL). Visually, the terminal slopes appear identical and rate of absorption similar. In support of this observation, reductions in TFV exposure are not a result of inhibition of renal transporters during excretion, and are likely attributable to decreased absorption, as supported by a significant decrease in TFV Cmax with LDMTX. Within the placebo arm, TFV Cmax averaged 315 ng/mL, consistent with published TFV exposure18.
Within PI subgroups, comparisons between study arms showed that TFV AUC6, AUC24i, and Cmax trended toward higher values in participants on PIs, consistent with a previous report19,20. This potential PI based-TFV interaction is likely driven by increased absorption by PI-related inhibition of P-glycoprotein (P-gp) and intestinal esterase20–22, resulting in higher TFV exposure estimates in the context of PIs which partially compensates for the overall lower TFV exposure measured during MTX co-administration. This trend toward lower TFV exposure is likely driven by inclusion of five participants not receiving PIs in the LDMTX group who exhibited particularly low TFV exposure. These participants were demographically similar to the PK substudy population with consistent body mass indices, age, creatinine clearance and mixed with regards to race. Overall when the TFV PK parameters are stratified by use of concomitant PIs, a modest shift is seen in the distributions in the absence of PIs; however, this shift is not apparent in the co-administration of concomitant PIs and LDMTX.
MTX levels were also quantified using a precise analytical method but no statistical comparisons were possible due to lack of a control group not receiving TFV. However, comparisons were made to parameters published previously for low dose MTX (Table 3). One would anticipate lower peak concentrations for intramuscular (IM) versus oral (PO) dosing and higher peak concentrations for 10 mg versus 7.5 mg PO doses. The MTX Cmax observed in this study was about 40–60 and 34% lower than published studies of weekly 10 mg IM dosing 23–25 and weekly 7.5 mg PO dosing 26, respectively. Of note, studies among individuals with rheumatoid arthritis report a wide range of bioavailability of oral methotrexate ranging from 20–100% 27,28. It is also possible that that TFV and/or HIV disease may decrease MTX exposure. The earlier studies used florescence polarization immunoassays (FPIA) or radioimmunoassay; a recent comparison of FPIA to LC-MS/MS indicates FPIA overestimated concentrations in this range29, which may explain the discrepancy with prior PK estimates. One study, using a homogenous enzymatic immunoassay, reported no effect of TFV on MTX elimination17. However, that study examined high doses of intravenous MTX, supporting that a potential interaction is likely driven by an absorption process, rather than elimination. Further studies with a non-TFV control are needed to confirm the effect of TFV on MTX exposure.
Table 3.
Comparison of methotrexate exposure in this study and published literature. Data represent Geometric mean (90% CIs)
One of the limitations of study A5314 was that the targeted LDMTX was on the low end of the dose range that had been effective in reducing cardiovascular events in larger cohort studies of MTX30–34. While exposure-response analysis was not the focus of the primary study, it is possible that if the 57/86 participants assigned to LDMTX who were taking TDF had lower than anticipated exposure to MTX, its effects on the primary efficacy and safety endpoints may have been reduced, contributing to decreased power to detect differences between arms.
Understanding the underlying mechanisms of TFV- and MTX-mediated drug-drug interactions and toxicities, particularly the role of renal and intestinal transporters, continues to evolve and is a rich area of research and scientific debate. TFV is a substrate of renal transporters (OAT1, OAT3, multidrug resistance protein[MRP] 4) and TDF is subject to intestinal transport via P-gp, and likely MRP4. MRP2’s role in TFV PK has been debated in the literature21,35. More recently, TFV was identified as a substrate of MRP8 for which MTX is also a known substrate36. Other transporters, such as MRP7, have been associated with renal injury but mechanistic studies are lacking37. MTX is a substrate for numerous transporters38; those shared with known or potential TFV or TDF pathways include OAT1 and OAT32,3, MRP239, MRP439, MRP836, and P-gp40.
Of these pathways MRP4, mechanistically, has the most potential for a TDF-MTX PK interaction. MRP4 is an efflux transporter; although since it’s located on the basolateral side of enterocytes, it ultimately facilitates drug entry to the blood and serves to mediate intestinal basolateral influx of TDF and MTX. The observed results of this study would be supported by this mechanism given lower Cmax for TFV in the presence of MTX and for MTX as compared to prior studies.
While the role of intestinal MRP4 in TDF absorption has not been well described, it was found to be a major contributor to adefovir dipivoxil uptake41, a compound similar structurally and pharmacokinetically to TDF. Furthermore, Vitamin D3, a known inducer of MRP4 expression, enhanced adefovir exposure in rats42. Mouse models have also demonstrated the importance of MRP4 in cefadroxil absorption43 and dasatinib absorption and efficacy44. In vitro and in situ interaction studies of TDF on atazanavir absorption supported clinical findings that TDF decreases atazanavir bioavailability; the authors hypothesized that OATP or MRP-mediated absorption pathways were likely involved45.
While MRP4 present in proximal renal cells is considered important in TFV-mediated kidney injury46, intestinal MRP4 may be more so subject to saturation by MTX and TDF, given its lower expression frequency47. Although preclinical experiments in MRP4 knockout mice failed to show importance of MRP4 in MTX absorption48, in vitro interaction assays demonstrated MRP4-mediated interactions between MTX and nonsteroidal anti-inflammatory drugs39. There are known inconsistencies in drug transporter expression in in vitro and preclinical models versus in human tissues49 limiting clinical translation of preclinical findings. MRP4 is expressed in the human jejunum49, the likely site of MTX absorption5,50.
Further, genetic polymorphisms in transporter expression may have contributed to observed between-subject PK variability. Both MTX and TFV have documented polymorphisms that contribute to differences in PK, efficacy, or toxicity in varying patient populations and indications51. For instance, genotype differences in MRP4 have been linked to altered TFV clearance in PWH and differential MTX plasma exposure in pediatric patients being treated for acute lymphoblastic leukemia19,52.
Aside from transporters, an emerging field, the gastrointestinal microbiome, which may also play a key role in HIV disease pathogenesis53, has been shown to metabolize and/or be altered by exposure to both TDF54 and MTX 55,56. Future studies may investigate the interplay between the microbiome and pharmacokinetics of multiple coadministered drugs.
The clinical implications of these results on the use of tenofovir alafenamide (TAF) are unclear. Unlike TDF, TAF is relatively stable in plasma and exhibits lower TFV plasma levels and higher intracellular tenofovir diphosphate levels than TDF. While sharing some transport pathways with TDF (i.e. P-gp and breast cancer resistance protein [BCRP]), TAF is additionally a substrate of OATP1B1 and OATP1B3, but not subject to OAT1 or OAT3 transport57,58. Current literature does not indicate whether TAF is a substrate of MRP4. Given these differences, separate preclinical and/or clinical PK investigations of TAF and its potential in transporter-mediated interactions with MTX are warranted.
In summary, LDMTX resulted in modest reductions in TFV exposure that are not expected to be clinically significant for maintenance of HIV-1 viral suppression. However, these decreases appear driven by a subset of participants who were not on PIs, which indicates a change in TFV dosing is not warranted for those taking PIs. Further confirmatory and mechanistic studies of TFV PK in the setting of LDMTX without PI co-administration are warranted. Although MTX exposure was only characterized in the context of TFV co-administration, exposure was lower than anticipated from prior reports, which may have impacted effects of LDMTX on cardiovascular outcomes. This study will help inform dosing during MTX-TFV co-administration particularly for PWH who have rheumatoid arthritis or psoriasis.
Supplementary Material
ACKNOWLEDGMENTS
A.N.D. was supported by NIGMS training grant T32GM007546.
Participating AIDS Clinical Trials Group Units. 101 - Massachusetts General Hospital Clinical Research Site (CRS); 107 - Brigham and Women’s Hospital Therapeutics CRS; 201 - Johns Hopkins University CRS; 601 - University of California, Los Angeles CARE Center CRS; 603 - Harbor University of California Los Angeles Center CRS; 701 - University of California, San Diego AntiViral Research Center CRS; 801 - University of California, San Francisco HIV/AIDS CRS; 1001 - University of Pittsburgh CRS; 1201 - University of Southern California CRS; 2101 - Washington University Therapeutics CRS; 2301 - Ohio State University CRS; 2401 - Cincinnati CRS; 2501 - Case Western Reserve University CRS; 2701 - Northwestern University CRS; 2951 - The Miriam Hospital CRS; 3201 - Chapel Hill CRS; 3203 - Greensboro CRS; 3652 - Vanderbilt Therapeutics CRS; 6101 - University of Colorado Hospital CRS; 6201 - Penn Therapeutics CRS; 31473 - Houston AIDS Research Team CRS; 31786 - New Jersey Medical School Clinical Research Center CRS; 31788 - Alabama CRS.
A5314 Acknowledgments. Eva Whitehead, RN, and Elaine M. Urbina, MD, MS - Cincinnati CRS (Site 2401) Grant UM1-AI069501. Eric Daar and Sadia Shaik - Harbor-UCLA (Site 603) Grant AI 069424, UCLA CTSI Grant UL1 TR000124. Annie Luetkemeyer, MD, and Jay Dwyer, RN - UCSF AIDS CRS (Site 801) CTU Grant UM1AI069502. Kristen Allen, RN, and Jane Baum, RN - Case CRS (Site 2501) Grant AI069501. Nina Lambert and Babafemi Taiwo - Northwestern University CRS (Site 2701) Grant 2UM1 AI069471, UL1TR001422. Michael Messer and Dana Green - Alabama CRS (Site 31788) Grant UM1 AI069452. Joan Gottesman, BSN, RN, and JoAnn A. Gottlieb - Vanderbilt Therapeutics CRS (Site 3652) Grant UM1AI 069439, NIH TR000445. Paul Sax, MD, and Cheryl Keenan, RN, BC - Brigham and Women’s Hospital (Site 107) Grant R01HL117713. Shobha Swaminathan and Baljinder Singh - New Jersey Medical School Clinical Research Center (Site 31786) Grant 5R01 HL117713. Dr Pablo Tebas and Ro Kappes, MPH - Philadelphia HIV Therapeutics and Prevention CTU (Site 6201) Grant UM1AI068636, UM1AI069534. Lisa Kessels and Teresa Spitz - Washington University Therapeutics CRS (Site 2101) Grant UM1AI068619. Sana Majid and Arezou Sadighi Akha - UCLA Care Center (Site 601) Grant AI069424, UCLA CTSI UL-1TR000124, CFAR P30-AI028697. Renee Weinman, MPPM, and Lisa Klevens, RN, BSN - University of Pittsburgh (Site 1001) Grant UM1 AI069494. Cornelius Van Dam, MD, and Timothy Lane, MD - Greensboro CRS (Site 3203) Grant 5UM1AI068636. Cathi Basler and Christine Griesmer - UCH CRS (Site 6101) Grant 2UM1AI069432, UL1 TR001082. Andrea Weiss and Ilene Wiggins - Johns Hopkins University CRS (Site 201) Grant TBD. Dr Susan Koletar and Kathy Watson, RN - Ohio State University (Site 2301) Grant UM1AI069494. Christopher Evans, MSN, and David Currin, AAS - Chapel Hill CRS (Site 3201) Grant UM1 AI069423, CTSA: 1UL1TR001111, CFAR: P30 AI50410. Michael Phillip Dube, MD, and Frances Canchola, RN - University of Southern California CRS (Site 1201) Grant 2UM1AI069432. Dee Dee Pacheco and Michael Connor - UCSD CRS (Site 701) Grant AI069432. Karen Tashima, MD, and Pamela Poethke, RN - The Miriam Hospital (Site 2951) Grant 2UM1A1069412-08. Dr Roberto C. Arduino and Dr Aristoteles E. Villamil - HART (Site 31473) Grant 5 UM1 AI069503, 5 UM1 AI068636.
Source of funding:
The project described was supported by the NIAID (award numbers U01AI068636, UM1AI068636, UM1AI068634) and the NHLBI (award number R01HL1177131).
References
- 1.Hsue PY, Ribaudo HJ, Deeks SG, et al. Safety and Impact of Low-dose Methotrexate on Endothelial Function and Inflammation in Individuals With Treated Human Immunodeficiency Virus: AIDS Clinical Trials Group Study A5314. Clin Infect Dis. 2019;68(11):1877–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fujita T, Brown C, Carlson EJ, et al. Functional analysis of polymorphisms in the organic anion transporter, SLC22A6 (OAT1). Pharmacogenet Genomics. 2005;15(4):201–209. [DOI] [PubMed] [Google Scholar]
- 3.Cha SH, Sekine T, Fukushima JI, et al. Identification and characterization of human organic anion transporter 3 expressing predominantly in the kidney. Mol Pharmacol. 2001;59(5):1277–1286. [DOI] [PubMed] [Google Scholar]
- 4.LABEL: XATMEP- methotrexate solution. Silvergate Pharmaceuticals Inc. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=aec9984e-34c5-481b-b6bf-9bb5caf1daf8. Updated June 5, 2019. Accessed April 14, 2020. [Google Scholar]
- 5.Bannwarth B, Pehourcq F, Schaeverbeke T, Dehais J. Clinical pharmacokinetics of low-dose pulse methotrexate in rheumatoid arthritis. Clin Pharmacokinet. 1996;30(3):194–210. [DOI] [PubMed] [Google Scholar]
- 6.Shen DD, Azarnoff DL. Clinical pharmacokinetics of methotrexate. Clin Pharmacokinet. 1978;3(1):1–13. [DOI] [PubMed] [Google Scholar]
- 7.Zarychanski R, Wlodarczyk K, Ariano R, Bow E. Pharmacokinetic interaction between methotrexate and piperacillin/tazobactam resulting in prolonged toxic concentrations of methotrexate. J Antimicrob Chemother. 2006;58(1):228–230. [DOI] [PubMed] [Google Scholar]
- 8.Titier K, Lagrange F, Pehourcq F, Moore N, Molimard M. Pharmacokinetic interaction between high-dose methotrexate and oxacillin. Ther Drug Monit. 2002;24(4):570–572. [DOI] [PubMed] [Google Scholar]
- 9.Dalle JH, Auvrignon A, Vassal G, Leverger G. Interaction between methotrexate and ciprofloxacin. J Pediatr Hematol Oncol. 2002;24(4):321–322. [DOI] [PubMed] [Google Scholar]
- 10.Methotrexate. https://www.micromedexsolutions.com/. Accessed June 2, 2019.
- 11.Ray AS, Cihlar T, Robinson KL, et al. Mechanism of active renal tubular efflux of tenofovir. Antimicrob Agents Chemother. 2006;50(10):3297–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uwai Y, Ida H, Tsuji Y, Katsura T, Inui K. Renal transport of adefovir, cidofovir, and tenofovir by SLC22A family members (hOAT1, hOAT3, and hOCT2). Pharm Res. 2007;24(4):811–815. [DOI] [PubMed] [Google Scholar]
- 13.Barditch-Crovo P, Deeks SG, Collier A, et al. Phase I/II trial of the pharmacokinetics, safety, and antiretroviral activity of tenofovir disoproxil fumarate in human immunodeficiency virus-infected adults. Antimicrob Agents Chemother. 2001;45(10):2733–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dailly E, Tribut O, Tattevin P, et al. Influence of tenofovir, nevirapine and efavirenz on ritonavir-boosted atazanavir pharmacokinetics in HIV-infected patients. Eur J Clin Pharmacol. 2006;62(7):523–526. [DOI] [PubMed] [Google Scholar]
- 15.Wenning LA, Friedman EJ, Kost JT, et al. Lack of a significant drug interaction between raltegravir and tenofovir. Antimicrob Agents Chemother. 2008;52(9):3253–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moss DM, Kwan WS, Liptrott NJ, et al. Raltegravir is a substrate for SLC22A6: a putative mechanism for the interaction between raltegravir and tenofovir. Antimicrob Agents Chemother. 2011;55(2):879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dalla Pria A, Bendle M, Ramaswami R, Boffito M, Bower M. The pharmacokinetics of high-dose methotrexate in people living with HIV on antiretroviral therapy. Cancer Chemother Pharmacol. 2016;77(3):653–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chittick GE, Zong J, Blum MR, et al. Pharmacokinetics of tenofovir disoproxil fumarate and ritonavir-boosted saquinavir mesylate administered alone or in combination at steady state. Antimicrob Agents Chemother. 2006;50(4):1304–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kiser JJ, Carten ML, Aquilante CL, et al. The effect of lopinavir/ritonavir on the renal clearance of tenofovir in HIV-infected patients. Clinical pharmacology and therapeutics. 2008;83(2):265–272. [DOI] [PubMed] [Google Scholar]
- 20.Kearney BP, Mathias A, Mittan A, Sayre J, Ebrahimi R, Cheng AK. Pharmacokinetics and safety of tenofovir disoproxil fumarate on coadministration with lopinavir/ritonavir. J Acquir Immune Defic Syndr. 2006;43(3):278–283. [DOI] [PubMed] [Google Scholar]
- 21.Tong L, Phan TK, Robinson KL, et al. Effects of human immunodeficiency virus protease inhibitors on the intestinal absorption of tenofovir disoproxil fumarate in vitro. Antimicrob Agents Chemother. 2007;51(10):3498–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murphy RA, Valentovic MA. Factors Contributing to the Antiviral Effectiveness of Tenofovir. J Pharmacol Exp Ther. 2017;363(2):156–163. [DOI] [PubMed] [Google Scholar]
- 23.Combe B, Edno L, Lafforgue P, et al. Total and free methotrexate pharmacokinetics, with and without piroxicam, in rheumatoid arthritis patients. Br J Rheumatol. 1995;34(5):421–428. [DOI] [PubMed] [Google Scholar]
- 24.Anaya JM, Fabre D, Bressolle F, et al. Effect of etodolac on methotrexate pharmacokinetics in patients with rheumatoid arthritis. J Rheumatol. 1994;21(2):203–208. [PubMed] [Google Scholar]
- 25.Edelman J, Biggs DF, Jamali F, Russell AS. Low-dose methotrexate kinetics in arthritis. Clinical pharmacology and therapeutics. 1984;35(3):382–386. [DOI] [PubMed] [Google Scholar]
- 26.Sinnett MJ, Groff GD, Raddatz DA, Franck WA, Bertino JS, Jr. Methotrexate pharmacokinetics in patients with rheumatoid arthritis. J Rheumatol. 1989;16(6):745–748. [PubMed] [Google Scholar]
- 27.Herman RA, Veng-Pedersen P, Hoffman J, Koehnke R, Furst DE. Pharmacokinetics of low-dose methotrexate in rheumatoid arthritis patients. J Pharm Sci. 1989;78(2):165–171. [DOI] [PubMed] [Google Scholar]
- 28.Bello AE, Perkins EL, Jay R, Efthimiou P. Recommendations for optimizing methotrexate treatment for patients with rheumatoid arthritis. Open Access Rheumatol. 2017;9:67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gunther V, Mueller D, von Eckardstein A, Saleh L. Head to head evaluation of the analytical performance of two commercial methotrexate immunoassays and comparison with liquid chromatography-mass spectrometry and the former fluorescence polarization immunoassay. Clin Chem Lab Med. 2016;54(5):823–831. [DOI] [PubMed] [Google Scholar]
- 30.Choi HK, Hernan MA, Seeger JD, Robins JM, Wolfe F. Methotrexate and mortality in patients with rheumatoid arthritis: a prospective study. Lancet. 2002;359(9313):1173–1177. [DOI] [PubMed] [Google Scholar]
- 31.Naranjo A, Sokka T, Descalzo MA, et al. Cardiovascular disease in patients with rheumatoid arthritis: results from the QUEST-RA study. Arthritis Res Ther. 2008;10(2):R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prodanovich S, Ma F, Taylor JR, Pezon C, Fasihi T, Kirsner RS. Methotrexate reduces incidence of vascular diseases in veterans with psoriasis or rheumatoid arthritis. J Am Acad Dermatol. 2005;52(2):262–267. [DOI] [PubMed] [Google Scholar]
- 33.Solomon DH, Avorn J, Katz JN, et al. Immunosuppressive medications and hospitalization for cardiovascular events in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54(12):3790–3798. [DOI] [PubMed] [Google Scholar]
- 34.van Halm VP, Nurmohamed MT, Twisk JW, Dijkmans BA, Voskuyl AE. Disease-modifying antirheumatic drugs are associated with a reduced risk for cardiovascular disease in patients with rheumatoid arthritis: a case control study. Arthritis Res Ther. 2006;8(5):R151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ray AS, Cihlar T. Unlikely association of multidrug-resistance protein 2 single-nucleotide polymorphisms with tenofovir-induced renal adverse events. J Infect Dis. 2007;195(9):1389–1390; author reply 1390–1381. [DOI] [PubMed] [Google Scholar]
- 36.Tun-Yhong W, Chinpaisal C, Pamonsinlapatham P, Kaewkitichai S. Tenofovir Disoproxil Fumarate Is a New Substrate of ATP-Binding Cassette Subfamily C Member 11. Antimicrob Agents Chemother. 2017;61(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pushpakom SP, Liptrott NJ, Rodriguez-Novoa S, et al. Genetic variants of ABCC10, a novel tenofovir transporter, are associated with kidney tubular dysfunction. J Infect Dis. 2011;204(1):145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morrissey KM, Wen CC, Johns SJ, Zhang L, Huang SM, Giacomini KM. The UCSF-FDA TransPortal: a public drug transporter database. Clinical pharmacology and therapeutics. 2012;92(5):545–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El-Sheikh AA, van den Heuvel JJ, Koenderink JB, Russel FG. Interaction of nonsteroidal anti-inflammatory drugs with multidrug resistance protein (MRP) 2/ABCC2- and MRP4/ABCC4-mediated methotrexate transport. J Pharmacol Exp Ther. 2007;320(1):229–235. [DOI] [PubMed] [Google Scholar]
- 40.Jia Y, Liu Z, Wang C, et al. P-gp, MRP2 and OAT1/OAT3 mediate the drug-drug interaction between resveratrol and methotrexate. Toxicol Appl Pharmacol. 2016;306:27–35. [DOI] [PubMed] [Google Scholar]
- 41.Ming X, Thakker DR. Role of basolateral efflux transporter MRP4 in the intestinal absorption of the antiviral drug adefovir dipivoxil. Biochem Pharmacol. 2010;79(3):455–462. [DOI] [PubMed] [Google Scholar]
- 42.Yoon IS, Son JH, Kim SB, Choi MK, Maeng HJ. Effects of 1alpha,25-Dihydroxyvitamin D3 on Intestinal Absorption and Disposition of Adefovir Dipivoxil and Its Metabolite, Adefovir, in Rats. Biol Pharm Bull. 2015;38(11):1732–1737. [DOI] [PubMed] [Google Scholar]
- 43.de Waart DR, van de Wetering K, Kunne C, Duijst S, Paulusma CC, Oude Elferink RP. Oral availability of cefadroxil depends on ABCC3 and ABCC4. Drug Metab Dispos. 2012;40(3):515–521. [DOI] [PubMed] [Google Scholar]
- 44.Furmanski BD, Hu S, Fujita KI, et al. Contribution of ABCC4-mediated gastric transport to the absorption and efficacy of dasatinib. Clin Cancer Res. 2013;19(16):4359–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kis O, Zastre JA, Hoque MT, Walmsley SL, Bendayan R. Role of drug efflux and uptake transporters in atazanavir intestinal permeability and drug-drug interactions. Pharm Res. 2013;30(4):1050–1064. [DOI] [PubMed] [Google Scholar]
- 46.Kohler JJ, Hosseini SH, Green E, et al. Tenofovir renal proximal tubular toxicity is regulated by OAT1 and MRP4 transporters. Lab Invest. 2011;91(6):852–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab Pharmacokinet. 2005;20(6):452–477. [DOI] [PubMed] [Google Scholar]
- 48.Kitamura Y, Hirouchi M, Kusuhara H, Schuetz JD, Sugiyama Y. Increasing systemic exposure of methotrexate by active efflux mediated by multidrug resistance-associated protein 3 (mrp3/abcc3). J Pharmacol Exp Ther. 2008;327(2):465–473. [DOI] [PubMed] [Google Scholar]
- 49.Taipalensuu J, Tornblom H, Lindberg G, et al. Correlation of gene expression of ten drug efflux proteins of the ATP-binding cassette transporter family in normal human jejunum and in human intestinal epithelial Caco-2 cell monolayers. J Pharmacol Exp Ther. 2001;299(1):164–170. [PubMed] [Google Scholar]
- 50.Songsiridej N, Furst DE. Methotrexate--the rapidly acting drug. Baillieres Clin Rheumatol. 1990;4(3):575–593. [DOI] [PubMed] [Google Scholar]
- 51.Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clinical pharmacology and therapeutics. 2012;92(4):414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lopez-Lopez E, Ballesteros J, Pinan MA, et al. Polymorphisms in the methotrexate transport pathway: a new tool for MTX plasma level prediction in pediatric acute lymphoblastic leukemia. Pharmacogenet Genomics. 2013;23(2):53–61. [DOI] [PubMed] [Google Scholar]
- 53.Dillon SM, Frank DN, Wilson CC. The gut microbiome and HIV-1 pathogenesis: a two-way street. AIDS. 2016;30(18):2737–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dube MP, Park SY, Ross H, Love TMT, Morris SR, Lee HY. Daily HIV pre-exposure prophylaxis (PrEP) with tenofovir disoproxil fumarate-emtricitabine reduced Streptococcus and increased Erysipelotrichaceae in rectal microbiota. Sci Rep. 2018;8(1):15212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nayak R, O’Loughlin C, Fischbach M, Turnbaugh P. Methotrexate Is an Antibacterial Drug Metabolized By Human Gut Bacteria [abstract]. Arthritis Rheumatol. 2016;68(suppl 10). [Google Scholar]
- 56.Valerino DM, Johns DG, Zaharko DS, Oliverio VT. Studies of the metabolism of methotrexate by intestinal flora. I. Identification and study of biological properties of the metabolite 4-amino-4-deoxy-N 10 -methylpteroic acid. Biochem Pharmacol. 1972;21(6):821–831. [DOI] [PubMed] [Google Scholar]
- 57.LABEL: VEMLIDY- tenofovir alafenamide tablet. Gilead Sciences, Inc. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=72e6b33c-0351-4070-9172-eeaa186c01d2. Updated February 11, 2020. Accessed July 8, 2020. [Google Scholar]
- 58.LABEL: VIREAD- tenofovir disoproxil fumarate tablet, coated. Gilead Sciences, Inc. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=33fd6418-fbdc-42ca-a50d-ce2a476a5418. Updated April 29, 2019. Accessed July 8, 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.