

Abstract
Hepatocytes are highly polarized epithelia. Loss of hepatocyte polarity is associated with various liver diseases, including cholestasis. However, the molecular underpinnings of hepatocyte polarization remain poorly understood. Loss of β-catenin at adherens junctions is compensated by γ-catenin and dual loss of both catenins in double knockouts (DKOs) in mice liver leads to progressive intrahepatic cholestasis. However, the clinical relevance of this observation, and further phenotypic characterization of the phenotype, is important. Herein, simultaneous loss of β-catenin and γ-catenin was identified in a subset of liver samples from patients of progressive familial intrahepatic cholestasis and primary sclerosing cholangitis. Hepatocytes in DKO mice exhibited defects in apical-basolateral localization of polarity proteins, impaired bile canaliculi formation, and loss of microvilli. Loss of polarity in DKO livers manifested as epithelial-mesenchymal transition, increased hepatocyte proliferation, and suppression of hepatocyte differentiation, which was associated with up-regulation of transforming growth factor-β signaling and repression of hepatocyte nuclear factor 4α expression and activity. In conclusion, concomitant loss of the two catenins in the liver may play a pathogenic role in subsets of cholangiopathies. The findings also support a previously unknown role of β-catenin and γ-catenin in the maintenance of hepatocyte polarity. Improved understanding of the regulation of hepatocyte polarization processes by β-catenin and γ-catenin may potentially benefit development of new therapies for cholestasis.
A hallmark of epithelial cells is polarization, which is achieved by the orchestration of external cues, such as cellular contact, extracellular matrix, signal transduction, growth factors, and spatial organization.1 Hepatocytes in the liver show a unique polarity by forming several apical and basolateral poles within a cell.2 The apical poles of adjacent hepatocytes form a continuous network of bile canaliculi into which bile is secreted, whereas the basolateral membrane domain forms the sinusoidal pole, which secretes various components, such as proteins or drugs, into the blood circulation.3 Loss of hepatic polarity has been associated with several cholestatic and developmental disorders, including progressive familial intrahepatic cholestasis (PFIC) and primary sclerosing cholangitis (PSC).4,5 Although the molecular mechanisms governing hepatocyte polarity have been extensively studied in the in vitro systems, there is still a significant gap in our understanding of how polarity is established within the context of tissue during development or maintained during homeostasis.6,7 Similarly, the molecular pathways contributing to hepatic polarity are not entirely understood, and a better comprehension of hepatic polarity regulation is thus warranted.
Previous studies have confirmed the role of hepatocellular junctions, such as tight and gap junctions, in the maintenance of hepatocyte polarity.8,9 Studies done in vitro and in vivo have shown that loss of junctional proteins, such as zonula occludens protein (ZO)-1, junctional adhesion molecule-A, and claudins, lead to impairment of polarity and distorted bile canaliculi formation.10, 11, 12, 13 In addition, proteins involved in tight junction assembly, such as liver kinase B1, are also involved in polarity maintenance.14 Among adherens junction proteins, various in vitro cell culture models have confirmed the role of E-cadherin in the regulation of hepatocyte polarity, possibly through its interaction with β-catenin.15,16 However, there is a lack of an in vivo model to study the role of adherens junction proteins in hepatocyte polarity and their misexpression contributing to various liver diseases.
β-Catenin plays diverse functions in the liver during development, regeneration, zonation, and tumorigenesis.17, 18, 19 The relative contribution of β-catenin as part of the adherens junction is challenging to study because like in other tissues, γ-catenin compensates for the β-catenin loss in the liver.20,21 To address this redundancy, we previously reported a hepatocyte-specific -catenin and γ-catenin double-knockout (DKO) mouse model was reported.22 Simultaneous deletion of β-catenin and γ-catenin in mice livers led to cholestasis, partially through the breach of cell-cell junctions. However, more comprehensive understanding of the molecular underpinnings of the phenotype is needed.
In the current study, prior preclinical findings of dual β-catenin and γ-catenin loss were extended to a subset of PFIC and PSC patients. In vivo studies using the murine model with hepatocyte-specific dual loss of β-catenin and γ-catenin showed complete loss of hepatocyte polarity compared to the wild-type controls (CONs). Loss of polarity in DKO liver was accompanied by epithelial-mesenchymal transition (EMT), activation of transforming growth factor (TGF)-β signaling, and reduced expression of hepatocyte nuclear factor 4α (HNF4α). Our findings suggest that β-catenin and γ-catenin and in turn adherens junction integrity, are critical for the maintenance of hepatocyte polarity, and any perturbations in this process can contribute to the pathogenesis of cholestatic liver disease.
Materials and Methods
Animals, Viruses, and Infections
Previously reported homozygous β-catenin–floxed mice and γ-catenin–floxed mice were interbred to generate double-floxed mice.22 All animal experiments and procedures were performed according to the NIH Guide for the Care and Use of Laboratory Animals23 under an animal protocol approved by the Institutional Animal Use and Care Committee at University of Pittsburgh.
Adeno-associated virus (AAV8)-TBG-Cre or AAV8-TBG-GFP (control) were obtained from Penn Vector Core at the University of Pennsylvania (Philadelphia, PA). β-Catenin;γ-catenin double-floxed mice, aged ≥4 weeks, were given a single i.p. injection of 2.5 × 1011 genome copies of AAV8-TBG-Cre or AAV8-TBG-GFP, as described previously.22 At least three mice per time point, ranging from 7 to 14 days, were used for the studies.
Transmission Electron Microscopy and Scanning Electron Microscopy
For transmission electron microscopy, whole liver was perfused and fixed in glutaraldehyde. The surgical and perfusion techniques have been described previously.22 Slides were examined at a JEM 1011 transmission electron microscope at 80 kV.
For scanning electron microscopy, whole liver was collected from wild-type and DKO mice. Liver samples were fixed in 2.5% glutaraldehyde in phosphate-buffered saline (pH 7.4) for 10 minutes. Tissue samples were washed thoroughly in phosphate-buffered saline for 15 minutes. Tissues were then fixed in 1% OsO4 in phosphate-buffered saline for 60 minutes. Samples were dehydrated with different concentration of ethanol (30%, 50%, 70%, and 90%) for 15 minutes and then samples were critical point dried. Samples were visualized using Field Emission Scanning Electron Microscope (JEOL JSM6335F) at the magnification of ×10,000 to ×30,000.
All imaging was performed at the Advanced Cell and Tissue Imaging Core of the Pittsburgh Liver Research Center.
RNA Sequencing
Livers were pelleted and lysed with RLT Buffer (Qiagen, Hilden, Germany). RNA was isolated from the lysate using RNEasy kits (Qiagen). Library preparation, sequencing, and bioinformatics analysis were performed by Medgenome Foster City, (CA). Total RNA was processed for next-generation sequencing, and sequencing was performed with a HiSeq 2500 (Illumina, San Diego, CA). Clontech SMARTer UltraTM Low Input RNA Kits (Mountain View, CA) and NexteraXT (San Diego, CA) kits were used for library preparation.
The bioinformatics analysis was performed by the Genomics and Systems Biology Core of the Pittsburgh Liver Research Center. Briefly, pairwise analysis was performed between the control and DKO mice liver mRNA expression of a total of 23,851 genes, and differentially expressed genes in each pairwise analysis were identified (false discovery rate <0.01; fold change ≥5). The RNA-sequencing data have been submitted to the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query, accession number GSE166087.
siRNA, Cell Culture, and TGF-β Inhibitor
The Hep3B human hepatoma cell line, which was obtained from ATCC (Manassas, VA), was transfected as previously described.21 Briefly, Hep3Bs grown in Eagle's minimal essential medium (ATCC) with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) were seeded onto 6-well plates and transiently transfected with validated human β-catenin (CTNNB1) and γ-catenin (jupiter) siRNA or negative control siRNA 1 (Ambion, Inc., Austin, TX) at a final concentration of 25 nmol/L in the presence of Lipofectamine MAX reagent (Invitrogen, Carlsbad, CA), as per the manufacturer's instructions. The cells were harvested 72 hours after transfection for RNA or protein extraction. For TGF-β inhibition, we have used TGF-β receptor I kinase inhibitor (alias HTS466284), which was synthesized by the Chemical Synthesis Core of Vanderbilt University.
Patients
All patient samples used in the study were approved by the Institutional Review Board. The PFIC cases (n = 8), for which the demographic and pertinent histologic and analytical information is included in Supplemental Tables S1 and S2, were obtained under an Institutional Review Board approval number PRO17090320, from the Children's Hospital, University of Pittsburgh Medical Center (Pittsburgh, PA). For the PSC cases (n = 11), frozen liver samples from the patients were obtained from the Pittsburgh Liver Research Center's Clinical Biospecimen Repository and Processing Core (Institutional Review Board approval number PRO08010372). The demographic and pertinent histologic information on PSC cases is included in Supplemental Table S3.
Histology, IHC, and IF
Tissue sections (4 to 6 μm thick) were stained with hematoxylin and eosin. Immunohistochemistry (IHC) on paraffin-embedded sections was performed on livers, as described elsewhere.22 IHC and immunofluorescence (IF) were done, as previously described.22 Primary antibodies used were against Ki-67, phosphorylated SMAD3, CD10 (Thermo Scientific, Freemont, CA), β-catenin (Cell Signaling, Danvers, MA), SRY-related HMG-box gene-9 (Invitrogen), matrix metalloproteinase 9, α-smooth muscle actin, HNF4α, bile salt export pump, and ezrin (Abcam, Cambridge, UK). Secondary antibodies were horse anti-mouse (Vector Laboratories, Inc., Burlingame, CA), goat anti-rabbit, and donkey anti-goat (Chemicon, Temecula, CA), all used at a 1:400 dilution. Sections were stained, and Nikon (Melville, NY) A1 Spectral Confocal microscopes were used to capture images. Nikon NIS software was used to analyze the data.
Western Blot and Immunoprecipitation Studies
Western blot analysis was performed as described elsewhere.22,24 Immunoprecipitation was performed with 1 mg of protein and as described previously.22,24 The following primary antibodies were used: β-catenin (1:1000; Santa Cruz Biotechnology, Dallas, TX), phosphorylated SMAD2/3 (1:1000; Santa Cruz Biotechnology), occludin (1:100; BD Biosciences [San Jose, CA] and Abcam), γ-catenin (1:300; Abcam), E-cadherin (1:300), claudin-2 (1:50; Invitrogen), glutamine synthetase (1:1000; Invitrogen), ZO-2 (1:100; Life Tech, Carlsbad, CA), glyceraldehyde-3-phosphate dehydrogenase (1:1000; Invitrogen), ZO-1 (1:500; MAB), claudin-3 (1:50; Abcam), claudin-5 (1:50; Millipore Sigma, Burlington, MA), HNF4α, bile salt export pump, and ezrin (Abcam). Membranes were washed five times for 5 minutes each in tris-buffered saline with Tween 20 before being probed with horseradish peroxidase–conjugated secondary antibodies (1:5000 diluted in tris-buffered saline with Tween 20; Santa Cruz Biotechnology) for 1.5 hours at room temperature. Membranes were washed three times for 10 minutes each in tris-buffered saline with Tween 20 and visualized using the Enhanced Chemiluminescence System (GE Healthcare, Marlborough, MA).
mRNA Isolation and Real-Time PCR
mRNA was isolated and purified from livers of CON and DKO mice (n = 3 per group). mRNA was isolated using Trizol (Invitrogen). Real-time PCR was performed as described elsewhere.22 Changes in target mRNA were normalized to glyceraldehyde-3-phosphate dehydrogenase mRNA for each sample and presented as fold-change over the average of the respective control group. Each sample was run in triplicate. Sequences of primers used in this study are provided in Table 1.
Table 1.
Sequences of Primers Used in the Study
| Gene (mouse) | Forward primer | Reverse primer |
|---|---|---|
| Gapdh | 5′-AACTTTGGCATTGTGGAAGG-3′ | 5′-ACACATTGGGGGTAGGAACA-3′ |
| Sox9 | 5′-TCGTGTGTGTGTGTTTATAG-3′ | 5′-ATTCCTATTGCTACACTCAG-3′ |
| Ki67 | 5′-AGAGCCTTAGCAATAGCAACG-3′ | GTCTCCCGCGATTCCTCTG-3′ |
| Timp1 | 5′-AGGTGGTCTCGTTGATTTCT-3′ | 5′-GTAAGGCCTGTAGCTGTGCC-3′ |
| Tgfb1 | 5′-TACCATGCCAACTTCTGTCTGGGA-3′ | 5′-ATGTTGGACAACTGCTCCACCTTG-3′ |
| Vim | 5′-GCTGCGAGAGAAATTGCAGGA-3′ | 5′-CCACTTTCCGTTCAAGGTCAAG-3′ |
| 18s | 5′-CGGCTACCACATCCAAGGAA-3′ | 5′-GCTGGAATTACCGCGGCT-3′ |
| Sdc1 | 5′-ATGAGACGCGCGGCGCTCTG-3′ | 5′-CTGATTGGCAGTTCCATCCT-3′ |
| Zona occludens-1 | 5′-CCACCTCTGTCCAGCTCTTC-3′ | 5′-CACCGGAGTGATGGTTTTCT-3′ |
| Cdh2 | 5′-AGGGTGGACGTCATTGTAGC-3′ | 5′-CTGTTGGGGTCTGTCAGGAT-3′ |
| Fsp1 | 5′-GAGGAGGCCCTGGATGTAAT-3′ | 5′-CTTCATTGTCCCTGTTGCTG-3′ |
| Snail | 5′-CCACTGCAACCGTGCTTTT-3′ | 5′-CACATCCGAGTGGGTTTGG-3′ |
| Slug | 5′-CTCACCTCGGGAGCATACAGC-3′ | 5′-TGAAGTGTCAGAGGAAGGCGGG-3′ |
| Zeb1 | 5′-ACAAGACACCGCCGTCATTT-3′ | 5′-GCAGGTGAGCAACTGGGAAA-3′ |
| Zeb2 | 5′-CACCCAGCTCGAGAGGCATA-3′ | 5′-CACTCCGTGCACTTGAACTTG-3′ |
| Twist1 | 5′-CGGGTCATGGCTAACGTG-3′ | 5′-CAGCTTGCCATCTTGGAGTC-3′ |
| Hsp47 | 5′-GCAGCAGCAAGCAACACTACAACT-3′ | 5′-AGAACATGGCGTTCACAAGCAGTG-3′ |
| G6pc | 5′-TCCCCAGAATTCTCCACTTG-3′ | 5′-AACATCGGAGTGACCTTTGG-3′ |
| Pcsk9 | 5′-TTGCAGCAGCTGGGAACTT-3′ | 5′-CCGACTGTGATGACCTCTGGA-3′ |
| Cmyc | 5′-AGCTGTTTGAAGGCTGGATTTC-3′ | 5′-TCGAGGTCATAGTTCCTGTTGGT-3′ |
| Cdh1 | 5′-CACCTGGAGAGAGGCCATGT-3′ | 5′-TGGGAAACATGAGCAGCTCT-3′ |
| Tgfb2 | 5′-GGAGGTGATTTCCATCTACAAC-3′ | 5′-AGCGGACGATTCTGAAGTA-3′ |
| Tgfbr1 | 5′-TGCAATCAGGACCACTGCAATAA-3′ | 5′-GTGCAATGCAGACGAAGCAGA-3′ |
| Tgfbr2 | 5′-AAATTCCCAGCTTCTGGCTCAAC-3′ | 5′-TGTGCTGTGAGACGGGCTTC-3′ |
| Smad2 | 5′-AACCCGAATGTGCACCATAAGAA-3′ | 5′-GCGAGTCTTTGATGGGTTTACGA-3′ |
| Smad3 | 5′-GTCAACAAGTGGTGGCGTGTG-3′ | 5′-GCAGCAAAGGCTTCTGGGATAA-3′ |
| Smad4 | 5′-TGACGCCCTAACCATTTCCAG-3′ | 5′-CTGCTAAGAGCAAGGCAGCAAA-3′ |
| Smad7 | 5′-AGAGGCTGTGTTGCTGTGAATC-3′ | 5′-CCATTGGGTATCTGGAGTAAGGA-3′ |
| Mmp2 | 5′-CACCACCACAACTGAACCAC-3′ | 5′-CTCAGAAGAGCCCGCAGTAG-3′ |
| Krt19 | 5′-CCGGACCCTCCCGAGATTA-3′ | 5′-CTCCACGCTCAGACGCAAG-3′ |
| Timp2 | 5′-GTAGTGATCAGGGCCA-3′ | 5′-CCTTCIGCCTTYCCTGC-3′ |
| Dio1 | 5′-GTTTGTCCTGAAGGTCCGCT-3′ | 5′-GCCTGCTGCCTTGAATGAAATC-3′ |
| Egr1 | 5′-GAGCGAACAACCCTATGAG-3′ | 5′-GTCGTTTGGCTGGGATAA-3′ |
| Acot3 | 5′-GCTCAGTCACCCTCAGGTAA-3′ | 5′-AAGTTTCCGCCGATGTTGGA-3′ |
| Ces3 | 5′-AAGCTCCTAGCAAACAAGCAA-3′ | 5′-TGGGCTAATAAGGGCCTTGAA-3′ |
| Ugt2B1 | 5′-GTGCTGGTGTGGCCTACAG-3′ | 5′-ATTGCTCGGCCCAATGAGG-3′ |
| Ect2 | 5′-AGAAGGTGCTGGACATCCGAGA-3′ | 5′-CCTTTGGAAGCTCCTCTGACGT-3′ |
| Akr1b7 | 5′-CAGATTGAGAGCCACCCTTA-3′ | 5′-TGGGAATCTCCATTACTACG-3′ |
|
Tgfb3 |
5′-GAGAATTGAGCTCTTCCAGATAC-3′ |
5′-GAAAGGTGTGACATGGACAG-3′ |
| Gene (human) |
Forward primer |
Reverse primer |
| CTNNB1 | 5′-GAAACGGCTTTCAGTTGAGC-3′ | 5′-CTGGCCATATCCACCAGAGT-3′ |
| JUP | 5′-AAGGTGCTATCCGTGTGTCC-3′ | 5′-GACGTTGACGTCATCCACAC-3′ |
| GADPH | 5′-CTTCACCACCATGGAGAAGGC-3′ | 5′-GGCATGGACTGTGGTCATGAG-3′ |
Statistical Analysis
All comparisons between two groups were deemed statistically significant by unpaired two-tailed t-test if P < 0.05 or P < 0.001. When more than two groups were compared, statistical analysis was performed with Prism version 7.0a (GraphPad Software, San Diego, CA) using one- and two-way analysis of variance with Bonferroni correction. All in vitro studies are either compilation of three independent experiments or presented data are representative of at least three independent experiments.
Image Analysis
Movies were processed using Nikon's NIS Elements (Nikon Elements 3.10). A median filter with a kernel size of 3 was applied over each video frame to improve signal/noise ratio. Signal contrast in each channel of a multicolor image was further enhanced by adjusting the maxima and minima of the intensity histogram of that channel.
Motif Analysis in DKO Mice
To discover the upstream transcription factors (TFs) that potentially regulate the detected differentially expressed genes (DEGs) in DKO livers, we explored two pipelines described herein. For the first pipeline, we aimed to discover the binding motif. We collected the DEGs by the RNA-sequencing analysis and extracted their 1000-bp upstream DNA sequences.25,26 These sequences were then used to discover common motif by tool MEME.27 As a result, four high-occurrence motifs were detected across the promoter sequences of the DEGs. Then, we explored the JASPAR TF database to check the similarity between these motifs and known TF binding sites. Peroxisome proliferator activated receptor-gamma and HNF4A were among the top TF candidates that had high similarity with the detected motifs and strong correlation with the DKO model.28 For the second pipeline, we directly employed TF database and performed the Fisher exact test to check the enrichment between TF regulated genes and DEGs detected in DKO livers.28 Eventually, the top 20 significant TFs and then most relevant two were selected, which showed high correlation to our model. A heat map was generated, which showed the overlapping TF regulated genes and DEGs in DKO livers. This analysis was performed by the Genomics and Systems Biology Core of the Pittsburgh Liver Research Center.
Results
β-Catenin and γ-Catenin Are Concomitantly Absent in Liver Samples of a Subset of PFIC Cases and Patients with Cholangiopathy
Simultaneous deletion of β-catenin and γ-catenin leads to cholestasis.22 Because the phenotype in DKO mice was reminiscent of PFIC, as shown by enhanced hepatic and serum bile acids and notable ductular reaction,22 the status of β-catenin and γ-catenin was assessed in eight patient samples for which frozen livers were available (Supplemental Table S1). Of these eight samples, one did not belong to PFIC-1/PFIC-2/PFIC-3.4 Western blot analysis showed loss of both β-catenin and γ-catenin in sample 34, whereas sample 38 showed the absence of β-catenin and a decrease in γ-catenin (Figure 1A). Glutamine synthetase, a downstream target of the Wnt/β-catenin pathway, was absent in samples 34 and 38 (Figure 1A). RT-PCR also revealed decreased mRNA levels of β-catenin and γ-catenin mRNA in samples 34 and 38 (Figure 1B). IHC for β-catenin showed predominantly membranous β-catenin in all PFIC cases (shown 34, 38, 45, and 46) (Figure 1C). However, overall decreased membranous localization along with weak but diffuse cytoplasmic β-catenin staining was observed in case 34 (Figure 1C). Areas of liver from case 38 also showed similar decrease in membranous β-catenin. Immunoprecipitation studies showed intact association of E-cadherin to β-catenin and γ-catenin in all samples (shown samples 34, 38, and 45), except sample 34 (Figure 1D). Patient 34 also showed unique aberrations in other tight junction (TJ) and transporter protein expression profiles (Figure 1E), as summarized in Supplemental Table S2.
Figure 1.

β-Catenin and γ-catenin are absent in subset of progressive familial intrahepatic cholestasis (PFIC) patients. A: Representative Western blot images from liver lysates of available PFIC patients showing loss of β-catenin, γ-catenin, Wnt target glutamine synthetase (GS), occludin, and E-cadherin in available PFIC patient samples. B: Real-time PCR analysis of PFIC liver lysates shows lowest expression of β-catenin and γ-catenin mRNA in cases 34 and 38. C: Immunohistochemical analysis of β-catenin in explanted livers from PFIC patients indicating β-catenin staining in patients. D: Immunoprecipitation studies indicating association of β-catenin and γ-catenin to E-cadherin in patient sample 34 compared with others (shown 38 and 45). E: Western blot analysis using liver lysates from PFIC patients reveals investigating zonula occludens protein (ZO)-2, bile salt export pump, and claudin-2. junctional adhesion molecule-A protein levels. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to verify comparable loading. F: Western blot analysis using liver lysates from primary sclerosing cholangitis patients indicating β-catenin and γ-catenin. GAPDH was used to verify comparable loading. Original magnification, ×50 (C, main images); ×200 (C, insets). IB, immunoblot; IHC, immunohistochemistry; IP, immunoprecipitation; NHL, normal human liver.
Dysregulation of β-catenin and γ-catenin was investigated in other liver diseases with prominent cholestatic component, such as PSC. In the 11 PSC cases analyzed (Supplemental Table S3), a generalized decrease in β-catenin and an increase in γ-catenin was observed in most samples compared with the two normal human liver controls (tumor-free margin of livers from metastatic disease to the liver) (Figure 1F). However, a more profound decrease in β-catenin was observed in six cases (1, 2, 3, 6, 7, and 8), of which two cases showed a notable increase in γ-catenin (1 and 6). Intriguingly, cases 2, 3, 7, and 8 continued to show low to absent γ-catenin despite β-catenin decrease (Figure 1F).
Thus, our findings suggest that concomitant decreases in β-catenin and γ-catenin can occur in a subset of pediatric and adult cholestatic liver diseases, requiring a more in-depth analysis of the process and consequences.
Loss of β-Catenin and γ-Catenin Leads to Misexpression of Several Cell Polarity Proteins in Mouse Liver
Inspired by the findings in PFIC and PSC patients, a more in-depth examination of the basis of the cholestasis phenotype in the dual absence of β-catenin and γ-catenin was conducted. β-catenin and γ-catenin DKO mice using adeno-associated virus 8 expressing Cre recombinase were previously generated under a hepatocyte-specific thyroid-binding globulin promoter (AAV8-TBG-Cre).4 AAV8-TBG-Cre or AAV8-TBG-GFP (CON) was injected into 4-week–old β-catenin; γ-catenin double-floxed mice, followed by blood and liver pathology analysis starting at day 7 after injection (Supplemental Figure S1A). As reported previously, at day 12 after injection, DKO but not CON mice started showing a significant increase in total and direct bilirubin, and alkaline phosphatase, in serum (Supplemental Figure S1B).22 IHC of liver tissue supported a complete loss of β-catenin (Supplemental Figure S1C) and γ-catenin (not shown) from the hepatocytes on day 12. Because day 12 after injection was the first time point that exhibited liver injury, DKO mice at day 12 after injection were used for further phenotypic characterization.
To identify the pathways associated with cholestatic injury in DKO mice liver, RNA-sequencing analysis was performed using day 12 postinjection DKO and CON livers. Pairwise analysis between control and DKO mice liver identified mRNA expression of a total of 23,851 genes. A comparison of the total number of differentially expressed genes in each pairwise analysis (false discovery rate <0.01; fold change ≥5) indicated a change in the expression of 3223 genes (Supplemental Figure S1D and Figure 2A). Among them, 3157 genes were up-regulated and 166 genes were down-regulated. Interestingly, the maximum fold change in DKO livers was observed in genes that encode for proteins that are highly polarized in their location (Figure 2B), extracellular matrix proteins (Figure 2C), and cell membrane–associated proteins (Figure 2D). Furthermore, genes involved in cell adhesion (Figure 2E), and EMT (Figure 2F), also showed significant misexpression in the livers of DKO versus CON mice. Overall, dual loss of β-catenin and γ-catenin from hepatocytes led to changes in the expression of genes encoding for proteins that are highly polarized in their location, specifically at cell membranes, and that may play a role in the maintenance of epithelial cell identity.
Figure 2.

RNA sequence analysis of double-knockout (DKO) liver exhibits misexpression of genes involved in polarity and fibrosis. A: Heat map of hierarchical clustering indicates differentially expressed genes (rows) between control (adeno-associated virus-GFP) and DKO (DKO-12 day) mice; false discovery rate = 0.05, and fold change = 5. B–F: Pathway analysis revealed altered expression of genes involved in polarity proteins (B), extracellular matrix (ECM)–associated proteins (C), cell membrane (D), cell adhesion (E), and EMT-associated genes (F) in the livers of the DKO mice, shown as heat maps. n = 3 mice per group (A). EMT, epithelial-mesenchymal transition.
Simultaneous Ablation of β-Catenin and γ-Catenin Leads to Loss of Hepatocyte Polarity
Given that DKO mice show significant misexpression of genes encoding for proteins that are highly polarized in location, a loss of hepatocyte polarity was hypothesized in the DKO mice.29,30 Hepatocytes in the liver exhibit a unique polarity by forming apical and basolateral domains or poles (Figure 3A). The apical (canalicular) poles of adjacent hepatocytes form the bile canaliculi, whereas the basolateral domain is in contact with the sinusoidal blood and hence also called the sinusoidal pole. Localization studies of both apical (bile salt export pump) (Figure 3B) and basolateral (CD10) proteins (Figure 3C) showed these proteins to be reduced and abnormally localized in the DKO livers, suggestive of aberrant polarity. Intercellular adhesion also requires proper polarization of cells.31, 32, 33, 34, 35 We found that DKO hepatocytes in cell culture were devoid of cellular contact (Figure 3D), and this manifested as an impairment of formation of normal bile canalicular structures in vitro. Transmission electron microscopy analysis of DKO mice livers confirmed the presence of misshapen bile canaliculi (Figure 3E). Many of the canaliculi had a few or no microvilli when compared to the wild type (Figure 3E). Scanning electron micrographs further confirmed the complete loss of microvilli in the liver of DKO mice (Figure 3E). This alteration in the canalicular structure was also associated with misexpression of microvilli-associated proteins, as shown by the IF staining for ezrin. Wild-type ezrin staining showed a mixed pattern of puncta and fine lines and was polarized along the hepatocyte membrane (Figure 3F). However, in DKO livers, the staining for ezrin was overall reduced and more diffuse, lacking specific polarity (Figure 3F). These results suggest that simultaneous ablation of β-catenin and γ-catenin leads to misexpression of polarized proteins, aberrant bile canalicular structure, and loss of hepatic microvilli.
Figure 3.

Absence of β-catenin and γ-catenin leads to loss of hepatocyte polarity. A: Schematic exhibiting hepatocyte polarization across the apical and basolateral poles. B: Confocal images of control (CON; adeno-associated virus-GFP) and double-knockout (DKO; DKO-12 day) liver showing bile salt export pump from the membrane in DKO and control (CON) mice. Inset shows higher magnification of staining. C: Immunohistochemistry images of control and DKO liver show basolateral marker CD10 in DKO liver. D: Phase contrast microscopy showing DKO hepatocytes in culture do not form cellular contact or bile canalicular structures when compared with CON hepatocytes (arrows). E: Top and middle rows: Transmission electron micrographs (TEMs) of control and DKO liver sections reveal aberrant bile canalicular structure with loss of microvilli. Dotted lines represents the bile canalicular structure. Arrows represent the microvilli. Bottom row: Scanning electron micrographs (SEMs) of control and DKO showing microvilli in biliary canaliculi (boxed areas) in the DKO liver at day 12 after adeno-associated virus-Cre injection. F: Confocal images of control and DKO liver showing ezrin in the membrane in DKO. Scale bars: 10 μm (B, C, E, bottom row, and F); 20 μm (D); 1 μm (E, top row); 0.5 μm (E, middle row). Original magnification, ×60 (B). AJ, adherens junction; BC, biliary canaliculi; Mt, mitochondria; TJ, tight junction.
Hepatocyte-Specific Loss of β-Catenin and γ-Catenin Contributes to EMT
Loss of hepatocyte polarity is frequently associated with loss of epithelial identity and transition to a more mesenchymal characteristic, also described as EMT.32, 33, 34, 35 We hypothesized that loss of hepatocyte polarity is associated with EMT in the DKO liver. Indeed, our RNA-sequencing data had shown alterations in EMT genes in the DKO by pathway analysis (Figure 2F). Therefore, we next analyzed the expression of specific EMT markers in the liver of DKO mice. Using real-time PCR, we found that loss of β-catenin and γ-catenin led to a significant decrease in the expression of E-cadherin (Figure 4A) and a significant up-regulation of mesenchymal markers, such as vimentin and fibroblast specific protein-1 (Figure 4B). Moreover, expression of EMT inducers, such as snail, slug, TGF-β, and twist, showed a significant increase in their mRNA expression in DKO livers compared with CON (Figure 4C). Several of these changes were verified on agarose gel electrophoresis and by Western blot analysis (Figure 4, D and E). Further validation of some of these observations came from immunofluorescence assay, which revealed a significant reduction in pan-cadherin and ATP-binding cassette super-family G member (Figure 4F) and an increase in ZO-1 in the DKO livers compared with CON (Figure 4G). Altogether, key molecules that convey epithelial identity through their proper expression and localization were aberrant in DKO liver, whereas mesenchymal markers were overexpressed, suggesting induction of an EMT-like phenotype in the absence of β-catenin and γ-catenin in the hepatocytes.
Figure 4.

Double-knockout (DKO) mice show misexpression of epithelial and mesenchymal markers and increase in epithelial-mesenchymal (EMT) inducers. A: RT-Real-time PCR analysis showing relative mRNA expression of epithelial markers in DKO liver compared to control (CON) livers, such as E-cadherin, cytokeratin, zonula occludens protein (ZO)-1, and syndecan-1. B: Real-time PCR analysis showing relative mRNA expression of mesenchymal markers, like vimentin, fibroblast specific protein-1, and N-cadherin. In DKO livers. C: Real-time PCR analysis showing EMT inducers in DKO livers, including snail, slug, twist, Transforming growth factor (TGF)-b, and zeb-2. D: Representative gel electrophoresis images supporting the real-time PCR observations of epithelial and mesenchymal markers, and EMT inducers. E: Western blot analysis to verify real-time PCR observations. GAPDH was used to indicate comparable protein loading. F: Confocal images of control and DKO liver showing pan-cadherin and ATP-binding cassette super-family G member from the membranes in DKO. G: Confocal images of XZ section showing ZO-1 in the DKO liver compared with the control (CON). ∗P < 0.05 versus WT. Scale bars = 10 μm (F). WT, wild type.
Loss of β-Catenin and γ-Catenin Is Associated with Increased Hepatocyte Proliferation and Reduced HNF4α Expression
EMT can play a major role in wound healing, regeneration, and fibrosis through alterations in basic cellular functions, including cell identity, proliferation, and migration.35, 36, 37 Loss of epithelial cell characteristic and gain of mesenchymal markers in the DKO livers, prompted analysis of whether this manifested as altered cellular identity or proliferation. IHC of Sox9, a marker that identifies only biliary epithelial cells in a CON liver, was notably mislocalized to the hepatocytes in the DKO livers, demonstrating a disruption in hepatocyte identity or maturation (Figure 5A). Likewise, Ki-67 staining also revealed significantly more positivity in the DKO liver, showing more cells in cell cycle (Figure 5B). Quantitation of both Sox9- and Ki-67–positive hepatocytes confirmed a significant enrichment of these markers in the DKO liver (Figure 5B).
Figure 5.

Loss of β-catenin and γ-catenin is associated with increased hepatocyte proliferation and reduced hepatocyte nuclear factor 4α (HNF4α) expression in the liver. A: Immunohistochemistry showing Sox9 staining and Ki-67 staining in hepatocytes of double-knockout (DKO) and control (CON) livers. B: Quantification of Ki-67– and Sox9-positive hepatocytes in control and DKO mouse liver tissue from 20 random fields. C: HNF4α in hepatocyte-positive HNF4α in DKO mouse liver. D: Heat map showing altered expression of various negative and positive target genes of HNF4α pathway in DKO livers compared with CON. E: Analysis of mRNA expression by real-time quantitative PCR showing expression of HNF4α-positive targets and mRNA expression of HNF4α-negative targets in DKO livers compared with CON. F: Western blot analysis of HNF4α knockout (KO) liver showing epithelial marker, epithelial-mesenchymal transition inducer, and β-catenin target, mimicking the DKO mice phenotype. ∗P < 0.05 versus CON; †P < 0.05 versus WT. Scale bars: 100 μm (A, top left panel); 50 μm (A, bottom left panel); 20 μm (A, right column, and C). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
The key molecule responsible for the maintenance of hepatocyte differentiation and curbing cell proliferation in an adult liver is HNF4α, a key liver-specific member of hepatocyte nuclear factor family of transcription factors.38,39 Indeed, HNF4α protein levels were overall reduced in DKO liver (Figure 5C), as seen by IHC. More importantly, an evaluation of HNF4α target genes by RNA sequencing further confirmed the suppression of HNF4α activity in DKO liver (Figure 5D). In fact, significant decreases in the positive targets of HNF4α, including Acot3, Ces3, Ugt2b1, and Dio1, and increased expression of negative targets of HNF4α, including Akr1b7, Ect2, Egr1, and Myc, were evident in the DKO livers compared with CON livers by real-time PCR (Figure 5E).
Finally, the effect of HNF4α knockdown were investigated on hepatocyte polarity and EMT induction. We hypothesized that HNF4α-deficient mice may themselves exhibit a similar phenotype, as seen in the DKO livers. Western blot analysis of the liver of HNF4α-deficient mice showed a significant reduction in the protein level of junctional protein claudin-2, occludin, ERM protein family (ezrin, radixin, moesin), and E-cadherin (Figure 5F). In addition, EMT inducers snail and twist were also up-regulated in the liver of HNF4α-deficient mice (Figure 5E). Interestingly, no significant changes in β-catenin expression in the liver of HNF4α-deficient mice were seen (Figure 5E). Together, these observations suggest that loss of β-catenin and γ-catenin in hepatocytes leads to EMT-like phenotype, which is associated with a notable reduction in HNF4α activity, which, in turn, leads to impairment in hepatocyte differentiation and promotes proliferation.
TGF-β Signaling Is Activated in the Liver of DKO Mice
Given that DKO livers displayed some EMT characteristics, which were also associated with a decrease in HNF4α activity, the state of TGF-β pathway was assessed. TGF-β was recently shown to directly impact hepatocyte differentiation in preclinical and clinical scenarios by inhibiting HNF4α activation.40 An evaluation of TGF-β and target genes by RNA sequencing indicated TGF-β activation in the DKO mice livers (Figure 6A). Further validation using qRT-PCR confirmed significant up-regulation of the mRNA expression of TGF-β1 and SMAD4 (Figure 6B). Among other members of the TGF-β pathway, the receptors TGF-β R1 and R2 and SMAD2/3 did not show any significant change in mRNA expression (Figure 6B). Phosphorylated SMAD2 protein expression was found significantly enriched in the nucleus of the hepatocytes of DKO mice by IHC, which was absent in CON (Figure 6C). TGF-β activation was further validated with IF using antibodies against matrix metalloproteinase 9, a TGF-β pathway target, which showed notable up-regulation by both RNA sequencing (Figure 6A) and by IF (Figure 6D) in the DKO livers when compared with CON.41 Remarkably, when two different strategies (details in Materials and Methods) were utilized to computationally identify TF candidates that might be regulating DEGs in DKO, SMAD4 and HNF4A were identified among the top TF candidates (Supplemental Figure S2).
Figure 6.

Double-knockout (DKO) mice exhibit activation of transforming growth factor (TGF)-β pathway in the liver. A: Heat maps exhibiting differential expression of several selective TGF-β pathway-related genes in the DKO livers compared with control (CON). B: Real-time quantitative PCR analysis showing relative mRNA expression of several components of the TGF-β pathway signaling in the DKO versus CON livers. C: Immunohistochemistry showing phosphorylated SMAD2 in hepatocytes in DKO and not CON livers. D: TGF-β pathway target matrix metalloproteinase (MMP) 9 in hepatocytes in the DKO livers compared with CON. E: Western blot analysis showing hepatocyte nuclear factor 4α (HNF4α) expression in Hep3B cells on siRNA-mediated knockdown of β-catenin and γ-catenin, in the presence of varying doses of TGF-β inhibitor. ∗P < 0.05 versus WT. Scale bars = 10 μm (C and D). GAPDH, glyceraldehyde-3-phosphate dehydrogenase; G6PC, glucose-6-phosphatase, catalytic subunit; TIMP, tissue inhibitor of metalloproteinases; WT, wild type.
Eventually, to understand the mechanistic regulation of TGF-β in the DKO model, an in vitro cell culture system consisting of Hep3B cells was used, which were subjected to β-catenin and γ-catenin knockdown by siRNA. Consistent with the in vivo data, we found dual catenin loss to yield a notable decrease in HNF4α (Figure 6E). Remarkably, treating Hep3B cells with a TGF-β inhibitor at concentrations that inhibit both TGF-β R1 and TGF-β R2 rescued the loss of HNF4α in a dose-dependent manner (Figure 6E). Interestingly, no change in β-catenin or γ-catenin levels on treatment with a TGF-β inhibitor was observed (Figure 6E). Altogether, these findings indicate that loss of β-catenin and γ-catenin causes activation of TGF-β, which, in turn, down-regulates HNF4α levels and activity to contribute to changes in epithelial cell identity, acquisition of mesenchymal phenotype, altered cell proliferation, and a loss of hepatocyte polarity, which all contribute to the overall phenotype observed in the DKO mice.
Discussion
Loss of hepatocyte polarity is observed in many types of acquired and inherited cholestatic liver diseases.3,42 However, the molecular mechanisms linking loss of polarity to cholestasis are not well understood. Herein, β-catenin and γ-catenin were lost in a small subset of uncharacterized PFIC patient liver samples and a subset of PSC cases. It was intriguing to note that in a subset of these clinical cases that showed decrease in β-catenin, γ-catenin was up-regulated. However, others lacked this compensation. It would be important to address in future the basis of β-catenin loss as well as the mechanism that allows for γ-catenin to compensate in such scenarios. Needless to say, elucidating such mechanisms would allow development of novel therapies for cholestatic disorders through stabilization of adherens junctions.
Further analysis using a murine model containing a hepatocyte-specific deletion of β-catenin and γ-catenin genes showed that cholestasis was associated with the loss of hepatocyte polarity and misexpression of epithelial proteins that mediate cell-cell and cell-matrix contacts, as well as the cytoskeletal reorganization. Although previous studies have confirmed the role of β-catenin and γ-catenin in cell growth, differentiation, and proliferation, an association of β-catenin and γ-catenin in hepatocyte polarization process was not known. Similarly, neither the loss of HNF4α nor the activation of TGF-β is known to cause impairment of hepatic polarity13,18. Herein, the drastic phenotypes seen in DKO mice occur due to collective changes brought about by loss of β-catenin and γ-catenin, leading to impairment in HNF4α activity secondary to TGF-β activation. Mechanistically, the loss of hepatocyte polarity in DKO liver occurs in a sequential manner (Figure 7). The absence of β-catenin and γ-catenin in the hepatocytes induces TGF-β signaling, which has been shown to decrease HNF4α activation,40 and also known to increase expression of EMT inducers snail, slug, and twist, and in reducing expression of E-cadherin,22 all contributing to hepatocyte EMT-like phenotype. Both losses of E-cadherin and an increase in EMT have also been previously shown to inhibit HNF4α.43,44 In addition, E-cadherin loss is also known to cause cell adhesion and cytoskeletal defects, which, together with HNF4α inactivation, lead to hepatocyte dedifferentiation and loss of hepatocyte polarity, also contributing to the features of EMT phenotype.45
Figure 7.

The β-catenin–γ-catenin–transforming growth factor (TGF)-β–hepatocyte nuclear factor 4α (HNF4α) nexus promotes hepatocyte polarization. Schematic diagram depicting the mechanism of cholestasis injury in double knockout (DKO) through regulation of hepatocyte polarity. In a control liver, adherens junction (AJ) proteins β-catenin (and in its absence γ-catenin) maintain hepatocyte polarity by inhibiting TGF-β activation and maintaining HNF4α activation. Hepatocyte-specific dual loss of β-catenin and γ-catenin induces TGF-β signaling in the liver, which leads to down-regulation of HNFα activity and expression of epithelial-mesenchymal transition (EMT) inducers snail, slug, and twist, which collectively lead to loss of hepatocyte polarity, thus contributing to the pathology of cholestatic liver disease. BC, biliary canaliculi; TJ, tight junction; WT, wild type.
Previous studies have shown that Hnf4α mutant hepatocytes exhibit reduced cell contacts and misexpression of cell junctional proteins, such as E-cadherin and ZO-1, respectively.43 However, little is known about mechanistic regulation of Hnf4α in hepatocyte polarity. Similarly, inactivation of Hnf4α during early development specifically blocks hepatoblast transition into a mature hepatocyte.46 The DKO model shows a reduction in Hnf4α activity, which may directly contribute to some of the overall observed defects. It would be interesting to determine in the future if forced re-expression of HNF4α could rescue some or all of the observed aberrations in the DKO. Indeed, in other models, such as that of the end-stage liver disease, HNFα re-expression rescues these animals and even reverses advanced fibrosis and cirrhosis.47
PFIC patients are characterized by specific genetic mutations that lead to perturbations in bile flow, eventually leading to cholestasis. Role of cell-cell junctions in biliary homeostasis has been deemed relevant because loss-of-function mutations in TJP2 have been identified in a novel subgroup of PFIC cases.9 One case did not belong to a known category of PFIC genotype, but showed concomitant β-catenin and γ-catenin loss. Additional studies with more patients will be needed to substantiate this finding and address the mechanism of concomitant loss of the two catenins in such cases. Because PFIC cases can be only symptomatically treated and eventually may require liver transplantation as disease worsens, it is likely that a subset of PFIC diseases may benefit from ways to stimulate β-catenin and γ-catenin expression at cell surface. Besides this approach, overexpression of HNF4α or knocking down TGF-β might be achievable and may serve to alter the disease course. This study highlights a higher-order function of adherens junction as master regulator of TGF-β signaling, EMT induction, and differentiation, and eventually hepatocyte polarity in mice, patients, and cell lines. Anomalies in the expression of β-catenin, γ-catenin, and TGF-β may thus be important molecular determinants of cholestatic liver diseases, and may serve as disease modifiers, with potential prognostic and/or therapeutic implications. Indeed, in future, directly testing these hypotheses and studying if blocking TGF-β or HNF4α re-expression may cause amelioration of disease phenotype in the DKO mice.
PSC, like PFIC, remains a disease of poor prognosis, and with limited therapies.48 The molecular basis of PSC also remains unknown, and the clinical course of this disease is highly variable.49 Several preclinical models are being utilized to understand disease biology and have yielded potential therapeutic insights.50 Whether simultaneous loss of both β-catenin and γ-catenin observed in a subset of PSC cases could be a driver of disease pathology will need careful future studies. Likewise, it will be pertinent to investigate if improper compensation by γ-catenin in the event of β-catenin decrease could be a prognostic indicator of the variable clinical course seen in PSC cases. Perturbations in TJ proteins and in-cell polarity are being noted in PSC cases.51,52 Even mouse models such as multidrug resistance protein-2 knockout, which have been used to study diseases like PSC, have been shown to have defects in cell polarity.53 Similarly, EMT has been shown to play an important role in preclinical models of PSC, and knockdown of vimentin itself was shown to have therapeutic benefit.54 Hence, more careful analysis of junctional integrity and cell polarity in cholangiopathies may provide novel insights into disease pathogenesis.
Among the weaknesses of the study include the need for additional characterization of β-catenin and γ-catenin in PFIC, PSC, and other cholestatic conditions in patients. A more direct mechanism of TGF-β activation by the dual loss of β-catenin and γ-catenin in hepatocytes is also needed. Because high concentration of TGF-β inhibitor was used in vitro, some of its effect on HNF4α rescue could be through inhibition of other kinases, such as p38 mitogen-activated protein kinase and mixed-lineage kinase 7.55,56 Although the cross talk between β-catenin and γ-catenin, TGF-β, and Hnf4α seems to play a role in the hepatocyte polarity and the overall DKO phenotype, the present findings do not exclude role for other molecules in the process.
In conclusion, losses of β-catenin and γ-catenin, followed by TGF-β pathway activation, and/or reduced expression of HNF4α, can all lead to the disruption of hepatocyte polarity. In this study, a previously unknown function of β-catenin and γ-catenin was identified in regulating TGF-β and Hnf4α signaling. These findings also lay the groundwork for future investigations aimed at understanding the molecular interactions involved in hepatocyte polarization.
Footnotes
Supported in part by NIH grants 1R01DK62277, 1R01DK116993, 1R01CA204586, and 1R01CA251155, Endowed Chair for Experimental Pathology (S.P.M.), Pilot and Feasibility grant 1P30DK120531 (Pittsburgh Liver Research Center), and the University of Pittsburgh Center for Research Computing (S.L.). Pittsburgh Liver Research Center provided tissue samples through the Biospecimen Repository and Processing Core, imaging through the Advanced Cell and Tissue Imaging Core, and analysis of genomic data through the Genomics and Systems Biology Core.
Disclosures: None declared.
Supplemental material for this article can be found at http://doi.org/10.1016/j.ajpath.2021.02.008.
Contributor Information
Tirthadipa Pradhan-Sundd, Email: tip9@pitt.edu.
Satdarshan P. Monga, Email: smonga@pitt.edu.
Authors Contributions
T.P.-S. and S.P.M. developed the study concept and design; T.P.-S., S.L., S.S., and J.K. acquired data; T.P.-S. and S.P.M. analyzed and interpreted data; T.P.-S. and S.P.M. drafted the manuscript; T.P.-S., S.P.M., and S.L. performed critical revision of the manuscript; T.P.-S. performed statistical analysis; S.P.M. obtained funding; M.P., S.K., A.B., I.H., D.S., U.A., S.R., and K.N.-B. provided technical or material support.
Supplemental Data
Supplemental Figure S1.
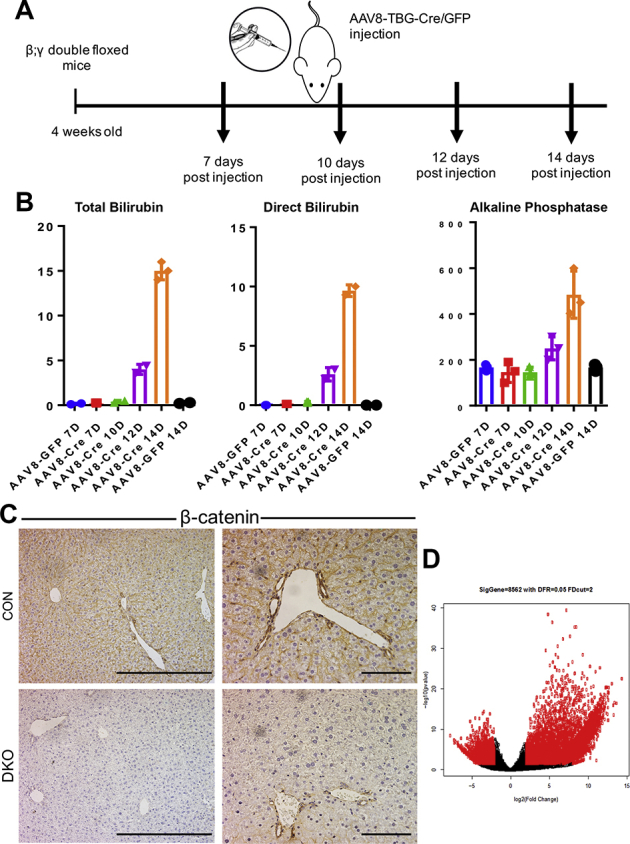
AAV8-TBG-Cre–driven hepatocyte-specific knockdown of β-catenin and γ-catenin [double knockout (DKO)] causes liver injury. A: Schematic showing delivery of AAV8-TBG-Cre or AAV8-TBG-GFP to β-catenin; γ-catenin double-floxed mice to generate β-catenin; γ-catenin DKO or control (CON), respectively. B: Serum analysis from control (AAV8-GFP) and AAV8 Cre β-catenin; γ-catenin mice indicates persistent increases in direct and total bilirubin and alkaline phosphatase, starting from day 12 after injection. C: Immunohistochemistry for β-catenin, depicting its intact expression in both hepatocytes and cholangiocytes in CON livers, whereas in DKO livers, hepatocytes are completely devoid of β-catenin staining, and cholangiocytes are β-catenin positive. D: Scatterplot of the RNA-sequencing data analysis between DKO and CON livers harvested from β-catenin; γ-catenin double-floxed mice 12 days after AAV8-TBG-Cre or AAV8-TBG-GFP injection. Scale bars: 5 μm (C, left column); 10 μm (C, right column).
Supplemental Figure S2.
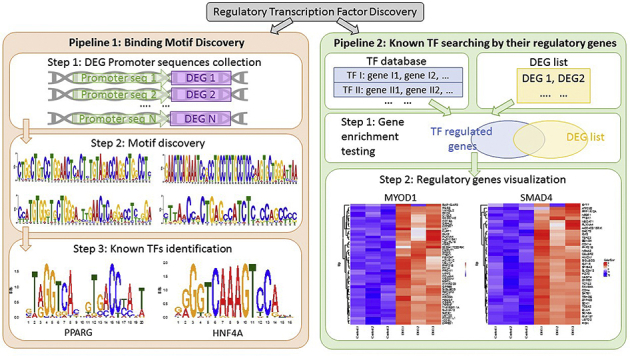
Transcription factor (TF) binding site and motif analysis in double-knockout (DKO) liver. Two strategies to identify TF regulating differentially expressed genes (DEGs) in DKO liver and results from these analyses. The left-hand side depicts strategy to detect common TFs by motif discovery pipeline, where we find PPARG and hepatocyte nuclear factor 4α (HNF4α) are the top TFs regulating differentially expressed genes in the DKO liver. The right-hand side is an alternate strategy used to detect TFs with known regulated target genes, where we find TF MYOD1 and SMAD4 are among the top upstream regulators for the DKO differentially expressed genes. Overall, the analysis illustrates that SMAD4 and HNF4α are among the top TF candidates that are upstream of the differentially expressed genes in DKO detected by two different pipelines.
References
- 1.Wodarz A., Nathke I. Cell polarity in development and cancer. Nat Cell Biol. 2007;9:1016–1024. doi: 10.1038/ncb433. [DOI] [PubMed] [Google Scholar]
- 2.Musch A. The unique polarity phenotype of hepatocytes. Exp Cell Res. 2014;328:276–283. doi: 10.1016/j.yexcr.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gissen P., Arias I.M. Structural and functional hepatocyte polarity and liver disease. J Hepatol. 2015;63:1023–1037. doi: 10.1016/j.jhep.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morotti R.A., Suchy F.J., Magid M.S. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: a review of the liver pathology findings. Semin Liver Dis. 2011;31:3–10. doi: 10.1055/s-0031-1272831. [DOI] [PubMed] [Google Scholar]
- 5.Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol. 2014;4:25–36. doi: 10.1016/j.jceh.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Decaens C., Durand M., Grosse B., Cassio D. Which in vitro models could be best used to study hepatocyte polarity? Biol Cell. 2008;100:387–398. doi: 10.1042/BC20070127. [DOI] [PubMed] [Google Scholar]
- 7.Ihrke G., Neufeld E.B., Meads T., Shanks M.R., Cassio D., Laurent M., Schroer T.A., Pagano R.E., Hubbard A.L. WIF-B cells: an in vitro model for studies of hepatocyte polarity. J Cell Biol. 1993;123:1761–1775. doi: 10.1083/jcb.123.6.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlton V.E., Harris B.Z., Puffenberger E.G., Batta A.K., Knisely A.S., Robinson D.L., Strauss K.A., Shneider B.L., Lim W.A., Salen G., Morton D.H., Bull L.N. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet. 2003;34:91–96. doi: 10.1038/ng1147. [DOI] [PubMed] [Google Scholar]
- 9.Sambrotta M., Strautnieks S., Papouli E., Rushton P., Clark B.E., Parry D.A., Logan C.V., Newbury L.J., Kamath B.M., Ling S., Grammatikopoulos T., Wagner B.E., Magee J.C., Sokol R.J., Mieli-Vergani G., University of Washington Center for Mendelian Genomics. Smith J.D., Johnson C.A., McClean P., Simpson M.A., Knisely A.S., Bull L.N., Thompson R.J. Mutations in TJP2 cause progressive cholestatic liver disease. Nat Genet. 2014;46:326–328. doi: 10.1038/ng.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cereijido M., Contreras R.G., Shoshani L., Flores-Benitez D., Larre I. Tight junction and polarity interaction in the transporting epithelial phenotype. Biochim Biophys Acta. 2008;1778:770–793. doi: 10.1016/j.bbamem.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Ebnet K., Aurrand-Lions M., Kuhn A., Kiefer F., Butz S., Zander K., Meyer zu Brickwedde M.K., Suzuki A., Imhof B.A., Vestweber D. The junctional adhesion molecule (JAM) family members JAM-2 and JAM-3 associate with the cell polarity protein PAR-3: a possible role for JAMs in endothelial cell polarity. J Cell Sci. 2003;116:3879–3891. doi: 10.1242/jcs.00704. [DOI] [PubMed] [Google Scholar]
- 12.Nemeth Z., Szasz A.M., Tatrai P., Nemeth J., Gyorffy H., Somoracz A., Szijarto A., Kupcsulik P., Kiss A., Schaff Z. Claudin-1, -2, -3, -4, -7, -8, and -10 protein expression in biliary tract cancers. J Histochem Cytochem. 2009;57:113–121. doi: 10.1369/jhc.2008.952291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pradhan-Sundd T., Monga S.P. Blood-bile barrier: morphology, regulation, and pathophysiology. Gene Expr. 2019;19:69–87. doi: 10.3727/105221619X15469715711907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maillet V., Boussetta N., Leclerc J., Fauveau V., Foretz M., Viollet B., Couty J.P., Celton-Morizur S., Perret C., Desdouets C. LKB1 as a gatekeeper of hepatocyte proliferation and genomic integrity during liver regeneration. Cell Rep. 2018;22:1994–2005. doi: 10.1016/j.celrep.2018.01.086. [DOI] [PubMed] [Google Scholar]
- 15.Taddei A., Giampietro C., Conti A., Orsenigo F., Breviario F., Pirazzoli V., Potente M., Daly C., Dimmeler S., Dejana E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10:923–934. doi: 10.1038/ncb1752. [DOI] [PubMed] [Google Scholar]
- 16.Theard D., Steiner M., Kalicharan D., Hoekstra D., van Ijzendoorn S.C. Cell polarity development and protein trafficking in hepatocytes lacking E-cadherin/beta-catenin-based adherens junctions. Mol Biol Cell. 2007;18:2313–2321. doi: 10.1091/mbc.E06-11-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Behari J., Yeh T.H., Krauland L., Otruba W., Cieply B., Hauth B., Apte U., Wu T., Evans R., Monga S.P. Liver-specific beta-catenin knockout mice exhibit defective bile acid and cholesterol homeostasis and increased susceptibility to diet-induced steatohepatitis. Am J Pathol. 2010;176:744–753. doi: 10.2353/ajpath.2010.090667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monga S.P. beta-Catenin signaling and roles in liver homeostasis, injury, and tumorigenesis. Gastroenterology. 2015;148:1294–1310. doi: 10.1053/j.gastro.2015.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeh T.H., Krauland L., Singh V., Zou B., Devaraj P., Stolz D.B., Franks J., Monga S.P., Sasatomi E., Behari J. Liver-specific beta-catenin knockout mice have bile canalicular abnormalities, bile secretory defect, and intrahepatic cholestasis. Hepatology. 2010;52:1410–1419. doi: 10.1002/hep.23801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wickline E.D., Awuah P.K., Behari J., Ross M., Stolz D.B., Monga S.P. Hepatocyte gamma-catenin compensates for conditionally deleted beta-catenin at adherens junctions. J Hepatol. 2011;55:1256–1262. doi: 10.1016/j.jhep.2011.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wickline E.D., Du Y., Stolz D.B., Kahn M., Monga S.P. gamma-Catenin at adherens junctions: mechanism and biologic implications in hepatocellular cancer after beta-catenin knockdown. Neoplasia. 2013;15:421–434. doi: 10.1593/neo.122098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pradhan-Sundd T., Zhou L., Vats R., Jiang A., Molina L., Singh S., Poddar M., Russell J., Stolz D.B., Oertel M., Apte U., Watkins S., Ranganathan S., Nejak-Bowen K.N., Sundd P., Monga S.P. Dual catenin loss in murine liver causes tight junctional deregulation and progressive intrahepatic cholestasis. Hepatology. 2018;67:2320–2337. doi: 10.1002/hep.29585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Committee for the Update of the Guide for the Care and Use of Laboratory Animals; National Research Council . National Academies Press; Washington, DC: 2011. Guide for the Care and Use of Laboratory Animals: Eighth Edition. [Google Scholar]
- 24.Zhou L., Pradhan-Sundd T., Poddar M., Singh S., Kikuchi A., Stolz D.B., Shou W., Li Z., Nejak-Bowen K.N., Monga S.P. Mice with hepatic loss of the desmosomal protein gamma-catenin are prone to cholestatic injury and chemical carcinogenesis. Am J Pathol. 2015;185:3274–3289. doi: 10.1016/j.ajpath.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quinlan A.R. BEDTools: the Swiss-army tool for genome feature analysis. Curr Protoc Bioinformatics. 2014;47 doi: 10.1002/0471250953.bi1112s47. 11.12.1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranasic D., Santana-Garcia W., Tan G., Cheneby J., Ballester B., Parcy F., Sandelin A., Lenhard B., Wasserman W.W., Mathelier A. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48:D87–D92. doi: 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luedde T., Schwabe R.F. NF-kappaB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:108–118. doi: 10.1038/nrgastro.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rovillain E., Mansfield L., Caetano C., Alvarez-Fernandez M., Caballero O.L., Medema R.H., Hummerich H., Jat P.S. Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene. 2011;30:2356–2366. doi: 10.1038/onc.2010.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Desai R.A., Gao L., Raghavan S., Liu W.F., Chen C.S. Cell polarity triggered by cell-cell adhesion via E-cadherin. J Cell Sci. 2009;122:905–911. doi: 10.1242/jcs.028183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ozdamar B., Bose R., Barrios-Rodiles M., Wang H.R., Zhang Y., Wrana J.L. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307:1603–1609. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- 34.Pei D., Shu X., Gassama-Diagne A., Thiery J.P. Mesenchymal-epithelial transition in development and reprogramming. Nat Cell Biol. 2019;21:44–53. doi: 10.1038/s41556-018-0195-z. [DOI] [PubMed] [Google Scholar]
- 35.Thiery J.P. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 36.Gressner A.M., Weiskirchen R., Breitkopf K., Dooley S. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7:d793–d807. doi: 10.2741/A812. [DOI] [PubMed] [Google Scholar]
- 37.Xue Z.F., Wu X.M., Liu M. Hepatic regeneration and the epithelial to mesenchymal transition. World J Gastroenterol. 2013;19:1380–1386. doi: 10.3748/wjg.v19.i9.1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayhurst G.P., Lee Y.H., Lambert G., Ward J.M., Gonzalez F.J. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol. 2001;21:1393–1403. doi: 10.1128/MCB.21.4.1393-1403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parviz F., Matullo C., Garrison W.D., Savatski L., Adamson J.W., Ning G., Kaestner K.H., Rossi J.M., Zaret K.S., Duncan S.A. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet. 2003;34:292–296. doi: 10.1038/ng1175. [DOI] [PubMed] [Google Scholar]
- 40.Argemi J., Latasa M.U., Atkinson S.R., Blokhin I.O., Massey V., Gue J.P. Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat Commun. 2019;10:3126. doi: 10.1038/s41467-019-11004-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moore-Smith L.D., Isayeva T., Lee J.H., Frost A., Ponnazhagan S. Silencing of TGF-beta1 in tumor cells impacts MMP-9 in tumor microenvironment. Sci Rep. 2017;7:8678. doi: 10.1038/s41598-017-09062-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Treyer A., Musch A. Hepatocyte polarity. Compr Physiol. 2013;3:243–287. doi: 10.1002/cphy.c120009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Battle M.A., Konopka G., Parviz F., Gaggl A.L., Yang C., Sladek F.M., Duncan S.A. Hepatocyte nuclear factor 4alpha orchestrates expression of cell adhesion proteins during the epithelial transformation of the developing liver. Proc Natl Acad Sci U S A. 2006;103:8419–8424. doi: 10.1073/pnas.0600246103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cicchini C., Amicone L., Alonzi T., Marchetti A., Mancone C., Tripodi M. Molecular mechanisms controlling the phenotype and the EMT/MET dynamics of hepatocyte. Liver Int. 2015;35:302–310. doi: 10.1111/liv.12577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gonzalez-Sanchez E., Vaquero J., Fouassier L., Chignard N. E-cadherin, guardian of liver physiology. Clin Res Hepatol Gastroenterol. 2015;39:3–6. doi: 10.1016/j.clinre.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 46.Walesky C., Gunewardena S., Terwilliger E.F., Edwards G., Borude P., Apte U. Hepatocyte-specific deletion of hepatocyte nuclear factor-4alpha in adult mice results in increased hepatocyte proliferation. Am J Physiol Gastrointest Liver Physiol. 2013;304:G26–G37. doi: 10.1152/ajpgi.00064.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishikawa T., Bell A., Brooks J.M., Setoyama K., Melis M., Han B., Fukumitsu K., Handa K., Tian J., Kaestner K.H., Vodovotz Y., Locker J., Soto-Gutierrez A., Fox I.J. Resetting the transcription factor network reverses terminal chronic hepatic failure. J Clin Invest. 2015;125:1533–1544. doi: 10.1172/JCI73137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tabibian J.H., Ali A.H., Lindor K.D. Primary sclerosing cholangitis, part 1: epidemiology, etiopathogenesis, clinical features, and treatment. Gastroenterol Hepatol (N Y) 2018;14:293–304. [PMC free article] [PubMed] [Google Scholar]
- 49.Fabris L., Fiorotto R., Spirli C., Cadamuro M., Mariotti V., Perugorria M.J., Banales J.M., Strazzabosco M. Pathobiology of inherited biliary diseases: a roadmap to understand acquired liver diseases. Nat Rev Gastroenterol Hepatol. 2019;16:497–511. doi: 10.1038/s41575-019-0156-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sato K., Glaser S., Kennedy L., Liangpunsakul S., Meng F., Francis H., Alpini G. Preclinical insights into cholangiopathies: disease modeling and emerging therapeutic targets. Expert Opin Ther Targets. 2019;23:461–472. doi: 10.1080/14728222.2019.1608950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roehlen N., Roca Suarez A.A., El Saghire H., Saviano A., Schuster C., Lupberger J., Baumert T.F. Tight junction proteins and the biology of hepatobiliary disease. Int J Mol Sci. 2020;21:825. doi: 10.3390/ijms21030825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tam P.K.H., Yiu R.S., Lendahl U., Andersson E.R. Cholangiopathies - towards a molecular understanding. EBioMedicine. 2018;35:381–393. doi: 10.1016/j.ebiom.2018.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pradhan-Sundd T., Kosar K., Saggi H., Zhang R., Vats R., Cornuet P., Green S., Singh S., Zeng G., Sundd P., Nejak-Bowen K. Wnt/beta-catenin signaling plays a protective role in the Mdr2 knockout murine model of cholestatic liver disease. Hepatology. 2020;71:1732–1749. doi: 10.1002/hep.30927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou T., Kyritsi K., Wu N., Francis H., Yang Z., Chen L., O'Brien A., Kennedy L., Ceci L., Meadows V., Kusumanchi P., Wu C., Baiocchi L., Skill N.J., Saxena R., Sybenga A., Xie L., Liangpunsakul S., Meng F., Alpini G., Glaser S. Knockdown of vimentin reduces mesenchymal phenotype of cholangiocytes in the Mdr2(-/-) mouse model of primary sclerosing cholangitis (PSC) EBioMedicine. 2019;48:130–142. doi: 10.1016/j.ebiom.2019.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H.Y., Wang Y., Heap C.R., King C.H., Mundla S.R., Voss M., Clawson D.K., Yan L., Campbell R.M., Anderson B.D., Wagner J.R., Britt K., Lu K.X., McMillen W.T., Yingling J.M. Dihydropyrrolopyrazole transforming growth factor-beta type I receptor kinase domain inhibitors: a novel benzimidazole series with selectivity versus transforming growth factor-beta type II receptor kinase and mixed lineage kinase-7. J Med Chem. 2006;49:2138–2142. doi: 10.1021/jm058209g. [DOI] [PubMed] [Google Scholar]
- 56.Sawyer J.S., Anderson B.D., Beight D.W., Campbell R.M., Jones M.L., Herron D.K., Lampe J.W., McCowan J.R., McMillen W.T., Mort N., Parsons S., Smith E.C., Vieth M., Weir L.C., Yan L., Zhang F., Yingling J.M. Synthesis and activity of new aryl- and heteroaryl-substituted pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. J Med Chem. 2003;46:3953–3956. doi: 10.1021/jm0205705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.