

CLINICAL HISTORY AND BACKGROUND:
A previously healthy 12-month-old boy presented with recurrent episodes of emesis for a week after eating his birthday cake. He had multiple visits to the emergency department for poor feeding and sleepiness, where he received dextrose-containing intravenous fluids (IVF) with mild improvement. He was lethargic with normal vital signs. Plasma ammonia (NH3) was increased at 156 µmol/L (reference interval (RI): 9-33) and he was started on 10% dextrose-containing IVF at 1.5 maintenance rate, IV loading dose of nitrogen scavenger (NS), Ammonul (250 mg/Kg over 2 hr) followed with 24-hr infusion (250 mg/Kg/24 hr). Plasma glucose, total CO2 and anion gap were within reference intervals; while plasma hepatic enzymes, alanine aminotransferase (ALT) (64 U/L; RI: 10-30) and aspartate aminotransferase (AST) (83 U/L; RI: 20-60) were increased, and prothrombin time (PT) was prolonged at 17.7 seconds (s)(RI: 10.5 – 13.0). Plasma amino acids (PAA) revealed undetectable citrulline (<2 µmol/L) (RI: 7-47), increased glutamine (1,635 µmol/L) (RI: 285-832). Organic acids revealed increased urinary uracil (UU, 36 mg/g Cr)(RI:0-21) concerning for ornithine transcarbamylase deficiency (OTCD). Subsequently quantitative urine orotic acid (OA) was increased at 21.09 mmol/mol Cr (RI: 1.15-3.09) and OTC gene sequencing revealed a hemizygous pathogenic mutation: c.386G>A (p.Arg129His), previously reported in both neonatal and late-onset OTCD (1,2). He was managed with IVF, low protein diet (6 grams/day), oral citrulline (240 mg/Kg) and oral NS. At discharge on day 4, hyperammonemia (NH3 <9 μmol/L) and coagulopathy (PT = 11.6 s) had resolved; however, ALT (2964 U/L) and AST (643 U/L) were increased and worsened after discharge, before normalizing at 4 weeks.
DIAGNOSIS AND SUMMARY:
OTCD is an X-linked recessive urea cycle disorder (UCD) with a prevalence of 1 in 77,000 people. The presentation of OTCD can range from life-threatening hyperammonemic crises in neonates to a subclinical disease with late-onset (partial OTCD) in about 50% of patients. Untreated late-onset UCDs have a high mortality rate (11%). Therefore, a low clinical threshold to check NH3 as a standard of care for any patient presenting with altered mental status and immediate treatment is crucial to prevent mortality and irreversible neurological damage.
The major biochemical hallmarks of OTCD include increased plasma NH3 and glutamine, decreased citrulline and increased urine OA. Increased urine OA distinguishes OTCD from other proximal UCDs such N-acetylglutamate synthase or carbamoyl-phosphate synthetase 1 deficiencies.
Uracil is an endogenously synthesized pyrimidine. Increased UU is characteristic of enzyme deficiencies of pyrimidine metabolism. UU can accumulate in UCDs, due to shunting of carbamoylphosphate to pyrimidine synthesis (Fig. 1). Increased UU has been reported in OTCD, hyperornithinemia-hyperammonemia-homocitrullinuria syndrome and in errors of creatine metabolism (3). Mildly increased OA may not be identified by urine organic acids analysis by conventional GC-MS as it co-elutes with aconitic acid, but UU can be readily semi-quantified (Fig. 2). UU is a known biomarker for symptomatic and asymptomatic OTCD females with or without OA elevation (4); however, it has only been occasionally reported in males (3–5). Importantly two reported males had normal NH3 and PAA (4); increased UU and OA were the exclusive biomarkers that led to the diagnosis of OTCD.
Figure 1. Illustration of urea cycle.

OTCD results in accumulation of carbamoyl phosphate in the mitochondria, which then leaks into the cytoplasm to be used as a substrate for pyrimidine biosynthesis resulting in increase in uracil. CPS1: carbamoyl phosphate synthetase 1. NAGS: N-acetylglutamate synthetase. OTC: ornithine transcarbamylase. ASA: argininosuccinic acid.
Figure 2. Organic acids analysis of patient urine using gas chromatography-mass spectrometry.
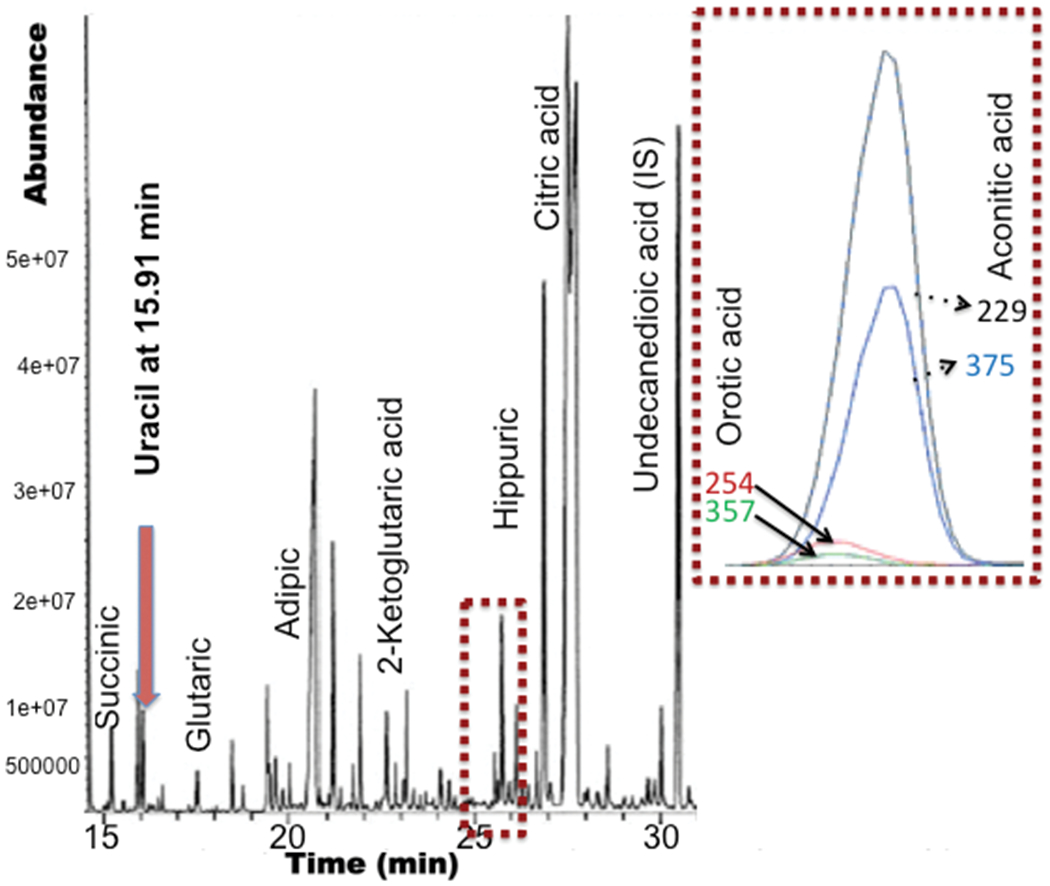
Urine organic acids were extracted into ethyl acetate and converted to trimethylsilyl derivatives prior to analysis using a GC 7890B/MS 5977B system equipped with a HP-5MS column (Agilent). Chromatogram illustrates uracil and orotic acid at 15.91 and 25.7 minutes respectively. Orotic acid co-elutes with aconitic acid as shown by the overlay of compound specific ions (m/z).
Thus, UU may be more reliable than OA for late-onset OTCD during acute and quiescent phases. In our patient, both OA and UU were increased. OA is typically normal in female carriers and could be normal in OTCD, highlighting the importance of UU as an adjuvant marker for the diagnosis.
In conclusion, UU may not only be a useful marker for OTCD in females, but also is a sensitive and rapidly semi-quantifiable marker applicable to males with partial OTCD. Therefore, it should be considered more often in the workup of UCDs.
Abbreviations:
- OTC
ornithine transcarbamylase
- IVF
intravenous fluids
- NH3
ammonia
- IV
intravenous
- RI
reference interval
- hr
hour
- NS
nitrogen scavenger
- H2CO3
bicarbonate
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- PT
prothrombin time
- s
seconds
- PAA
plasma amino acids
- OTCD
ornithine transcarbamylase deficiency
- OA
orotic acid
- UCD
urea cycle disorder
- UU
urinary uracil
- HHH
hyperornithinemia-hyperammonemia-homocitrullinuria
- CPS1
carbamoyl phosphate synthetase 1
- NAGS
N-acetylglutamate synthetase
- GC
gas chromatography
- MS
mass spectrometry
Footnotes
Human Genes:
OTC: HGNC approved gene symbol is OTC.
No previous presentations of the manuscript
References
- 1.Tuchman M, Plante R, McCann M, and Qureshi A. Seven new mutations in the human ornithine transcarbamylase gene. Hum Mutat 1994; 4:57–60. [DOI] [PubMed] [Google Scholar]
- 2.Mastuura T, Hoshide R, Kiwaki K, Koike E, Endo F, Oyanagi K, Suzuki Y, Kato I, Ishikawa et al. Four newly identified ornithine transcarbamylase (OTC) mutation (D126G, R129H, I172M and W332X) in Japanese male patients with early onset OTC deficiency. Hum Mutat 1994;3:402–6. [DOI] [PubMed] [Google Scholar]
- 3.Auray-Blais C, Maranda B, Lavoie P. High-throughput tandem mass spectrometry multiplex analysis for newborn urinary screening of creatine synthesis and transport disorders, Triple H syndrome and OTC deficiency. Clinic` Chimca Acta 436 (2014) 249–255. [DOI] [PubMed] [Google Scholar]
- 4.Burlina AB, Peduto A, Di Palma A, Bellizzi A, Sperlì D, Morrone A, Burlina AP. An unusual clinical and biochemical presentation of ornithine transcarbamylase deficiency in a male patient. J Inherit Metab Dis 2006;29:179–81. [DOI] [PubMed] [Google Scholar]
- 5.Mak CM, Siu T, Lam C, Chan GC, Poon GW, Wong K et al. Complete recovery from acute encephalopathy of late-onset ornithine transcarbamylase deficiency in a 3-year-old boy. J Inherit Metab Dis 2007;30, 981. [DOI] [PubMed] [Google Scholar]