

Abstract
Two new diprenylated coumaric acid isomers (1a/b) and two known congeners, capillartemisin A (2) and B (3), were isolated from Artemisia scoparia as bioactive markers using bioactivity-guided HPLC fractionation. Their structures were determined by spectroscopic means, including 1D and 2D NMR methods, LC-MS, with their purity assessed by 1D 1H pure shift qNMR spectroscopic analysis. The bioactivity of compounds was assessed by enhanced accumulation of lipids, as measured using Oil Red O staining, and by increased expression of several adipocyte marker genes, including adiponectin, in 3T3-L1 adipocytes relative to untreated negative controls. Compared to the plant 80% EtOH extract, these purified compounds showed significant but still weaker inhibition of TNFα-induced lipolysis in 3T3-L1 adipocytes. This suggests that additional bioactive substances are responsible for the multiple metabolically favorable effects on adipocytes observed with Artemisia scoparia extract.
Graphical Abstract
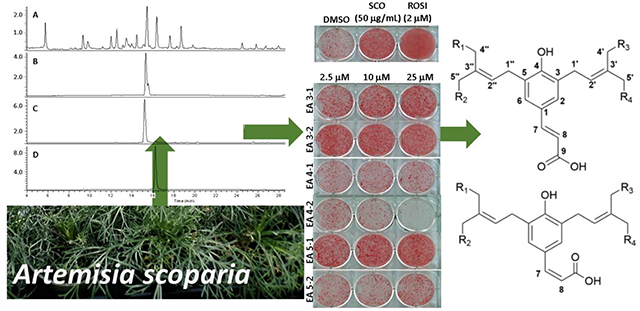
Artemisia is one of the largest and most diverse genera of the family Asteraceae and comprised of over 500 species, which contain a diverse array of phytochemicals that are used widely as traditional medicines and sources of pharmaceutical agents. Artemisia scoparia Waldst. & Kit is an annual herb distributed from Central Europe to Western Asia that has been studied for the biological activity of its essential oil as well as for other phytochemical preparations.1 We previously determined the adipogenic activity of an EtOH extract of A. scoparia (SCO) as the result of screening hundreds of plants for bioactivity related to metabolic syndrome.2 Several bioactivities of the extract have been identified, including the promotion of adipogenesis in vitro, as measured by both lipid accumulation and the expression of adipogenic genes), as well as the enhancement of insulin sensitivity in high-fat diet-induced obese mice.3,4 This extract also showed distinct inhibitory effects on lipolysis (the release of lipids) in both cultured murine adipocytes and in mice fed a high-fat diet supplemented with SCO.5,6 In an effort to better understand how the constituents of the extract contribute to the observed adipogenic bioactivity, a series of the bioactivity-guided fractionation studies were conducted. This previously conducted research led to the putative identification of the principal bioactive constituents in SCO as prenylated coumaric acids, coumarin monoterpene ethers, 6-demethoxycapillarisin, and two polymethoxyflavones.6
A study by Yahagi et al. identified some compounds isolated from an aqueous EtOH extract of A. scoparia that inhibited triglyceride accumulation in adipocytes.1 The inhibition of lipid accumulation and adipocyte differentiation was once considered an appropriate strategy for combating obesity-related metabolic dysfunction. However, it is well established that impairments in adipogenesis and adipose tissue expansion can promote the metabolic dysregulation associated with insulin resistance.7–10 This is underscored by the potent insulin-sensitizing effects of the thiazolidinedione (TZD) drugs, which act by enhancing adipocyte differentiation.11 The compounds identified as inhibitors of triglyceride accumulation were primarily chromane derivatives that were active at higher concentrations than those tested.1 In the present investigation, compounds 1–3 were purified from SCO using bioassay guided fractionation and characterized by spectroscopic methods including NMR spectroscopy, and validated as bioactive markers for this extract by a panel of in vitro assays.
RESULTS AND DISCUSSION
An 80% EtOH extract of A. scoparia, code-named SCO, was shown previously to have discernible adipogenic properties in vitro and in vivo, which were maintained through the process of the bioactivity-guided fractionation and present in distinct fractions comprised of prenylated coumarins and related compounds.6 Further separation of the fractions led to the isolation of six compounds that subsequently were structurally characterized and subjected to bioassays. Three of those compounds retained some, i.e., a statistically significant portion, but not all the biological activity of their parent fractions. The structures of compounds 1–3 are shown in Figures 1 and 2. Compounds 1a/b are prenylated cinnamic acid isomers identified as new natural products, while 2 and 3 are isomers of the prenylated coumaric acids, capillartemisin A and B.
Figure 1.
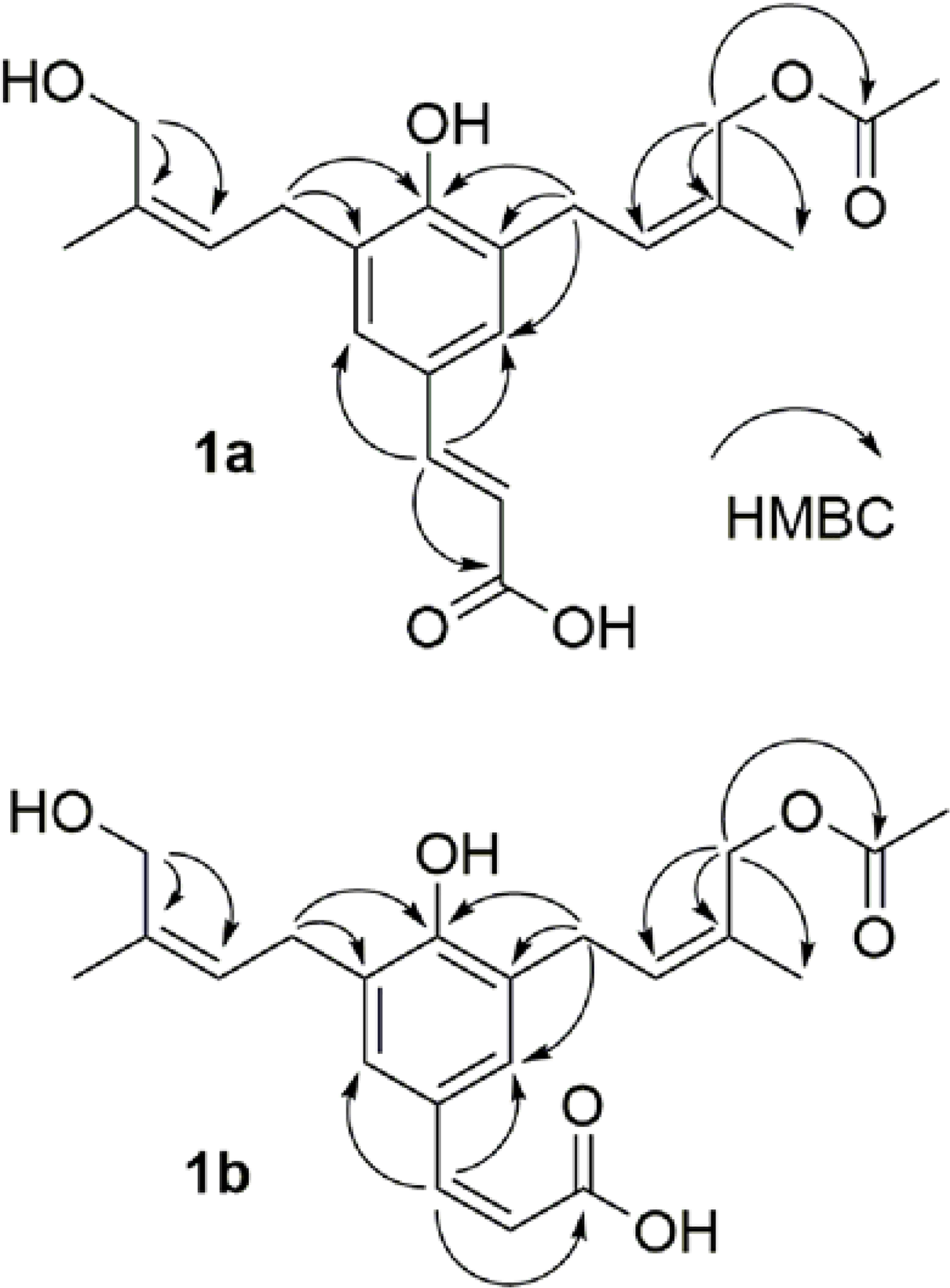
Key HMBC correlations of compounds 1a and 1b, shown as H to C arrows.
Figure 2.
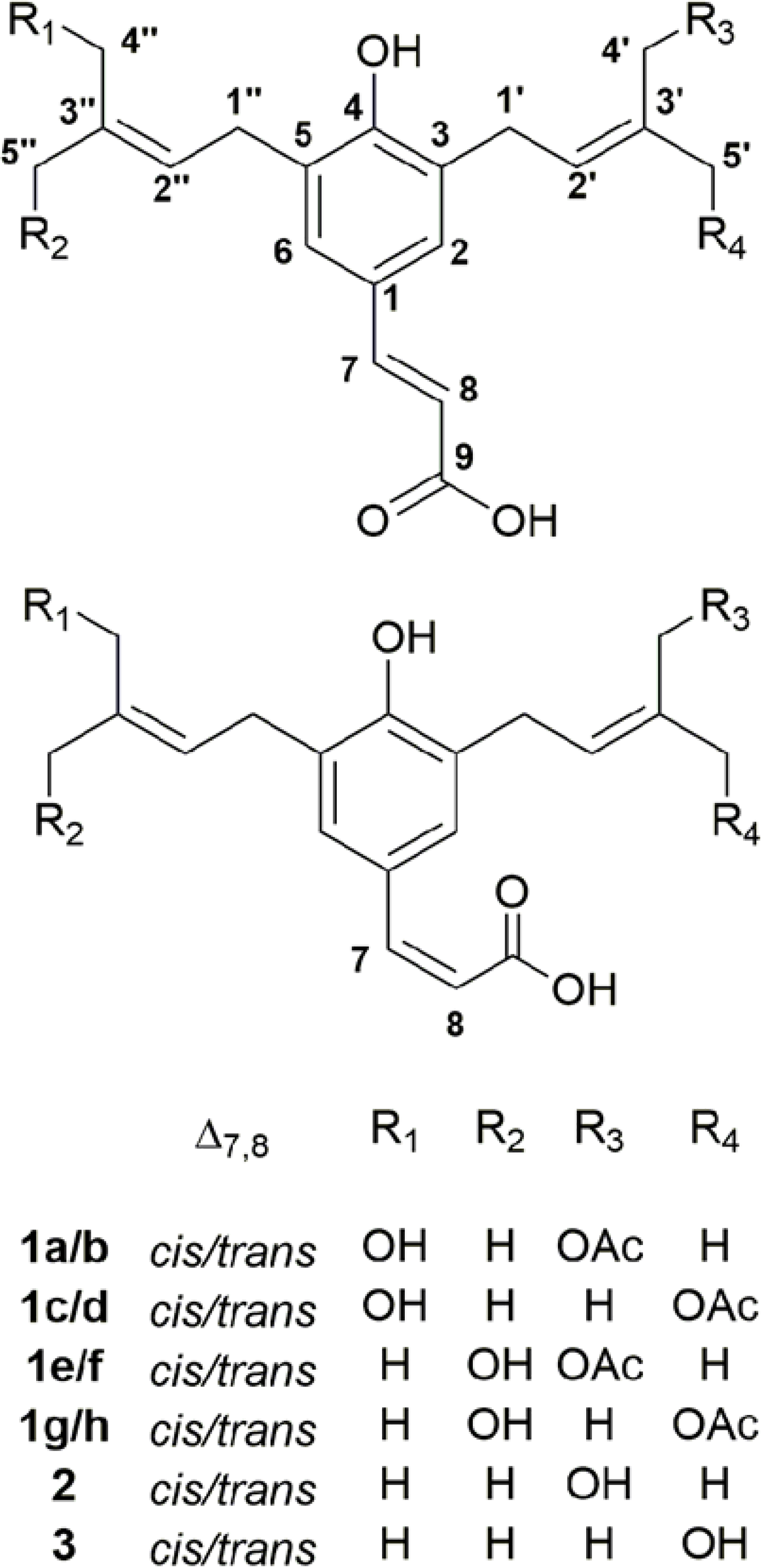
The cis-trans isomerism of the diprenylated acetoxy coumaric acids allows for eight stereoisomers of 1. The presence of such a complex diastereomeric mixture is supported by pure shift 1H NMR and LCHRMS measurements. Compounds 2 and 3 are capillartemisin A and capillartemisin B, respectively.
Bioactivity-guided fractionation is a method commonly used to identify the active constituents of complex mixtures of compounds and relies on the use of appropriate in vitro assays where small amounts of separated materials can be tested. While it can lead to bioactive markers, the inability to assess “synergistic” bioactivity is its major and widely recognized limitation. Compared to the previously published process,6 the separation and extraction process was optimized for the present study in terms of compound yield and minimization of samples to be tested. Extraction with EtOAc of the dried A. scoparia plants replaced the broader polarity EtOH extraction and its subsequent fractionation by CPC. HPLC separation of the EtOAc extract produced three fractions that contained the bioactive compounds previously described. Individual HPLC methods utilizing a chiral column were then used to produce nearly pure compounds (see discussion below about purity) from each of those fractions.6 Compounds 1–3 were obtained from fractions III and V. While fraction IV was also separated further, no resulting component exhibited adipogenic activity and, thus, no structural studies were performed on these components.
Structure Elucidation of Diprenylated Coumaric Acid Derivatives.
The crude ethyl acetate partition of the aerial parts of A. scoparia was purified by HPLC column chromatography (CC), which led to the separation of two diprenylated coumaric acids, 1a and 1b, isolated as a mixture, along with two known compounds, 2 and 3, which were characterized as capillartemisins A (2) and B (3) by 1D and 2D NMR spectroscopic data and comparison with reported spectroscopic data.12–15
Compound 1a was isolated as an amorphous solid and as a mixture with 1b. The HRESIMS (m/z 397.1616 [M+Na]+, calcd for C21H26O6Na+, 397.1622) indicated a molecular formula of C21H26O6 associated with an LC retention time of 5.8 min (Figures S1–2A and 2B, Supporting Information). The 1H NMR spectrum showed the characteristic resonances arising from a trans double bond [δH 7.453 (1H, d, J = 15.8 Hz, H-7) and 6.280 (1H, d, J = 15.8 Hz, H-8)], meta coupling in an aromatic ring [δH 7.174 (1H, d, J = 2.3 Hz, H-6) and 7.164 (1H, d, J = 2.3 Hz, H-2)], olefinic hydrogens in a diprenylated moiety [δH 5.560 (1H, br t, J = 7.7 Hz, H-2ꞌ) and 5.400 (1H, br t, J = 7.6 Hz, H-2ꞌꞌ)], and an acetyl group [δH 2.045 (3H, s, OAc-4ꞌ)]. The DEPTQ-135 spectrum exhibited 21 well-resolved signals assigned to two carbonyls C-9 (δC 173.0), COO-4’ (δC 173.1), twelve sp2 carbons C-1 (δC 128.1), C-2 (δC 128.6), C-3 (δC 129.4), C-4 (δC 155.9), C-5 (δC 129.2), C-6 (δC 129.1), C-7 (δC 144.9), C-8 (δC 118.5), C-2’ (δC 129.5), C-3’ (δC 132.2), C-2” (δC 126.8) and C-3” (δC 136.5), two methylene carbons C-1’ (δC 29.5), C-1” (δC 29.9), two hydroxylated methylene carbons C-4’ (δC 64.2), C-4” (δC 61.9), and three methyl carbons C-5’ (δC 21.7), C-5” (δC 22.1), OAc-4’ (δC 20.8). All of these data indicated that 1a is a diprenylated coumaric acid derivative. The 1D and 2D spectra (Figure S1–1, Supporting Information) confirmed key elements of the structure of 1a via key HMBC correlations (Figure 1) as follows: from H-4ꞌ to C-2’ (δC 129.5), C-3’ (δC 132.2) and C-5’ (δC 21.7), H-4ꞌꞌ to C-2” (δC 126.8), C-3” (δC 136.5) and C-5” (δC 22.1), H-1ꞌ to C-2 (δC 128.6), C-3 (δC 129.4), C-4 (δC 155.9), C-2’ (δC 129.5) and C-3’ (δC 132.2), H-1ꞌꞌ to C-4 (δC 155.9), C-5 (δC 129.2), C-6 (δC 129.1), C-2” (δC 126.8) and C-3” (δC 136.5), H-7 to C-1 (δC 128.1), C-2 (δC 128.6), C-6 (δC 129.1), C-8 (δC 118.5) and C-9 (δC 173.1). The HMBC correlation (Figure 1) from H-4ꞌ to C=O-4’ (δC 173.1) confirmed the location of the acetyl group at C-4ꞌ. The E/Z configuration of the double bond in the isoprenyl group was deduced via comparison of the 1H and 13C NMR chemical shifts of the oxygenated methylene groups with reference data,12,13 taking into account additional high-frequency shifts from acetylation. The location of the resonance of the oxygenated methine proton, H-4’, at 4.700 ppm confirmed the Z configuration.13 In the other prenyl moiety, C/H-4” also confirmed a Z configuration via their 1H and 13C NMR chemical shifts of 4.200 ppm and 62.0 ppm, respectively.12 On the basis of this collective evidence (Table 1), 1a was determined as 3-[4ꞌ-acetoxyprenyl]-5-[4ꞌꞌ-hydroxyprenyl]-7(E)-p-coumaric acid and named cis-scopa-trans-coumaricin.
Table 1.
1H and 13C NMR Spectroscopic Data of Compound 1a and 1b in CD3ODa
| 1ab |
1bb |
|||
|---|---|---|---|---|
| position | δH, (J in Hz) | δC, type | δH, (J in Hz) | δC, type |
| 1 | 128.1, C | 129.2, C | ||
| 2 | 7.164, d (2.3) | 128.6, CH | 7.213, d (1.8) | 130.5, CH |
| 3 | 129.4, C | 128.3, C | ||
| 4 | 155.9, C | 154.1, C | ||
| 5 | 129.2, C | 128.5, C | ||
| 6 | 7.174, d (2.3) | 129.1, CH | 7.319, d (1.8) | 130.5, CH |
| 7 | 7.453, d (15.8) | 144.9, CH | 6.437, d (12.6) | 136.2, CH |
| 8 | 6.280, d (15.8) | 118.5, CH | 5.814, d (12.6) | 122.6, CH |
| 9 | 173.0, C | 173.0, C | ||
| 1′ | 3.412, d (8.3) | 29.5, CH2 | 3.379, d (7.2) | 29.6, CH2 |
| 2′ | 5.560, t (7.7) | 129.5, CH | 5.544, t (7.2) | 129.9, CH |
| 3′ | 132.2, C | 131.7, C | ||
| 4′ | 4.733, s | 64.2, CH2 | 4.729, s | 64.4, CH2 |
| 5′ | 1.796, s | 21.7, CH3 | 1.771, s | 21.7, CH3 |
| 1″ | 3.421, d (8.3) | 29.9, CH2 | 3.379, d (7.2) | 29.6, CH2 |
| 2″ | 5.400, t (7.6) | 126.8, CH | 5.392, t (7.2) | 127.1, CH |
| 3″ | 136.5, C | 136.2, C | ||
| 4″ | 4.206, s | 61.9, CH2 | 4.198, s | 61.9, CH2 |
| 5″ | 1.809, s | 22.1, CH3 | 1.796, s | 22.0, CH3 |
| CO-4′ | 173.1, C | 173.1, C | ||
| OAc-4′ | 2.045, s | 20.8, CH3 | 2.045, s | 20.8, CH3 |
Assignments supported by HSQC and HMBC experiments.
900 and 225 MHz, in CD3OD.
The molecular formula of 1b (C21H26O6), which was the second component in the mixture along with 1a, was also deduced from the HRESIMS (m/z 397.1627 [M+Na]+), exhibiting the same molecular formula of C21H26O6 ([M+Na]+, calcd for C21H26O6Na+, 397.1622) as 1a, but it exhibited an LC retention time of 6.0 min. The 1H and 13C NMR data of 1b were almost identical to those of 1a. The key difference of 1b arose from a cis double bond of coumaric acid, with its olefinic hydrogens exhibiting a cis-characteristic J coupling and characteristic chemical shifts [δH 5.81 (d, J = 12.6 Hz, H-7) and δC 122.6 for H-/C-7; δH 6.44 (d, J = 12.6 Hz, H-8) and δC 136.2 for H-/C-8]. The diprenylated group could be located via the HMBC correlation from H-1’ to C-2 and C-3 (δC 130.5 and 128.3 ppm, respectively), and from H-1” to C-5 and C-6 (δC 128.5 and 130.5, respectively). The HMBC correlation (Figure 1) from H-4ꞌ to COO-4ꞌ at δC 173.1 ppm confirmed the location of the acetyl group. Further analysis of 1D and 2D NMR data (Figure S1, Supporting Information) indicated that 1b otherwise shares the identical molecular scaffold as 1a. On the basis of all of the spectroscopic evidence obtained, 1b (Table 1) was determined as 3-[4ꞌ-acetoxyprenyl]-5-[4ꞌꞌ-hydroxyprenyl]-7(Z)-p-coumaric acid and named cis-scopa-cis-coumaricin.
cis-trans Isomerism of Diprenylated Coumaric Acid.
Photochemical mechanisms are known to generate cis-trans isomerization of olefins. Other potential mechanisms are induced thermally, by acid or base catalysis, or by reaction with molecules that contain an odd number of electrons.16,17 Among these isomerism factors, photoisomerization is likely to occur spontaneously in the plant due to exposure to sunlight. As the diprenylated coumaric acids possess an α,β-unsaturation, absorption of longer wavelengths can promote trans-to-cis isomerization in these compounds.18
A general assumption in natural products is that the trans (E) isomer tends to be more stable than the cis (Z) isomer.19,20 Notably, the diprenylated coumaric acid skeleton contains a total of three α,β-unsaturated olefinic hydrogens, and each can undergo cis-trans isomerization. This leads to the plausible hypothesis that, besides the presence of the diastereomeric species 1a and 1b, additional cis-trans isomers exist in the plant, the mother fraction that contained 1a and 1b, and co-purified 1a/1b samples. The pure shift 1H NMR experiments confirmed the presence of the isomeric compounds in the sample containing the mixture of 1a/1b, exhibiting singlet peaks at the characteristic chemical shift of each isomer. The existence of these isomers was also supported by extracting isotopic ion traces from the LC-MS chromatogram (Figures 2–3 and S1, Supporting Information). The common names of the isolated new compounds describes the demonstrated cis-/trans- isomerism on the diprenylated coumaric acids. Therefore, the nomenclature of isomers of this class of diprenylated compounds follows the proposed rational naming scheme, taking into account their cis/trans isomerization properties (Figure S1, Supporting Information).
Figure 3.
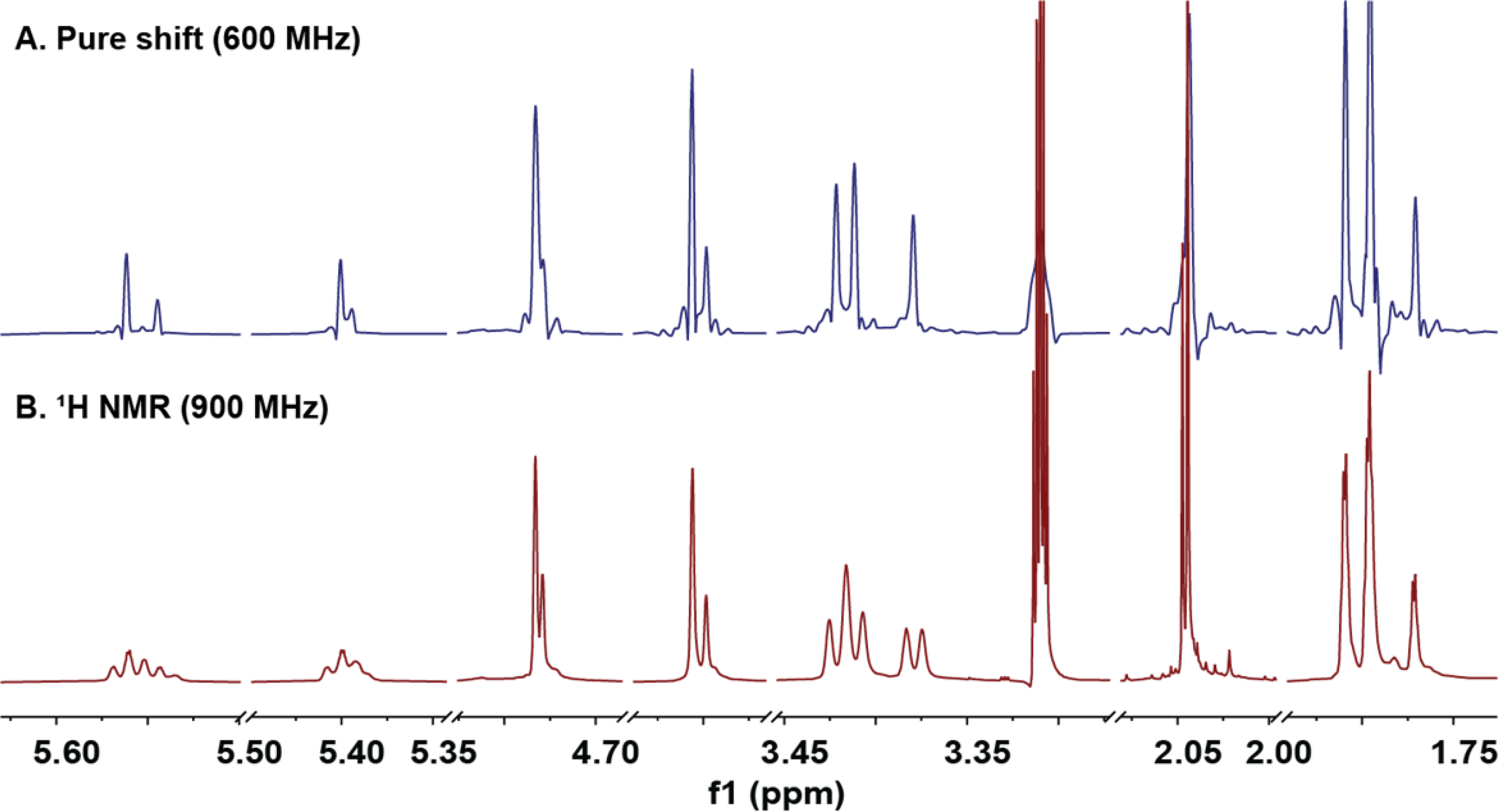
The pure shift 1D 1H NMR spectra showed the presence of the diastereomeric components within the purified mixture of 1a and 1b.
Spectroscopic Detection of Isomeric Patterns.
Representing a versatile structural tool, NMR spectroscopy is utilized widely to elucidate the structures of isolated (“pure”) natural products. While the classical 1D 1H NMR spectrum contributes essential information (number of hydrogens, chemical shifts and spin-spin coupling constants), the spectra of pure compounds are often already (over) crowded, which explains why spectra of materials that contain other, near-identical structures as mixtures often challenge NMR data interpretation. One potential solution to the signal overlap challenge is the pure shift NMR spectrum,21 which removes spin-spin coupling and, thereby, reduces the resonance of each hydrogen to a singlet. This improves the signal resolution and simplifies spectral analysis and interpretation.22 In the present case of a mixture of closely related, diastereomeric congeners (1a and 1b), the ability to distinguish molecular species is a valuable addition to the structural information obtained from coupling patterns to determine cis-trans isomerization.
As shown in Figure 3, in the regular 1D 1H NMR spectra, isomeric species of low abundance were detectable, but only as shoulders of the major signals of H-1a/b. More compelling NMR evidence for the presence of isomeric species required an increase in spectral resolution and/or dispersion. The pure shift 1H NMR spectrum of 1a and 1b thus, was obtained to suppress the spin-spin coupling patterns in the crowded spectral regions between δH 1.70 and 5.70 ppm (Figure 3). Due to the singlet nature of all resonances in pure shift spectra, distinct single peaks were observed instead of complex shoulders of the already complex, fully coupled major resonances. The results provided evidence for the presence of additional minor isomers involving diprenylated coumaric acid moieties. As shown in Figure S1–2A, Supporting Information, LC-MS analysis verified these findings via the detection of additional low-abundance isomers of 1a/1b in the extracted ion chromatogram by filtering out ions of 375±0.30 amu. The use of a window function when extracting the ion chromatogram allowed this selective filtering of ions of a specific molecular weight in the LC-MS chromatogram (Figure S1, Supporting Information). The results showed that the theoretically plausible isomers of 1a, 1b, 2, and 3 can be observed in these samples and constitutes a case of Residual Complexity (go.uic.edu/residualcomplexity). These insights could be gained despite the obvious limitation of their quantities due to significant differences in their LC retention time (as shown for 1a vs. 1b, vide infra), confirmed by their identical molecular formula (Figure S1, Supporting Information).
Purity Analysis Using the 100% PF-qNMR Method.
The pure shift NMR data confirmed the presence of other isomeric diprenylated coumaric acids, which gave rise to additional minor NMR signals resonating close to those of 1a and 1b. The pure shift experiments also revealed that the presence of minor isomers were obscured under the “shoulders” of the main resonances of 1a and 1b. Applying the peak-fitting (PF) approach using peak deconvolution, PF-qNMR enabled the disentangling of the overlapped resonances for the purpose of their relative quantitation (Figure S1, Supporting Information).23 Subsequent application of the 100% qNMR method led to the measurement of the relative ratio of 1a and 1b to be calculated as 71.6% ± 5.70 and 28.4% ± 1.86, respectively (Table 2). The error of relative quantitation is due to the presence of residual amounts of the minor cis-trans isomers, which could be confirmed by peak deconvolution.
Table 2.
Relative Ratios of 1a and 1b, Calculated Using the 100% Quantitative NMR Method with Normalized Integration Values.
| hydrogen | 1a | 1b |
|---|---|---|
| 7 | 100.00 | 38.56 |
| 6 | 98.95 | 38.13 |
| 2 | 106.38 | 40.35 |
| 8 | 94.54 | 38.74 |
| 4′ | 85.57 | 42.18 |
| 4″ | 88.62 | 35.38 |
| OAc-4′ | 104.00 | 35.16 |
| normalized integral value | 96.87 | 38.36 |
| calculated purity (%) | 71.63 | 28.37 |
| ±SD | 5.70 | 1.86 |
Compounds from A. scoparia Promote Adipogenesis.
Bioactivity-guided fractionation studies were previously used to identify several fractions from SCO capable of promoting adipocyte differentiation, as assessed by enhanced accumulation of lipid and increased expression of adipogenic genes in differentiating 3T3-L1 adipocytes.5,6 Six distinct sub-fractions were isolated from these fractions as “single” compounds in order to assign the bioactivity to individual compounds (Figure 4). Oil Red O staining was used to examine neutral lipid accumulation in 3T3-L1 cells treated with the purified sub-fractions (Figures 4A and 4B) relative to the total extract SCO (50 μg/mL), rosiglitazone (ROSI)(2 μM) as the positive control, and DMSO as the negative control. The sub-fractions III-1 (2), III-2 (1a/b) and V-1 (3) showed enhanced lipid accumulation similar to SCO and rosiglitazone over most of the concentrations tested (2.5, 10, and 25 μM). The lowest dose for the sub-fractions was chosen to be similar to the concentration of the rosiglitazone positive control. Compound 2 was active in a dose-dependent manner, while 1a/b and 3 showed the best activity over the tested dose range.
Figure 4.
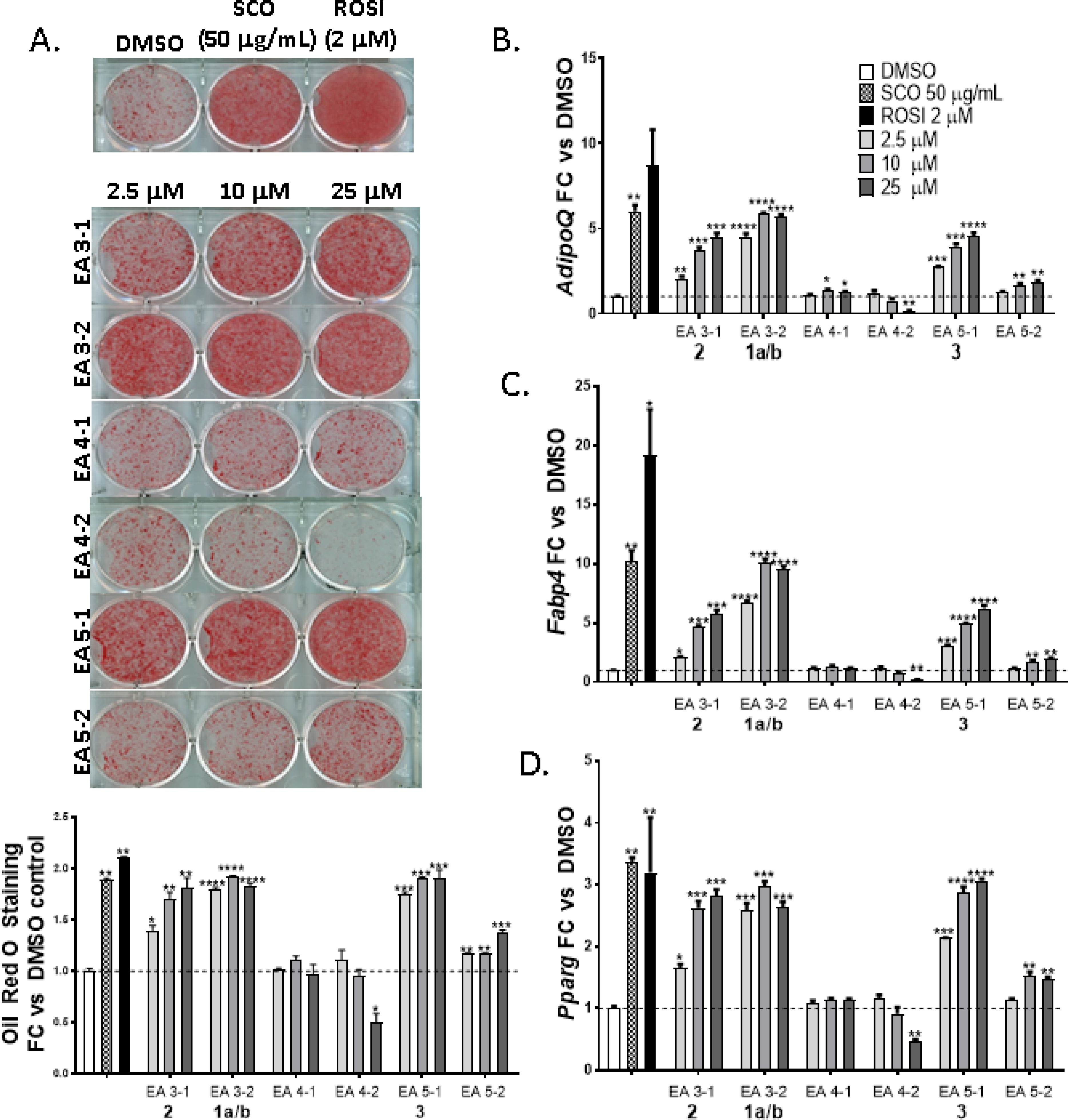
Compounds 1a/b, 2 and 3 from A. scoparia promote adipogenesis: 3T3-L1 cells were induced to differentiate using half-strength MDI cocktail containing DMSO vehicle, 50 μg/mL SCO, 2 μM ROSI, or one of three doses of test compounds (2.5, 10, or 25 μM). Cells were then either fixed and stained with Oil Red O (4 days post-differentiation) (panel A), or harvested (3 days post differentiation) for RNA isolation and gene expression analysis of adiponectin (AdipoQ), fatty acid binding protein 4 (Fabp4), or Pparγ (panels B-D). All treatments were compared to their respective DMSO controls. The effects of SCO and ROSI were analyzed by t-tests, while the effects of individual test compounds were analyzed by one-way ANOVA. *p >0.05, ** p <0.01, *** p <0.001, **** p < 0.0001 vs. DMSO controls.
Compounds 1a/b-3 also increased the expression of adipogenic genes for adiponectin (AdipoQ), fatty acid binding protein 4 (Fabp4), and peroxisome proliferation activated-receptor gamma (PPARγ) in 3T3-L1 adipocytes relative to the negative controls (Figures 4C–4E). As expected, the rosiglitazone control had much higher activity for Fabp4 than any A. scoparia compounds or the SCO extract, as rosiglitazone is a potent PPARγ agonist drug used for treating insulin resistance. Sub-fraction V-2 showed enhanced adipogenic activity for each of these assays, but was consistently weak relative to 1a/b-3, and barely above the DMSO control. Sub-fractions IV-1 and IV-2 had no effect on any of the measured parameters for adipogenesis, while IV-2 inhibited lipid accumulation and adipogenic gene expression at the highest dose.
Compounds from A scoparia Inhibit TNFα-induced Lipolysis.
The chromatographic sub-fractions were also tested in 3T3-L1 adipocytes relative to the parent extract for their ability to inhibit TNFα-induced lipolysis. This extract was previously shown to reduce lipolysis in vitro (TNFα-induced lipolysis in cultured adipocytes) and in vivo (mice fed a high-fat diet with SCO supplementation) bioassays.4 In order to determine whether the SCO extract constituents with pro-adipogenic activity in differentiating adipocytes also have anti-lipolytic effects in mature adipocytes, mature 3T3-L1 adipocytes were pretreated with each of the six sub-fractions and then with TNFα to induce lipolysis. Lipolysis was measured on the basis of glycerol release, which is enhanced by insulin resistance and TNFα treatment of cell cultures. The SCO extract was able to reduce TNFα-induced lipolysis to basal levels as demonstrated previously, but only at highest concentration did 1a/b show any statistically significant inhibitory activity (Figure 5). Compounds 2 and 3 and the other sub-fractions that demonstrated adipogenic activity were not able to significantly mitigate TNFα-induced lipolysis at the concentrations tested. The low lipolysis inhibitory activity of the pure compounds relative to the robust activity of the parent plant extract SCO (as well as the EtOAc extract, EA, not shown) suggests that the antilipolytic activity of the parent extract results not only from the current isolates, but also from other compound(s) that did not fractionate with the adipogenic activity followed here. In fact, the observation of this kind of “bioactivity gap” is a common outcome of bioassay-guided botanical studies and a strong indicator of botanical polypharmacology, as shown recently for hops (Humulus lupulus).24,25 While the process of bioactivity-guided fractionation was not guided by antilipolytic activity, it was used to characterize the parent extract.
Figure 5.
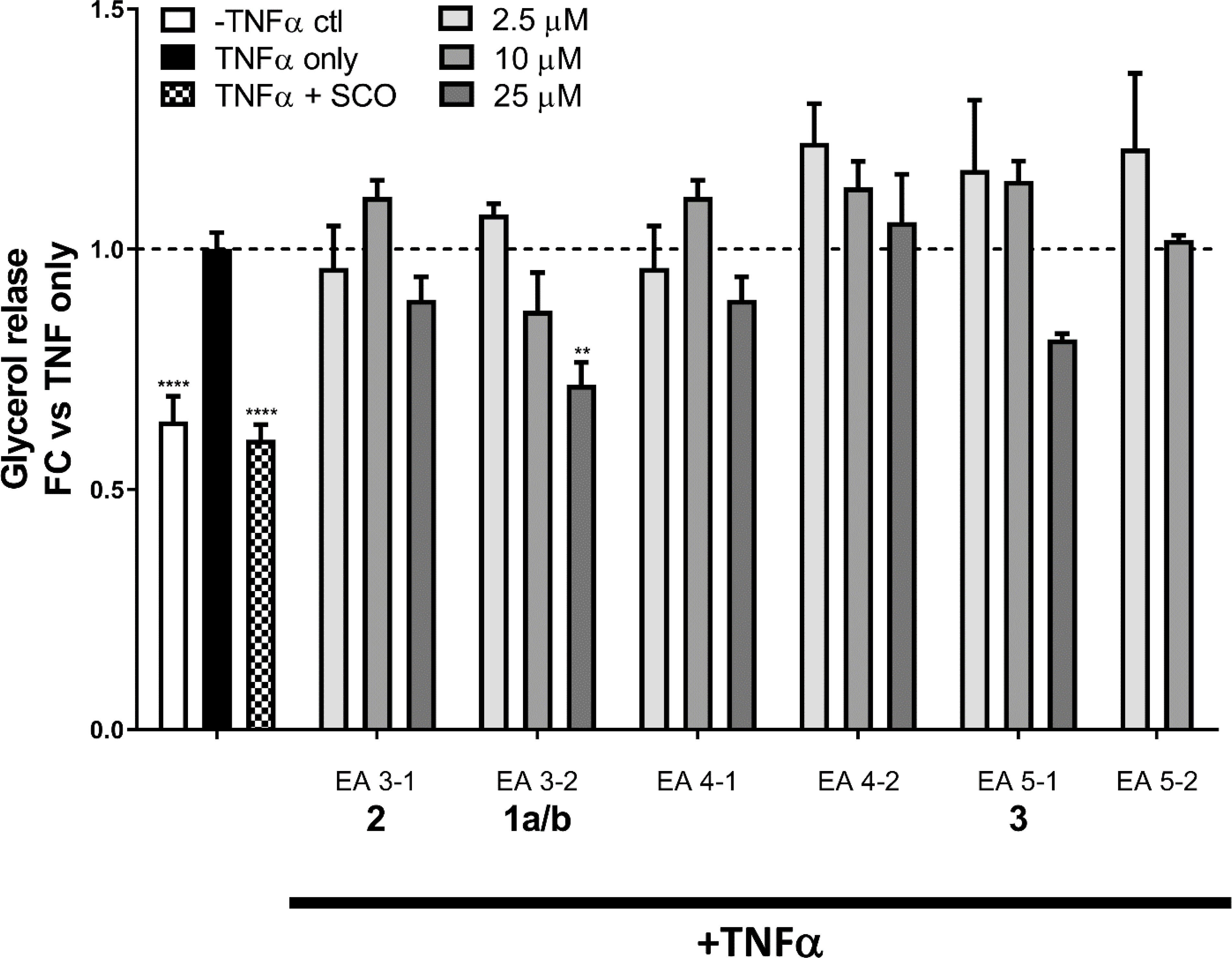
Compounds from A. scoparia inhibit TNFα-induced lipolysis. Fully differentiated 3T3-L1 adipocytes pretreated for three days with 50 μg/mL the plant crude extract (SCO) or varying doses of test compounds (2.5, 10, or 25 μM), then overnight with 0.75 nM TNFα (or vehicle). The next morning, the culture medium was replaced with a lipolysis incubation medium containing 0.75 nM TNFα (or vehicle). After 4 h, the conditioned medium was collected and assayed for glycerol. Data are expressed as fold-change vs. TNFα-only treatment. Data from no-TNFα controls and each test compound were analyzed by one-way ANOVA. ** p <0.01, **** p <0.0001 vs. TNFα-only condition.
In conclusion, this study led to the characterization of bioactive marker compounds that are associated with the adipogenic bioactivity observed from an 80% EtOH extract of A. scoparia (SCO). One of the compounds was identified as a new pair of prenylated cinnamic acid isomers, cis-scopa-trans-coumaricin and cis-scopa-cis-coumaricin (1a/b). The others were prenylated coumaric acid isomers previously designated as capillartemisin A (2) and B (3). These compounds share the cinnamic acid core and diprenyl substitution in both meta positions, which could suggest this motif as being the core of the pharmacophore. The adipogenic bioactivities used for the initial characterization of the extract could be replicated for the isolates. However, these compounds were unable to reduce lipolysis like the SCO extract, which indicates the presence of other bioactive compounds within the extract. Potential compound synergy is a plausible hypothesis and could be addressed in the future, e.g., by using biological testing of combinations of the compounds or perhaps via DESIGNER knock-out extracts for the selective removal of putative bioactives from the complex mixture, as has recently been demonstrated with another Artemisia extract.26 Collectively, despite the success in linking a new chemical entity to the adipogenic activity of SCO, the present outcomes underscore the complexity of botanical bioactive principles, the likelihood of polypharmacological action, and the limitations of reductionist approaches in botanical research.
EXPERIMENTAL SECTION
General Experimental Procedures.
The samples were dissolved in 200 μL of methanol-d4, then transferred into a 3 mm NMR tube. 1D and 2D NMR spectra were acquired at 298 K using a Bruker AVANCE I 900/225 NMR spectrometer. The spectrometer was equipped with a 5-mm Bruker TCI triple resonance, inverse-detection cryoprobe with a z-axis pulse field gradient. The pure shift spectroscopic data were acquired at 297.9 K using an AVANCE III NMR spectrometer equipped with an inverse-detection C/H Cryoprobe Bruker 600 MHz. Processing was accomplished using the Mnova software package (v.14.1.1, Mestrelab Research S.L., A Coruña, Spain). 1H NMR spectra were processed with the following parameters: GM (LB −0.3 Hz, GB 0.5), zero filling to SI = 256 k and with the application of automatic phasing, the pure shift spectra were processed with the following parameter: GM (LB −0.3 Hz, GB 0.5, zero fillings to SI = 64k with a linear prediction of MIST method application of automatic phasing, using Mnova software. The integral values were obtained after using a fifth order polynomial fit baseline correction.
Mass spectrometric analyses were carried out using a Bruker Impact II, quadrupole time-of-flight (Bremen, Germany) coupled to a Shimadzu Nexera X2 UHPLC system (Kyoto, Japan). Data analyses were performed on the Compass Data Analysis software (Bruker Version 4.4). The instrument was equipped with an electrospray source, using the positive-ion mode with a capillary voltage at 4.5 kV. The nebulizer and drying gas (N2) at 3.0 bar and 12.0 L/min, respectively, and a dry temperature of 225 °C, was used with the mass scan range set from m/z 200 to 800. Also, the negative ion electrospray ionization mode was employed, with a capillary voltage at −2.5 kV, with the nebulizer and drying gas (N2) at 4.0 bar and 12.0 L/min, respectively, a dry temperature of 225 °C, was used with the mass scan range set from m/z 200 to 800. The separation was performed on a CORTECS C18 (100 × 3.0 mm, 2.7 μm) UHPLC column.
Plant Material.
Artemisia scoparia was grown in a Rutgers University greenhouse facility in New Brunswick, NJ (40°28′41.9″ N 74°26′15.7″ W) and the whole aerial parts of the plant at the flowering stage were periodically harvested for the production of extract. Some plants were left to produce seed to maintain the seed source for future cultivation. Voucher specimens are retained at Rutgers University Chrysler Herbarium under the CHRB accession number 154645.
Extraction and Isolation.
A. scoparia extract (SCO) was prepared from greenhouse-grown plants at Rutgers University as described previously.3–5 Briefly, the plant was freeze-dried and extracted in 80% EtOH (1∶20 w/v). The preparation of distinct chromatographic fractions from SCO was previously described in detail. Each of the fractions consisted of compounds that were not sufficiently purified for structural characterization but maintained adipogenic activities were originally characterized from the SCO extract. The fractions were created using multiple chromatographic techniques including solvent partitioning, CPC and HPLC.6
The chromatographic response of the active compounds obtained from the enriched fractions of the SCO extract was used to enhance the purification process. Thus, the A. scoparia plants were freeze-dried, ground into a powder and extracted directly with EtOAc (1:20 w/v), sonicated for 1 h, and incubated at 22 ˚C for 24 h. Solids were removed by filtration, and the extract was dried by rotary evaporation. The yield of this crude extract was 4.6% from the dried plant.
The freeze-dried extract was dissolved in 90% EtOH (25 mg/mL) for initial HPLC separation. The sample was purified on a semi-preparative HPLC system consisting of Waters™ Alliance e2695 separations module and 2998 photodiode array detector with a Phenomenex Synergi 4 μm 80 Å Hydro-RP column, 250 × 21.2 mm. The mobile phases consisted of two components: Solvent A (0.1% ACS grade acetic acid in double-distilled deionized H2O) and Solvent B (CH3CN). The separation was completed using a linear gradient run of 40% B in A to 60% B over 20 min at a flow rate of 15 mL/min, followed by reconditioning of the column with 100% B for 5 min and return to initial conditions. Fractions III, IV and V were collected manually at 14–16 min, 16–17 min and 17–18 min, respectively. The yields of fractions III and V were 9.3% and 5.3%, respectively, from the total extract injected. Each fraction was dried by rotary evaporation.
Fraction III was separated and purified using the same HPLC system described above with a Phenomenex Lux® 5 μM Cellulose-2 chiral column, 250 × 10 mm. The mobile phases consisted of: Solvent A (0.1% ACS grade formic acid in double-distilled deionized H2O), and Solvent B (CH3CN). The separation was completed using a linear gradient run of 35% B in A to 65% B over 60 min at a flow rate of 5 mL/min, followed by reconditioning of the column with 100% B for 10 min and return to initial conditions. Fractions III-1 and III-2 were collected at 14–18 min and 21 −25 min, respectively. Fractions III-1 and III-2 were analyzed by LC-MS for exact mass and identified using NMR spectroscopy.
Fraction IV was separated using the same HPLC system as fraction III. However, the separation was completed using a different linear gradient as 45% B in A to 60% B over 30 min, then to 100% B at 35 min at a flow rate of 5 mL/min, followed by reconditioning of the column with 100% B for 10 min and return to initial conditions. Fractions IV-1 and IV-2 were collected at 36–39 min and 39–41 min, respectively.
Fraction V was separated using the same HPLC system as fraction III with a linear gradient 40% B in A to 50% B over 20 min at a flow rate of 5 mL/min, followed by reconditioning of the column with 100% B for 10 min and return to initial conditions. Fractions V-1 and V-2 were collected at 12–14 min and 29–30 min.
UPLC/MS Analysis of A. scoparia Fractions.
Compounds in samples were separated and analyzed during the process of fractionation by a UPLC/MS system (Figures S1–S5, Supporting Information) including the Dionex® UltiMate 3000 RSLC ultra-high pressure liquid chromatography system, consisting of a workstation with the ThermoFisher Scientific’s Xcalibur version 4.0 software package combined with Dionex® SII LC control software, a solvent rack/degasser SRD-3400, a pulseless chromatography pump HPG-3400RS, an autosampler WPS-3000RS, a column compartment TCC-3000RS, and a photodiode array detector DAD-3000RS. After the photodiode array detector, the eluent flow was guided to a Q Exactive Plus Orbitrap high-resolution high-mass-accuracy mass spectrometer (MS). Mass detection was full MS scan with low-collision energy-induced dissociation (CID) from m/z 100 to 1000 in either the positive-ion or negative-ion ionization mode with an electrospray ionization (ESI) interface. The sheath gas flow rate was 30 arbitrary units, the auxiliary gas flow rate was 7, and the sweep gas flow rate was 1. The spray voltage was 3500 volts (−3500 for negative ESI) with a capillary temperature of 275 °C. The mass resolution was 70,000 or higher. Substances were separated on a Phenomenex™ Kinetex C8 reversed-phase column (100 × 2 mm, 2.6 μm particle size, and 100 Å pore size). The mobile phase consisted of two components: Solvent A (0.5% ACS grade acetic acid in LCMS grade water, pH 3–3.5), and Solvent B (100% acetonitrile, LCMS grade). The mobile phase flow was 0.20 mL/min, and a gradient mode was used for all analyses. The initial conditions of the gradient were 95% A and 5% B; for 30.0 min the proportion reached 5% A and 95% B, which was kept for the next 8.0 min, and during the following 4 min the ratio was brought to initial conditions. An equilibration interval of 8.0 min was included between subsequent injections. The average pump pressure using these parameters was typically around 3900 psi for the initial conditions. Putative formulas of natural products were determined by performing isotope abundance analysis on the high-resolution mass spectrometric data with Xcalibur v. 4.0 software and reporting the best fitting empirical formula. Database searches were performed using www.reaxys.com (Elsevier RELX Intellectual Properties SA) and SciFinder (scifinder.cas.org, American Chemical Society).
Compound 1a and 1b:
amorphous solid; UV (MeOH) λmax 220 (2.49), 238 (2.48), 303 (2.22), 314 (2.55) nm; 1H NMR (900 MHz, methanol-d4) and 13C NMR (225 MHz, methanol-d4) data, see Table 1; HRMS m/z 397.1616 for 1a and 397.1627 for 1b [M+Na]+ (calcd for C21H25O6Na+, 397.1622) (Figures S1 and S2, Supporting information).
Cell Culture and Treatments.
3T3-L1 preadipocytes (murine) were cultured as previously described,4 and induced to differentiate two days after reaching confluence. For experiments using mature adipocytes, cells were induced with a standard methylxanthine-dexamethasone-insulin (MDI) cocktail, which contained 0.5 mM isobutylmethylxanthine (IBMX), 1 μM dexamethasone (DEX), and 1.72 μM insulin in high-glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). IBMX, DEX, insulin, and DMEM were obtained from Sigma-Aldrich (St. Louis, MO, USA), and FBS from Hyclone (GE Healthcare Life Sciences, Logan, UT, USA). For differentiation experiments, cells were induced with a half-strength MDI cocktail. In both cases, cells were fed with high-glucose DMEM + 10% FBS with 0.43 μM insulin three days following MDI induction. Test compounds were dissolved in dimethylsulfoxide (DMSO) as 1,000 × stocks. For differentiation experiments, cells were treated with the test compounds (or DMSO vehicle as a control) at the time of induction and at the first feeding thereafter (3 days post-MDI). For lipolysis experiments on mature adipocytes, cells were fed with high-glucose DMEM + 10% FBS 6 days after MDI and treated with test compounds or DMSO vehicle for three days. On the third day, cells were also treated overnight with 0.75 nM tumor necrosis factor alpha (TNFα) (Life Technologies, Carlsbad, CA, USA) or its vehicle [0.1% bovine serum albumin (BSA) in phosphate buffered saline (PBS)]. The following morning, the culture medium was replaced with lipolysis incubation medium (low-glucose DMEM + 2% BSA) and 0.75 nM TNFα (or vehicle). After 4 h, the conditioned medium and cell lysates were collected.
Lipid Accumulation Assay.
Five days after MDI induction, cell monolayers were fixed in 10% neutral buffered formalin (ThermoFisher, Waltham, MA, USA) and stained with the neutral lipid stain, Oil Red O (ORO). Plates were scanned to generate images of staining. The ORO was then eluted in isopropyl alcohol, and absorbance of eluates was measured at 520 nm for quantitation.
RNA Purification and Gene Expression.
Four days after MDI induction, cells were harvested and RNA purified using the RNeasy Mini kit (Qiagen, Hilden, Germany). Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s protocol. Gene expression assays using primers from Integrated DNA Technologies (Skokie, IL). Primer sequences are shown in Table 1. SYBR Premix (Takara Bio, Mountain View, CA, USA) was used for quantitative polymerase chain reaction (qPCR), performed on the Applied Biosystems 7900HT system. Data analysis used SDS 2.3 software. Target gene data were normalized to the reference gene, non-POU-domain-containing, octamer binding protein (Nono). All primer sequences are shown in Table 3.
Table 3.
Primer Sequences for Gene Expression Analysis.
| gene name and symbol | forward primer, 3′-5′ | reverse primer, 3′-5′ |
|---|---|---|
| adiponectin (Adipoq) | TGTCTGTACGATTGTCAGTGG | GCAGGATTAAGAGGAACAGGAG |
| adipocyte protein 2 (aP2)/Fatty acid binding protein 3 (Fabp4) | CCCTCCTGTGCTGCAGCCTTTC | GTGGCAAAGCCCACTCCCACTT |
| peroxisome proliferator-activated receptor gamma (Ppparg) | TCACAAGAAATTACCATGGTTGACA | CGAGTGGTCTTCCATCACGG |
| non-POU-domain-containing, octamer binding protein (Nono) | CATCATCAGCATCACCACCA | TCTTCAGGTCAATAGTCAAGCC |
Glycerol Assay.
Aliquots of 50 μL of conditioned medium from treated adipocytes were analyzed for glycerol content using the Free Glycerol Reagent (Sigma-Aldrich), following the manufacturer’s protocol. Glycerol concentrations were normalized to protein concentrations of the samples’ respective cell lysates, as measured by bicinchoninic acid (BCA) assay (Sigma-Aldrich).
Statistics.
All statistics were performed using GraphPad Prism 6 software (La Jolla, CA, USA). Tests performed are described in the figure legends.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Office of Dietary Supplements and the National Center for Complementary and Integrative Health, both of the National Institutes of Health (NIH), under the two awards; P50AT002776, funding the Botanical Dietary Supplements Research Center of Pennington Biomedical Research Center and the Department of Plant Biology and Pathology in the School of Environmental and Biological Sciences (SEBS) of Rutgers University, and U41 AT008706, supporting the CENAPT at the University of Illinois at Chicago. The authors are also grateful for support by the New Jersey Agricultural Experiment Station (NJAES) at Rutgers University. Additional support was provided by the Fogarty International Center of NIH award U01 TW006674.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at the ACS publications website at DOI and include: 1H NMR spectrum of compounds 1a and 1b in CD3OD at 900 MHz; DEPTQ-135 spectrum of compounds 1a and 1b in CD3OD at 900 MHz; COSY spectrum of compounds 1a and 1b in CD3OD at 900MHz; HSQC spectrum of compounds 1a and 1b in CD3OD at 900 MHz; HMBC spectrum of compounds 1a and 1b in CD3OD at 900 MHz, LC-MS chromatogram of compounds 1a and 1b; LC-MS chromatogram of capillartemisin A; LC-MS chromatogram of capillartemisin B; table of a proposed rational nomenclature for the prospective naming scheme of diprenylated coumaric acids; deconvolution of pure shift spectroscopic data of compounds 1a and 1b.
Contributor Information
David Ribnicky, Department of Plant Biology, Rutgers University, New Brunswick, New Jersey 08901, United States.
Seon Beom Kim, Center for Natural Product Technologies, Pharmacognosy Institute, and Department of Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Illinois 60612, United States.
Alexander Poulev, Department of Plant Biology, Rutgers University, New Brunswick, New Jersey 08901, United States.
Yang Wang, Department of Plant Biology, Rutgers University, New Brunswick, New Jersey 08901, United States.
Anik Boudreau, Pennington Biomedical Research Center, Louisiana State University System, Baton Rouge, Louisiana 70808, United States.
Ilya Raskin, Department of Plant Biology, Rutgers University, New Brunswick, New Jersey 08901, United States.
Jonathan Bisson, Center for Natural Product Technologies, Pharmacognosy Institute, and Department of Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Illinois 60612, United States.
G. Joseph Ray, Pharmacognosy Institute and Department of Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Illinois 60612, United States.
Shao-Nong Chen, Center for Natural Product Technologies, Pharmacognosy Institute, and Department of Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Illinois 60612, United States.
Allison Richard, Pennington Biomedical Research Center, Louisiana State University System, Baton Rouge, Louisiana 70808, United States.
Jacqueline M. Stephens, Pennington Biomedical Research Center, Louisiana State University System, Baton Rouge, Louisiana 70808, United States.
Guido F. Pauli, Center for Natural Product Technologies, Pharmacognosy Institute, and Department of Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Illinois 60612, United States.
REFERENCES
- (1).Yahagi T; Yakura N; Matsuzaki K; Kitanaka S J. Nat. Med. 2014, 68, 414–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Dey M; Ripoll C; Pouleva R; Dorn R; Aranovich I; Zaurov D; Kurmukov A; Eliseyeva M; Belolipov I; Akimaliev A; Sodombekov I; Akimaliev D; Lila MA; Raskin I Phytother. Res. 2008, 22, 929–934. [DOI] [PubMed] [Google Scholar]
- (3).Richard AJ; Burris TP; Sanchez-Infantes D; Wang Y; Ribnicky DM; Stephens JM Nutrition 2014, 30, S31–S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Richard AJ; Fuller S; Fedorcenco V; Beyl R; Burris TP; Mynatt R; Ribnicky DM; Stephens JM PLoS One 2014, 9, e98897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Boudreau A; Richard AJ; Burrell JA; King WT; Dunn R; Schwarz J-M; Ribnicky DM; Rood J; Salbaum JM; Stephens JM Am. J. of Physiol.-Endocrin. Metab. 2018, 315, E1053–E1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Boudreau A; Poulev A; Ribnicky DM; Raskin I; Rathinasabapathy T; Richard AJ; Stephens JM Front. Nutr. 2019, 6, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gustafson B; Hedjazifar S; Gogg S; Hammarstedt A; Smith U Trends Endocrinol. Metab. 2015, 26, 193–200. [DOI] [PubMed] [Google Scholar]
- (8).Smith U; Kahn BB J. Int. Med. 2016, 280, 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Danforth E Nat. Genet. 2000, 26, 13–13. [DOI] [PubMed] [Google Scholar]
- (10).Kim J-Y; van de Wall E; Laplante M; Azzara A; Trujillo ME; Hofmann SM; Schraw T; Durand JL; Li H; Li G; Jelicks LA; Mehler MF; Hui DY; Deshaies Y; Shulman GI; Schwartz GJ; Scherer PE J. Clin. Invest. 2007, 117, 2621–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cignarelli A; Giorgino F; Vettor R Arch. Physiol. Biochem. 2013, 119, 139–150. [DOI] [PubMed] [Google Scholar]
- (12).Kitagawa I; Fukuda Y; Yoshihara M; Yamahara J; Yoshikawa M Chem. Pharm. Bull. 1983, 31, 352–355. [DOI] [PubMed] [Google Scholar]
- (13).Jakupovic J; Tan RX; Bohlmann JZJ; Huneck S Phytochemistry 1991, 30, 1645–1648. [Google Scholar]
- (14).Huneck S; Zdero C; Bohlmann F Phytochemistry 1986, 25, 883–889. [Google Scholar]
- (15).Okuno I; Uchida K; Nakamura M; Sakurawi K Chem. Pharm. Bull. 1988, 36, 769–775. [DOI] [PubMed] [Google Scholar]
- (16).Simmler C; Lankin DC; Nikolić D; van Breemen RB; Pauli GF Fitoterapia 2017, 121, 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sanchez AM; Barra M; de Rossi RH J. Org. Chem. 1999, 64, 1604–1609. [DOI] [PubMed] [Google Scholar]
- (18).Dugave C; Demange L Chem. Rev. 2003, 103, 2475–2532. [DOI] [PubMed] [Google Scholar]
- (19).Caccamese S; Azzolina R; Davino M Chromatographia 1979, 12, 545–547. [Google Scholar]
- (20).Kort R; Vonk H; Xu X; Hoff WD; Crielaard W; Hellingwerf KJ FEBS Lett. 1996, 382, 73–78. [DOI] [PubMed] [Google Scholar]
- (21).Zangger K Prog. Nucl. Magn. Reson. Spectrosc. 2015, 86–87, 1–20. [DOI] [PubMed] [Google Scholar]
- (22).Castañar L Mag. Reson. Chem. 2018, 56, 874–875. [DOI] [PubMed] [Google Scholar]
- (23).Phansalkar RS; Simmler C; Bisson J; Chen S-N; Lankin DC; McAlpine JB; Niemitz M; Pauli GF J. Nat. Prod. 2017, 80, 634–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bolton JL; Dunlap TL; Hajirahimkhan A; Mbachu O; Chen S-N; Chadwick L; Nikolic D; van Breemen RB; Pauli GF; Dietz BM Chem. Res. Toxicol. 2019, 32, 222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dietz BM; Chen S-N; Alvarenga RFR; Dong H; Nikolić D; Biendl M; van Breemen RB; Bolton JL; Pauli GF J. Nat. Prod. 2017, 80, 2284–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yu Y; Simmler C; Kuhn P; Poulev A; Raskin I; Ribnicky D; Floyd ZE; Pauli GF J. Nat. Prod. 2019, 82, 3321–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.