

Abstract
To define the role of glutathione peroxidase (GPx) in modulating the oxygen radical-related cytotoxicity of doxorubicin and H2O2 in cells that overexpress P-glycoprotein (Pgp), the GPx activity of NCI/ADR-RES cancer cells was altered by growth in 0.5% serum with (MR-30 subline) or without (MR-0 subline) selenium supplementation. GPx activity increased from 2.2 nmol/min/mg (MR-0) to 22.5 nmol/min/mg (MR-30) when cells were grown in 30-nM selenium, p < .01; the activities of other antioxidant enzymes were unchanged by selenium. By reverse transcriptase polymerase chain reaction, MR-30 and MR-0 cells expressed similar levels of the MDR1, GPx-1, BCL2 and TOP2A mRNA. The IC50 concentration for H2O2 in MR-0 cells was 10-fold lower than in the MR-30 subline, p < .01. Despite identical anthracycline accumulation and efflux in these two lines that expressed equivalent levels of Pgp, the doxorubicin IC50 decreased fivefold in MR-0 versus MR-30 cells, p < .01. Log-linear tumour cell killing by doxorubicin was observed only in selenium-deficient MR-0 cells. Doxorubicin exposure also produced substantially more apoptosis in MR-0 than MR-30 cells; this was not related to the presence of selenium per se. MR-0 cells generated ≈5-times more methane from dimethyl sulfoxide (a measure of reactive oxygen metabolism) than MR-30 cells in the presence of equimolar doxorubicin concentrations (p < .05). These studies suggest that GPx-mediated detoxification of peroxides can modulate the antitumor activity of doxorubicin in the presence of high levels of Pgp.
Keywords: Cancer, doxorubicin, glutathione peroxidase, hydrogen peroxide, reactive oxygen
Introduction
Molecular mechanisms of doxorubicin sensitivity and resistance that have been defined for tumour cells propagated in tissue culture include overexpression of the MDR1 gene, overexpression of antiapoptotic genes and alterations in the function or level of topoisomerase II [1]. In addition to the interaction of the anthracycline antibiotics with these cellular constituents, the doxorubicin quinone moiety undergoes futile cycles of flavin dehydrogenase-catalyzed reduction and oxidation in essentially every cellular compartment that generate H2O2, lipid hydroperoxides and strong oxidant species, such as the hydroxyl radical [2], both in vitro and in vivo [3].
While the biochemistry of doxorubicin redox cycling has been well-described, and its role in the mechanism of doxorubicin cardiac toxicity has been accepted [1], the question remains whether enhanced oxygen radical detoxification could contribute to acquired tumour cell resistance for this drug [4,5]. Most of the evidence addressing this issue has been derived from analysis of tumour cell lines developed by stepwise selection in vitro with increasing doxorubicin concentrations. In such experiments, MDR1 overexpression or alterations in topoisomerase II have not infrequently been associated with increases in cytoplasmic glutathione (GSH) or glutathione peroxidase (GPx) levels [6,7].
Because the selenoprotein GPx is a critical intracellular enzyme capable of utilising reduced glutathione to detoxify H2O2 or lipid hydroperoxides to water or lipid alcohols, respectively [8], it plays a central role in decreasing the production of strong oxidant species with the chemical characteristics of the hydroxyl radical by modulation of the Fenton reaction: Fe2+ + H2O2 → Fe3+ + OH− + •OH. Since reactive oxygen species produce a multiplicity of pathophysiologic effects, including altered mitochondrial energy metabolism and impaired calcium homeostasis [9], and appear to play a critical role in several signal transduction and apoptotic pathways [10], enhanced levels of intracellular antioxidant proteins, such as GPx, could complement transport-dependent (MDR1) or DNA replication-related (topoisomerase II) mechanisms of doxorubicin resistance.
In the experiments reported here, we examined whether decreased GPx activity produced by selenium supplementation could alter tumour cell killing, apoptosis and oxygen radical production by doxorubicin in the NCI/ADR-RES cell line. Because these cells overexpress both MDR1 and GPx-1 [11] and possess altered topoisomerase II activity [12], they provide an appropriate model system with which to evaluate whether changing GPx specific activity could affect doxorubicin-induced tumour cell killing. We found that decreasing GPx levels, without producing other changes in antioxidant defense, doxorubicin accumulation and efflux, or apoptosis-related gene expression, significantly sensitised NCI/ADR-RES cells to both the cytotoxic and apoptotic effects of doxorubicin-enhanced oxygen radical metabolism.
Materials and methods
Materials
Doxorubicin hydrochloride of clinical grade was purchased from commercial sources; 5-iminodaunorubicin was supplied by the Drug Synthesis and Chemistry Branch, Division of Cancer Treatment and Diagnosis, National Cancer Institute, Bethesda, MD. In all experiments, anticancer quinones were protected from light during use. Reduced glutathione (GSH), glutathione reductase type III, 5′−5′-dithiobis-(2-nitrobenzoic acid), 1-chloro-2,4-dinitrobenzene (CDNB), NADPH, superoxide dismutase, glucose oxidase, propidium iodide and avidin-FITC were obtained from Sigma Chemical Co (St. Louis, MO). Hydrogen peroxide (30% solution) and paraformaldehyde were supplied by Mallinckrodt, Inc (St. Louis, MO). DME/F12 minimal essential medium and heat-inactivated fetal calf serum (FCS) were purchased from Life Technologies Inc (Grand Island, NY). Monoclonal antibody to the P-glycoprotein (4E3) was obtained from Signet Laboratories, Inc (Dedham, MA); fluorescein-conjugated goat antimouse IgG (H + L) F(ab’)2 was purchased from Jackson Immuno Research Laboratories, Inc (Westgrove, PA). 14C-doxorubicin was from Amersham Life Science Corp (Arlington Heights, IL); and Nyosil silicone oil was from W.F. Nye, Inc, New Bedford, MA, USA. RNeasy kits were obtained from Qiagen, Inc (Valencia, CA); Moloney murine leukaemia virus reverse transcriptase and QPCR buffers were purchased from Life Technologies Inc (Grand Island, NY); human primers and probes as QPCR TaqMan assays were obtained from Life Technologies/ABI (Foster City, CA). Terminal transferase (TdT) and related materials for determinations of apoptosis were obtained in kit form from Boehringer Mannheim, Corp (Indianapolis, IN).
Cell culture
NCI/ADR-RES cancer cells (with a known IC50 for doxorubicin under standard conditions in a 1-h clonogenic assay of ≈500 μM) were originally obtained from the National Cancer Institute, Bethesda, MD and were maintained in 1 μM doxorubicin during routine cell culture. These cells were adapted to growth in DME/F12 media with 0.5% fetal calf serum to optimise the potential for expression of GPx by decreasing the binding of selenium to serum proteins; cells were passaged at least twice in the absence of doxorubicin before use in these experiments. Maximal enzyme activity was observed in the presence of ≥30-nM selenium (as sodium selenite) and 5 μg/ml each of transferrin and insulin. Two new lines were developed from the parental cells: NCI/ADR-RES-30 (grown in 30-nM selenium) which was renamed MR-30; and NCI/ADR-RES-0 (maintained in 0.5% serum without added selenium) which was renamed MR-0. GPx activity had a half-life of ≈48 h after withdrawal of exogenous selenium in the MR-30 cells, as had been described previously for the parental NCI/ADR-RES tumour cell line [13]. The doubling times of MR-30 and MR-0 cells were both 38–42 h; plating efficiencies in clonogenic assays were ≈35–40% for both cell lines.
Antioxidant enzyme activities and glutathione levels
MR-30 or MR-0 cells in logarithmic-phase growth were dislodged with trypsin/EDTA, washed twice in phosphate-buffered physiologic saline (PBS) and then disrupted on ice by sonication with 5–7 1-second bursts of a Branson Sonifier Cell Disrupter 250 (Branson Sonic Power Co., Danbury, CT). The cell homogenates were then centrifuged at 4 °C for 20 min at 16,000 × g; the resulting supernatants were assayed for antioxidant enzyme levels or glutathione. Experiments were performed, at minimum, in triplicate; each experimental sample was prepared from a separate tissue culture flask containing approximately 2–4 × 106 MR-30 or MR-0 cells. Selenium-dependent glutathione peroxidase activity, with hydrogen peroxide as substrate, superoxide dismutase and catalase activities were measured as described previously [14]; glutathione S-transferase (GST) activity with 1-chloro-2,4-dinitrobenzene as substrate was measured as described [15]. Glucose-6-phosphate dehydrogenase and glutathione reductase were assayed by the method of Beutler [16,17]. The level of nonprotein sulfhydryls (principally GSH) in MR-30 and MR-0 cells was determined using 5,5′-dithiobis-2-nitro benzoic acid [18].
RNA Preparation, cDNA synthesis and reverse transcriptase polymerase chain reaction
Total cellular RNA was prepared from ≈3 × 106 tumour cells using the Qiagen RNeasy kit following the manufacturer’s instructions. Real time reverse transcriptase polymerase chain reaction (RT-PCR) was carried out as previously described [19]. Expression of the target genes GPx-1, MDR1, BCL2 and TOP2A, and the reference gene, β-actin, were evaluated by QPCR using the following primers: β-actin (Hs01060665_g1), MDR1 (Hs00184500_m1), GPX1 (Hs00829989_g1), (Hs00699698_m1), BCL2 (Hs0060 8023_m1), TOP2A (Hs01032137_m1).
Cytotoxicity assays
The cytotoxic effects of doxorubicin, 5-iminodaunorubicin (an anthracycline antibiotic that does not redox cycle) and H2O2 were examined using logarithmically-growing MR-30 or MR-0 cells (96 h after plating). The cytotoxic effect of H2O2 was determined by generating the oxidant continuously following the addition of glucose oxidase to the glucose-containing tissue culture medium [20]. The drug under examination or glucose oxidase was added directly to the tissue culture flasks in triplicate for a 1-hr treatment period. The constant rate of H2O2 production, determined with a hydrogen peroxide electrode, increased linearly with concentration, ranging from 11 nmol/min at 1-mU glucose oxidase/ml to 62 nmol/min at 25-mU glucose oxidase/ml. Cells were then harvested with trypsin-EDTA, washed with PBS and dilutions of 1000 and 2500 cells were plated in 60 × 15 mm tissue culture dishes in triplicate. Tumour cell colonies of ≥ 40 cells were counted after incubation for 6–8 days at 37 °C in a humidified atmosphere of 5% CO2 in air. The per cent clonogenic survival has been expressed as the number of colonies produced by drug-treated cells divided by the number of colonies produced by control cells × 100.
Terminal deoxynucleotidyltransferase-mediated dUTP nick-end labelling (TUNEL) assay for apoptosis, and cell cycle status using flow cytometric analysis
MR-0 and MR-30 cells in logarithmic-phase growth were exposed to 450 μM doxorubicin (or an equal volume of media for control experiments) in tissue culture flasks for 4 h; after drug exposure, the tumour cells were washed twice with PBS, their growth medium was replaced and the cells were returned to the tissue culture incubator. For certain experiments, 30 nM sodium selenite was added to MR-0 cells 2 h before doxorubicin exposure and maintained at that level following introduction of the anthracycline. Ninety-six hours following treatment with doxorubicin or media, the cells were washed twice with PBS, trypsinized, washed again in PBS and centrifuged at 800 × g for 5 min. Tumour cells were resuspended in 0.5 ml complete medium and then fixed in 5 ml of freshly prepared, ice-cold 4% paraformaldehyde in PBS, pH 7.2, added dropwise. After a 30-min fixation period on ice, the cells were centrifuged at 4 °C for 5 min at 600 × g; the cell pellet was resuspended in 10 ml of ice-cold PBS and the centrifugation repeated. After complete removal of the PBS, the cell pellet was resuspended in 2 ml of 70% ethanol. Tumour cells were either processed immediately after fixation or frozen in ethanol for up to 1 week at −20 °C prior to TUNEL assay. The cell suspension in ethanol, which contained 1–2 × 106 tumour cells, was then centrifuged for 5 min at 800 × g; the cell pellet was washed twice with 1 ml of 0.1% bovine serum albumin (BSA) in Hanks’ balanced salt solution (HBSS) and centrifuged in an Eppendorf microcentrifuge for 5 min at 8000 × g. The cell pellet was resuspended in 25 μl PBS to which was added 8.5 μl of the TUNEL reaction mixture: 5 μl of 5 × TdT reaction buffer, 2.5 μl cochloride, 0.5 μl TdT enzyme (all from Boehringer kit # 220 582) and 0.5-μl biotin-16-dUTP. Cells were then thoroughly mixed and incubated for 30 min at 37 °C. Following incubation, the cell suspension was centrifuged for 1 min at 10,000 × g, the supernatant carefully removed and the pellet resuspended in 0.1% BSA in HBSS. This cell suspension was centrifuged for 5 min at 8000 × g; after aspiration of the supernatant, 100 μl avidin-FITC staining solution was added to the pellet which was then incubated for 30 min at room temperature in the dark. After incubation, the reaction mixture was centrifuged for 2 min at 8000 × g; the supernatant was aspirated, and the pellet was resuspended in 1 ml of 0.1% Triton X-100 in HBSS and then centrifuged for 5 min at 8000 × g. Following removal of the supernatant, the cell pellet was resuspended in 1 ml of propidium iodide-RNase nuclear staining solution in HBSS (0.5 ml propidium iodide [100 μg/ml], 9.5 ml HBSS containing 500 units RNase). The cells were allowed to stain for ≥ 30 min before flow cytometric analysis. MR-0 and MR-30 cell samples were analysed on a MoFlo flow cytometer (Cytomation, Fort Collins, CO); data (50,000 events) were acquired using dual laser excitation. Scatter signals were acquired with an HeNe laser (Spectra Physics Co., Inc, Mountain View, CA). All fluorescence excitation was performed at a wavelength of 488 nm using an Innova-90 argon laser (Coherrent Inc, Santa Clara, CA) at 500 mW. FITC emission was measured through a 530-nm cut-off filter (530DF30 filter; Omega Optical Co., Inc, Brattleboro, VT). Cell cycle parameters were determined using propidium iodide with emission measured through a 640-nm cut-off filter (640EFLP filter; Omega Optical Co., Inc). A 580DRLP dichroic mirror was used to split the two signals.
Oxygen radical production
Oxygen radical species with the chemical characteristics of the hydroxyl radical (•OH) were evaluated by measurement of the release of methane from dimethyl sulfoxide using gas chromatography with flame ionisation detection as previously described for intact tumour cells [2]. In brief, the final, 1-ml reaction volume, which was incubated in a siliconized 2-ml gas-tight reaction vessel for 4 h with shaking at 37 °C, contained 5 mM NADPH, 0.1% Triton X-100, 7.5 × 106 tumour cells, 100 mM DMSO and the indicated doxorubicin concentration in DME/F12 medium; 500 μl of the headspace gas was taken for measurement of methane. The sensitivity limit of this assay is 20–30 pmol of methane. The data have been expressed as pmol methane/4-h/107 cells ± SE.
Flow cytometry for assessment of P-glycoprotein expression
To examine the effect of selenium and GPx on Pgp expression, flow cytometry was performed on MR-30 and MR-0 cells as previously described [21]. The primary antibody recognises an external epitope of the MDR1 gene product, Pgp. The secondary antibody was fluorescein-conjugated goat antimouse IgG (H + L) F(ab’)2.
Doxorubicin accumulation and efflux
To determine the functional effect of both GPx expression and selenium on the action of Pgp in MR-30 and MR-0 cells, doxorubicin accumulation and efflux were examined by measurement of total cell-associated radioactivity of 106 MR-30 or MR-0 cells treated for 120 min with 2.5 μM, [14C]-labeled doxorubicin in DME/F12 medium containing 0.5% fetal calf serum. Immediately after the 2-h drug accumulation phase, cells were washed twice in PBS and harvested with trypsin. Cells were then immediately layered onto 0.3 ml of Nyosil silicone oil and centrifuged for 6 min at 13,500 × g; the trypsin-Nyosil supernatant was decanted, and the pellet was resuspended in 0.5 ml of 2% SDS, pH 10.0. The cells were mixed and allowed to stand overnight before total radioactivity in the cell pellets was assessed by scintillation spectrometry. For efflux studies, after 2 h of drug accumulation as outlined above, cells were washed in PBS, resuspended in drug-free DME/F12 containing 0.5% FCS and incubated for 1 or 2 h at 37 °C in 5% CO2 in air. At the end of the specified incubation times, cells were washed in PBS, harvested with trypsin and processed as above. “Zero” time controls were also processed as described. All accumulation and efflux experiments were performed at least three times.
Statistical analyses
Cytotoxicity assays, antioxidant levels, reactive oxygen production and doxorubicin accumulation and efflux were analysed using Student’s two-tailed t-test for independent means; significance required p < .05.
Results
Antioxidant enzyme and GSH levels in NCI/ADR-RES-derived cell lines adapted to growth in low serum
Because incorporation of selenocysteine molecules into the active site of GPx is a critical prerequisite for enzymatic activity [22], we performed preliminary studies attempting to optimise conditions for the production of active GPx in tissue culture. Since more than 75% of the elemental selenium in tissue culture medium is found in a nondialyzable, protein-bound state [23], we first adapted the NCI/ADR-RES line which had been developed by step-wise exposure of parental cells to increasing concentrations of doxorubicin [24] to growth under low serum conditions. We found that NCI/ADR-RES cells grow well in 0.5% FCS supplemented with insulin and transferrin, with no change in morphology, doubling time or ability to generate colonies on a plastic support (data not shown). This observation provided the basis for our examination of the effect of GPx and selenium on the toxicity of doxorubicin and H2O2 in human NCI/ADR-RES tumour cells expressing high levels of Pgp.
When NCI/ADR-RES cells were adapted to growth in 0.5% serum without supplemental selenium (MR-0 cells, Table 1), GPx activity was at the lower limit of detectability. We have previously demonstrated that NCI/ADR-RES cells passaged under standard conditions with 5% FCS and no supplemental selenium have a GPx activity level of 8–12 nmol/min/mg [13]. Passage in 30-nM selenium under low serum conditions produced the MR-30 cell line with levels of GPx ≈ 10-fold greater than MR-0 cells, (Table 1, p < .01). The addition of sodium selenite, however, did not result in any significant effect on the specific activities of other antioxidant enzymes capable of detoxifying H2O2 or superoxide anion, or those required to generate reducing equivalents for the glutathione-glutathione peroxidase cycle (Table 1).
Table 1.
Effect of selenium on antioxidant activities in MR-30 and MR-0 human cancer cells.
| Antioxidant activitya | MR-0 | MR-30 |
|---|---|---|
| Glutathione Peroxidase (nmol/min/mg) | 2.2 ± 0.9b | 22.5 ± 1.5 |
| Catalase (μmol/min/mg) | 1.55 ± 0.14 | 1.39 ± 0.04 |
| Glutathione S-Transferase (nmol/min/mg) | 91.1 ± 4.2 | 81.9 ± 3.5 |
| Glucose-6-Phosphate Dehydrogenase (nmol/min/mg) | 255 ± 33 | 204 ± 37 |
| Glutathione Reductase (nmol/min/mg) | 7.6 ± 1.7 | 6.4 ± 2.5 |
| Superoxide Dismutase (μg superoxide dismutase/mg) | 0.14 ± 0.01 | 0.12 ± 0.01 |
| Glutathione (nmol/106 cells) | 7.7 ± 0.4 | 7.7 ± 0.4 |
Antioxidant activities were determined in the two cell lines after multiple passages without (MR-0) or with (MR-30) supplemental sodium selenite (30 nM) in the media.
Antioxidant activities obtained using cytosolic preparations; data presented as the mean ± SE.
p < .01 versus MR-30 cells.
Expression of GPx-1, MDR1 and apoptosis-related genes in MR-30 and MR-0 cells
GPx-1 mRNA levels in NCI/ADR-RES cells passaged in 5% FCS without supplemental selenium are significantly increased compared to the parental line [11]. Because the specific enzymatic activity of GPx was significantly enhanced in MR-30 versus MR-0 cells, the mRNA levels of GPx-1 as well as a panel of resistance genes (MDR1, BCL-2 and TOP2A) were examined in these cells by RT-PCR; as shown in Table 2, expression ratios (vs. β-actin) of genes involved in doxorubicin-related tumour cell killing demonstrated that the relative amount of GPx-1 mRNA in both MR-30 and MR-0 cells was equivalent. Selenium supplementation did not alter GPx-1 mRNA expression per se; this is consistent with our previous observation that modulation of GPx activity in MCF-7 cells occurs largely at the translational level [13]. As expected, MDR1 overexpression was observed in MR-30 and MR-0 cells which is consistent with prior studies; there was no apparent effect of selenium on MDR1 mRNA levels. Selenium supplementation also did not affect BCL-2 or TOP2A expression.
Table 2.
Effect of selenium on gene expression in MR-0 and MR-30 cancer cells.
| Gene of interesta | MR-0 | MR-30 |
|---|---|---|
| GPX1 | 102,217 ± 23,012 | 78,085 ± 11,422 |
| MDR1 | 78,210 ± 6133 | 106,399 ± 30,421 |
| BCL-2 | 724 ± 140 | 932 ± 109 |
| TOP2A | 27,049 ± 2902 | 23,375 ± 2811 |
mRNA expression was determined in triplicate by real time RT-PCR and has been shown as gene of interest/β-actin × 10−6; data presented as mean ± SE.
Effect of GPx activity on the cytotoxicity of H2O2 and doxorubicin
To examine the effect of an approximate 10-fold variation in GPx activity on the cytotoxicity of H2O2 in cells expressing high levels of MDR1 mRNA, H2O2 was generated by treating cells with glucose oxidase to produce a continuous flux of the oxidant. The results shown in Figure 1(A) demonstrate that the IC50 for glucose oxidase increased from 1–2 mU/ml in MR-0 cells to 15–20 mU/ml in MR-30 cells, p < .01, paralleling the GPx activities of the two cell lines. These results are not surprising in light of the observations that Pgp does not transport H2O2, and that the only significant difference in antioxidant Defence found between MR-0 and MR-30 cells was in the level of GPx (Table 1).
Figure 1.
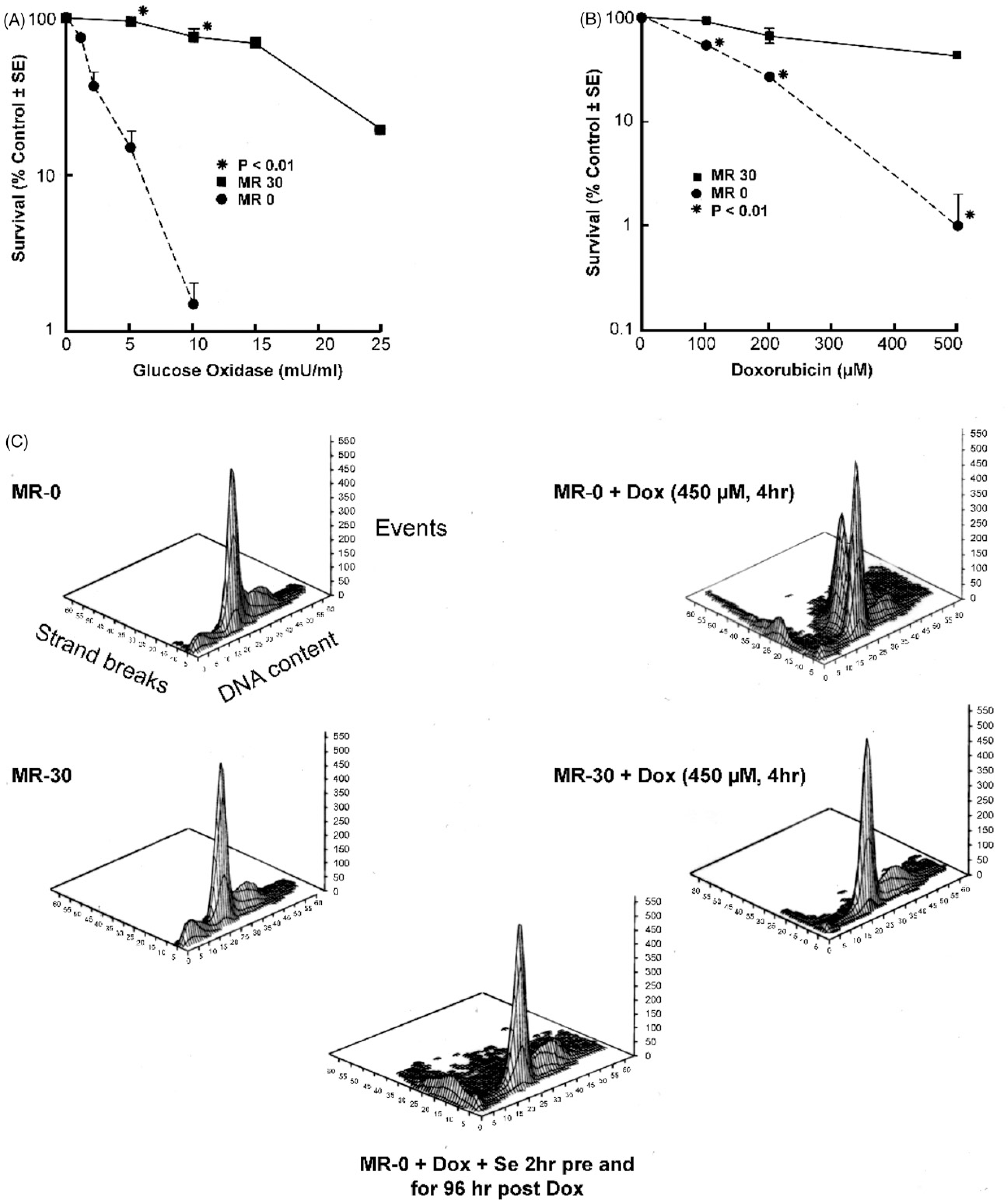
Effect of glutathione peroxidase activity on the toxicity of hydrogen peroxide and doxorubicin in MR-30 and MR-0 cells. (A) Hydrogen peroxide was generated in the tissue culture medium continuously by addition of glucose oxidase and cytotoxicity evaluated by clonogenic assay; (B) doxorubicin cytotoxicity in MR-30 and MR-0 cells was determined by clonogenic assay; (C) the effect of glutathione peroxidase activity and selenium on doxorubicin-induced apoptosis in MR-30 and MR-0 cells was determined by TUNEL assay. As shown in the upper left panel, cell cycle progression is demonstrated on the x axis, extent of apoptosis (TUNEL positivity) on the y axis and the number of events on the z axis. Apoptosis is represented both as the extent of TUNEL positive low molecular weight DNA and as TUNEL positive cells present in various phases of the cell cycle. In the bottom panel, MR-0 cells were treated with 30 nM sodium selenite beginning 2 h prior to doxorubicin treatment and continuing both during and for the 96 h following drug exposure.
Another major goal of these studies was to determine whether GPx activity contributed to doxorubicin, as well as H2O2, resistance in cells that overexpressed both the MDR1 and GPx-1 genes. Using a clonogenic assay, MR-30 cells were found to be extraordinarily resistant to high concentrations of doxorubicin (Figure 1(B)). The IC50 for doxorubicin was ≈500 μM in the MR-30 subline, and it was difficult to produce further cytotoxicity on a logarithmic scale for these cells within the solubility limits of the drug (1–2 mM). P-glycoprotein expression, in part, explains the need to employ high levels of doxorubicin in these experiments (far above the 1–2-μM concentrations necessary to inhibit tumour cell proliferation in the parental line). On the other hand, as seen in Figure 1(B), the IC50 for doxorubicin in the MR-0 subline decreased fivefold to ≈100 μM, p < .01. Furthermore, log-linear tumour cell cytotoxicity was observed in MR-0 cells where GPx activity was significantly decreased. In control experiments with the MR-0 line, the addition of 30 nM sodium selenite to cells that had not been passed regularly in selenium-containing medium, for the 2 h prior to treatment with doxorubicin, did not change the GPx activity of the cells (data not shown). We also found that exposure of MR-30 and MR-0 cells for 1 h to 2, 5 or 25 μM 5-iminodaunorubicin, a doxorubicin analogue that does not generate reactive oxygen species [25], decreased clonogenic survival to an equivalent level in either cell line (Table 3).
Table 3.
Effect of a 1-h exposure to 5-iminodaunorubicin on the clonogenic survival of MR-0 and MR-30 cells.
| 5-Iminodaunorubicin concentration | MR-0 | MR-30 |
|---|---|---|
| 2 μM | 95 ± 26a | 91 ± 2.5 |
| 5 μM | 86 ± 2.2 | 81 ± 7.5 |
| 25 μM | 68 ± 4.4 | 68 ± 2.1 |
Percent of control ± SE.
Doxorubicin-related apoptosis in MR-30 and MR-0 cells
As shown in Figure 1(C), MR-0 cells were substantially more sensitive to doxorubicin-induced apoptosis than the MR-30 line. When studied at the IC50 concentration determined by clonogenic assay (450 μM), a small amount of TUNEL positivity and low molecular weight DNA was observed for MR-30 cells after doxorubicin exposure compared to untreated control cells. However, a much more substantial degree of TUNEL positivity was observed in MR-0 cells treated with the identical concentration of doxorubicin. Apoptotic MR-0 cells were derived from both G1/S and G2/M boundaries. When MR-0 cells were exposed to 30 nM sodium selenite 2 h before, during, and for 96 h after doxorubicin treatment, an extensive degree of apoptosis continued to be observed.
Effect of GPx activity on hydroxyl radical formation
Because GPx can modulate hydrogen peroxide-induced apoptosis [26], and our finding that doxorubicin-related apoptosis and tumour cell killing were significantly increased in MR-0 cells, we evaluated the effect of altered GPx activity on the generation of strong oxidant species with the chemical characteristics of •OH. As shown in Table 4, doxorubicin exposure led to the generation of easily quantifiable amounts of methane from dimethyl sulfoxide only in MR-0 cells that contain low levels of GPx activity. Methane production required doxorubicin and NADPH, was inhibited by the hydrogen peroxide detoxifying enzyme catalase and the •OH scavenger thiourea, but not by its redox inactive analogue urea, and was also inhibited by the iron chelator deferoxamine. The concentration-response relationship for doxorubicin and •OH formation is shown in Figure 2(A); minimal amounts of methane (at the lower limit of detection) were produced in this system by MR-30 cells that contained substantial levels of GPx.
Table 4.
Effect of glutathione peroxidase activity on doxorubicin-stimulated hydroxyl radical production by MR-0 and MR-30 human cancer cells.
| Reaction conditions | MR-0 | MR-30 |
|---|---|---|
| Initial | 276 ± 64a,b | 66 ± 29 |
| Minus doxorubicin | 0c | 0 |
| Minus NADPH | 0c | 0 |
| Plus Catalase (1500 U/ml) | 0c | 0 |
| Plus Superoxide Dismutase (20 μg/ml) | 84 ± 30c | 0 |
| Plus Deferoxamine (100 μM) | 89 ± 49d | 0 |
| Plus Thiourea (100 mM) | 14 ± 14c | 0 |
| Plus Urea (100 mM) | 177 ± 50 | 106 ± 60 |
Hydroxyl radical production in MR-0 and MR-30 cells was determined by gas chromatography as the release of methane from dimethyl sulfoxide in a closed reaction system.
Methane production from dimethyl sulfoxide in a 1-ml reaction volume containing 2.5 mM doxorubicin, 5 mM NADPH, 0.1% Triton X-100, DME/F12 medium and 7.5 × 106 tumour cells incubated for 4 h at 37°C; data have been expressed as pmol methane/4 h/107 cells ± SE.
p < .02 versus MR-30 cells.
p < .02 versus initial reaction system in MR-0 cells.
p < .05 versus initial reaction system in MR-0 cells.
Figure 2.
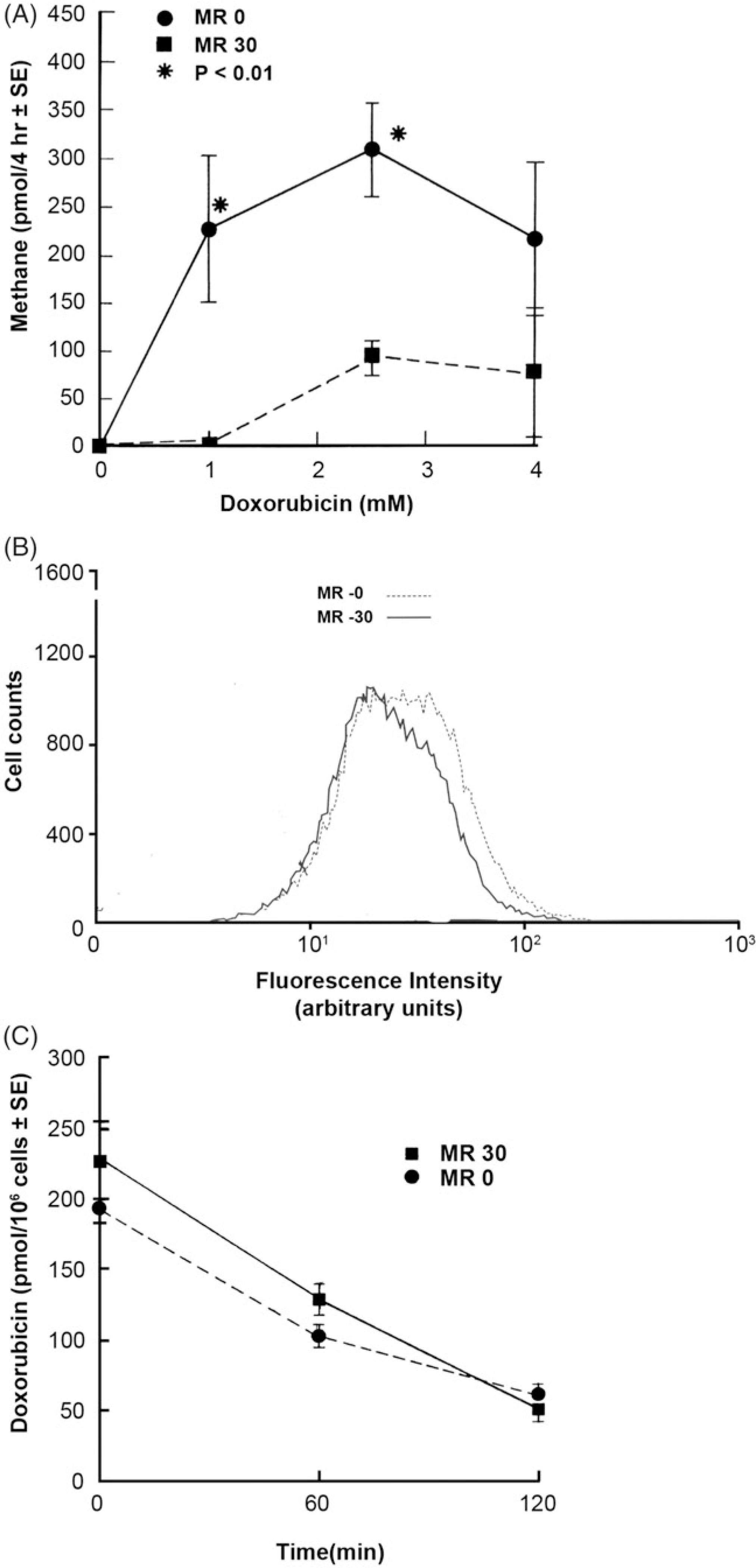
Effect of glutathione peroxidase activity on doxorubicin-enhanced hydroxyl radical production, P-glycoprotein expression and anthracycline uptake and efflux in MR-30 and MR-0 cells. (A) Hydroxyl radical (or a related strong oxidant species) has been quantitated by gas chromatographically as the release of CH4 from DMSO in a closed reaction system. Results represent the mean ± SE of at least three separate determinations. (B) Effect of glutathione peroxidase activity and selenium on P-glycoprotein expression in MR-30 and MR-0 cells. Representative fluorescence histograms are presented from duplicate experiments for these cell lines. (C) Effect of glutathione peroxidase activity on 14C-doxorubicin accumulation and efflux in MR-30 and MR-0 cells. The results represent the mean ± SE of experiments performed in triplicate.
Pgp expression in MR-30 and MR-0 cells
To determine whether Pgp levels on the cell surface of the MR sublines were influenced by their GPx or selenium status, we performed flow cytometric analysis using a monoclonal antibody which binds to a cell-surface epitope of human Pgp. Figure 2(B) shows that Pgp was expressed to a substantial degree on both MR-30 and MR-0 cells, and that the level of Pgp was similar irrespective of selenium status.
Doxorubicin accumulation and efflux in MR-30 and MR-0 cells
To ensure that enhanced sensitivity toward doxorubicin in the presence of diminished GPx activity was not due to an alteration in the function of the Pgp multidrug transporter, the accumulation and efflux of doxorubicin were evaluated in MR-0 and MR-30 cells. After the tumour cells were loaded for 2 h with 2.5 μM 14C-doxorubicin, cell-associated doxorubicin levels were (mean ± SE) 195 ± 5 pmol/106 cells in MR-0 cells compared to 225 ± 10 pmol/106 cells in the MR-30 subline, (Figure 2(C); p > .05; not significant). Doxorubicin concentrations were examined after 1 and 2 h of drug efflux in fresh media. The results shown in Figure 2(C) demonstrate that the presence of exogenous selenium did not significantly alter the level of residual cell-associated doxorubicin. After 2 h in drug-free media, >70% of the anthracycline had been effluxed from both sublines irrespective of selenium status.
Discussion
Several molecular mechanisms have been advanced to explain doxorubicin resistance in vitro, including overexpression of MDR1, BCL-2 and alterations in the function of topoisomerase II or antioxidant proteins [1]. Although increased GPx activity has been described previously for tumour cells selected in doxorubicin [7,27], the functional significance of these observations has been questioned in the past [5]. Furthermore, it has been observed that several drug resistance mechanisms may be selected simultaneously during stepwise exposure to increasing doxorubicin concentrations [27,28]. To evaluate the role of H2O2 detoxification in acquired doxorubicin resistance further, we examined the contribution of GPx activity to the modulation of doxorubicin-related cytotoxicity, apoptosis and hydroxyl radical formation in a human cancer cell line (NCI/ADR-RES) overexpressing the P-glycoprotein.
To perform these experiments, we developed the MR-0 and MR-30 cell lines that have an identical complement of antioxidant enzymes except for GPx activities that vary by ≈10-fold. The mRNA expression of GPx-1, MDR1, BCL-2 and TOP2A did not vary based on the GPx activity of the cells. Furthermore, we found no difference in Pgp level or doxorubicin accumulation or efflux in the MR-0 line compared with MR-30 cells. Finally, as previously documented for the parental NCI/ADR-RES line [13], the GPx activity of MR-0 cells was unchanged after short-term (< 24 h) exposure to 30 nM sodium selenite. Although selenium itself has antioxidant properties [29], our results suggest that differences in GPx per se, and not inorganic selenium, were responsible for the observed alterations in doxorubicin and H2O2 toxicity.
The GSH-GPx cycle plays a critical role in regulating intracellular hydrogen and lipid hydroperoxide tone [30]. Since H2O2 is not a substrate for Pgp, the enhanced sensitivity of MR-0 cells to a continuous flux of H2O2 that we observed was not surprising. However, a significant (≈fivefold) difference in doxorubicin IC50 between cell lines varying only in GPx activity, under experimental conditions in which the accumulation and efflux of doxorubicin and the expression of Pgp were equivalent, has not previously been demonstrated. In light of the observation that GPx activity had no effect on the toxicity of the nonredox cycling anthracycline 5-iminodaunorubicin, and that hydroxyl radical production was undetectable in MR-30 cells, it is reasonable to suggest that enhanced GPx activity contributed to a decrease in the intracellular level of hydrogen peroxide, with a consequent decrease in Fenton chemistry, and diminished tumour cell killing by doxorubicin. These results are supported by previous studies in cells not demonstrating the multidrug resistance phenotype which suggested that overexpression of GPx-1 produces doxorubicin resistance in the T47D breast cancer line [31].
NCI/ADR-RES cells display a complex phenotype that includes, among other changes, overexpression of MDR1 and glutathione S-transferase π, and alterations in the function of topoisomerase II, as well as increased GPx activity. It is remarkable, in the context of the pleiotropic nature of the acquired resistance to doxorubicin exhibited by these cells, that decreasing GPx activity should, in itself, sensitise them to the anthracycline. In the presence of a multiplicity of resistance mechanisms, it is also understandable that decreasing GPx alone would not completely reverse the multidrug-resistance phenotype. Taken together, these data suggest that Pgp-mediated drug efflux and GPx-related oxidant detoxification play complementary roles in defending tumour cells against the cytotoxic effects of doxorubicin.
In conclusion, these experiments indicate that depletion of intracellular GPx, without concomitant changes in other antioxidant proteins, Pgp or the expression of the apoptosis-related genes leads to a significant increase in doxorubicin-induced hydroxyl radical formation, clonogenic tumour cell killing and programmed cell death in the face of equivalent levels of cell-associated anthracycline. Our results suggest that detoxification of peroxides by GPx contributes to doxorubicin resistance even in the presence of high levels of P-glycoprotein expression.
Funding
This study was supported by the US National Cancer Institute and by the City of Hope Comprehensive Cancer Center.
Footnotes
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Doroshow JH. Topoisomerase II inhibitors: anthracyclines. In: Chabner BA, Longo DL, editor. Cancer chemotherapy and biotherapy: principles and practice. Philadelphia: Lippincott Williams & Wilkins Publishers; 2011. p. 356–391. [Google Scholar]
- [2].Doroshow JH. Role of hydrogen peroxide and hydroxyl radical formation in the killing of Ehrlich tumor cells by anticancer quinones. Proc Natl Acad Sci USA.1986;83:4514–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].McCormick PN, Greenwood HE, Glaser M, et al. Assessment of tumor redox status through (S)-4-(3-[18F]fluoropropyl)-L-glutamic acid PET imaging of system xC− activity. Cancer Res. 2019;79:853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Di X, Shiu RP, Newsham IF, Gewirtz DA. Apoptosis, autophagy, accelerated senescence and reactive oxygen in the response of human breast tumor cells to adriamycin. Biochem Pharmacol. 2009;77:1139–1150. [DOI] [PubMed] [Google Scholar]
- [5].Ortiz C, Caja L, Sancho P, et al. Inhibition of the EGF receptor blocks autocrine growth and increases the cytotoxic effects of doxorubicin in rat hepatoma cells: role of reactive oxygen species production and glutathione depletion. Biochem Pharmacol. 2008;75: 1935–1945. [DOI] [PubMed] [Google Scholar]
- [6].Szwed M, Kania KD, Jozwiak Z. Changes in the activity of antioxidant barrier after treatment of K562 and CCRF-CEM cell lines with doxorubicin-transferrin conjugate. Biochimie. 2014;107:358–366. [DOI] [PubMed] [Google Scholar]
- [7].Ozkan A, Fiskin K. Protective effect of antioxidant enzymes against drug cytotoxicity in MCF-7 cells. Exp Oncol. 2006;28:86–88. [PubMed] [Google Scholar]
- [8].Cohen G, Hochstein P. Glutathione peroxidase: the primary agent for the elimination of hydrogen peroxide in erythrocytes. Biochemistry. 1963;2:1420–1428. [DOI] [PubMed] [Google Scholar]
- [9].Inoue T, Suzuki-Karasaki Y. Mitochondrial superoxide mediates mitochondrial and endoplasmic reticulum dysfunctions. Free Radic Biol Med. 2013;61:273–284. [DOI] [PubMed] [Google Scholar]
- [10].Wold LE, Aberle NS, Ren J. Doxorubicin induces cardiomyocyte dysfunction via a p38 MAP kinase-dependent oxidative stress mechanism. Cancer Detect Prev. 2005;29:294–299. [DOI] [PubMed] [Google Scholar]
- [11].Akman SA, Forrest G, Chu FF, et al. Antioxidant and xenobiotic-metabolizing enzyme gene expression in doxorubicin-resistant MCF-7 breast cancer cells. Cancer Res. 1990;50:1397–1402. [PubMed] [Google Scholar]
- [12].Sinha BK, Haim N, Dusre L, et al. DNA strand breaks produced by etoposide (vp-16,213) in sensitive and resistant human breast tumor cells: implications for the mechanism of action. Cancer Res. 1988;48:5096–5100. [PubMed] [Google Scholar]
- [13].Chu F-F, Esworthy RS, Akman S, Doroshow JH. Modulation of glutathione peroxidase expression by selenium: effect on human MCF-7 breast cancer cell transfectants expressing a cellular glutathione peroxidase cDNA and doxorubicin-resistant MCF-7 cells. Nucl Acids Res. 1990;18:1531–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Doroshow JH, Locker GY, Myers CE. Enzymatic defenses of the mouse heart against reactive oxygen metabolites: alterations produced by doxorubicin. J Clin Invest. 1980;65:128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Habig WH, Jakoby WB. Glutathione S-transferases (rat and human). Meth Enzymol. 1981;77:218–231. [DOI] [PubMed] [Google Scholar]
- [16].Beutler E, Glucose-6-phosphate dehydrogenase (G-6-PD) and 6-phosphogluconate dehydrogenase (6-PGD). In: Beutler E, editor. Red cell metabolism: a manual of biochemical methods. New York (NY): Grune & Stratton; 1975. p. 66–69. [Google Scholar]
- [17].Beutler E, Glutathione reductase (GR). In: Beutler E, editor. Red cell metabolism: a manual of biochemical methods. New York (NY): Grune & Stratton; 1975. p. 69–71. [Google Scholar]
- [18].Sedlak J, Lindsay RH. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal Biochem. 1968;25: 192–205. [DOI] [PubMed] [Google Scholar]
- [19].Juhasz A, Markel S, Gaur S, et al. NADPH oxidase 1 supports proliferation of colon cancer cells by modulating reactive oxygen species-dependent signal transduction. J Biol Chem. 2017;292:7866–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kyle ME, Nakae D, Sakaida I, et al. Protein thiol depletion and the killing of cultured hepatocytes by hydrogen peroxide. Biochem Pharmacol. 1989;38:3797–3805. [DOI] [PubMed] [Google Scholar]
- [21].Metz MZ, Best DM, Kane SE. Harvey murine sarcoma virus/MDR1 retroviral vectors: efficient virus production and foreign gene transduction using MDR1 as a selectable marker. Virology. 1995;208:634–643. [DOI] [PubMed] [Google Scholar]
- [22].Shen Q, Wu R, Leonard JL, et al. Identification and molecular cloning of a human selenocysteine insertion sequence-binding protein. A bifunctional role for DNA- binding protein. J Biol Chem. 1998;273:5443–5446. [DOI] [PubMed] [Google Scholar]
- [23].Sandström BER, Carlsson J, Marklund SL. Variations among cultured cells in glutathione peroxidase activity in response to selenite supplementation. Biochim Biophys Acta. 1987;929:148–153. [DOI] [PubMed] [Google Scholar]
- [24].Fairchild CR, Ivy SP, Kao-Shan CS, et al. Isolation of amplified and overexpressed DNA sequences from adriamycin-resistant human breast cancer cells. Cancer Res. 1987;47:5141–5148. [PubMed] [Google Scholar]
- [25].Pollakis G, Goormaghtigh E, Ruysschaert JM. Role of the quinone structure in the mitochondrial damage induced by antitumor anthracyclines. Comparison of adriamycin and 5-iminodaunorubicin. FEBS Lett. 1983; 155:267–272. [DOI] [PubMed] [Google Scholar]
- [26].Wang S, Konorev EA, Kotamraju S, et al. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms. Intermediacy of H2O2-and p53-dependent pathways. J Biol Chem. 2004;279: 25535–25543. [DOI] [PubMed] [Google Scholar]
- [27].Ye C-G, Yeung JH-K, Huang G-L, et al. Increased glutathione and mitogen-activated protein kinase phosphorylation are involved in the induction of doxorubicin resistance in hepatocellular carcinoma cells. Hepatol Res. 2013;43:289–299. [DOI] [PubMed] [Google Scholar]
- [28].Zeng R, Tang Y, Zhou H, et al. STAT3 mediates multidrug resistance of Burkitt lymphoma cells by promoting antioxidant feedback. Biochem Biophys Res Commun. 2017;488:182–188. [DOI] [PubMed] [Google Scholar]
- [29].Barceloux DG, Barceloux D. Selenium. J Toxicol Clin Toxicol. 1999;37:145–172. [DOI] [PubMed] [Google Scholar]
- [30].Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830:3289–3303. [DOI] [PubMed] [Google Scholar]
- [31].Gouazé V, Mirault ME, Carpentier S, et al. Glutathione peroxidase-1 overexpression prevents ceramide production and partially inhibits apoptosis in doxorubicin-treated human breast carcinoma cells. Mol Pharmacol. 2001;60:488–496. [PubMed] [Google Scholar]