

Abstract
Mercaptobenzamide thioesters and thioethers are chemically simple HIV-1 maturation inhibitors with a unique mechanism of action, low toxicity, and a high barrier to viral resistance. A structure-activity relationship (SAR) profile based on 39 mercaptobenzamide prodrug analogs exposed divergent activity/toxicity roles for the internal and terminal amides. To probe the relationship between antiviral activity and toxicity, we generated an improved computational model for the binding of mercaptobenzamide thioesters (SAMTs) to the HIV-1 NCp7 C-terminal zinc finger, revealing the presence of a second low-energy binding orientation, hitherto undisclosed. Finally, using NMR-derived thiol–thioester exchange equilibrium constants, we propose that thermodynamics plays a role in determining the antiviral activity observed in the SAR profile.
Keywords: HIV, NCp7, Antiviral, Mercaptobenzamide prodrug, SAMT, Activity limit, Molecular dynamics
1. Introduction
By many metrics, the global community has made significant progress against human immunodeficiency virus (HIV). According to the latest (2018) statistics from the World Health Organization (WHO), new HIV infections have decreased by 47% to 1.8 million (compared to 3.4 million in 1996), AIDS-related deaths have dropped to 940,000 (a 51% reduction from 2004), and 20 new drugs/combinations have gained FDA approval in the past decade [1]. Much of this progress is due to HAART (highly active antiretroviral therapy), a combination of antiretroviral drugs targeting the virus at multiple stages of its replication cycle [2]. HAART has effectively transformed HIV into a manageable chronic condition [3]. However, as a consequence of its routine use, HAART has become susceptible to multi-drug resistance and is associated with delayed, adverse side-effects, including bone and renal toxicity, dyslipidemia, insulin resistance, and cardiovascular disease [4]. Moreover, due to the cost and logistical challenge of administering HAART to the third world [5], only 54% of adults and 43% of children with HIV are currently receiving treatment [1a]. It is therefore crucial to continue the development of novel antivirals, particularly those that are inexpensive, nontoxic, and which are unlikely to result in viral resistance [6,7].
Mercaptobenzamide thioesters (SAMTs), such as SAMT-247 (1), are an attractive class of HIV maturation inhibitors due to their structural simplicity, synthetic scalability [8], high barrier to development of viral resistance [9], and very low toxicity in cellular, murine, and primate models (Fig. 1) [9,10]. On the basis of kinetic and pKa studies, our group has recently suggested that in cellulo, SAMTs exist in a complex equilibrium of reactive and inert states [8]. SAMT-247 (1) will rapidly acetylate intracellular thiols such as glutathione (GSH) and nucleophilic proteinogenic cysteines at a rate (k) of 84 ± 11 M−1s−1 to form the thiolate 2 (pKa = 5.83 ± 0.04), which we believe is the resting-state of the molecule in cells. Upon exposure to reactive oxygen species or cytoplasmic disulfides, the thiolate 2 is readily oxidized to a mixed disulfide 3 that may disproportionate to the benzisothiazolinone 4. Both 3 and 4 are competent oxidation catalysts, promoting disulfide formation between proximal thiols.
Fig. 1.

Proposed mechanism of action of mercaptobenzamide prodrugs.
As a consequence of their dual acetylative and oxidative reactivity, SAMTs have a complex, multi-step mechanism of action that ultimately prevents the proper maturation of HIV viral particles. During the maturation process of HIV, 1 likely acetylates Cys-36 of the highly-conserved C-terminal CCHC zinc finger of HIV-1 nucleocapsid (NCp7) [11], a 55-amino acid portion of the HIV polyprotein Gag which also contains the matrix and capsid domains. NMR studies by Deshmukh et al. suggest that a sparsely populated state (1.2%) of the C-terminal zinc finger has the Cys-36 sulfhydryl unbound from zinc, enhancing its nucleophilicity [12]. Initial intermolecular acylation of Cys-36 is followed by intramolecular transacetylation to proximal Lys-38, disruption of zinc-finger integrity, and complete loss of zinc ion coordination by NCp7 [11]. As a consequence of zinc finger structural disruption, exposed cysteine sulfhydryls become free to engage in oxidative inter- and intra-Gag disulfide formation, a process that can be catalyzed by both the mixed disulfide 3 and the benzisothiazolinone 4 [8]. The formation of disulfide–cross-linked Gag appears to ultimately be responsible for the production of non-infectious viral particles [9].
While SAMTs have shown promise in a preclinical setting, including in topical microbicide and slow-release formulations [13], their micromolar IC50 values for antiviral activity [9] limit their potential as future therapeutics. Our group has previously investigated the structure-activity relationship (SAR) of mercaptobenzamide esterase-sensitive prodrugs, such as 5, focusing on tuning both the electronics of the aromatic ring, as well as the steric-bulk of the β-alaninamide side-chain (Fig. 2) [14]. Several modifications were found to completely eliminate antiviral activity, such as ortho-substitution (6) or positioning the side-chain meta to the sulfur (7) [9], while others, such as introducing electron-withdrawing groups on the aromatic ring (8), increased cellular toxicity. Electron-donating substituents on the aromatic ring (9) were tolerated, as were side-chain modifications such as dehomologation [β-alaninamide to glycinamide (10)] and α-(di)substitution (11). Despite these efforts, we were unable to substantially improve upon the low micromolar IC50 values for antiviral activity of the original molecule 5. As a consequence of NCp7 flexibility [15] and numerous potential on-target and off-target reactivity modes, optimization of mercaptobenzamide structure is largely unassisted by traditional computer-aided and structure-based drug design strategies [16] and continues to be challenging. Herein, we report a thorough exploration of mercaptobenzamide SAR, focused primarily on side-chain modification. These studies were instrumental in identifying critical features required for activity, structural trends resulting in toxicity, and tolerated sites for modification that may enable future mechanistic experiments. Moreover, NMR-derived thermodynamic parameters provide insight into the possibility that the equilibrium between mercaptobenzamide species controls the limits of antiviral activity.
Fig. 2.

Summary of results from previous mercaptobenzamide structure-activity relationship (SAR) studies.
2. Results and discussion
2.1. Synthesis of compounds
The majority of mercaptobenzamide prodrugs were synthesized using the two-step, one-pot general sequence shown in Scheme 1A [14]. To avoid thiol oxidation, thiosalicylic acid (12, n = 0) or 2-mercaptophenylacetic acid (12, n = 1) were first treated with chloromethyl butyrate and di-iso-propylethylamine overnight, providing an intermediate thioether. Without workup, the carboxylic acid moiety was activated using coupling reagents such as N,N′-carbonyldiimidazole (CDI) or HATU. Subsequent addition of the nucleophilic amine (free base or hydrochloride salt) or alcohol accomplished amide/ester bond formation, furnishing the desired mercaptobenzamide 13 (n = 0), the mercaptophenylacetamide 13 (n = 1), or the mercaptobenzoate 14. Four general classes of amines were utilized, all either commercially available or synthetically accessible in 1–2 steps (Scheme 1B). Amines of type 15 were prepared in one step from the α-chloroamide 16 via aminolysis, while amines of type 17 and 18 were attained from the Boc-protected amino acid 19 via CDI-mediated amide bond formation, followed by Boc deprotection (HCl). Lastly, amines of type 20 were synthesized from the mono-Boc protected diamines 21 via acylation (acetic anhydride, triethylamine) and subsequent Boc deprotection (HCl). The synthesis of selenobenzamide prodrugs with general structure 22 followed a different synthetic sequence (Scheme 1C) [17]. The intermediate diselenide 23, prepared via treatment of diazotized anthranilic acid (24; sodium nitrite, HCl) with sodium diselenide generated in situ (selenium powder, sodium borohydride, aqueous sodium hydroxide), followed by HATU-mediated amide bond formation, was subjected to one-pot reduction (sodium borohydride), followed by capping (acetic acid, chloromethyl butyrate), furnishing the desired selenobenzamide prodrug 22.
Scheme 1.

A) General synthesis of mercaptobenzamide, mercaptophenylacetamide, and mercaptobenzoate prodrugs. B) Synthesis of four general amine classes used for the preparation of mercaptobenzamide prodrugs. C) General synthesis of selenobenzamide prodrugs.
2.2. Determination of structure-activity relationships (SAR)
2.2.1. Linker length modification and conformational restriction
As our previous investigation had indicated the tolerability of side-chain dehomologation from β-alaninamide to glycinamide, we began by exploring the effects of homologation via the progressive addition of methylene units to 5 (Table 1). While EC50 values remained largely unchanged (low micromolar), the introduction of a single CH2 increased the toxicity from >100 μM for the β-alaninamide 5 to 56.6 ± 0.5 μM for the butanamide 25. Each additional methylene appeared to progressively worsen the toxicity (26–28), with the heptanamide 28 displaying a TC50 of 10.4 ± 5.3 μM. A similar trend was observed for the inverted terminal acetamides 29–33; while 29, containing an ethylene, was well tolerated, all longer side chains were found to be highly toxic.
Table 1.
Antiviral activity (EC50), toxicity (TC50), and selectivity index (SI; TC50/EC50) data for traditional and inverted-amide mercaptobenzamide prodrugs with variable side-chain length.
 | ||||
|---|---|---|---|---|
| Compound | R | EC50 [μM] | TC50 [μM] | SI |
| 25 |  |
1.2 ± 0.9 | 56.6 ± 0.5 | 47.2 |
| 26 |  |
3.7 ± 2.8 | 51.7 ± 2.7 | 14.0 |
| 27 | 3.1 ± 2.5 | 16.3 ± 3.3 | 5.26 | |
| 28 | 1.0 ± 0.8 | 10.4 ± 5.3 | 10.4 | |
| 29 |  |
2.6 ± 2.3 | 91.9 ± 11.5 | 35.3 |
| 30 |  |
1.1 ± 0.7 | 21.1 ± 7.3 | 19.2 |
| 31 |  |
0.57 ± 0.01 | 17.4 ± 0.8 | 30.5 |
| 32 | 3.9 ± 3.9 | 9.2 ± 3.7 | 2.36 | |
| 33 | 1.5 ± 1.4 | 8.0 ± 6.6 | 5.33 | |
We hypothesized that as the side chain was lengthened, a desirable arrangement of amide hydrogen bonds became increasingly disfavored entropically. To test this theory, we studied the effects of conformationally restricting the side-chain backbone (Table 2). Mercaptobenzamides containing 2-, 3-, and 4-aminobenzamide side chains were prepared and tested in HIV-infected CEM-SS cells [18]. Although all three derivatives displayed some toxicity, the aminobenzamide 34, containing an ortho arrangement between internal and terminal amides, was noticeably less toxic (TC50 = 50.1 ± 3.3 μM) than the meta derivative 35 and the para derivative 36. We also explored a vicinal arrangement of amides constrained within a cyclopentane ring. As such derivatives have two stereocenters, we prepared both racemic (37–38) and enantiopure stereoisomers (39–42) for analysis. Although significant differences in toxicity were observed between cis (TC50 ~ 90 μM) and trans (TC50 ~ 40 μM) arrangements, toxicity differences within each enantiomeric pair were surprisingly negligible.
Table 2.
Antiviral activity (EC50), toxicity (TC50), and selectivity index (SI; TC50/EC50) data for mercaptobenzamide prodrugs with conformationally constrained internal and terminal amides.
 | ||||
|---|---|---|---|---|
| Compound | R | EC50 [μM] | TC50 [μM] | SI |
| 34 |  |
5.1 ± 7.0 | 50.1 ± 3.3 | 9.82 |
| 35 |  |
1.2 ± 0.7 | 17.5 ± 5.9 | 14.6 |
| 36 | 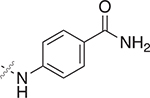 |
1.6 ± 0.7 | 39.0 ± 4.9 | 24.4 |
| 37 (rac) |  |
0.9 ± 0.7 | 31.2 ± 21.9 | 34.7 |
| 38 (rac) |  |
5.32 ± 0.01 | 89.9 ± 14.5 | 16.9 |
| 39 (1R,2R) |  |
1.7 ± 1.3 | 43.7 ± 1.1 | 25.7 |
| 40 (1S,2S) |  |
2.9 ± 2.3 | 43.3 ± 11.5 | 15.0 |
| 41 (1R,2S) |  |
1.8 ± 2.1 | 90.0 ± 17.3 | 50.0 |
| 42 (1S,2R) |  |
3.0 ± 1.4 | 84.8 ± 1.6 | 28.3 |
2.2.2. Heteroatom substitution
We next explored the effect of heteroatom substitution on the activity and toxicity profile of the mercaptobenzamide prodrugs (Table 3). Using the glycinamide side-chain as a scaffold, each amide NH was systematically replaced with an oxygen atom, yielding the internal ester 43 and terminal methyl ester 44. Both derivatives completely lost antiviral activity (EC50 > 100 μM), while simultaneously gaining toxicity. The lower toxicity of 43 (TC50 = 82.1 ± 18.1 μM) relative to 44 (TC50 = 27.3 ± 2.8 μM) may be related to the relative importance of hydrogen bond donor capability at the terminal position, a notion corroborated by the vastly different toxicities obtained by varying the position of the terminal amide (vide supra). Alternatively, one or both ester derivatives may be a substrate for endogenous esterases, thus affecting the interpretation of activity and toxicity trends. We also investigated the effect of substituting sulfur with selenium, using both the glycinamide and β-alaninamide side chains. Both selenobenzamide prodrugs (45 and 46) proved to be highly toxic (TC50 ~ 10 μM), a surprising result given the high tolerability of ebselen derivatives [19]. As selenium is both more redox active and nucleophilic than sulfur, the toxicity observed herein may reflect the subtle disruption of one or multiple mercaptobenzamide reactivity modes.
Table 3.
Antiviral activity (EC50), toxicity (TC50), and selectivity index (SI; TC50/EC50) data for heteroatom-substituted mercaptobenzamide prodrug analogs.
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | X | Y | Z | n | EC50 [μM] | TC50 [μM] | SI |
| 43 | S | O | NH2 | 1 | >100 | 82.1 ± 18.1 | – |
| 44 | S | NH | OCH3 | 1 | >100 | 27.3 ± 2.8 | – |
| 45 | Se | NH | NH2 | 1 | >100 | 6.8 ± 0.7 | – |
| 46 | Se | NH | NH2 | 2 | 8.6 ± 1.6 | 10.5 ± 9.7 | 1.22 |
2.2.3. Methyl scan, homologation, and dehomologation
Given the apparent importance of both the internal and terminal amides, we wished to investigate the relevance of hydrogen bond donor and acceptor ability at each moiety. To this end, each amide nitrogen was systematically methylated, producing the series of derivatives 47–51 (Table 4). While mono-methylation of the terminal amide (47) was tolerated, the N,N-dimethylated derivative 48 gained considerable toxicity yet maintained low micromolar antiviral activity. Strikingly, derivatives containing a tertiary amide at the internal position (49–52) completely lost activity, regardless of terminal amide substitution. Except for the prolinamide 52, these analogs were also moderately toxic, with TC50 values of ~60 μM. The activity/toxicity profile of the methylated series strongly suggests that the internal and terminal amides play different roles within the cell. Terminal amide substitution appears to exclusively influence toxicity, while modification of the internal amide profoundly affects antiviral activity. Further corroborating evidence was obtained from the highly toxic derivatives 53 and 54, which completely lack a terminal amide. As a consequence of low micromolar TC50 values, the assay could not reliably gauge the antiviral properties of these analogs. Conversely, the homologated derivative 55, which maintains the terminal amide in a desirable position but loses aromatic conjugation to the internal amide, was both inactive and non-toxic. Collectively, these results suggest that the internal amide is intricately involved in the mechanism of the mercaptobenzamides.
Table 4.
Antiviral activity (EC50), toxicity (TC50), and selectivity index (SI; TC50/EC50) data for “methyl-scanned”, homologated, and dehomologated mercaptobenzamide prodrugs.
 | ||||
|---|---|---|---|---|
| Compound | R | EC50 [μM] | TC50 [μM] | SI |
| 47 |  |
1.4 ± 1.3 | >100 | 71.4 |
| 48 |  |
4.7 ± 2.6 | 38.3 ± 0.6 | 8.15 |
| 49 |  |
>100 | 55.3 ± 8.8 | – |
| 50 |  |
>100 | 62.5 ± 10.0 | – |
| 51 |  |
>100 | 57.3 ± 8.6 | – |
| 52 | 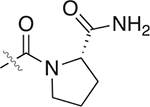 |
>100 | >100 | – |
| 53 |  |
>100 | 5.7 ± 0.8 | – |
| 54 |  |
>100 | 12.4 ± 7.6 | – |
| 55 |  |
>100 | >100 | – |
2.2.4. Terminus modification
Since mono-substitution of the terminal amide appeared to be well tolerated, we explored the possibility of using this position as a handle for further derivatization. In this vein, we synthesized a series of analogs containing a variety of sterically and electronically demanding substituents for biological testing (Table 5). Unsurprisingly, all analogs within this series had similar, low micromolar EC50 values, consistent with the observation that the antiviral activity of the mercaptobenzamides does not appear to be affected by substitution of the terminal amide. Toxicity trends, however, varied widely. While small aliphatic substituents, such as isopropyl (56) or propargyl (57) were tolerated, larger increases in hydrophobicity, such as benzyl (58), pentyl (59), and tert-butyl (60) were not. Polar substituents, on the other hand, displayed limited toxicity, regardless of cationic (61), anionic (62, after exposure to esterases), or neutral (63) character. Importantly, the tolerability of alkynes enables future click-derivatization, particularly with highly polar azide-containing fragments.
Table 5.
Antiviral activity (EC50), toxicity (TC50), and selectivity index (SI; TC50/EC50) data for terminus-modified mercaptobenzamide prodrugs.
 | ||||
|---|---|---|---|---|
| Compound | R | EC50 [μM] | TC50 [μM] | SI |
| 56 |  |
5.0 ± 3.1 | 92.0 ± 11.4 | 18.4 |
| 57 |  |
4.7 ± 0.2 | 95.4 ± 6.6 | 20.3 |
| 58 | 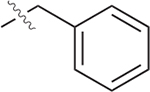 |
4.8 ± 0.8 | 29.3 ± 10.4 | 6.10 |
| 59 | 10.3 ± 7.0 | 24.8 ± 19.5 | 2.41 | |
| 60 |  |
9.1 ± 6.7 | 46.8 ± 11.2 | 5.14 |
| 61 |  |
6.6 ± 3.4 | 76.9 ± 10.8 | 11.6 |
| 62 |  |
4.9 ± 1.1 | >100 | 20.4 |
| 63 |  |
27.5 ± 25.7 | >100 | 3.64 |
2.2.5. SAR Conclusions
Of the 39 mercaptobenzamides examined in this manuscript, the mono-methylated terminal amide 47 had the greatest selectivity index (SI; 71.4). Consistent with our SAR, 47 uniquely possessed the following structural features: 1) a secondary internal amide, 2) a terminal amide with at least one hydrogen-bond donor, and 3) a minimally hydrophobic skeleton. Despite the favorable SI, the EC50 value of mercaptobenzamide 47 was not statistically different than that of the original prodrug 5, nor that of the majority of active compounds explored previously or herein (all low single-digit micromolar). While we chose not to explore 47 computationally due to its resemblance to 5 (both in terms of structure and activity/toxicity profile), we were inspired to further investigate this apparent limit in antiviral activity (see section 2.4).
2.3. Computational modeling and docking
The surprising activity/toxicity profile of the cyclopentane-constrained stereoisomers (39–42) inspired us to pursue a computational model that could provide some insight into the empirical data. We hypothesized that the greater toxicity of the trans isomers could be associated with lower NCp7 C-terminal zinc finger binding affinity, potentially leading to an increase in off-target effects and therefore increases in toxicity.
Improving upon our previous model [14] of SAMT-247 (1) “manually” docked to an NMR-derived ensemble structure of the NCp7 C-terminal zinc finger [20], 500-ns molecular dynamics trajectories were generated for three different starting orientations of 1 confined at the thioester carbonyl carbon to within 3.75 Å of the Cys36 sulfur atom. Snapshots were saved every 10 ps, and each snapshot was scored using the AutoDock-Vina energy function [21], revealing two trajectories of similarly low energy (lowest energy conformations displayed in Fig. 3). The first trajectory led to “conformation A,” which was similar to that previously obtained using “manual” docking. Within this conformation, SAMT-247 (1) is stabilized via π-stacking with W37, hydrophobic interactions with the aliphatic portion of K34, and hydrogen bonding to Q45 via the terminal amide. The second trajectory led to “conformation B,” which has not been previously observed. As can be seen from Fig. 3, 1 has lost interactions with K34 and Q45, but gains hydrogen bonds to R52 and the backbone of W37, while maintaining π-stacking with the tryptophan moiety. Using conformations A and B as starting points, scored trajectories were generated for the acetyl thioester of all four cyclopentane-derived stereoisomers. The lowest energy 10 ps snapshot for each isomer is presented below (trajectories starting from conformation A – Fig. 4A; trajectories starting from conformation B – Fig. 4B).
Fig. 3.

Lowest energy snapshots of 500-ns molecular dynamics simulation of SAMT-247 (1) binding to the NMR-ensemble structure of the C-terminal zinc finger of NCp7 in previously explored conformation A (left) and newly discovered conformation B (right). The AutoDock-Vina energy scores are shown in the upper left of each conformation window.
Fig. 4.

Lowest energy snapshots of 500-ns molecular dynamics simulation of cyclopentane-constrained mercaptobenzamide thioesters binding to the NMR-ensemble structure of the C-terminal zinc finger of NCp7 in (A) previously explored conformation A and (B) newly discovered conformation B. The AutoDock-Vina energy scores are shown in the upper right of each conformation window.
Although the lowest energy snapshot for each stereoisomer indicated a distinct conformation, the stabilizing interactions made by each compound appeared to depend less on stereochemistry and more on starting orientation. The lowest energy snapshots for all trajectories indicated some degree of π-stacking with W37; trajectories starting from conformation A achieved a hydrogen bond with Q45, while those starting from conformation B maintained a hydrogen bond to K38 or R52 instead. Notably, these contacts are essentially identical to those formed by the parent mercaptobenzamide thioester, SAMT-247 (1), which was calculated to have similar energies for both conformations A and B. As a result, the AutoDock-Vina score for all cyclopentane stereoisomers, regardless of conformation, is approximately −5.0. To demonstrate that the choice of scoring function was inconsequential, the calculated trajectories were also scored using a Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA) protocol [22]. Although the lowest energy snapshots indicated far greater variation than what was obtained with the AutoDock-Vina score function, these differences disappeared upon averaging across 100–500 ns; within a standard deviation, all cyclopentane stereoisomers were of comparable affinity (see Supporting Information).
Since all cyclopentane isomers bind the C-terminal zinc finger with nearly identical affinity, our initial hypothesis that antiviral activity and cytotoxicity are correlated through the strength of zinc finger binding appears incorrect. While the origin of cellular toxicity remains unknown, the models described herein provide insight into the shallow NCp7 binding landscape, potentially inspiring synthetic efforts to span both conformations A and B.
2.4. Limits in antiviral activity suggest thermodynamic control
The consistent EC50 values observed across current and previous mercaptobenzamide analogs suggest that this class of molecules may have reached a thermodynamically pre-determined activity limit that cannot advance beyond low micromolar values. Given the complexities of the mercaptobenzamide mechanism, we suspect that a low equilibrium concentration of active thioester (SAMT) may be responsible for this activity limit. Upon entering the cell, mercaptobenzamide prodrugs such as 5 are cleaved by endogenous esterases to the thiolate 2 (Fig. 5A). As 2 appears to be an inert resting-state for the mercaptobenzamide activity cycle, reaction with acetyl-CoA to generate the SAMT 1 is critical for antiviral activity [9]. The SAMT is then primed for acetyl transfer to cytosolic and proteinogenic thiols, regenerating the thiolate 2. To a first approximation, the equilibrium concentration of active thioester 1 can be calculated from the concentrations of acetyl-CoA, glutathione (the most abundant cytosolic thiol), and the equilibrium constant for thiol–thioester exchange.
Fig. 5.

A) First-approximation activity limit calculations using the following assumptions: mercaptobenzamide prodrugs are quantitatively cleaved by esterases to the thiolate 2, which can either passively diffuse across the cell membrane or accumulate within cells. Intracellularly, an equilibrium is established between the thiolate 2 and the mercaptobenzamide thioester 1 which depends solely on the concentration of glutathione (acetyl sink) and acetyl-CoA (acetyl source). B) 13C NMR spectrum of mercaptobenzamide 2 (SH) and acetyl-1,2–13C2 CoA (10:1 stoichiometric ratio) after incubation in degassed 0.1 M pH 7.0 phosphate buffer (4:1 H2O/D2O) with 2% N,N-dimethylformamide for 12 h at 37 °C.
To determine the equilibrium constant (Fig. 5B), a 10:1 stoichiometric ratio of the thiol 2 and acetyl-1,2–13C2 CoA were incubated in degassed 0.1 M pH 7.0 phosphate buffer (4:1 H2O/D2O) with 2% N,N-dimethylformamide for 12 h at 37 °C (monitoring by 13C NMR indicated that 12 h was sufficient for equilibrium to be established). The 13C NMR spectrum was integrated and the equilibrium concentrations thus attained were used to calculate a thiol–thioester exchange equilibrium constant of 0.0023 (see experimental section for more details).
Using the calculated equilibrium constant and additional information about the antiviral assay, we hoped to approximate the amount of SAMT formed within cells. During the course of an antiviral experiment, the prodrug 5 is added to a 96-well plate where each well contains 2500 cells (where an individual cell volume is 282.9 fL) [23] in a total well volume of 200 μL. Each cell contains approximately 2.6 mM glutathione [24] and 10 μM acetyl-CoA [25]. After 5 enters a cell, it is cleaved by esterases to the thiolate 2, which is likely the major species within the cell. To form the active species 1, 2 must react with acetyl CoA. Under thermodynamic control, the concentration of 1 will depend on the intracellular concentrations of glutathione, acetyl-CoA, and 2. As we are unable to quantify the precise intracellular concentration of 2, we considered two potential theoretical scenarios at opposite extremes: (A) both the prodrug 5 and the thiolate 2 passively diffuse across the cell membrane, resulting in an intracellular concentration of 2 that is identical to the initial concentration of 5, or (B) the thiolate 2 is unable to passively diffuse across the cell membrane, causing 2 to completely concentrate within the cell after esterases remove the prodrug.
Using the parameters above and assuming that 2 is able to passively diffuse between cells and solution, an initial concentration of 1 μM of 5 would result in an intracellular concentration of 1 μM of 2 after esterase cleavage. In this scenario, the equilibrium constant between 2 and acetyl-CoA predicts the formation of approximately one molecule of SAMT-247 (1) per cell. As it is improbable that one molecule of 1 per cell can account for EC50 values in the low micromolar range, it is therefore likely that the thiolate 2 concentrates to some degree within the cell.
At the other extreme, if 2 is completely unable to diffuse across cell membranes, it would accumulate within cells after its formation via esterase cleavage of 5. In this scenario, an initial concentration of 1 μM of 5 would afford a 283mM solution of the thiolate 2 within each cell. This value corresponds to the maximum theoretical thiolate concentration as it requires the assumption that all thiolate is trapped and concentrated within the cells and undergoes no reactions aside from acetyl transfer. In this scenario, our NMR-derived equilibrium constant predicts the steady-state intracellular concentration of 1 to be 2 μM. This value is comparable to the cytosolic Gag concentration of >500 nM [26] and is similar to the EC50 values for the molecules in this study (see experimental section for more details). Prodrug well concentrations lower than 1 μM would further reduce the concentration of 1 available in the cell, limiting the inhibition of virus-induced cytopathic effects to beneath detectable levels. These approximations are consistent with observed antiviral data. Moreover, given the high concentration of cellular glutathione and the thermodynamic stability of alkyl thioesters relative to aryl thioesters, structural changes to either the ring or sidechain of the mercaptobenzamides are unlikely to significantly modify the available concentration of active SAMTs. Since all EC50 values for the molecules in this study are in the low micromolar range, we propose that the thiolates are likely concentrated to some degree within the cells and that the antiviral activity is closely correlated with thermodynamic control over formation of the active SAMT.
3. Conclusion
In summary, a thorough exploration of mercaptobenzamide side-chain SAR exposed divergent activity/toxicity roles for the internal and terminal amides and revealed viable handles for further derivatization. To probe the relationship between antiviral activity and toxicity, we generated an improved computational model for the transient binding of SAMTs to the NCp7 C-terminal zinc finger, revealing a second viable binding mode. Finally, we proposed a thermodynamically plausible explanation for the low micromolar activity of mercaptobenzamides, using NMR-derived thiol–thioester exchange equilibrium constants. While further SAR may not improve the activity limit, such efforts are invaluable for constructing an activity/toxicity profile for this therapeutically-promising class of molecules, and for enabling future mechanistic and NCp7-targeting experiments.
4. Experimental section
4.1. General experimental methods
4.1.1. Anti-HIV cytoprotection assay
Inhibition of virus-induced cytopathic effects (CPE) and cell viability following HIV replication in CEM-SS cells was measured by XTT tetrazolium dye. CEM-SS cells (2.5 × 103 cells per well) were seeded in 96-well U-bottomed tissue culture plates in RPMI medium and supplemented with 10% FBS, 2mM L-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin. Serially diluted compounds (6 concentrations) and HIV-1RF diluted to a pre-determined titer to yield 85–95% cell killing at 6 days post-infection were added to the plate. AZT was evaluated in parallel as a positive control. Following incubation at 37 °C, 5% CO2 for six days, cell viability was measured by XTT staining. The optical density of the cell-culture plate was determined spectrophotometrically at 450 and 650 nm using Softmax Pro 4.6 software. Percent CPE reduction of the virus-infected wells and the percent cell viability of the uninfected drug control wells was calculated to define the EC50, TC50 and the selectivity index (SI) using Microsoft Excel Xlfit4.
4.1.2. Computer analyses of complexes
Molecular dynamics simulations were used to sample the interactions of SAMT-247 (1) and its analog cyclopentane enantiomers to the C-terminal zinc finger of NCp7. To concentrate on the state just prior to the initial acetylation, the acetyl carbonyl carbon of 1 and the other ligands was harmonically constrained to a distance of 3.75 Å from the sulfur atom of the Cys36 residue ligand of the zinc ion. To minimize bias, two simulations were run for each ligand with its “side-chain” starting in alternate positions relative to the zinc finger (vide supra). Because it has been observed that the structure of the zinc finger remains relatively constant during reaction [12], the alpha carbons of the peptides were also harmonically constrained to their starting structures throughout the simulations. The Molecular Dynamic simulations were conducted with the NAMD 2.12 software [27]. Topologies, parameters and 3D coordinates for 1 and the four cyclopentane enantiomers (i.e., RR, RS, SR & SS) were generated with Charmm-GUI [28]. The 3D structure of the C-terminal zinc finger was derived from Model 2 of PDB ID: 1MFS from the RCSB Protein Databank [29], truncating to residues 32–52 (standard nucleocapsid numbering) [20]. The Force Field was CHARMM36m [30]. Each complex was centered in a box of initial dimensions 50.0 × 44.0 × 38.0 Å, and VMD was used to solvate with TIP3P water molecules and add Na+ and Cl− ions to neutralize and form a concentration of 150 mM [31]. Nonbonded real and image interactions were calculated with a switching distance of 10.0 Å and a cutoff of 12.0 Å. All covalent bonds were kept rigid to allow for a 2.0 fs integration step. All simulations were run for a minimum of 500.0 ns with constant pressure at 1 atm and temperature at 300 K, saving coordinate snapshots every 10.0 ps. Subsequently, each snapshot of the trajectories was scored for ligand binding affinity using two methods: the AutoDock Vina software [21] and a Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA) protocol [22] implemented with the CHARMM v. 39 software [32]. For the latter, the free energy of binding was determined from the difference in calculated free energy of the complex minus that of the protein and ligand individually. The “single-trajectory” approach was used, which ignores possible conformational changes of the protein and ligand upon binding. This causes the Molecular Mechanics component of the energy to reduce to just the in vacuo interaction energy (electrostatic and VDW). The polar component of the solvation energy was calculated with the PBEQ module of CHARMM, with a grid-scale of 0.4 Å/unit, a minimum of 6 Å from the protein or ligand to the boundary, an ionic strength of 0.15 M, an ion and water radius of 1.4 Å, a protein and ligand dielectric constant of 4.0, and external dielectric constants of 80.0 and 1.0 for the solvent and vacuum calculations, respectively. The nonpolar component of the solvation energy was calculated from the Solvent-Accessible Surface Area using the COOR SURFACE command in CHARMM, with a probe radius of 1.4 Å. The following equation was used: ΔG = γ x ΔArea(complex – protein – ligand) + b, where γ = 0.00542 kcal/mol/Å2 and b = 0.92 kcal/mol [33]. No attempt was made to account for the entropic component of the binding energy, such as the change in conformational ensembles for each upon complex formation.
4.1.3. Equilibrium constant determination [13C2-AcCoA–mercaptobenzamide (2, SH)]
A solution of the mercaptobenzamide 2 (SH; 4.0 mg, 18.0 μmol, 1 equiv) in N,N-dimethylformamide (12 μL) and a solution of acetyl-1,2–13C2 coenzyme A lithium salt (1.5 mg, 1.80 μmol, 0.10 equiv) in 0. 1 M phosphate buffer (pH 7.0, 4:1 H2O/D2O, 12 μL; deoxygenated by sparging with helium for 15 min) were sequentially added to 0.1 M phosphate buffer (pH 7.0, 4:1 H2O/D2O, 576 μL; deoxygenated by sparging with helium for 15 min) in an NMR tube. The NMR tube was sealed with a standard cap, and the reaction mixture was incubated at 37 °C for 12 h. A13C NMR spectrum was obtained of the product mixture, and the acetyl carbonyl carbon resonances were integrated (the acetyl carbonyl carbon of 13C2-247 (1) was set to 1.000). Using the following expressions: [CoASH]eqm = [13C2-247 (1)]eqm; [13C2-AcCoA]initial = [13C2-AcCoA]eqm + [CoASH]eqm; and [247-SH (2)]eqm = (10 × [13C2-AcCoA]initial) – [13C2-247 (1)]eqm, the equilibrium constant was determined using the following equation: . Notably, an incubation time of 48 h did not noticeably affect the 13C NMR spectrum, indicating that equilibrium had been achieved by 12 h. Furthermore, while some thiol oxidation to the insoluble disulfide was observed, rigorous deoxygenation techniques, such as longer sparge times, freeze-pump-thaw, argon-filled glove bags, and the use of Schlenk NMR tubes, were found to be unnecessary; while such techniques decreased the amount of visible precipitation, their effect on the calculated equilibrium constant was negligible.
4.1.4. Activity limit calculations (assumption – passive diffusion of the thiolate 2)
The following standard parameters were used: 96-well plate well volume = 200 μL; cells/well = 2500; PBMC volume = 282.9 fL [23]; prodrug concentration (in well) = 1 μM; glutathione concentration = 2.6 mM [24]; acetyl-CoA concentration = 10 μM [25]. Activity limit calculations required the following assumptions: 1) The prodrug passively diffuses into cells, where it is quantitatively cleaved by esterases to the thiolate. The thiolate is then able to passively diffuse across the cell membrane, causing the thiolate concentration within cells to become equal to the starting concentration of the prodrug; 2) Within each cell, the equilibrium between the mercaptobenzamide thiolate and the mercaptobenzamide thioester is dictated exclusively by the experimentally determined equilibrium constant from the reaction of 13C2-acetyl CoA and mercaptobenzamide 2 (SH), and the concentrations of the most abundant acetyl source (acetyl-CoA) and acetyl sink (glutathione). Given this set of assumptions, the concentration of thiolate becomes 1 μM. Using the empirically determined equilibrium constant, the following expression can be set up: . Solving for x, an equilibrium concentration of 8.8 pM is obtained for the mercaptobenzamide thioester. Using the aforementioned cell volume, the number of thioester molecules/cell is calculated to be ~1.
4.1.5. Activity limit calculations (assumption – no diffusion of the thiolate 2)
The following standard parameters were used: 96-well plate well volume = 200 μL; cells/well = 2500; PBMC volume = 282.9 fL [23]; prodrug concentration (in well) = 1 μM; glutathione concentration = 2.6mM [24]; acetyl-CoA concentration = 10 μM [25]. Activity limit calculations required the following assumptions: 1) The prodrug passively diffuses into cells, where it is quantitatively cleaved by esterases to the thiolate. As the thiolate is charged, it is unable to passively diffuse out of the cells, and all mercaptobenzamide molecules are eventually concentrated within the total volume of all cells; 2) Within each cell, the equilibrium between the mercaptobenzamide thiolate and the mercaptobenzamide thioester is dictated exclusively by the experimentally determined equilibrium constant from the reaction of 13C2-acetyl CoA and mercaptobenzamide 2 (SH), and the concentrations of the most abundant acetyl source (acetyl-CoA) and acetyl sink (glutathione). Given the prodrug well concentration and the well volume, the amount of prodrug per well is calculated to be 0.2 nmol. Once concentrated within the total volume of all cells (0.71 nL), the concentration of thiolate becomes 283 mM. Using the empirically determined equilibrium constant, the following expression can be set up: . Solving for x, an equilibrium concentration of 2 μM is obtained for the mercaptobenzamide thioester. It is worth noting that given the assumptions being used, this value approximates the theoretical maximum concentration of active SAMT.
4.2. General experimental procedures
All reactions were performed in single-neck, oven-dried (130 °C for 12 h), round-bottomed flasks fitted with rubber septa under a positive pressure of argon, unless otherwise specified. Organic solutions were concentrated by rotary evaporator at 30–50 °C, as noted. Analytical thin-layered chromatography (TLC) was performed using plates pre-coated with silica gel (Sigma Aldrich; 60 Å, 17 μm particle size), impregnated with a fluorescent indicator (254 nm). TLC plates were visualized by exposure to ultraviolet (UV) light, submersion in butanolic ninhydrin solution, or basic potassium permanganate solution, followed by brief heating with a Master-Mite heat gun (hot setting, 30 s). Flash-column chromatography was performed using a Biotage Isolera One automated flash purification system with Biotage SNAP KP-Sil cartridges.
4.3. Materials
Commercial solvents were obtained from Millipore Sigma. Commercial reagents were obtained from Combi-Blocks, Inc. with the following exceptions: 7M ammonia solution in methanol (Millipore Sigma), 2M methylamine solution in tetrahydrofuran (Millipore Sigma), sodium bicarbonate (Millipore Sigma), sodium chloride (Millipore Sigma), sodium sulfate (Millipore Sigma), ammonium chloride (Millipore Sigma), acetic anhydride (Millipore Sigma), N,N-di-iso-propylethylamine (Millipore Sigma), triethylamine (Millipore Sigma), thiosalicylic acid (Millipore Sigma), HATU (Millipore Sigma), sodium borohydride (Millipore Sigma), acetic acid (Millipore Sigma), 2-mercaptophenylacetic acid (Millipore Sigma), 2-chloroacetamide (Millipore Sigma), l-prolinamide (Millipore Sigma), glycine methyl ester hydrochloride (Millipore Sigma), anthranilic acid (Millipore Sigma), benzylamine (Millipore Sigma), 1,1′-carbonyldiimidazole (Oakwood Products, Inc.), 4M hydrogen chloride solution in dioxane (Oakwood Products, Inc.), N,N′-dimethylethylenediamine (Oakwood Products, Inc.), 2-aminobenzamide (Oakwood Products, Inc.), 3-aminobenzamide (Oakwood Products, Inc.), 4-aminobenzamide (Oakwood Products, Inc.), 2-hydroxyacetamide (Chem-Impex International, Inc.), trans-2-((tert-butoxycarbonyl)amino)cyclopentane-1-carboxylic acid (Chem-Impex International, Inc.), cis-2-((tert-butoxycarbonyl) amino)cyclopentane-1-carboxylic acid (Chem-Impex International, Inc.), 200 mesh selenium powder (Acros Organics), β -alaninamide hydrochloride (Pharmaron, Inc.), propan-2-amine (Fisher Scientific), pentan-1-amine (Fisher Scientific), 2-methylpropan-2-amine (Fisher Scientific), (1S,2S)-2-((tert-butoxycarbonyl)amino)cyclopentane-1-carboxylic acid (4DrugDiscovery, LLC), (1R,2R)-2-((tert-butoxycarbonyl) amino)cyclopentane-1-carboxylic acid (4DrugDiscovery, LLC), (1S,2R)-2-((tert-butoxycarbonyl)amino)cyclopentane-1-carboxylic acid (4DrugDiscovery, LLC), (1R,2S)-2-((tert-butoxycarbonyl) amino)cyclopentane-1-carboxylic acid (4DrugDiscovery, LLC). Anhydrous commercial solvents were dispensed from a JC Meyer solvent drying system. Commercial reagents were used as received.
4.4. Instrumentation
1H NMR spectra were recorded at 25 °C at 400 MHz or 500 MHz. Chemical shifts are expressed in parts per million (ppm, δ scale) downfield from tetramethylsilane and are referenced to residual protium in the NMR (DMSO-d6, δ 2.50; CD3OD, δ 3.31). Data are represented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, sex = sextet, sep = septet, oct = octet, m = multiplet and/or multiple resonances, br = broad, app = apparent), coupling constant in Hertz, integration, and major/minor rotamer, designated A/B, if applicable. Proton-decoupled carbon nuclear magnetic resonance spectra (13C NMR) were recorded at 100 MHz or 125 MHz at 25 °C. Chemical shifts are expressed in parts per million (ppm, δ scale) downfield from tetramethylsilane and are referenced to the carbon resonances of the solvent (DMSO-d6, δ 39.5; CD3OD, δ 49.0). Proton-decoupled selenium nuclear magnetic resonance spectra (77Se NMR) were recorded at 76 MHz at 25 °C. Chemical shifts are expressed in part per million (ppm, δ scale) relative to diphenyldiselenide (δ 364.0). High-resolution mass spectrometry (HRMS) data were obtained using a Waters Xevo-G2 XS qTOF™ instrument. Optical rotations were measured on a Molecular Devices SpectraMax Plus384 polarimeter equipped with a sodium (589 nm, D) lamp. Optical rotation data are represented as follows: specific rotation ([α]DT, concentration (g/100 mL), and solvent).
4.5. Synthetic procedures
4.5.1. General procedure A for the synthesis of amine hydrochlorides of general structure 15
A 20-mL vial was charged with α-chloroamide (specified below; 1 equiv) and a solution of ammonia in methanol (7.00 M, 5.00 mL, 18.8–21.3 equiv). The reaction mixture was stirred for 24 h at 23 °C. The product mixture was concentrated, providing the amine hydrochloride of general structure 15. The product so obtained was used without further purification.
4.5.2. General procedure B for the synthesis of amine hydrochlorides of general structure 15
A 20-mL vial was charged with α-chloroamide (specified below; 1 equiv) and a solution of methylamine in tetrahydrofuran (2.00 M, 5.00 mL, 4.68–6.08 equiv). The reaction mixture was stirred for 24 h at 23 °C. The product mixture was concentrated, providing the amine hydrochloride of general structure 15. The product so obtained was used without further purification.
4.5.3. General procedure C for the synthesis of amine hydrochlorides of general structure 17
A 20-mL vial was charged with Boc-protected amino acid (specified below; 1 equiv) and tetrahydrofuran (0.15 M). 1,1′-Carbonyldiimidazole (1.50 equiv) was added and the resulting mixture was stirred for 1 h at 23 °C. A solution of ammonia in methanol (7.00 M, 5.00 equiv) was added to the reaction mixture, and the resulting mixture was stirred for 12 h at 23 °C. The product mixture was concentrated, and the residue obtained was diluted with ethyl acetate (120 mL). The diluted product mixture was transferred to a separatory funnel and sequentially washed with saturated aqueous ammonium chloride solution (1 × 50 mL), saturated aqueous sodium bicarbonate solution (1 × 50 mL), and saturated aqueous sodium chloride solution (1 × 50 mL). The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The product thus obtained was transferred to a 20-mL vial. A solution of hydrogen chloride in dioxane (4.00 M, 5.00 equiv) was added and the resulting mixture was stirred at 23 °C for 1 h. The product mixture was concentrated, providing the amine hydrochloride of general structure 17. The product so obtained was used without further purification.
4.5.4. General procedure D for the synthesis of amine hydrochlorides of general structure 18
A 20-mL vial was charged with Boc-Gly-OH (1 equiv) and N,N-dimethylformamide (0.20 M). The reaction mixture was cooled to 0 °C. 1,1′-Carbonyldiimidazole (1.20 equiv) was added and the resulting mixture was warmed to 23 °C over 30 min. The warmed reaction mixture was stirred at 23 °C for 1 h. Amine (specified below; 1.50 equiv) and N,N-di-iso-propylethylamine (2.00 equiv) were sequentially added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 1 h. The product mixture was concentrated, and the residue obtained was diluted with ethyl acetate (120 mL). The diluted product mixture was transferred to a separatory funnel and sequentially washed with saturated aqueous sodium bicarbonate solution (1 × 50 mL), water (1 × 50 mL), and saturated aqueous sodium chloride solution (1 × 50 mL). The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The product thus obtained was transferred to a 50-mL round bottomed flask. A solution of hydrogen chloride in ethyl acetate (1.00 M, 5.00 equiv) was added and the resulting mixture was stirred at 23 °C for 48 h. The product mixture was concentrated, providing the amine hydrochloride of general structure 18. The product so obtained was used without further purification.
4.5.5. General procedure E for the synthesis of amine hydrochlorides of general structure 20
A 20-mL vial was charged with Boc-protected amino acid (specified below; 1 equiv) and dichloromethane (0.20 M). Acetic anhydride (1.10 equiv) and triethylamine (1.20 equiv) were sequentially added dropwise via syringe to the reaction mixture, and the resulting mixture was stirred at 23 °C for 68 h. The product mixture was concentrated, and the residue obtained was diluted with ethyl acetate (120 mL). The diluted product mixture was transferred to a separatory funnel and sequentially washed with saturated aqueous ammonium chloride solution (1 × 50 mL), saturated aqueous sodium bicarbonate solution (1 × 50 mL), and saturated aqueous sodium chloride solution (1 × 50 mL). The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The product thus obtained was transferred to a 20-mL vial. A solution of hydrogen chloride in dioxane (4.00 M, 10.0 equiv) was added and the resulting mixture was stirred at 23 °C for 20 h. The product mixture was concentrated, providing the amine hydrochloride of general structure 20. The product so obtained was used without further purification.
4.5.6. General procedure F for the synthesis of mercaptobenzamides of general structure 13
Chloromethyl butyrate (1.00 equiv) and N,N-di-iso-propylethylamine (3.50 equiv) were sequentially added to a solution of thiosalicylic acid (1 equiv) in N,N-dimethylformamide (0.20 M) and the resulting mixture was stirred at 23 °C for 20 h. HATU (1.00 equiv) was added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 1 h. Amine (specified below; 1.00 equiv) was added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 24 h. The product mixture was concentrated, and the residue obtained was diluted with ethyl acetate (120 mL). The diluted product mixture was transferred to a separatory funnel and sequentially washed with saturated aqueous sodium bicarbonate solution (1 × 50 mL), water (1 × 50 mL), and saturated aqueous sodium chloride solution (1 × 50 mL). The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue thus obtained was purified by flash-column chromatography (as noted) to provide the mercaptobenzamide of general structure 13.
4.5.7. General procedure G for the synthesis of mercaptobenzamides of general structure 13
Chloromethyl butyrate (1.00 equiv) and N,N-di-iso-propylethylamine (1.20 equiv) were sequentially added to a solution of thiosalicylic acid (1 equiv) in N,N-dimethylformamide (0.20 M) and the resulting mixture was stirred at 23 °C for 12 h. 1,1′-Carbonyldiimidazole (1.00 equiv) was added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 1 h. A solution of amine (specified below; 1.00 equiv) in N,N-dimethylformamide (4.0 mL) and N,N-di-iso-propylethylamine (2.30 equiv) were sequentially added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 12 h. The product mixture was concentrated, and the residue obtained was diluted with ethyl acetate (120 mL). The diluted product mixture was transferred to a separatory funnel and sequentially washed with saturated aqueous sodium bicarbonate solution (1 × 50 mL), water (1× 50 mL), and saturated aqueous sodium chloride solution (1 × 50 mL). The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue thus obtained was purified by flash-column chromatography (as noted) to provide the mercaptobenzamide of general structure 13.
4.5.8. General procedure H for the synthesis of diselenobenzamides of general structure 23
HATU (2.20 equiv) and N,N-di-iso-propylethylamine (2.50 equiv) were sequentially added to a solution of 2,2′-diselanediyldibenzoic acid (S1; 1 equiv) in N,N-dimethylformamide (0.10 M). The resulting mixture was stirred at 23 °C for 20 min. Amine hydrochloride (specified below; 2.50 equiv) and N,N-di-iso-propylethylamine (3.50 equiv) were sequentially added to the reaction mixture and the resulting mixture was stirred at 23 °C for 12 h. The product mixture was concentrated, and the residue obtained was purified by flash-column chromatography (eluting with dichloromethane initially, grading to 15% methanol–dichloromethane, linear gradient) to provide the diselenobenzamide of general structure 23.
4.5.9. General orocedure I for the synthesis of selenobenzamides of general structure 22
Sodium borohydride (2.00 equiv) was added to a solution of the diselenobenzamide (specified below; 1.00 equiv) in ethyl acetate (deoxygenated by sparging with argon for 15 min; 5.00 mL). The resulting mixture heated to 40 °C and stirred for 30 min. The reaction mixture was cooled to 23 °C. Acetic acid (8.00 equiv) was added to the cooled reaction mixture and the resulting mixture was stirred at 23 °C for 30 min. Chloromethyl butyrate (2.10 equiv) and N,N-di-iso-propylethylamine (10.0 equiv) were sequentially added to the reaction mixture and the resulting mixture was stirred at 23 °C for 12 h. The product mixture was concentrated, and the residue obtained was diluted with ethyl acetate (30 mL). The diluted product mixture was transferred to a separatory funnel and sequentially washed with water (1 × 20 mL) and saturated aqueous sodium chloride solution (1 × 20 mL). The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue thus obtained was purified by flash-column chromatography (eluting with 10% methanol–dichloromethane) to provide the selenobenzamide of general structure 22.
4.5.10. Synthesis of ((2-((4-amino-4-oxobutyl)carbamoyl)phenyl)thio)methyl butyrate (25)
The mercaptobenzamide 25, a white solid (35.5 mg, 18% over 3 steps), was synthesized by general procedure F using the amine hydrochloride obtained from 4-((tert-butoxycarbonyl)amino)butanoic acid via general procedure C. Flash-column chromatography: 1) eluted with dichloromethane initially, grading to 10% methanol–dichloromethane, linear gradient; 2) eluted with ethyl acetate initially, grading to 15% ethanol–ethyl acetate, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.38 (br t, J = 5.0 Hz, 1H), 7.58 (d, J = 7.7 Hz, 1H), 7.45 (t, J = 7.5 Hz, 1H), 7.42 (d, J = 7.5 Hz, 1H), 7.31 (t, J = 7.5 Hz, 1H), 7.28 (br s, 1H), 6.77 (br s, 1H), 5.46 (s, 2H), 3.19 (br q, J = 6.5 Hz, 2H), 2.32 (t, J = 7.3 Hz, 2H), 2.11 (t, J = 7.4 Hz, 2H), 1.70 (app quin, J = 7.3 Hz, 2H), 1.54 (app sex, J = 7.4 Hz, 2H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 174.0, 172.3, 167.4, 137.7, 133.6, 130.2, 128.8, 127.7, 126.3, 66.3, 38.8, 35.4, 32.6, 25.1, 17.9, 13.4. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 361.1198; found, 361.1204.
4.5.11. Synthesis of ((2-((5-amino-5-oxopentyl)carbamoyl)phenyl)thio)methyl butyrate (26)
The mercaptobenzamide 26, a white solid (41.0 mg, 20% over 3 steps), was synthesized by general procedure F using the amine hydrochloride obtained from 5-((tert-butoxycarbonyl)amino)pentanoic acid via general procedure C. Flash-column chromatography: 1) eluted with dichloromethane initially, grading to 10% methanol–dichloromethane, linear gradient; 2) eluted with ethyl acetate initially, grading to 15% ethanol–ethyl acetate, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.36 (br t, J = 5.4 Hz, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.45 (t, J = 7.6 Hz, 1H), 7.40 (d, J = 7.5 Hz, 1H), 7.31 (t, J = 7.4 Hz, 1H), 7.25 (br s, 1H), 6.72 (br s, 1H), 5.45 (s, 2H), 3.18 (br q, J = 6.6 Hz, 2H), 2.32 (t, J = 7.3 Hz, 2H), 2.07 (t, J = 6.9 Hz, 2H), 1.59–1.43 (m, 6H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 174.2, 172.3, 167.3, 137.8, 133.6, 130.2, 128.8, 127.6, 126.3, 66.3, 38.8, 35.4, 34.7, 28.7, 22.6, 17.9, 13.4. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 375.1354; found, 375.1356.
4.5.12. Synthesis of ((2-((6-amino-6-oxohexyl)carbamoyl)phenyl)thio)methyl butyrate (27)
The mercaptobenzamide 27, a white solid (66.2 mg, 27% over 3 steps), was synthesized by general procedure F using the amine hydrochloride obtained from 6-((tert-butoxycarbonyl)amino)hexanoic acid via general procedure C. Flash-column chromatography: 1) eluted with dichloromethane initially, grading to 10% methanol–dichloromethane, linear gradient; 2) eluted with ethyl acetate initially, grading to 15% ethanol–ethyl acetate, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.35 (t, J = 5.3 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.45 (t, J = 7.3 Hz, 1H), 7.39 (d, J = 7.5 Hz, 1H), 7.31 (t, J = 7.5 Hz, 1H), 7.23 (br s, 1H), 6.70 (br s, 1H), 5.45 (s, 2H), 3.17 (br q, J = 6.7 Hz, 2H), 2.32 (t, J = 7.3 Hz, 2H), 2.04 (t, J = 7.3 Hz, 2H), 1.58–1.44 (m, 6H), 1.34–1.25 (m, 2H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 174.3, 172.3, 167.3, 137.8, 133.5, 130.2, 128.8, 127.6, 126.3, 66.3, 38.9, 35.4, 35.1, 28.8, 26.2, 24.9, 17.9, 13.4. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 389.1511; found, 389.1506.
4.5.13. Synthesis of ((2-((7-amino-7-oxoheptyl)carbamoyl)phenyl)thio)methyl butyrate (28)
The mercaptobenzamide 28, a white solid (45.7 mg, 20% over 3 steps), was synthesized by general procedure F using the amine hydrochloride obtained from 7-((tert-butoxycarbonyl)amino)heptanoic acid via general procedure C. Flash-column chromatography: 1) eluted with dichloromethane initially, grading to 10% methanol–dichloromethane, linear gradient; 2) eluted with ethyl acetate initially, grading to 15% ethanol–ethyl acetate, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.34 (br t, J = 5.4 Hz, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.45 (t, J = 7.7 Hz, 1H), 7.39 (d, J = 7.4 Hz, 1H), 7.31 (t, J = 7.4 Hz, 1H), 7.23 (br s, 1H), 6.69 (br s, 1H), 5.45 (s, 2H), 3.18 (q, J = 6.7 Hz, 2H), 2.32 (t, J = 7.3 Hz, 2H), 2.03 (t, J = 7.4 Hz, 2H), 1.59–1.44 (m, 6H), 1.35–1.22 (m, 4H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 174.3, 172.3, 167.3, 137.9, 133.5, 130.1, 128.8, 127.6, 126.4, 66.3, 39.0, 35.4, 35.1, 28.9, 28.5, 26.3, 25.1, 17.9, 13.4. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 403.1667; found, 403.1663.
4.5.14. Synthesis of ((2-((2-acetamidoethyl)carbamoyl)phenyl)thio)methyl butyrate (29)
The mercaptobenzamide 29, a white solid (156 mg, 44% over 3 steps), was synthesized by general procedure G using the amine hydrochloride obtained from tert-butyl (2-aminoethyl)carbamate via general procedure E. Flash-column chromatography: eluted with ethyl acetate initially, grading to 50% ethanol–ethyl acetate, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.39 (br t, J = 5.3 Hz, 1H), 7.92 (br t, J = 5.3 Hz, 1H), 7.58 (dd, J = 8.3, 1.1 Hz, 1H), 7.48–7.44 (m, 2H), 7.31 (dt, J = 7.5, 0.9 Hz, 1H), 5.46 (s, 2H), 3.27–3.22 (m, 2H), 3.20–3.14 (m, 2H), 2.32 (t, J = 7.2 Hz, 2H), 1.81 (s, 3H), 1.54 (app sex, J = 7.3 Hz, 2H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.3, 169.4, 167.5, 137.2, 133.9, 130.4, 128.6, 127.8, 126.3, 66.2, 39.0, 38.2, 35.3, 22.7, 17.9, 13.4. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 361.1198; found, 361.1197.
4.5.15. Synthesis of ((2-((3-acetamidopropyl)carbamoyl)phenyl)thio)methyl butyrate (30)
The mercaptobenzamide 30, a white solid (115 mg, 32% over 3 steps), was synthesized by general procedure G using the amine hydrochloride obtained from tert-butyl (3-aminopropyl)carbamate via general procedure E. Flash-column chromatography: 1) eluted with ethyl acetate initially, grading to 50% ethanol–ethyl acetate, linear gradient; 2) eluted with dichloromethane initially, grading to 10% methanol–dichloromethane, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.35 (bt, J = 5.6 Hz, 1H), 7.85 (bt, J = 5.3 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.45 (td, J = 7.6 Hz, 1.5 Hz, 1H), 7.41 (dd, J = 7.6 Hz, 1.3 Hz, 1H), 7.31 (td, J = 7.5 Hz, 0.7 Hz, 1H), 5.46 (s, 2H), 3.20 (app q, J = 6.4 Hz, 2H), 3.09 (app q, J = 6.6 Hz, 2H), 2.32 (t, J = 7.2 Hz, 2H), 1.80 (s, 3H), 1.61 (app quin, J = 7.0 Hz, 2H), 1.54 (app sex, J = 7.3 Hz, 2H), 0.87 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.80, 169.61, 167.91, 138.18, 134.02, 130.72, 129.38, 128.11, 126.88, 66.79, 37.39, 36.86, 35.83, 29.68, 23.15, 18.34, 13.83. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 375.1354; found, 375.1349.
4.5.16. Synthesis of ((2-((4-acetamidobutyl)carbamoyl)phenyl)thio)methyl butyrate (31)
The mercaptobenzamide 31, a white solid (228 mg, 41%), was synthesized by general procedure G using N-(4-aminobutyl)acetamide. Flash-column chromatography: 1) eluted with ethyl acetate initially, grading to 50% ethanol–ethyl acetate, linear gradient; 2) eluted with dichloromethane initially, grading to 10% methanol–dichloromethane, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.36 (bt, J = 5.5 Hz, 1H), 7.82 (bt, J = 5.5 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.45 (td, J = 7.6 Hz, 1.5 Hz), 7.40 (dd, J = 7.6 Hz, 1.5 Hz), 7.30 (td, J = 7.6 Hz, 0.9 Hz, 1H), 5.45 (s, 2H), 3.19 (app q, J = 6.3 Hz, 2H), 3.03 (app q, J = 6.2 Hz, 2H), 2.32 (t, J = 7.3 Hz, 2H), 1.54 (app sex, J = 7.4 Hz, 2H), 1.49–1.42 (m, 4H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.78, 169.41, 167.84, 138.22, 134.02, 130.65, 129.30, 128.09, 126.80, 66.76, 39.18, 38.71, 35.81, 27.13, 27.00, 23.12, 18.32, 13.81. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 389.1511; found, 389.1515.
4.5.17. Synthesis of ((2-((5-acetamidopentyl)carbamoyl)phenyl)thio)methyl butyrate (32)
The mercaptobenzamide 32, a white solid (68.5 mg, 55%), was synthesized by general procedure G using N-(5-aminopentyl)acetamide. Flash-column chromatography: 1) eluted with ethyl acetate initially, grading to 20% ethanol–ethyl acetate, linear gradient; 2) eluted with dichloromethane initially, grading to acetone, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.34 (bt, J = 5.5 Hz, 1H), 7.80 (bt, J = 5.1 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.45 (td, J = 7.7 Hz, 1.4 Hz, 1H), 7.39 (dd, J = 7.5 Hz, 1.3 Hz, 1H), 7.30 (td, J = 7.5 Hz, 0.9 Hz, 1H), 5.45 (s, 2H), 3.18 (app q, J = 6.4 Hz, 2H), 3.01 (app q, J = 6.4 Hz, 2H), 2.32 (t, J = 7.2 Hz, 2H), 1.78 (s, 3H), 1.54 (app sex, J = 7.3 Hz, 2H), 1.48 (app quin, J = 7.4 Hz, 2H), 1.40 (app quin, J = 7.3 Hz, 2H), 1.33–1.28 (m, 2H), 0.87 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.80, 169.40, 167.85, 138.33, 133.99, 130.65, 129.34, 128.12, 126.84, 66.79, 39.37, 38.95, 35.84, 29.34, 29.20, 24.34, 23.14, 18.35, 13.84. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 403.1667; found, 403.1664.
4.5.18. Synthesis of ((2-((6-acetamidohexyl)carbamoyl)phenyl)thio)methyl butyrate (33)
The mercaptobenzamide 33, a pale yellow solid (133 mg, 50% over 3 steps), was synthesized by general procedure G using the amine hydrochloride obtained from tert-butyl (6-aminohexyl) carbamate via general procedure E. Flash-column chromatography: eluted with ethyl acetate initially, grading to 50% ethanol–ethyl acetate, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.34 (bt, J = 5.5 Hz, 1H), 7.79 (bt, J = 5.1 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.44 (td, J = 7.6 Hz, 1.6 Hz, 1H) 7.39 (dd, J = 7.7 Hz, 1.5 Hz, 1H), 7.30 (td, J = 7.5 Hz, 1.0 Hz, 1H), 5.45 (s, 2H), 3.18 (app q, J = 6.7 Hz, 2H), 3.00 (app q, J = 6.4 Hz, 2H), 2.31 (t, J = 7.3 Hz, 2H), 1.77 (s, 3H), 1.54 (app sex, J = 7.4 Hz, 2H), 1.48 (app quin, J = 7.0 Hz, 2H), 1.38 (app quin, J = 7.2 Hz, 2H) 1.33–1.24 (m, 4H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.80, 169.37, 167.83, 138.36, 133.98, 130.64, 129.32, 128.10, 126.85, 66.77, 39.40, 38.94, 35.84, 29.65, 29.47, 26.65, 23.14, 18.35, 13.84. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 417.1824; found, 417.1829.
4.5.19. Synthesis of ((2-((2-carbamoylphenyl)carbamoyl)phenyl)thio)methyl butyrate (34)
The mercaptobenzamide 34, a white solid (155 mg, 26%), was synthesized by general procedure F using 2-aminobenzamide. Flash-column chromatography: 1) eluted with hexanes initially, grading to ethyl acetate, linear gradient; 2) eluted with 15% ethyl acetate–hexanes initially, grading to 75% ethyl acetate–hexanes, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 12.42 (s, 1H), 8.59 (d, J = 8.4 Hz, 1H), 8.38 (s, 1H), 7.87 (dd, J = 8.0 Hz, 1.3 Hz, 1H), 7.78 (s, 1H), 7.70 (d, J = 8.1 Hz, 1H), 7.65 (dd, J = 7.7 Hz, 1.3 Hz, 1H), 7.59–7.55 (m, 2H), 7.41 (td, J = 7.5 Hz, 0.9 Hz, 1H), 7.19 (td, J = 7.6 Hz, 1.0 Hz, 1H), 5.50 (s, 2H), 2.29 (t, J = 7.3 Hz, 2H), 1.50 (app sex, J = 7.3 Hz, 0.82 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.79, 171.32, 166.07, 140.18, 137.25, 135.19, 133.03, 131.81, 129.67, 129.22, 127.80, 127.26, 123.46, 120.49, 119.95, 66.65, 35.78, 18.33, 13.77. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 395.1041; found, 395.1040.
4.5.20. Synthesis of ((2-((3-carbamoylphenyl)carbamoyl)phenyl)thio)methyl butyrate (35)
The mercaptobenzamide 35, a white solid (180 mg, 30%), was synthesized by general procedure F using 3-aminobenzamide. Flash-column chromatography: 1) eluted with hexanes initially, grading to ethyl acetate, linear gradient; 2) eluted with 35% ethyl acetate–hexanes initially, grading to ethyl acetate, linear gradient; 3) eluted with hexanes initially, grading to 80% acetone–hexanes, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 10.52 (s, 1H), 8.21 (s, 1H), 7.96 (s, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.67 (d, J = 8.1 Hz, 1H), 7.59–7.58 (m, 2H), 7.53 (td, J = 7.7 Hz, 1.5 Hz, 1H), 7.42–7.38 (m, 3H), 5.48 (s, 2H), 2.29 (t, J = 7.2 Hz, 2H), 1.51 (app sex, J = 7.3 Hz, 2H), 0.84 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.78, 168.36, 166.81, 139.55, 138.51, 135.62, 133.86, 131.20, 130.25, 129.01, 128.45, 127.32, 122.93, 119.88, 67.08, 35.80, 18.31, 13.82. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 395.1041; found, 395.1046.
4.5.21. Synthesis of ((2-((4-carbamoylphenyl)carbamoyl)phenyl)thio)methyl butyrate (36)
The mercaptobenzamide 36, a white solid (53.6 mg, 9%), was synthesized by general procedure F using 4-aminobenzamide. Flash-column chromatography: 1) eluted with ethyl acetate initially, grading to 15% methanol–ethyl acetate, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 10.61 (s, 1H), 7.90 (s, 1H), 7.87 (d, J = 8.7 Hz, 2H), 7.76 (d, J = 8.6 Hz, 2H), 7.68 (d, J = 8.0 Hz, 1H), 7.58 (d, J = 7.5 Hz, 1H), 7.54 (td, J = 7.7 Hz, 1.3 Hz, 1H), 7.42 (t, J = 7.5 Hz, 1H), 7.29 (s, 1H), 5.47 (s, 2H), 2.29 (t, J = 7.3 Hz, 2H), 1.51 (app sex, J = 7.3 Hz, 2H), 0.84 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.77, 167.82, 166.96, 142.15, 138.55, 133.76, 131.28, 130.38, 129.71, 128.83, 128.46, 127.40, 119.23, 67.10, 35.79, 18.30, 13.83. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 395.1041; found, 395.1044.
4.5.22. Synthesis of ((2-(trans-2-carbamoylcyclopentylcarbamoyl)phenyl)thio)methyl butyrate (37)
A 20-mL vial was charged with trans-2-((tert-butoxycarbonyl) amino)cyclopentane-1-carboxylic acid (224 mg, 978 μmol, 1 equiv) and tetrahydrofuran (4.9 mL). 1,1′-Carbonyldiimidazole (159 mg, 978 μmol, 1.00 equiv) was added and the resulting mixture was stirred for 1 h at 23 °C. A solution of ammonia in methanol (7.00 M; 1.40 mL, 9.78 mmol, 10.0 equiv) was added to the reaction mixture, and the resulting mixture was stirred for 12 h at 23 °C. The product mixture was concentrated, and the residue obtained was transferred to a 20-mL vial. A solution of hydrogen chloride in dioxane (4.00 M; 4.90 mL, 19.6 mmol, 20.0 equiv) was added and the resulting mixture was stirred at 23 °C for 1 h. The product mixture was concentrated. In parallel, chloromethyl butyrate (124 μL, 978 μmol, 1.00 equiv) and N,N-di-iso-propylethylamine (170 μL, 978 μmol, 1.00 equiv) were sequentially added to a solution of thiosalicylic acid (151 mg, 978 μmol, 1.00 equiv) in N,N-dimethylformamide (4.9 mL) and the resulting mixture was stirred at 23 °C for 12 h. 1,1′-Carbonyldiimidazole (159 mg, 978 μmol, 1.00 equiv) was added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 1 h. A solution of the previously prepared amine hydrochloride (978 μmol, 1 equiv) in N,N-dimethylformamide (3.0 mL) and N,N-di-iso-propylethylamine (426 μL, 2.45 mmol, 2.50 equiv) were sequentially added to the reaction mixture, and the resulting mixturewas stirred at 23 °C for 12 h. The product mixture was concentrated, and the residue obtained was diluted with ethyl acetate (120 mL). The diluted product mixture was transferred to a separatory funnel and sequentially washed with saturated aqueous sodium bicarbonate solution (1 × 50 mL), water (1 × 50 mL), and saturated aqueous sodium chloride solution (1 × 50 mL). The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue thus obtained was purified by flash-column chromatography (eluting with hexanes initially, grading to ethyl acetate, linear gradient) to provide the mercaptobenzamide 37 as a white solid (44.5 mg, 12% over 3 steps).
1H NMR (500 MHz, DMSO-d6) δ 8.38 (d, J = 7.5 Hz, 1H), 7.5 (d, J = 8.0 Hz, 1H), 7.45 (t, J = 7.7 Hz, 1H), 7.41 (d, J = 7.8 Hz, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.27 (s, 1H), 6.83 (s, 1H), 5.45 (s, 2H), 4.29 (app quin, J = 7.0 Hz, 1H), 2.60 (app q, J = 7.6 Hz, 1H), 2.32 (t, J = 7.3 Hz, 2H), 1.93 (app sex, J = 6.5 Hz, 1H), 1.89–1.82 (m, 1H), 1.73–1.59 (m, 3H), 1.54 (app sex, J = 7.2 Hz, 3H), 0.87 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 176.12, 172.81, 167.66, 138.25, 133.99, 130.70, 129.53, 128.32, 126.87, 66.89, 54.75, 50.80, 35.84, 33.17, 29.35, 24.14, 18.35, 13.85. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 387.1354; found, 387.1348.
4.5.23. Synthesis of ((2-(cis-2-carbamoylcyclopentylcarbamoyl)phenyl)thio)methyl butyrate (38)
A 20-mL vial was charged with cis-2-((tert-butoxycarbonyl) amino)cyclopentane-1-carboxylic acid (249 mg, 1.08 mmol, 1 equiv) and tetrahydrofuran (5.4 mL). 1,1′-Carbonyldiimidazole (176 mg, 1.08 mmol, 1.00 equiv) was added and the resulting mixture was stirred for 1 h at 23 °C. A solution of ammonia in methanol (7.00 M; 1.55 mL, 10.8 mmol, 10.0 equiv) was added to the reaction mixture, and the resulting mixture was stirred for 12 h at 23 °C. The product mixture was concentrated, and the residue obtained was transferred to a 20-mL vial. A solution of hydrogen chloride in dioxane (4.00 M; 7.40 mL, 29.6 mmol, 27.4 equiv) was added and the resulting mixture was stirred at 23 °C for 1 h. The product mixture was concentrated. In parallel, chloromethyl butyrate (138 μL, 1.08 mmol, 1.00 equiv) and N,N-di-iso-propylethylamine (189 μL, 1.08 mmol, 1.00 equiv) were sequentially added to a solution of thiosalicylic acid (167 mg, 1.08 mmol, 1.00 equiv) in N,N-dimethylformamide (5.4 mL) and the resulting mixture was stirred at 23 °C for 12 h. 1,1′-Carbonyldiimidazole (176 mg, 1.08 mmol, 1.00 equiv) was added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 1 h. A solution of the previously prepared amine hydrochloride (1.08 mmol, 1 equiv) in N,N-dimethylformamide (3.0 mL) and N,N-di-iso-propylethylamine (473 μL, 2.71 mmol, 2.51 equiv) were sequentially added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 12 h. The product mixture was concentrated, and the residue obtained was diluted with ethyl acetate (120 mL). The diluted product mixture was transferred to a separatory funnel and sequentially washed with saturated aqueous sodium bicarbonate solution (1 × 50 mL), water (1 × 50 mL), and saturated aqueous sodium chloride solution (1 × 50 mL). The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue thus obtained was purified by flash-column chromatography (eluting with hexanes initially, grading to ethyl acetate, linear gradient) to provide the mercaptobenzamide 38 as a white solid (63.9 mg, 16% over 3 steps).
1H NMR (500 MHz, DMSO-d6) δ 8.04 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 8.0 Hz, 1H)7.46–7.41 (m, 2H), 7.29 (m, 2H), 6.87 (s, 1H), 5.48 (d, J = 12.2 Hz, 1H), 5.40 (d, J = 12.2 Hz, 1H), 4.42 (app quin, J = 6.9 Hz, 1H), 2.83 (app q, J = 8.0 Hz, 1H), 2.32 (t, J = 7.3 Hz, 2H), 1.93–1.89 (m, 1H), 1.87–1.81 (m, 1H), 1.80–1.70 (m, 3H), 1.57–1.46 (m, 3H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 175.26, 172.84, 167.30, 137.91, 134.23, 130.78, 129.29, 128.54, 126.73, 66.80, 52.73, 47.36, 35.84, 32.41, 28.03, 22.95, 18.35, 13.84. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 387.1354; found, 387.1359.
4.5.24. Synthesis of ((2-(((1R,2R)-2-carbamoylcyclopentyl)carbamoyl)phenyl)thio)methyl butyrate (39)
A 20-mL vial was charged with (1R,2R)-2-((tert-butoxycarbonyl) amino)cyclopentane-1-carboxylic acid (252 mg, 1.10 mmol, 1 equiv) and N,N-dimethylformamide (5.5 mL). 1,1′-Carbonyldiimidazole (178 mg, 1.10 mmol, 1.00 equiv) was added and the resulting mixture was stirred for 1 h at 23 °C. A solution of ammonia in methanol (7.00 M; 1.57 mL, 11.0 mmol, 10.0 equiv) was added to the reaction mixture, and the resulting mixture was stirred for 12 h at 23 °C. The product mixture was concentrated, and the residue obtained was transferred to a 20-mL vial. A solution of hydrogen chloride in dioxane (4.00 M; 7.48 mL, 29.9 mmol, 27.2 equiv) was added and the resulting mixture was stirred at 23 °C for 1 h. The product mixture was concentrated. In parallel, chloromethyl butyrate (140 μL, 1.10 mmol, 1.00 equiv) and N,N-di-iso-propylethylamine (191 μL, 1.10 mmol, 1.00 equiv) were sequentially added to a solution of thiosalicylic acid (169 mg, 1.10 mmol, 1.00 equiv) in N,N-dimethylformamide (5.5 mL) and the resulting mixture was stirred at 23 °C for 12 h. 1,1′-Carbonyldiimidazole (178 mg, 1.10 mmol, 1.00 equiv) was added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 1 h. A solution of the previously prepared amine hydrochloride (1.10 mmol, 1 equiv) in N,N-dimethylformamide (3.0 mL) and N,N-di-iso-propylethylamine (478 μL, 2.74 mmol, 2.49 equiv) were sequentially added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 12 h. The product mixture was concentrated, and the residue obtained was diluted with ethyl acetate (60 mL). The diluted product mixture was transferred to a separatory funnel and sequentially washed with saturated aqueous sodium bicarbonate solution (1 × 30 mL), water (1× 30 mL), and saturated aqueous sodium chloride solution (1 × 30 mL). The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue thus obtained was purified by flash-column chromatography (eluting with hexanes initially, grading to ethyl acetate, (linear gradient), followed by grading to 15% methanol–ethyl acetate (linear gradient)) to provide the mercaptobenzamide 39 as a white solid (22.2 mg, 6% over 3 steps).
1H NMR (400 MHz, CD3OD) δ 7.65 (d, J = 7.8 Hz, 1H), 7.46–7.41 (m, 2H), 7.35 (t, J = 7.4 Hz, 1H), 5.43 (s, 2H), 4.48 (app q, J = 7.0 Hz, 1H), 2.76 (app q, J = 7.7 Hz, 1H), 2.33 (t, J = 7.4 Hz, 2H), 2.12 (app sex, J = 6.5 Hz, 1H), 2.06–1.97 (m, 2H), 1.93–1.84 (m, 1H), 1.79 (app quin, J = 7.1 Hz, 2H), 1.73–1.59 (m, 3H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ 179.62, 174.36, 171.11, 140.30, 134.12, 132.53, 131.48, 128.77, 128.50, 68.78, 56.58, 52.62, 36.93, 33.47, 30.13, 24.87, 19.28, 13.85. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 387.1354; found, 387.1355. −24.4 (c 1.4, CH3OH).
4.5.25. Synthesis of ((2-(((1S,2S)-2-carbamoylcyclopentyl)carbamoyl)phenyl)thio)methyl butyrate (40)
A 20-mL vial was charged with (1S,2S)-2-((tert-butoxycarbonyl) amino)cyclopentane-1-carboxylic acid (265 mg, 1.16 mmol, 1 equiv) and N,N-dimethylformamide (5.8 mL). 1,1′-Carbonyldiimidazole (187 mg, 1.16 mmol, 1.00 equiv) was added and the resulting mixture was stirred for 1 h at 23 °C. A solution of ammonia in methanol (7.00 M; 1.65 mL, 11.6 mmol, 10.0 equiv) was added to the reaction mixture, and the resulting mixture was stirred for 12 h at 23 °C. The product mixture was concentrated, and the residue obtained was transferred to a 20-mL vial. A solution of hydrogen chloride in dioxane (4.00 M; 7.88 mL, 31.5 mmol, 27.2 equiv) was added and the resulting mixture was stirred at 23 °C for 1 h. The product mixture was concentrated. In parallel, chloromethyl butyrate (147 μL, 1.16 mmol, 1.00 equiv) and N,N-di-iso-propylethylamine (201 μL, 1.15 mmol, 0.99 equiv) were sequentially added to a solution of thiosalicylic acid (178 mg, 1.15 mmol, 0.99 equiv) in N,N-dimethylformamide (5.8 mL) and the resulting mixture was stirred at 23 °C for 12 h. 1,1′-Carbonyldiimidazole (187 mg, 1.16 mmol, 1.00 equiv) was added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 1 h. A solution of the previously prepared amine hydrochloride (1.16 mmol, 1 equiv) in N,N-dimethylformamide (3.0 mL) and N,N-di-iso-propylethylamine (503 μL, 2.89 mmol, 2.49 equiv) were sequentially added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 12 h. The product mixture was concentrated, and the residue obtained was diluted with ethyl acetate (60 mL). The diluted product mixture was transferred to a separatory funnel and sequentially washed with saturated aqueous sodium bicarbonate solution (1 × 30 mL), water (1× 30 mL), and saturated aqueous sodium chloride solution (1 × 30 mL). The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue thus obtained was purified by flash-column chromatography (eluting with hexanes initially, grading to ethyl acetate, (linear gradient), followed by grading to 15% methanol–ethyl acetate (linear gradient)) to provide the mercaptobenzamide 40 as a white solid (31.3 mg, 8% over 3 steps).
1H NMR (400 MHz, CD3OD) δ. 7.65 (d, J = 7.6 Hz, 1H), 7.46–7.41 (m, 2H), 7.35 (t, J = 7.2 Hz, 1H), 5.43 (s, 2H), 4.48 (app q, J = 7.0 Hz, 1H), 2.76 (app q, J = 7.8 Hz, 1H), 2.34 (t, J = 7.3 Hz, 2H), 2.12 (app sex, J = 6.6 Hz, 1H), 2.06–1.98 (m, 1H), 1.93–1.84 (m, 1H), 1.79 (app quin, J = 7.0 Hz, 2H), 1.73–1.59 (m, 3H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ 179.61, 174.36, 171.08, 140.25, 134.13, 132.48, 131.48, 128.77, 128.48, 68.76, 56.56, 52.59, 36.92, 33.47, 30.12, 24.87, 19.27, 13.86. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 387.1354; found, 387.1359. +25.1 (c 1.9, CH3OH).
4.5.26. Synthesis of ((2-(((1R,2S)-2-carbamoylcyclopentyl)carbamoyl)phenyl)thio)methyl butyrate (41)
A 20-mL vial was charged with (1R,2S)-2-((tert-butoxycarbonyl) amino)cyclopentane-1-carboxylic acid (252 mg, 1.10 mmol, 1 equiv) and N,N-dimethylformamide (5.5 mL). 1,1′-Carbonyldiimidazole (178 mg, 1.10 mmol, 1.00 equiv) was added and the resulting mixture was stirred for 1 h at 23 °C. A solution of ammonia in methanol (7.00 M; 1.57 mL, 11.0 mmol, 10.0 equiv) was added to the reaction mixture, and the resulting mixture was stirred for 12 h at 23 °C. The product mixture was concentrated, and the residue obtained was transferred to a 20-mL vial. A solution of hydrogen chloride in dioxane (4.00 M; 7.51 mL, 30.0 mmol, 27.3 equiv) was added and the resulting mixture was stirred at 23 °C for 1 h. The product mixture was concentrated. In parallel, chloromethyl butyrate (140 μL, 1.10 mmol, 1.00 equiv) and N,N-di-iso-propylethylamine (192 μL, 1.10 mmol, 1.00 equiv) were sequentially added to a solution of thiosalicylic acid (170 mg, 1.10 mmol, 1.00 equiv) in N,N-dimethylformamide (5.5 mL) and the resulting mixture was stirred at 23 °C for 12 h. 1,1′-Carbonyldiimidazole (178 mg, 1.10 mmol, 1.00 equiv) was added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 1 h. A solution of the previously prepared amine hydrochloride (1.10 mmol, 1 equiv) in N,N-dimethylformamide (3.0 mL) and N,N-di-iso-propylethylamine (480 μL, 2.76 mmol, 2.51 equiv) were sequentially added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 12 h. The product mixture was concentrated, and the residue obtained was diluted with ethyl acetate (60 mL). The diluted product mixture was transferred to a separatory funnel and sequentially washed with saturated aqueous sodium bicarbonate solution (1 × 30 mL), water (1 × 30 mL), and saturated aqueous sodium chloride solution (1 × 30 mL). The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue thus obtained was purified by flash-column chromatography (eluting with hexanes initially, grading to ethyl acetate, (linear gradient), followed by grading to 15% methanol–ethyl acetate (linear gradient)) to provide the mercaptobenzamide 41 as a white solid (58.6 mg, 15% over 3 steps).
1H NMR (400 MHz, CD3OD) δ 7.65 (d, J = 7.7 Hz, 1H), 7.46–7.42 (m, 2H), 7.32 (t, J = 7.5 Hz, 1H), 5.47 (d, J = 12.0 Hz, 1H), 5.43 (d, J = 12.0 Hz, 1H), 4.56 (app q, J = 7.1 Hz, 1H), 3.05 (app q, J = 7.8 Hz, 1H), 2.34 (t, J = 7.3 Hz, 2H), 2.05–1.81 (m, 6H), 1.68–1.59 (m, 3H), 0.94 (t, J = 7.5 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ 178.45, 174.33, 170.69, 139.52, 134.58, 131.95, 131.59, 129.03, 128.23, 68.54, 54.43, 48.40, 36.91, 33.13, 29.10, 23.81, 19.28, 13.86. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 387.1354; found, 387.1358. −21.0 (c 3.7, CH3OH).
4.5.27. Synthesis of ((2-(((1S,2R)-2-carbamoylcyclopentyl)carbamoyl)phenyl)thio)methyl butyrate (42)
A 20-mL vial was charged with (1S,2R)-2-((tert-butoxycarbonyl) amino)cyclopentane-1-carboxylic acid (241 mg, 1.05 mmol, 1 equiv) and N,N-dimethylformamide (5.2 mL). 1,1′-Carbonyldiimidazole (171 mg, 1.05 mmol, 1.00 equiv) was added and the resulting mixture was stirred for 1 h at 23 °C. A solution of ammonia in methanol (7.00 M; 1.50 mL, 10.5 mmol, 10.0 equiv) was added to the reaction mixture, and the resulting mixture was stirred for 12 h at 23 °C. The product mixture was concentrated, and the residue obtained was transferred to a 20-mL vial. A solution of hydrogen chloride in dioxane (4.00 M; 7.18 mL, 28.7 mmol, 27.3 equiv) was added and the resulting mixture was stirred at 23 °C for 1 h. The product mixture was concentrated. In parallel, chloromethyl butyrate (134 μL, 1.05 mmol, 1.00 equiv) and N,N-di-iso-propylethylamine (183 μL, 1.05 mmol, 1.00 equiv) were sequentially added to a solution of thiosalicylic acid (162 mg, 1.05 mmol, 1.00 equiv) in N,N-dimethylformamide (5.2 mL) and the resulting mixture was stirred at 23 °C for 12 h. 1,1′-Carbonyldiimidazole (171 mg, 1.05 mmol, 1.00 equiv) was added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 1 h. A solution of the previously prepared amine hydrochloride (1.10 mmol, 1 equiv) in N,N-dimethylformamide (3.0 mL) and N,N-di-iso-propylethylamine (459 μL, 2.63 mmol, 2.50 equiv) were sequentially added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 12 h. The product mixture was concentrated, and the residue obtained was diluted with ethyl acetate (60 mL). The diluted product mixture was transferred to a separatory funnel and sequentially washed with saturated aqueous sodium bicarbonate solution (1 × 30 mL), water (1 × 30 mL), and saturated aqueous sodium chloride solution (1 × 30 mL). The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue thus obtained was purified by flash-column chromatography (eluting with hexanes initially, grading to ethyl acetate, (linear gradient), followed by grading to 15% methanol–ethyl acetate (linear gradient)) to provide the mercaptobenzamide 42 as a white solid (55.9 mg, 15% over 3 steps).
1H NMR (400 MHz, CD3OD) δ 7.64 (d, J = 7.9 Hz, 1H), 7.46–7.42 (m, 2H), 7.33 (t, J = 7.5 Hz, 1H)., 5.46 (d, J = 12.0 Hz, 1H), 5.42 (d, J = 12.0 Hz, 1H), 4.56 (app q, J = 7.1 Hz, 1H), 3.05 (app q, J = 7.8 Hz, 1H), 2.34 (t, J = 7.3 Hz, 2H), 2.06–1.81 (m, 6H), 1.68–1.59 (m, 3H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ 178.48, 174.36, 170.75, 139.57, 134.59, 132.00, 129.11, 128.25, 68.56, 54.46, 48.43, 36.92, 33.14, 29.10, 23.82, 19.29, 13.86. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 387.1354; found, 387.1359. +21.4 (c 3.6, CH3OH).
4.5.28. Synthesis of 2-amino-2-oxoethyl 2-(((butyryloxy)methyl)thio)benzoate (43)
Chloromethyl butyrate (177 μL, 1.30 mmol, 1.00 equiv) and N,N-di-iso-propylethylamine (91 μL, 4.54 mmol, 3.50 equiv) were sequentially added to a solution of thiosalicylic acid (200 mg, 1.30 mmol, 1 equiv) in N,N-dimethylformamide (6.5 mL) and the resulting mixture was stirred at 23 °C for 20 h. HATU (493 mg, 1.30 mmol, 1.00 equiv) was added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 1 h. 2-Hydroxyacetamide (97.4 mg, 1.30 mmol, 1.00 equiv) was added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 24 h. The product mixture was concentrated, and the residue obtained was diluted with ethyl acetate (120 mL). The diluted product mixture was transferred to a separatory funnel and sequentially washed with saturated aqueous sodium bicarbonate solution (1 × 50 mL), water (1 × 50 mL), and saturated aqueous sodium chloride solution (1 × 50 mL). The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue thus obtained was purified by flash-column chromatography (first: eluted with 30% ethyl acetate–hexanes initially, grading to ethyl acetate, linear gradient; second: eluted with dichloromethane initially, grading to 25% acetone–dichloromethane, linear gradient) to provide the mercaptobenzoate 43 as a white solid (112 mg, 28%).
1H NMR (500 MHz, DMSO-d6) δ 8.05 (d, J = 7.8 Hz, 1H), 7.69–7.62 (m, 2H), 7.56 (br s, 1H), 7.39–7.34 (m, 1H), 7.31 (br s, 1H), 5.57 (s, 2H), 4.65 (s, 2H), 2.32 (t, J = 7.2 Hz, 2H), 1.53 (app sex, J = 7.4 Hz, 2H), 0.85 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.3, 168.4, 164.9, 139.0, 133.3, 131.4, 127.0, 126.5, 125.4, 64.4, 62.8, 35.3, 17.9, 13.3. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 334.0725; found, 334.0724.
4.5.29. Synthesis of ((2-((2-methoxy-2-oxoethyl)carbamoyl)phenyl)thio)methyl butyrate (44)
The mercaptobenzamide 44, an orange oil (288 mg, 68%), was synthesized by general procedure F using glycine methyl ester hydrochloride. Flash-column chromatography: eluted with 30% ethyl acetate–hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 10% methanol–ethyl acetate (linear gradient).
1H NMR (500 MHz, DMSO-d6) δ 8.86 (br t, J = 5.7 Hz, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.53–7.47 (m, 2H), 7.34 (t, J = 7.5 Hz, 1H), 5.47 (s, 2H), 3.99 (d, J = 5.9 Hz, 2H), 3.67 (s, 3H), 2.32 (t, J = 7.2 Hz, 2H), 1.54 (app sex, J = 7.4 Hz, 2H), 0.87 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.3, 170.2, 167.8, 135.8, 134.5, 130.8, 128.5, 128.0, 126.2, 66.0, 51.8, 41.0, 35.3, 17.9, 13.3. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 348.0882; found, 348.0886.
4.5.30. Synthesis of 2,2′-diselanediyldibenzoic acid (S1)
2,2′-Diselanediyldibenzoic acid (S1) was synthesized according to a modified literature procedure [17b] as follows: A 250-mL round bottomed flask was charged with selenium powder (200 mesh; 1.15 g, 14.6 mmol, 2.00 equiv) and a solution of aqueous sodium hydroxide (3.50 M, 50 mL). The resulting suspension was cooled to 0 °C. Sodium borohydride (68.9 mg, 1.82 mmol, 0.25 equiv) was added to the reaction mixture in one portion, and the resulting mixture was stirred at 100 °C for 1 h. The reaction mixture was cooled to 23 °C over 30 min. In parallel, a solution of sodium nitrite (654 mg, 9.48 mmol, 1.30 equiv) in water (2.0 mL) was added to a solution of anthranilic acid (1.00 g, 7.29 mmol, 1 equiv) in 5% aqueous hydrochloric acid (10 mL) at 0 °C, and the resulting mixture was stirred for 40 min at 0 °C. The resulting diazonium salt solution was added dropwise to the round bottomed flask containing the freshly prepared solution of sodium diselenide. Upon completion of addition, the round bottomed flask was equipped with a reflux condenser, and the reaction mixture was heated at refluxing temperature (~100 °C) for 12 h. The reaction mixture was cooled to 23 °C and acidified with 10% aqueous hydrochloric acid to pH 1. The red precipitate thus obtained was filtered, and the filtered solid was resuspended in saturated aqueous sodium bicarbonate solution (250 mL). The resulting suspension was heated at refluxing temperature (~100 °C) for 12 h. The product mixture was cooled to 23 °C and acidified with 10% aqueous hydrochloric acid to pH 1. The yellow precipitate thus obtained was filtered and the filtered solid was recrystallized from methanol to provide 2,2′-diselanediyldibenzoic acid (S1) as a yellow solid (718 mg, 49%).
1H NMR (500 MHz, DMSO-d6) δ 8.02 (dd, J = 7.8 Hz, 1.5 Hz, 1H), 7.66 (d, J = 8.2 Hz, 1H), 7.47 (td, J = 7.5 Hz, 1.3 Hz, 1H), 7.35 (t, J = 7.5 Hz, 1H). 13C NMR (125 MHz, DMSO-d6) δ 169.10, 134.01, 133.98, 132.06, 129.96, 129.51, 127.01. 77Se NMR (76 MHz, DMSO-d6) δ 439.95. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 422.8651; found 422.8649.
4.5.31. Synthesis of 2,2′-diselanediylbis(N-(2-amino-2-oxoethyl)benzamide) (S2)
The diselenobenzamide S2, a white solid (42.0 mg, 22%), was synthesized according to the general procedure H using glycinamide hydrochloride.
1H NMR (500 MHz, DMSO-d6) δ 8.98 (bt, 5.7 Hz, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.68 (d, J = 8.05 Hz, 1H), 7.47 (bs, 1H), 7.40 (t, J = 7.5 Hz, 1H), 7.34 (t, J = 7.4 Hz, 1H), 7.11 (bs, 1H), 3.86 (d, J = 6.0 Hz, 2H). 13C NMR (125 MHz, DMSO-d6) δ 171.18, 168.01, 132.76, 132.65, 130.35, 128.87, 126.63, 43.02. 77Se NMR (76 MHz, DMSO-d6) δ 445.43. HRMS-ESI+ (m/z): [M]+ calcd for , 514.9737; found 514.9745.
4.5.32. Synthesis of ((2-((2-amino-2-oxoethyl)carbamoyl)phenyl)selanyl)methyl butyrate (45)
The selenobenzamide 45, a white solid (30.0 mg, 47%), was synthesized according to the general procedure I using the diselenobenzamide S2.
1H NMR (500 MHz, DMSO-d6) δ 8.72 (bt, J = 5.7 Hz, 1H), 7.77 (dd, J = 7.7 Hz, 1.2 Hz, 1H), 7.71 (d, J = 7.9 Hz, 1H), 7.49 (dt, J = 7.7 Hz, 1.2 Hz, 1H), 7.39 (bs, 1H), 7.35 (t, J = 7.5 Hz, 1H), 7.10 (bs, 1H), 5.60 (t, J = 9.7 Hz, 2H), 3.79 (d, J = 6.0 Hz, 2H), 2.33 (t, J = 7.2 Hz, 2H), 1.55 (app sex, J = 7.4 Hz, 2H), 0.88 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 173.02, 171.21, 168.14, 134.56, 133.35, 131.83, 129.48, 128.87, 126.19, 59.93, 42.82, 35.90, 18.40, 13.81. 77Se NMR (76 MHz, DMSO-d6) δ 411.48. HRMS-ESI+ (m/z): [M + Na]+ calcd for C14H18N2O4Na80Se, 381.0329; found, 381.0323.
4.5.33. Synthesis of 2,2′-diselanediylbis(N-(3-amino-3-oxopropyl)benzamide) (S3)
The diselenobenzamide S3, a white solid (66.0 mg, 33%), was synthesized according to the general procedure H using β -alaninamide hydrochloride.
1H NMR (500 MHz, DMSO-d6) δ 8.83 (bt, J = 5.5 Hz, 1H), 7.79 (d, J = 7.7 Hz, 1H), 7.67 (d, J = 7.7 Hz, 1H), 7.42 (bs, 1H), 7.39 (td, J = 7.7 Hz, 1.2 Hz, 1H), 7.32 (td, J = 7.6 Hz, 1.0 Hz, 1H), 6.90 (bs, 1H), 3.47 (app q, J = 6.7 Hz, 2H), 2.40 (t, J = 7.1 Hz, 2H). 13C NMR (125 MHz, DMSO-d6) δ 172.91, 167.82, 133.30, 132.45, 130.33, 128.50, 126.63, 36.72, 35.31. 77Se NMR (76 MHz, DMSO-d6) δ 443.13. HRMS-ESI+ (m/z): [M]+ calcd for , 543.0050; found 543.0056.
4.5.34. Synthesis of ((2-((3-amino-3-oxopropyl)carbamoyl)phenyl)selanyl)methyl butyrate (46)
The selenobenzamide 46, a white solid (65.1 mg, 92%), was synthesized according to the general procedure I using the diselenobenzamide S3.
1H NMR (500 MHz, DMSO-d6) δ 8.54 (bt, J = 5.2 Hz, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 7.38 (bs, 1H), 7.31 (t, J = 7.4 Hz, 1H), 6.86 (bs, 1H), 5.58 (t, J = 9.5 Hz, 2H), 3.38 (app q, J = 6.8 Hz, 2H), 2.32 (app q, J = 8.0 Hz, 4H), 1.53 (app sex, J = 7.4 Hz, 2H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 173.01, 172.90, 168.03, 135.66, 132.75, 131.56, 129.71, 128.45, 126.30, 60.10, 36.49, 35.90, 35.36, 18.40, 13.81. 77Se NMR (76 MHz, DMSO-d6) δ 407.58. HRMS-ESI+ (m/z): [M + Na]+ calcd for C15H20N2O4Na80Se, 395.0486; found, 395.0480.
4.5.35. Synthesis of ((2-((2-(methylamino)-2-oxoethyl)carbamoyl)phenyl)thio)methyl butyrate (47)
The mercaptobenzamide 47, a white solid (110 mg, 35% over 2 steps), was synthesized by general procedure F using the amine hydrochloride obtained from 2-chloro-N-methylacetamide via general procedure A. Flash-column chromatography: eluted with 30% ethyl acetate–hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 10% methanol–ethyl acetate (linear gradient).
1H NMR (500 MHz, DMSO-d6) δ 8.61 (br t, J = 5.6 Hz, 1H), 7.81–7.74 (m, 1H), 7.59 (app d, J = 7.6 Hz, 2H), 7.48 (t, J = 7.4 Hz, 1H), 7.33 (t, J = 7.6 Hz, 1H), 5.46 (s, 2H), 3.79 (d, J = 5.8 Hz, 2H), 2.62 (d, J = 4.4 Hz, 3H), 2.32 (t, J = 7.3 Hz, 2H), 1.54 (app sex, J = 7.3 Hz, 2H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.3, 169.0, 167.7, 136.2, 134.3, 130.6, 128.6, 128.2, 126.2, 66.1, 42.4, 35.3, 25.6, 17.9, 13.3. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 347.1041; found, 347.1042.
4.5.36. Synthesis of ((2-((2-(dimethylamino)-2-oxoethyl)carbamoyl)phenyl)thio)methyl butyrate (48)
The mercaptobenzamide 48, a yellow oil (169 mg, 51% over 2 steps), was synthesized by general procedure F using the amine hydrochloride obtained from 2-chloro-N,N-dimethylacetamide via general procedure A. Flash-column chromatography: eluted with 30% ethyl acetate–hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 10% methanol–ethyl acetate (linear gradient).
1H NMR (500 MHz, DMSO-d6) δ 8.39 (br t, J = 5.4 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 7.5 Hz, 1H), 7.48 (t, J = 7.5 Hz, 1H), 7.33 (t, J = 7.3 Hz, 1H), 5.47 (s, 2H), 4.07 (d, J = 5.6 Hz, 2H), 3.00 (s, 3H), 2.85 (s, 3H), 2.32 (t, J = 7.2 Hz, 2H), 1.54 (app sex, J = 7.3 Hz, 2H), 0.87 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.3, 168.0, 167.5, 136.5, 134.1, 130.6, 128.6, 128.1, 126.2, 66.1, 40.8, 35.7, 35.3, 35.1, 17.9, 13.3. HRMS-ESI+ (m/z): [M + H]+ calcd for , 339.1379; found, 339.1384.
4.5.37. Synthesis of ((2-((2-amino-2-oxoethyl)(methyl)carbamoyl)phenyl)thio)methyl butyrate (49)
The mercaptobenzamide 49, a clear oil (121 mg, 38% over 2 steps), was synthesized by general procedure F using the amine hydrochloride obtained from 2-chloroacetamide via general procedure B. Flash-column chromatography: eluted with 30% ethyl acetate–hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 10% methanol–ethyl acetate (linear gradient).
1H NMR (500 MHz, DMSO-d6) (1:1 mixture of rotamers) δ 7.63 (d, J = 7.9 Hz, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.48–7.37 (m, 3H), 7.36–7.28 (m, 4H), 7.22 (d, J = 7.5 Hz, 1H), 7.15 (br s, 1H), 7.09 (br s, 1H), 5.48 (s, 2H), 5.46 (s, 2H), 4.02 (s, 2H), 3.62 (s, 2H), 2.95 (s, 3H), 2.75 (s, 3H), 2.33 (t, J = 7.2 Hz, 2H), 2.31 (t, J = 7.2 Hz, 2H), 1.53 (app sex, J = 7.2 Hz, 4H), 0.87 (t, J = 7.2 Hz, 3H), 0.86 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.3, 172.3, 169.6, 169.4, 168.9, 168.9, 138.5, 138.3, 130.7, 130.6, 130.6, 129.6, 127.6, 127.4, 126.9, 126.7, 52.7, 49.2, 37.5, 35.3, 33.4, 17.8, 13.3. HRMS-ESI+ (m/z): [M + H]+ calcd for , 325.1222; found, 325.1223.
4.5.38. Synthesis of ((2-(methyl(2-(methylamino)-2-oxoethyl)carbamoyl)phenyl)thio)methyl butyrate (50)
The mercaptobenzamide 50, a yellow oil (170 mg, 52% over 2 steps), was synthesized by general procedure F using the amine hydrochloride obtained from 2-chloro-N-methylacetamide via general procedure B. Flash-column chromatography: eluted with 30% ethyl acetate–hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 10% methanol–ethyl acetate (linear gradient).
1H NMR (500 MHz, DMSO-d6) (1:1 mixture of rotamers) δ 7.85–7.78 (m, 2H), 7.64 (d, J = 7.8 Hz, 1H), 7.60 (d, J = 7.8 Hz, 1H), 7.48–7.38 (m, 3H), 7.35–7.28 (m, 2H), 7.24 (d, J = 7.4 Hz, 1H), 5.48 (s, 2H), 5.46 (s, 2H), 4.05 (s, 2H), 3.64 (s, 2H), 2.95 (s, 3H), 2.75 (s, 3H), 2.64 (d, J = 4.5 Hz, 3H), 2.55 (d, J = 4.4 Hz, 3H), 2.32 (t, J = 7.4 Hz, 2H), 2.31 (t, J = 7.4 Hz, 2H), 1.53 (app sep, J = 7.1 Hz, 4H), 0.87 (t, J = 7.2 Hz, 3H), 0.85 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.3, 169.0, 169.0, 168.0, 167.8, 138.4, 138.2, 130.7, 130.6, 130.6, 129.7, 129.6, 127.6, 127.4, 126.9, 126.7, 66.6, 66.6, 52.9, 49.3, 37.5, 35.3, 33.4, 25.5, 17.8, 13.3. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 361.1198; found, 361.1201.
4.5.39. Synthesis of ((2-((2-(dimethylamino)-2-oxoethyl)(methyl)carbamoyl)phenyl)thio)methyl butyrate (51)
The mercaptobenzamide 51, a yellow oil (149 mg, 43% over 2 steps), was synthesized by general procedure F using the amine hydrochloride obtained from 2-chloro-N,N-dimethylacetamide via general procedure B. Flash-column chromatography: eluted with 30% ethyl acetate–hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 10% methanol–ethyl acetate (linear gradient).
1H NMR (500 MHz, DMSO-d6) (1.7:1 mixture of rotamers; A: major, B: minor) δ 7.63 (d, J = 7.8 Hz, 1H A), 7.59 (d, J = 7.9 Hz, 1H B), 7.48–7.37 (m, 2H A, 1H B), 7.30 (t, J = 7.4 Hz, 1H B), 7.25 (d, J = 7.3 Hz, 1H A), 7.11 (d, J = 7.4 Hz, 1H B), 5.49 (s, 2H A), 5.47 (s, 2H B), 4.32 (s, 2H A), 3.93 (br s, 2H B), 3.00 (s, 3H A), 2.94 (s, 3H B), 2.87 (s, 3H A), 2.78 (s, 3H B), 2.72 (s, 3H A), 2.67 (s, 3H B), 2.35–2.28 (m, 2H A, 2H B), 1.58–1.48 (m, 2H A, 2H B), 0.89–0.82 (m, 3H A, 3H B). 13C NMR (125 MHz, DMSO-d6) δ 172.3, 172.3, 169.0, 168.9, 167.2, 166.9, 138.5, 138.3, 130.7, 130.6, 129.6, 127.6, 127.3, 126.9, 126.5, 66.6, 66.5, 51.5, 48.0, 37.4, 35.7, 35.5, 35.3, 35.2, 35.1, 33.7, 17.8, 13.3. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 375.1354; found, 375.1355.
4.5.40. Synthesis of (S)-((2-(2-carbamoylpyrrolidine-1-carbonyl)phenyl)thio)methyl butyrate (52)
The mercaptobenzamide 52, a yellow oil (238 mg, 42%), was synthesized by general procedure F using l-prolinamide. Flash-column chromatography: eluted with ethyl acetate initially, grading to 15% methanol–ethyl acetate, linear gradient.
1H NMR (500 MHz, DMSO-d6) (1.9:1 mixture of rotamers; A: major, B: minor) δ 7.63 (d, J = 7.9 Hz, 1H A), 7.59 (d, J = 7.9 Hz, 1H B), 7.48–7.35 (m, 3H A, 1H B), 7.34–7.28 (m, 1H A, 1H B), 7.21 (br s, 1H B), 7.18 (d, J = 7.5 Hz, 1H B), 7.06 (br s, 1H A), 6.91 (br s, 1H B), 5.56–5.41 (m, 2H A, 2H B), 4.36 (dd, J = 8.1, 3.9 Hz, 1H A), 3.82 (br d, J = 7.5 Hz, 1H B), 3.60–3.52 (m, 2H B), 3.27–3.20 (m, 1H A), 3.13–3.05 (m, 1H A), 2.33 (t, J = 7.2 Hz, 2H B), 2.30 (t, J = 7.2 Hz, 2H A), 2.19–2.00 (m, 1H A, 1H B), 1.93–1.68 (m, 3H A, 3H B), 1.59–1.48 (m, 2H A, 2H B), 0.90–0.82 (m, 3H A, 3H B). 13C NMR (125 MHz, DMSO-d6) δ 173.2, 172.3, 172.2, 167.3, 167.1, 139.2, 139.1, 130.5, 130.3, 129.7, 129.5, 127.6, 127.3, 126.9, 126.8, 66.6, 66.6, 61.1, 59.3, 48.7, 46.0, 35.3, 31.7, 29.7, 24.5, 22.4, 17.8, 13.3. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 373.1198; found, 373.1198. [α]D23–86.1 (c 0.7, CH3OH).
4.5.41. Synthesis of ((2-(methylcarbamoyl)phenyl)thio)methyl butyrate (53)
The mercaptobenzamide 53, a yellow solid (252 mg, 73%), was synthesized by general procedure F using a solution of methylamine in tetrahydrofuran (2.00 M). Flash-column chromatography: eluted with 30% ethyl acetate–hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 10% methanol–ethyl acetate (linear gradient).
1H NMR (500 MHz, DMSO-d6) δ 8.33–8.27 (m, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.45 (t, J = 7.5 Hz, 1H), 7.41 (d, J = 7.5 Hz, 1H), 7.30 (t, J = 7.5 Hz, 1H), 5.45 (s, 2H), 2.73 (d, J = 4.5 Hz, 3H), 2.32 (t, J = 7.2 Hz, 2H), 1.54 (app sex, J = 7.4 Hz, 2H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.3, 167.9, 137.3, 133.8, 130.3, 128.7, 127.6, 126.3, 66.2, 35.3, 26.1, 17.9, 13.3. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 290.0827; found, 290.0828.
4.5.42. Synthesis of ((2-carbamoylphenyl)thio)methyl butyrate (54)
The mercaptobenzamide 54, a pale yellow solid (17.2 mg, 21%), was synthesized by general procedure F using a solution of ammonia in methanol (7.00 M). Flash-column chromatography: 1) eluted with dichloromethane initially, grading to 8% methanol–dichloromethane, linear gradient; 2) eluted with ethyl acetate initially, grading to 7% methanol–ethyl acetate, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 7.85 (br s, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.50–7.43 (m, 3H), 7.29 (t, J = 7.5 Hz, 1H), 5.47 (s, 2H), 2.32 (t, J = 7.2 Hz, 2H), 1.54 (app sex, J = 7.4 Hz, 2H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.3, 169.4, 136.7, 134.2, 130.4, 128.3, 127.8, 126.0, 66.0, 35.4, 17.9, 13.4. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 276.0670; found, 276.0671.
4.5.43. Synthesis of ((2-(2-((2-amino-2-oxoethyl)amino)-2-oxoethyl)phenyl)thio)methyl butyrate (55)
Chloromethyl butyrate (30.4 μL, 238 μmol, 1.00 equiv) and N,N-di-iso-propylethylamine (104 μL, 595 μmol, 2.50 equiv) were sequentially added to a solution of 2-mercaptophenylacetic acid (40.0 mg, 238 μmol, 1 equiv) in N,N-dimethylformamide (1.2 mL) and the resulting mixture was stirred at 23 °C for 19 h. HATU (90.4 mg, 238 μmol, 1.00 equiv) was added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 15 min. Glycinamide hydrochloride (28.9 mg, 262 μmol, 1.00 equiv) was added to the reaction mixture, and the resulting mixture was stirred at 23 °C for 12 h. The product mixture was concentrated, and the residue obtained was diluted with ethyl acetate (60 mL). The diluted product mixture was transferred to a separatory funnel and sequentially washed with saturated aqueous sodium bicarbonate solution (1 × 30 mL), water (1 × 30 mL), and saturated aqueous sodium chloride solution (1 × 30 mL). The organic layer was dried over sodium sulfate. The dried solution was filtered and the filtrate was concentrated. The residue thus obtained was purified by flash-column chromatography (eluting with ethyl acetate initially, grading to 20% methanol–ethyl acetate, linear gradient) to provide the mercaptophenylacetamide 55 as a white solid (30.0 mg, 39%).
1H NMR (500 MHz, DMSO-d6) δ 8.17 (br t, J = 5.4 Hz, 1H), 7.56–7.51 (m, 1H), 7.34–7.24 (m, 4H), 7.05 (br s, 1H), 5.40 (s, 2H), 3.68 (s, 2H), 3.64 (d, J = 5.7 Hz, 2H), 2.31 (t, J = 7.2 Hz, 2H), 1.53 (app sex, J = 7.4 Hz, 2H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.3, 170.9, 169.8, 137.4, 134.1, 131.5, 130.7, 127.7, 127.6, 67.4, 42.0, 40.3, 35.3, 17.8, 13.4. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 347.1041; found, 347.1043.
4.5.44. Synthesis of ((2-((2-(isopropylamino)-2-oxoethyl)carbamoyl)phenyl)thio)methyl butyrate (56)
The mercaptobenzamide 56, a clear oil (61.8 mg, 25% over 3 steps), was synthesized by general procedure F using the amine hydrochloride obtained from propan-2-amine via general procedure D. Flash-column chromatography: 1) eluted with hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 15% methanol–ethyl acetate (linear gradient); 2) eluted with hexanes initially, grading to ethyl acetate, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.53 (bt, J = 5.6 Hz, 1H), 7.67 (bd, J = 7.4 Hz, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.52 (d, J = 7.8 Hz, 1H) 7.48 (t, J = 7.5 Hz, 1H), 7.33 (t, J = 7.5 Hz, 1H), 5.46 (s, 2H), 3.87 (app oct, J = 6.5 Hz, 1H), 3.77 (d, J = 5.9 Hz, 2H), 2.32 (t, J = 7.2 Hz, 2H), 1.53 (app sex, J = 7.4 Hz, 2H), 1.08 (s, 3H), 1.07 (s, 3H), 0.86 (t, J = 7.4 Hz). 13C NMR (125 MHz, DMSO-d6) δ 172.82, 168.11, 167.94, 137.07, 134.47, 131.07, 129.24, 128.51, 126.80, 66.66, 42.84, 41.04, 35.82, 22.92, 18.34, 13.83. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 375.1354; found, 375.1349.
4.5.45. Synthesis of ((2-((2-oxo-2-(prop-2-yn-1-ylamino)ethyl) carbamoyl)phenyl)thio)methyl butyrate (57)
The mercaptobenzamide 57, a white solid (56.1 mg, 27% over 3 steps), was synthesized by general procedure F using the amine hydrochloride obtained from prop-2-yn-1-amine via general procedure D. Flash-column chromatography: 1) eluted with hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 15% methanol–ethyl acetate (linear gradient); 2) eluted with hexanes initially, grading to ethyl acetate, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.63 (bt, J = 5.8 Hz, 1H), 8.33 (bt, J = 5.3 Hz, 1H), 7.58 (t, J = 7.3 Hz, 2H), 7.48 (t, J = 7.7 Hz, 1H), 7.34 (t, J = 7.5 Hz, 1H), 5.46 (s, 2H), 3.90 (dd, J = 5.1 Hz, 2.2 Hz, 2H), 3.83 (d, J = 6.0 Hz, 2H), 3.14 (s, 1H), 2.32 (t, J = 7.3 Hz, 2H), 1.53 (app sex, J = 7.3 Hz, 2H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.83, 168.95, 168.16, 136.65, 134.78, 131.14, 129.05, 128.65, 126.64, 81.72 (HMBC), 73.03 (HSQC), 66.61, 42.64, 35.82, 28.44, 18.34, 13.83. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 371.1041; found, 371.1042.
4.5.46. Synthesis of ((2-((2-(benzylamino)-2-oxoethyl)carbamoyl)phenyl)thio)methyl butyrate (58)
The mercaptobenzamide 58, a pale yellow solid (18.4 mg, 9% over 3 steps), was synthesized by general procedure F using the amine hydrochloride obtained from benzyl amine via general procedure D. Flash-column chromatography: 1) eluted with hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 15% methanol–ethyl acetate (linear gradient); 2) eluted with hexanes initially, grading to ethyl acetate, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.67 (bt, J = 5.7 Hz, 1H), 8.37 (bt, J = 5.8 Hz, 1H), 7.60–7.58 (m, 2H), 7.48 (t, J = 7.7 Hz, 1H), 7.35–7.27 (m, 4H), 7.23 (t, J = 7.1 Hz, 1H), 5.45 (s, 2H), 4.32 (d, J = 5.8 Hz, 2H), 3.88 (d, J = 5.9 Hz, 2H), 2.31 (t, J = 7.2 Hz, 2H), 1.53 (app sex, J = 7.3 Hz, 2H), 0.86 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ172.83, 169.18, 168.22, 139.82, 136.71, 134.78, 131.14, 129.07, 128.76, 128.66, 127.72, 127.28, 126.66, 66.62, 43.01, 42.58, 35.82, 18.35, 13.84. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 423.1354; found, 423.1348.
4.5.47. Synthesis of ((2-((2-oxo-2-(pentylamino)ethyl)carbamoyl)phenyl)thio)methyl butyrate (59)
The mercaptobenzamide 59, a pale yellow solid (79.6 mg, 34% over 3 steps), was synthesized by general procedure F using the amine hydrochloride obtained from pentan-1-amine via general procedure D. Flash-column chromatography: 1) eluted with hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 15% methanol–ethyl acetate (linear gradient); 2) eluted with hexanes initially, grading to ethyl acetate, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.57 (bt, J = 5.8 Hz, 1H), 7.78 (bt, J = 5.1 Hz, 1H), 7.59 (d, J = 7.6 Hz, 1H), 7.55 (t, J = 7.6 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.33 (t, J = 7.5 Hz, 1H), 5.46 (s, 2H), 3.79 (d, J = 5.9 Hz, 2H), 3.07 (app q, J = 6.5 Hz, 2H), 2.31 (t, J = 7.3 Hz, 2H), 1.53 (app sex, J = 7.2 Hz, 2H), 1.41 (app quin, J = 7.1 Hz, 2H), 1.32–1.21 (m, 4H), 0.86 (t, J = 7.1 Hz, 6H). 13C NMR (125 MHz, DMSO-d6) Δ172.82, 168.82, 168.14, 136.91, 134.60, 131.10, 129.17, 128.59, 126.73, 66.65, 42.91, 39.03, 35.82, 29.32, 29.08, 22.38, 18.34, 14.45, 13.83. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 403.1667; found, 403.1669.
4.5.48. Synthesis of ((2-((2-(tert-butylamino)-2-oxoethyl)carbamoyl)phenyl)thio)methyl butyrate (60)
The mercaptobenzamide 60, a clear oil (55.9 mg, 23% over 3 steps), was synthesized by general procedure F using the amine hydrochloride obtained from 2-methylpropan-2-amine via general procedure D. Flash-column chromatography: 1) eluted with hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 15% methanol–ethyl acetate (linear gradient); 2) eluted with hexanes initially, grading to ethyl acetate, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.47 (bt, J = 5.8 Hz, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.48 (t, J = 7.7 Hz, 1H), 7.40 (s, 1H), 7.33 (t, J = 7.5 Hz, 1H), 5.46 (s, 2H), 3.76 (d, J = 6.0 Hz, 2H), 2.32 (t, J = 7.2 Hz, 2H), 1.53 (app sex, J = 7.4 Hz, 2H), 1.27 (s, 9H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.80, 167.23, 168.07, 137.20, 134.36, 131.05, 129.27, 128.45, 126.83, 66.66, 50.71, 43.16, 35.81, 29.03, 18.33, 13.83. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 389.1511; found, 389.1506.
4.5.49. Synthesis of ((2-((2-((2-(dimethylamino)ethyl)amino)-2-oxoethyl)carbamoyl)phenyl)thio)methyl butyrate (61)
The mercaptobenzamide 61, a yellow oil (86.2 mg, 8% over 3 steps), was synthesized by general procedure F using the amine hydrochloride obtained from N,N-dimethylethylenediamine via general procedure D. Flash-column chromatography: 1) eluted with hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 15% methanol–ethyl acetate (linear gradient); 2) eluted with 1% triethylamine–dichloromethane initially, grading to 1% triethylamine–10% methanol–dichloromethane, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.61 (bt, J = 5.8 Hz, 1H), 7.73 (bt, J = 5.3 Hz, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.48 (t, J = 7.5 Hz, 1H), 7.34 (t, J = 7.6 Hz, 1H), 5.46 (s, 2H), 3.80 (d, J = 6.0 Hz, 2H), 3.18 (q, J = 6.3 Hz, 2H), 2.31 (app q, J = 6.4 Hz, 4H), 2.15 (s, 6H), 1.53 (app sex, J = 7.3 Hz, 2H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.82, 168.95, 168.18, 137.00, 134.52, 131.10, 129.28, 128.57, 126.78, 58.58, 45.64, 42.92, 37.14, 35.82, 18.35, 13.84. HRMS-ESI+ (m/z): [M]+ calcd for , 382.1801; found, 382.1799.
4.5.50. Synthesis of ((2-((2-((2-methoxy-2-oxoethyl)amino)-2-oxoethyl)carbamoyl)phenyl)thio)methyl butyrate (62)
The mercaptobenzamide 62, a pale yellow solid (127.9 mg, 12% over 3 steps), was synthesized by general procedure F using the amine hydrochloride obtained from glycine methyl ester hydrochloride via general procedure D. Flash-column chromatography: 1) eluted with hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 15% methanol–ethyl acetate (linear gradient); 2) eluted with dichloromethane initially, grading to 10% methanol–dichloromethane, linear gradient; 3) eluted with 50% ethyl acetate–hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 15% methanol–ethyl acetate (linear gradient).
1H NMR (500 MHz, DMSO-d6) δ 8.67 (bt, J = 5.9 Hz, 1H), 8.32 (bt, J = 5.9 Hz, 1H), 7.59 (t, J = 6.5 Hz, 2H), 7.48 (t, J = 7.6 Hz, 1H), 7.33 (t, J = 7.5 Hz, 1H), 5.46 (s, 2H), 3.89 (app t, J = 7.9 Hz, 4H), 3.64 (s, 3H), 2.32 (t, J = 7.2 Hz, 2H), 1.53 (app sex, J = 7.3 Hz, 2H), 0.87 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.84, 170.79, 169.76, 168.17, 136.64, 134.80, 131.16, 129.06, 128.66, 126.64, 66.62, 42.57, 41.09, 35.82, 18.35, 13.83. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 405.1096; found, 405.1098.
4.5.51. Synthesis of ((2-((2-((2-amino-2-oxoethyl)amino)-2-oxoethyl)carbamoyl)phenyl)thio)methyl butyrate (63)
The mercaptobenzamide 63, a white solid (77.0 mg, 7% over 3 steps), was synthesized by general procedure G using the amine hydrochloride obtained from glycinamide hydrochloride via general procedure D. Flash-column chromatography: 1) eluted with hexanes initially, grading to ethyl acetate (linear gradient), followed by grading to 15% methanol–ethyl acetate (linear gradient); 2) eluted with 50% acetone–dichloromethane initially, grading to acetone, linear gradient.
1H NMR (500 MHz, DMSO-d6) δ 8.68 (bt, J = 5.6 Hz, 1H), 8.11 (bt, J = 5.6 Hz, 1H), 7.58 (t, J = 8.2 Hz, 2H), 7.48 (t, J = 7.7 Hz, 1H), 7.34 (t, J = 7.6 Hz, 1H), 7.27 (s, 1H), 7.12 (s, 1H), 5.45 (s, 2H), 3.86 (d, J = 5.9 Hz, 2H), 3.66 (d, J = 5.9 Hz, 2H), 2.31 (t, J = 7.3 Hz, 2H), 1.53 (app sex, J = 7.3 Hz, 2H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.83, 171.37, 169.41, 168.36, 136.71, 134.76, 131.17, 129.19, 128.64, 126.71, 66.69, 43.10, 42.40, 35.83, 18.35, 13.84. HRMS-ESI+ (m/z): [M + Na]+ calcd for , 390.1100; found, 390.1094.
Supplementary Material
Acknowledgements
This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). Support for this work comes from the Intramural Research Program of NIDDK, NIH and the Office of AIDS Research at NIH.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2019.06.020.
References
- [1].a) UNAIDS, Global HIV & AIDS statistics – 2018 fact sheet. http://www.unaids.org/en/resources/fact-sheet (24 September 2018);; b) U.S. Department of Health and Human Services, FDA-approved HIV medicines. https://aidsinfo.nih.gov/understanding-hiv-aids/fact-sheets/21/58/fda-approved-hiv-medicines (24 September 2018).
- [2].a) National Institute on Drug Abuse, What is HAART?. https://www.drugabuse.gov/publications/research-reports/hivaids/what-haart (24 September 2018);; b) Moore RD, Chaisson RE, AIDS 13 (1999) 1933–1942. [DOI] [PubMed] [Google Scholar]
- [3].a) Palella FJ, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD, Engl N. J. Med. 338 (1998) 853–860; [DOI] [PubMed] [Google Scholar]; b) Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, Hakim JG, Kumwenda J, Grinsztejn B, Pilotto JHS, Godbole SV, Mehendale S, Chariyalertsak S, Santos BR, Mayer KH, Hoffman IF, Eshleman SH, Piwowar-Manning E, Wang L, Makhema J, Mills LA, de Bruyn G, Sanne I, Eron J, Gallant J, Havlir D, Swindells S, Ribaudo H, Elharrar V, Burns D, Taha TE, Nielsen-Saines K, Celentano D, Essex M, Fleming TR, Engl N. J. Med. 365 (2011) 493–505. [Google Scholar]
- [4].DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents – A Working Group of the Office of AIDS Research Advisory Council (OARAC), Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf (September 24, 2018).
- [5].Loubiere S, Meiners C, Sloan C, Freedberg KA, Yazdanpanah Y, Curr. Opin. HIV AIDS 5 (2010) 225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Oldfield E, Feng X, Trends Pharmacol. Sci. 35 (2014) 664–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].For a review of recent strategies used in the development of next-generation HIV antivirals, see: a) Zuo X, Huo Z, Kang D, Wu G, Zhou Z, Liu X, Zhan P, Expert Opin. Ther. Pat. 28 (2018) 299–316; [DOI] [PubMed] [Google Scholar]; b) Zhan P, Pannecouque C, Clercq ED, Liu X, J. Med. Chem. 59 (2016) 2849–2878. [DOI] [PubMed] [Google Scholar]
- [8].Nikolayevskiy H, Scerba MT, Deschamps JR, Appella DH, Chem. Eur J. 24 (2018) 9485–9489. [DOI] [PubMed] [Google Scholar]
- [9].Jenkins LMM, Ott DE, Hayashi R, Coren LV, Wang D, Xu Q, Schito ML, Inman JK, Appella DH, Appella E, Nat. Chem. Biol. 6 (2010) 887–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Schito ML, Goel A, Song Y, Inman JK, Fattah RJ, Rice WG, Turpin JA, Sher A, Appella E, AIDS Res. Hum. Retrovir. 19 (2003) 91–101; [DOI] [PubMed] [Google Scholar]; b) Schito ML, Soloff AC, Slovitz D, Trichel A, Inman JK, Appella E, Turpin JA, Barratt-Boyes SM, Curr. HIV Res. 4 (2006) 379–386. [DOI] [PubMed] [Google Scholar]
- [11].Jenkins LMM, Hara T, Durell SR, Hayashi R, Inman JK, Piquemal J-P, Gresh N, Appella E, J. Am. Chem. Soc. 129 (2007) 11067–11078. [DOI] [PubMed] [Google Scholar]
- [12].Deshmukh L, Tugarinov V, Appella DH, Clore GM, Angew. Chem. Int. Ed. 57 (2018) 2687–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Ugaonkar SR, Clark JT, English LB, Johnson TJ, Buckheit KW, Bahde RJ, Appella DH, Buckheit RW, Kiser PF, J. Pharm. Sci. 104 (2015) 3426–3439; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang Y, Zhu J, Hassink M, Jenkins LMM, Wan Y, Appella DH, Xu J, Appella E, Zhang X, Emerg. Microb. Infect. 6 (2017) e40; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wallace GS, Cheng-Mayer C, Schito ML, Fletcher P, Miller Jenkins LM, Hayashi R, Neurath AR, Appella E, Shattock RJ, J. Virol. 83 (2009) 9175–9182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Saha M, Scerba MT, Shank NI, Hartman TL, Buchholz CA, Buckheit RW, Durell SR, Appella DH, ChemMedChem 12 (2017) 714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Morellet N, Jullian N, De Rocquigny H, Maigret B, Darlix JL, Roques BP, EMBO J. 11 (1992) 3059–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Adriano DA, Lívia BS, Donald JA, Curr. Top. Med. Chem. 9 (2009) 771–790.19754394 [Google Scholar]
- [17].a) Temperini A, Piazzolla F, Minuti L, Curini M, Siciliano C, J. Org. Chem. 82 (2017) 4588–4603; [DOI] [PubMed] [Google Scholar]; b) Nascimento V, Ferreira NL, Canto RFS, Schott KL, Waczuk EP, Sancineto L, Santi C, Rocha JBT, Braga AL, Eur. J. Med. Chem. 87 (2014) 131–139. [DOI] [PubMed] [Google Scholar]
- [18].Nara PL, Fischinger PJ, Nature 332 (1988) 469–470. [DOI] [PubMed] [Google Scholar]
- [19].Schewe T, Gen. Pharmacol. Vasc. Syst. 26 (1995) 1153–1169. [DOI] [PubMed] [Google Scholar]
- [20].Lee BM, De Guzman RN, Turner BG, Tjandra N, Summers MF, J. Mol. Biol. 279 (1998) 633–649. [DOI] [PubMed] [Google Scholar]
- [21].Trott O, Olson AJ, J. Comput. Chem. 31 (2010) 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang C, Nguyen PH, Pham K, Huynh D, Le T-BN, Wang H, Ren P, Luo R, J. Comput. Chem. 37 (2016) 2436–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Simiele M, D’Avolio A, Baietto L, Siccardi M, Sciandra M, Agati S, Cusato J, Bonora S, Di Perri G, Antimicrob. Agents Chemother. 55 (2011) 2976–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Utsugi M, Dobashi K, Koga Y, Shimizu Y, Ishizuka T, Iizuka K, Hamuro J, Nakazawa T, Mori M, J. Leukoc. Biol. 71 (2002) 339–347. [PubMed] [Google Scholar]
- [25].Leonardi R, Rock CO, Jackowski S, Zhang Y-M, Proc. Natl. Acad. Sci. U. S. A 104 (2007) 1494–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fogarty KH, Berk S, Grigsby IF, Chen Y, Mansky LM, Mueller JD, J. Mol. Biol. 426 (2014) 1611–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K, J. Comput. Chem. 26 (2005) 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].a) Jo S, Kim T, Iyer VG, Im W, J. Comput. Chem. 29 (2008) 1859–1865; [DOI] [PubMed] [Google Scholar]; b) Kim S, Lee J, Jo S, Brooks CL, Lee HS, Im W, J. Comput. Chem. 38 (2017) 1879–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE, Nucleic Acids Res. 28 (2000) 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, MacKerell AD Jr., Nat. Methods 14 (2016) 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Humphrey W, Dalke A, Schulten K, J. Mol. Graph. 14 (1996) 33–38. [DOI] [PubMed] [Google Scholar]
- [32].Brooks BR, Brooks CL III, Mackerell AD Jr., Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M, J. Comput. Chem. 30 (2009) 1545–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kumari R, Kumar R, Lynn A, J. Chem. Inf. Model. 54 (2014) 1951–1962. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.