

Abstract
BACKGROUND:
MicroRNAs (miRs) play critical roles in regulation of numerous biological events, including cardiac electrophysiology and arrhythmia, through canonical RNA interference (RNAi) mechanism. However, it remains unknown if endogenous miRs modulate the physiological homeostasis of the heart through noncanonical mechanisms.
METHODS:
We focused on the predominant miR of the heart––miR1 and investigated if miR1 could physically bind with ion channels in cardiomyocytes by electrophoretic mobility shift assay (EMSA), in situ proximity ligation assay (PLA), RNA pull down and RNA Immunoprecipitation (RIP) assays. The functional modulations of cellular electrophysiology were evaluated by inside-out and whole-cell patch clamp. Mutagenesis of miR1 and the ion channel was utilized to understand the underlying mechanism. The effect on the ex vivo heart was demonstrated through investigating arrhythmia-associated human single nucleotide-polymorphisms (hSNPs) with miR1-deficient mice.
RESULTS:
We found that endogenous miR1 could physically bind with cardiac membrane proteins, including an inward-rectifier potassium channel Kir2.1. The miR1-Kir2.1 physical interaction was observed in mouse, guinea pig, canine and human cardiomyocytes. miR1 quickly and significantly suppressed IK1 at sub-pmol/L concentration, which is close to endogenous miR-expression level. Acute presence of miR1 depolarized resting membrane potential (RMP) and prolonged final repolarization of the action potential in cardiomyocytes. We identified three miR1-binding residues on the C-terminus of Kir2.1. Mechanistically, miR1 binds to the pore-facing G-loop of Kir2.1 through the core sequence AAGAAG, which is outside its RNAi seed region. This biophysical modulation is involved in the dysregulation of gain-of-function Kir2.1-M301K mutation in short QT or atrial fibrillation. We found that an arrhythmia-associated human single nucleotide polymorphism of miR1––hSNP14A/G specifically disrupts the biophysical modulation while retaining the RNAi function. Remarkably, miR1 but not hSNP14A/G relieved the hyperpolarized RMP in miR1-deficient cardiomyocytes, improved the conduction velocity, and eliminated the high inducibility of arrhythmia in miR1-deficient hearts ex vivo.
CONCLUSIONS:
Our study reveals a novel evolutionarily-conserved biophysical action of endogenous miRs in modulating cardiac electrophysiology. Our discovery of miRs’ biophysical modulation provides a more comprehensive understanding of ion-channel dysregulation and may provide new insights into the pathogenesis of cardiac arrhythmias.
Keywords: microRNA, biophysical modulation, ion channel, cardiac electrophysiology, arrhythmia
INTRODUCTION
Cardiac arrhythmias, due to profound dysregulation of numerous ion channels and transporters,1 dramatically increase the death risk of patients.2 MicroRNAs (miRs) have a remarkable influence on the physiology of the heart and the remodeling of diseased hearts through canonical RNA-interference (RNAi) mechanism.3–6 miR1, the most predominant miR in the heart,7 controls cardiac development and functions.8 Depletion of miR1 causes incompletely penetrant lethality;9, 10 miR1 upregulation exacerbates arrhythmogenesis by post-transcriptionally targeting various ion channels, such as KCNJ2 (encodes Kir2.1) and GJA1 (encodes Cx43).6 miR1 downregulation and Kir2.1 upregulation were consistently observed in human atrial fibrillation (AF).11 Meanwhile, ion channel could also regulate miR expression in developing electrical dysfunction through a KChIP2/miR34 regulatory axis.4 Current studies of the crosstalk between cardiac ion channels and miRs are limited to the canonical RNAi mechanism.
It has been recently recognized that miRs play broad roles in regulating biological processes beyond being interfering RNAs.12 Extracellular let-7 exhibits ligand-like roles by interacting with Toll-like receptors (TLRs) in neurodegeneration13 or pain sensing,14 although the direct physical binding has not been proven yet. Tumor-secreted miR-21 and miR1-29a could also activate TLR7/TLR8 of immune cells;15 extracellular miR-711 mediates itch sense in neurons by activating TRPA1.16 However, a >μmol/L concentration of miRs, applied in those extracellular miR studies, is much higher than the expression level of endogenous miRs,17 raising an artifact concern. Particularly, there are no reports that abundant intracellular miRs can elicit biological responses via a mechanism of physical interaction with non-classical RNA binding proteins.
The action potential (AP) of cardiomyocyte is generated by highly-coordinated ion movements through sarcolemmal voltage-gated ion channels.18 Genetic mutation and toxic modulator are two major epidemiological factors of arrhythmia and cause dysregulations of ion channels, including gene transcription and translation, trafficking and biophysical modulation.18, 19 For example, the inward rectifier K+ (Kir) channels are tetrameric and contain two transmembrane helix domains, the ion-selective P-loop and cytoplasmic N- and C-terminal domains, and have an unusual dependence of rectification on extracellular K+.20, 21 The C-terminal domain contains an intrinsically flexible pore-facing G-loop structure, which forms the narrowest portion and acts as the critical regulatory element for gating.22 Kir2.1 is the most abundant Kir channel in the ventricle and is responsible for the inward rectifying potassium current IK1 that stabilizes the resting membrane potential (RMP), controls cardiac excitability, and shapes the final repolarization phase of AP.23, 24 Mutations in KCNJ2 have been associated with multiple channelopathies.25 IK1 deficiency leads to abnormal automaticity of heart-muscle cells or long-QT syndromes (LQTS).26, 27 A gain-of-function mutation of Kir2.1––M301K, in which the methionine at 301 is mutated to lysine, disrupts the biophysical modulation of voltage-dependent rectification and causes short-QT syndrome and AF.28, 29 In spite of a large diversity of ion-channel modulators,30 to date, it is still unknown whether RNA molecules serve as a biophysical modulator of ion-channels to regulate cardiac electrophysiology.
Herein, we investigated if miR1 specifically binds with cardiac plasma membrane proteins, and revealed an evolutionarily-conserved direct binding between miR1 and Kir2.1, which endogenously exists in cardiomyocytes. Inside-out and whole-cell patch clamp recordings exhibited the biophysical modulation of cardiac electrophysiology by miR1. We further studied the mechanism of this physical interaction, and investigated its pathophysiological relevance by using miR1-deficient transgenic mice. Our studies demonstrated a novel mechanism of miR-ion channel biophysical modulation that regulates cardiac arrhythmic risk.
METHODS
Animal models, cell lines information and all experimental approaches are detailed in the online-only Data Supplement. IRB approval was obtained for the study, and all animal studies were performed in accordance with institutional guidelines.
Quantification and Statistical Analysis
Data are expressed as mean±S.E.M, with n indicating the number of distinct biological samples studied. Statistical significance of differences in means was estimated by ANOVA and subsequent contrasts test with multiple comparison correction. I-V curve data was assessed using separate independent t-tests, and count data were assessed by one-sided Fisher’s exact test (R statistical software version 3.6.1). p<0.05 was considered statistically significant.
Data Availability
The data, methods, and study materials, including a primer list in Table I in the Supplement, that support the findings of this work are available from the corresponding author upon reasonable request.
RESULTS
A Physical Binding of miR1 and Kir2.1 Exists Endogenously in Cardiomyocytes
To determine whether intracellular miRs could act as direct modulators of cardiac ion channels, we extracted membrane proteins (M-proteins) from neonatal mouse hearts, performed electrophoretic mobility shift assay (EMSA) and assessed if miR1 or miR451a, both of which are increasingly expressed in human ventricle during the development and maturation of the heart,31 can bind with cardiac M-proteins. We found that biotinylated (Bio)-miR1 yielded several shifted bands with different intensities (Figure 1A), which was not observed with Bio-miR451a. Especially, non-biotinylated miR1, but not miR451a, competitively inhibited some specific shifted bands. These results suggested that miR1 can specifically bind with cardiac M-proteins.
Figure 1. An evolutionarily-conserved physical binding of miR1 and Kir2.1 exists endogenously in cardiomyocytes.
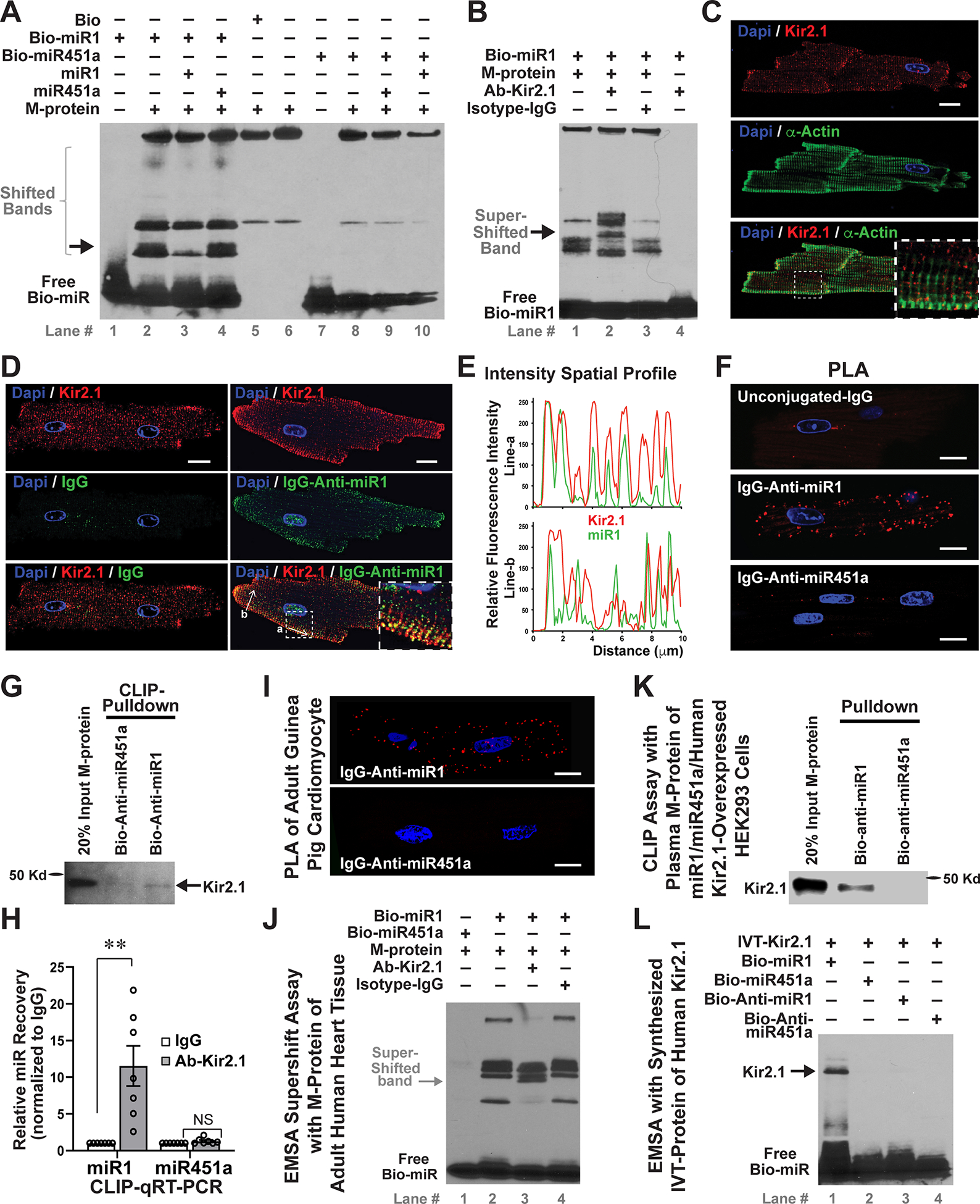
A, A representative image of in vitro EMSA of biotinylated (Bio)-miR1 and membrane protein (M-protein), extracted from neonatal mouse hearts, showed several shifted bands (lane #2) that were specifically suppressed by non-biotinylated miR1 (lane #3), but not by miR451a (lane #4); while no specific shifted bands were observed in EMSA of Bio-miR451a. B, EMSA supershift assays with a rabbit antibody of Kir2.1 (Ab-Kir2.1), but not isotype IgG, yielded specific further-shifted bands (arrow, lane #2). C, Immunostaining of Kir2.1 and sarcomere α-actin of adult mouse ventricular cardiomyocytes. The inserted panel is an amplified region to show that Kir2.1 was localized on the sarcolemmal membrane and T-tubules between sarcomeres. D and E, Immunostaining (D) and traces of fluorescence intensity spatial profiles (E) of Kir2.1 (Red) and miR1 (Green) through two white arrow-lines in panel D, showed that endogenous miR1 and Kir2.1 were co-localized in adult mouse ventricular cardiomyocytes. The inserted panel is an amplified region. F, An in situ PLA of IgG-Anti-miR1 and Ab-Kir2.1 yielded discrete fluorescence spots (Red), demonstrating the miR1-Kir2.1 interaction at single molecular level in adult mouse ventricular cardiomyocytes. G, Cross-linking immunoprecipitation (CLIP) assays showed that Kir2.1 protein was pulled down by Bio-Anti-miR1, but not by Bio-Anti-miR451a, from plasma M-protein of adult mouse heart tissue. An input (20%) was used as the loading control. H, CLIP qRT-PCR of adult heart plasma M-protein by using Ab-Kir2.1 or IgG revealed a preferential interaction of Kir2.1 with miR1 but not miR451a. n=7; **p<0.01 vs. IgG group; NS, not significant. I, In situ PLA assays proved that miR-Kir2.1 interaction exists in guinea pig ventricular cardiomyocytes. J, EMSA supershift assays with adult human heart M-protein revealed that miR1 specifically binds with human Kir2.1. K, CLIP pulldown assays of plasma M-proteins, extracted from HEK293 cells that were co-transfected with human Kir2.1 and miR1 / miR451a mimics, showed that Kir2.1 was successfully pulled down by Bio-Anti-miR1, but not by Bio-Anti-miR451a. 20% input was used as the loading control. L, EMSA of IVT-synthesized Kir2.1 (IVT-Kir2.1) showed that only Bio-miR1 produced shifted band, while no bands were generated by Bio-Anti-miR1, -miR451a or –Anti-miR451a, confirming the specific binding of miR1 with human Kir2.1. The nuclei are DAPI stained in all images, and all bars indicate 10μm. Statistical significance was estimated by subsequent Student’s t test.
We asked if miR1-bound M-proteins contain ion channels and thus conducted the EMSA supershift assay by using various ion-channels antibodies (Abs). The Ab-Kir2.1 dramatically attenuated a shifted band and generated another further-shifted band with heavier molecular weight, while the other ion-channel antibodies and isotype-IgG control showed no obvious influence on those EMSA-shifted bands (Figure IA in the Supplement and Figure 1B). This miR1-Kir2.1 interaction was replicated with adult mouse ventricle tissues (Figure IB in the Supplement), in which Kir2.1 is highly expressed.32 Kir2.1 was successfully pulled down from cardiac M-proteins by Bio-miR1, but not by Bio-miR451a in cross-linking and immunoprecipitation (CLIP) assays (Figure IC in the Supplement). Taken together, these results demonstrate that Kir2.1 is one of cardiac M-proteins to which miR1 binds.
We next studied whether the miR1-Kir2.1 physical interaction occurs endogenously in cardiomyocytes. Consistent with previous study,33 we observed that Kir2.1 is highly enriched on sarcolemma and T-tubules, between α-actinin-stained sarcomeres in ventricular cardiomyocytes (Figure 1C). To detect endogenous miR1, we conjugated complementary antisense oligonucleotide of miR1 (Anti-miR1) with mouse IgG to form an IgG-Anti-miR1 complex. IgG-Anti-miR1 yielded specific positive staining in cardiomyocytes, including a particular cell nuclei area, which was not observed in unconjugated IgG staining (Figure 1D). Importantly, much of IgG-Anti-miR1 staining overlapped with Kir2.1 staining on the sarcolemmal and T-tubule membrane (Figure 1E), indicating the endogenous co-localization of miR1 with Kir2.1. By using In situ proximity ligation assay (PLA), which can detect protein-RNA direct interaction with single-molecule sensitivity,34 we observed that IgG-Anti-miR1, but not unconjugated-IgG or IgG-Anti-miR451a, produced discrete fluorescence spots in mouse ventricular cardiomyocytes (Figure 1F and Movie I in the Supplement). Moreover, Kir2.1 was specifically pulled down by Bio-Anti-miR1 from cardiac plasma M-proteins (Figure 1G), and miR1 was significantly recovered by Ab-Kir2.1 (Figure 1H). Collectively, these results reveal that the physical binding of miR1 with Kir2.1 exists endogenously in cardiomyocytes.
The miR1-Kir2.1 Physical Binding Is Evolutionarily Conserved across Species
We asked whether this novel miR1-Kir2.1 direct interaction is evolutionarily conserved in mammals, which implies a broad functional significance of this miR-ion channel biophysical action. Indeed, PLA assays showed that endogenous miR1-Kir2.1 binding exists in both guinea pig (Figure 1I) and canine (Figure ID in the Supplement) ventricular myocytes. Furthermore, Bio-miR1 generated obvious bands with M-proteins of human heart tissues and produced a specific supershift band by Ab-Kir2.1, but not by Isotype-IgG (Figure 1J). We extracted plasma M-proteins from HEK293 cells (Figure IE in the Supplement), in which human Kir2.1, miR1 and miR451a were co-expressed, and expectedly observed that Kir2.1 was specifically pulled down by Bio-Anti-miR1, but not by Bio-Anti-miR451a (Figure 1K), confirming that the miR1-Kir2.1 physical interaction occurs endogenously in living cells. We also synthesized and purified human Kir2.1 by using in vitro translation (IVT) system (Figure IF and IG in the Supplement). EMSA showed that miR451a, anti-miR451a and anti-miR1 didn’t bind with Kir2.1, and only miR1 bound to IVT-Kir2.1 and produced a specific shifted band (Figure 1L), which was confirmed by EMSA supershift assays (Figure IH in the Supplement). In contrast to cell extractions, assays with IVT-Kir2.1 protein suggest that the miR1-Kir2.1 affinity is a direct RNA-protein contact and doesn’t require a third factor mediating the binding.
The G-loop Region of Kir2.1 Contains Three Necessary miR-Binding Residues
To pinpoint the miR1-binding region(s) of Kir2.1, we synthesized IVT-proteins of Kir2.1-fragments, including the N-terminus, transmembrane and C-terminus (CT) domains (Figure 2A), and performed EMSA and pulldown assays. The specifically shifted and pulled-down bands in the CT group indicated that miR1 only binds to the CT. We broke the CT into three domains (i.e. CT1, CT2 and CT3) and synthesized IVT-proteins of fragments with N-terminal Flag epitope. Only CT2 fragment produced a specific shifted band in EMSA and was pulled down by Bio-miR1 (Figure 2B), demonstrating that miR1 binds to the middle region CT2. We designed and synthesized CT-2.1, −2.2, 2.3 and −2.4, which cover different CT2 domains with partial overlapped regions. The pulldown assays demonstrated that Bio-miR1 obviously binds to CT2.3 with high affinity, while negative binding of CT2.1, CT2.2 and CT2.4 suggests that the miR1-binding region is partially cleft at the CT2.2-CT2.4 junction (Figure 2C). We further synthesized a series of peptides, in which three by three amino acids (AAs) were either truncated from CT2 (T1–T9) or extended from CT2.4 (E1–E7). RNA pulldown assays showed that three-AA truncations from CT2 fragments had no significant influence on the affinity efficiency (Figure IIA in the Supplement) until 324H-326Y were removed from CT2-T6, which abolished miR1’s binding with CT2-T6 (15.37±2.98%, n=3) and resulted in a significantly decreased pulldown efficiency of CT2-T7 (1.87±0.67%, n=3, p=0.0397 vs. CT2-T6) (Figure 2D). These data suggest that 324H-326Y is an essential residue for miR1-Kir2.1 interaction. Meanwhile, CT2.4-E1 and -E2 showed no enrichment (Figure IIB in the Supplement) and CT2.4-E3 generated a suspicious pulldown band (0.53±0.23%, n=4), while CT2.4-E4 was obviously immunoprecipitated by Bio-miR1 with a 3.71±0.67% pulldown efficiency (n=4, p=0.0296 vs. CT2.4-E3) (Figure 2E and 2F), suggesting that 306A-308T is another critical site for miR1-Kir2.1 binding. Moreover, three extra AAs on CT2.4-E6 significantly increased the pulldown efficiency (7.43±0.36%, n=4) comparing to CT2.4-E4 or –E5 (4.17±0.40%, n=4, p=0.0106 vs. CT2.4-E6) (Figure 2F), demonstrating that 300G-302V contains the third important miR1-binding residue. Noticeably, 300G-302V and 306A-308T are the arms of the G-loop, the critical regulatory element of Kir2.1 gating,22 indicating a potential functional modulation of miR1 on Kir2.1. In 3D model of Kir2.1 structure with highlights of identified miR1-binding residues (Figure 2G), the electrostatic surface potential analysis revealed a large patch of positive charges on Kir2.1 surface and G-loop, suggesting a binding potential with naturally negatively-charged RNA. Overall, our studies demonstrated that the 300G-326Y region of Kir2.1 contains three separated residues that are critical for miR1-Kr2.1 physical binding.
Figure 2. The G-loop region of Kir2.1 contains three necessary miR1-binding residues.
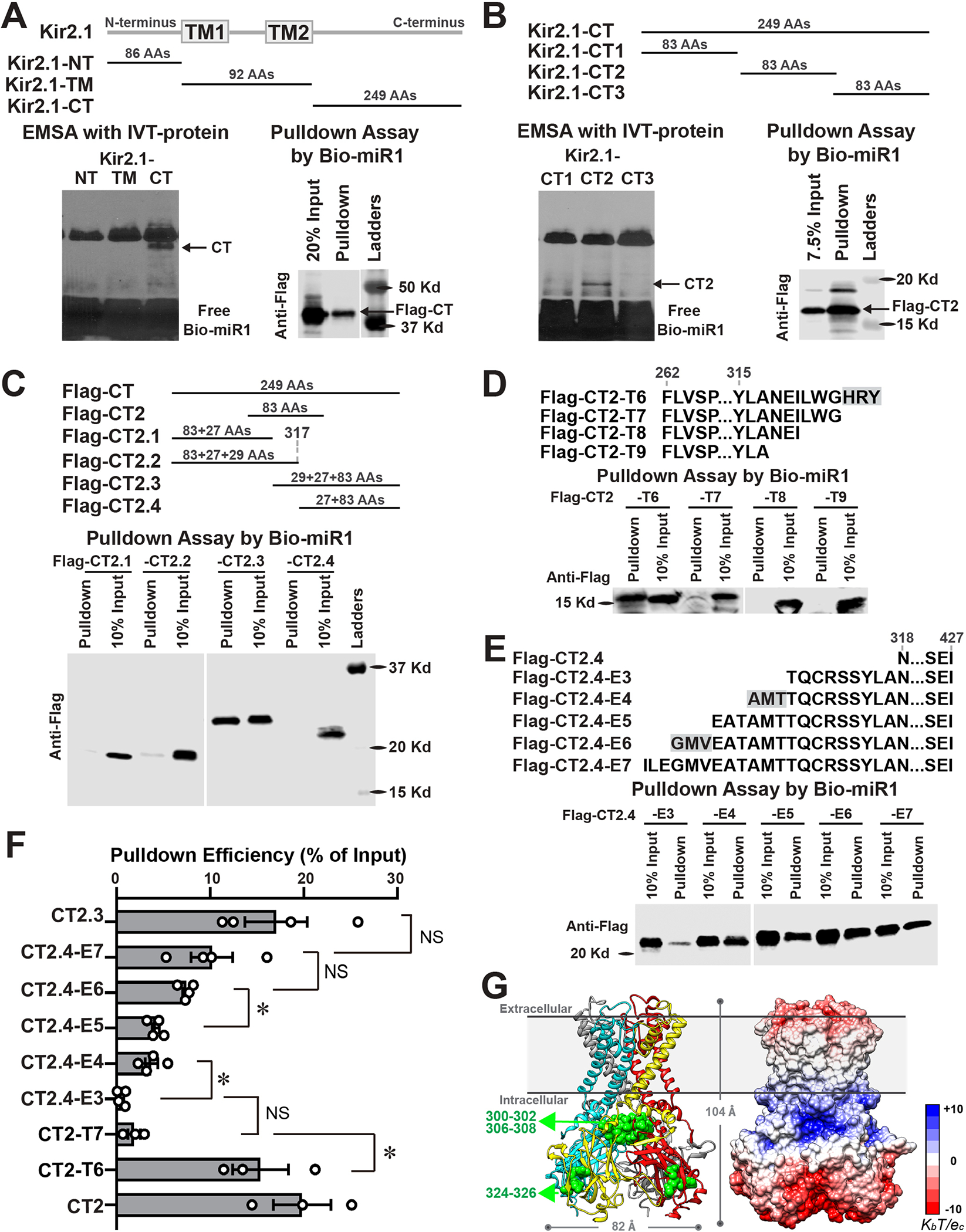
A, EMSA (left panel) and pulldown assays (right panel) of IVT-proteins of Kir2.1 fragments demonstrated that miR1 specifically binds with the C-terminus (CT), but not with N-terminus (NT) and transmembrane (TM) regions. B, EMSA with three subdivided Kir2.1-CT fragments (left panel) showed that miR1 specifically binds with CT2, which was confirmed by pulldown assay (right panel). C, Pulldown assays with four designed CT fragments revealed that CT2.3 was specifically immunoprecipitated by Bio-miR1. The negative enrichment of CT2.2 and CT2.4 demonstrated that miR1-binding region was partially cleft at their junction. D, Pulldown assays with Flag-CT2 truncated fragments (T6–T9) showed that remove of 324H-326Y (highlighted with gray background) from CT2-T6 abolished the binding between miR1 and CT2-T7. E, Pulldown assays with Flag-CT2.4 extended fragments (E3–E7) showed that an extension of 306A-308T produced obvious enrichment of CT2.4-E4 by Bio-miR1 and the extension of 300G-302V further increased the pulldown efficiency of CT2.4-E6 than that of CT2.4-E5. F, A quantification summary of Bio-miR1 pulldown assay revealed that the immunoprecipitation efficiency significantly changed three times while three AAs were added or truncated, demonstrating three essential miR1-binding residues on the C-terminus of Kir2.1 as highlighted in panel D and E. n≥3 in all groups. *p <0.05 vs. indicated groups; NS, not significant. Statistical significance was estimated by one-way ANOVA and subsequent Student’s t test with multiple testing adjustments. G, Front view of 3D modelled structure with each subunit in different colors (left) and electrostatic potential surface (right) of human Kir2.1 at a physiological pH. Three miR1-binding residues are indicated in green; the scale of electrostatic potential is indicated as ± KbT/ec, where Kb is the Boltzmann constant, T is the temperature and ec is the charge of one electron.
miR1 Suppresses IK1 through Direct Binding to Kir2.1
To investigate the functional consequence of miR1-Kir2.1 physical interaction, we performed inside-out patch clamping in human Kir2.1-overexpressed HEK293 (Kir2.1-HEK293) cells. The inside-out patch exposes the intracellular domains of Kir2.1 and enables to directly study the acute effect of miR on IK1 (Figure 3A). We found that miR1 significantly suppressed IK1, including inward and outward currents, while miR451a, miR133a, and miR499 had no influence on IK1 (Figures 3A–3C and Figure IIC and IID in the Supplement). A dose-response curve showed that miR1 significantly suppressed IK1 even at 0.01 pmol/L level and reached the maximum ~35% suppression with >100 pmol/L (Figure 3D). The time-course recording of miR1 administration showed that IK1 suppression occurred within seconds and reached the maximum level at ~2.5 minutes (Figure IIE in the Supplement), demonstrating a quick modulation of miR1 on Kir2.1 dynamic through a physical interaction. We also studied the biophysical modulation of miR1 in Kir2.1-HEK293 cells by whole-cell patch clamp. To bypass the conventional RNAi function in living cells, we delivered miR1 or miR451a through the recording pipette and measured IK1 within 5 minutes after rupture to allow diffusion of miRs (Figure 3E). As a result, IK1 was significantly smaller with acute-presence of miR1 (e.g. 6.22±1.07 pA/pF at −40mV, n=18, p=0.019 vs. control) than that in the control (12.52±2.42 pA/pF at −40mV, n=17) or miR451a (10.61±2.05 pA/pF at −40mV, n=11) groups (Figure 3F).
Figure 3. miR1 suppresses IK1 through directly binding to Kir2.1.

A, Representative traces of IK1, recorded from inside-out patch clamp (inserted box) from Kir2.1- HEK293 cells before and after miR1 or miR451a treatment. B and C, IK1 current-voltage (I-V) curve from inside-out patch clamp recordings showed that miR1 (100pmol/L, n=19, ***p<0.001) significantly suppressed IK1 (B), while miR451a (n=13) had no significant influence on IK1 (C). D, A dose-response curve of miR1 on IK1, measured from inside-out patch clamping at both +80mV and −80mV, demonstrated that miR1 at sub-pmol/L level significantly suppressed IK1. n=8. E and F, Representative traces (E) and I-V curves (F) of IK1, measured by whole-cell patch clamp with Kir2.1- HEK293 cells in the absence (control) and presence of miR1 or miR451a, demonstrated that miR1 acutely inhibited IK1 (*p <0.05 vs. control). miRs were delivered through the patch clamp recording pipette as shown in the inserted box. G and H, Representative traces (G) and I-V curves (H) of IK1 from inside-out patch clamp recordings of Kir2.1-HEK293 cells before and after miR1 treatment combined with the presence of 27AA (300G-326Y) or mutated-peptide, showed that the presence of 27AA-peptid, but not mutated-peptide, completely blocked the suppression of IK1 by miR1. Three miR1-binding residues are highlighted with gray background; the mutated AAs are underlined. *p <0.05 vs. before miR1 treatment. I, HEK293 cells were transfected with wildtype Kir2.1 DNA (WT-Homo) or co-transfected with WT and M301K-Kir2.1 DNA (WT/M301K-Hetero). Representative traces (left panel) of IK1 from inside-out patch-clamp recordings before (control) and after miR1 treatment and a summary bar graph of suppressed IK1 (at +80mV and −80mV) (right panel) demonstrated that miR1suppressed WT-Homo (n=9) but not WT/M301K-Hetero IK1 (n=7). J, IK1 I-V curve from whole-cell patch-clamp recordings consistently showed that miR1 had no effect on WT/M301K-Hetero IK1. Statistical significance of I-V curve data was estimated by separate independent t test.
To confirm if IK1 suppression by miR1 requires a physical binding with Kir2.1, we synthesized two peptides, a 27-AA peptide of 300G-326Y that binds with miR1 (Figure IIF in the Supplement) and a mutated peptide in which eight AAs of miR1-binding residues were mutated to alanine, yielding no binding with miR1. Functionally, miR1 failed to suppress IK1 in the presence of the 27-AA peptide, but the mutated peptide had no effect on miR1’s biophysical modulation (Figure 3G and 3H), demonstrating that the 27-AA peptide competitively bound with miR1, prevented miR1-Kir2.1 interaction and subsequently abolished the miR1-induced suppression of IK1.
Since the gain-of-function M301K mutation, reported in short-QT syndrome and AF patients,28, 29 is located at one of miR1-binding residues on Kir2.1, we asked if miR1’s biophysical modulation is involved in the dysregulation of arrhythmogenic diseases. While no IK1 was measured in M301K alone-transfected HEK293 cells (M301K-homo), IK1 in HEK293 cells co-transfected with wildtype (WT)-Kir2.1 and M301K had significantly larger outward current at potentials ≧−30mV (e.g. 15.09±3.63 pA/pF at −20mV, n=5) compared with WT alone-transfected cells (WT-homo, 1.33±1.99 pA/pF at −20mV, n=4) (Figure IIIA and IIIB in the Supplement), which is consistent with previous reports.28, 29 Intriguingly, the acute presence of miR1 significantly weakened IK1 of WT-homo group, but showed no effect on IK1 of Kir2.1-WT/M301K heterotetramer channels (Figure IIIC and IIID in the Supplement, Figure 3I and 3J), suggesting that the loss of biophysical suppression of Kir2.1 by miR1 contributes to the ion-channel dysregulation of this gain-of-function arrhythmogenic mutation. Noticeably, Bio-miR1 had similar binding affinity with Kir2.1 and M301K (Figure IIIE and IIIF in the Supplement), indicating that, in addition to physical binding, more complex mechanisms are involved in the biophysical modulation of ion channels by miRs.
miR1 Biophysically Modulates the Cellular Electrophysiology of Cardiomyocytes
Could the miR1-Kir.2.1 binding modulate cardiac electrophysiology? We recorded APs of guinea pig ventricular cardiomyocytes with or without miRs in the recording pipette (Figure 4A) at 0.5 and 5 minutes after rupture, which allows diffusion of miRs into cardiomyocytes for the physical interaction but bypasses the RNAi regulation of gene expression. Analysis of electrophysiological parameters (Table II in the Supplement) showed that the acute 5-minute presence of miR1 significantly depolarized RMP of ventricular cardiomyocytes from −73.06±0.52mV (control, n=39) to −70.77±0.69mV (n=37, p=0.015) (Figure 4B), while miR451a (−72.90±0.41mV, n=33, p=0.999 vs. control) and Anti-miR1 (−73.63±0.54mV, n=35, p=0.895 vs. control) showed no effect. None of miRs induced significant changes on the amplitude and duration of APs (APD) within 5 minutes (Figure 4C–4F); noticeably, both APD90 and APD50 were significantly longer in the miR1 group than in the Anti-miR1 group, indicating an opposite effect of miR1 and Anti-miR1 on the APD. Importantly, acute miR1 significantly reduced the maximum derivative of membrane potential (dV/dTMax) from −6.49±0.23 mV/ms (control) to −5.80±0.22 mV/ms (p=0.048) during repolarization (Figure 4G) and prolonged the time of 70%-to-90% repolarization (5.70±0.23 ms, p=0.034 vs. 5.04±0.18 ms in control) (Figure 4C insert panel and 4H). Next, we assessed the dynamic change of AP from 0.5 minutes to 5 minutes after patch rupture and noticed that all groups showed faster repolarization at 5 minutes than at 0.5 minutes (Table II in the Supplement), likely due to dilution of intracellular compositions, such as polyamines that suppress the outward current of IK1.21 Then, we normalized AP parameters at 5 minutes to those at 0.5 minutes, and observed that both normalized dV/dTMax and final-repolarization time showed a significantly faster acceleration of repolarization in the Anti-miR1 group compared to all other groups (Figure 4I and 4J). All dynamic changes of AP parameters in the presence of miR1 or Anti-miR1 consistently indicate a suppression of IK1 by miR1. Indeed, compared to the control (−24.10±1.42 pA/pF at −120mV, n=27) and miR451a (−23.66±1.78 pA/pF, n=21) groups, IK1 was significantly smaller in the acute presence of miR1 (−19.98±1.27 pA/pF, n=22, p=0.039 vs. control), while acute Anti-miR1 enhanced IK1 (−28.14±1.28 pA/pF, n=29, p=0.046 vs. control) of guinea pig cardiomyocytes (Fig 4K and 4L). In summary, our results demonstrate that miR1 rapidly modulates cardiac electrophysiology through a physical interaction with Kir2.1.
Figure 4. miR1 biophysically modulates the cellular electrophysiology of cardiomyocytes.

A, Representative traces of action potential (AP) of guinea pig ventricular cardiomyocytes in absence (control, n=39) or in acute presence of miR1 (100pmol/L, n=37), miR451a (100pmol/L, n=33), or anti-miR1 (100pmol/L, n=35). miRs were delivered through the recording pipette as shown in the inserted box. B, A summarized bar graph showed that the acute presence of miR1 significantly depolarized the RMP of ventricular cardiomyocytes, which was not observed with miR451a and Anti-miR1. C, AP traces of all groups were aligned at the 60% repolarization to directly compare the final repolarization of APs. The inserted panel is an amplified region of final repolarization. D, miRs had minimal influence on the amplitude of APs. E and F, Compared to control, the individual miRs had small effect on the duration of APs (APD); however, 90% (APD90) (E) and 50% repolarization (APD50) (F) of APD were significantly longed in miR1 group than that in Anti-miR1 group, indicating an opposite function of miR1 and Anti-miR1. G, Acute presence of miR1 significantly reduced the maximum derivative of membrane potential repolarization (dV/dTMax), while miR451a and Anti-miR1 showed no effect. H, Acute presence of miR1, but not miR451 and Anti-miR1, significantly prolonged the time of AP final repolarization (from 70% to 90% repolarization). I and J, The dV/dTMax and final-repolarization time of APs measured at 5 minutes were normalized to that at 0.5 minutes. Anti-miR1 group demonstrated dramatically faster normalized dV/dTMax (I) and shorter final-repolarization time (J) in comparison with all other groups, indicating faster acceleration of repolarization that was driven by Anti-miR1. K and L, Representative traces (K) and I-V curves (L) of IK1 from whole-cell patch clamping recordings in presence of miR1 (n=21), or miR451a (n=22), or Anti-miR1 (n=29) or without miRs (control, n=27), showed that acute presence of miR1 (p=0.027) significantly suppressed and Anti-miR1 (p=0.043) enhanced IK1 in guinea pig ventricular cardiomyocytes. *p <0.05, **p <0.01, ***p <0.001 vs. control or as indicated. Statistical significance was estimated by one-way ANOVA and subsequent contrasts tests with multiple testing adjustments.
The Core Sequence of AAGAAG Is Critical for the Biophysical Function of miR1
To identify the essential nucleotides of miR1 for miR1-Kir2.1 modulation, we designed various miR1-mutants (Mu1-Mu5), in which two or three nucleotides were mutated to uridine or adenine (Figure 5A), and evaluated their biophysical actions. EMSA of IVT-synthesized Kir2.1 with Bio-mutants showed that Mu2 and Mu5 bind to Kir2.1, yielding specific shifted bands, but Mu1, Mu3 and Mu4 clearly impaired or lost the binding capability with Kir2.1 (Figure 5B). Accordingly, Mu1, Mu3 and Mu4 failed to modulate IK1 (Figure IVA in the Supplement and Figure 5C), while Mu2 and Mu5 still suppressed IK1.
Figure 5. Core sequence of AAGAAG is critical for the biophysical modulation of miR1.
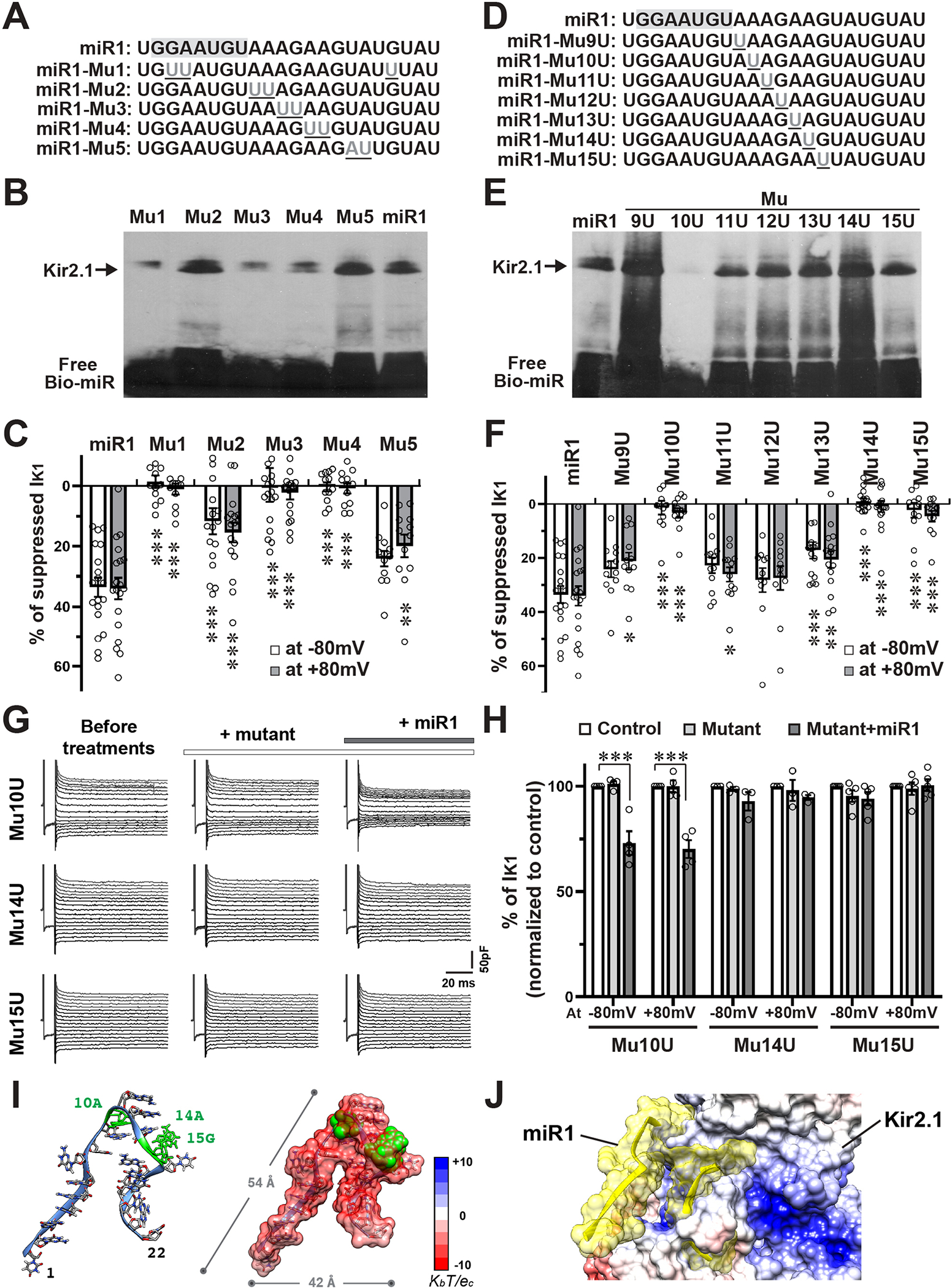
A, Nucleotide sequences of designed miR1-mutants (Mu1- Mu5). The mutated nucleotides were underlined. The seed region for RNAi mechanism was highlighted with gray background. B, EMSA of IVT-Kir2.1 protein and miR1 mutants showed that Mu2 and Mu5 still bind with Kir2.1 and generated specific shifted bands (arrow), while Mu1, Mu3, and Mu4 clearly impaired the binding with Kir2.1. C, Summarized bar graph of IK1 suppression (at +80mV and −80mV) by miR1 and mutants showed that Mu1 (n=12), Mu3 (n=17), and Mu4 (n=12) completely lost, while Mu2 (n=17) and Mu5 (n=12) showed significantly weaker suppression on IK1, in comparison with miR1 (n=19). D, Nucleotide sequences of single-nucleotide mutants (Mu9U- Mu15U). E, EMSA of IVT-Kir2.1 protein and miR1 mutants showed that only Mu10U lost the binding capacity, while other mutants bind with Kir2.1 and generated specific shifted bands (arrow). F, Summarized bar graph of IK1 suppression (at +80mV and −80mV) by miR1 and mutants demonstrated that Mu10U (n=12), Mu14U (n=18), and Mu15U (n=12) failed to repress IK1, while Mu9U (n=12), Mu11U (n=12), and Mu13U (n=15) weakly decreased IK1, and Mu12U (n=12) showed no significant difference from miR1 in suppression of IK1. G and H, Representative traces (G) and summarized bar graph (H) of IK1 from inside-out patch-clamp recordings indicated that the presence of Mu14U (n=3) or Mu15U (n=5) abolished, while Mu10 (n=4) showed no effect on miR1’s suppression on IK1. I, A modelled 3D structure (Left) and an electrostatic potential surface (right) of miR1 at a physiological pH. The 10A, 14A and 15G nucleotides are highlighted in green. The scale of electrostatic potential is indicated as ± KbT/ec, where Kb is the Boltzmann constant, T is the temperature and ec is the charge of one electron. J, A computational modeling shows that negatively-charged miR1 binds on the positively-charged G-loop domain through the external surface of Kir2.1. *p <0.05, **p <0.01, ***p <0.001 vs. miR1 group or as indicated. Statistical significance was estimated by two-way ANOVA and subsequent contrasts tests with multiple testing adjustments.
Considering the importance of seed sequence (2G-8U) for miR’s RNAi role, we focused on nucleotides outside the seed region that are specifically dedicated to the unconventional function. We designed single-nucleotide mutants of miR1 (Mu9U-15U) (Figure 5D), and found that Mu10U lost the binding affinity to Kir2.1 and had no biophysical suppression on IK1 (Figures 5E and 5F and Figure IVB in the Supplement). Although Mu14U and Mu15U still bound with Kir2.1, neither of them suppressed IK1; while the other mutants still suppressed IK1 and some of them showed significantly weaker suppression than miR1. Altogether, these results demonstrated that 10A, 14A and 15G are the most essential nucleotides, whereas the AAGAAG (10A-15G) might be the core sequence that are responsible for the proper biophysical function of miR1. Next, we preincubated inside-out patches with Mu10U, 14U or 15U, respectively, and then treated the patch with miR1. A preincubation of Mu14U or Mu15U completely blocked miR1’s suppression on IK1, while Mu10U didn’t affect miR1’s function (Figure 5G and 5H), meaning that Mu14U- or Mu15U-binding with Kir2.1 competitively prevents miR1’s access to Kir2.1 for its biophysical modulation. These results also confirmed the requirement of physical binding to IK1 suppression by miR1.
It is noteworthy that the miR1-Mu1 has the full AAGAAG core sequence but still lost the physical interaction and function, and Mu5 and Mu9U had significantly weaker suppression on IK1 than miR1, suggesting that nucleotides outside this core sequence also have influence on its biophysical modulation, probably by modifying the structure of miR1. We utilized the RNAstructure35 and ROSIE36 webservers to computationally predict the possible secondary and 3D structure of miR1 (Figure 5I and Figure VA in the Supplement). Inspection of the electrostatic surface potential revealed that miR1 is highly negative at physiological pH, thus enabling the interaction on the positively-charged G-loop region of Kir2.1 through the external surface of the C-terminus (Figure 5J, Figure VC and movie II in the Supplement). The 3D models of miR1 and mutants (Figure V in the Supplement), combining with all above functional data, suggest that miR1-mutants without an intact AAGAAG in a loop structure couldn’t biophysically modulate the function of Kir2.1.
Arrhythmia-Associated miR1 hSNP Specifically Disrupts the Biophysical Function
We studied the NCBI SNP database and found seven reported hSNPs within the mature sequence of miR1 genes (Figure 6A and Figure VIA in the Supplement). We focused on five with SNP outside the seed sequence, and observed that they could bind with Kir2.1 (Figure 6B). However, hSNP14A/G, identified in AF populations, and hSNP15G/A didn’t suppress IK1, while hSNP16U/C, 18U/C and 22U/G significantly suppressed IK1 (Figure 6C and 6D, Figure VIB–VIE in the Supplement). Consistently, hSNP14A/G and Mu10U couldn’t significantly affect APs and IK1 of guinea pig cardiomyocytes (Figures 6E and 6F, Figure VIF–VIK in the Supplement). Expectedly, both hSNP14A/G and hSNP15G/A competitively abolished miR1’s suppression on IK1 (Figure 6G and 6H, Figure VIL–VIM in the Supplement). Therefore, the loss-of-function hSNP with competitive binding ability to Kir2.1 could be one critical mechanism by which miR1 biophysically dysregulates the function of ion channels.
Figure 6. Arrhythmia-associated miR1 hSNPs specifically disrupt the biophysical function.

A, Nucleotide sequences of miR1 human single nucleotide polymorphisms (hSNPs), in which the mutation is outside seed-sequence, reported in the NCBI SNP database. B, miR1 hSNPs bound with Kir2.1 and produced specific shifted bands in EMSA assays with IVT-Kir2.1 protein. C, I-V curves of IK1 showed that hSNP14A/G (100pmol/L, n=13) lost the ability to biophysically modulate IK1 in inside-out patch clamping of Kir2.1-HEK293 cells. D, Summarized bar graph of IK1 suppression (at +80mV and −80mV) revealed that, similar to hSNP14A/G, hSNP15G/A (n=14) also lost the capability to modulate IK1, and that hSNP18U/G (n=11) and hSNP22U/G (n=7) weakly repressed IK1, while hSNP16U/C (n=14) showed equal suppression on IK1, in comparison with miR1. *p <0.05, and ***p <0.001 vs. miR1 group. E, Representative traces of APs of guinea pig ventricular cardiomyocytes in presence of hSNP14A/G, or miR1-Mu10U, or without miRs (Control) in the recording pipette solution. AP traces were aligned at the 60% repolarization for a direct comparison of the final repolarization. The inserted panel is an amplified region of final repolarization. F, IK1 I-V curves measured in guinea pig ventricular cardiomyocytes without (control, n=14) or with miRs, showed that neither Mu10U (n=16) nor hSNP14A/G (n=10) significantly affect IK1 in cardiomyocytes. G and H, Representative traces (G) and summarized bar graph (H) of IK1 from inside-out patch-clamp recordings showed that the presence of hSNP14A/G (n=5) abolished miR1’s suppression on IK1. I and J, Luciferase reporter assays, performed in NIH3T3 cells, showed that miR1-Mu10U, hSNP14A/G and hSNP15G/A significantly decreased reporter genes activities of miR1 targets including mouse (I) and human (J) Twf1, Kcnj2, Gja1, Stx6. Firefly luciferase activity was normalized to control Renilla Luciferase activity. n=3. K and L, Representative images (K) and summarized bar graph (L) of Western blot assays in neonatal mouse cardiomyocytes (P2.5), transfected with individual miR mimics, demonstrated that Mu10U, hSNP14A/G, and hSNP15G/A act as miR1 and significantly suppressed protein levels of miR1’s RNAi targets, including Ptk9, Cx43 and Kir2.1. n=4; *p <0.05, **p <0.01, and ***p <0.001 vs. Ctrl mimic in panels I-L. Statistical significance of differences was estimated by ANOVA and contrasts tests with multiple testing adjustments.
To determine if a defect of biophysical function affects the canonical RNAi mechanism, we investigated the RNAi function of Mu10U, hSNP14A/G and hSNP15G/A. We cloned 3’UTR of miR1’s target genes, including mouse and human Twf1, Kcnj2, Gja1, and Stx6, into the pmirGLO luciferase reporter vector. Luciferase assay in NIH3T3 cells showed that, similar to miR1, Mu10U, hSNP14A/G and hSNP15G/A significantly suppressed the luciferase activities of all miR1 targets (Figure 6I and 6J), demonstrating the maintained RNAi function. Consistent results were observed in miR-mimic transfected neonatal mouse cardiomyocytes, as miR1, Mu10U, hSNP14A/G and hSNP15G/A all significantly decreased the protein levels of Ptk9 (encoded by Twf1), Cx43 and Kir2.1 (Figures 6K and 6L and Figure VII in the Supplement). Notably, the RNAi functions of miR1 and hSNPs had no difference, suggesting that the biophysical modulation of miR1 is sequence-dependent but is separated from the RNAi mechanism. Importantly, arrhythmia-associated hSNP14A/G only disrupts miR1’s biophysical function.
miR1 But Not hSNP14A/G Eliminates the High Arrhythmia Inducibility of miR1-Deficient Heart
We then utilized 50%-miR1-knockdown (50%KD) mice to investigate if the biophysical modulation of miR1 is physiologically relevant to the functional homeostasis of the heart. We confirmed that mature miR1 in 50%KD hearts was decreased to approximately half amount of the level in WT, accompanied by significant upregulation of Kir2.1 protein (Figure VIII in the Supplement). Electrophysiological recordings (Figure 7A and 7B) disclosed significantly hyperpolarized RMP in 50%KD cardiomyocytes (−69.45±1.21 mV, n=11, p=0.047) compared to WT cells (−62.86±1.64 mV, n=7). Then, we acutely restored mature miR1 or hSNP14G (≤5 minutes) through the patch-clamp recording pipette and observed that miR1 relieved the hyperpolarized RMP of 50%KD cardiomyocytes (−58.71±2.26 mV, n=7), while hSNP14G (−69.80±1.75 mV, n=10) showed no significant effect on the RMP (Figure 7B).
Figure 7. miR1 but not hSNP14A/G eliminates the high arrhythmia inducibility of miR1-deficient hearts ex vivo.

A and B, Representative traces of APs (A) and summarized bar graph of RMP (B) recorded in wildtype (WT) and 50%-miR1-knockdown (50%KD) ventricular cardiomyocytes and in 50% KD cells with acute restoration of miR1 or hSNP14A/G (100pmol/L). miRs were delivered through the patch clamp recording pipette. C, Experiment schema of miR1 or hSNP14A/G mimics delivery into the heart of 50%KD mice through left ventricle intraluminal injection. D, qRT-PCR analysis confirmed the success of miR1 and hSNP14A/G overexpression in 50%KD mouse hearts. E and F, Images of Western blot (E) and quantification assay of protein intensities (F) demonstrated that both miR1 and hSNP14A/G decreased the protein expression of Kir2.1 in 50%KD mouse hearts. G, Representative ECG traces of Langendorff-perfused ex vivo hearts showed that ventricular fibrillation was induced in 50%KD hearts, while WT hearts maintained normal heart rhythm after programmed electrical stimulations (▼). H, A summary bar graph revealed that 50%KD hearts had significantly higher arrhythmia inducibility than the WT group, which was eliminated by overnight-transfection of miR1 but not by hSNP14A/G mimics. I and J, Representative contour map of ventricular activation time in WT (I) and 50%KD (J) hearts during pacing. Pacing sites are indicated by ▼. K, A summary bar graph of conduction velocity, measured longitudinal and transverse to fiber direction, showed that 50%KD hearts had isochrone crowding and slower action potential propagation compared to WT. Delivery of miR1 but not hSNP14A/G significantly enhanced conduction velocity in 50%KD hearts. L, miRs are involved in cardiac ion channel dysregulation and arrhythmia through two different mechanisms: 1) canonical post-transcriptional RNAi mechanism of ion-channel expression that needs hours-days to take effect and 2) the newly-discovered biophysical modulation that miR1 acts as an ion-channel modulator to directly and quickly (seconds to minutes) change the dynamics of ion channels. A disruption of biophysical modulation (i.e. hSNP14A/G) predisposes the heart to an increased risk of arrhythmia. *p <0.05, **p <0.01 vs. WT group or as indicated. Statistical significance of differences was estimated by ANOVA and subsequent Student’s t tests with multiple testing adjustments, and 7H was by one-side Fisher’s exact test.
To explore the physiological significance of miR1’s biophysical function in the whole heart, we delivered miR1 or hSNP14A/G mimics into 50%-KD mouse hearts by left-ventricle intraluminal injection (Figure 7C) and performed optocardiography of Langendorff-perfused ex- vivo heart within 24 hours after miR-mimic delivery. qPCR results showed that an overnight miR-mimic delivery significantly increased miR1 level in 50%KD hearts (Figure 7D), which was validated by Kir2.1 downregulation in both miR1- and hSNP14A/G-transfected hearts (Figure 7E and 7F). Ventricular tachycardia (VT) or fibrillation (VF) were frequently induced in ex-vivo 50%-KD hearts (6 out of 10 animals, p=0.005 vs. 0 out of 10 WT animals) by programmed electrical stimulations (Figure 7G and 7H). Remarkably, miR1 administration eliminated the high inducibility of arrhythmia in 50%KD hearts; only one out of six miR1-transfected 50%KD hearts demonstrated induced VT/VF. In contrast, hSNP14A/G-transfected 50%KD hearts still showed high arrhythmia inducibility (3 out of 6 animals, p=0.036 vs. WT). Furthermore, the activation time contours of optical mapping revealed isochrone crowding and slower propagation of APs in 50% KD hearts (Figure 7I–7K); conduction velocity was significantly slower in 50%KD hearts compared to WT group in both longitudinal (0.539±0.013 m/s, p=0.005 vs. 0.607±0.021 m/s of WT, n=10) and transversal (0.389±0.005 m/s, p=0.005 vs. 0.434±0.018 m/s of WT) directions. Strikingly, the restoration of miR1 sufficiently improved the conduction velocity (longitudinal: 0.667±0.036, n=6, p<0.001; transversal: 0.441±0.015, p<0.001 vs. 50%KD counterparts); however, hSNP14A/G-transfected 50%KD hearts still showed slow AP propagation (longitudinal: 0.563±0.014; transversal: 0.398±0.016, n=6), despite an observed small improvement without statistical significance compared to 50%KD groups. Collectively, this significant difference of rescue effect between miR1 and hSNP14A/G at both cellular and organ levels provided strong proof of the pathophysiological relevance that the biophysical modulation of miR contributes to the functional homeostasis of the heart, and a disruption of biophysical modulation predisposes the heart to an increased risk of arrhythmogensis (Figure 7L).
DISCUSSION
Here, we reveal an unconventional function of endogenous miRs where miR1 acts as an ion-channel modulator, directly binds to Kir2.1, and biophysically modulates cardiac electrophysiology. Importantly, the biophysical action of miR1 is evolutionarily conserved and plays a vital physiological role in maintaining the functional homeostasis of the heart. This biophysical suppression of IK1 by miR1 is functionally consistent with, while mechanistically distinct from the canonical RNAi role, by which miR1 post-transcriptionally represses Kir2.1 protein output. Our discoveries expand the biological significance of miRs and demonstrate that miR’s noncanonical biophysical modulation, together with its classic RNAi effect, cooperatively manages cardiac physiology.
As an essential regulator, intracellular miRs appear to target about 60% of mammalian genes through RNAi mechanism,37, 38 by forming miR-induced silencing complex (miRISC) with Ago proteins. It has been noticed that the amount of intracellular miRs is 13 times higher than the amount of Ago proteins39 and only a fraction of each miRs possibly bind to Ago proteins in human cells.40, 41 The large amounts of intracellular Ago-free miRs indicate the possibility of RNAi-independent mechanisms.12 While extracellular miRs were found to physically interact with cell-surface proteins, such as TLR and TRPA1,13, 16 this biophysical action should be true for intracellular miRs in cytoplasm, which is the original biogenesis site of mature miRs and contains much more naked miRs than extracellular fluid. Here, our study firstly provides evidence for an intracellular biophysical action of miRs in physiological regulation beyond RNAi mechanism.
miRISC has been found to localize in distinct subcellular compartments, such as rough endoplasmic reticulum, mitochondria, golgi, multivesicular bodies, messenger ribonucleoprotein granules and cytoskeleton-bound polysomes.42–46 Interestingly, we found that miR1 binds on plasma membrane, more specifically on the intracellular C-terminus of Kir2.1, suggesting a novel RISC-independent function of miR1. Indeed, acute presence (within seconds/minutes) of miR1 immediately suppressed IK1 and modulated APs in cardiomyocytes. This rapidly-occurred effect is obviously not resulted from the conventional RNAi mechanism, providing direct evidence that miRs biophysically modulate the function of proteins that they bind to. Not like previous studies that applied >μmol/L concentration of miRs,13–16 importantly, our studies revealed that miR1 functions at a sub-pmol/L concentration that is close to endogenous intracellular miRs level,17 indicating the physiology significance of endogenous miRs’ biophysical actions. MiR1 accounts for 40% transcripts of all the miRs in the heart7 and plays a critical role in cardiac electrophysiology.9, 10, 31 miR1-deficient heart shows pathological phenotypes of abnormal electrophysiology, including hyperpolarized RMP, slow conduction, and high inducibility of arrhythmia. Remarkably, an acute recovery of miR1 corrected the hyperpolarized RMP of 50%KD cardiomyocytes, rescued the conduction in ventricular tissues, and eliminated the high inducibility of arrhythmia in 50%KD hearts. However, hSNP14A/G, which specifically disrupts the biophysical modulation of miR1 with normal RNAi function, couldn’t act as miR1 to eliminate those arrhythmic phenotypes of miR1-deficient cells and hearts, demonstrating that intracellular endogenous miRs indeed biophysically regulate cardiac electrophysiology through directly binding to ion channels.
In addition, the miR1-Kir2.1 physical interaction is involved in the functional disruption of arrhythmia-associated hSNPs, which are the most common type of genetic variation among people.47 SNPs within miR have been reported to associate with coronary artery disease48 and congenital heart disease,49, 50 through mechanisms of aberrant miR processing51–54 or disrupted miR-mRNA interaction.55 Here, we reveal a new dysfunctional mechanism for miR hSNPs and find that hSNP14A/G and hSNP15G/A maintain the canonical RNAi function but specifically lack their biophysical modulation. In addition, the Kir2.1-M301K hSNP not only disrupts the biophysical modulation of intracellular polyanimes/Mg2+ on M301K/WT heterotetramer channels,28, 29 but also relieves the biophysical suppression of miR1, resulting in a gain-of-function with larger outward current of IK1 in short-QT syndrome and AF patients. These findings indicate a pathological significance that the biophysical modulation of miRs is virtually involved in the ion-channel dysregulation of cardiac arrhythmogenesis. Since hSNP14A/G retains the RNAi function, our findings also provide a basis for guiding the future research to separate the specific physiological significance of a new action of miRs in living cells and organs from the powerful canonical RNAi mechanism.
Mechanistically, the biophysical modulation of ion channels by miRs requires a physical binding with sequence dependence. No physical binding resulted in no functional modulation of Kir2.1 by MiR1-Mu1, Mu3, Mu4 and Mu10U. Furthermore, miR1’s biophysical modulation was competitively blocked by the 27AA peptide (300G-326Y) of Kir2.1 or miR1-mutants (i.e. Mu14U, Mu15U, hSNP14A/G and hSNP15G/A). However, binding is necessary but not sufficient to modulate IK1, as evidenced by the 14th and 15th mutations of miR1 and Kir2.1-M301K, which showed no impairment on miR1/Kir2.1 binding, but lost the biophysical suppression on IK1. Meanwhile, the consistence of three essential nucleotides (i.e. 10A, 14A, 15G) with three pinpointed miR1-binding residues on Kir2.1 justifies a direct physical interaction between miR1 and Kir2.1; although more sophisticated studies are needed to understand how the binding of miR1 to Kir2.1 modifies the dynamics of ion-channel gating. RNA binding domains are generally enriched in intrinsically disordered regions that are characterized by a low content of bulky hydrophobic amino acids or a high proportion of polar and/or charged amino acids.56 Computational assays showed that negatively-charged miR1 could dock on the positively-charged G-loop region through the external surface of Kir2.1 and subsequently prevent intrinsic large conformational changes associated with channel gating.57 Our mechanistic studies have demonstrated that intracellular miRs are one type of ion-channel biophysical modulators in regulation of cardiac electrophysiology.
MiR1 has been recognized as a key regulator of ion-channel expressions in cardiac arrhythmogenesis.11 We unveiled that plasma-membrane localized miR1 directly and rapidly modulates the dynamics of Kir2.1 channel, while cytosolic miR1 will be loaded into the RISC, which is relatively slower but in long-term inhibits the protein expression of Kir2.1. The biophysical-modulation discovery not only reveals a novel action for miRs with spatial and temporal differences from its RNAi mechanism, but also suggests that miRs could prevent or trigger arrhythmias through biophysically dysregulating ion channel before a significant change of protein expression in diseased hearts. Our studies provide a more comprehensive understanding of ion-channel dysregulation during arrhythmogenesis, and suggest that miR-induced arrhythmogenic risk should be studied as one of important safety assessments in the development of miR-based therapeutic approaches.
In conclusion, miRs maintain the stability of cardiac electrophysiology by two different mechanisms (Figure 7L): first, the canonical RNAi mechanism of post-transcriptional regulation of gene expression, which requires hours to days to take effect; second, the newly-discovered biophysical mechanism of physical interaction with target proteins, which quickly (seconds to minutes) modulates their functions. This biophysical modulation enables miRs to rapidly respond to environmental and genetic perturbations. Understanding miR-protein (i.e. ion channel) physical interaction will guide the development of more effective anti-arrhythmic therapeutic approaches for cardiovascular disease. We have also observed that miR1 binds with calcium channel protein (Data not shown), and speculate that this biophysical modulation by miRs might generally exists in various biological processes. Therefore, our discovery expands the biological significance of miRs as a protein modulator and has broad implication for overall biology where miRs play crucial roles.
Supplementary Material
CLINICAL PERSPECTIVE.
What Is New?
MiR1 acts as a modulator of ion channels, directly binds to and biophysically modulates the function of an inward rectifier potassium channel—Kir2.1 and this miR1-Kir2.1 physical binding occurs endogenously in cardiomyocytes and is evolutionarily conserved across species.
An arrhythmia-associated human single nucleotide polymorphism (SNP) of miR1, hSNP14A/G, specifically disrupts the biophysical function but maintains the canonical RNAi function.
MiR1 biophysically modulates the cellular electrophysiology of cardiomyocytes and maintains the functional homeostasis of the heart.
What Are the Clinical Implications?
Cardiac electrophysiology is regulated by miRs through two different mechanisms: the canonical RNAi mechanism that needs hours to days to regulate gene expression and the newly-discovered biophysical mechanism that quickly (seconds to minutes) modulates the function of ion channels.
MiRs could prevent or trigger arrhythmias through biophysically modulating ion channels before its RNAi regulation of protein expressions in diseased hearts.
The biophysical modulation of miRs is involved in the dysregulation of ion channels in arrhythmogenesis, such as in atrial fibrillation patients with Kir2.1-M301K or hSNP14A/G mutation and its defect might predispose patients to arrhythmias.
ACKNOWLEDGMENTS
We are grateful to Dr. Jill Dunham for editorial assistance and thankful to Dr. Deepak Srivastava at the Gladstone Institutes and Dr. Lance D. Wilson at the MetroHealth System for scientific discussion and advices. We also thank Dr. Deepak Srivastava for generously sharing miR1-1 homozygous-null (1−/−2+/+) and miR1-2 homozygous-null (1+/+2−/−) mice and thank Dr. Christine S. Moravec at Cleveland Clinic for generously sharing human cardiac biopsy tissues.
SOURCES OF FUNDING
This research was supported by the Start-up Fund from The MetroHealth System and from The Ohio State University (to J.D.F.) and grants from the American Heart Association- 13SDG14580035 (to J.D.F.) and the National Institutes of Health (NIH)-R01HL139006 (to I.D. and J.D.F.), NIH-R01HL132520 (to I.D.), NIH-R01HL096962 (to I.D.), NIH-R56HL142754 (to K.R.L.), NIH-R01HL149369 (to K.R.L.), NIH-R01HL135096 (to T.J.H.), NIH-R01HL134824 (to P.J.M. and T.J.H.). D.Y was funded by the Kenneth M. Rosen Fellowship from the Heart Rhythm Society.
NONSTANDARD ABBREVIATIONS AND ACRONYMS
- 50%KD
50%-miR1-knockdown
- AP
Action potential
- Anti-miR
Antisense oligonucleotide of mircoRNA
- AF
Atrial fibrillation
- Bio-miR
Biotinylated mircoRNA
- CLIP
Cross-linking and immunoprecipitation
- CT
C-terminus
- APD
Duration of action potential
- EMSA
Electrophoretic mobility shift assay
- I-V curve
IK1 current-voltage characteristic curve
- PLA
In situ proximity ligation assay
- IVT
In vitro translation
- IK1
Inward rectifier current
- Kir
Inward rectifier potassium channel
- Kir2.1-HEK293 cell
Kir2.1-overexpressed HEK293 cell
- LQTS
Long-QT syndromes
- M301K-Homo
M301K alone-transfected HEK293 cell
- dV/dTMax
Maximum derivative of membrane potential
- M-protein
Membrane protein
- miR
MicroRNA
- Mu
MiR1-mutant
- miRISC
MiR-induced silencing complex
- NT
N-terminus
- qPCR
Real-Time quantitative reverse transcription PCR
- RMP
Resting membrane potential
- RIP
RNA immunoprecipitation
- RNAi
RNA interference
- TM
Transmembrane
- VF
Ventricular fibrillation
- VT
Ventricular tachycardia
- WT
Wildtype
- WT/M301K-Hetero
Wildtype Kir2.1 and M301K co-transfected HEK293 cell
- WT-Homo
WT alone-transfected cell
Footnotes
REFERENCES
- 1.Wang Y, Hill JA. Electrophysiological remodeling in heart failure. J Mol Cell Cardiol. 2010;48:619–632. doi: 10.1016/j.yjmcc.2004.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019;139:e56–e528. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 3.Myers R, Timofeyev V, Li N, Kim C, Ledford HA, Sirish P, Lau V, Zhang Y, Fayyaz K, Singapuri A, et al. Feedback mechanisms for cardiac-specific microRNAs and cAMP signaling in electrical remodeling. Circ Arrhythm Electrophysiol. 2015;8:942–950. doi: 10.1161/CIRCEP.114.002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nassal DM, Wan X, Liu H, Maleski D, Ramirez-Navarro A, Moravec CS, Ficker E, Laurita KR, Deschenes I. KChIP2 is a core transcriptional regulator of cardiac excitability. Elife. 2017;6: e17304. doi: 10.7554/eLife.17304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinti MV, Hathaway QA, Hollander JM. Role of microRNA in metabolic shift during heart failure. Am J Physiol Heart Circ Physiol. 2017;312:H33–H45. doi: 10.1152/ajpheart.00341.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, Zhang Y, Xu C, Bai Y, Wang H, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 7.Rao PK, Toyama Y, Chiang HR, Gupta S, Bauer M, Medvid R, Reinhardt F, Liao R, Krieger M, Jaenisch R, et al. Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ Res. 2009;105:585–594. doi: 10.1161/CIRCRESAHA.109.200451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liao C, Gui Y, Guo Y, Xu D. The regulatory function of microRNA-1 in arrhythmias. Mol Biosyst. 2016;12:328–333. doi: 10.1039/c5mb00806a. [DOI] [PubMed] [Google Scholar]
- 9.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1–2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 10.Heidersbach A, Saxby C, Carver-Moore K, Huang Y, Ang YS, de Jong PJ, Ivey KN, Srivastava D. microRNA-1 regulates sarcomere formation and suppresses smooth muscle gene expression in the mammalian heart. Elife. 2013;2:e1323. doi: 10.7554/eLife.01323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Girmatsion Z, Biliczki P, Bonauer A, Wimmer-Greinecker G, Scherer M, Moritz A, Bukowska A, Goette A, Nattel S, Hohnloser SH, et al. Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm. 2009;6:1802–1809. doi: 10.1016/j.hrthm.2009.08.035. [DOI] [PubMed] [Google Scholar]
- 12.Dragomir MP, Knutsen E, Calin GA. SnapShot: Unconventional miRNA Functions. Cell. 2018;174:1038. doi: 10.1016/j.cell.2018.07.040. [DOI] [PubMed] [Google Scholar]
- 13.Lehmann SM, Kruger C, Park B, Derkow K, Rosenberger K, Baumgart J, Trimbuch T, Eom G, Hinz M, Kaul D, et al. An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat Neurosci. 2012;15:827–835. doi: 10.1038/nn.3113. [DOI] [PubMed] [Google Scholar]
- 14.Park CK, Xu ZZ, Berta T, Han Q, Chen G, Liu XJ, Ji RR. Extracellular microRNAs activate nociceptor neurons to elicit pain via TLR7 and TRPA1. Neuron. 2014;82:47–54. doi: 10.1016/j.neuron.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, Lovat F, Fadda P, Mao C, Nuovo GJ, et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci USA. 2012;109:E2110–E2116. doi: 10.1073/pnas.1209414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han Q, Liu D, Convertino M, Wang Z, Jiang C, Kim YH, Luo X, Zhang X, Nackley A, Dokholyan NV, et al. miRNA-711 Binds and Activates TRPA1 Extracellularly to Evoke Acute and Chronic Pruritus. Neuron. 2018;99:449–463. doi: 10.1016/j.neuron.2018.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bissels U, Wild S, Tomiuk S, Holste A, Hafner M, Tuschl T, Bosio A. Absolute quantification of microRNAs by using a universal reference. RNA. 2009;15:2375–2384. doi: 10.1261/rna.1754109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moreau A, Gosselin-Badaroudine P, Chahine M. Biophysics, pathophysiology, and pharmacology of ion channel gating pores. Front Pharmacol. 2014;5:53. doi: 10.3389/fphar.2014.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balse E, Boycott HE. Ion Channel Trafficking: Control of Ion Channel Density as a Target for Arrhythmias? Front Physiol. 2017;8:808. doi: 10.3389/fphys.2017.00808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuda H, Saigusa A, Irisawa H. Ohmic conductance through the inwardly rectifying K channel and blocking by internal Mg2+. Nature. 1987;325:156–159. doi: 10.1038/325156a0. [DOI] [PubMed] [Google Scholar]
- 21.Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 1994;372:366–369. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- 22.Pegan S, Arrabit C, Zhou W, Kwiatkowski W, Collins A, Slesinger PA, Choe S. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci. 2005;8:279–287. doi: 10.1038/nn1411. [DOI] [PubMed] [Google Scholar]
- 23.Kubo Y, Adelman JP, Clapham DE, Jan LY, Karschin A, Kurachi Y, Lazdunski M, Nichols CG, Seino S, Vandenberg CA. International Union of Pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol Rev. 2005;57:509–526. doi: 10.1124/pr.57.4.11. [DOI] [PubMed] [Google Scholar]
- 24.Grunnet M Repolarization of the cardiac action potential. Does an increase in repolarization capacity constitute a new anti-arrhythmic principle? Acta Physiol (Oxf). 2010;198 Suppl 676:1–48. doi: 10.1111/j.1748-1716.2009.02072.x. [DOI] [PubMed] [Google Scholar]
- 25.Anumonwo JM, Lopatin AN. Cardiac strong inward rectifier potassium channels. J Mol Cell Cardiol. 2010;48:45–54. doi: 10.1016/j.yjmcc.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miake J, Marban E, Nuss HB. Biological pacemaker created by gene transfer. Nature. 2002;419:132–133. doi: 10.1038/419132b. [DOI] [PubMed] [Google Scholar]
- 27.Plaster NM, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001;105:511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 28.Hattori T, Makiyama T, Akao M, Ehara E, Ohno S, Iguchi M, Nishio Y, Sasaki K, Itoh H, Yokode M, et al. A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of Kir2.1 currents. Cardiovasc Res. 2012;93:666–673. doi: 10.1093/cvr/cvr329. [DOI] [PubMed] [Google Scholar]
- 29.Hasegawa K, Ohno S, Ashihara T, Itoh H, Ding WG, Toyoda F, Makiyama T, Aoki H, Nakamura Y, Delisle BP, et al. A novel KCNQ1 missense mutation identified in a patient with juvenile-onset atrial fibrillation causes constitutively open IKs channels. Heart Rhythm. 2014;11:67–75. doi: 10.1016/j.hrthm.2013.09.073. [DOI] [PubMed] [Google Scholar]
- 30.Dilly S, Lamy C, Marrion NV, Liegeois JF, Seutin V. Ion-channel modulators: more diversity than previously thought. Chembiochem. 2011;12:1808–1812. doi: 10.1002/cbic.201100236. [DOI] [PubMed] [Google Scholar]
- 31.Fu JD, Rushing SN, Lieu DK, Chan CW, Kong CW, Geng L, Wilson KD, Chiamvimonvat N, Boheler KR, Wu JC, et al. Distinct roles of microRNA-1 and −499 in ventricular specification and functional maturation of human embryonic stem cell-derived cardiomyocytes. Plos One. 2011;6:e27417. doi: 10.1371/journal.pone.0027417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giles WR, Imaizumi Y. Comparison of potassium currents in rabbit atrial and ventricular cells. J Physiol. 1988;405:123–145. doi: 10.1113/jphysiol.1988.sp017325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clark RB, Tremblay A, Melnyk P, Allen BG, Giles WR, Fiset C. T-tubule localization of the inward-rectifier K(+) channel in mouse ventricular myocytes: a role in K(+) accumulation. J Physiol. 2001;537:979–992. doi: 10.1111/j.1469-7793.2001.00979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zurla C, Jung J, Blanchard EL, Santangelo PJ. A Novel Method to Quantify RNA-Protein Interactions In Situ Using FMTRIP and Proximity Ligation. Methods Mol Biol. 2017;1468:155–170. doi: 10.1007/978-1-4939-4035-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bellaousov S, Reuter JS, Seetin MG, Mathews DH. RNAstructure: Web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res. 2013;41:W471–W474. doi: 10.1093/nar/gkt290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyskov S, Chou FC, Conchuir SO, Der BS, Drew K, Kuroda D, Xu J, Weitzner BD, Renfrew PD, Sripakdeevong P, et al. Serverification of molecular modeling applications: the Rosetta Online Server that Includes Everyone (ROSIE). Plos One. 2013;8:e63906. doi: 10.1371/journal.pone.0063906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 38.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janas MM, Wang B, Harris AS, Aguiar M, Shaffer JM, Subrahmanyam YV, Behlke MA, Wucherpfennig KW, Gygi SP, Gagnon E, et al. Alternative RISC assembly: binding and repression of microRNA-mRNA duplexes by human Ago proteins. RNA. 2012;18:2041–2055. doi: 10.1261/rna.035675.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flores O, Kennedy EM, Skalsky RL, Cullen BR. Differential RISC association of endogenous human microRNAs predicts their inhibitory potential. Nucleic Acids Res. 2014;42:4629–4639. doi: 10.1093/nar/gkt1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stalder L, Heusermann W, Sokol L, Trojer D, Wirz J, Hean J, Fritzsche A, Aeschimann F, Pfanzagl V, Basselet P, et al. The rough endoplasmatic reticulum is a central nucleation site of siRNA-mediated RNA silencing. EMBO J. 2013;32:1115–1127. doi: 10.1038/emboj.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sen GL, Blau HM. Argonaute 2/RISC resides in sites of mammalian mRNA decay known as cytoplasmic bodies. Nat Cell Biol. 2005;7:633–636. doi: 10.1038/ncb1265. [DOI] [PubMed] [Google Scholar]
- 43.Makarova JA, Shkurnikov MU, Wicklein D, Lange T, Samatov TR, Turchinovich AA, Tonevitsky AG. Intracellular and extracellular microRNA: An update on localization and biological role. Prog Histochem Cytochem. 2016;51:33–49. doi: 10.1016/j.proghi.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Zuo X, Yang B, Li Z, Xue Y, Zhou Y, Huang J, Zhao X, Zhou J, Yan Y, et al. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell. 2014;158:607–619. doi: 10.1016/j.cell.2014.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat Cell Biol. 2005;7:719–723. doi: 10.1038/ncb1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalantari R, Chiang CM, Corey DR. Regulation of mammalian transcription and splicing by Nuclear RNAi. Nucleic Acids Res. 2016;44:524–537. doi: 10.1093/nar/gkv1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kruglyak L, Nickerson DA. Variation is the spice of life. Nat Genet. 2001;27:234–236. doi: 10.1038/85776. [DOI] [PubMed] [Google Scholar]
- 48.Sung JH, Kim SH, Yang WI, Kim WJ, Moon JY, Kim IJ, Cha DH, Cho SY, Kim JO, Kim KA, et al. miRNA polymorphisms (miR146a, miR149, miR196a2 and miR499) are associated with the risk of coronary artery disease. Mol Med Rep. 2016;14:2328–2342. doi: 10.3892/mmr.2016.5495. [DOI] [PubMed] [Google Scholar]
- 49.Guo R, Feng Z, Yang Y, Xu H, Zhang J, Guo K, Bi Y. Association of a MiR-499 SNP and risk of congenital heart disease in a Chinese population. Cell Mol Biol (Noisy-Le-Grand). 2018;64:108–112. [PubMed] [Google Scholar]
- 50.Yu K, Ji Y, Wang H, Xuan QK, Li BB, Xiao JJ, Sun W, Kong XQ. Association of miR-196a2, miR-27a, and miR-499 polymorphisms with isolated congenital heart disease in a Chinese population. Genet Mol Res. 2016;15. doi: 10.4238/gmr15048929. [DOI] [PubMed] [Google Scholar]
- 51.Gottwein E, Cai X, Cullen BR. A novel assay for viral microRNA function identifies a single nucleotide polymorphism that affects Drosha processing. J Virol. 2006;80:5321–5326. doi: 10.1128/JVI.02734-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu Z, Liang J, Wang Z, Tian T, Zhou X, Chen J, Miao R, Wang Y, Wang X, Shen H. Common genetic variants in pre-microRNAs were associated with increased risk of breast cancer in Chinese women. Hum Mutat. 2009;30:79–84. doi: 10.1002/humu.20837. [DOI] [PubMed] [Google Scholar]
- 53.Jazdzewski K, Murray EL, Franssila K, Jarzab B, Schoenberg DR, de la Chapelle A. Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc Natl Acad Sci USA. 2008;105:7269–7274. doi: 10.1073/pnas.0802682105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duan R, Pak C, Jin P. Single nucleotide polymorphism associated with mature miR-125a alters the processing of pri-miRNA. Hum Mol Genet. 2007;16:1124–1131. doi: 10.1093/hmg/ddm062. [DOI] [PubMed] [Google Scholar]
- 55.He S, Ou H, Zhao C, Zhang J. Clustering Pattern and Functional Effect of SNPs in Human miRNA Seed Regions. Int J Genomics. 2018;2018:2456076. doi: 10.1155/2018/2456076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Castello A, Fischer B, Frese CK, Horos R, Alleaume AM, Foehr S, Curk T, Krijgsveld J, Hentze MW. Comprehensive Identification of RNA-Binding Domains in Human Cells. Mol Cell. 2016;63:696–710. doi: 10.1016/j.molcel.2016.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fagnen C, Bannwarth L, Oubella I, Forest E, De Zorzi R, de Araujo A, Mhoumadi Y, Bendahhou S, Perahia D, Venien-Bryan C. New Structural insights into Kir channel gating from molecular simulations, HDX-MS and functional studies. Sci Rep. 2020;10:8392. doi: 10.1038/s41598-020-65246-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wan X, Laurita KR, Pruvot EJ, Rosenbaum DS. Molecular correlates of repolarization alternans in cardiac myocytes. J Mol Cell Cardiol. 2005;39:419–428. doi: 10.1016/j.yjmcc.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 59.Mikami S, Masutani M, Sonenberg N, Yokoyama S, Imataka H. An efficient mammalian cell-free translation system supplemented with translation factors. Protein Expr Purif. 2006;46:348–357. doi: 10.1016/j.pep.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 60.Gagnon KT, Maxwell ES. Electrophoretic mobility shift assay for characterizing RNA-protein interaction. Methods Mol Biol. 2011;703:275–291. doi: 10.1007/978-1-59745-248-9_19. [DOI] [PubMed] [Google Scholar]
- 61.Grotzky A, Manaka Y, Kojima T, Walde P. Preparation of catalytically active, covalent alpha-polylysine-enzyme conjugates via UV/vis-quantifiable bis-aryl hydrazone bond formation. Biomacromolecules. 2011;12:134–144. doi: 10.1021/bm101074s. [DOI] [PubMed] [Google Scholar]
- 62.Yoon JH, Gorospe M. Cross-Linking Immunoprecipitation and qPCR (CLIP-qPCR) Analysis to Map Interactions Between Long Noncoding RNAs and RNA-Binding Proteins. Methods Mol Biol. 2016;1402:11–17. doi: 10.1007/978-1-4939-3378-5_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lukas A Electrophysiology of Myocardial Cells in the Epicardial, Midmyocardial, and Endocardial Layers of the Ventricle. J Cardiovasc Pharmacol Ther. 1997;2:61–72. doi: 10.1177/107424849700200108. [DOI] [PubMed] [Google Scholar]
- 64.Sicouri S, Quist M, Antzelevitch C. Evidence for the presence of M cells in the guinea pig ventricle. J Cardiovasc Electrophysiol. 1996;7:503–511. doi: 10.1111/j.1540-8167.1996.tb00557.x. [DOI] [PubMed] [Google Scholar]
- 65.Bryant SM, Wan X, Shipsey SJ, Hart G. Regional differences in the delayed rectifier current (IKr and IKs) contribute to the differences in action potential duration in basal left ventricular myocytes in guinea-pig. Cardiovasc Res. 1998;40:322–331. doi: 10.1016/s0008-6363(98)00133-3. [DOI] [PubMed] [Google Scholar]
- 66.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 67.Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018;27:293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Popenda M, Szachniuk M, Antczak M, Purzycka KJ, Lukasiak P, Bartol N, Blazewicz J, Adamiak RW. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012;40:e112. doi: 10.1093/nar/gks339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sondergaard CR, Olsson MH, Rostkowski M, Jensen JH. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J Chem Theory Comput. 2011;7:2284–2295. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 71.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 73.Mills WR, Mal N, Forudi F, Popovic ZB, Penn MS, Laurita KR. Optical mapping of late myocardial infarction in rats. Am J Physiol Heart Circ Physiol. 2006;290:H1298–H1306. doi: 10.1152/ajpheart.00437.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data, methods, and study materials, including a primer list in Table I in the Supplement, that support the findings of this work are available from the corresponding author upon reasonable request.