

Abstract
Advanced cancer patients exhibit cachexia, a condition characterized by a significant reduction in the body weight predominantly from loss of skeletal muscle and adipose tissue. Cachexia is one of the major causes of morbidity and mortality in cancer patients. Decreased food intake and multi-organ energy imbalance in cancer patients worsen the cachexia syndrome. Cachectic cancer patients have a low tolerance for chemo- and radiation therapies and also have a reduced quality of life. The presence of tumors and the current treatment options for cancer further exacerbate the cachexia condition, which remains an unmet medical need. The onset of cachexia involves crosstalk between different organs leading to muscle wasting. Recent advancements in understanding the molecular mechanisms of skeletal muscle atrophy/hypertrophy and adipose tissue wasting/browning provide a platform for the development of new targeted therapies. Therefore, a better understanding of this multifactorial disorder will help to improve the quality of life of cachectic patients. In this review, we summarize the metabolic mediators of cachexia, their molecular functions, affected organs especially with respect to muscle atrophy and adipose browning and then discuss advanced therapeutic approaches to cancer cachexia.
Keywords: Cachexia, Cancer, Skeletal muscle wasting, Adipose tissue browning, Mediators, Cytokines
1. Introduction
The majority of cancer patients with advanced metastatic disease exhibit cachexia, a condition characterized by the loss of body weight due to extensive loss of skeletal muscle and adipose tissue. Besides cancer, cachexia is prominent in other diseases such as chronic obstructive pulmonary disease, acquired immunodeficiency syndrome (AIDS), chronic renal failure, diabetes, chronic heart failure, and rheumatoid arthritis [1]. Cancer cachexia accounts for one-fourth of cancer-related deaths, and the degree of cachexia depends on both tumor type and stage. Although cachexia predominantly affects skeletal muscle, which comprises almost half of the body weight, it also damages other organs such as adipose tissue, liver, brain, gut, pancreas, bone, and heart. Several factors, such as negative energy balance due to metabolic dysregulation, increased catabolic processes in muscle and fats and neurohormonal dysregulation, worsen this multifactorial syndrome [2].
Anorexia or loss of appetite is commonly seen in cancer patients. Cachexia, in combination with anorexia, reduces the quality of life (QOL) and overall survival of patients [3]. While tumor and host-derived factors play an essential role in the advancement of cachexia, chemotherapy and radiotherapy exacerbate cachectic syndrome [4,5]. A decrease in skeletal muscle index or increase in muscle loss during chemotherapy or surgery is associated with poor survival in advanced-stage ovarian cancer [6]. Furthermore, cachexia-mediated muscle wasting significantly increases the toxicity of chemotherapy [7]. The clinical management of cachexia is challenging due to the complexity of this multifactorial metabolic disorder and lack of definitive therapies. Here in this article, we provide an overview of the underlying molecular mechanism and pathophysiology of muscle wasting and adipose browning in cancer cachexia and explore therapeutic strategies that could combat and offer more effective care for this devastating multifactorial syndrome.
2. Cancer cachexia prevalence and stages
Cancer cachexia is well-defined as a multifactorial syndrome characterized by an ongoing loss of skeletal muscle mass and body weight, with or without loss of fat mass, which cannot be fully reversed by conventional supplementation of nutrition [8,9]. Another condition of involuntary loss of skeletal muscle mass and strength is known as sarcopenia, which is prevalent in the elderly population. Although wasting disorders such as cachexia (50%–80%) and sarcopenia (15%–50%) are both prevalent in cancer patients, the underlying mechanism of muscle loss in cachexia and sarcopenia differ [10]. Sarcopenia is an age-associated decrease in muscle mass due to reductions in muscle synthesis, while the rates of muscle protein degradation are unchanged. Besides reduced muscle mass, sarcopenia is often associated with an increase in fat mass that accompanies advanced age in older adults [11]. In contrast, cachexia results from increases in muscle protein degradation, total energy expenditure, basal metabolic rates, and either unaltered or a decreased adipose mass. While an estimated 50–80% of cancer patients exhibit cachexia, it is more prevalent in patients with advanced lethal cancers, like pancreatic, gastrointestinal, and non-small cell lung cancers (NSCLC) [12]. Cachectic syndrome causes significant morbidity and mortality in both localized and metastatic cancer patients, and by itself contributes to almost 20% of deaths in cancer patients [13]. The reported deaths associated with cachexia represent cancer patients with observed weight loss of more than 30–40%; however, due to lack of proper diagnostic techniques, cachexia-associated deaths of cancer patients remain underreported.
Weight loss is the first sign of cachexia and serves as an independent predictor of mortality in cancer patients [14]. Multiple factors such as systemic inflammation, cancer type and stage, low food intake, and treatment-associated toxicities can contribute to the progression of cachexia; concomitant anorexia contributes to lower caloric intake and further worsens the patient’s condition. Cachexia may progress even in the absence of anorexia [15]. Recently, the cachectic syndrome was classified into three stages: pre-cachexia, cachexia, and refractory cachexia, based on the degree of reduction of energy stores and body protein or body mass index, along with the extent of ongoing weight loss [9]. Less than 5% weight loss, along with anorexia and glucose tolerance is referred to as a pre-cachectic stage. Clinically, cachectic patients are characterized by an involuntary weight loss of > 5% within six months that cannot be entirely reversed by conventional nutritional support. Finally, in refractory cachexia, due to the higher level of active catabolism, weight loss management is no longer possible and patients are unlikely to benefit from any treatment. At this stage, patients usually have less than three months of survival. The stages of cachexia and the amount of weight loss are directly correlated to survival in cancer patients [16]. At the stage of refractory cachexia, the only goal of therapy is palliative to reduce distress in patients (Fig. 1).
Fig. 1.
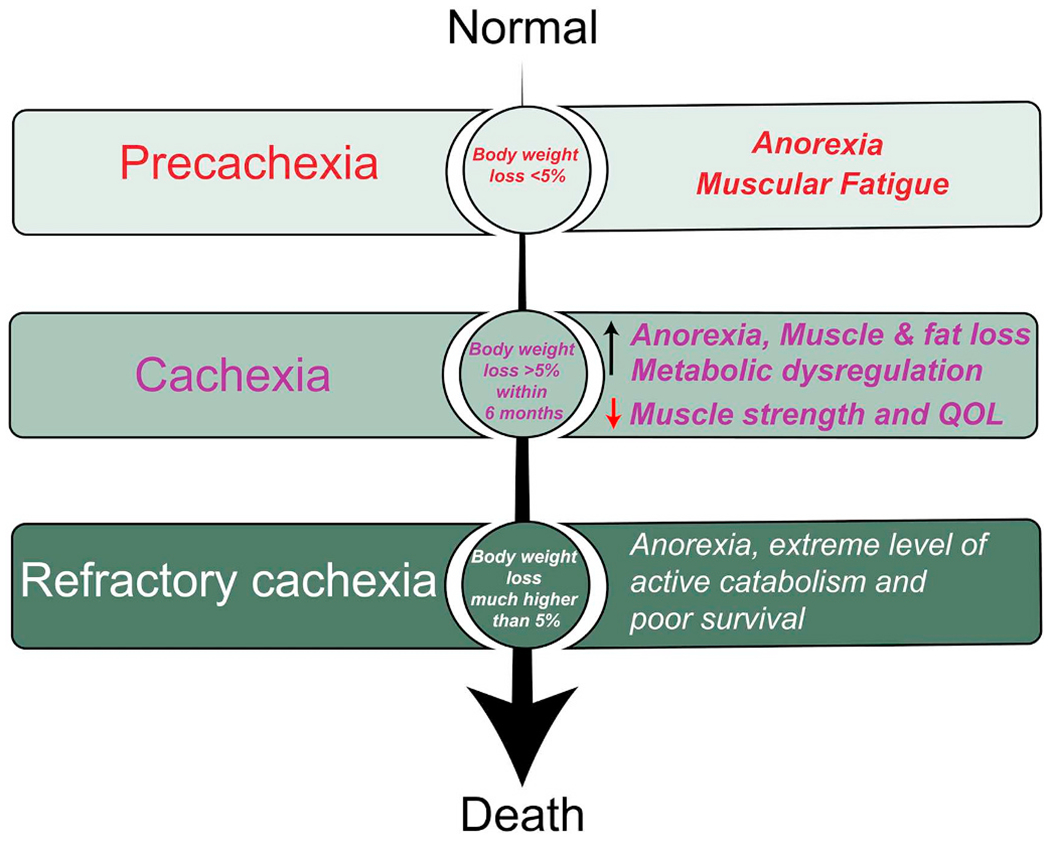
Different stages of cancer cachexia. Based on the severity of disease and degree of loss of body weight, cachexia syndrome is classified as pre-cachexia, cachexia, and refractory cachexia. Pre-cachexia: Pre-cachexia is a condition with less than 5% of weight loss accompanied by anorexia and muscular fatigue. Cachexia: Patients with an involuntary weight loss of > 5% within six months that cannot be entirely reversed by conventional nutritional support and leads to progressive functional impairment. An increase in anorexia, weight loss (due to loss of muscle and fat), inflammation, metabolic dysregulation, and a decrease in muscle strength, mobility, and quality of life are the clinical symptoms of cachexia. Refractory cachexia: Weight loss is variable. Due to the higher level of active catabolism and increased expression of ubiquitin ligases, weight loss management is no longer possible, and patients are unlikely to benefit from any treatment, with very poor chances of survival. A severe reduction in skeletal and respiratory muscle strength leads to impaired mobility and inadequate respiration.
3. Energy imbalance in cancer cachexia
It is important to understand the role of energy balance in the development of cancer cachexia. Cancer cachexia is often associated with a negative energy balance driven by anorexia along with increased energy expenditure or abnormal metabolism. Almost half of cancer patients exhibit a high metabolism or high resting energy expenditure (REE), which indicates development of cachexia. The degree of REE is highly dependent on the type of cancer, pathological stage, and duration of disease [17]. Elevated REE has been reported in the majority of patients with gastric, esophageal, pancreatic, and lung cancers (NSCLC) and typically exacerbates the disease [17]. Cachexia results from the concurrent hyperactivation of numerous metabolic pathways that subsequently leads to energy dissipation in the form of heat, and systemic inflammation, which contribute to shorter survival in patients with metastatic cancer [18]. Tumors have an abnormally high glucose requirement compared to normal tissue, and upregulated glycolysis is a hallmark of cancer cells [19]. This high demand for glucose even in the presence of sufficient oxygen, known as the Warburg effect, results in elevated production of lactic acid by the tumor cells to compensate for their inefficient energy production [20,21]. Warburg estimated that highly glycolytic tumors generate as much as 50% of their energy from glycolysis [22]. The amount of energy produced by tumors is based on the degree of utilization of the glycolytic pathway (an anaerobic process) versus oxidative phosphorylation (an aerobic process). The Cori cycle, which is responsible for energy dissipation in the form of heat lipolysis in adipose tissue, and protein catabolism in the muscle tissue both contribute to increased liver gluconeogenesis in cancer patients. Further, increased expression of mitochondrial uncoupling proteins (UCPs) can generate heat as an alternative of ATP and speed up the hypermetabolic processes in cancer cachexia. Recently, it has been reported that ER stress and unfolded protein response are elevated in the skeletal muscle of cachectic patients [23–25]. Despite accelerated glycolysis and lactate production, numerous circulating factors, such as tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), interferon-gamma (IFN-γ), myostatin, and parathyroid hormone-related peptide (PTHrP) are believed to be responsible for the hypermetabolic state in cachectic patients. Altogether, these metabolic factors could help to identify the pre-cachexia stage, which would be highly beneficial for high-risk patients.
4. Muscle wasting in cancer cachexia
Skeletal muscle comprises a significant proportion of body weight in the human body and is necessary for numerous biological activities such as movement, support to soft tissue, and respiration. Skeletal muscle is one of the most metabolically active tissues in the body and serves as a storehouse of protein and free amino acids. During normal physiological conditions, adult muscle mass remains constant due to a balance in protein synthesis and degradation. A well-coordinated network of various hypertrophic (anabolic) and atrophic (catabolic) signals within the skeletal muscle is required to ensure a balance between synthesis and wasting of the muscle, a function that is commonly disrupted during the tumor progression. Indeed, extensive loss of skeletal muscle represents a key manifestation of cancer-associated cachexia. Cachexia primarily results from an acceleration of protein degradation, often combined with reduced protein synthesis in skeletal muscle. Muscle progenitor cells with disrupted metabolism contribute to the impaired muscle generation in cachexia [26]. Several factors such as fasting, exercise, aging, and disease, affect the homeostatic balance of muscle by decreasing muscle protein synthesis or increasing the degradation of muscle, which worsens the cachexic condition [27]. Overall, muscle wasting due to the cachexic condition reduces QOL and contributes to mortality (Fig. 2).
Fig. 2.
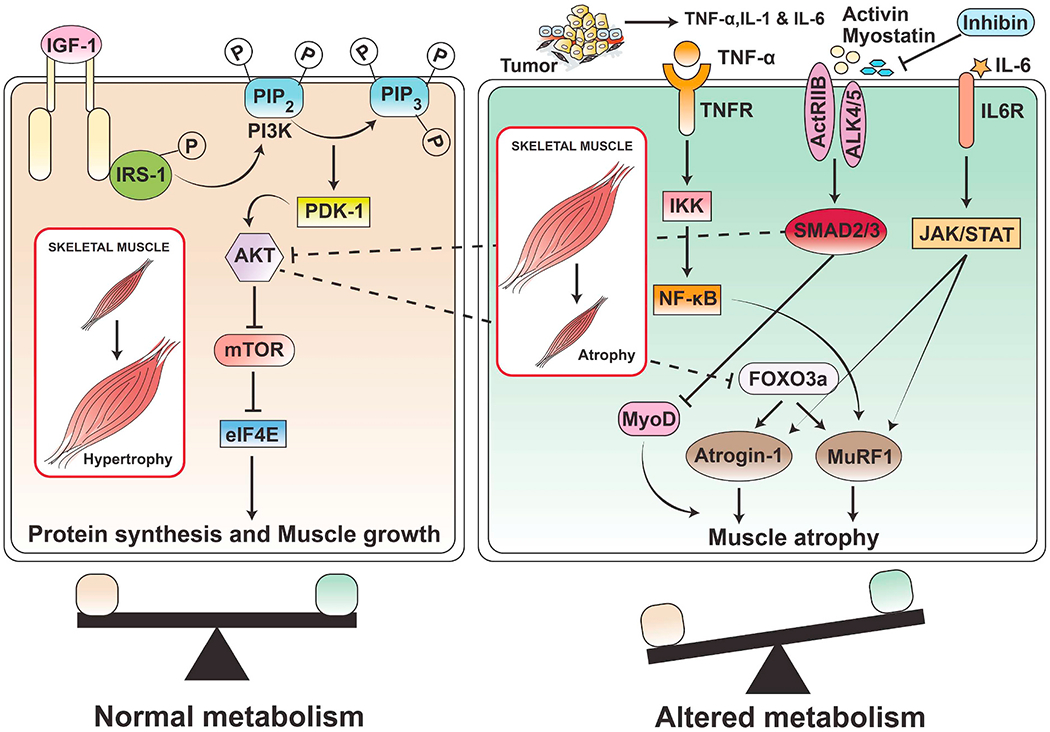
Muscle anabolic and catabolic signaling and regulation of muscle wasting in cancer cachexia. Reduced muscle anabolic signaling: IGF-1 acts as an anabolic growth factor that stimulates muscle protein synthesis as well as proliferation and differentiation of muscle stem cells (satellite cells). IGF-1 binds with its receptor IGF1R, resulting in phosphorylation of insulin receptor substrate (IRS) and activation of PI3K/AKT signaling. Activation of PI3K/AKT signaling leads to the activation of downstream targets required for protein synthesis. Cancer cachexia causes impaired IGF-1 signaling, which leads to muscle atrophy. In addition to protein synthesis, AKT signaling also phosphorylates and inactivates the FoxO transcription factor, which is a negative regulator of myogenesis, and finally inhibits the expression of the muscle-specific ubiquitin ligases atrogin-1 and MuRF1. Increased muscle catabolism signaling: Tumor and immune cells induce inflammatory cytokines IL-1 and TNF-α, which activate transcription factor NF-κB via IKK. NF-κB signaling causes muscle wasting in cachexia through increased activity of Murf-1 and atrogin-1. Myostatin and activin bind to activin type II receptor (ActRIIB) and ALK4/5 and subsequently phosphorylate Smad2/3. Phosphorylated Smad2/3 makes a complex with Smad4 that translocates to the nucleus and induces muscle wasting. In addition, myostatin/activin signaling inhibits FoxO phosphorylation via reducing AKT activity, which increases the expression of ubiquitin ligases (Murf-1 and atrogin-1) as well as activates autophagy, subsequently increasing muscle protein breakdown. Another cytokine IL-6 binds to its receptors IL-6R and activates JAK/STAT3 signaling, which is implicated in muscle protein wasting.
4.1. Increased protein degradation or muscle atrophy in cachexia
In patients with cancer cachexia, activation of the ATP-dependent ubiquitin-proteasome proteolytic pathway (UPP) causes the breakdown of myofibrillar proteins and plays a crucial role in muscle wasting [28,29]. This pathway involves the hyperactivation of muscle-specific E3 ubiquitin ligases (muscle atrophy F box protein, MAFbx/atrogin-1) and muscle RING finger-containing protein 1 (MuRF1), which are responsible for the selective polyubiquitination of proteins targeted for degradation [30,31]. The upregulation of MAFbx/atrogin-1 has been reported in the majority of cancer cachexia cases and is considered as a marker of acute muscle atrophy [32]. Genetic deletion of muscle-specific E3 ligases MAFbx/atrogin-1 or Murf1 protected skeletal muscle from atrophy, suggesting their role in skeletal muscle breakdown [33]. Protein degradation via activation of the UPP was observed in a pre-clinical tumor-bearing mouse model of cancer cachexia [34]. Ubiquitin ligases of the N-end rule pathway (UBRs) control the ubiquitination of muscle protein and have a role in muscle wasting in cachectic mice [35]. Recently, loss of UBR4 was shown to induce hypertrophy via decreased ubiquitination of target proteins, especially the histone-binding complex (HAT1/RBBP4/RBBP7), which suggests a role for UBR4 in myofiber hypertrophy [36]. A recent report suggested that the autophagic lysosomal system along with the UPP coordinate cachexia-induced muscle loss in gastric patients [37]. Hsp70 and Hsp90 may also drive cancer cachexia, as they were highly secreted by Lewis lung carcinoma (LLC) cells and induced muscle catabolism via activation of Toll-like receptor (TLR4) [38]. Further, cachectic muscle showed mitochondrial dysfunction, which may alter amino acid metabolism via decreasing cationic amino acid transporter (CAT1) expression as well as degrading mitochondrial proteins [39].
TGF-β family members, such as myostatin and activin A, have also been shown to promote muscle loss through the myostatin/activin receptor type IIB (ActRIIB), and overaction of the ActRIIB pathway has been observed in many cancers [40–42]. Myostatin, commonly known as growth/differentiation factor 8 (GDF8), inhibits myoblast differentiation and facilitates Forkhead box O (FoxO) activation and the expression of ubiquitin ligases in response to inflammatory signals [43]. Transgenic overexpression of FoxO3 in the skeletal muscle caused skeletal muscle wasting, and inhibition of FoxO prevented skeletal muscle atrophy in a mouse model of cancer cachexia [44]. However, deletion of myostatin in mice showed an increase in muscle mass, suggesting myostatin is a negative regulator of muscle growth [45]. Similarly, inhibition of bone morphogenetic protein signaling abolished the hypertrophic phenotype observed in myostatin-deficient mice and caused muscle atrophy via the upregulation of muscle ubiquitin ligase of the SCF complex in atrophy-1 [46]. However, overexpression of the myostatin gene in adult mice showed profound muscle loss and fat wasting similar to human cachexia, suggesting that myostatin is a potential pharmacologic target for managing cachexia [47]. Another report suggested that myostatin can inhibit the activation of satellite cells (muscle stem cells) [45].
Interestingly, transgenic mice with dominant-negative ActRIIB exhibited hypertrophy of skeletal muscle, and blockade of ActRIIB improved cachexia in tumor-bearing mice [48,49]. In addition to the prevention of muscle wasting, blockade of ActRIIB also reversed prior loss of skeletal muscle and cancer-induced cardiac atrophy via inhibition of the atrophy-specific ubiquitin ligases and stimulation of muscle stem cell growth in muscle. ActRIIB blockade significantly prolonged survival, even in the absence of a beneficial effect on tumor growth [48]. In addition, activin A induced the secretion of IL-6 from ovarian cancer cells in an autocrine manner, and blocking ActRIIB with antibody reduced serum levels of IL-6 and reversed cachexia in cachectic tumor-bearing mice, suggesting the therapeutic potential of targeting activin A and IL-6 signaling pathways [50].
Metabolic changes in the adipose tissue can also regulate the muscle wasting. PGC-1α, a transcriptional coactivator of PPARγ in brown adipose tissue, regulates the expression of genes involved in oxidative metabolism during exercise. High expression of PGC-1α4 (an isoform of PGC1α) prevents skeletal muscle atrophy by activating the expression of IGF1 and repressing myostatin activity. Transgenic mice that overexpress PGC-1α4 and bear LLC tumors showed a dramatic resistance to muscle wasting, suggesting PGC-1α4 protects against muscle atrophy [51].
4.2. Muscle wasting through pro-inflammatory cytokines and NF-kB signaling/pathway
Treatment of mice with TNF-α and IL-1 can cause skeletal muscle atrophy similar to that observed in cachectic cancer patients [52]. TNF-α activates the NF-κB pathway and subsequently inhibits the differentiation of muscle cells via downregulation of MyoD, suggesting NF-κB is activated in skeletal muscle wasting [53]. NF-κB signaling not only augments the expression of UPP proteins that target specific muscle proteins for degradation, but also interferes with myogenic differentiation during muscle atrophy [54]. UPP-mediated proteolytic processing of NF-κB and IκB family proteins is a prerequisite for NF-κB activation [55,56]. Transgenic mice with constitutively active IKKβ exhibited profound muscle wasting similar to cachectic patients. Muscle atrophy was reduced, however, in IKKβ-expressing mice crossed with MuRF1-knockout mice, suggesting that IKKβ-induced muscle wasting is facilitated by the upregulation of the MuRF1 gene [57].
NF-κB signaling can also promote muscle wasting by increasing the expression of inflammation-related molecules, including cytokines, chemokines, and matrix metalloproteinases (MMPs) [58]. Several of these pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 activate NF-κB in a positive feedback loop, causing over-stimulation of the NF-κB pathway and enhanced muscle wasting [58]. Overall, therapeutic approaches to inhibit the NF-κB pathway can augment muscle performance in cachectic cancer patients. The pro-inflammatory cytokines induced by NF-κB contribute to muscle wasting and cachexia through additional mechanisms such as activation of JAK/STAT signaling [59]. Indeed, in an experimental model of cancer cachexia, STAT3 activation was both necessary and sufficient for muscle wasting. Inhibition of JAK/STAT signaling attenuated cancer cachexia in colorectal and lung cancer [60,61]. However, the outcome of a randomized, double-blind phase II clinical trial of ruxolitinib, a JAK1/JAK2 inhibitor, in combination with capecitabine, did not improve the survival of patients with metastatic pancreatic cancer [62].
4.3. Decrease in anabolic signals (insulin-like growth factor 1)
Although most of the studies involving cachexia research focused on the factors responsible for skeletal muscle wasting, studies examining relevant muscle hypertrophy factors have provided detailed insights into the mechanisms underlying cachexia-induced muscle wasting. Insulinopenic rats exhibit skeletal muscle atrophy, suggesting that insulin is the key anabolic factor opposing the catabolic effects of glucocorticoids [63]. Insulin-like growth factor 1 (IGF1) is the most studied anabolic factor, and the transgenic mice overexpressing IGF1 exhibit skeletal muscle hypertrophy [64]. IGF1 promotes anabolic signaling in muscle via binding with its tyrosine kinase receptor, IGF1 receptor (IGF1R) [65]. The binding of ligands to IGF1R induces transphosphorylation of the dimeric receptor and facilitates the recruitment of insulin receptor substrate 1 (IRS1) [66]. IRS1 phosphorylation is crucial for the downstream signaling of IGF1 and leads to the activation of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, as well as other downstream signals. Prolonged insulin stimulation can cause ubiquitin-mediated degradation of IRS1 through the PI3K pathway and inactivate IGF1 signaling [67–69]. The IGF1-PI3K-AKT pathway induces muscle growth primarily by stimulating protein synthesis through mTOR kinase [67]. The activation of PI3K is required for IRS1 degradation independent of mTOR signaling [69]. Studies from transgenic mice suggest that acute activation of Akt in the skeletal muscle is sufficient to induce rapid and significant muscle hypertrophy by activation of the downstream Akt/p70S6 kinase protein synthesis pathway [70]. The IGF-I/PI3K/Akt pathway can suppress transcription of atrophy-related E3 atrogin-1 and reduce the degradation of muscle protein [71]. IRS1 is rapidly degraded after IGF1 stimulation by several E3 ubiquitin ligases, and this limits the downstream hyperactivation of the IRS1/PI3K/Akt pathway. It has been shown that SOCS1 or SOCS3 targets IRS1 and IRS2 for ubiquitin-mediated degradation, respectively [72]. Another ubiquitin ligase, Cbl-b, facilitates the degradation of IRS-1 and acts as a dual mediator of both increased protein degradation and reduced protein synthesis in unloading-induced muscle atrophy [73]. Muscle-specific mitsugumin 53 (MG53), also known as TRIM72, mediates the degradation of IRS1 and insulin receptors and also inhibits myogenesis [74,75]. F-box-containing protein Fbxo40 is another physiologic regulator of IGF1 signaling, and Fbxo40 null mice have enhanced muscle size. Overall, a decrease in circulating IGF-1 levels and insulin resistance has been reported in cancer cachectic patients, and mouse models of cachexia suggest that an IGF-1-mediated decrease in anabolic signal contributes to muscle wasting [27,64,76].
5. Mediators of muscle atrophy in cancer cachexia
Tumors secrete a diverse array of potential mediators, which can cause cachexia, and their production and secretion vary from one type of tumor to another. Increased expression of these mediators has been reported in different cancer cell lines and tumor tissue, suggesting that tumors are the potential source of these mediators. Although the mechanisms of action of these mediators have not been elucidated completely, pro-inflammatory cytokines are thought to be critical in inducing muscle and adipose wasting [77].
5.1. Glucocorticoids
Glucocorticoids (GC) are released either endogenously in response to stress and disease, or are administered exogenously as drugs to treat inflammation. GC alter protein and glucose metabolism in skeletal muscles [78]. GC increase the rate of skeletal protein catabolism and decrease the rate of protein synthesis, eventually leading to skeletal muscle atrophy via increases in FoxO-dependent transcription of E3 ubiquitin ligases [79,80]. Elevated levels of GC have also been observed in cancer patients [81]. Further, intact GC signaling in skeletal muscle is required for cancer-induced cachexia [82]. However, in rodent models of cancer cachexia, treatment with GC antagonist RU38486 failed to prevent the loss of skeletal muscle, thereby raising doubts about the direct role of GC in cancer cachexia [83].
5.2. Tumor necrosis factor-alpha
Tumor or immune cell-derived tumor necrosis factor-alpha (TNF-α) is one of the most prominent pro-inflammatory cytokines and mediators of tumor-induced adipose and skeletal muscle wasting [84–86]. TNF-α activates the ubiquitin E3 ligase pathway to promote muscle protein breakdown [87–89]. TNF-α was reported to stimulate the degradation of muscle proteins, including adult myosin heavy chain (MHC) in muscle rather than altering the rate of protein synthesis [84]. Along with myofibrillar breakdown, TNF-α was found to inhibit skeletal myocyte differentiation [53]. Acute treatment with recombinant TNF-α in rats resulted in increased degradation and decreased synthesis of muscle protein, suggesting that TNF-α causes muscle wasting via activating the ubiquitin E3 ligase pathway as well as diminishing the formation of new muscle protein [90]. In addition to adipose and muscle wasting, TNF-α exerts pleiotropic effects to alter carbohydrate, protein, and lipid metabolism, induce insulin resistance and mediate systemic inflammation in cancer-cachexia. Enhanced TNF-α expression in skeletal muscle is associated with insulin resistance in cancer patients in part by stimulation of stress-related protein kinases (JNK) and inhibitor kappa beta kinase beta (IKKβ)/NF-κB [91,92]. In cachexia, increased TNF-α and high energy-wasting conditions enhance the content of mitochondrial cardiolipin, which leads to reduction in mitochondrial oxidative phosphorylation [93].
5.3. Interleukin-6
Interleukin-6 (IL-6) is a well-studied cytokine that impacts several biological functions such as immune response, metabolism, hematopoiesis, and tumorigenesis. IL-6 exhibits both pro and anti-inflammatory functions, depending on context and tissue type. The role of IL-6 is well established in both rodent and human muscle wasting, and elevated levels of serum IL-6 are associated with cancer cachexia [94]. Transgenic mice overexpressing IL-6 showed an increase in ubiquitin-related proteins, suggesting the role of IL-6 in accelerating proteolysis [95]. However, only supra-physiological doses of IL-6 induced muscle atrophy in tumor-free animals [96]. Over-expression of IL-6 accelerated muscle and fat wasting, while deletion of the IL-6 gene prevented the development of cachexia in an ApcMin/+ mouse model of colorectal cancer [97]. Similarly, IL-6 administration reduced skeletal muscle protein synthesis, a condition similar to what is observed in patients with cachexia [98]. Furthermore, IL-6 levels corresponded to the magnitude of muscle wasting in APCMin/+ mice [99]. In this model, suppression of protein synthesis is one of the early events in progression of cachexia, while elevated ATP-independent protein degradation occurs in the later stages [100]. Treatment with IL-6 receptor antibody prevented the progression of cancer cachexia via suppression of muscle protein degradation [100]. Further, IL-6 can stimulate pro-inflammatory cytokines (e.g., IL-1) to enhance muscle atrophy. IL-6 has also been shown to regulate food intake and metabolism via signaling through neural gp130 receptors, suggesting that IL-6 could play a role in anorexia [101,102]. In a clinical study, treatment of NSCLC cachectic patients with anti-IL-6 antibody (ALD518) improved anorexia, but failed to reverse the loss of lean body mass [96]. The mechanisms by which IL-6 directly induces muscle atrophy are not well known. However, activated macrophages secrete IL-6 to initiate the acute phase response, which may eventually mediate cancer cachexia. Altogether, IL-6 is a potent mediator for muscle wasting in cancer, while its direct role in normal muscle is uncertain. Another interleukin, IL-8 released from cancer and tumor-associated stromal cells, has been reported to induce muscle atrophy in pancreatic cancer [103].
5.4. Growth differentiation factor 15
Growth differentiation factor 15 (GDF15), also recognized as macrophage inhibitory cytokine-1 (MIC-1), is a member of the TGF-β superfamily, which is expressed broadly in several tissues. The circulatory levels of GDF15 rise in several pathological conditions such as inflammation, injury, cardiovascular and kidney disease, and many malignancies where it often correlates with poor prognosis [104]. Elevated GDF15 levels were observed in patients with anorexia-related loss of appetite and weight [105]. In patients with cancer cachexia, circulating levels of GDF15 were elevated in early onset of cachexia and remained elevated in the advanced stages of cachexia [106]. Further, tumor-derived GDF15 was sufficient to trigger the cachexia phenotype in an animal model, and treatment with anti-GDF15 antibody reversed the bodyweight loss and restored muscle and fat tissue mass in cachectic mice [106]. GDF15 caused its anorectic effect by directly acting on the feeding center of the brainstem [107]. Recently, several studies have shown that mice treated with recombinant GDF15 exhibit a decrease in food intake and body weight, and these effects are mediated by the receptor GFRAL (glial-cell-line-derived neurotrophic, a GDNF family receptor-a like) [108–112]. Overexpression of GDF15 in skeletal muscle initiates muscle atrophy, although the mechanism for the direct effect of GDF15 on muscle cells remains poorly understood. In vitro, GDF15 increased the expression of atrogin-1 and MuRF-1 in myotubes [113,114]. Altogether, these reports suggest that along with its strong anorectic effects, GDF15 might directly induce skeletal muscle wasting in cancer cachexia, and the level of circulating GDF15 could be a biomarker of cancer cachexia.
5.5. Growth differentiation factor 11
Growth differentiation factor 11 (GDF11) is another member of the TGF-β superfamily and is homologous to myostatin (Mstn). Mstn is a known negative regulator of skeletal muscle growth [115]. Both GDF11 and myostatin bind to ActRIIB and activate the SMAD2/3 pathway. However, circulating GDF11 levels are associated with skeletal and cardiac muscle wasting, a function that is distinct from Mstn. Further, deletion of GDF11 in mice caused skeletal abnormalities, suggesting its role in skeletal development. Supraphysiological levels of GDF11 can cause skeletal and cardiac muscle wasting [116], and high doses of GDF11 cause severe cachexia and death in mice [117]. Due to its low serum concentration and structural similarity with Mstn, existing techniques to measure GDF11 need to be validated.
5.6. Parathyroid hormone-related peptide
Parathyroid hormone (PTH) and parathyroid hormone-related protein (PTHrP) cause hypercalcemia by stimulating renal calcium reabsorption and bone resorption [118]. Cancer patients with high PTHrP levels exhibit humoral hypercalcemia of malignancy (HHM), a condition characterized by increased osteoclastic bone resorption. Although primary myotubes treated with PTHrP did not show signs of atrophy, PTHrP treatment in tumor-bearing mice increased expression of muscle-atrophy-associated genes [119]. Anti-PTHrP antibody treatment decreased circulating PTHrP levels in a mouse model of cancer cachexia and somewhat reversed skeletal muscle wasting. However, the main beneficial effect was seen in preventing adipose tissue loss and energy expenditure. Since PTHrP does not have direct effects on primary muscle cells, it probably promotes skeletal muscle wasting in collaboration with other factors or by facilitating crosstalk between adipose and skeletal muscle tissues [119,120]. Further, patients with high circulating PTHrP exhibited higher REE and a lower lean mass [121]. In cancer patients, PTHrP predicts body weight loss, suggesting that circulating PTHrP could be used as a biomarker of cancer cachexia [119].
6. Adipose browning in cancer cachexia
Loss of skeletal muscle and adipose tissue (AT) are both symptoms of cachexia [122]. While much is known about muscle wasting, information about the mediators and mechanism of AT loss in cancer patients with cachexia is scarce. As shown in Fig. 3, the tumor secretes several mediators such as TNF-α, IL-6, IFN-γ and LIF during cachexia. TNF-α, also known as cachectin [123], is also released from the AT and may mediate cancer cachexia by reducing the expression of glucose transporter 4 (GLUT4), which in turn inhibits glucose transport and lipogenesis [124]. In cancer cachexia, TNF-α activates expression of monocyte chemoattractant protein 1, which attracts monocytes to the AT that subsequently promote inflammation in AT [123]. AT-infiltrating macrophages are responsible for the activation of other cytokines (IL-6 and IL-1β), which results in further recruitment of macrophages [125]. Activated macrophages (a major source of TNF-α) in the AT facilitate the mobilization of lipids and dysregulation of metabolic pathways, both of which contribute to body weight loss in patients [14]. Interferon-gamma (IFN-γ) is a pleiotropic cytokine that plays a vital role in deregulated fat metabolism in cancer cachexia. In rodent tumor models, elevated IFN-γ correlated with anorexia and weight loss during tumor development [126]. Additionally, in cachexia patients, IFN-γ mediates insulin resistance via reduction of glucose uptake; this leads to enhanced lipid breakdown in white adipose tissue (WAT) [127]. Another cytokine that cooperates with TNF-α to promote cancer cachexia is IL-6. During cachexia, IL-6 levels were further upregulated by pro-inflammatory cytokine IL-1, resulting in weight loss and reduced survival in cancer patients [128,129]. Chronic inflammation mediated by IL-6 promotes early cancer cachexia by regulating WAT lipolysis and late cachexia by WAT browning [130]. Scant evidence is available regarding the mechanisms by which IL-6 facilitates lipid mobilization in both mice and humans [14].
Fig. 3.
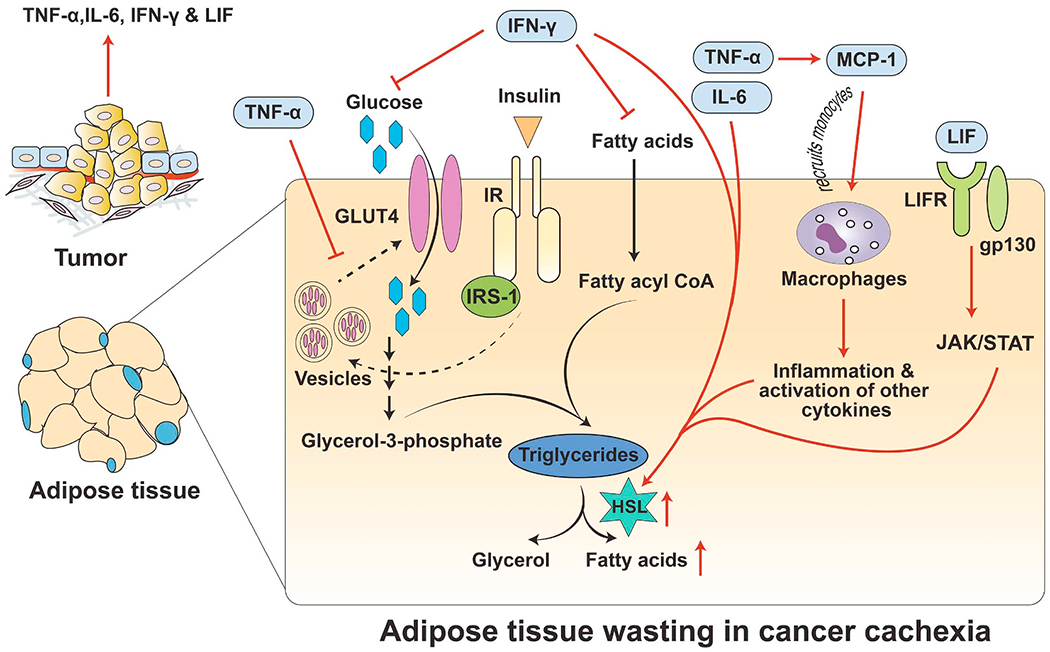
Regulation of adipose wasting in cancer cachexia. During cancer cachexia, the tumor secretes several inflammatory cytokines such as TNF-α, IL-6, IFN-γ, and LIF. After secretion from the tumor, TNF-α inhibits the recycling of GLUT4 from the cytoplasm to the plasma membrane. In addition, it also activates monocyte chemotactic protein (MCP-1), which recruits monocytes followed by activated macrophages in the adipose tissue (AT), leading to inflammation. Inside the AT, macrophages also secrete TNF-α and IL-6, which induce lipolysis by activating hormone-sensitive lipase (HSL). Similarly, IFN-γ prevents uptake of glucose as well as free fatty acids, which results in insulin resistance along with lipid breakdown in the AT. Finally, LIF binds to its receptor LIFR and co-receptor gp130 and activates the JAK/STAT pathway. Activation of the JAK/STAT pathway further upregulates HSL activity. Overall, cytokines released from the tumor during cachexia lead to AT wasting, which may contribute to body weight loss in cachectic patients.
In addition to TNF-α, activated macrophages also secrete IL-6 during cancer cachexia, which activates the acute-phase response from the liver. The first step in this pathway is binding of IL-6 to the membrane-bound receptor gp130, which activates the downstream JAK/STAT pathway with translocation of STAT into the nucleus, thereby inducing transcription of acute-phase proteins [131]. In a recent study, tumor humoral factor (i.e., leukemia inhibitory factor [LIF], a member of the IL-6 cytokine family from cancer cells), was found to induce AT lipolysis [132] by binding receptor LIFR-α and co-receptor gp130, which activates JAK/STAT signaling. Activated JAK/STAT resulted in the upregulation of adipose triglyceride lipase (ATGL) for fat degradation. In addition, in vivo administration of rLIF into Balb/c mice resulted in a decrease in body weight and AT loss, but little change in overall food intake. Similarly, in genetically hyperphagic mouse models [leptin deficient (ob/ob) and its signal deficient (db/db)], administration of rLIF decreased food intake and promoted AT loss along with > 10% reduction in animal body weight [132]. Overall, these studies suggest that inflammatory cytokines TNF-α and IL-6 are involved in the depletion of AT in cancer cachexia. Moreover, the function of cytokines in cancer cachexia depends on the type of tumor and the complex network mediators within it rather than a single cytokine alone [133].
In humans, AT is classified as either WAT, which stores triglycerides in the form of lipid droplets, or brown adipose tissue (BAT), which is the primary site of non-shivering thermogenesis [134]. Although WAT dysfunction is not directly implicated in cachexia, the switching of WAT to a more BAT-like phenotype is associated with cancer cachexia [135]. This phenomenon called WAT “browning” occurs when WAT acquire brown fat characteristics, including high mitochondrial content [136,137]. The transformation of WAT into “beige” or “brite” cells requires a sophisticated transcriptional machinery, including the transcriptional coregulator PR domain-containing 16 (PRDM16). Mice deficient for fat-specific PRDM16 exhibit reductions in browning, thermogenesis, and WAT atrophy [119]. Beige cells are phenotypically distinct from both WAT and BAT, and can emerge in response to extensive cold exposure [138], adrenergic stimulation [139], and prostaglandin synthesis enzyme (cyclooxygenase2) [140]; these cells can significantly contribute to total energy expenditure and promote fat reduction [141]. WAT browning in mice with cancer cachexia resulted in increased energy expenditure in interscapular BAT. Tumors directly activate thermogenesis in beige cells via secretion of PTHrP, which induces UCP1 [119]. UCP1 stimulates thermogenesis at the cost of ATP synthesis, leading to increased lipid mobilization and energy expenditure in cachectic mice. Moreover, cancer cachexia patients have higher expression of UCP1 in AT compared with cancer patients without cachexia, a change that presumably results in atrophy and subsequently leads to more thermogenesis [142]. Targeting upstream PTHrP with neutralizing antibodies reduced the intensity of cancer cachexia and atrophy [119]. WAT browning can also be inhibited by β3-andrenergic receptor antagonists or non-steroidal anti-inflammatory drugs, which improved wasting in mice. Therefore, targeting WAT browning is a promising approach for cancer cachexia patients.
7. Cachexia as a multi-organ metabolic syndrome
Cachexia is a multi-organ syndrome. Although the skeletal muscle and AT are the primary targets of cachexia, other tissues such as brain, liver, heart, bone, pancreas, cardiac muscle, and gut are directly involved in the cachectic process. Alterations in other organs also influence the degree of muscle wasting in cachectic patients (Fig. 4).
Fig. 4.
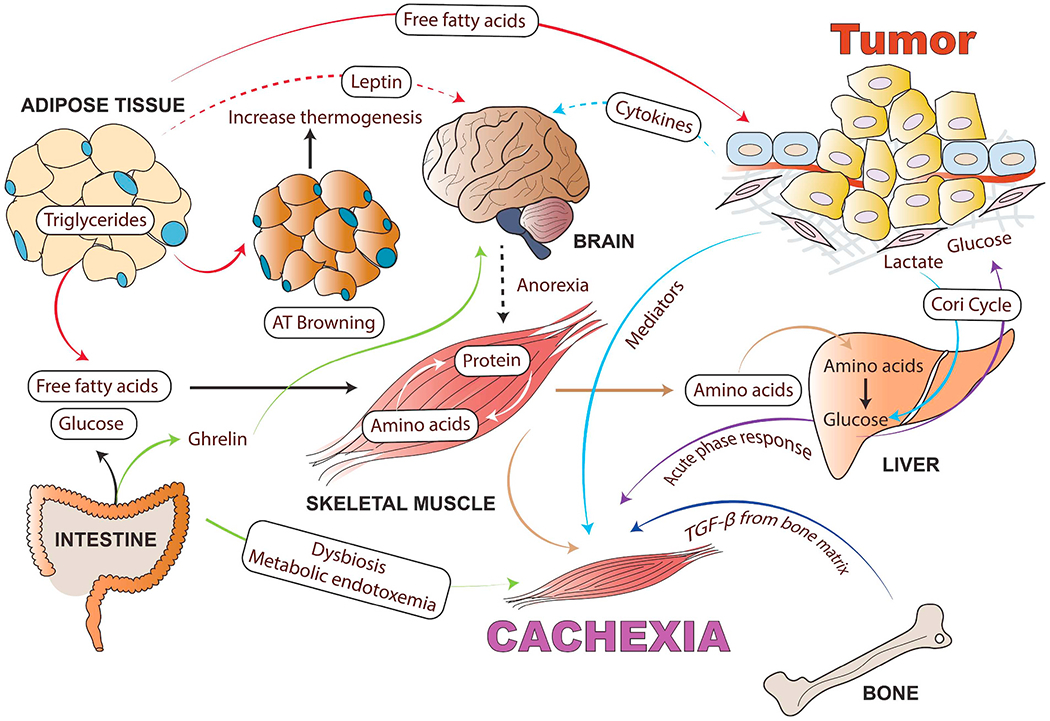
Cancer cachexia as a multi-organ metabolic syndrome: Although muscle and adipose tissue wasting are the main characteristics of cancer cachexia, tumor and host-derived factors and systemic inflammation also influence many other organs such as brain, liver, heart, bone, pancreas, cardiac muscle, and gut, which are involved in the cachectic process. Tumor-derived cytokines cause systemic inflammation in the hypothalamus and stimulate neuropeptides involved in the regulation of food intake. Anorexia exacerbates body wasting in cancer cachexia. In addition to tumor-derived mediators, adipokines, muscle-derived cytokines (myokines) and brain-derived anorexia factors also influence the wasting of skeletal muscle and AT as well as increase inflammation in the tumor microenvironment. This inter-tissue communication directly or indirectly influences tissue metabolism and the severity of the cachexia syndrome. Inflammatory cytokines can increase lipolysis in white adipose tissue (WAT), which releases free fatty acids that further fuel the tumor and facilitate muscle wasting. Cachexia can cause adipose tissue browning (WAT switches to brown adipose tissue) and increased thermogenesis. In turn, the myokines activate pathogenic mechanisms in the skeletal muscle and adipose tissue. Muscle wasting releases free amino acids resulting from active protein degradation during cancer cachexia, which drives the acute phase response in the liver. Cancer-associated gut microbiota dysfunction alters mitochondrial energy metabolism in skeletal muscle, contributing to the negative energy balance in cachectic cancer patients. In the case of cancer-mediated bone metastasis, increased activity of the osteoclasts results in the activation of TGF-β from the bone matrix, which can facilitate muscle wasting and reduce muscle strength. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
7.1. Brain and food intake (anorexia)
The hypothalamus controls energy expenditure and food intake by coordinating the neuropeptide Y (NPY)/agouti-related peptide (AgRP) and pro-opiomelanocortin (POMC)/cocaineamphetamine-regulated transcript (CART) neurons that stimulate energy production and inhibit food intake, respectively. These functions in the CNS are regulated by release of the protein leptin by adipose tissue. Leptin is part of a regulatory feedback loop that stimulates and inhibits several hypothalamic neuropeptides, including NPY and corticotropin-releasing factor (CRF). Administration of inflammatory cytokines, such as TNF-α and IL-1, in cancer patients and a mouse model of inflammation increased the expression of leptin in adipocytes and caused an increase in plasma levels of leptin despite starvation, suggesting a mechanism by which leptin levels contribute to cancer anorexia [143,144]. Tumor-induced cytokines impair hypothalamus functions by persistent stimulation of anorexigenic pathways and inhibition of orexigenic pathways [145]. Several pro-inflammatory cytokines, such as IL-1, hyperactivated the POMC/CART pathway in cancer cachexia [146,147]. Further, hypothalamic inflammation influenced anorexia-cachexia syndrome via the hypothalamic-pituitary-adrenal axis and serotonin pathway activation. It has been reported that serotonin is associated with the development of cancer anorexia by activating the melanocortin system, and blocking of melanocortin receptors ultimately reversed cancer-mediated anorexia in rats [148]. In addition, IL-1 may also increase the secretion of hypothalamic serotonin, which activates POMC/CART neurons, resulting in decreased appetite and anorexia [149]. These findings suggest the significance of the hypothalamic serotonin-melanocortin axis in cancer cachexia pathogenesis and its relevance as a potential therapeutic target for cancer cachexia.
7.2. Liver
The liver is a highly metabolic organ that contributes to energy wasting in cancer cachexia though futile metabolic cycles. In cancer patients, tumors were capable of renewing glucose from lactate via hepatic gluconeogenesis, which leads to high amounts of energy loss [150]. Further, in the case of cachexia, free amino acids resultant from muscle protein degradation can drive the liver acute phase response (APR) and energy wasting [151]. An increase in macrophage infiltration due to hepatic APR has reported in cachectic pancreatic cancer patients compared to non-cachectic patients, suggesting a significant role of liver macrophages in cancer cachexia [152]. Activated macrophages secrete IL-6, which in turn stimulates the synthesis of hepatic acute-phase proteins [153]. Further, hyperlipidemia was reported in a rodent model of cachexia due to increased liver lipogenesis with tumor burden [151]. Hepatic steatosis, commonly seen in nonalcoholic fatty liver disease, has also been documented in patients with cancer cachexia and is associated with muscle loss, further suggesting crosstalk between the liver and muscle under energy-wasting conditions [154]. The mechanism may involve enhanced expression of transcription factor TGFβ1-stimulated clone 22 D4 (TSC22D4) and nuclear receptor cofactor receptor-interacting protein 140 (RIP140), which could lead to hepatic steatosis by preventing the mobilization of hepatic triglyceride stores [155,156]. Additional hepatic alterations were reported during progression of cachexia including increased deposition of collagen and fibrosis in the Lewis lung carcinoma mouse model of cachexia [157]. Finally, the changes in the liver directly or indirectly contribute to energy wasting and cachexia in cancer patients.
7.3. Pancreas
Metabolic deregulation, such as impaired glucose tolerance and decreased insulin sensitivity, is the hallmark of the majority of cancer patients [2]. Insulin resistance is a risk factor for cancer development and seen in the majority of cancer patients during the onset of cachexia [127]. Further, mice bearing colon-26 tumors can develop insulin resistance before the onset of cachexia, suggesting the role of insulin resistance in cachexia pathogenesis [158]. Islets of Langerhans isolated from the pancreas of Walker 256 tumor-bearing rats showed resistance to glucose stimulation [159]. Furthermore, this decrease in insulin sensitivity was not associated with structural changes in pancreatic islets, but instead with changes in insulin signaling [160]. Rosiglitazone (insulin sensitizers) treatment in mice bearing colon-26 tumors improved insulin sensitivity and diminished the early markers of cachexia [158]. In the clinical setting, daily insulin treatment for weight-losing cancer patients diminished the progression of cancer cachexia, suggesting a role for insulin in palliative care for cachectic cancer patients [161].
7.4. Gut
Several alterations in gut composition and function have a role in cancer cachexia including gut barrier dysfunction, altered ghrelin production, and microbiota dysbiosis [162]. Cancer patients receiving radiotherapy and/or chemotherapy often develop gut barrier dysfunction, mainly leakage of the gut epithelial barrier. The barrier becomes more permeable due to decreased expression of tight junction proteins (ZO1 and occludin) and macrophage infiltration. The leaky barrier facilitates entry of either bacterial cell wall components (endotoxin or lipopolysaccharide) or intact bacteria into the circulation and causes inflammation. Apc(Min/+) mice with colorectal cancer exhibited compromised intestinal gut barrier integrity, elevated plasma endotoxin concentration, and cachexia symptoms [163]. Interestingly, anti-IL-6 antibody treatment restored gut integrity in a C26 cachexia model, which suggested inflammation may contribute to gut dysfunction in cancer [164]. Further, cancer-associated gut barrier dysfunction can cause malabsorption of nutrients, diarrhea, and other complications that contribute to the negative energy balance in patients with cancer.
The gut-derived hormone ghrelin, also known as the hunger hormone, is elevated in multiple cancers [165]. In the case of cachexia, even increased levels of ghrelin fail to induce the appetite, known as ghrelin resistance. The ghrelin receptor, growth hormone secretagogue receptor 1α, is expressed in the hypothalamic and pituitary regions and mediates growth hormone release and enhances appetite; however, the mechanism of action remains elusive [166,167]. Besides controlling hormonal release, energy homeostasis, and appetite, ghrelin suppresses inflammation via the release of IL-10, an anti-inflammatory cytokine. Ghrelin-mediated induction of IL-10 reduced levels of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α [168]. Furthermore, ghrelin reduced muscle catabolism via suppression of the NFκB-mediated ubiquitin-proteasome pathway [169]. Ghrelin administration in mice reduced atrogin-1, MuRF-1 and dexamethasone-induced skeletal muscle atrophy [170].
Gut microbiota play an important role in the context of cancer cachexia during chemotherapy/radiotherapy regimen. Gut microbiota influences the host’s immunity and metabolism, and decreased levels of bacteria were found in an experimental model of cachexia [164]. Another study provided evidence that administration of exogenous bacteria restores intestinal homeostasis and prolongs survival in leukemic mice [171]. The amino acid bioavailability and metabolites generated by the gut microbiota can influence energy expenditure in the muscle cells, highlighting the role of the gut microbiota - skeletal muscle axis in cancer cachexia. Gut microbiota release lipopolysaccharide and peptidoglycan, which can stimulate the Toll-like receptors and subsequently activate muscle-specific NF-κB to facilitate muscle wasting [172]. In addition to muscle wasting, gut microbiota influence the accumulation of triacylglycerol into the adipose tissues [173].
7.5. Bone
The skeletal muscle and bone are well connected, and functions of both organs are tightly coupled during growth and development [174,175]. Recent evidence suggests that several cytokines secreted from muscle cells (myokines), such as insulin-like growth factor 1 (IGF-1), fibroblast growth factor 2 (FGF-2), myostatin/GDF8, and IL-6, not only regulate skeletal muscle development and maintenance, but also have a role in bone cell function [176]. For example, IGF-1 and FGF-2 both stimulate bone formation [177,178]. Myostatin negatively regulates muscle growth and development, and is upregulated in atrophic conditions. When administered to mice, myostatin induces cachexia, likely by activating the ubiquitin proteolytic system in the muscle through a FoxO1-dependent mechanism [179,180]. Several growth factors embedded in the bone matrix, primarily involved in normal bone physiology, can also modulate the functions of the skeletal muscle. Cancer cells disrupt the normal bone remodeling cycle during bone metastasis and facilitate the release of growth factors. One of the bone-derived factors Indian hedgehog (Ihh) promotes myoblast survival and myogenesis in mice [174]. Preclinical mouse models of breast cancer bone metastases and multiple myeloma both showed osteolytic bone destruction, reduced forelimb grip strength, and systemic muscle dysfunction, further suggesting the impact of bone-derived factors on muscle wasting [181]. A recent observation showed that osteoclast-mediated release of latent TGF-β from the bone matrix could affect intracellular calcium signaling and skeletal muscle functions in mice with bone metastases [182]. Further, inhibition of sclerostin, a soluble Wnt inhibitor that represses osteoblast differentiation and bone formation, has been shown to alleviate bone metastasis burden, muscle weakness, and overall survival in a mouse model of breast cancer [183]. Altogether, this evidence suggests that bone microenvironment can modulate muscle wasting, and identification and characterization of factors involved in this bone-muscle crosstalk will provide new possibilities for therapeutic intervention in cancer cachexia-associated muscle wasting.
7.6. Cardiac muscle
In addition to losing skeletal muscle mass and function, many cachectic patients have shown heart muscle wasting and dysfunction. Preclinical studies showed that potential signaling pathways contributing to cardiac atrophy and muscle wasting overlap, such as activation of NF-kB. The ApcMin/+ mouse model of colorectal cancer showed a decline in cardiac mass during cachexia progression by affecting AMPK, Akt, and mTOR signaling in the heart. Other mechanisms for loss of cardiac mass in this model include Akt-independent suppression of anabolic signaling and increased levels of Beclin1 or autophagy [184]. The cancer itself and its treatment can further worsen underlying heart disease in cancer cachexia. Most of the anticancer drugs, such as docetaxel, anthracycline, and fluorouracil, are associated with cardiotoxicity in cancer patients [185]. Further, chemo and/or radiotherapy increase the occurrence of cachexia in cancer patients. Cardiac muscle wasting is exceptionally lethal, and thus screening for it and managing it may improve the response to anticancer treatments, QOL, and chances of survival. Taken altogether, early detection of cardiac atrophy, personalized therapies, and mechanistic studies are needed, so that cardiac muscle wasting can be better managed in cancer cachexia patients.
7.7. Neural invasion
Tumor cells can invade in, around, and through nerves in a process known as perineural invasion, which most commonly occurs in pancreatic cancer patients. Recent studies suggest that perineuronal invasion is associated with cachexia and can include nerve damage, as well as astrocyte activation and microglia stimulation in the spinal cord [186]. However, the mechanisms by which perineuronal invasion could contribute to cachexia are not well known. One possibility is that damaged peripheral nerves activate astrocytes in the spinal cord which may induce muscle atrophy as well as lipolysis, perhaps via stimulating the sympathetic nervous system [186]. However, despite significant muscle loss, neuromuscular junctions remain structurally intact in cancer cachexia patients, suggesting that denervation of skeletal muscle is not a major driver of cachexia [187].
8. Current and emerging pharmacological management of cancer cachexia
The present existing treatment options for cachexia are limited, and most of the available therapies are palliative in nature. Because cancer cachexia is a multi-organ syndrome, multimodal treatment approaches, including exercise, appetite stimulation, nutrient supplementation, and pharmacological intervention may prove beneficial [1]. Most cancer patients show symptoms of anorexia, which is strongly associated with development of cachexia. Therefore, calorie supplementation and appetite stimulants are primarily used as treatment for this disease. In addition, control of appetite and food intake requires precise balance between prophagic and anorexigenic signals to the hypothalamus.
8.1. Nutritional interventions and appetite stimulants to combat cachexia
A large-scale meta-analysis discovered that nutritional interventions are beneficial in increasing energy intake, body weight, and improving QOL in malnourished patients with cancer [188]. Some beneficial effects of anti-inflammatory nutrient supplementations, such as eicosapentaenoic acid (EPA) or fish oil, on muscle loss have been reported [1]. In mice bearing MAC-16 tumors, EPA prevented the development of cancer cachexia [189]. In pancreatic cancer cachectic patients, EPA can downregulate the production of pro-inflammatory cytokines (mainly suppression of IL-6 production) and the acute phase protein response [190]. Further, EPA has shown some beneficial effects on muscle mass and weight in patients with NSCLC receiving chemotherapy [191]. Overall nutritional interventions to combat the cancer cachexia have been listed in Table 1.
Table 1.
Appetite enhancing agents investigated for combating cancer cachexia
| Therapy/Class | Effects | Possible mode of action | Reference |
|---|---|---|---|
| Eicosapentaenoic acid or fish oil | Improves muscle mass and appetite | Downregulates the production of pro-inflammatory cytokines (Mainly IL-6) | [189,190] |
| Acute-phase protein response | |||
| Megestrol acetate or Megace (Synthetic progestin) | Improves appetite, Increases body weight due to the increase in fat mass and water retention | Unknown, Probably by antagonizing the effects of catabolic cytokines, Neuropeptide Y release, Reduction of serum levels of IL-1, IL-2, IL-6, and TNF-a | [191–193, 195, 196, 198, 199 |
| Dronabinol (Marijuana) A derivative of cannabinol |
Stimulates the appetite and weight gain, fails to restore the weight loss | Activates the cannabinoid receptors (CB1 and CB2) in CNS | [203,204] |
| Metformin (Oral type 2 diabetes mellitus drug) | Improves the appetite | Minimized the tumor-induced muscle wasting | [225] |
| OHR118 (Immunomodulator) | Improves the appetite, dyspepsia, and depression | Modulates the synthesis of chemokines and cytokines including TNF-α | [207] |
| Anamorelin or Ghrelin, (Stomach released neuropeptide) | Increases food intake and body weight due to an increase in body fat composition and reduces the muscle catabolism | Binds with growth hormone secretagogue receptor 1α (GHSR-1α) expressed in the hypothalamus and pituitary and mediates growth hormone release | [208–211] |
Megestrol acetate (Megace), a synthetic progestin, is widely used as an appetite stimulant and is approved for the treatment of weight loss and severe malnutrition in patients with AIDS [192]. Megace improves appetite and is associated with slight weight gain in cancer and AIDS patients with anorexia-cachexia syndrome [193,194]. A high dose of Megace resulted in a significant improvement in appetite and body weight in some patients with cancer cachexia [195–197]. However, the increase in body weight was primarily due to an increase in fat mass rather than an increase in lean body mass [198,199]. Recent in vivo studies showed that Megace improved survival and reduced skeletal and heart muscle wasting through the downregulation of autophagy, leading to a significant improvement of cardiac function; thus, this therapy may have a role in cancer cachexia-induced cardiomyopathy [200]. Although Megace is well tolerated, the use of Megace as an appetite stimulant has a red box warning as it has been associated with an increased risk of thromboembolic events and inadequate response to chemotherapy [201,202]. Dronabinol (marijuana) is a synthetic oral derivative of cannabinol and has limited FDA approval for anorexia-associated weight loss in AIDS patients. In cachectic cancer patients, dronabinol is known to stimulate appetite and weight gain but is not able to restore weight loss. Although dronabinol has a beneficial effect on cancer-related anorexia/cachexia syndrome, dronabinol is less effective than Megace against anorexia in clinical trials for advanced cancer [203,204].
Other appetite stimulants include corticosteroids such as dexamethasone and prednisolone, which are among the earliest drugs evaluated for cancer cachexia. In a randomized clinical trial, dexamethasone increased appetite but did not affect body weight due to its catabolic effects on muscle and bone [205]. One preliminary study showed dexamethasone treatment significantly improved patient-reported cluster scores for fatigue/anorexia-cachexia/depression (FAD) symptoms after two weeks in advanced cancer patients [206]. Another novel immunomodulatory, OHR118, affects the synthesis of chemokines and cytokines, including TNF-α. In a phase 2 study, OHR118 significantly improved appetite, dyspepsia, and depression in cancer patients [207].
Ghrelin, a neuropeptide released from the stomach, is known to stimulate energy intake in cachectic cancer patients [208]. In a pre-clinical study, ghrelin was shown to increase food intake and body weight due to an increase in body fat composition. In a clinical trial, RC-1291, a ghrelin mimetic, increased muscle strength and lean body mass but did not affect body weight and QOL [170,209,210]. In a multicenter clinical trial, ghrelin receptor agonist anamorelin (ONO-7643) improved appetite, lean body mass, and body weight in advanced unresectable gastrointestinal cancer cachectic patients [211].
Advanced stages of malignancy, which account for many cachectic patients, eventually acquire resistance to cancer therapy and are hard to cure. Nutritional status is a key consideration for the initiation of cancer therapy, and patients with poor nutritional status should be carefully monitored throughout the treatment regimen [212]. In obese cancer patients, chronic inflammation may influence the relationship between cancer and host immunity; therefore, the patient’s inflammatory status, metabolism, and weight, should be taken into consideration before starting cancer immunotherapy [213]. Although nutritional supplements and appetite stimulants are currently being used extensively for the management of cachexia, these interventions are often inadequate. The benefits of nutritional supplementation may be limited because they have minimal impact on the underlying catabolic processes responsible for skeletal muscle wasting. Data from at least 40 prospective randomized clinical trials in cancer patients showed that nutritional interventions had minimal clinical efficacy, particularly in improving muscle mass, QOL, and long-term survival [214].
8.2. Direct targeting of the mediators of cachexia
Although appetite stimulants have proven beneficial in improving QOL, they do not improve functional outcomes. Recent pre-clinical and clinical studies have explored targeted therapies in cancer-induced muscle wasting and have shown encouraging results. In a preclinical study, a monoclonal antibody against fibroblast growth factor-inducible 14 (Fn14), which targets the receptor for the TWEAK cytokine (TNF receptor superfamily), prevented cachexia and prolonged survival in a C26 tumor-bearing mouse model of cachexia [215]. However, adding infliximab, a recombinant anti-TNF-α antibody, to gemcitabine therapy had no effect on efficacy or safety in pancreatic cancer patients with cachexia [216]. In a clinical trial, ALD518, a humanized monoclonal antibody against human IL-6, significantly improved handgrip strength, fatigue, and cancer-related cachexia in advanced NSCLC patients [217,218]. Another humanized monoclonal antibody, MABp1, targeting cytokine IL-1a, prolonged median overall survival compared to Megace in advanced colorectal cancer (CRC) patients with cachexia [219,220]. Treatment with anti-inflammatory drugs like cyclooxygenase (COX) inhibitors (indomethacin, ibuprofen) had beneficial metabolic effects and prolonged survival in weight-losing cancer patients by attenuation of resting metabolism; it also improved appetite due to decreased systemic inflammation [221]. Further, the novel histone deacetylase (HDAC) inhibitor AR-42 suppressed the production of multiple inflammatory cytokines and proteins related to cancer cachexia (IL-6, IL-6Rα, leukemia inhibitory factor, Foxo1, Atrogin-1, MuRF1, adipose triglyceride lipase, uncoupling protein 3, and myocyte enhancer factor 2c) and ultimately showed therapeutic potential against loss of muscle and AT in C-26 colon adenocarcinoma and LLC murine models of cancer cachexia [222]. Non-steroidal selective androgen receptor modulator GTx-024 (enobosarm) showed a promising effect on lean body mass improvement and muscle function in a pre-clinical study [223]. Furthermore, the combination of GTx-024 with AR-42 showed a synergistic effect on cachexia improvement in the C-26 murine model of cachexia [224].
Since diabetes and cancer cachexia share certain metabolic features, an oral type 2 diabetes mellitus drug, metformin, was evaluated in tumor-bearing rats and found to minimize tumor-induced muscle wasting in rats [225]. Similarly, glucose intolerance in cachexia patients prompted evaluation of insulin treatment, which significantly increased food intake, increased whole-body fat, and improved survival; however, it did not affect the lean body mass of weight-losing cancer patients [161]. Further, a combination of metformin and insulin treatment attenuated the progression of cancer cachexia and improved the metabolism of weight-losing cancer patients via increasing their total body fat.
Cachectic patients showed a better response to adjuvant therapy with early intervention rather than at late-stage or refractory cachexia. In some cancer patients, effective treatment of the primary tumor may compromise one’s appetite, weight loss, and muscle wasting [226]. Thus, a major obstacle in evaluating anti-cachexia therapy is how to address the confounding effect of systemic chemotherapy on muscle wasting. Comprehensive metabolic profiling suggests that cachexia induced by cancer is associated with different metabolic derangements than cachexia induced by chemotherapy; thus, effective therapeutic interventions should take into account different drivers of cachexia [227]. Integrative strategies to target multiple cachexia mechanisms have been tested in preclinical and clinical investigations in recent years with limited success. In an early phase II study, combined treatment with polyphenol-enriched diet, antioxidants, medroxyprogesterone acetate, and anti-cyclooxygenase-2 inhibitor (celecoxib), was shown to be effective and safe for cancer cachexia [228]. Bimagrumab (BYM338), a human dual-specific anti-ActRIIA/ActRIIB antibody, showed better efficacy in improving muscle mass compared to blockade of a single receptor [229]. Recently, a HASPIN kinase (a nuclear Ser/Thr kinase) inhibitor (CHR-6494) reduced cancer cachexia in APCmin/+ mice [230]. In a preclinical study, the steroidal lactone, Withaferin-A ameliorated ovarian cancer-induced cachexia via reduced production of NF-kB-mediated pro-inflammatory cytokines [231]. Toll-like receptor (TLR7/8) agonist R848 improved cachexia in the PDAC mouse model of cachexia [232].
Taken together, although several promising agents are in development that target tumor-induced inflammation, muscle wasting, and adipose browning associated with cancer cachexia, to date, most of the therapeutic strategies to manage cachexia are palliative (Table 2).
Table 2.
Direct targeting the mediators of cancer cachexia
| Therapy/Class | Effects | Possible mode of action | Reference |
|---|---|---|---|
| Fibroblast growth factor-inducible 14 (Fn14, monoclonal antibody) | Inhibits cachexia and prolonged survival in C26 tumor-bearing mice | Via targeting the receptor for the TWEAK cytokine (TNF receptor superfamily) | [215] |
| ALD518 (Humanized monoclonal antibody) | Improves muscle strength and fatigue | A humanized monoclonal antibody against human IL-6 | [217,218] |
| MABp1 (Humanized monoclonal antibody) | Stabilize the symptom of cachexia via anti-inflammatory activity | Targeting cytokine IL-1α | [219,220] |
| COX inhibitors (indomethacin, ibuprofen) | Attenuation of resting metabolism, improved appetite and prolonged survival | Decreased systemic inflammation | [221] |
| AR-42 (Histone Deacetylase inhibitor) | Protects loss of muscle and adipose tissue | Suppressed the production of inflammatory cytokine and protein (IL-6, IL-6Rα, leukemia inhibitory factor) | [222] |
| GTx-024 (Enobosarm) | Increases lean body mass | Androgen receptor modulator | [223] |
| Bimagrumab (BYM338),(Dual anti-ActRIIA/IIB antibody) | Inhibits muscle wasting | Activin type-2 receptor (ActRIIA/ActRIIB) | [229] |
| CHR-6494 (HASPIN kinase inhibitor) | Inhibits the loss of body weight | HASPIN kinase (a nuclear Ser/Thr kinase) inhibitors | [230] |
| Withaferin-A (Steroidal lactone) | Rescue the loss of muscle strength | Reduces NF-kB-mediated pro-inflammatory cytokines production | [231] |
| R848 (Toll-like receptors (TLR7/8) agonist) | Decreases cardiac and lean mass catabolism | Reduces the Hypothalamic inflammatory gene expression | [232] |
9. Summary and future directions
Cachexia is a multifactorial syndrome best characterized by a severe loss of body weight and increased protein catabolism. Pathologically, negative energy and protein imbalance due to systemic inflammation, muscle wasting, and impaired metabolism contribute to cachexia. The prevalence of cancer cachexia is high in advanced cancer. Cachexia research has made a significant advancement in the understanding of this multifactorial disorder. However, the available therapeutic options are limited, and the mechanisms underlying cachexia development are not well defined. Most cachexia research has focused on the involvement of pro-inflammatory cytokines in inadequate food intake or anorexia, improper energy expenditure, altered hepatic function, skeletal muscle wasting, and lipolysis/browning in adipocytes. Several pre-clinical studies have revealed potential mediators, signaling pathways, and novel therapeutic targets, but cancer cachexia is still the leading cause of morbidity and mortality in cancer patients. Several tumor- and host-derived pro-inflammatory cytokines (i.e., TNFα, IL-6 and IL-1, and PTHrP) are the key mediators of cancer cachexia. These molecules play an essential role in cancer growth, inhibition of apoptosis, tumor angiogenesis, and metastasis, along with other factors. Given the complex multifactorial nature of the cachexia secretome, targeting any single dominant circulating factor is unlikely to reverse cachexia. In the array of tumor- and host-derived chemokines and cytokines, which mediators are most responsible for driving cachexia requires further investigation.
The use of appropriate diagnostic criteria among different stages of cachexia may be beneficial to halt the advancement of this disorder. Generally, cancer cachexia is diagnosed at the terminal stage of cancer patients, but it can arise in any phase of cancer progression. Therefore, continuous weight loss should be taken into account for early assessment of cachexia symptoms; this would enable timely treatment decisions. In obese patients, the assessment of lean muscle mass versus bodyweight loss is essential for proper diagnosis of cachexia. Further, the accurate diagnosis of cachexia is challenging due to the growing prevalence of obesity. Weight loss in obese cancer patients may get overlooked because patients appear normally nourished; however, unnoticed severe muscle wasting may exist, and muscle degradation is one of the most critical consequences of cancer cachexia. Screening and monitoring for muscle degradation currently requires muscle biopsies, which are intrusive. Therefore, the identification of more sensitive and specific mediators of muscle atrophy may support development of promising biomarkers for early detection of disease, before the evident signs of active muscle wasting or loss of body weight.
Acknowledgments
The authors on this manuscript are supported, in part, by grants from the National Institutes of Health (P01 CA217798, U01 CA185148, U01 CA200466, U01 CA210240, U01 CA1213862, R01 CA195586, and R01 CA218545). We also would like to thank Dr. Jessica Mercer and Jeffrey Patterson for editing this manuscript.
Abbreviations:
- ActRIIB
Activin type-2 receptor
- CART
Cocaine-amphetamine-regulated transcript
- COX
Cyclooxygenase
- IGF-1
Insulin-like growth factor
- IL
Interleukin
- IFN-γ
Interferon-gamma
- IRS1
Insulin Receptor Substrate 1
- LCC
Lewis lung carcinoma
- MAFbx
Muscle atrophy F box protein
- MIC-1/GDF15
Macrophage inhibitory cytokine/growth differentiation factor-15
- MuRF-1
Muscle RING finger protein
- PTHrP
Parathyroid hormone-related peptide
- NF-κB
Nuclear factor κB
- NSCLC
Non-small cell lung cancer
- POMC
Pro-opiomelanocortin
- QOL
Quality of life
- REE
Resting energy expenditure
- TGF-β
Transforming growth factor-beta
- STAT
Signal transducers and activators of transcription
- TNFR
TNF-receptor
- TNF-α
tumor necrosis factor-alpha
- TWEAK
TNF receptor superfamily
- UCP
Uncoupling protein
- UPP
Ubiquitin-dependent proteasome pathway
Footnotes
Declaration of competing interest
SKB is one of the co-founders of Sanguine Diagnostics and Therapeutics, Inc. The other authors declare no competing interests. The authors have declared that the submitted manuscript is original, is not under consideration by another journal, and has not been published previously in any form. Every author is aware of and has agreed to the content of this paper.
References
- [1].Fearon K, Arends J, Baracos V, Understanding the mechanisms and treatment options in cancer cachexia, Nat. Rev. Clin. Oncol 10 (2013) 90–99. [DOI] [PubMed] [Google Scholar]
- [2].Porporato PE, Understanding cachexia as a cancer metabolism syndrome, Oncogenesis 5 (2016) e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mattox TW, Cancer Cachexia: Cause, Diagnosis, and Treatment, Nutr. Clin. Pract 32 (2017) 599–606. [DOI] [PubMed] [Google Scholar]
- [4].Aversa Z, Costelli P, Muscaritoli M, Cancer-induced muscle wasting: latest findings in prevention and treatment, Ther. Adv. Med. Oncol 9 (2017) 369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Imai K, Takai K, Miwa T, Taguchi D, Hanai T, Suetsugu A, Shiraki M, Shimizu M, Rapid Depletions of Subcutaneous Fat Mass and Skeletal Muscle Mass Predict Worse Survival in Patients with Hepatocellular Carcinoma Treated with Sorafenib, Cancers (Basel) 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Huang CY, Yang YC, Chen TC, Chen JR, Chen YJ, Wu MH, Jan YT, Chang CL, Lee J, Muscle loss during primary debulking surgery and chemotherapy predicts poor survival in advanced-stage ovarian cancer, J. Cachexia. Sarcopenia Muscle (2020), 10.1002/jcsm.12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vermaete N, Wolter P, Verhoef G, Gosselink R, Physical activity and physical fitness in lymphoma patients before, during, and after chemotherapy: a prospective longitudinal study, Ann. Hematol 93 (2014) 411–424. [DOI] [PubMed] [Google Scholar]
- [8].Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D, Jatoi A, Kalantar-Zadeh K, Lochs H, Mantovani G, Marks D, Mitch WE, Muscaritoli M, Najand A, Ponikowski P, Rossi Fanelli F, Schambelan M, Schols A, Schuster M, Thomas D, Wolfe R, Anker SD, Cachexia: a new definition, Clin. Nutr 27 (2008) 793–799. [DOI] [PubMed] [Google Scholar]
- [9].Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G, Davis M, Muscaritoli M, Ottery F, Radbruch L, Ravasco P, Walsh D, Wilcock A, Kaasa S, Baracos VE, Definition and classification of cancer cachexia: an international consensus, Lancet Oncol. 12 (2011) 489–495. [DOI] [PubMed] [Google Scholar]
- [10].Vigano A, Kasvis P, Di Tomasso J, Gillis C, Kilgour R, Carli F, Pearls of optimizing nutrition and physical performance of older adults undergoing cancer therapy, J. Geriatr. Oncol 8 (2017) 428–436. [DOI] [PubMed] [Google Scholar]
- [11].Dunne RF, Loh KP, Williams GR, Jatoi A, Mustian KM, Mohile SG, Cachexia and Sarcopenia in Older Adults with Cancer: A Comprehensive Review, Cancers (Basel) 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].von Haehling S, Anker MS, Anker SD, Prevalence and clinical impact of cachexia in chronic illness in Europe, USA, and Japan: facts and numbers update 2016, J. Cachexia. Sarcopenia Muscle 7 (2016) 507–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Arthur ST, Noone JM, Van Doren BA, Roy D, Blanchette CM, One-year prevalence, comorbidities and cost of cachexia-related inpatient admissions in the USA, Drugs Context 3 (2014) 212265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fearon KC, Glass DJ, Guttridge DC, Cancer cachexia: mediators, signaling, and metabolic pathways, Cell Metab. 16 (2012) 153–166. [DOI] [PubMed] [Google Scholar]
- [15].Al-Zoughbi W, Huang J, Paramasivan GS, Till H, Pichler M, Guertl-Lackner B, Hoefler G, Tumor macroenvironment and metabolism, Semin. Oncol 41 (2014) 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tisdale MJ, Mechanisms of cancer cachexia, Physiol. Rev 89 (2009) 381–410. [DOI] [PubMed] [Google Scholar]
- [17].Cao DX, Wu GH, Zhang B, Quan YJ, Wei J, Jin H, Jiang Y, Yang ZA, Resting energy expenditure and body composition in patients with newly detected cancer, Clin. Nutr 29 (2010) 72–77. [DOI] [PubMed] [Google Scholar]
- [18].Vazeille C, Jouinot A, Durand JP, Neveux N, Boudou-Rouquette P, Huillard O, Alexandre J, Cynober L, Goldwasser F, Relation between hypermetabolism, cachexia, and survival in cancer patients: a prospective study in 390 cancer patients before initiation of anticancer therapy, Am. J. Clin. Nutr 105 (2017) 1139–1147. [DOI] [PubMed] [Google Scholar]
- [19].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell 144 (2011) 646–674. [DOI] [PubMed] [Google Scholar]
- [20].Liberti MV, Locasale JW, The Warburg Effect: how does it benefit Cancer Cells? Trends Biochem. Sci. 41 (2016) 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vander Heiden MG, DeBerardinis RJ, Understanding the Intersections between Metabolism and Cancer Biology, Cell 168 (2017) 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Warburg O, On the origin of cancer cells, Science 123 (1956) 309–314. [DOI] [PubMed] [Google Scholar]
- [23].Afroze D, Kumar A, ER stress in skeletal muscle remodeling and myopathies, FEBS J. 286 (2019) 379–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Barreiro E, Salazar-Degracia A, Sancho-Munoz A, Gea J, Endoplasmic reticulum stress and unfolded protein response profile in quadriceps of sarcopenic patients with respiratory diseases, J. Cell. Physiol 234 (2019) 11315–11329. [DOI] [PubMed] [Google Scholar]
- [25].Roy A, Kumar A, ER Stress and Unfolded Protein Response in Cancer Cachexia, Cancers (Basel) 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Arneson PC, Doles JD, Impaired Muscle Regeneration in Cancer-Associated Cachexia, Trends Cancer 5 (2019) 579–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cohen S, Nathan JA, Goldberg AL, Muscle wasting in disease: molecular mechanisms and promising therapies, Nat. Rev. Drug Discov 14 (2015) 58–74. [DOI] [PubMed] [Google Scholar]
- [28].Baracos VE, DeVivo C, Hoyle DH, Goldberg AL, Activation of the ATP-ubiquitin-proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma, Am. J. Phys 268 (1995) E996–1006. [DOI] [PubMed] [Google Scholar]
- [29].Solomon V, Baracos V, Sarraf P, Goldberg AL, Rates of ubiquitin conjugation increase when muscles atrophy, largely through activation of the N-end rule pathway, Proc. Natl. Acad. Sci. U. S. A 95 (1998) 12602–12607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, Rakhilin SV, Stitt TN, Patterson C, Latres E, Glass DJ, The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle, Cell Metab. 6 (2007) 376–385. [DOI] [PubMed] [Google Scholar]
- [31].Glass DJ, Signaling pathways perturbing muscle mass, Curr. Opin. Clin. Nutr. Metab. Care 13 (2010) 225–229. [DOI] [PubMed] [Google Scholar]
- [32].Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL, Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy, Proc. Natl. Acad. Sci. U. S. A 98 (2001) 14440–14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ, Identification of ubiquitin ligases required for skeletal muscle atrophy, Science 294 (2001) 1704–1708. [DOI] [PubMed] [Google Scholar]
- [34].Temparis S, Asensi M, Taillandier D, Aurousseau E, Larbaud D, Obled A, Bechet D, Ferrara M, Estrela JM, Attaix D, Increased ATP-ubiquitin-dependent proteolysis in skeletal muscles of tumor-bearing rats, Cancer Res. 54 (1994) 5568–5573. [PubMed] [Google Scholar]
- [35].Lecker SH, Solomon V, Mitch WE, Goldberg AL, Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states, J. Nutr 129 (1999) 227S–237S. [DOI] [PubMed] [Google Scholar]
- [36].Hunt LC, Stover J, Haugen B, Shaw TI, Li Y, Pagala VR, Finkelstein D, Barton ER, Fan Y, Labelle M, Peng J, Demontis F, A Key Role for the Ubiquitin Ligase UBR4 in Myofiber Hypertrophy in Drosophila and Mice, Cell Rep. 28 (2019) 1268–1281 e1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang Y, Wang J, Wang X, Gao T, Tian H, Zhou D, Zhang L, Li G, Wang X, The autophagic-lysosomal and ubiquitin proteasome systems are simultaneously activated in the skeletal muscle of gastric cancer patients with cachexia, Am. J. Clin. Nutr 111 (3) (2020) 570–579. [DOI] [PubMed] [Google Scholar]
- [38].Zhang G, Liu Z, Ding H, Zhou Y, Doan HA, Sin KWT, Zhu ZJ, Flores R, Wen Y, Gong X, Liu Q, Li YP, Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90, Nat. Commun 8 (2017) 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kunzke T, Buck A, Prade VM, Feuchtinger A, Prokopchuk O, Martignoni ME, Heisz S, Hauner H, Janssen KP, Walch A, Aichler M, Derangements of amino acids in cachectic skeletal muscle are caused by mitochondrial dysfunction, J. Cachexia. Sarcopenia Muscle 11 (2020) 226–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tse MT, Muscle disorders: preventing wastage, Nat. Rev. Drug Discov 9 (2010) 763. [DOI] [PubMed] [Google Scholar]
- [41].Otani T, Minami S, Yamoto M, Umesaki N, Production of activin A in hyperplasia and adenocarcinoma of the human endometrium, Gynecol. Oncol 83 (2001) 31–38. [DOI] [PubMed] [Google Scholar]
- [42].Wildi S, Kleeff J, Maruyama H, Maurer CA, Buchler MW, Korc M, Overexpression of activin A in stage IV colorectal cancer, Gut 49 (2001) 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ, Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size, Am. J. Phys. Cell Physiol 296 (2009) C1258–C1270. [DOI] [PubMed] [Google Scholar]
- [44].Reed SA, Sandesara PB, Senf SM, Judge AR, Inhibition of FoxO transcriptional activity prevents muscle fiber atrophy during cachexia and induces hypertrophy, FASEB J. 26 (2012) 987–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ, Myostatin mutation associated with gross muscle hypertrophy in a child, N. Engl. J. Med 350 (2004) 2682–2688. [DOI] [PubMed] [Google Scholar]
- [46].Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, Stantzou A, Mouisel E, Toniolo L, Ferry A, Strieker S, Goldberg AL, Dupont S, Piccolo S, Amthor H, Sandri M, BMP signaling controls muscle mass, Nat. Genet 45 (2013) 1309–1318. [DOI] [PubMed] [Google Scholar]
- [47].Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, McPherron AC, Wolfman NM, Lee SJ, Induction of cachexia in mice by systemically administered myostatin, Science 296 (2002) 1486–1488. [DOI] [PubMed] [Google Scholar]
- [48].Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, Lacey DL, Goldberg AL, Han HQ, Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival, Cell 142 (2010) 531–543. [DOI] [PubMed] [Google Scholar]
- [49].Lee SJ, McPherron AC, Regulation of myostatin activity and muscle growth, Proc. Natl. Acad. Sci. U. S. A 98 (2001) 9306–9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pettersen K, Andersen S, van der Veen A, Nonstad U, Hatakeyama S, Lambert C, Lach-Trifilieff E, Moestue S, Kim J, Gronberg BH, Schilb A, Jacobi C, Bjorkoy G, Autocrine activin A signaling in ovarian cancer cells regulates secretion of interleukin 6, autophagy, and cachexia, J. Cachexia. Sarcopenia Muscle 11 (1) (2019) 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ruas JL, White JP, Rao RR, Kleiner S, Brannan KT, Harrison BC, Greene NP, Wu J, Estall JL, Irving BA, Lanza IR, Rasbach KA, Okutsu M, Nair KS, Yan Z, Leinwand LA, Spiegelman BM, A PGC-1 alpha isoform induced by resistance training regulates skeletal muscle hypertrophy, Cell 151 (2012) 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fong Y, Moldawer LL, Marano M, Wei H, Barber A, Manogue K, Tracey KJ, Kuo G, Fischman DA, Cerami A, et al. , Cachectin/TNF or IL-1 alpha induces cachexia with redistribution of body proteins, Am. J. Phys 256 (1989) R659–R665. [DOI] [PubMed] [Google Scholar]
- [53].Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS Jr., NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia, Science 289 (2000) 2363–2366. [DOI] [PubMed] [Google Scholar]
- [54].Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, Guttridge DC, Cancer cachexia is regulated by selective targeting of skeletal muscle gene products, J. Clin. Invest 114 (2004) 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hayden MS, Ghosh S, Shared principles in NF-kappaB signaling, Cell 132 (2008) 344–362. [DOI] [PubMed] [Google Scholar]
- [56].Li Q, Verma IM, NF-kappaB regulation in the immune system, Nat. Rev. Immunol 2 (2002) 725–734. [DOI] [PubMed] [Google Scholar]
- [57].Cai D, Frantz JD, Tawa NE Jr., Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE, IKKbeta/NF-kappaB activation causes severe muscle wasting in mice, Cell 119 (2004) 285–298. [DOI] [PubMed] [Google Scholar]
- [58].Kumar A, Takada Y, Boriek AM, Aggarwal BB, Nuclear factor-kappaB: its role in health and disease, J. Mol. Med. (Berl) 82 (2004) 434–448. [DOI] [PubMed] [Google Scholar]
- [59].Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, Koniaris LG, Zimmers TA, JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia, Am. J. Physiol. Endocrinol. Metab 303 (2012) E410–E421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Miller A, McLeod L, Alhayyani S, Szczepny A, Watkins DN, Chen W, Enriori P, Ferlin W, Ruwanpura S, Jenkins BJ, Blockade of the IL-6 trans-signaling/STAT3 axis suppresses cachexia in Kras-induced lung adenocarcinoma, Oncogene 36 (2017) 3059–3066. [DOI] [PubMed] [Google Scholar]
- [61].Guo D, Wang C, Wang Q, Qiao Z, Tang H, Pantoprazole blocks the JAK2/STAT3 pathway to alleviate skeletal muscle wasting in cancer cachexia by inhibiting inflammatory response, Oncotarget 8 (2017) 39640–39648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hurwitz H, Van Cutsem E, Bendell J, Hidalgo M, Li CP, Salvo MG, Macarulla T, Sahai V, Sama A, Greeno E, Yu KH, Verslype C, Dawkins F, Walker C, Clark J, O’Reilly EM, Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies, Investig. New Drugs 36 (2018) 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Price SR, Bailey JL, Wang X, Jurkovitz C, England BK, Ding X, Phillips LS, Mitch WE, Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription, J. Clin. Invest 98 (1996) 1703–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Costelli P, Muscaritoli M, Bossola M, Penna F, Reffo P, Bonetto A, Busquets S, Bonelli G, Lopez-Soriano FJ, Doglietto GB, Argiles JM, Baccino FM, Rossi Fanelli F, IGF-1 is downregulated in experimental cancer cachexia, Am. J. Phys. Regul. Integr. Comp. Phys 291 (2006) R674–R683. [DOI] [PubMed] [Google Scholar]
- [65].Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ, Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways, Nat. Cell Biol 3 (2001) 1009–1013. [DOI] [PubMed] [Google Scholar]
- [66].Bohni R, Riesgo-Escovar J, Oldham S, Brogiolo W, Stocker H, Andruss BF, Beckingham K, Hafen E, Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4, Cell 97 (1999) 865–875. [DOI] [PubMed] [Google Scholar]
- [67].Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, Olefsky JM, Kobayashi M, A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1, Mol. Endocrinol 14 (2000) 783–794. [DOI] [PubMed] [Google Scholar]
- [68].Lee AV, Gooch JL, Oesterreich S, Guler RL, Yee D, Insulin-like growth factor I-induced degradation of insulin receptor substrate 1 is mediated by the 26S proteasome and blocked by phosphatidylinositol 3’-kinase inhibition, Mol. Cell. Biol 20 (2000) 1489–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhande R, Mitchell JJ, Wu J, Sun XJ, Molecular mechanism of insulin-induced degradation of insulin receptor substrate 1, Mol. Cell. Biol 22 (2002) 1016–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lai KM, Gonzalez M, Poueymirou WT, Kline WO, Na E, Zlotchenko E, Stitt TN, Economides AN, Yancopoulos GD, Glass DJ, Conditional activation of akt in adult skeletal muscle induces rapid hypertrophy, Mol. Cell. Biol 24 (2004) 9295–9304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sacheck JM, Ohtsuka A, McLary SC, Goldberg AL, IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1, Am. J. Physiol. Endocrinol. Metab 287 (2004) E591–E601. [DOI] [PubMed] [Google Scholar]
- [72].Rui L, Yuan M, Frantz D, Shoelson S, White MF, SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2, J. Biol. Chem 277 (2002) 42394–42398. [DOI] [PubMed] [Google Scholar]
- [73].Nakao R, Hirasaka K, Goto J, Ishidoh K, Yamada C, Ohno A, Okumura Y, Nonaka I, Yasutomo K, Baldwin KM, Kominami E, Higashibata A, Nagano K, Tanaka K, Yasui N, Mills EM, Takeda S, Nikawa T, Ubiquitin ligase Cbl-b is a negative regulator for insulin-like growth factor 1 signaling during muscle atrophy caused by unloading, Mol. Cell. Biol 29 (2009) 4798–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Song R, Peng W, Zhang Y, Lv F, Wu HK, Guo J, Cao Y, Pi Y, Zhang X, Jin L, Zhang M, Jiang P, Liu F, Meng S, Zhang X, Jiang P, Cao CM, Xiao RP, Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders, Nature 494 (2013) 375–379. [DOI] [PubMed] [Google Scholar]
- [75].Yi JS, Park JS, Ham YM, Nguyen N, Lee NR, Hong J, Kim BW, Lee H, Lee CS, Jeong BC, Song HK, Cho H, Kim YK, Lee JS, Park KS, Shin H, Choi I, Lee SH, Park WJ, Park SY, Choi CS, Lin P, Karunasiri M, Tan T, Duann P, Zhu H, Ma J, Ko YG, MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signaling, Nat. Commun 4 (2013) 2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Rofe AM, Bourgeois CS, Coyle P, Taylor A, Abdi EA, Altered insulin response to glucose in weight-losing cancer patients, Anticancer Res. 14 (1994) 647–650. [PubMed] [Google Scholar]
- [77].Seruga B, Zhang H, Bernstein LJ, Tannock IF, Cytokines and their relationship to the symptoms and outcome of cancer, Nat. Rev. Cancer 8 (2008) 887–899. [DOI] [PubMed] [Google Scholar]
- [78].Bodine SC, Furlow JD, Glucocorticoids and Skeletal Muscle, Adv. Exp. Med. Biol 872 (2015) 145–176. [DOI] [PubMed] [Google Scholar]
- [79].Schakman O, Gilson H, Thissen JP, Mechanisms of glucocorticoid-induced myopathy, J. Endocrinol 197 (2008) 1–10. [DOI] [PubMed] [Google Scholar]
- [80].Schakman O, Kalista S, Barbe C, Loumaye A, Thissen JP, Glucocorticoid-induced skeletal muscle atrophy, Int. J. Biochem. Cell Biol 45 (2013) 2163–2172. [DOI] [PubMed] [Google Scholar]
- [81].Pereira RM, Freire de Carvalho J, Glucocorticoid-induced myopathy, Joint Bone Spine 78 (2011) 41–44. [DOI] [PubMed] [Google Scholar]
- [82].Braun TP, Grossberg AJ, Krasnow SM, Levasseur PR, Szumowski M, Zhu XX, Maxson JE, Knoll JG, Barnes AP, Marks DL, Cancer- and endotoxin-induced cachexia require intact glucocorticoid signaling in skeletal muscle, FASEB J. 27 (2013) 3572–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Llovera M, Garcia-Martinez C, Costelli P, Agell N, Carbo N, Lopez-Soriano FJ, Argiles JM, Muscle hypercatabolism during cancer cachexia is not reversed by the glucocorticoid receptor antagonist RU38486, Cancer Lett. 99 (1996) 7–14. [DOI] [PubMed] [Google Scholar]
- [84].Li YP, Reid MB, NF-kappaB mediates the protein loss induced by TNF-alpha in differentiated skeletal muscle myotubes, Am. J. Phys. Regul. Integr. Comp. Phys 279 (2000) R1165–R1170. [DOI] [PubMed] [Google Scholar]
- [85].Li YP, Schwartz RJ, Waddell ID, Holloway BR, Reid MB, Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-kappaB activation in response to tumor necrosis factor alpha, FASEB J. 12 (1998) 871–880. [DOI] [PubMed] [Google Scholar]
- [86].Oliff A, Defeo-Jones D, Boyer M, Martinez D, Kiefer D, Vuocolo G, Wolfe A, Socher SH, Tumors secreting human TNF/cachectin induce cachexia in mice, Cell 50 (1987) 555–563. [DOI] [PubMed] [Google Scholar]
- [87].Frost RA, Nystrom GJ, Jefferson LS, Lang CH, Hormone, cytokine, and nutritional regulation of sepsis-induced increases in atrogin-1 and MuRF1 in skeletal muscle, Am. J. Physiol. Endocrinol. Metab 292 (2007) E501–E512. [DOI] [PubMed] [Google Scholar]
- [88].Moylan JS, Smith JD, Chambers MA, McLoughlin TJ, Reid MB, TNF induction of atrogin-1/MAFbx mRNA depends on Foxo4 expression but not AKT-Foxo1/3 signaling, Am. J. Phys. Cell Physiol 295 (2008) C986–C993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Sishi BJ, Engelbrecht AM, Tumor necrosis factor alpha (TNF-alpha) inactivates the PI3-kinase/PKB pathway and induces atrophy and apoptosis in L6 myotubes, Cytokine 54 (2011) 173–184. [DOI] [PubMed] [Google Scholar]
- [90].Garcia-Martinez C, Lopez-Soriano FJ, Argiles JM, Acute treatment with tumor necrosis factor-alpha induces changes in protein metabolism in rat skeletal muscle, Mol. Cell. Biochem 125 (1993) 11–18. [DOI] [PubMed] [Google Scholar]
- [91].Noguchi Y, Yoshikawa T, Marat D, Doi C, Makino T, Fukuzawa K, Tsuburaya A, Satoh S, Ito T, Mitsuse S, Insulin resistance in cancer patients is associated with enhanced tumor necrosis factor-alpha expression in skeletal muscle, Biochem. Biophys. Res. Commun 253 (1998) 887–892. [DOI] [PubMed] [Google Scholar]
- [92].Shoelson SE, Lee J, Goldfine AB, Inflammation and insulin resistance, J. Clin. Invest 116 (2006) 1793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Peyta L, Jarnouen K, Pinault M, Coulouarn C, Guimaraes C, Goupille C, de Barros JP, Chevalier S, Dumas JF, Maillot F, Hatch GM, Loyer P, Servais S, Regulation of hepatic cardiolipin metabolism by TNFalpha: Implication in cancer cachexia, Biochim. Biophys. Acta 1851 (2015) 1490–1500. [DOI] [PubMed] [Google Scholar]
- [94].Narsale AA, Carson JA, Role of interleukin-6 in cachexia: therapeutic implications, Curr. Opin. Support Palliat. Care 8 (2014) 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Tsujinaka T, Fujita J, Ebisui C, Yano M, Kominami E, Suzuki K, Tanaka K, Katsume A, Ohsugi Y, Shiozaki H, Monden M, Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice, J. Clin. Invest 97 (1996) 244–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Bayliss TJ, Smith JT, Schuster M, Dragnev KH, Rigas JR, A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer, Expert. Opin. Biol. Ther 11 (2011) 1663–1668. [DOI] [PubMed] [Google Scholar]
- [97].Baltgalvis KA, Berger FG, Pena MM, Davis JM, Muga SJ, Carson JA, Interleukin-6 and cachexia in ApcMin/+ mice, Am. J. Phys. Regul. Integr. Comp. Phys 294 (2008) R393–R401. [DOI] [PubMed] [Google Scholar]
- [98].van Hall G, Steensberg A, Fischer C, Keller C, Moller K, Moseley P, Pedersen BK, Interleukin-6 markedly decreases skeletal muscle protein turnover and increases nonmuscle amino acid utilization in healthy individuals, J. Clin. Endocrinol. Metab 93 (2008) 2851–2858. [DOI] [PubMed] [Google Scholar]
- [99].Baltgalvis KA, Berger FG, Pena MM, Davis JM, White JP, Carson JA, Muscle wasting and interleukin-6-induced atrogin-I expression in the cachectic Ape (Min/+) mouse, Pflugers Arch. 457 (2009) 989–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, Carson JA, The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse, PLoS ONE 6 (2011) e24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Benrick A, Scheie E, Pinnock SB, Wernstedt-Asterholm I, Dickson SL, Karlsson-Lindahl L, Jansson JO, Interleukin-6 gene knockout influences energy balance regulating peptides in the hypothalamic paraventricular and supraoptic nuclei, J. Neuroendocrinol 21 (2009) 620–628. [DOI] [PubMed] [Google Scholar]
- [102].Bossola M, Luciani G, Giungi S, Tazza L, Anorexia, fatigue, and plasma interleukin-6 levels in chronic hemodialysis patients, Ren. Fail 32 (2010) 1049–1054. [DOI] [PubMed] [Google Scholar]
- [103].Callaway CS, Delitto AE, Patel R, Nosacka RL, D’Lugos AC, Delitto D, Deyhle MR, Trevino JG, Judge SM, Judge AR, IL-8 Released from Human Pancreatic Cancer and Tumor-Associated Stromal Cells Signals through a CXCR2-ERK1/2 Axis to Induce Muscle Atrophy, Cancers (Basel) 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Mimeault M, Batra SK, Divergent molecular mechanisms underlying the pleiotropic functions of macrophage inhibitory cytokine-1 in cancer, J. Cell. Physiol 224 (2010) 626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Dostalova I, Kavalkova P, Papezova H, Domluvilova D, Zikan V, Haluzik M, Association of macrophage inhibitory cytokine-1 math nutritional status, body composition and bone mineral density in patients with anorexia nervosa: the influence of partial realimentation, Nutr. Metab. (Lond.) 7 (2010) 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Lerner L, Tao J, Liu Q, Nicoletti R, Feng B, Krieger B, Mazsa E, Siddiquee Z, Wang R, Huang L, Shen L, Lin J, Vigano A, Chiu MI, Weng Z, Winston W, Weiler S, Gyuris J, MAP3K11/GDF15 axis is a critical driver of cancer cachexia, J. Cachexia. Sarcopenia Muscle 7 (2016) 467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Tsai VW, Zhang HP, Manandhar R, Schofield P, Christ D, Lee-Ng KKM, Lebhar H, Marquis CP, Husaini Y, Brown DA, Breit SN, GDF15 mediates adiposity resistance through actions on GFRAL neurons in the hindbrain AP/NTS, Int. J. Obes 43 (12) (2019) 2370–2380. [DOI] [PubMed] [Google Scholar]
- [108].Emmerson PJ, Wang F, Du Y, Liu Q, Pickard RT, Gonciarz MD, Coskun T, Hamang MJ, Sindelar DK, Ballman KK, Foltz LA, Muppidi A, Alsina-Fernandez J, Barnard GC, Tang JX, Liu X, Mao X, Siegel R, Sloan JH, Mitchell PJ, Zhang BB, Gimeno RE, Shan B, Wu X, The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL, Nat. Med 23 (2017) 1215–1219. [DOI] [PubMed] [Google Scholar]
- [109].Hsu JY, Cramdey S, Chen M, Ayupova DA, Lindhout DA, Higbee J, Kutach A, Joo W, Gao Z, Fu D, To C, Mondal K, Li B, Kekatpure A, Wang M, Laird T, Horner G, Chan J, McEntee M, Lopez M, Lakshminarasimhan D, White A, Wang SP, Yao J, Yie J, Matern H, Solloway M, Haldankar R, Parsons T, Tang J, Shen WD, Alice Chen Y, Tian H, Allan BB, Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15, Nature 550 (2017) 255–259. [DOI] [PubMed] [Google Scholar]
- [110].Mullican SE, Lin-Schmidt X, Chin CN, Chavez JA, Furman JL, Armstrong AA, Beck SC, South VJ, Dinh TQ, Cash-Mason TD, Cavanaugh CR, Nelson S, Huang C, Hunter MJ, Rangwala SM, GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates, Nat. Med 23 (2017) 1150–1157. [DOI] [PubMed] [Google Scholar]
- [111].Yang L, Chang CC, Sun Z, Madsen D, Zhu H, Padkjaer SB, Wu X, Huang T, Hultman K, Paulsen SJ, Wang J, Bugge A, Frantzen JB, Norgaard P, Jeppesen JF, Yang Z, Secher A, Chen H, Li X, John LM, Shan B, He Z, Gao X, Su J, Hansen KT, Yang W, Jorgensen SB, GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand, Nat. Med 23 (2017) 1158–1166. [DOI] [PubMed] [Google Scholar]
- [112].Mullican SE, Rangwala SM, Uniting GDF15 and GFRAL: therapeutic opportunities in Obesity and Beyond, Trends Endocrinol. Metab 29 (2018) 560–570. [DOI] [PubMed] [Google Scholar]
- [113].Loumaye A, de Barsy M, Nachit M, Lause P, van Maanen A, Trefois P, Gruson D, Thissen JP, Circulating Activin A predicts survival in cancer patients, J. Cachexia. Sarcopenia Muscle 8 (2017) 768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Loumaye A, Thissen JP, Biomarkers of cancer cachexia, Clin. Biochem 50 (2017) 1281–1288. [DOI] [PubMed] [Google Scholar]
- [115].Parsons SA, Millay DP, Sargent MA, McNally EM, Molkentin JD, Age-dependent effect of myostatin blockade on disease severity in a murine model of limb-girdle muscular dystrophy, Am. J. Pathol 168 (2006) 1975–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Hammers DW, Merscham-Banda M, Hsiao JY, Engst S, Hartman JJ, Sweeney HL, Supraphysiological levels of GDF11 induce striated muscle atrophy, EMBO Mol. Med 9 (2017) 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Harper SC, Johnson J, Borghetti G, Zhao H, Wang T, Wallner M, Kubo H, Feldsott EA, Yang Y, Joo Y, Gou X, Sabri AK, Gupta P, Myzithras M, Khalil A, Franti M, Houser SR, GDF11 Decreases Pressure Overload-Induced Hypertrophy, but Can Cause Severe Cachexia and Premature Death, Circ. Res 123 (2018) 1220–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Martin TJ, Osteoblast-derived PTHrP is a physiological regulator of bone formation, J. Clin. Invest 115 (2005) 2322–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, Spiegelman BM, Tumor-derived PTH-related protein triggers adipose tissue browning and cancer cachexia, Nature 513 (2014) 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Kir S, Komaba H, Garcia AP, Economopoulos KP, Liu W, Lanske B, Hodin RA, Spiegelman BM, PTH/PTHrP Receptor Mediates Cachexia in Models of Kidney Failure and Cancer, Cell Metab. 23 (2016) 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Thomas SS, Mitch WE, Parathyroid hormone stimulates adipose tissue browning: a pathway to muscle wasting, Curr. Opin. Clin. Nutr. Metab. Care 20 (2017) 153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Agustsson T, Ryden M, Hoffstedt J, van Harmelen V, Dicker A, Laurencikiene J, Isaksson B, Permert J, Arner P, Mechanism of increased lipolysis in cancer cachexia, Cancer Res. 67 (2007) 5531–5537. [DOI] [PubMed] [Google Scholar]
- [123].Uversky VN, El-Baky NA, El-Fakharany EM, Sabry A, Mattar EH, Uversky AV, Redwan EM, Functionality of intrinsic disorder in tumor necrosis factor-alpha and its receptors, FEBS J. 284 (2017) 3589–3618. [DOI] [PubMed] [Google Scholar]
- [124].Choe SS, Huh JY, Hwang IJ, Kim JI, Kim JB, Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders, Front. Endocrinol. (Lausanne) 7 (2016) 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Jeanson Y, Carriere A, Casteilla L, A New Role for Browning as a Redox and Stress Adaptive Mechanism? Front. Endocrinol. (Lausanne) 6 (2015) 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Petruzzelli M, Wagner EF, Mechanisms of metabolic dysfunction in cancer-associated cachexia, Genes Dev. 30 (2016) 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Honors MA, Kinzig KP, The role of insulin resistance in the development of muscle wasting during cancer cachexia, J. Cachexia. Sarcopenia Muscle 3 (2012) 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Scott HR, McMillan DC, Crilly A, McArdle CS, Milroy R, The relationship between weight loss and interleukin 6 in non-small-cell lung cancer, Br. J. Cancer 73 (1996) 1560–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Moses AG, Maingay J, Sangster K, Fearon KC, Ross JA, Pro-inflammatory cytokine release by peripheral blood mononuclear cells from patients with advanced pancreatic cancer: relationship to acute phase response and survival, Oncol. Rep 21 (2009) 1091–1095. [DOI] [PubMed] [Google Scholar]
- [130].Han J, Meng Q, Shen L, Wu G, Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning, Lipids Health Dis. 17 (2018) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L, Interleukin-6-type cytokine signaling through the gp130/Jak/STAT pathway, Biochem. J 334 (Pt 2) (1998) 297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Arora GK, Gupta A, Narayanan S, Guo T, Iyengar P, Infante RE, Cachexia-associated adipose loss induced by tumor-secreted leukemia inhibitory factor is counterbalanced by decreased leptin, JCI Insight 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Ebadi M, Mazurak VC, Potential Biomarkers of Fat Loss as a Feature of Cancer Cachexia, Mediat. Inflamm 2015 (2015) 820934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Pothuraju R, Rachagani S, Junker WM, Chaudhary S, Saraswathi V, Kaur S, Batra SK, Pancreatic cancer associated with obesity and diabetes: an alternative approach for its targeting, J. Exp. Clin. Cancer Res 37 (2018) 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Daas SI, Rizeq BR, Nasrallah GK, Adipose tissue dysfunction in cancer cachexia, J. Cell. Physiol 234 (2018) 13–22. [DOI] [PubMed] [Google Scholar]
- [136].Wu J, Cohen P, Spiegelman BM, Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev. 27 (2013) 234–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Harms M, Seale P, Brown and beige fat: development, function and therapeutic potential, Nat. Med 19 (2013) 1252–1263. [DOI] [PubMed] [Google Scholar]
- [138].Loncar D, Bedrica L, Mayer J, Cannon B, Nedergaard J, Afzelius BA, Svajger A, The effect of intermittent cold treatment on the adipose tissue of the cat. Apparent transformation from white to brown adipose tissue, J. Ultrastruct. Mol. Struct. Res 97 (1986) 119–129. [DOI] [PubMed] [Google Scholar]
- [139].Cao L, Choi EY, Liu X, Martin A, Wang C, Xu X, During MJ, White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamic-adipocyte axis, Cell Metab. 14 (2011) 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Vegiopoulos A, Muller-Decker K, Strzoda D, Schmitt I, Chichelnitskiy E, Ostertag A, Berriel Diaz M, Rozman J, Hrabe de Angelis M, Nusing RM, Meyer CW, Wahli W, Klingenspor M, Herzig S, Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes, Science 328 (2010) 1158–1161. [DOI] [PubMed] [Google Scholar]
- [141].Yoneshiro T, Aita S, Matsushita M, Kayahara T, Kameya T, Kawai Y, Iwanaga T, Saito M, Recruited brown adipose tissue as an antiobesity agent in humans, J. Clin. Invest 123 (2013) 3404–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J, Swarbrick M, Rose-John S, Rincon M, Robertson G, Zechner R, Wagner EF, A switch from white to brown fat increases energy expenditure in cancer-associated cachexia, Cell Metab. 20 (2014) 433–447. [DOI] [PubMed] [Google Scholar]
- [143].Janik JE, Curti BD, Considine RV, Rager HC, Powers GC, Alvord WG, Smith JW 2nd, Gause BL, Kopp WC, Interleukin 1 alpha increases serum leptin concentrations in humans, J. Clin. Endocrinol. Metab 82 (1997) 3084–3086. [DOI] [PubMed] [Google Scholar]
- [144].Sarraf P, Frederich RC, Turner EM, Ma G, Jaskowiak NT, Rivet DJ 3rd, Flier JS, Lowell BB, Fraker DL, Alexander HR, Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in inflammatory anorexia, J. Exp. Med 185 (1997) 171–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Tuca A, Jimenez-Fonseca P, Gascon P, Clinical evaluation and optimal management of cancer cachexia, Crit. Rev. Oncol. Hematol 88 (2013) 625–636. [DOI] [PubMed] [Google Scholar]
- [146].Marks DL, Butler AA, Turner R, Brookhart G, Cone RD, Differential role of melanocortin receptor subtypes in cachexia, Endocrinology 144 (2003) 1513–1523. [DOI] [PubMed] [Google Scholar]
- [147].Scarlett JM, Jobst EE, Enriori PJ, Bowe DD, Batra AK, Grant WF, Cowley MA, Marks DL, Regulation of central melanocortin signaling by interleukin-1 beta, Endocrinology 148 (2007) 4217–4225. [DOI] [PubMed] [Google Scholar]
- [148].Wisse BE, Frayo RS, Schwartz MW, Cummings DE, Reversal of cancer anorexia by blockade of central melanocortin receptors in rats, Endocrinology 142 (2001) 3292–3301. [DOI] [PubMed] [Google Scholar]
- [149].Heisler LK, Cowley MA, Tecott LH, Fan W, Low MJ, Smart JL, Rubinstein M, Tatro JB, Marcus JN, Holstege H, Lee CE, Cone RD, Elmquist JK, Activation of central melanocortin pathways by fenfluramine, Science 297 (2002) 609–611. [DOI] [PubMed] [Google Scholar]
- [150].Friesen DE, Baracos VE, Tuszynski JA, Modeling the energetic cost of cancer as a result of altered energy metabolism: implications for cachexia, Theor. Biol. Med. Model 12 (2015) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Narsale AA, Enos RT, Puppa MJ, Chatterjee S, Murphy EA, Fayad R, Pena MO, Durstine JL, Carson JA, Liver inflammation and metabolic signaling in ApcMin/+ mice: the role of cachexia progression, PLoS ONE 10 (2015) e0119888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Martignoni ME, Dimitriu C, Bachmann J, Krakowski-Rosen H, Ketterer K, Kinscherf R, Friess H, Liver macrophages contribute to pancreatic cancer-related cachexia, Oncol. Rep 21 (2009) 363–369. [PubMed] [Google Scholar]
- [153].Castell JV, Gomez-Lechon MJ, David M, Andus T, Geiger T, Trullenque R, Fabra R, Heinrich PC, Interleukin-6 is the major regulator of acute phase protein synthesis in adult human hepatocytes, FEBS Lett. 242 (1989) 237–239. [DOI] [PubMed] [Google Scholar]
- [154].Bhanji RA, Narayanan P, Allen AM, Malhi H, Watt KD, Sarcopenia in hiding: The risk and consequence of underestimating muscle dysfunction in nonalcoholic steatohepatitis, Hepatology 66 (2017) 2055–2065. [DOI] [PubMed] [Google Scholar]
- [155].Berriel Diaz M, Krones-Herzig A, Metzger D, Ziegler A, Vegiopoulos A, Klingenspor M, Muller-Decker K, Herzig S, Nuclear receptor cofactor receptor interacting protein 140 controls hepatic triglyceride metabolism during wasting in mice, Hepatology 48 (2008) 782–791. [DOI] [PubMed] [Google Scholar]
- [156].Jones A, Friedrich K, Rohm M, Schafer M, Algire C, Kulozik P, Seibert O, Muller-Decker K, Sijmonsma T, Strzoda D, Sticht C, Gretz N, Dallinga-Thie GM, Leuchs B, Kogl M, Stremmel W, Diaz MB, Herzig S, TSC22D4 is a molecular output of hepatic wasting metabolism, EMBO Mol. Med 5 (2013) 294–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Rosa-Caldwell ME, Brown JL, Lee DE, Wiggs MP, Perry RA Jr., Haynie WS, Caldwell AR, Washington TA, Lo WJ, Greene NP, Hepatic alterations during the development and progression of cancer cachexia, Appl. Physiol. Nutr. Metab (2019), 10.1139/apnm-2019-0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Asp ML, Tian M, Wendel AA, Belury MA, Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice, Int. J. Cancer 126 (2010) 756–763. [DOI] [PubMed] [Google Scholar]
- [159].Fernandes LC, Machado UF, Nogueira CR, Carpinelli AR, Curi R, Insulin secretion in Walker 256 tumor cachexia, Am. J. Phys 258 (1990) E1033–E1036. [DOI] [PubMed] [Google Scholar]
- [160].el Razi Neto S, Zorn TM, Curi R, Carpinelli AR, Impairment of insulin secretion in pancreatic islets isolated from Walker 256 tumor-bearing rats, Am. J. Phys 271 (1996) C804–C809. [DOI] [PubMed] [Google Scholar]
- [161].Lundholm K, Korner U, Gunnebo L, Sixt-Ammilon P, Fouladiun M, Daneryd P, Bosaeus I, Insulin treatment in cancer cachexia: effects on survival, metabolism, and physical functioning, Clin. Cancer Res 13 (2007) 2699–2706. [DOI] [PubMed] [Google Scholar]
- [162].Rohm M, Zeigerer A, Machado J, Herzig S, Energy metabolism in cachexia, EMBO Rep, 20, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Puppa MJ, White JP, Sato S, Cairns M, Baynes JW, Carson JA, Gut barrier dysfunction in the Apc(Min/+) mouse model of colon cancer cachexia, Biochim. Biophys. Acta 1812 (2011) 1601–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Bindels LB, Neyrinck AM, Loumaye A, Catry E, Walgrave H, Cherbuy C, Leclercq S, Van Hul M, Plovier H, Pachikian B, Bermudez-Humaran LG, Langella P, Cani PD, Thissen JP, Delzenne NM, Increased gut permeability in cancer cachexia: mechanisms and clinical relevance, Oncotarget 9 (2018) 18224–18238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Shimizu Y, Nagaya N, Isobe T, Imazu M, Okumura H, Hosoda H, Kojima M, Kangawa K, Kohno N, Increased plasma ghrelin level in lung cancer cachexia, Clin. Cancer Res. 9 (2003) 774–778. [PubMed] [Google Scholar]
- [166].Lin TC, Hsiao M, Ghrelin and cancer progression, Biochim. Biophys. Acta Rev. Cancer 1868 (2017) 51–57. [DOI] [PubMed] [Google Scholar]
- [167].Esposito A, Criscitiello C, Gelao L, Pravettoni G, Locatelli M, Minchella I, Di Leo M, Liuzzi R, Milani A, Massaro M, Curigliano G, Mechanisms of anorexia-cachexia syndrome and rational for treatment with selective ghrelin receptor agonist, Cancer Treat. Rev 41 (2015) 793–797. [DOI] [PubMed] [Google Scholar]
- [168].Dixit VD, Schaffer EM, Pyle RS, Collins GD, Sakthivel SK, Palaniappan R, Lillard JW Jr., Taub DD, Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells, J. Clin. Invest 114 (2004) 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Chen JA, Splenser A, Guillory B, Luo J, Mendiratta M, Belinova B, Halder T, Zhang G, Li YP, Garcia JM, Ghrelin prevents tumor- and cisplatin-induced muscle wasting: characterization of multiple mechanisms involved, J. Cachexia. Sarcopenia Muscle 6 (2015) 132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Porporato PE, Filigheddu N, Reano S, Ferrara M, Angelino E, Gnocchi VF, Prodam F, Ronchi G, Fagoonee S, Fornaro M, Chianale F, Baldanzi G, Surico N, Sinigaglia F, Perroteau I, Smith RG, Sun Y, Geuna S, Graziani A, Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice, J. Clin. Invest 123 (2013) 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Bindels LB, Neyrinck AM, Claus SP, Le Roy CI, Grangette C, Pot B, Martinez I, Walter J, Cani PD, Delzenne NM, Synbiotic approach restores intestinal homeostasis and prolongs survival in leukaemic mice with cachexia, ISME J. 10 (2016) 1456–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Bindels LB, Delzenne NM, Muscle wasting: the gut microbiota as a new therapeutic target? Int. J. Biochem. Cell Biol 45 (2013) 2186–2190. [DOI] [PubMed] [Google Scholar]
- [173].Delzenne NM, Neyrinck AM, Backhed F, Cani PD, Targeting gut micro biota in obesity: effects of prebiotics and probiotics, Nat. Rev. Endocrinol 7 (2011) 639–646. [DOI] [PubMed] [Google Scholar]
- [174].Bren-Mattison Y, Hausburg M, Olwin BB, Growth of limb muscle is dependent on skeletal-derived Indian hedgehog, Dev. Biol 356 (2011) 486–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Rauch F, Bailey DA, Baxter-Jones A, Mirwald R, Faulkner R, The ‘muscle-bone unit’ during the pubertal growth spurt, Bone 34 (2004) 771–775. [DOI] [PubMed] [Google Scholar]
- [176].DiGirolamo DJ, Kiel DP, Esser KA, Bone and skeletal muscle: neighbors with close ties, J. Bone Miner. Res 28 (2013) 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Liang H, Pun S, Wronski TJ, Bone anabolic effects of basic fibroblast growth factor in ovariectomized rats, Endocrinology 140 (1999) 5780–5788. [DOI] [PubMed] [Google Scholar]
- [178].Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL, Ooi GT, Setser J, Frystyk J, Boisclair YR, LeRoith D, Circulating levels of IGF-1 directly regulate bone growth and density, J. Clin. Invest 110 (2002) 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Hamrick MW, Samaddar T, Pennington C, McCormick J, Increased muscle mass with myostatin deficiency improves gains in bone strength with exercise, J. Bone Miner. Res 21 (2006) 477–483. [DOI] [PubMed] [Google Scholar]
- [180].McFarlane C, Plummer E, Thomas M, Hennebry A, Ashby M, Ling N, Smith H, Sharma M, Kambadur R, Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-kappaB-independent, FoxO1-dependent mechanism, J. Cell. Physiol 209 (2006) 501–514. [DOI] [PubMed] [Google Scholar]
- [181].Waning DL, Guise TA, Cancer-associated muscle weakness: What’s bone got to do with it? Bonekey Rep. 4 (2015) 691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S, Chiechi A, Wright LE, Umanskaya A, Niewolna M, Trivedi T, Charkhzarrin S, Khatiwada P, Wronska A, Haynes A, Benassi MS, Witzmann FA, Zhen G, Wang X, Cao X, Roodman GD, Marks AR, Guise TA, Excess TGF-beta mediates muscle weakness associated with bone metastases in mice, Nat. Med 21 (2015) 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Hesse E, Schroder S, Brandt D, Pamperin J, Saito H, Taipaleenmaki H, Sclerostin inhibition alleviates breast cancer-induced bone metastases and muscle weakness, JCI Insight 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Manne ND, Lima M, Enos RT, Wehner P, Carson JA, Blough E, Altered cardiac muscle mTOR regulation during the progression of cancer cachexia in the ApcMin/+ mouse, Int. J. Oncol 42 (2013) 2134–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Yusuf SW, Razeghi P, Yeh ET, The diagnosis and management of cardiovascular disease in cancer patients, Curr. Probl. Cardiol 33 (2008) 163–196. [DOI] [PubMed] [Google Scholar]
- [186].Imoto A, Mitsunaga S, Inagaki M, Aoyagi K, Sasaki H, Ikeda M, Nakachi K, Higuchi K, Ochiai A, Neural invasion induces cachexia via astrocytic activation of neural route in pancreatic cancer, Int. J. Cancer 131 (2012) 2795–2807. [DOI] [PubMed] [Google Scholar]
- [187].Boehm I, Miller J, Wishart TM, Wigmore SJ, Skipworth RJ, Jones RA, Gillingwater TH, Neuromuscular junctions are stable in patients with cancer cachexia, J. Clin. Invest. 130 (3) (2020) 1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Baldwin C, Spiro A, Ahern R, Emery PW, Oral nutritional interventions in malnourished patients with cancer: a systematic review and meta-analysis, J. Natl. Cancer Inst 104 (2012) 371–385. [DOI] [PubMed] [Google Scholar]
- [189].Beck SA, Smith KL, Tisdale MJ, Anticachectic and antitumor effect of eicosapentaenoic acid and its effect on protein turnover, Cancer Res. 51 (1991) 6089–6093. [PubMed] [Google Scholar]
- [190].Wigmore SJ, Fearon KC, Maingay JP, Ross JA, Down-regulation of the acute-phase response in patients with pancreatic cancer cachexia receiving oral eicosapentaenoic acid is mediated via suppression of interleukin-6, Clin. Sci. (Lond.) 92 (1997) 215–221. [DOI] [PubMed] [Google Scholar]
- [191].Murphy RA, Mourtzakis M, Chu QS, Baracos VE, Reiman T, Mazurak VC, Nutritional intervention with fish oil provides a benefit over standard of care for weight and skeletal muscle mass in patients with nonsmall cell lung cancer receiving chemotherapy, Cancer 117 (2011) 1775–1782. [DOI] [PubMed] [Google Scholar]
- [192].Von Roenn JH, Randomized trials of megestrol acetate for AIDS-associated anorexia and cachexia, Oncology 51 (Suppl. 1) (1994) 19–24. [DOI] [PubMed] [Google Scholar]
- [193].Ruiz Garcia V, Lopez-Briz E, Carbonell Sanchis R, Gonzalvez Perales JL, Bort-Marti S, Megestrol acetate for treatment of anorexia-cachexia syndrome, Cochrane Database Syst. Rev (2013) CD004310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Argiles JM, Anguera A, Stemmier B, A new look at an old drug for the treatment of cancer cachexia: megestrol acetate, Clin. Nutr 32 (2013) 319–324. [DOI] [PubMed] [Google Scholar]
- [195].Tchekmedyian NS, Tait N, Moody M, Aisner J, High-dose megestrol acetate. A possible treatment for cachexia, JAMA 257 (1987) 1195–1198. [PubMed] [Google Scholar]
- [196].Maltoni M, Nanni O, Scarpi E, Rossi D, Serra P, Amadori D, High-dose progestins for the treatment of cancer anorexia-cachexia syndrome: a systematic review of randomized clinical trials, Ann. Oncol 12 (2001) 289–300. [DOI] [PubMed] [Google Scholar]
- [197].Feliu J, Gonzalez-Baron M, Berrocal A, Artal A, Ordonez A, Garrido P, Zamora P, Garcia de Paredes ML, Montero JM, Usefulness of megestrol acetate in cancer cachexia and anorexia. A placebo-controlled study, Am. J. Clin. Oncol 15 (1992) 436–440. [DOI] [PubMed] [Google Scholar]
- [198].Lesniak W, Bala M, Jaeschke R, Krzakowski M, Effects of megestrol acetate in patients with cancer anorexia-cachexia syndrome–a systematic review and meta-analysis, Pol. Arch. Med. Wewn 118 (2008) 636–644. [PubMed] [Google Scholar]
- [199].Schmid I, Stachel DK, Freudenberg S, Schmitt M, Schuster F, Haas RJ, Megestrol acetate to correct the nutritional status in an adolescent with growth hormone deficiency: Increase of appetite and body weight but only by increase of body water and fat mass followed by profound cortisol and testosterone depletion, Klin. Padiatr 214 (2002) 54–57. [DOI] [PubMed] [Google Scholar]
- [200].Musolino V, Palus S, Tschirner A, Drescher C, Gliozzi M, Carresi C, Vitale C, Muscoli C, Doehner W, von Haehling S, Anker SD, Mollace V, Springer J, Megestrol acetate improves cardiac function in a model of cancer cachexia-induced cardiomyopathy by autophagic modulation, J. Cachexia. Sarcopenia Muscle 7 (2016) 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Koller E, Gibert C, Green L, Mann M, Bernstein B, Thrombotic events associated with megestrol acetate in patients with AIDS cachexia, Nutrition 15 (1999) 294–298. [DOI] [PubMed] [Google Scholar]
- [202].Thomas DR, Incidence of venous thromboembolism in megestrol acetate users, J. Am. Med. Dir. Assoc 5 (2004) 65–66 author reply 66–67. [DOI] [PubMed] [Google Scholar]
- [203].Cannabis GIn Cachexia Study, Strasser F, Luftner D, Possinger K, Ernst G, Ruhstaller T, Meissner W, Ko YD, Schnelle M, Reif M, Cerny T, Comparison of orally administered cannabis extract and delta-9-tetrahydrocannabinol in treating patients with cancer-related anorexia-cachexia syndrome: a multicenter, phase III, randomized, double-blind, placebo-controlled clinical trial from the Cannabis-In-Cachexia-Study-Group, J. Clin. Oncol 24 (2006) 3394–3400. [DOI] [PubMed] [Google Scholar]
- [204].Jatoi A, Windschitl HE, Loprinzi CL, Sloan JA, Dakhil SR, Mailliard JA, Pundaleeka S, Kardinal CG, Fitch TR, Krook JE, Novotny PJ, Christensen B, Dronabinol versus megestrol acetate versus combination therapy for cancer-associated anorexia: a North Central Cancer Treatment Group study, J. Clin. Oncol 20 (2002) 567–573. [DOI] [PubMed] [Google Scholar]
- [205].Loprinzi CL, Kugler JW, Sloan JA, Mailliard JA, Krook JE, Wilwerding MB, Rowland KM Jr., Camoriano JK, Novotny PJ, Christensen BJ, Randomized comparison of megestrol acetate versus dexamethasone versus fluoxymesterone for the treatment of cancer anorexia/cachexia, J. Clin. Oncol 17 (1999) 3299–3306. [DOI] [PubMed] [Google Scholar]
- [206].Yennurajalingam S, Williams JL, Chisholm G, Bruera E, Effects of Dexamethasone and Placebo on Symptom Clusters in Advanced Cancer Patients: A Preliminary Report, Oncologist 21 (2016) 384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Chasen M, Hirschman SZ, Bhargava R, Phase II study of the novel peptide-nucleic acid OHR118 in the management of cancer-related anorexia/cachexia, J. Am. Med. Dir. Assoc 12 (2011) 62–67. [DOI] [PubMed] [Google Scholar]
- [208].Asakawa A, Inui A, Kaga T, Yuzuriha H, Nagata T, Ueno N, Makino S, Fujimiya M, Niijima A, Fujino MA, Kasuga M, Ghrelin is an appetite-stimulatory signal from stomach with structural resemblance to motilin, Gastroenterology 120 (2001) 337–345. [DOI] [PubMed] [Google Scholar]
- [209].Molfino A, Gioia G, Muscaritoli M, The hunger hormone ghrelin in cachexia, Expert. Opin. Biol. Ther 13 (2013) 465–468. [DOI] [PubMed] [Google Scholar]
- [210].Pietra C, Takeda Y, Tazawa-Ogata N, Minami M, Yuanfeng X, Duus EM, Northrup R, Anamorelin HC1 (ONO-7643), a novel ghrelin receptor agonist, for the treatment of cancer anorexia-cachexia syndrome: preclinical profile, J. Cachexia. Sarcopenia Muscle 5 (2014) 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Hamauchi S, Furuse J, Takano T, Munemoto Y, Furuya K, Baba H, Takeuchi M, Choda Y, Higashiguchi T, Naito T, Muro K, Takayama K, Oyama S, Takiguchi T, Komura N, Tamura K, A multicenter, open-label, singlearm study of anamorelin (ONO-7643) in advanced gastrointestinal cancer patients with cancer cachexia, Cancer 125 (23) (2019) 4294–4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Chowdhry SM, Chowdhry VK, Cancer cachexia and treatment toxicity, Curr. Opin. Support Palliat. Care 13 (4) (2019) 292–297. [DOI] [PubMed] [Google Scholar]
- [213].Brocco D, Di Marino P, Grassandonia A, From cachexia to obesity: the role of host metabolism in cancer immunotherapy, Curr. Opin. Support Palliat. Care 13 (4) (2019) 305–310. [DOI] [PubMed] [Google Scholar]
- [214].Klein S, Kinney J, Jeejeebhoy K, Alpers D, Hellerstein M, Murray M, Twomey P, Nutrition support in clinical practice: review of published data and recommendations for future research directions. Summary of a conference sponsored by the National Institutes of Health, American Society for Parenteral and Enteral Nutrition, and American Society for Clinical Nutrition, Am. J. Clin. Nutr 66 (1997) 683–706. [DOI] [PubMed] [Google Scholar]
- [215].Johnston AJ, Murphy KT, Jenkinson L, Laine D, Emmrich K, Faou P, Weston R, Jayatilleke KM, Schloegel J, Talbo G, Casey JL, Levina V, Wong WW, Dillon H, Sahay T, Hoogenraad J, Anderton H, Hall C, Schneider P, Tanzer M, Foley M, Scott AM, Gregorevic P, Liu SY, Burkly LC, Lynch GS, Silke J, Hoogenraad NJ, Targeting of Fn14 Prevents Cancer-Induced Cachexia and Prolongs Survival, Cell 162 (2015) 1365–1378. [DOI] [PubMed] [Google Scholar]
- [216].Wiedenmann B, Malfertheiner P, Friess H, Ritch P, Arseneau J, Mantovani G, Caprioni F, Van Cutsem E, Richel D, DeWitte M, Qi M, Robinson D Jr., Zhong B, De Boer C, Lu JD, Prabhakar U, Corringham R, Von Hoff D, A multicenter, phase II study of infliximab plus gemcitabine in pancreatic cancer cachexia, J. Support Oncol 6 (2008) 18–25. [PubMed] [Google Scholar]
- [217].Rigas J, Schuster M, Orlov S, Milovanovic B, Prabhash K, Smith J, A.s. group, Efect of ALD518, a humanized anti-IL-6 antibody, on lean body mass loss and symptoms in patients with advanced non-small cell lung cancer (NSCLC): Results of a phase II randomized, double-blind safety and efficacy trial, J. Clin. Oncol 28 (2010) 7622, 10.1200/jco.2010.28.15_suppl.7622. [DOI] [Google Scholar]
- [218].Clarke S, Smith J, Gebbie C, Sweeney C, Olszewski N, A phase I, pharmacokinetic (PK), and preliminary efficacy assessment of ALD518, a humanized anti-IL-6 antibody, in patients with advanced cancer, J. Clin. Oncol 27 (2009) 3025. [Google Scholar]
- [219].Hong DS, Hui D, Bruera E, Janku F, Naing A, Falchook GS, Piha-Paul S, Wheler JJ, Fu S, Tsimberidou AM, Stecher M, Mohanty P, Simard J, Kurzrock R, MABp1, a first-in-class true human antibody targeting interleukin-1alpha in refractory cancers: an open-label, phase 1 dose-escalation and expansion study, Lancet Oncol. 15 (2014) 656–666. [DOI] [PubMed] [Google Scholar]
- [220].Dinarello CA, Interleukin-1 alpha neutralisation in patients with cancer, Lancet Oncol. 15 (2014) 552–553. [DOI] [PubMed] [Google Scholar]
- [221].Lundholm K, Daneryd P, Korner U, Hyltander A, Bosaeus I, Evidence that long-term COX-treatment improves energy homeostasis and body composition in cancer patients with progressive cachexia, Int. J. Oncol 24 (2004) 505–512. [PubMed] [Google Scholar]
- [222].Tseng YC, Kulp SK, Lai IL, Hsu EC, He WA, Frankhouser DE, Yan PS, Mo X, Bloomston M, Lesinski GB, Marcucci G, Guttridge DC, Bekaii-Saab T, Chen CS, Preclinical Investigation of the Novel Histone Deacetylase Inhibitor AR-42 in the Treatment of Cancer-Induced Cachexia, J. Natl. Cancer Inst 107 (2015) djv274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Dalton JT, Barnette KG, Bohl CE, Hancock ML, Rodriguez D, Dodson ST, Morton RA, Steiner MS, The selective androgen receptor modulator GTx-024 (enobosarm) improves lean body mass and physical function in healthy elderly men and postmenopausal women: results of a double-blind, placebo-controlled phase II trial, J. Cachexia. Sarcopenia Muscle 2 (2011) 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Liva SG, Tseng YC, Dauki AM, Sovic MG, Vu T, Henderson SE, Kuo YC, Benedict JA, Zhang X, Remaily BC, Kulp SK, Campbell M, Bekaii-Saab T, Phelps MA, Chen CS, Coss CC, Overcoming resistance to anabolic SARM therapy in experimental cancer cachexia with an HDAC inhibitor, EMBO Mol. Med. 12 (2) (2020) e9910, 10.15252/emmm.201809910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Oliveira AG, Gomes-Marcondes MC, Metformin treatment modulates the tumor-induced wasting effects in muscle protein metabolism minimising the cachexia in tumor-bearing rats, BMC Cancer 16 (2016) 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Behl D, Jatoi A, Pharmacological options for advanced cancer patients with loss of appetite and weight, Expert. Opin. Pharmacother. 8 (2007) 1085–1090. [DOI] [PubMed] [Google Scholar]
- [227].Pin F, Barreto R, Couch ME, Bonetto A, O’Connell TM, Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism, J. Cachexia. Sarcopenia Muscle 10 (2019) 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Mantovani G, Maccio A, Madeddu C, Gramignano G, Lusso MR, Serpe R, Massa E, Astara G, Deiana L, A phase II study with antioxidants, both in the diet and supplemented, pharmaconutritional support, progestagen, and anti-cyclooxygenase-2 showing efficacy and safety in patients with cancer-related anorexia/cachexia and oxidative stress, Cancer Epidemiol. Biomark. Prev 15 (2006) 1030–1034. [DOI] [PubMed] [Google Scholar]
- [229].Morvan F, Rondeau JM, Zou C, Minetti G, Scheufler C, Scharenberg M, Jacobi C, Brebbia P, Ritter V, Toussaint G, Koelbing C, Leber X, Schilb A, Witte F, Lehmann S, Koch E, Geisse S, Glass DJ, Lach-Trifilieff E, Blockade of activin type II receptors with a dual anti-ActRIIA/IIB antibody is critical to promote maximal skeletal muscle hypertrophy, Proc. Natl. Acad. Sci. U. S. A 114 (2017) 12448–12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Tanaka H, Wada M, Park J, HASPIN kinase inhibitor CHR-6494 suppresses intestinal polyp development, cachexia, and hypogonadism in Apcmin/+ mice, Eur. J. Cancer Prev (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Straughn AR, Kakar SS, Withaferin A ameliorates ovarian cancer-induced cachexia and proinflammatory signaling, J. Ovarian Res. 12 (2019) 115, 10.1097/CEJ.0000000000000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Michaelis KA, Norgard MA, Zhu X, Levasseur PR, Sivagnanam S, Liudahl SM, Burfeind KG, Olson B, Pelz KR, Angeles Ramos DM, Maurer HC, Olive KP, Coussens LM, Morgan TK, Marks DL, The TLR7/8 agonist R848 remodels tumor and host responses to promote survival in pancreatic cancer, Nat. Commun 10 (2019) 4682. [DOI] [PMC free article] [PubMed] [Google Scholar]