

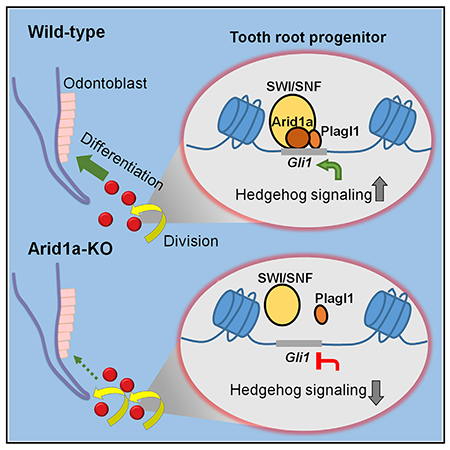
SUMMARY
Chromatin remodelers often show broad expression patterns in multiple cell types yet can elicit cell-specific effects in development and diseases. Arid1a binds DNA and regulates gene expression during tissue development and homeostasis. However, it is unclear how Arid1a achieves its functional specificity in regulating progenitor cells. Using the tooth root as a model, we show that loss of Arid1a impairs the differentiation-associated cell cycle arrest of tooth root progenitors through Hedgehog (Hh) signaling regulation, leading to shortened roots. Our data suggest that Plagl1, as a co-factor, endows Arid1a with its cell-type/spatial functional specificity. Furthermore, we show that loss of Arid1a leads to increased expression of Arid1b, which is also indispensable for odontoblast differentiation but is not involved in regulation of Hh signaling. This study expands our knowledge of the intricate interactions among chromatin remodelers, transcription factors, and signaling molecules during progenitor cell fate determination and lineage commitment.
In brief
Du et al. show that Arid1a promotes the transition of root progenitors from proliferation to differentiation through Hh signaling regulation. Plagl1 endows Arid1a with cell-type/spatial functional specificity. Loss of Arid1a leads to increase of Arid1b, which is also indispensable for odontoblast differentiation but is not involved in Hh signaling.
Graphical Abstract
INTRODUCTION
Stem/progenitor cells typically reside in a specialized, restricted niche environment. They can self-renew the progenitor pool through proliferation and differentiate into mature, functional cells. The regulation and fate decisions of stem/progenitor cells are crucial during tissue development, homeostasis, and regeneration. A precise combination of signaling pathways and transcription factors (TFs) is a prerequisite for the dynamic transcription of stage-specific genes during lineage commitment of stem/progenitor cells. Previous studies have shown that several signaling cascades, such as Notch and Wnt signaling, participate in the regulation of stem/progenitor cell fate determination in mammary and spermatogonial development (Dontu et al., 2004; Golestaneh et al., 2009). The critical role of TFs during the fate determination of stem/progenitor cells has been increasingly appreciated, particularly following the discovery that forced expression of four specific genes encoding TFs (Myc, Oct3/4, Sox2, and Klf4) can convert somatic cells into pluripotent stem cells (Takahashi and Yamanaka, 2006).
In addition to the network of diverse signaling pathways and TFs, emerging evidence suggests that epigenetic modifications–including DNA methylation, histone modification, RNA-mediated regulation, and chromatin remodeling–are indispensable for transcriptional regulation during the fate determination of stem/progenitor cells (Cakouros and Gronthos, 2020; Wu and Sun, 2006). The switch/sucrose nonfermentable (SWI/SNF) chromatin remodeling complex is one important family of ATP-dependent chromatin remodelers comprising multiple protein subunits that translocate nucleosomes and regulate gene transcription with DNA binding factors (Wilson and Roberts, 2011). Multiple lines of evidence indicate that the SWI/SNF complex plays key roles in tissue development, alongside its better-known function in suppressing a variety of cancers in humans (Helming et al., 2014). Several mutations in SWI/SNF subunits can cause Coffin-Siris syndrome (CSS) (MIM135900), which is associated with intellectual disability, hair and digital abnormalities, and patterning defects of the heart and craniofacial complex (Santen et al., 2013). Arid1a and Arid1b, which are mutually exclusive core components of the SWI/SNF complex, each contain a DNA-binding domain and mediate the chromatin remodeling function of the SWI/SNF complex (Chandler et al., 2013; Helming et al., 2014). Recent studies have found that Arid1a plays a critical role during pluripotency reprogramming, development, and homeostasis (Hota and Bruneau, 2016). For example, Arid1a regulates the fate commitment and differentiation of embryonic stem cells (Lei et al., 2015), hematopoietic stem cells (Han et al., 2019; Krosl et al., 2010), cardiac progenitor cells (Lei et al., 2012), and intestinal stem cells (Hiramatsu et al., 2019) by modulating nucleosome occupancy and the poised chromatin configuration at specific loci. Cranial neural crest (CNC) cells, which exhibit stem cell properties, contribute to the development of bone, teeth, cartilage, and neural tissue during craniofacial development (Chai et al., 2000). A previous study has shown that loss of Arid1a in CNC cells leads to cranial bone defects, recapitulating CSS-like phenotypes in mice (Chandler and Magnuson, 2016). However, the cellular and molecular mechanisms involved in Arid1a’s regulation of CNC fate determination and lineage commitment are not yet clear.
The tooth root is an ideal model to facilitate the investigation of the molecular and cellular regulation of organogenesis in CNC-derived tissues. The formation of a functional molar tooth root in both humans and rodents requires reciprocal interactions between the dental epithelium and mesenchyme, as well as tightly regulated proliferation and differentiation of stem and progenitor cells (Feng et al., 2017; Kumakami-Sakano et al., 2014; Li et al., 2017; Yao et al., 2008). Previous studies using lineage tracing and cell ablation approaches have established that Gli1+ cells, which are restricted to the most apical region of the tooth when molar root formation initiates, represent progenitor cells that give rise to root pulp, dentin, and periodontium (Feng et al., 2017; Liu et al., 2015). It has also been established that ARID1A mutations in humans lead to delayed development of the primary and permanent dentition, suggesting these mutations may be associated with defective tooth root development (Santen et al., 2013).
Here, we have used the tooth root as a model to investigate the role of Arid1a in the fate commitment and differentiation of progenitor cells during postnatal development. Arid1a is expressed in the dental mesenchyme including the Gli1+ root apical progenitor cells and their derivatives during postnatal development. We have found that loss of Arid1a in Gli1+ root progenitor cells leads to shortened roots with defective dentin and periodontal tissue development. Our results show that Arid1a promotes the differentiation-associated cell cycle arrest of tooth root progenitors during root development through Hedgehog (Hh) signaling regulation. Furthermore, we have identified that Plagl1, a critical TF in the tooth, serves as a co-factor that endows Arid1a with its cell-type/spatial functional specificity during the transition of root progenitors from proliferation to differentiation. In addition, we show that loss of Arid1a leads to increased expression of Arid1b, which is also indispensable for odontoblast (OD) differentiation through a distinct epigenetic regulatory mechanism. Our findings further expand our knowledge of the delicate interactions among chromatin remodelers, TFs, and signaling pathways during the fate determination and lineage commitment of stem/progenitor cells.
RESULTS
Loss of Arid1a in tooth root progenitor cells leads to a shortened tooth root
Arid1a plays an essential role in CNC cells during embryonic development, but it is unclear whether Arid1a functions in CNC-derived cells during postnatal development (Chandler and Magnuson, 2016). We examined its expression pattern and found that from postnatal day (PN) 4.5 to PN21.5 (Figures 1A–1D), Arid1a is expressed ubiquitously in both dental epithelium and mesenchyme (Figures 1E–1H). Moreover, our previous studies have shown that Gli1+ cells are tooth root progenitors that contribute to the entire root complex, including ODs, pulp cells, periodontal ligament (PDL), and surrounding alveolar bone (Feng et al., 2017; Liu et al., 2015). To evaluate the role of Arid1a in Gli1+ root progenitor cells, we assessed the expression pattern of Arid1a in the Gli1+ population. We found that Arid1a is expressed in Gli1+ cells in the apical part of the mouse molar at PN4.5 (Figures 1I–1L), as well as the progeny of Gli1+ cells at PN18.5 (Figures 1M–1P), during tooth root development. To test whether Arid1a plays a crucial role in the cell fate commitment of root progenitor cells, we generated Gli1-CreER;Arid1afl/fl mice. We induced Cre activity with tamoxifen at PN3.5, at the onset of tooth root development. Following the deletion of Arid1a (Figure S1A), we observed shortened roots in Gli1-CreER;Arid1afl/fl mice compared to those of controls at PN14.5 (Figures 2A–2D) and PN21.5 (Figures 2E–2H). Quantitative analysis revealed that the root length of first mandibular molar was significantly shorter in Gli1-CreER;Arid1afl/fl mice compared to control samples (Figure 2I). This result demonstrates the indispensability of Arid1a during tooth root development.
Figure 1. Colocalization of Arid1a and Gli1+ cells and their progeny in developing roots.
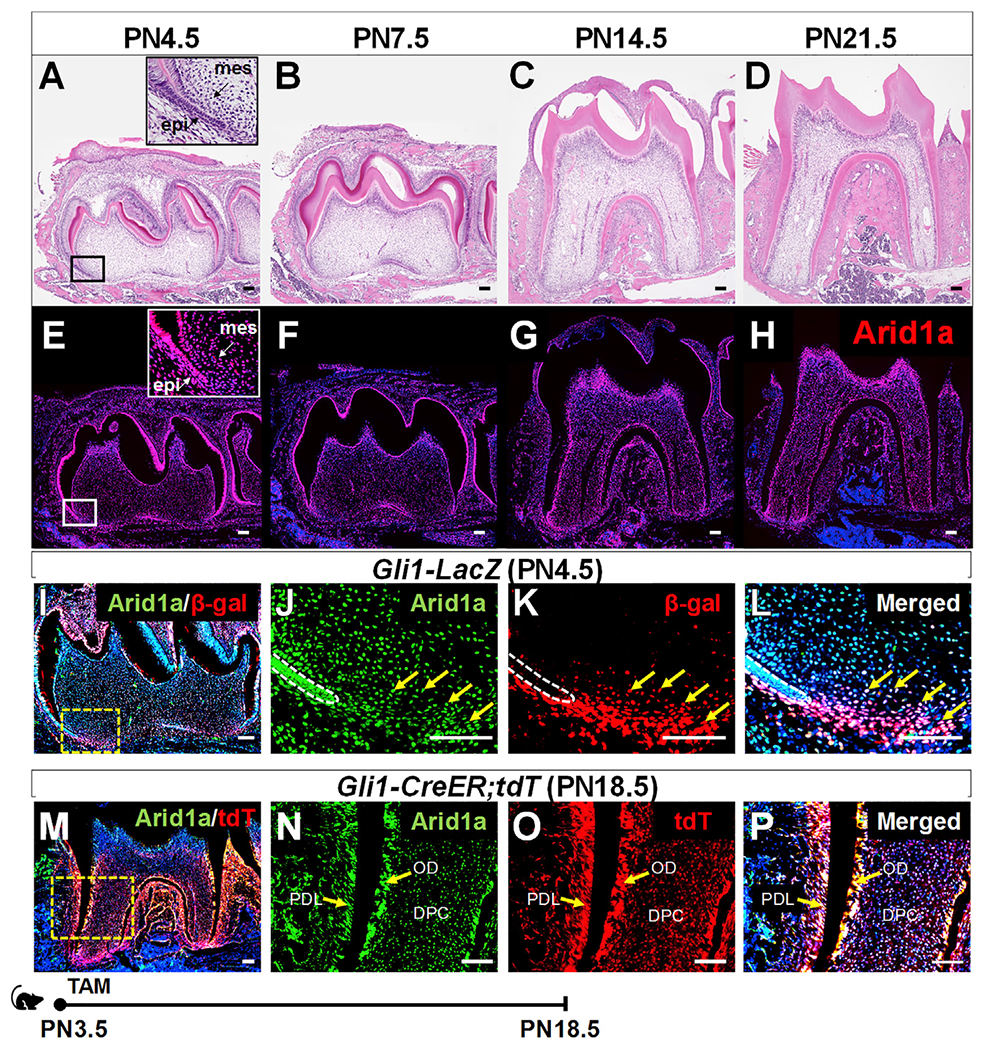
(A–H) H&E staining (A–D) and Arid1a immunofluorescence (red) (E–H) of the first mandibular molar of a wild-type (WT) mouse from PN4.5 to PN21.5. Mes, dental mesenchyme; epi, dental epithelium. (I–L) Co-immunofluorescence of Arid1a (green) and Gli1 (stained by β-gal in red) in the first mandibular molar of a Gli1-LacZ mouse at PN4.5. Box in (I) is shown at higher magnification at the right. Arrows indicate positive signals. White dashed lines outline Hertwig’s epithelial root sheath (HERS).
(M–P) Arid1a immunofluorescence (green) and visualization of tdTomato (red) of the first mandibular molar of a Gli1-CreER;tdTomato mouse at PN18.5 after induction at PN3.5. The progeny of the Gli1+ lineage show red signal. Arrows indicate positive signals. TdT, tdTomato; DPC, dental pulp cell; OD, odontoblast; PDL, periodontal ligament. Schematic at the bottom indicates induction protocol. TAM, tamoxifen. Scale bars: 100 μm.
Figure 2. Loss of Arid1a in Gli1+ root progenitor cells leads to shortened roots.
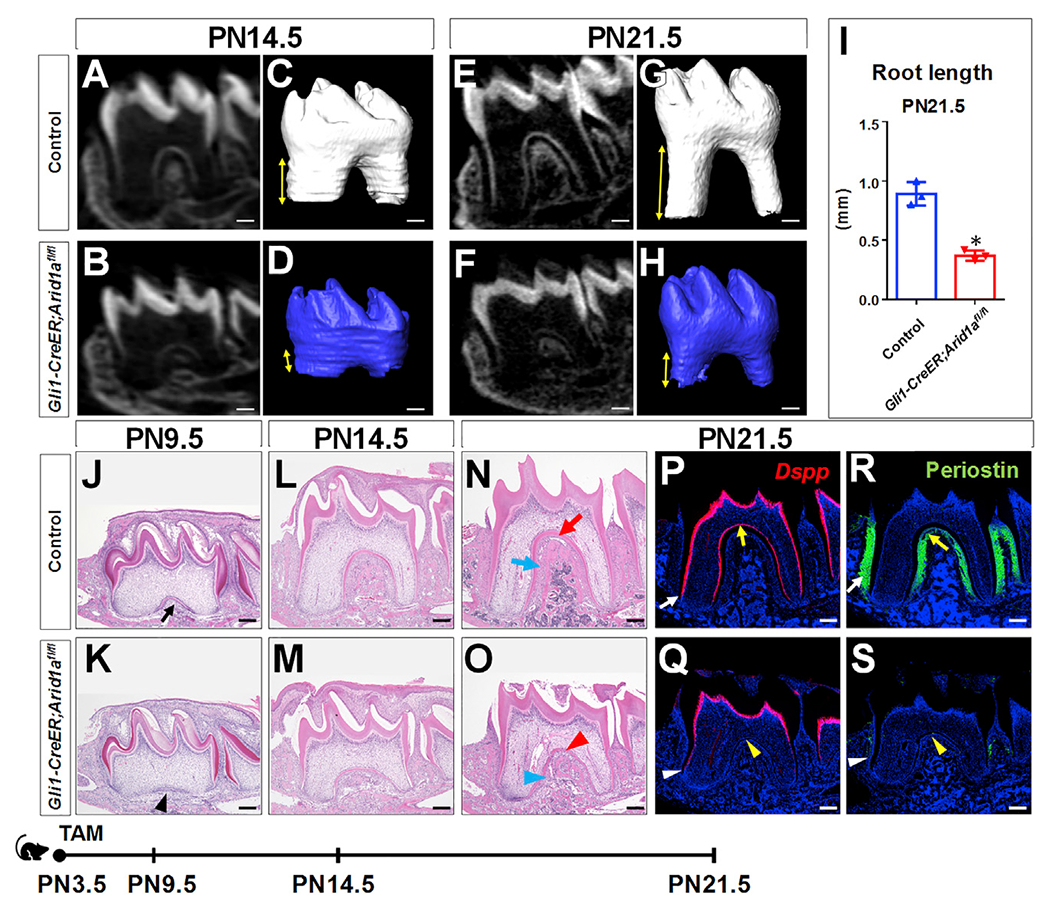
(A–H) MicroCT 2D and 3D images of the first mandibular molars of control and Gli1-CreER;Arid1afl/fl mice at PN14.5 (A–D) and PN21.5 (E–H). Distance between arrowheads represents tooth root length.
(I) Quantitative analysis of tooth root length at PN21.5. n = 3, *p < 0.05.
(J–O) H&E staining of first mandibular molars of control and Gli1-CreER;Arid1afl/fl mice at PN9.5 (J and K), PN14.5 (L and M), and PN21.5 (N and O). Red and blue arrowheads in (O) indicate the compromised dentin and PDL in Gli1-CreER;Arid1afl/fl mice compared to control mice, respectively.
(P–S) In situ hybridization of Dspp (red; P and Q) and immunofluorescence of periostin (green; R and S) of first mandibular molars of control and Gli1-CreER;Arid1afl/fl mice at PN21.5. Yellow and white arrowheads in (Q) and (S), respectively, indicate absence of signal or compromised signal in the furcation region and the lateral region of the root in Gli1-CreER;Arid1afl/fl mice compared to control mice.
Schematic at the bottom indicates induction protocol. Data are represented as mean ± SD. Scale bars: 200 μm. See also Figures S1 and S2A.
We then analyzed various stages of the root development to investigate the pathogenesis of root development in Gli1-CreER;Arid1afl/fl mice. Although Arid1a was efficiently ablated in the apical portion of the first mandibular molar roots as early as PN7.5, the morphology of the apical part of the tooth was comparable in Arid1a mutants and controls at this stage (Figure S1B). At PN9.5, root defects started to appear in Gli1-CreER;Arid1afl/fl mice (Figure 2K), and the delayed development and tissue defects were increasingly apparent at PN14.5 (Figure 2M) and PN21.5 (Figure 2O). Specifically, impaired OD differentiation (Figure 2Q) and PDL formation (Figure 2S) in both the furcation region (yellow arrowhead) and the lateral region (white arrowhead) of the root were visible in Gli1-CreER;Arid1afl/fl mice at PN21.5.
Since Gli1-CreER targets both dental epithelium and mesenchyme, Gli1-CreER;Arid1afl/fl mice lose Arid1a in both of these tissues. To evaluate the effect of epithelial loss of Arid1a on molar roots, we generated K14-rtTA;tetO-Cre;Arid1afl/fl mice. However, we did not observe any apparent defects of root length or dentin formation following loss of Arid1a in molar epithelium in K14-rtTA;tetO-Cre;Arid1afl/fl mice after inducing Cre activity with doxycycline from PN3.5 (Figure S2A). These data show that loss of Arid1a in tooth root mesenchymal progenitors is responsible for the shortened roots, defective dentin, and disrupted periodontal tissue development in Gli1-CreER;Arid1afl/fl mice.
Loss of Arid1a in tooth root progenitor cells leads to defective differentiation-associated cell cycle arrest
To further investigate the cellular mechanism underlying the root defects, we evaluated the cell fate of Gli1+ tooth root progenitor cells at several time points during root development. Using tdTomato (tdT) as a reporter to trace Gli1+ cells from PN3.5, we found that more tdT+ cells were in a mitotic state in Gli1-CreER;Arid1afl/fl mice compared with controls at PN7.5 (Figures 3A and 3B), even before root OD differentiation defects were apparent (Figures 3H and 3I). The continuously overactivated cell division in the apical region of the molars from Gli1-CreER;Arid1afl/fl mice led, over time, to a higher proportion of Gli1+ progeny than in control molars (Figures 3A–3G). However, the root OD differentiation and PDL formation of the Arid1a mutant Gli1+ progeny were compromised from PN9.5 (Figures 3J–3O). The cell cycle progression defect was further confirmed by examining mitotic exit. We labeled the cycling cells in DNA synthesis phase with 5-ethynyl-2′-deoxyuridine (EdU) in control and Gli1-CreER;Arid1afl/fl mouse molars at PN7.5 and found that apparently, more labeled cycling cells remained in division in mutant molars after 48 h, which contributed to the majority of the increase in the total number of EdU+ cells; the number of labeled cells not in mitosis was slightly reduced, although this reduction was not statistically significant (Figures 3P–3T). Thus, our results have shown that more Gli1+ progeny stay in dividing states without further differentiation following the loss of Arid1a, suggesting that Arid1a plays an indispensable function during the transition of Gli1+ root progenitors from proliferation to differentiation.
Figure 3. Tracing cell fate commitment of Gli1+ root progenitor cells in control and Gli1-CreER;Arid1afl/fl mice.
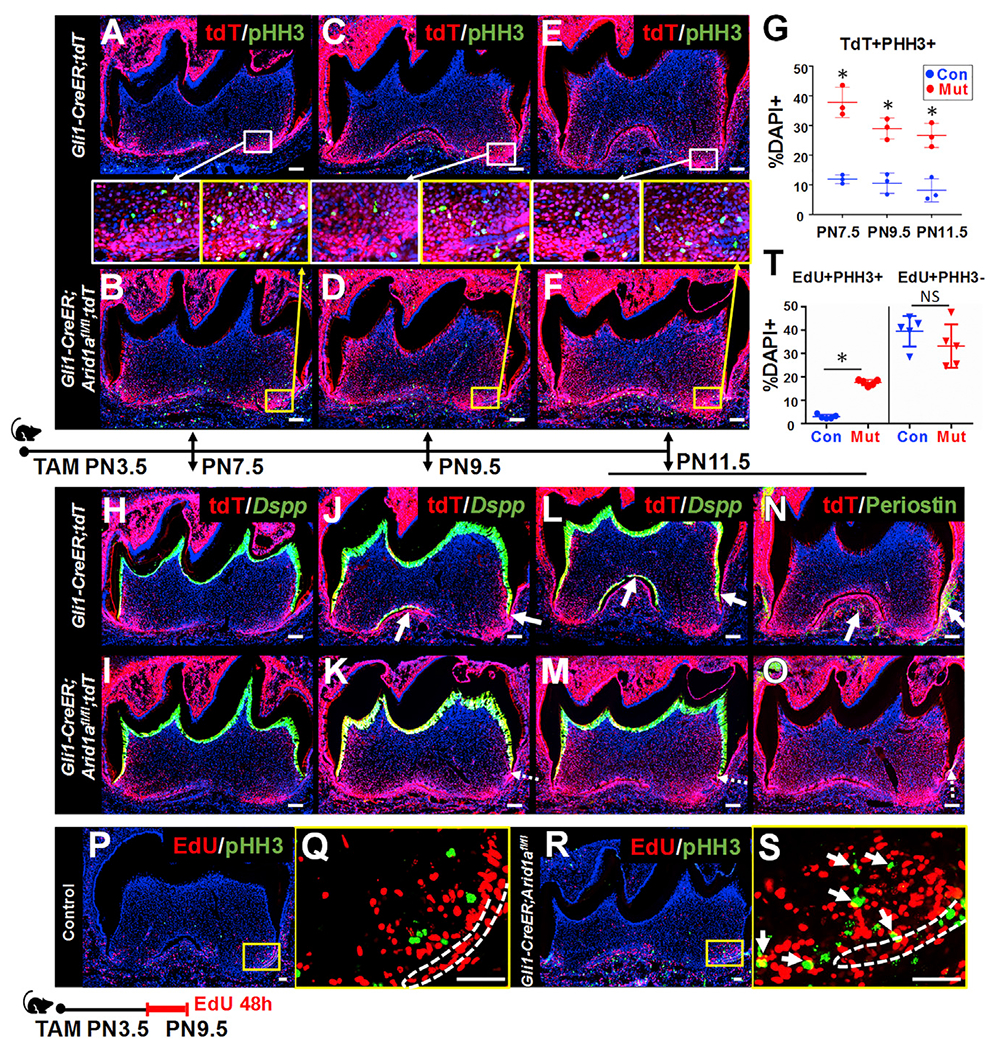
(A–F) Immunofluorescence of PHH3 (green) and visualization of tdT (red) of first mandibular molars from Gli1-CreER;tdT and Gli1-CreER;Arid1afl/fl;tdT mice at PN7.5, PN9.5, and PN11.5 after induction at PN3.5. The boxed areas in (A)–(F) are shown enlarged in the middle.
(G) Quantitative analysis of the ratio of tdT+pHH3+ cells in the root apical region in Gli1-CreER;tdT (Con) and Gli1-CreER;Arid1afl/fl;tdT (Mut) mouse molars. n = 3, *p < 0.05.
(H–O) In situ hybridization of Dspp (green) or immunofluorescence of periostin (green) and visualization of tdT (red) of first mandibular molars from Gli1-CreER;tdT and Gli1-CreER;Arid1afl/fl;tdT mice at PN7.5, PN9.5, and PN11.5 after induction at PN3.5. Arrows indicate positive signal. Dotted arrows indicate reduced signal.
(P–S) Immunofluorescence of pHH3 (green) and visualization of EdU (red) of first mandibular molars from control (P and Q) and Gli1-CreER;Arid1afl/fl (R and S) mice at PN9.5 after induction at PN3.5. The boxed areas in (P) and (R) are enlarged in (Q) and (S), respectively. White dashed lines outline HERS. Arrows indicate overlapping signal.
(T) Quantitative analysis of the ratio of EdU+PHH3+ and EdU+PHH3– cells in the root apical region in control (Con) and Gli1-CreER;Arid1afl/fl (Mut) mouse molars. n = 5, *p < 0.05. NS, no significant difference.
Schematic in the middle indicates induction protocol in (A)–(O). Schematic at the bottom indicates induction protocol in (P)–(T). Data are represented as mean ± SD. Scale bars: (A–O) 100 μm; (P–S) 50 μm. See also Figures S2B and S3.
We also noticed that there was a progressive decrease in the number of tdT+ proliferative cells in the apical region of the molars in Gli1-CreER;Arid1afl/fl mice from PN9.5, suggesting the possible disruption of the cell cycle following defective transition of tooth root progenitors from proliferation to differentiation after loss of Arid1a.
In addition, at the later stage of PN11.5, as defective cell cycle progression of tooth root progenitors in Gli1-CreER;Arid1afl/fl mice continued, we observed increased cell apoptosis in the apical region of Gli1-CreER;Arid1afl/fl mouse molars (Figures S3C, S3F, and S3G). Since cell apoptosis was not increased at earlier stages (Figures S3A, S3B, S3D, S3E, and S3G), we speculated that the later increase in apoptosis might be a secondary defect.
Given that Arid1a is expressed in ODs, to rule out the possibility that loss of Arid1a in ODs led directly to differentiation defects, we also generated Dmp1-Cre;Arid1afl/fl mice to evaluate the effect of loss of Arid1a in ODs. Using microCT analysis, we found that the length and mineralization of the tooth roots in these mice were comparable to those of control mouse molars (Figure S2B). We therefore concluded that loss of Arid1a in tooth root progenitor cells leads to defective differentiation-associated cell cycle arrest during tooth root development.
Loss of Arid1a in Gli1+ root progenitor cells leads to downregulated Hh signaling
To further investigate the mechanism of how Arid1a regulates the transition of Gli1+ root progenitor cells from proliferation to differentiation, we conducted RNA sequencing analysis to compare the gene transcription in the apical portion of the first mandibular molar roots in control and Gli1-CreER;Arid1afl/fl mice at PN7.5. Principal component analysis (PCA) of the RNA sequencing data showed well-separated gene expression patterns between the control and Gli1-CreER;Arid1afl/fl molars (Figure S4Aa), with a total of 215 downregulated and 193 upregulated genes (false discover rate [FDR] ≤ 0.1; fold change < −1.5 or > 1.5), as shown in the heatmap in Figure 4A. Among the differentially expressed genes, we noticed that Gli family zinc finger 1 (Gli1), which is a readout of Hh signaling pathway, was significantly downregulated in Gli1-CreER;Arid1afl/fl mice (Figure S4Ab). Hh signaling pathway controls cell growth, survival, and differentiation and plays an essential role during vertebrate embryonic development and adult tissue homeostasis (Jiang and Hui, 2008). Previous studies have shown that proper levels of Hh signaling activity are critical for the proliferation and differentiation of Gli1+ progenitor cells as the tooth root develops (Liu et al., 2015). In our present study, we found that in addition to Gli1 (Figures 4B–4E and S4Ba), the expression levels of other two target genes of Hh signaling, patched 1 (Ptch1) (Figures 4F–4I and S4Bb) and cyclin D1 (Ccnd1) (Figures 4J–4M and S4Bc), were both downregulated in the apical portion of the first mandibular molar roots in Gli1-CreER;Arid1afl/fl mice compared to controls at PN7.5. In summary, loss of Arid1a in Gli1+ root progenitor cells leads to downregulated Hh signaling activity during tooth root development.
Figure 4. Loss of Arid1a in Gli1+ root progenitor cells leads to downregulated Hh signaling activity.
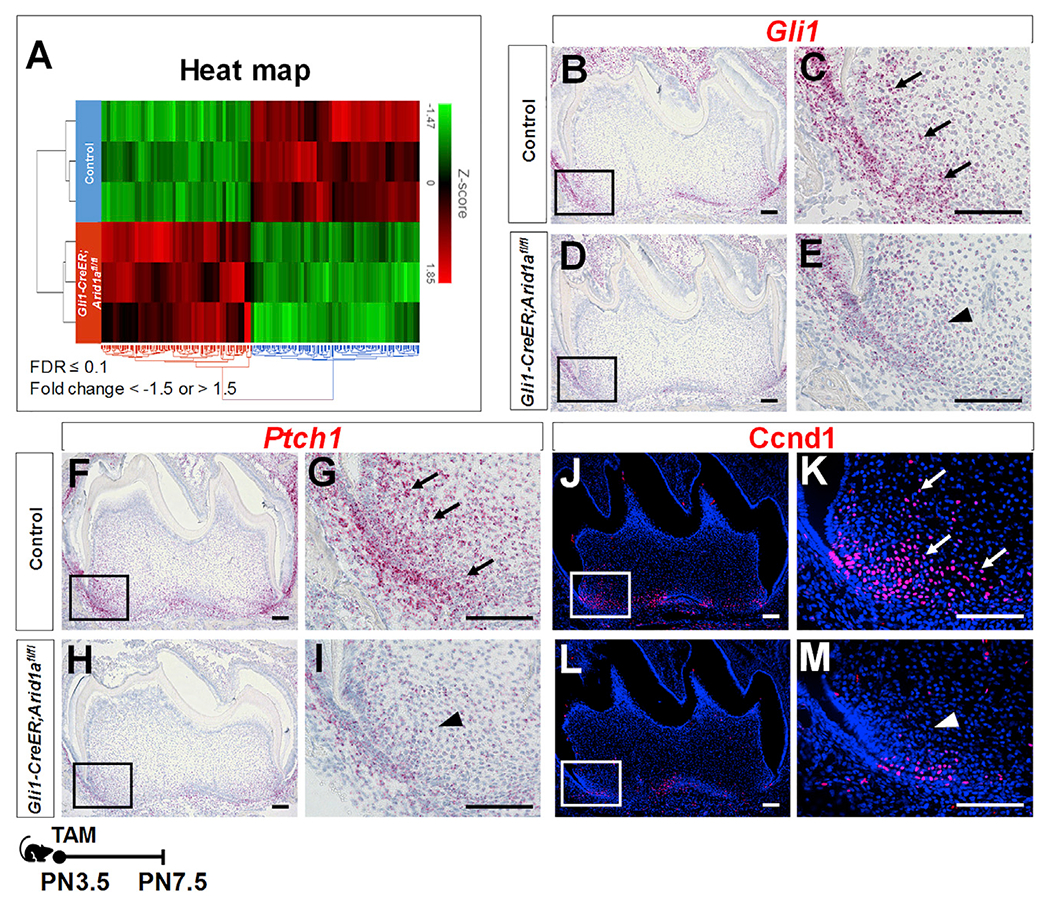
(A) Heatmap hierarchical clustering showing the gene expression profiles in the apical regions of control and Gli1-CreER;Arid1afl/fl mouse molars at PN7.5.
(B–M) RNAscope in situ hybridization (red) of Gli1 (B–E), Ptch1 (F–I), and immunofluorescence of Ccnd1 (red, J–M) of first mandibular molars of control and Gli1-CreER;Arid1afl/fl mice at PN7.5. The boxed areas are enlarged on the right. Arrows indicate positive signals in control group; arrowheads indicate reduced signal in targeted region of Gli1-CreER;Arid1afl/fl mouse molars.
Schematic at the bottom indicates induction protocol. Scale bars: 100 μm. See also Figures S4 and S5.
Interestingly, one recent study has shown that Arid1a regulates intestinal stem cells through Wnt signaling (Hiramatsu et al., 2019). However, we did not find any apparent change in Wnt signaling pathway after loss of Arid1a in tooth root progenitor cells, indicated by the unchanged gene expression levels of Axin2 and Lef1 (Figure S5A). To verify our finding in another CNC-derived tissue, we also evaluated the function of Arid1a in mandibular condyle development and found that loss of Arid1a leads to downregulated Hh signaling activity, overactivated mitosis, and defective osteogenesis (Figure S5B).
Upregulation of Hh signaling partially rescues differentiation-associated cell cycle arrest in Gli1-CreER;Arid1afl/fl mouse molars
To verify whether the downregulation of Hh signaling pathway is responsible for defective differentiation-associated cell cycle arrest of tooth root progenitors in Gli1-CreER;Arid1afl/fl mouse molars, we generated Gli1-CreER;Arid1afl/fl;SmoM2fl/+ mice with upregulated Hh signaling. At PN21.5, we found that upregulating Hh signaling partially rescued the shortened tooth root length and the defective dentin and PDL formation in Gli1-CreER;Arid1afl/fl mouse molars (Figure 5). We further confirmed that Hh signaling activity in the apical part of the molar was restored in PN7.5 Gli1-CreER;Arid1afl/fl;SmoM2fl/+ mice compared to Gli1-CreER;Arid1afl/fl mice, as indicated by the expression levels of Gli1 (Figures S6A, S6E, and S6I), Ptch1 (Figures S6B, S6F, and S6J), and Ccnd1 (Figures S6C, S6G, and S6K). The number of mitotic cells was reduced significantly in apical third of Gli1-CreER;Arid1afl/fl;SmoM2fl/+ molars compared to Gli1-CreER;Arid1afl/fl at PN7.5 (Figures S6D, S6H, S6L, and S6M). Taken together, these results demonstrate that during tooth root development, Arid1a is indispensable for differentiation-associated cell cycle arrest of tooth root progenitors, at least partially through Hh signaling.
Figure 5. Upregulation of Hh signaling partially rescues tooth root defects in Gli1-CreER;Arid1afl/fl mouse molars.
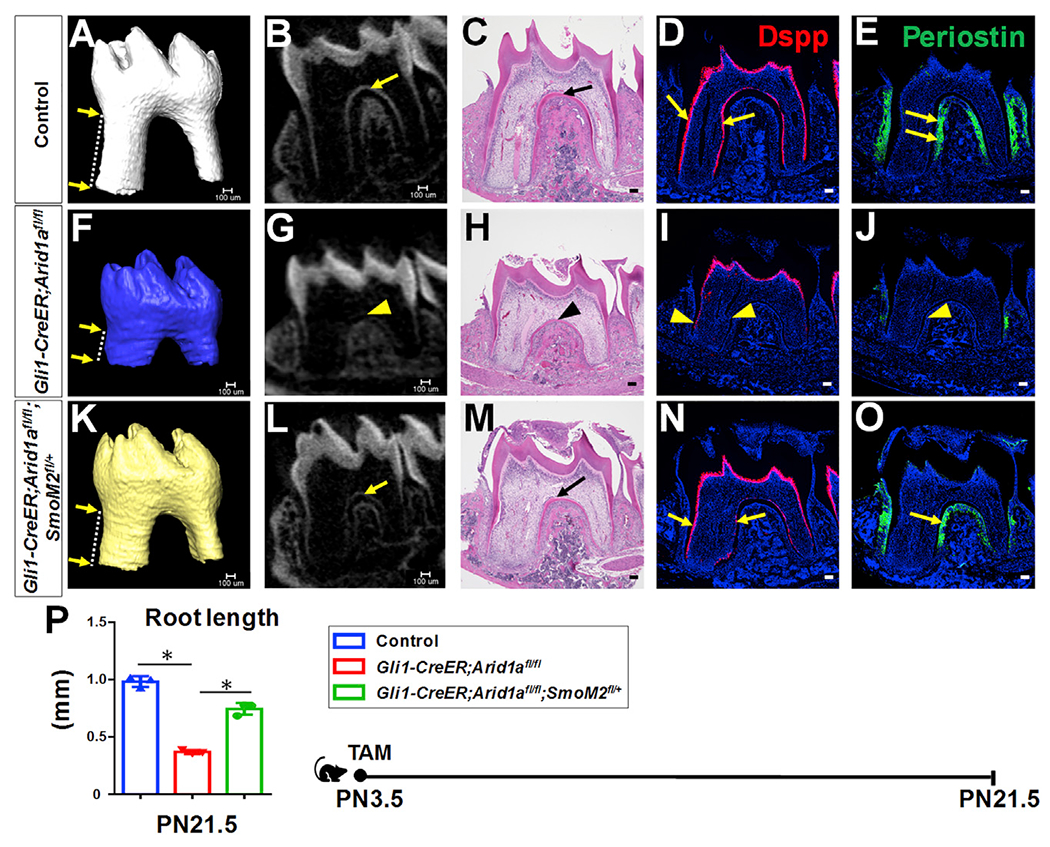
(A–O) MicroCT 3D and 2D images, H&E staining, RNAscope in situ hybridization of Dspp (red), and immunofluorescence of periostin (green) of first mandibular molars in control (A–E), Gli1-CreER; Arid1afl/fl (F–J), and Gli1-CreER;Arid1afl/fl;SmoM2fl/+ (K–O) mice at PN21.5 after induction at PN3.5. Distance between two arrows in (A), (F), and (K) indicates tooth root length. Arrows in (B)–(E) and (L)–(O) indicate positive signals in control and Gli1-CreER;Arid1afl/fl;SmoM2fl/+ mice; arrowheads in (G)–(J) indicate compromised signal in targeted region of Gli1-CreER;Arid1afl/fl mouse.
(P) Quantification of length of first mandibular molar roots from control, Gli1-CreER;Arid1afl/fl, and Gli1-CreER;Arid1afl/fl;SmoM2fl/+ mice at PN21.5 after induction at PN3.5. n = 3, *p < 0.05.
Schematic at the bottom indicates induction protocol. Data are represented as mean ± SD. Scale bars: 100 μm. See also Figure S6.
Arid1a interacts with Plagl1 to regulate Gli1 transcription during tooth root development
Arid1a, the core unit of the SWI/SNF complex, contains a DNA-binding domain and regulates gene transcription with coactivators and corepressors. In order to test how Arid1a regulates Hh/Gli1 signaling during tooth root development, we performed a chromatin immunoprecipitation (ChIP) assay with chromatin isolated from the apical regions of the mandibular first molars from control mice at PN3.5–PN7.5. A previous study has shown that the mouse Gli1 second intron, which is equivalent to the human GLI1 first intron, is a transcriptional regulatory region enriched with epigenetic marks (Taylor et al., 2019). We found that Arid1a could bind to the proximal promoter and second intronic region of Gli1 in the apical part of the mandibular first molar at PN7.5, suggesting Arid1a can directly regulate the transcription of Gli1 (Figure 6A).
Figure 6. Arid1a interacts with Plagl1 to regulate Gli1 transcription during tooth root development.
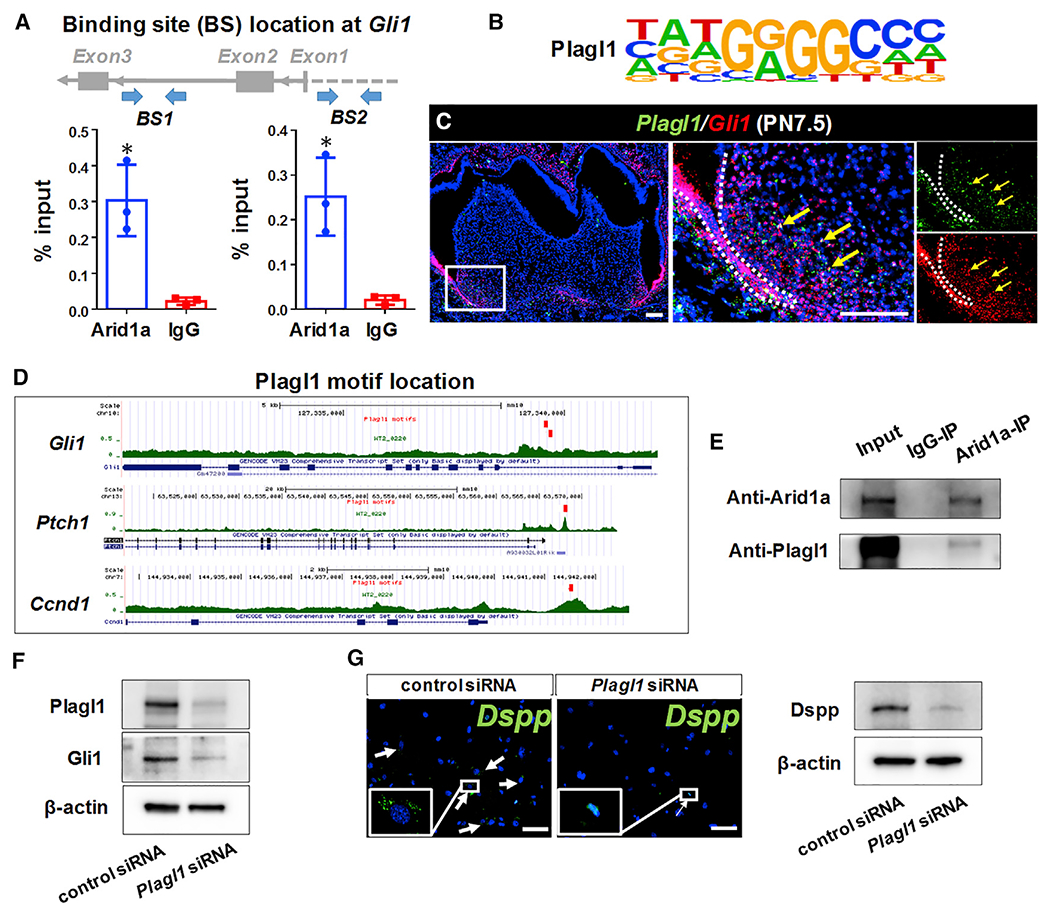
(A) Chromatin immunoprecipitation (ChIP) assay with Arid1a antibody (or immunoglobulin G [IgG]), followed by qPCR with two pairs of primers. n = 3, *p < 0.05.
(B) The Plagl1 motif identified based on the motif enrichment analysis on WT-specific ATAC-seq peaks.
(C) RNAscope in situ hybridization of Plagl1 (green) and Gli1 (red) of first mandibular molars of control mice at PN7.5.
(D) The Plagl1 motif hits are found on ATAC regions at the intron 2 of Gli1 and promoter region of Ptch1 and Ccnd1.
(E) CoIP assay with Arid1a antibody (or IgG), followed by immunoblotting of Arid1a and Plagl1.
(F) Western blot of Gli1 in cultured DPCs treated with control siRNA or Plagl1 siRNA.
(G) RNAscope in situ hybridization of Dspp (green) and the western blot of Dspp in cultured DPCs treated with control siRNA or Plagl1 siRNA after 6 days of odontogenic induction.
Data are represented as mean ± SD. Scale bars: 100 μm. See also Figure S7.
To find potential specific co-factors of Arid1a in the transcriptional regulation of Hh signaling during root development, we conducted ATAC (Assay for Transposase-Accessible Chromatin) sequencing and motif analysis of the apical portions of the first mandibular molar roots from control and Gli1-CreER;Arid1afl/fl mice at PN7.5. We observed 215 regions with increased accessibility in controls compared to Gli1-CreER;Arid1afl/fl mice (wild-type [WT]-specific regions; see Method details). We cross-referenced the top 13 TFs enriched in the WT-specific open chromatin regions (Figure S7A; see Method details) with our single-cell RNA sequencing data from first mandibular molars at PN7.5 (Wen et al., 2020) and identified Plagl1 (Figure 6B) as a potential binding partner of Arid1a.
Plagl1 is a zinc finger TF with expression induced during cell cycle exit and differentiation (Rraklli et al., 2016; Spengler et al., 1997; Varrault et al., 2017). Previous studies have shown that Plagl1 is critical for the development of multiple tissues, such as bone (Varrault et al., 2006), cardiac fibroblasts (Jonsson et al., 2016), and neurons (Adnani et al., 2015). However, its function in tooth development remains unclear. When we evaluated the expression pattern of Plagl1 at PN7.5, we found that it was highly expressed in the apical part of the tooth root, a region with active Hh signaling. Colocalization of Plagl1 with Gli1 (Figure 6C) suggested it may participate in transcriptional regulation of Arid1a as part of the Hh signaling pathway. To test this hypothesis, we used the Plagl1 motif to scan the ATAC peaks using HOMER (Heinz et al., 2010) and found that there are Plagl1 motif hits in intron 2 of Gli1 and the promoter regions of Ptch1 and Ccnd1 (Figure 6D). Given that the Plagl1 motif in intron 2 of Gli1 is close to the binding site of Arid1a on Gli1 mentioned above, we predicted that Arid1a may interact with Plagl1 and regulate Hh signaling synergistically. CoIP confirmed the interaction between Arid1a and Plagl1 in dental pulp mesenchymal cells (DPCs) (Figure 6E). To verify that Plagl1 regulates Hh signaling and OD differentiation, we knocked down Plagl1 in DPCs using small interfering RNA (siRNA) in vitro. We observed that the knockdown of Plagl1 in DPCs led to reduced expression of Gli1 (Figure 6F) and Dspp (Figure 6G) after 6 days of odontogenic induction, suggesting Plagl1 is a critical TF for Hh signaling and OD differentiation. Interestingly, however, we did not find a significant difference in Plagl1 expression between control and Gli1-CreER;Arid1afl/fl mice at the gene level using in situ RNA scope (Figures S7Ba–S7Bd) or at the protein level by western blot analysis (Figure S7Be). Therefore, we hypothesized that loss of Arid1a does not affect the expression level of Plagl1 but may instead affect its regulatory function during tooth root development through protein-protein interaction.
Arid1b is indispensable for OD differentiation but is not involved in Hh signaling regulation
Given that ARID1B, a paralog of ARID1A, preserves the genomic sites targeting function and physiological RNA polymerase II (RNAPII) activity of residual SWI/SNF complexes in human cancer cells (Mathur et al., 2017; Trizzino et al., 2018), we investigated the expression pattern and level of Arid1b in control and Gli1-CreER;Arid1afl/fl mouse molars. We found that Arid1b is broadly expressed in the mouse molar (Figures 7A and 7B), similar to the expression pattern of Arid1a, and that loss of Arid1a leads to an increased expression level of Arid1b in Gli1-CreER; Arid1afl/fl mouse molars (Figure 7C), suggesting that Arid1b may partially compensate for loss of Arid1a during tooth root development.
Figure 7. Arid1b is indispensable for OD differentiation but is not involved in Hh signaling regulation.
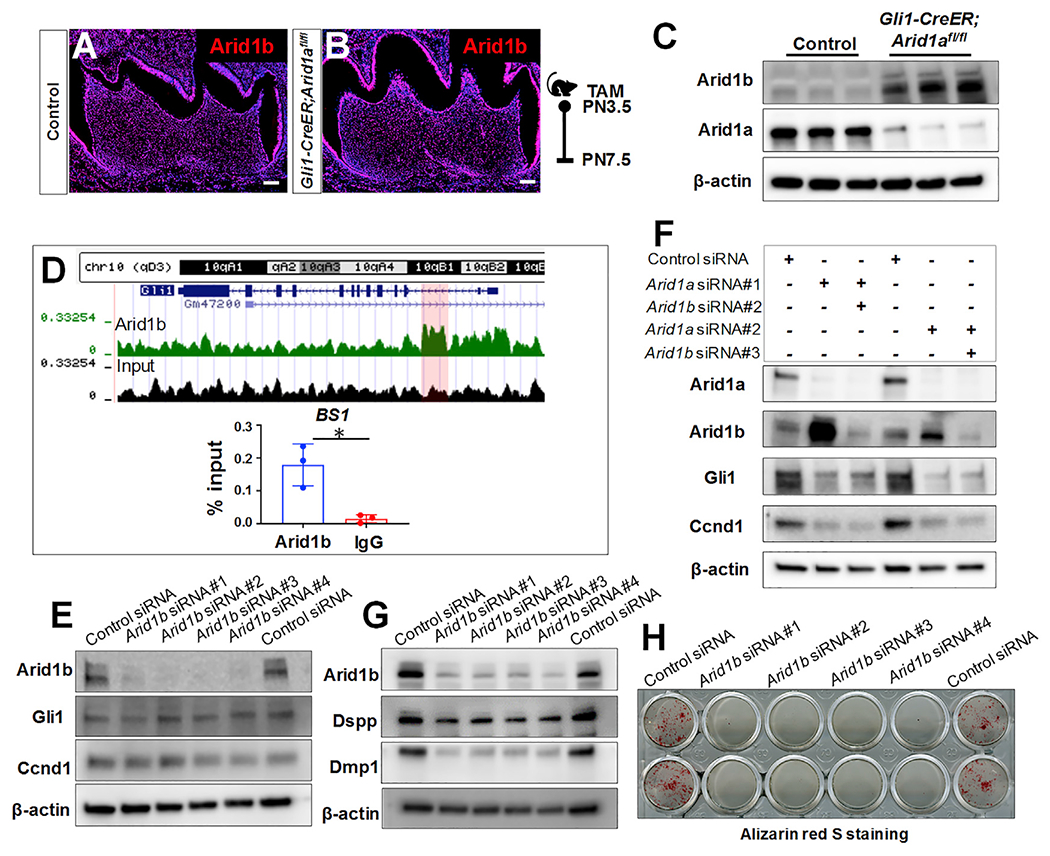
(A and B) Immunofluorescence of Arid1b of first mandibular molars of control and Gli1-CreER;Arid1afl/fl mice at PN7.5.
(C) Western blot of Arid1b in the apical region of the first mandibular molars from control and Gli1-CreER;Arid1afl/fl mice at PN7.5.
(D) UCSC Genome Browser screenshot of Arid1b at the Gli1 locus and ChIP assay with Arid1b antibody (or IgG), followed by qPCR. n = 3, *p < 0.05.
(E) Western blot of Gli1 and Ccnd1 in DPCs with control siRNA or four different Arid1b siRNA sets.
(F) Western blot of Gli1 and Ccnd1 in DPCs with control siRNA, Arid1a siRNA, or both Arid1a and Arid1b siRNA.
(G) Western blot of Dspp and Dmp1 in DPCs with control siRNA or four different Arid1b siRNA after odontogenic induction.
(H) Alizarin red S staining of DPCs with control siRNA or four different Arid1b siRNA after odontogenic induction. Schematic indicates induction protocol. Scale bars: 100 μm.
On the other hand, a previous study has found genomic co-occupancy of ARID1A and ARID1B and noted their distinct transcriptional regulatory functions achieved through interaction with associated co-factors in HepG2 cells (Raab et al., 2015). To investigate whether Arid1b is involved in regulating Hh signaling in our mouse molar root development model, we performed a ChIP assay of Arid1b and evaluated the change in Hh signaling activity after the knockdown of Arid1b using siRNA in vitro. The potential hit of Arid1b at Gli1 from the ChIP sequencing data was further validated using ChIP-qPCR. We found that Arid1b can bind to the second intron of Gli1, where there is an Arid1a binding site (Figure 7D). However, the knockdown of Arid1b led to no apparent change in the activity of Hh signaling (Figure 7E). Furthermore, we found that the activity of Hh signaling remained at a low level in DPCs with double Arid1a/b knockdown (Figure 7F), suggesting that the downregulated Hh signaling after loss of Arid1a is unlikely to be the result of elevated Arid1b.
Interestingly, our data have shown that knockdown of Arid1b leads to downregulated expression of OD differentiation markers Dspp and Dmp1 (Figure 7G) along with compromised mineralization ability of ODs indicated by alizarin red S staining (Figure 7H) in DPCs during odontogenic induction. These results suggested that although Hh signaling regulation is Arid1a dependent, Arid1b is also critical for OD differentiation, likely through a different mechanism, which is consistent with the delayed dentition phenotype in humans with ARID1B mutation (Wieczorek et al., 2013).
Taken together, our findings lead us to conclude that (1) loss of Arid1a leads to the increased expression of Arid1b; (2) although Arid1a and Arid1b can co-occupy the second intron of Gli1, Hh signaling regulation is Arid1a dependent; and (3) loss of Arid1a and knockdown of Arid1b can each lead to compromised OD differentiation, suggesting that both are indispensable for the cell fate determination of tooth root progenitors through distinct epigenetic mechanisms. The indispensability of both Arid1a/Arid1b and their ability to partially compensate for each other’s loss hint at the precision and complexity of chromatin remodeler functions during tissue development, which should be the topic of further exploration in the future.
DISCUSSION
Arid1a and Arid1b are mutually exclusive subunits in SWI/SNF complexes. In our study, loss of Arid1a in tooth root progenitors increased the expression of Arid1b, which is consistent with the finding that ARID1B is upregulated after depletion of ARID1A in ovarian clear cell carcinoma (Trizzino et al., 2018). According to previous studies, ARID1B is required for the maintenance of an intact SWI/SNF complex and compensates for the impaired physiological RNAPII activity in ARID1A mutant cancer cell lines (Helming et al., 2014; Trizzino et al., 2018), which may be a potential explanation for our finding that instead of leading to absolute root loss, molars still achieved a certain amount of tooth root growth after the loss of Arid1a, albeit with defective tissue components. Also, it has been proposed that Arid1b may compensate for loss of Arid1a in the intestine, although there is no apparent increase of gene expression of Arid1b in the intestine after loss of Arid1a at the mRNA level (Hiramatsu et al., 2019), which suggests that the compensatory mechanism involving Arid1a and Arid1b is context dependent.
Importantly, our findings that loss of Arid1a and knockdown of Arid1b can each lead to defective OD differentiation suggest that Arid1b is only partially redundant with Arid1a, and vice versa. Although we show both Arid1a and Arid1b can bind to the second intron of Gli1, Hh signaling regulation is Arid1a dependent. Distinct SWI/SNF complexes, as well as other chromatin remodeling proteins, share widespread overlap of genomic binding sites and achieve their gene-specific transcription regulation with associated co-factors (Morris et al., 2014; Raab et al., 2015). Our findings here are also consistent with a previous study showing that in ovarian clear cell carcinoma, the impaired ability to pause RNAPII after loss of ARID1A can be compensated by the upregulation of ARID1B, but a subset of ARID1A-dependent genes are not rescued by ARID1B (Trizzino et al., 2018). Interestingly, a previous study showed that ARID1B represses Wnt/β-Catenin signaling in neurodevelopment and cancer (Vasileiou et al., 2015). Although we have found that the levels of Wnt/β-Catenin signaling are comparable in control and Gli1-CreER;Arid1afl/fl mouse molars, we have detected increased expression of Arid1b following the loss of Arid1a. In addition, we noted that cultured DPCs with double Arid1a/b knockdown gradually under-went apoptosis, but this did not occur in cells with knockdown of only one Arid1 protein. This phenomenon is similar to the synthetic lethality seen with ARID1A and ARID1B in colorectal and ovarian cancer cells (Kelso et al., 2017). This phenomenon was attributed to ARID1B knockdown in ARID1A mutant cells, resulting in further up- or downregulation of accessibility at ARID1A-dependent and unique sites, particularly in enhancer regions (Kelso et al., 2017). Although the chromatin remodeling function of this pair of Arid1s has not been investigated in detail during tooth root development in the current study, this would be an interesting avenue of research for the future. Taken together, these results suggest that Arid1a and Arid1b have interdependent, partially redundant, yet also distinct and context-dependent roles during tissue development and carcinogenesis.
It has also been reported that in addition to the Arid1a-containing SWI/SNF complex (also called the BRG1-associated factors [BAF] complex), there is a non-canonical BAF complex (GBAF complex), which includes bromodomain-containing protein 9 (BRD9), in mouse embryonic stem cells (ESCs) (Gatchalian et al., 2018). The GBAF complex targets distinct genomic features from those targeted by Arid1a and maintains members of the transcriptional network associated with ESC pluripotency, such as Klf4 (Gatchalian et al., 2018). Interestingly, we have shown that Klf4 is a critical transcriptional factor for tooth root development (Feng et al., 2017). Therefore, we speculate that the GBAF complex may also have a distinct function during the fate commitment of root progenitors. Together, all these findings highlight the biological importance of heterogeneous BAF complexes in maintaining transcriptional networks in physiological and pathological conditions.
The accurate regulation of Hh signaling is essential for mammalian organogenesis, tissue homeostasis, and regeneration. Dysregulation of Hh activity can lead to a variety of developmental disorders and malignancies (Ruiz i Altaba et al., 2002; Varjosalo and Taipale, 2008). The function of Hh signaling in regulating cell proliferation is controversial. Some studies have found that aberrant Hh signaling increases proliferation, whereas inhibiting Hh signaling decreases it (Cayuso et al., 2006; Wang et al., 2017). However, other studies show the function of Hh signaling in promoting cell cycle exit of neuroblasts and demonstrate that loss of Hh signaling causes delayed cell cycle exit and excessive proliferation (Chai et al., 2013; Locker et al., 2006). The findings in our study are consistent with the latter set of findings mentioned above, suggesting that Hh signaling has multifaceted functions that are critical in different contexts.
Our study shows that Arid1a regulates the fate commitment of root progenitor cells by finetuning Hh signaling. Similar regulation of chromatin modification with an effect on Hh signaling has also been shown in the morphogenesis of other organs. For example, the molecular circuitry among SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A, member 4 (SMARCA4), and Hh signaling regulates neural development and hair follicle regeneration (Xiong et al., 2013; Zhan et al., 2011), and the scaffolding subunit of SWI/SNF, Srg3/mBaf155, is essential for patterning anteroposterior limb skeletal elements by transcriptional upregulation of Hh/Gli target genes in the posterior limb bud (Jeon and Seong, 2016). Interestingly, there is a recent report about the critical function of Arid1a in intestinal stem cells through Wnt signaling and Sox9 regulation (Hiramatsu et al., 2019). However, there is no apparent change in the Wnt signaling pathway after loss of Arid1a in tooth root progenitor cells (Figure S5A), which suggests that the regulatory function of Arid1a is tissue specific.
Interestingly, although Arid1a is widely expressed throughout the developing tooth germ prior to root formation, it exerts its functional specificity through regulating the fate of Gli1+ progenitor cells during root development, as loss of Arid1a in committed ODs does not cause root development defects. The tissue and stage specificity of Arid1a in regulating organogenesis are based on its chromatin remodeler role, through which it can mobilize nucleosomes and regulate gene transcription with DNA-sequence-specific binding functions such as TFs (Chandler et al., 2013). For example, our study shows that Arid1a regulates the transition of Gli1+ root progenitor cells from proliferation to differentiation with the help of Plagl1. This is an evolutionary outcome that accommodates the acquisition of a high degree of cellular complexity from high-density chromatin packaging in eukaryotes. This might be a common mechanism of tissue-specific regulation by chromatin remodelers, as similar modulation has also been found in the function of other chromatin remodelers; for example, the Nucleosome Remodeling and Deacetylase (NuRD) complex interacts physically with the multi-zinc finger protein FOG-2 and directly downregulates the cell cycle inhibitor Cdkn1a in cardiomyocytes during heart development (Garnatz et al., 2014).
Here, we have shown that Plagl1 motif hits in ATAC regions in the transcriptional regulatory region of Gli1 and the promoter regions of Ptch1 and Ccnd1. Furthermore, knockdown of Plagl1 in DPCs results in reduced expression of Gli1, phenocopying the loss of Arid1a, suggesting Plagl1 might participate in Arid1a’s regulation of Hh signaling in the apical part of the tooth root. Furthermore, Plagll co-localizes with Ki67, a marker labeling cycling cells, in the apical part of the developing tooth root (Figure S7C), suggesting that Plagl1 may contribute to the cell-type/spatial functional specificity of Arid1a during the transition of Gli1+ root progenitor cells from proliferation to differentiation. Consistent with our finding, previous studies have also shown that Plagl1 participates in the cell fate commitment of stem/progenitor cells during neurogenesis, chondrogenesis, and myogenesis (Valente et al., 2005). The pivotal function of Plagl1 in stem/progenitor cells is one of several roles it plays. As a TF, Plagl1 directly regulates genes that are critical for organogenesis or tissue homeostasis at their proximal promoter regions, such as Tcf4 in neuronal progenitors (Schmidt-Edelkraut et al., 2014) and Glu4 in cardiomyocytes (Czubryt et al., 2010). Furthermore, as a co-factor, Plagl1 interacts directly with Pax7 during myogenic differentiation (Yang et al., 2018), with Nkx2.5 to synergistically activate cardiac gene expression (Yuasa et al., 2010), and with p53 and p300/CREB-binding protein in embryonic stem cells differentiating into neurons (Hoffmann and Spengler, 2008). Our findings expand the understanding of its co-factor role by elucidating its interaction with the chromatin remodeler Arid1a in progenitor cells to regulate cell cycle exit and differentiation during tooth root formation.
Significantly, the cell cycle arrest defect after loss of Arid1a further leads to increased apoptosis at a later stage, which is consistent with findings in intestine (Hiramatsu et al., 2019). However, our finding of increased cell proliferation is distinct from what has been seen in other systems, such as intestine and blood (Han et al., 2019; Hiramatsu et al., 2019). The context-dependent functions of Arid1a and different downstream signaling pathways affected in different tissues may contribute to this discrepancy. We have also confirmed our findings of over-activated mitosis and downregulated Hh signaling after loss of Arid1a in the development of the mandibular condyle, which is another CNC-derived tissue, suggesting the function of Arid1a in the tooth root model might reflect regulatory role during the organogenesis of CNC-derived tissues more broadly.
In summary, our study has identified an Arid1a-Plagl1-Hh signaling cascade that is indispensable for differentiation-associated cell cycle arrest of tooth root progenitors. This study further expands our knowledge of the delicate interaction among signaling pathways, TFs, and chromatin remodelers during the specific fate determination and lineage commitment of stem/progenitor cells. These findings may also shed light on the etiology of dental abnormalities and are relevant for developing strategies for tooth regeneration.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yang Chai (ychai@usc.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and code availability
The bulk RNA-seq data, ATAC-seq data and ChIP-seq data from mouse molars in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE166360 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166360).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Arid1afl/fl (Gao et al., 2008), Gli1-CreER (Ahn and Joyner, 2004), tdTomato (Madisen et al., 2010), Dmp1-Cre (Lu et al., 2007), K14-rtTA (Xie et al., 1999), tetO-Cre (Perl et al., 2002), Gli1-LacZ (Bai et al., 2002), and SmoM2fl/fl (Jeong et al., 2004) mouse lines were used and cross-bred in this study. All mouse lines used in the present study are listed in the Key resources table. All mice were housed in pathogen-free conditions and euthanized by carbon dioxide overdose followed by cervical dislocation. All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Southern California. Both male and female mice were included in our experiments.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-ARID1A | Abcam | Cat# ab182561 |
| Rabbit anti-Ki67 | Abcam | Cat# ab15580, RRID:AB_443209 |
| Chicken anti-beta Galactosidase | Abcam | Cat# ab9361, RRID:AB_307210 |
| Rabbit anti-K14 | Abcam | Cat# ab181595, RRID:AB_2811031 |
| Rabbit anti-phospho-Histone H3 (Ser10) | Millipore | Cat# 06-570, RRID:AB_310177 |
| Rabbit anti-Cyclin D1 | Abcam | Cat# ab16663, RRID:AB_443423 |
| Rabbit polyclonal anti-Periostin | Abcam | Cat# ab14041, RRID:AB_2299859 |
| Rabbit anti-Arid1b | Abcam | Cat# ab244351 |
| Mouse monoclonal anti-ARID1A | Santa Cruz | Cat# sc-32761, RRID:AB_673396 |
| Mouse monoclonal anti-ZAC1(Plagl1) | Santa Cruz | Cat# sc-166944, RRID:AB_10613622 |
| Rabbit anti-Gli1 | Novus | Cat# NBP1-78259, RRID:AB_11030198 |
| Mouse monoclonal anti-DSPP | Santa Cruz | Cat# sc-73632, RRID:AB_2230660 |
| Sheep anti-DMP-1 | R&D | Cat# AF4386, RRID:AB_2091367 |
| Mouse monoclonal anti-cyclin D1 | Santa Cruz | sc-8396, RRID:AB_627344 |
| Mouse monoclonal anti-beta Actin | Abcam | Cat# ab20272, RRID:AB_445482 |
| Rabbit anti-ARID1A/BAF250A (D2A8U) | Cell Signaling Technology | Cat# 12354, RRID:AB_2637010 |
| Rabbit anti-ARID1B/BAF250B (E9J4T) | Cell Signaling Technology | Cat# 92964, RRID:AB_2800195 |
| Normal Rabbit IgG | Cell Signaling Technology | Cat# 2729, RRID:AB_1031062 |
| Mouse IgG HRP-conjugated antibody | R&D | Cat# HAF007, RRID:AB_357234 |
| Rabbit IgG HRP-conjugated antibody | R&D | Cat# HAF008, RRID:AB_357235 |
| Sheep IgG HRP-conjugated antibody | R&D | Cat# HAF016, RRID:AB_562591 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11008, RRID:AB_143165 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-21207, RRID:AB_141637 |
| Goat anti-Chicken IgY (H+L) Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11039, RRID:AB_142924 |
| Chemicals, peptides, and recombinant proteins | ||
| Tamoxifen | Sigma-Aldrich | Cat# T5648-5G |
| Rodent Diet (2018, 625 Dox, R) | ENVIGO | Cat# TD.08541 |
| Fetal Bovine Serum | Thermo Fisher Scientific | Cat# 12662029 |
| α-MEM | Thermo Fisher Scientific | Cat# 12571071 |
| Collagenase, Type 1 | Worthington | Cat# LS004194 |
| RIPA Buffer (10X) | Cell Signaling Technology | Cat# 9806s |
| Protease inhibitor | ThermoFisher Scientific | Cat# A32959 |
| DAPI Solution (1 mg/mL) | Thermo Fisher Scientific | Cat# 62248 |
| Alizarin Red S | ACROS Organics | Cat# 400480250 |
| Critical commercial assays | ||
| Click-iT Plus TUNEL Assay for In Situ Apoptosis Detection, Alexa Fluor 488 dye | Thermo Fisher Scientific | Cat# C10617 |
| Click-iT Plus EdU Cell Proliferation Kit for Imaging, Alexa Fluor 488 dye | Thermo Fisher Scientific | Cat# C10637 |
| RNeasy Micro Kit | QIAGEN | Cat# 74004 |
| RNAscope® Multiplex Fluorescent v2 | ACD | Cat# 323100 |
| RNAscope 2.5 HD Reagent Kit-RED assay | ACD | Cat# 322350 |
| Probe- Mm- Gli1 | ACD | Cat# 311001 |
| Probe- Mm- Gli1-C2 | ACD | Cat# 311001-C2 |
| Probe- Mm- Ptch1 | ACD | Cat# 402811 |
| Probe- Mm- Plagl1 | ACD | Cat# 462941 |
| Probe- Mm- Dspp | ACD | Cat# 448301 |
| Probe- Mm- Axin2 | ACD | Cat# 400331 |
| Probe- Mm- Lef1 | ACD | Cat# 441861 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat# 1708891 |
| SsoAdvanced Universal SYBR® Green Supermix | Bio-Rad | Cat# 1725270 |
| SimpleChIP® Plus Enzymatic Chromatin IP Kit | Cell Signaling Technology | Cat# 9005 |
| Deposited data | ||
| Bulk RNA-seq data | This paper | GEO: GSE166360 |
| ATAC-seq data | This paper | GEO: GSE166360 |
| Arid1b ChIP-Seq data | This paper | GEO: GSE166360 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | JAX:000664, RRID:IMSR_JAX:000664 |
| Mouse: Arid1afl/fl:STOCK Arid1atm1, 1Zhwa | Jackson Laboratory | JAX:027717, RRID:IMSR_JAX:027717 |
| Mouse: Gli1-CreER: STOCK Gli1tm3(cre/ERT2)Alj | Jackson Laboratory | JAX:007913, RRID:IMSR_JAX:007913 |
| Mouse: Gli1-LacZ:STOCK Gli1tm2Alj/J | Jackson Laboratory | JAX:008211, RRID:IMSR_JAX:008211 |
| Mouse: tdT:ROSA26loxp-STOP-loxp-tdTomato:B6;129S6-Gt(ROSA) 26Sortm9(CAG-tdTomato)Hze/J | Jackson Laboratory | JAX:007905, RRID:IMSR_JAX:007905 |
| Mouse: K14-rtTA:B6;SJL-Tg(KRT14-rtTA) 208Jek | Jackson Laboratory | JAX:007678, RRID:IMSR_JAX:007678 |
| Mouse: tetO-cre: B6.Cg-Tg(tetO-cre)1Jaw/J | Jackson Laboratory | JAX:006234, RRID:IMSR_JAX:006234 |
| Mouse: Dmp1-Cre: B6N.FVB-Tg (Dmp1-cre)1Jqfe/Bwd | Jackson Laboratory | JAX:023047, RRID:IMSR_JAX:023047 |
| Mouse: SmoM2fl/fl | (Jeong et al., 2004) | N/A |
| Oligonucleotides | ||
| Primer sequences | See Table S2 | N/A |
| Software and algorithms | ||
| ImageJ | NIH | RRID:SCR_003070 |
| GraphPad Prism | GraphPad Software | RRID:SCR_002798 |
METHOD DETAILS
Tamoxifen and doxycycline administration
Tamoxifen (Sigma, T5648) was dissolved in corn oil (Sigma, C8267) at 20 mg/ml. At PN3.5, control and Gli1-CreER;Arid1afl/fl mice were injected intraperitoneally at a dosage of 1.5 mg/10 g body weight. Doxycycline rodent diet (Envigo, TD.08541) was administered to the feeding dam every day beginning when the gestating pups were at PN3.5; the pups were also injected with doxycycline intraperitoneally at a dosage of 50 μg/g body weight every two days from PN3.5.
Cell culture and dentinogenic induction
Dental pulp mesenchyme cells (DPCs) was collected from the apical portion of first mandibular molars from wild-type mice at PN3.5-PN7.5. The tissues were cut into pieces and cultured in α-MEM (Thermo Fisher, 12571071) with 10% FBS (Thermo Fisher, 12662029) at 37°C in a 5% CO2 incubator. The cell culture medium was changed every two days. When the primary DPCs reached sub-confluence, they were passaged for siRNA transfection. In particular, Plagl1 siRNA (Thermo Fisher, 4390771, Assay ID s76196), Arid1b siRNA (Thermo Fisher, 4390771, #1 Assay ID s109133, #2 Assay ID s109134, #3 Assay ID s109135, #4 Assay ID s202727), Arid1a siRNA (QIAGEN, 1027418, #1 Mm_Arid1a_5 FlexiTube siRNA SI02676058; #2 Mm_Arid1a_6 FlexiTube siRNA SI02696771), control siRNA (Thermo Fisher, 4390844), lipofectamine™ RNAiMAX (Thermo Fisher, 13778075) and Opti-MEM I Reduced Serum Medium (Thermo Fisher, 31985062) were used in this study. The final concentration of each siRNA was 50 μM during reverse transfection. When the transfected DPCs reached sub-confluence, the medium was changed to odontogenic induction medium containing 1% FBS, β-glycerophosphate (β-GP) (5 mM), ascorbic acid (50 μg/ml), and dexamethasone (100 nM) in α-MEM. The odontogenic induction medium was changed every two days until protein extraction or mineralization evaluation. The mineralization ability of DPCs after odontogenic induction were evaluated using Alizarin red S staining (ACROS Organics, 400480250) as reported previously (Chen et al., 2020). In detail, at specific time points, the cultured cells were fixed with 4% PFA for 15 minutes on ice, then the fixed cells were incubated with 2% Alizarin red S solution for 30 minutes for visualization of mineralized nodules.
MicroCT analysis
MicroCT analysis of fixed mandibles was performed using a Skyscan 1174v1.2 (Bruker Corporation, USA) at 50 kVp, 800 μA and a resolution of 16.7 mm. Sagittal two-dimensional sections were obtained with NRecon version 1.6.9.8 (Bruker Corporation, USA) and three-dimensional reconstruction was done using Avizo/Amira 9.5.0 (FEI Visualization Sciences Group, France).
Histological analysis
Mouse mandibles were dissected and fixed in 4% PFA overnight, followed by decalcification in 10% EDTA in PBS for 1-3 weeks depending on the age of the samples. For hematoxylin and eosin (HE) staining, the decalcified mandibles were dehydrated with serial ethanol and xylene and embedded in paraffin. The paraffin-embedded samples were then cut into sections with a thickness of 4 μm using a microtome (Leica) and HE staining was performed according to standard protocol.
Immunofluorescence and in situ hybridization
The decalcified mandibles were dehydrated in serial sucrose/PBS solutions and embedded in OCT compound (Tissue-Tek, Sakura). OCT-embedded samples were cryosectioned at 8 μm using a cryostat (Leica CM1850) followed by staining. For immunofluorescence staining, cryosections were soaked in blocking solution (PerkinElmer, FP1012) for one hour at room temperature and then incubated with primary antibodies diluted in blocking solution at 4°C overnight. After washing three times in PBS, the sections were incubated with alexa-conjugated secondary antibody (Thermo Fisher) and counterstained with DAPI (Thermo Fisher Scientific, 62248). The primary antibodies are listed in Table S1. For in situ hybridization analysis, cryosections were stained with RNAscope Multiplex Fluorescent kit (Advanced Cell Diagnostics, 323100) or RNAscope 2.5 HD Reagent Kit-RED assay (Advanced Cell Diagnostics, 322350) according to the manufacturer’s instructions. All of the probes are listed in the Key resources table.
EdU incorporation, staining and TUNEL assays
Gli1-CreER;Arid1afl/fl mice and littermate controls were injected with EdU (10 μg/g body weight) intraperitoneally 48 hours before being euthanized. The mandibles were fixed and decalcified. Click-iT plus EdU cell proliferation kit (Thermo Fisher, C10637) was used on the cryosections for in situ EdU detection according to the manufacturer’s instructions.
Cell apoptosis was detected using a TUNEL assay (Click-iT Plus TUNEL Assay for In Situ Apoptosis Detection, Thermo Fisher, C10617) according to the manufacturer’s protocol.
RNA-sequencing analysis
At PN7.5, 4 days after tamoxifen induction, first mandibular molars from Gli1-CreER;Arid1afl/fl mice and littermate controls were dissected out. Then, the apical third of each first molar was removed and RNA was extracted with an RNeasy Micro Kit (QIAGEN, 74004). For RNA-sequencing analysis, cDNA library preparation and sequencing were performed at the Technology Center for Genomics & Bioinformatics at the University of California, Los Angeles (UCLA). Raw reads were trimmed and aligned with the mm10 genome. Differential analysis was performed by selecting transcripts that changed with an FDR cut-off of 0.1.
Quantitative reverse transcription PCR
RNA in the first mandibular molars was extracted as above. For quantitative reverse transcription PCR (qPCR) analysis, RNA was reverse-transcribed with an iScript cDNA Synthesis Kit (Bio-Rad, 1708891) and the relative amounts of each mRNA transcript were analyzed using the CFX96 Real-Time System (Bio-Rad, iCycler) with SsoAdvanced Universal SYBR® Green Supermix (Bio-Rad, 1725270). The relative expression levels of particular genes were compared across groups using the 2−ΔΔCt method, with the expression of beta-actin as an internal control. Primer sequences were obtained from PrimerBank (https://pga.mgh.harvard.edu/primerbank/) and are listed in the Table S2.
Western blot and co-immunoprecipitation
At PN7.5,4 days after tamoxifen induction, the apical thirds of first mandibular molars from Gli1-CreER;Arid1afl/fl mice and littermate controls were cut into pieces and homogenized in RIPA buffer (Cell Signaling, 9806s) supplemented with protease inhibitor (Thermo Fisher Scientific, A32959). Western blot was performed per standard protocol and signals were detected using Azure 300 (Azure biosystems). The primary antibodies are listed in Table S1. HRP-conjugated secondary antibodies (R&D, HAF007, HAF008, and HAF016) were used in the study. For co-immunoprecipitation (co-IP), DPCs cultured in vitro were harvested and lysed in lysis buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA (pH 8.0), 1mM PMSF, 1% NP-40, 5% glycerol]. Then lysates were subjected to immunoprecipitation with anti-Arid1a antibody (Abcam, ab182561) or normal Rabbit IgG (Cell Signaling Technology, 2729) and protein A-Sepharose (VWR, CA97067-898). Immune complexes were washed and subjected to immunoblotting with anti-Arid1a (Santa Cruz, sc-32761) or anti-Plagl1 (Santa Cruz, sc-166944) antibodies.
ATAC-seq and motif analysis
First mandibular molars from Gli1-CreER;Arid1afl/fl mice (MUT) and littermate controls (WT) were dissected out at PN7.5, 4 days after tamoxifen induction. The apical thirds of their first molars were collected and single cells were isolated with collagenase, type 1 (Worthington, LS004194). Then, 5000 cells from each group were used for preparation of ATAC-seq libraries according to the protocol described by Kaestner Lab (https://www.med.upenn.edu/kaestnerlab/protocols.html). The transposed DNA libraries were sequenced using 50 bp paired-end (50PE) sequencing on a Novaseq SP system at the Technology Center for Genomics & Bioinformatics at the University of California, Los Angeles (UCLA).
Library sequencing quality was assessed using FastQC v0.11.8 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and trimmed with Cutadapt v1.18 with default parameters (TrimGalore v0.6.5, https://www.bioinformatics.babraham.ac.uk). Trimmed libraries were aligned to the mm10 mouse genome using bowtie2 v2.3.5 (Langmead and Salzberg, 2012) (non-deterministic, pair-end mode and the rest of the settings left at their defaults). All unique alignments (MAPQ > = 10) were further filtered by Picard MarkDuplicates v2.21.4 (http://broadinstitute.github.io/picard) to remove duplicates. ATAC-Seq peaks were called by MACS2 (Cros et al., 2021; Zhang et al., 2008) v2.1.1 (callpeak -g ‘mm’, –format BAMPE) for each library with a stringent statistical criterion (fold enrichment > 4, p value < 1e-5 and FDR < 0.05). 9950, 8396 and 13342 high-confidence peaks were identified in WT replicate 1, WT replicate 2 and MUT groups, respectively. These peaks were merged using Bedtools (Quinlan and Hall, 2010) into 13500 chromatin accessibility regions of interest. Raw read counts from the three libraries on these regions were used as input for edgeR v3.20.7.2 (Robinson et al., 2010) (quasi-likelihood test, robust, fold-change 1.5 and FDR < 0.1) to identify differential ATAC-Seq peaks between WT and MUT. 1 MUT-specific peak and 215 WT-specific peaks were identified. The sequences of ± 100 bps flanking the peak summits were used for motif enrichment analysis with HOMER suite v4.11 (Heinz et al., 2010) with genome background.
ChIP assay
Primary DPCs, collected from the apical portion of first mandibular molars from wild-type mice at PN3.5-PN7.5, were fixed with formaldehyde after amplification for further ChIP-sequencing analysis. The apical portions of first mandibular molars from wild-type mice at PN3.5-PN7.5 were collected and fixed with formaldehyde for ChIP-qPCR analysis. The SimpleChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads; Cell Signaling Technology 9005), anti-Arid1a antibody (Cell Signaling, 12354), anti-Arid1b antibody (Cell Signaling, 92964), and rabbit IgG (Cell Signaling, 2729) were used following the manufacturer’s instructions. The enrichment of particular DNA sequences during IP was sequenced using 75 bp single-read sequencing on a NextSeq500 High Output system at the Technology Center for Genomics & Bioinformatics at UCLA. The sequencing quality of ChIP-Seq libraries was assessed by FastQC v0.11.8 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Bowtie2 v2.2.5 (Langmead and Salzberg, 2012) was used to align the sequencing reads to the mm10 mouse genome. All unique alignments (MAPQ > = 10) were further filtered by Picard Mark-Duplicates v2.21.4 (http://broadinstitute.github.io/picard) to remove duplicates. The Gli1 ChIP primers for binding site 1 (BS1) and binding site 2 (BS2) amplified a region at the second intron and proximal promoter, respectively, as listed in Table S2.
ImageJ image analysis
ImageJ was used to calculate the root length, the percentage of positive immunofluorescence signals in the apical regions of mouse molars. In particular, the root length of the mandibular first molar was measured along the mesial border of the mesial root up to the cemento-enamel junction based on microCT data (N = 3 for each group). The percentage of Tdt+PHH3+ (Figure 3G), Tdt+Tunel+ (Figure S3G), EdU+PHH3+ (Figure 3T) and EdU+PHH3− (Figure 3T) cells out of the total (%DAPI+) in the apical region of the mouse mandibular first molar (250 μm × 200 μm area surrounding Hertwig’s epithelial root sheath of mesial root) was quantified in discontinuous sagittal sections with about 50 μm intervals. Individual values in Figures 3G and S3G represent the average values from three sections of each sample, and three pairs of samples were included. The value in Figure 3T represents the average value from four sections of each sample, and five pairs of samples were included.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed with GraphPad Prism and are presented as mean ± standard deviation unless otherwise stated. Two-tailed Student’s t test or one-way analysis of variance (ANOVA) were applied for comparisons, with P values < 0.05 considered statistically significant. N ≥ 3 for all samples; all experiments were repeated in triplicate or more unless otherwise stated.
Supplementary Material
Highlights.
Arid1a promotes the proliferation-differentiation transition of root progenitors
Hh signaling participates in Arid1a’s regulation of tooth root progenitor cell fate
Plagl1 endows Arid1a with its cell-type/spatial functional specificity
Arid1b is crucial for odontoblast differentiation but not involved in Hh signaling
ACKNOWLEDGMENTS
We acknowledge Dr. Bridget Samuels and Ms. Linda Hattemer for critical editing of the manuscript, USC Libraries Bioinformatics Service for assisting with data analysis, and the USC Office of Research and the USC Libraries for supporting our access to bioinformatics software and computing resources. This study was supported by funding from the National Institute of Dental and Craniofacial Research, National Institutes of Health (R01 DE022503; U01 DE028729; R01 DE012711).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108964.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adnani L, Langevin LM, Gautier E, Dixit R, Parsons K, Li S, Kaushik G, Wilkinson G, Wilson R, Childs S, et al. (2015). Zac1 Regulates the Differentiation and Migration of Neocortical Neurons via Pac1. J. Neurosci 35, 13430–13447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S, and Joyner AL (2004). Dynamic changes in the response of cells to positive hedgehog signaling during mouse limb patterning. Cell 118, 505–516. [DOI] [PubMed] [Google Scholar]
- Bai CB, Auerbach W, Lee JS, Stephen D, and Joyner AL (2002). Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 129, 4753–4761. [DOI] [PubMed] [Google Scholar]
- Cakouros D, and Gronthos S (2020). Epigenetic Regulators of Mesenchymal Stem/Stromal Cell Lineage Determination. Curr. Osteoporos. Rep 18, 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayuso J, Ulloa F, Cox B, Briscoe J, and Martí E (2006). The Sonic hedgehog pathway independently controls the patterning, proliferation and survival of neuroepithelial cells by regulating Gli activity. Development 133, 517–528. [DOI] [PubMed] [Google Scholar]
- Chai Y, Jiang X, Ito Y, Bringas P Jr., Han J, Rowitch DH, Soriano P, McMahon AP, and Sucov HM (2000). Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development 127, 1671–1679. [DOI] [PubMed] [Google Scholar]
- Chai PC, Liu Z, Chia W, and Cai Y (2013). Hedgehog signaling acts with the temporal cascade to promote neuroblast cell cycle exit. PLoS Biol. 11, e1001494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RL, and Magnuson T (2016). The SWI/SNF BAF-A complex is essential for neural crest development. Dev. Biol 411, 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RL, Brennan J, Schisler JC, Serber D, Patterson C, and Magnuson T (2013). ARID1a-DNA interactions are required for promoter occupancy by SWI/SNF. Mol. Cell. Biol 33, 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Jing J, Yuan Y, Feng J, Han X, Wen Q, Ho TV, Lee C, and Chai Y (2020). Runx2+ Niche Cells Maintain Incisor Mesenchymal Tissue Homeostasis through IGF Signaling. Cell Rep. 32, 108007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cros J, Theou-Anton N, Gounant V, Nicolle R, Reyes C, Humez S, Hescot S, Thomas de Montpreville V, Guyetant S, Scoazec JY, et al. (2021). Specific genomic alterations in high grade pulmonary neuroendocrine tumours with carcinoid morphology. Neuroendocrinology 111, 158–169. [DOI] [PubMed] [Google Scholar]
- Czubryt MP, Lamoureux L, Ramjiawan A, Abrenica B, Jangamreddy J, and Swan K (2010). Regulation of cardiomyocyte Glut4 expression by ZAC1. J. Biol. Chem 285, 16942–16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, and Wicha MS (2004). Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 6, R605–R615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Jing J, Li J, Zhao H, Punj V, Zhang T, Xu J, and Chai Y (2017). BMP signaling orchestrates a transcriptional network to control the fate of mesenchymal stem cells in mice. Development 144, 2560–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Tate P, Hu P, Tjian R, Skarnes WC, and Wang Z (2008). ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc. Natl. Acad. Sci. USA 105, 6656–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnatz AS, Gao Z, Broman M, Martens S, Earley JU, and Svensson EC (2014). FOG-2 mediated recruitment of the NuRD complex regulates cardiomyocyte proliferation during heart development. Dev. Biol 395, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatchalian J, Malik S, Ho J, Lee DS, Kelso TWR, Shokhirev MN, Dixon JR, and Hargreaves DC (2018). A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat. Commun 9, 5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golestaneh N, Beauchamp E, Fallen S, Kokkinaki M, Uren A, and Dym M (2009). Wnt signaling promotes proliferation and stemness regulation of spermatogonial stem/progenitor cells. Reproduction 138, 151–162. [DOI] [PubMed] [Google Scholar]
- Han L, Madan V, Mayakonda A, Dakle P, Woon TW, Shyamsunder P, Nordin HBM, Cao Z, Sundaresan J, Lei I, et al. (2019). Chromatin remodeling mediated by ARID1A is indispensable for normal hematopoiesis in mice. Leukemia 33, 2291–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helming KC, Wang X, and Roberts CWM (2014). Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell 26, 309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiramatsu Y, Fukuda A, Ogawa S, Goto N, Ikuta K, Tsuda M, Matsumoto Y, Kimura Y, Yoshioka T, Takada Y, et al. (2019). Arid1a is essential for intestinal stem cells through Sox9 regulation. Proc. Natl. Acad. Sci. USA 116, 1704–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, and Spengler D (2008). A new coactivator function for Zac1’s C2H2 zinc finger DNA-binding domain in selectively controlling PCAF activity. Mol. Cell. Biol 28, 6078–6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hota SK, and Bruneau BG (2016). ATP-dependent chromatin remodeling during mammalian development. Development 143, 2882–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon S, and Seong RH (2016). Anteroposterior Limb Skeletal Patterning Requires the Bifunctional Action of SWI/SNF Chromatin Remodeling Complex in Hedgehog Pathway. PLoS Genet. 12, e1005915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Mao J, Tenzen T, Kottmann AH, and McMahon AP (2004). Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 18, 937–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, and Hui CC (2008). Hedgehog signaling in development and cancer. Dev. Cell 15, 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson MKB, Hartman RJG, Ackers-Johnson M, Tan WLW, Lim B, van Veen TAB, and Foo RS (2016). A Transcriptomic and Epigenomic Comparison of Fetal and Adult Human Cardiac Fibroblasts Reveals Novel Key Transcription Factors in Adult Cardiac Fibroblasts. JACC Basic Transl. Sci 1, 590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso TWR, Porter DK, Amaral ML, Shokhirev MN, Benner C, and Hargreaves DC (2017). Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. eLife 6, e30506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krosl J, Mamo A, Chagraoui J, Wilhelm BT, Girard S, Louis I, Lessard J, Perreault C, and Sauvageau G (2010). A mutant allele of the Swi/Snf member BAF250a determines the pool size of fetal liver hemopoietic stem cell populations. Blood 116, 1678–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumakami-Sakano M, Otsu K, Fujiwara N, and Harada H (2014). Regulatory mechanisms of Hertwig’s epithelial root sheath formation and anomaly correlated with root length. Exp. Cell Res 325, 78–82. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei I, Gao X, Sham MH, and Wang Z (2012). SWI/SNF protein component BAF250a regulates cardiac progenitor cell differentiation by modulating chromatin accessibility during second heart field development. J. Biol. Chem 287, 24255–24262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei I, West J, Yan Z, Gao X, Fang P, Dennis JH, Gnatovskiy L, Wang W, Kingston RE, and Wang Z (2015). BAF250a Protein Regulates Nucleosome Occupancy and Histone Modifications in Priming Embryonic Stem Cell Differentiation. J. Biol. Chem 290, 19343–19352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Parada C, and Chai Y (2017). Cellular and molecular mechanisms of tooth root development. Development 144, 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Feng J, Li J, Zhao H, Ho TV, and Chai Y (2015). An Nfic-hedgehog signaling cascade regulates tooth root development. Development 142, 3374–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locker M, Agathocleous M, Amato MA, Parain K, Harris WA, and Perron M (2006). Hedgehog signaling and the retina: insights into the mechanisms controlling the proliferative properties of neural precursors. Genes Dev. 20, 3036–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Xie Y, Zhang S, Dusevich V, Bonewald LF, and Feng JQ (2007). DMP1-targeted Cre expression in odontoblasts and osteocytes. J. Dent. Res 86, 320–325. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur R, Alver BH, San Roman AK, Wilson BG, Wang X, Agoston AT, Park PJ, Shivdasani RA, and Roberts CWM (2017). ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet 49, 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA, Baek S, Sung MH, John S, Wiench M, Johnson TA, Schiltz RL, and Hager GL (2014). Overlapping chromatin-remodeling systems collaborate genome wide at dynamic chromatin transitions (vol 21, pg 73, 2014). Nat. Struct. Mol. Biol 21, 1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl AK, Wert SE, Nagy A, Lobe CG, and Whitsett JA (2002). Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc. Natl. Acad. Sci. USA 99, 10482–10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab JR, Resnick S, and Magnuson T (2015). Genome-Wide Transcriptional Regulation Mediated by Biochemically Distinct SWI/SNF Complexes. PLoS Genet. 11, e1005748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rraklli V, Södersten E, Nyman U, Hagey DW, and Holmberg J (2016). Elevated levels of ZAC1 disrupt neurogenesis and promote rapid in vivo reprogramming. Stem Cell Res. (Amst.) 16, 1–9. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A, Sánchez P, and Dahmane N (2002). Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat. Rev. Cancer 2, 361–372. [DOI] [PubMed] [Google Scholar]
- Santen GW, Aten E, Vulto-van Silfhout AT, Pottinger C, van Bon BW, van Minderhout IJ, Snowdowne R, van der Lans CA, Boogaard M, Linssen MM, et al. ; Coffin-Siris consortium (2013). Coffin-Siris syndrome and the BAF complex: genotype-phenotype study in 63 patients. Hum. Mutat 34, 1519–1528. [DOI] [PubMed] [Google Scholar]
- Schmidt-Edelkraut U, Daniel G, Hoffmann A, and Spengler D (2014). Zac1 regulates cell cycle arrest in neuronal progenitors via Tcf4. Mol. Cell. Biol 34, 1020–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spengler D, Villalba M, Hoffmann A, Pantaloni C, Houssami S, Bockaert J, and Journot L (1997). Regulation of apoptosis and cell cycle arrest by Zac1, a novel zinc finger protein expressed in the pituitary gland and the brain. EMBO J. 16, 2814–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Taylor R, Long J, Yoon JW, Childs R, Sylvestersen KB, Nielsen ML, Leong KF, Iannaccone S, Walterhouse DO, Robbins DJ, and Iannaccone P (2019). Regulation of GLI1 by cis DNA elements and epigenetic marks. DNA Repair (Amst.) 79, 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trizzino M, Barbieri E, Petracovici A, Wu S, Welsh SA, Owens TA, Licciulli S, Zhang R, and Gardini A (2018). The Tumor Suppressor ARID1A Controls Global Transcription via Pausing of RNA Polymerase II. Cell Rep. 23, 3933–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente T, Junyent F, and Auladell C (2005). Zac1 is expressed in progenitor/stem cells of the neuroectoderm and mesoderm during embryogenesis: differential phenotype of the Zac1-expressing cells during development. Dev. Dyn 233, 667–679. [DOI] [PubMed] [Google Scholar]
- Varjosalo M, and Taipale J (2008). Hedgehog: functions and mechanisms. Genes Dev. 22, 2454–2472. [DOI] [PubMed] [Google Scholar]
- Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, Aknin C, Severac D, Chotard L, Kahli M, Le Digarcher A, et al. (2006). Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell 11, 711–722. [DOI] [PubMed] [Google Scholar]
- Varrault A, Dantec C, Le Digarcher A, Chotard L, Bilanges B, Parrinello H, Dubois E, Rialle S, Severac D, Bouschet T, and Journot L (2017). Identification of Plagl1/Zac1 binding sites and target genes establishes its role in the regulation of extracellular matrix genes and the imprinted gene network. Nucleic Acids Res. 45, 10466–10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasileiou G, Ekici AB, Uebe S, Zweier C, Hoyer J, Engels H, Behrens J, Reis A, and Hadjihannas MV (2015). Chromatin-Remodeling-Factor ARID1B Represses Wnt/β-Catenin Signaling. Am. J. Hum. Genet 97, 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DJ, Hu GH, Du Y, Zhang C, Lu QQ, Lv NH, and Luo SW (2017). Aberrant activation of hedgehog signaling promotes cell proliferation via the transcriptional activation of forkhead Box M1 in colorectal cancer cells. J. Exp. Clin. Cancer Res 36, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Q, Jing J, Han X, Feng J, Yuan Y, Ma Y, Chen S, Ho TV, and Chai Y (2020). Runx2 regulates mouse tooth root development via activation of WNT inhibitor Notum. J. Bone Miner. Res 35, 2252–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek D, Bögershausen N, Beleggia F, Steiner-Haldenstätt S, Pohl E, Li Y, Milz E, Martin M, Thiele H, Altmüller J, et al. (2013). A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum. Mol. Genet 22, 5121–5135. [DOI] [PubMed] [Google Scholar]
- Wilson BG, and Roberts CW (2011). SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 11, 481–492. [DOI] [PubMed] [Google Scholar]
- Wu H, and Sun YE (2006). Epigenetic regulation of stem cell differentiation. Pediatr. Res 59, 21R–25R. [DOI] [PubMed] [Google Scholar]
- Xie W, Chow LT, Paterson AJ, Chin E, and Kudlow JE (1999). Conditional expression of the ErbB2 oncogene elicits reversible hyperplasia in stratified epithelia and up-regulation of TGFalpha expression in transgenic mice. Oncogene 18, 3593–3607. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Li W, Shang C, Chen RM, Han P, Yang J, Stankunas K, Wu B, Pan M, Zhou B, et al. (2013). Brg1 governs a positive feedback circuit in the hair follicle for tissue regeneration and repair. Dev. Cell 25, 169–181. [DOI] [PubMed] [Google Scholar]
- Yang Q, Li Y, Zhang X, and Chen D (2018). Zac1/GPR39 phosphorylating CaMK-II contributes to the distinct roles of Pax3 and Pax7 in myogenic progression. Biochim. Biophys. Acta Mol. Basis Dis 1864, 407–419. [DOI] [PubMed] [Google Scholar]
- Yao S, Pan F, Prpic V, and Wise GE (2008). Differentiation of stem cells in the dental follicle. J. Dent. Res 87, 767–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa S, Onizuka T, Shimoji K, Ohno Y, Kageyama T, Yoon SH, Egashira T, Seki T, Hashimoto H, Nishiyama T, et al. (2010). Zac1 is an essential transcription factor for cardiac morphogenesis. Circ. Res 106, 1083–1091. [DOI] [PubMed] [Google Scholar]
- Zhan X, Shi X, Zhang Z, Chen Y, and Wu JI (2011). Dual role of Brg chromatin remodeling factor in Sonic hedgehog signaling during neural development. Proc. Natl. Acad. Sci. USA 108, 12758–12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The bulk RNA-seq data, ATAC-seq data and ChIP-seq data from mouse molars in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE166360 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166360).