

Abstract
ADAMTS proteases mediate biosynthesis and breakdown of secreted extracellular matrix (ECM) molecules in numerous physiological and disease processes. In addition to their catalytic domains, ADAMTS proteases contain ancillary domains, which mediate substrate recognition and ECM binding and confer distinctive properties and roles to individual ADAMTS proteases. Although alternative splicing can greatly expand the structural and functional diversity of ADAMTS proteases, it has been infrequently reported and functional consequences have been rarely investigated. Here, we characterize the structural and functional impact of alternative splicing of ADAMTS17, mutations in which cause Weill-Marchesani syndrome 4. Two novel ADAMTS17 splice variants, ADAMTS17A and ADAMTS17B, were investigated by structural modeling, mass spectrometry, and biochemical approaches. Our results identify a novel disulfide-bridged insertion in the ADAMTS17A spacer that originates from inclusion of a novel exon. This insertion results in differential autoproteolysis of ADAMTS17, and thus, predicts altered proteolytic activity against other substrates. The second variant, ADAMTS17B, results from an in-frame exon deletion and prevents ADAMTS17B secretion. Thus, alternative splicing of the ADAMTS spacer significantly regulates the physiologically relevant proteolytic activity of ADAMTS17, either by altering proteolytic specificity (ADAMTS17A) or by altering cellular localization (ADAMTS17B).
Keywords: ADAMTS proteases, fibrillin microfibrils, Weill-Marchesani syndrome, extracellular matrix, alternative splicing
1 |. INTRODUCTION
The disintegrin-like and metalloprotease with thrombospondin type 1 motifs (ADAMTS) family comprises 19 proteases that are secreted into the extracellular matrix (ECM). ADAMTS proteases execute a plethora of functions in tissue development and homeostasis.1,2 For example, ADAMTS2 and ADAMTS3 are procollagen propeptidases that are pivotal for collagen assembly and mediate vascular endothelial growth factor (VEGF)-C processing, respectively.3–5 Accordingly, mutations in ADAMTS2 and ADAMTS3 result in Ehlers-Danlos syndrome dermatosparactic type (or type VIIc) and Hennekam syndrome, respectively.2,4,6 Other ADAMTS proteases, known as aggrecanases (e.g. ADAMTS1, 4, and 5), are key mediators of aggrecan destruction in osteoarthritis and of aggrecan and versican turnover during development.7–10 Two other homologous proteases, ADAMTS9 and ADAMTS20 not only mediate ECM turnover, but also have an unexpected role in the formation of the primary cilium.11 Given their numerous roles in development and human disease, it is clear that ADAMTS proteases represent an important group of proteases shaping and reshaping the ECM in most, if not all, connective tissues.2,12
ADAMTS proteases are composed of a highly conserved N-terminal protease domain and a C-terminal ancillary domain, which mediates substrate binding and ECM localization. Accordingly, ADAMTS ancillary domains have a variable modular structure, albeit with strict conservation of canonical core domains.13 These core domains include a thrombospondin type 1 repeat (TSR), a cysteine-rich domain, and the spacer domain. Additional TSRs, mucin-rich domains, PLAC, or GON-1 domains and their specific arrangement following the spacer domain define the individual ADAMTS proteases and presumably underlie, at least in part, their individual biological functions.13 Functional diversity of ADAMTS proteases is further expanded by posttranscriptional and posttranslational processes, such as alternative splicing, glycosylation, or autoproteolytic processing.14–16
ADAMTS17 is one of the two ADAMTS proteases that are mutated in Weill-Marchesani syndrome (WMS), a rare inherited connective tissue and ocular disorder.17 Recessive ADAMTS17 mutations cause WMS 4 (MIM #613195), a multisystem disorder that presents with short stature, short digits (brachydactyly), joint contractures, tight skin, lens dislocation (ectopia lentis), and microspherophakia (small lens), which predisposes affected individuals to glaucoma.18–21 WMS can also be caused by recessive mutations in ADAMTS10 or LTBP2, or by dominant mutations in fibrillin-1 (FBN1), suggesting that these genes operate in a common molecular pathway.22–26 Recently, a mouse model where Adamts17 was deleted, showed features of human WMS, such as bone shortening and tighter skin.27 Reduced skeletal growth was attributed to alterations in the growth plate and aberrant bone morphogenetic protein signaling was identified as a possible molecular mechanism.
Although ADAMTS17 is clearly implicated in WMS, based on new findings in disease modeling of human mutations and findings from an Adamts17 knockout mouse model, little is known about ADAMTS17 regulation or its substrates.20,21,27,28 We previously found that ADAMTS17 undergoes autoproteolytic processing and that the recombinant catalytic domain of ADAMTS17 bound fibrillin-1 microfibrils in the ECM.28 However, ADAMTS17 did not appear to cleave fibrillin-1 or fibrillin-2 directly. Here, we describe two novel splice variants in the ADAMTS17 spacer domain, each of which has specific effects. One splice variant determines protein secretion and the other modulates the (auto)proteolytic activity of ADAMTS17. Differential expression of these splice variants in human tissues and cell lines, suggests that physiological regulation of ADAMTS17 activity is substantially more complex than previously anticipated.
2 |. MATERIALS AND METHODS
2.1 |. Polymerase chain reaction and ADAMTS17 sequence analysis
Human ADAMTS17 sequence variants were identified in the NCBI GenBank database (NIH) and sequence alignments were generated with ClustalW (EMBL-EBI, Hinxton, UK). The spliced exons were identified in the genomic ADAMTS17 DNA sequence (ENSG00000140470) deposited in the Ensembl database (EMBL-EBI, Hinxton, UK). Human cDNA MTC Panel I, MTC Panel II, and human fetal MTC Panel were purchased from Takara Bio USA and 5 μL cDNA was used for the polymerase chain reaction (PCR). To prepare cDNA from cells, total RNA was isolated using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. 1 μg of total RNA was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific) and 1 μL of cDNA was used as template for the PCR reaction. The PCR reaction was performed with Taq DNA polymerase (NEB) and the primers 5′-cacattggtgaagggcgac-3′ (F) and 5′-ccgcagtgcggtttacagg-3′ (R) flanking the alternatively spliced exons of the ADAMTS17 spacer (Figure 1A). The following PCR conditions were used: 95°C (5 minutes), 35 cycles of 95°C (30 seconds), 60°C (30 seconds), 72°C (45 seconds), followed by 72°C (5 minutes). PCR products were separated on a 2% agarose gel in the presence of ethidium bromide and visualized using a Gel Logic 100 imaging system (Kodak). Bands were cut out and DNA isolated for sequence analysis using the QIAquick Gel Extraction Kit (Qiagen). DNA sequences were analyzed by the Cleveland Clinic Lerner Research Institute Genomics Core or by Psomagen.
FIGURE 1.

Alternative splicing of the ADAMTS17 spacer domain. A, Sequence alignment of human ADAMTS17B (XM_139057.3), ADAMTS17 (XM005254872.3), and ADAMTS17A (XM_011521312.2) compared to primate and rodent ADAMTS17 sequences (top) and cartoon of ADAMTS17 domain organization showing the localization of the spacer domain (bottom). The positions of the forward (F) and reverse (R) primer used for amplification of the ADAMTS17 splice variants are indicated with arrows. B, Schematic representation of the alternative splicing events based on the genomic DNA sequence obtained from the Ensembldatabase (ENSG00000140470). The number of nucleotides for each exon and intron and the sequences at the exon/intron boundaries are indicated. C, PCR products obtained with the primer pair indicated in panel A using human cDNA from various sources as the template: HTB177 (lung epithelial cells), MG63 (osteosarcoma cells), NCEC (nonpigmented ciliary epithelial cells), BSMC (primary bronchial smooth muscle cells), CCL171 (lung fibroblasts). +, positive control (ADAMTS17B plasmid), -,negative control (water). The size of the expected PCR product for the different splice variants is indicated on the right. The sequence of bands representing the respective isoforms was confirmed by Sanger sequencing of the excised PCR products
2.2 |. Molecular modeling
The homology models for the cysteine-rich domain and the adjacent spacer domain of ADAMTS17 and ADAMTS17A were generated using I-Tasser without explicit specification of template models. I-Tasser identified crystal structures of ADAMTS13 as highly homologous templates and produced good quality models (C-scores of up to 0.7) for the overall domain folds.29–31 As expected, the resulting models were substantially similar in structure to ADAMTS13, with coordinate root mean square deviation of 0.8–1.1 over 92%−99% of the full sequences. The model of the amino acid sequence of exon 16 of the ADAMTS17A spacer domain was generated by Rosetta using the Robetta webserver (http://robetta.bakerlab.org), and manually connected to the I-Tasser-generated spacer domain using COOT.32,33 The composite model was further optimized to improve residue geometries and eliminate side-chain clashes in COOT and using fragment-guided molecular dynamics conformation sampling within fragment-guided molecular dynamics.34
2.3 |. Cloning of ADAMTS17 peptides
The cloning of full-length ADAMTS17, the active site mutant, ADAMTS17EA, the ADAMTS17 catalytic domain, (PCD), the ADAMTS17 ancillary domain (AD), and the constructs 1C and 25P were described recently.28 Additional ADAMTS17 constructs were cloned by generating the following PCR products using ADAMTS17B as template and the Phusion High Fidelity DNA polymerase (ThermoFisher Scientific) (forward/reverse primer pairs indicated in parentheses, HindIII and XhoI restriction sites are underlined): 1CS (5′-ataagctttggagcccgtggggcgcc-3′/5′-atctcgagcgatgaacaaagagtcctg-3′), S (5′-ataagcttcacttggtgaagggcgac-3′/5′-atctcgagcgatgaacaaagagtcctg-3′), and DC (5′-ataagcttttgctagtcacggacccc-3′/5′-atctcgagcgcaggtcttgccgtcccc-3′). The PCR products were digested with HindIII × XhoI and ligated in the HindIII × XhoI digested pSecTag 2B expression plasmid (ThermoFisher Scientific) using T4 DNA ligase (New England Biolabs). Plasmids were verified by DNA sequencing at the Cleveland Clinic Lerner Research Institute Genomics Core.
To clone the ADAMTS17 spacer, we used cDNA obtained from MG63 osteosarcoma cells as template in a PCR reaction with the primers 5′-ataagcttcacttggtgaagggcgac-3′ and 5′-atctcgagcgatgaacaaagagtcctg-3′ using Phusion High Fidelity DNA polymerase. The PCR product was digested with HindIII × XhoI and ligated in HindIII × XhoI digested pSecTag 2B using T4 DNA ligase. To clone the ADAMTS17A spacer domain, we purchased double-stranded synthetic DNA (gBlock) from Integrated DNA Technology (HindIII and XhoI sites are underlined): 5′- ccgtacgaagcttcacttggtgaagggcgacttcagccacgcccgggggacagttaagaatgatctctgtacgaaggtatccacatgtgtgatggcagaggctgttcccaagtgtttctcatgttatatcgaagctgccgtcattcctgctggagctcggaggatccgtgtggtggaggataaacctgcccacagctttctggctctcaaagactcgggtaaggggtccatcaacagtgactggaagatagagctccccggagagttccagattgcaggcacaactgttcgctatgtgagaagggggctgtgggagaagatctctgccaagggaccaaccaaactaccgctgcacttgatggtgttgttatttcacgaccaagattatggaattcattatgaatacactgttcctgtaaaccgcactgcggaaaatcaaagcgaaccagaaaaaccgcaggactctttgttcatcgagctcat-3′. The gBlock was digested with HindIII × XhoI and ligated in HindIII × XhoI digested pSecTag 2B using T4 DNA ligase.
To clone ADAMTS17A and ADAMTS17AEA, ADAMTS17 and ADAMTS17EA were sequentially digested with XcmI × EcoRI and the 7907 bp product was gel purified. In addition, a PCR product was generated overlapping with the XcmI site on the 5′ end and the beginning of the spacer domain on the 3′ end using ADAMTS17A as template and the primer pair 5′-catgcactacagtgccaacg-3′/5′-atgcgcccttcaccaagtg-3′. The vector and the PCR product were combined with a gBlock synthetic DNA fragment comprising the spacer domain with a 5′-end 15 bp overlap with the PCR product and a 3′-end 15 bp overlap with the EcoRI site using the Gibson Assembly Cloning Kit (NEB). To clone ADAMTS17AD-A, the ancillary domain of ADAMTS17A was amplified using Phusion High Fidelity DNA Polymerase with the primer pair 5′-ataagctttggagcccgtggggcgcc-3′/5′-atctcgagccgagttcggcggtggctg-3′ and the 1749 bp PCR product was purified. The PCR product was digested with HindIII × XhoI and cloned into HindIII × XhoI digested pSecTag 2B using T4 DNA ligase. All plasmids were verified by DNA sequencing at the Cleveland Clinic Lerner Research Institute Genomics Core.
2.4 |. Recombinant protein expression and purification
HEK293 cells (ATCC, Manassas, VA) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS, 100 units/mL penicillin, 100 μg/mL streptomycin in a 5% CO2 atmosphere in a humidified incubator at 37°C. To stably express ADAMTS17S-A, the ADAMTS17S-A plasmid was transfected in HEK293 cells using Lipofectamine 3000 (ThermoFisher Scientific). After 24h DMEM medium was replaced with fresh DMEM supplemented with 0.5 mg/mL zeocine (ThermoFisher Scientific) for selection. The stable cells were expanded on 150 mm cell culture dishes, rinsed once with 20 mL PBS when confluent, and maintained in serum-free DMEM, which was collected every 48–72h. Medium was cleared of cell debris, pooled, and stored at −20°C. 1 L conditioned medium was concentrated to ~25 mL using an Amicon Ultrafiltration Unit (MilliporeSigma) with a 10 kDa molecular-weight cut-off membrane. The concentrated medium was cleared by centrifugation for 10 minutes at 10 000 rpm in a Beckman J17 rotor and applied to a Capturem Maxiprep Nickel Column (Takara Bio USA) according to the manufacturer’s protocol. The filtrate was reapplied once. The resin was washed with 20 mmol/L imidazole diluted in the supplied wash buffer, and bound protein was eluted with 2 × 750 μL elution buffer (eluate 1 and 2). The quality of the eluted protein was evaluated by SDS-PAGE using a 10% polyacrylamide gel.
2.5 |. Mass spectrometry
For the tryptic digest, 19 μl (10 μg) of ADAMTS17S-A was diluted with 50 μL Tris-HCl buffer (pH 8.0) containing 6 mol/L urea. Tryptic digestion was performed by further diluting in 100 μL water and adding trypsin at a 1:20 ratio to protein. After initial digestion overnight at 37°C, a second aliquot of trypsin was added and the sample was incubated for an additional 6 hours. To determine the percentage of free cysteine residues, samples were (1) trypsinized or treated with GluC without prior reduction and iodoacetamide alkylation, or (2) alkylated but not reduced. The protein digest was desalted using the C18 SPE method and reconstituted in 50 μL 1% acetic acid for liquid chromatography-mass spectrometry (LC-MS) analysis using a Finnigan LTQ-Orbitrap Elite hybrid mass spectrometer system (ThermoFisher Scientific). The digest was analyzed using the data-dependent multitask capability of the instrument. In successive instrument scans, full scan mass spectra were acquired to determine peptide molecular weights and product ion spectra were used to determine peptide sequences.
All collision-induced dissociation (CID) spectra collected in the experiment were used in Mascot to search the human UniProtKB sequence databases. The LC-MS/MS data were searched using the program Mass Matrix (http://www.massmatrix.bio) considering disulfide crosslinking as a possible modification. The manual validation process included several steps. First, we compared the observed to the theoretical mass for the predicted cross-linked peptide. Since these experiments were performed on a high-resolution instrument, the difference between the predicted and the obtained mass should be less than 5 ppm. Next, we validated assignment of the fragment ions in the LC-MS/MS spectra. We expected to see product ions from both cross-linked peptide sequences, with the caveat that peptides shorter than four amino acids may not contribute fragment ions with appreciable abundance.
2.6 |. Western blotting
Proteins were separated by SDS-PAGE and transferred onto Immobilon P (chemiluminescence) or Immobilon F (infrared fluorescent detection) PVDF membranes (EMD Millipore) in 25 mmol/L Tris, 192 mmol/L glycine (pH 8.3), 20% methanol buffer for 1.5 hours at 70 V at 4°C. The membranes were blocked with 5% (w/v) milk in TBS (10 mmol/L Tris-HCl, pH 7.2, 0.15 mmol/L NaCl) for 1h at room temperature (RT) and incubated overnight at 4°C with primary antibodies diluted in 5% (w/v) milk in TBS + 0.1% Tween 20 (TBST). Anti-Myc clone 9E10 (ThermoFisher Scientific) was used at 1:500–1:1000 dilution and the mouse monoclonal antibody against human ADAMTS17 clone 3B7 (Novus Biologicals) was used at 1:500 dilution. Membranes were washed three times for 5 minutes at RT with TBST and incubated with the corresponding secondary horseradish peroxidase coupled goat-anti-mouse or goat-anti-rabbit antibodies (Jackson ImmunoResearch Laboratories) diluted 1:2500 in 5% milk in TBST for enhanced chemiluminescence or secondary IRDye conjugated goat-anti-mouse or goat-anti-rabbit antibodies (LI-COR Biosciences) diluted 1:10 000 in 5% milk in TBST + 0.01% SDS for infrared detection, for 1 hour at RT. For chemiluminescence detection, membranes were washed three times with TBST for 5 minutes at RT and the bound antibodies were visualized using enhanced chemiluminescence reagent (ECL Prime, GE Healthcare). For infrared detection, blots were washed three times with TBST for 5 minutes at RT, once with TBS for 5 minutes at RT, and were scanned wet on an Odyssey CLx scanner (LI-COR Biosciences).
2.7 |. Endoglucanase H/Peptide:N-glycosidase F digestion
50 000 HEK293 cells per well were seeded in a 12-well plate (Corning Inc) and grown to 70% confluency. Cells were then transfected with 1 μg of plasmid DNA using Lipofectamine 3000 reagent (Invitrogen). 3 days after transfection, cells were lysed with RIPA buffer (0.1% NP-40, 0.05% sodium deoxycholate, 0.01% sodium dodecyl sulfate in phosphate-buffered saline) for 10 minutes at RT. Samples were cleared at 12 000 rpm for 5 minutes and cell lysate was collected for the deglycosylation assay. Protein concentration was determined by Bradford assay (ThermoFisher Scientific). 100 μg of protein was denatured and digested with Endoglucanase H (EndoH) or Peptide:N-glycosidase F (PNGase F) (New England Biolabs) according to the manufacturer’s protocol. The proteins before and after deglycosylation were visualized by western blot analysis as described above.
2.8 |. Cell surface biotinylation
HEK293 cells stably expressing ADAMTS17B were grown to confluency in a 10 cm cell culture dish and the ancillary domains ADAMTS17ADB and ADAMTS17AD were produced by transient transfection of HEK293 cells. A Cell Surface Protein Isolation Kit (Abcam, ab206998) was used to biotinylate cell surface exposed proteins and to subsequently separate biotinylated and nonbiotinylated proteins from cell lysates according to the manufacturer’s instructions. Biotinylated and nonbiotinylated proteins were analyzed by western blotting.
2.9 |. Immunostaining for cell surface localization of recombinant ADAMTS17
HEK293 cells transiently or stably expressing ADAMTS17 isoforms were seeded in 8-well chamber slides (Celltreat), coated with 0.1% poly-L-lysine (Sigma), at a density of 50 000 cells per well and cultured in DMEM supplemented with 10% FBS, 100 units/mL penicillin, 100 μg/mL streptomycin in a 5% CO2 atmosphere in a humidified incubator at 37°C for 4 days. Cells were rinsed once with PBS and fixed with 4% (v/v) paraformaldehyde in PBS for 20 minutes. Cells were rinsed once with PBS and incubated with 10% normal goat serum (Jackson ImmunoResearch Laboratories) in PBS for 1h at RT, followed by incubation with the monoclonal ADAMTS17 clone 3B7 (1:500) or the polyclonal anti-Myc antibody (1:500, A21281, Invitrogen), diluted in 10% normal goat serum in PBS for 2 h at RT. Cells were rinsed 3 × 5 minutes with PBS, permeabilized with 0.1% Triton X-100 for 10 minutes, rinsed with PBS for 5 minutes, and incubated with fluorophore-labeled secondary goat anti-rabbit and goat anti-mouse antibodies (Jackson ImmunoResearch Laboratories, 1:300) for 1 h at RT. Cells were rinsed 3 × 5 minutes with PBS and slides were mounted with ProLong Diamond Antifade Mountant with DAPI (Invitrogen) and imaged using a Zeiss Axio Observer.Z1 Fluorescence Motorized Microscope w/ Definite Focus (Zeiss).
2.10 |. Co-culture system for ADAMTS17 ECM localization and immunostaining
HEK293 cells and human dermal fibroblasts (HDF) derived from foreskin explants were cultured in DMEM supplemented with 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin in a 5% CO2 atmosphere in a humidified incubator at 37°C. HEK293 cell lines stably expressing ADAMTS17 and ADAMTS17EA were described previously and pools of stably transfected ADAMTS17A and ADAMTS17AEA expressing cells were generated accordingly.28 For the co-culture system, 30 000 ADAMTS17 expressing cells and 30 000 HDFs per well of a 8-well chamber slide were combined, co-cultured for 4 days, and stained for fibrillin microfibrils and ADAMTS17 constructs.
Cells were rinsed once with PBS, fixed in ice-cold 70% methanol/30% acetone for 5 min at RT, rinsed once with PBS and blocked with 10% normal goat serum in PBS for 1 hour at RT. Cells were then incubated with the primary antibodies against fibrillin-1 (α-rFBN1C, 1:500) and ADAMTS17 (mAB-TS17, 1:200), diluted in blocking buffer. The polyclonal antiserum against the C-terminus of human fibrillin-1 was described previously.35 Cells were rinsed 3 × 5 minutes with PBS at RT and incubated with goat-anti-rabbit Alexa-564/goat-anti-mouse Alexa-488 diluted 1:350 in blocking buffer for 1.5 hours at RT. Cells were rinsed 3 × 5 minutes with PBS for 5 minutes at RT and mounted with ProLong Gold Antifade Mountant containing DAPI nuclear stain (ThermoFisher Scientific). The slides were imaged using a Zeiss Axio Observer.Z1 Fluorescence Motorized Microscope w/ Definite Focus (Zeiss).
3 |. RESULTS
3.1 |. ADAMTS17 isoforms are generated by alternative splicing of the spacer
Multiple ADAMTS17 mRNA sequence variants have been deposited in the NCBI GenBank database. ADAMTS17 (accession number XM_005254872.3; GenBank designation: ADAMTS17 isoform X5), contains a canonical spacer domain that is highly homologous and of comparable length to spacer domains in other ADAMTS proteases. We identified two additional human ADAMTS17 transcripts with sequence variations in the spacer domain (Figure 1A): XM_011521312.2 (ADAMTS17 X4, designated here as ADAMTS17A) and NM_139057.3 (ADAMTS17, designated here as ADAMTS17B). Comparison with the human genome sequence in the Ensembl Genome Browser suggested that these transcripts result from alternative splicing events with different consequences (Figure 1B). ADAMTS17A, which contains a longer form of the spacer, is generated by inclusion of a new exon 16, while ADAMTS17B represents a short isoform generated by bypassing of exons 16 and 17 (Figure 1B). The open reading frame is maintained in both transcripts. Exon 16 encodes a 23 amino acid extension in the spacer domain that contains four cysteine residues, which likely form inter- or intramolecular disulfide bonds (see below). Blast search with this extension sequence against the human nonredundant protein database returned only ADAMTS17 isoforms. Analysis of protein sequences deposited for other species indicated that ADAMTS17 from primates and rodents also have multiple isoforms (Figure 1A). In contrast to the three ADAMTS17 spacer isoforms reported in humans, only ADAMTS17A and ADAMTS17 were reported in chimpanzees. Among rodents, only the ADAMTS17 canonical spacer sequence was present in mouse, but ADAMTS17A and ADAMTS17 were both present in rats.
Semi-quantitative RT-PCR with primers that flank the spliced exons of the ADAMTS17 spacer domain and commercial fetal and adult human tissue cDNA libraries or cDNA prepared from mRNA isolated from human cell lines as templates demonstrated multiple bands in most tissues (Figure 1C). In some cell lines and tissues, such as lung fibroblasts (CCL171) or the pancreas, ADAMTS17A was the dominant splice variant. In other tissues, such as the heart, intestine, or testes, bands corresponding to ADAMTS17B and ADAMTS17 were present at similar intensity, but the intensity for the band representing ADAMTS17A was markedly reduced. The identity of the individual PCR products was confirmed by Sanger DNA sequencing. Thus, alternative splicing of ADAMTS17 transcripts results in two previously unrecognized ADAMTS17 isoforms with distinct spacer sequences that are differentially expressed in cell lines and tissues.
3.2 |. Alternative splicing of the ADAMTS17 spacer differentially regulates ADAMTS17 secretion and catalytic activity
To determine functional consequences of the splice variants in the ADAMTS17 spacer domain, we generated recombinant ADAMTS17B, ADAMTS17, and ADAMTS17A constructs for the expression of full-length and active site mutant forms of the alternatively spliced spacer transcripts (Figure 2A). Western blot analysis with α-Myc antibody detected neither full-length ADAMTS17B nor the corresponding active site mutant, ADAMTS17BEA, in conditioned medium from transiently transfected HEK293 cells (Figure 2B). However, robust expression of the zymogen of both recombinant proteins was detected in the cell lysate, suggesting that ADAMTS17B either was not secreted or retained at the cell surface (Figure 2B). In contrast, the active site mutant ADAMTS17EA was readily detected in the medium, and both ADAMTS17 and ADAMTS17EA were present in the cell lysate as previously reported (Figure 2B).28 Proteolytically active wild-type ADAMTS17 was only detected in the cell lysate but not in conditioned medium, consistent with previous results that showed that extensive autoproteolytic processing of ADAMTS17 results in the loss of the Myc-tag.28 To confirm that the ADAMTS17B spacer was responsible for the absence of ADAMTS17B in the medium, we expressed a panel of recombinant ADAMTS17B fragments (Figure 2A). None of the fragments that contained the ADAMTS17B spacer were detectable in the medium by a α-Myc antibody, despite their presence in the cell lysate (Figure 2C). However, when ADAMTS17S (S) or ADAMTS17A-S (SA) spacer domains were expressed individually or as part of the ADAMTS17AD construct (AD, ADA), they were detected in the medium (Figure 2D).
FIGURE 2.
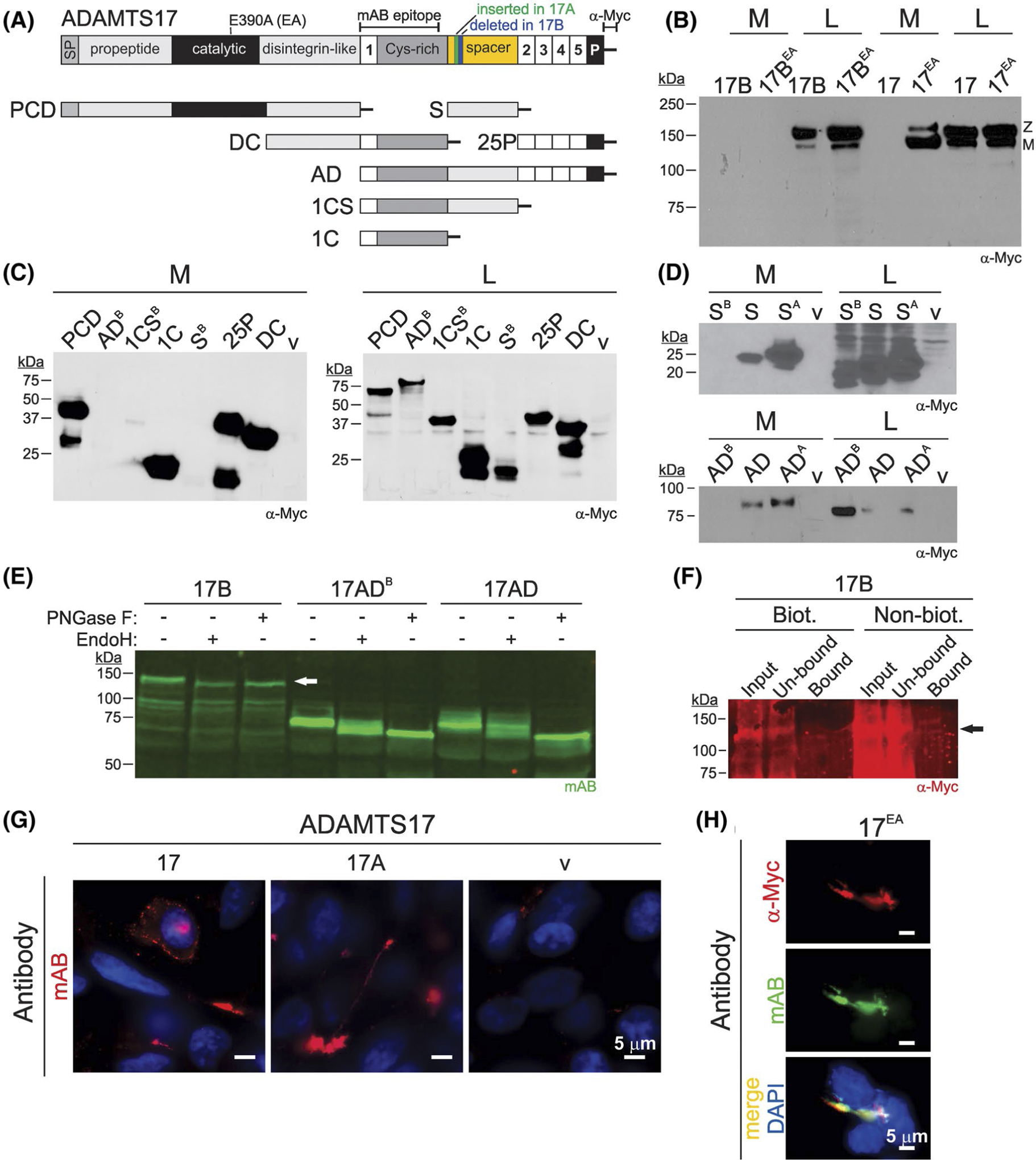
Alternative splicing regulates ADAMTS17 secretion. A, Cartoon depicting the ADAMTS17 domain organization and recombinant peptides generated for this study. Locations of the active site mutation (E390A), antibody epitopes, and alternatively spliced sequences are indicated on top. B, Western blot analysis of ADAMTS17B and ADAMTS17 showed no secretion of ADAMTS17B wild-type (17B) or the active site mutant (17BEA) into conditioned medium (M) but presence of both ADAMTS17B proteins in the cell lysate (L). In contrast, ADAMTS17 (due to autoproteolytic processing), but not ADAMTS17EA, was undetectable in conditioned medium, and both constructs were present in the cell lysate. Z, zymogen, M, mature enzyme. C, Western blot analysis of ADAMTS17B recombinant constructs expressed in HEK293F cells. ADAMTS17B-ancillary domain (ADB), -thrombospondin type 1 motive 1/cysteine-rich domain/spacer domain (1CSB), and -spacer domain (SB) constructs were absent in conditioned medium (M), but present in the cell lysate (L). 1CS, thrombospondin type 1 motive 1/cysteine-rich domain; 25P, thrombospondin type 1 motive 2–5/PLAC-domain; DC, disintegrin domain/cysteine-rich domain; PCD, propeptide/catalytic domain/disintegrin domain; v, empty vector. D, Western blot analysis of alternatively spliced ADAMTS17S (S) and ADAMTS17AD (AD) constructs. Note that the constructs S/AD and SA/ADA, but not SB/ADB, were secreted into the medium (M). All ADAMTS17 constructs were detectable in the cell lysate (L). v, empty vector. E, Western blot analysis of full-length ADAMTS17B, ADAMTS17ADB and ADAMTS17AD after digestion with EndoH or PNGase F. Note the size shifts to lower molecular weights indicating sensitivity to EndoH and PNGase F for ADAMTS17 and ADAMTS17ADB. White arrow indicates band representing full-length ADAMTS17. F, Western blot analysis of cell lysate for ADAMTS17 after cell surface proteins were biotinylated compared to nonbiotinylated control shows absence of ADAMTS17 in streptavidin-bound fraction (bound). Arrow indicates position of full-length ADAMTS17. Input, cell lysate; un-bound, supernatant after pulldown with streptavidin beads; bound, fraction containing biotinylated cell surface proteins that were precipitated with streptavidin beads. G, Cell surface localization of ADAMTS17 and ADAMTS17A in HEK293 cells. Cells were stained with mAB ADAMTS17 prior to cell membrane permeabilization. Pattern of deposition is consistent with cell surface deposition or deposition on the cell culture substratum. H, Costaining of ADAMTS17EA with a polyclonal α-Myc and the ADAMTS17 mAB antibody, indicating that both antibodies detect recombinant ADAMTS17
We further investigated, if ADAMTS17B was present at the cell surface by analyzing its sensitivity to EndoH or PNGase F digestion and by cell surface biotinylation. When cell lysate containing ADAMTS17B or its ancillary domain ADAMTS17ADB was digested with EndoH or PNGase F, we observed a shift to a lower molecular weight suggesting that ADAMTS17B was not fully N-glycosylated, and thus, did not complete transit through the Golgi apparatus (Figure 2E). In contrast, the ancillary domain of ADAMTS17 (AD) was sensitive to PNGase F digestion, but only partially sensitive to EndoH digestion, consistent with the observed secretion into the medium. In addition, we were unable to precipitate ADAMTS17B from the cell lysate where cell surface proteins were biotinylated prior to streptavidin pulldown (Figure 3F). ADAMTS17B could only be detected in the un-bound fraction at a similar intensity compared to the input. Collectively, these results suggest that the ADAMTS17B isoform with the shorter spacer is not present at the cell surface nor secreted into the medium.
FIGURE 3.
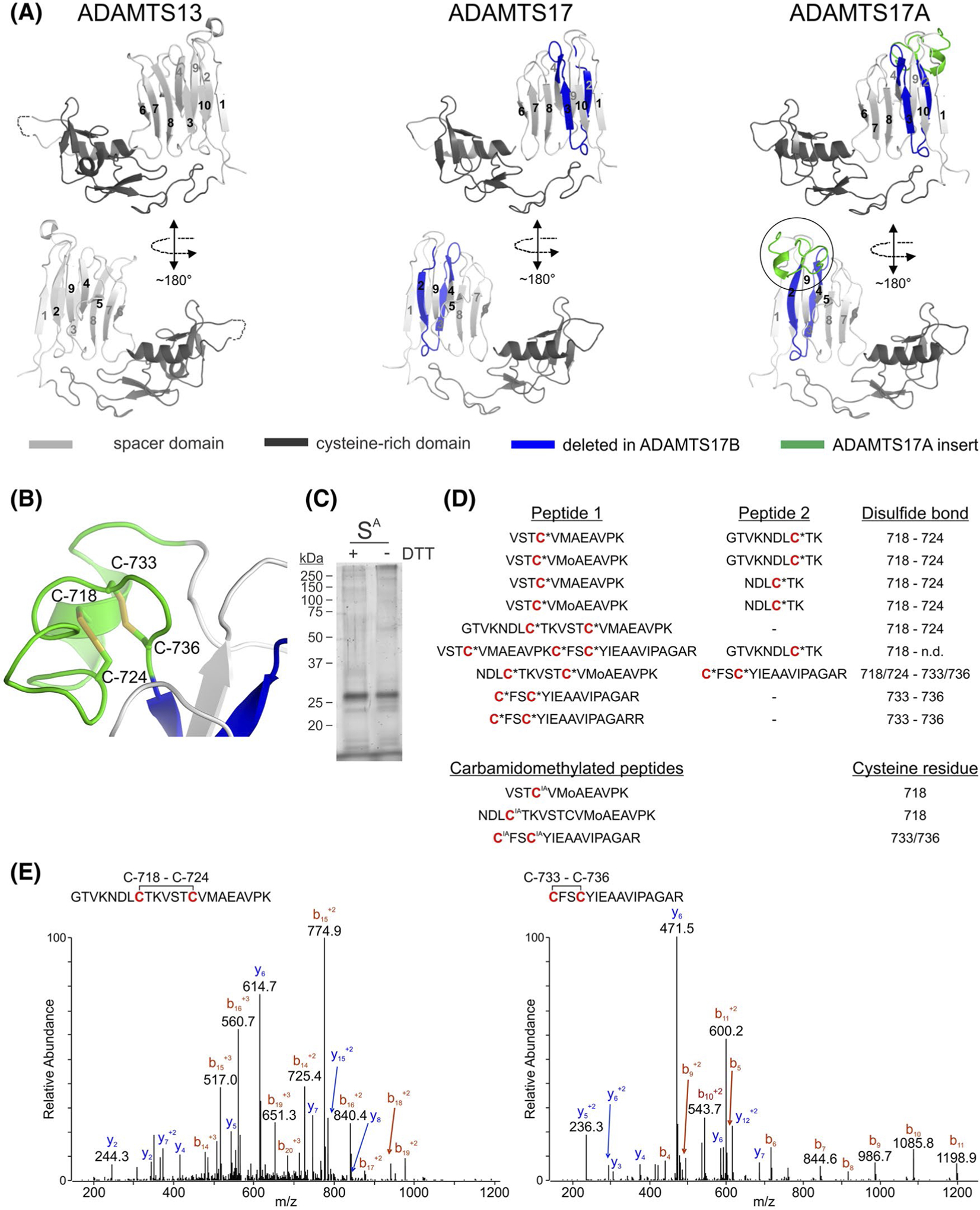
Structural modeling of the ADAMTS17A spacer domain and experimental disulfide bond assignment. A, Crystal structure of the ADAMTS13 cysteine-rich domain/spacer domain pair (left) and models of the respective spacer domains for ADAMTS17 (middle) and ADAMTS17A (right). Sequences spliced out in ADAMTS17B and the sequence included in ADAMTS17A are indicated in blue and green, respectively. Two views, rotated by 180° are shown. The circle in ADAMTS17A depicts the region magnified in B. B, Zoomed-in view of the ADAMTS17A spacer extension depicting predicted intramolecular disulfide bonds. Cysteine residues (C) are numbered. C, Coomassie blue-stained polyacrylamide gel of recombinant ADAMTS17A spacer domain (SA) in the presence (+) or absence (−) of DTT shows the purity of the protein preparation used for LC-MS/MS analysis. D, Disulfide-bonded peptides obtained from the LC-MS/MS analysis of nonreduced ADAMTS17A spacer protein, supporting the predicted disulfide bond patterning depicted in panel B. The bottom three peptides indicate the positions for the carbamidomethylation (IA) modification after iodoacetamide treatment of nonreduced ADAMTS17A spacer protein prior to tryptic digestion. E, Fragmentation and ionization patterns of two representative peptides containing disulfide bonds obtained by LC-MS/MS analysis. The MS/MS spectrum of a quadruple charged peptide identified at a m/z ratio of 573.791 Da is consistent with the peptide 711-GTVKNDLCTKVSTCVMAEAVPK-732 (left), containing an intrapeptide disulfide bond between Cys718 and Cys724. The MS/MS spectrum of a triply charged peptide identified at a m/z ratio of 556.934 Da is consistent with the peptide 733-CFSCYIEAAVIPAGAR-748 (right) containing an intrapeptide disulfide bond between C733 and C736. Amino acid numbering is based on Gene-Bank sequence accession number XM_011521312.2
To analyze the localization of the ADAMTS17 and ADAMTS17A isoforms, we stained HEK293 cells that transiently or stably express ADAMTS17 or ADAMTS17A with ADAMTS17 mAB prior to permeabilization of the cell membrane. We found ADAMTS17 or ADAMTS17A present in a pattern consistent with cell surface localization or deposition of ADAMTS17 on the cell culture substratum (Figure 2G). Most of the signal originated from punctate accumulations of the ADAMTS17 proteins, presumably located in-between cells. To demonstrate that ADAMTS17 mAB recognizes the recombinant ADAMTS17 proteins, we costained stable ADAMTS17EA-expressing HEK293 cells with a polyclonal α-Myc antibody and ADAMTS17 mAB and found substantial overlap of the signals origination from the two antibodies (Figure 2H).
3.3 |. Molecular modeling of ADAMTS17 splice variants predicts structural innovations in the spacer
We used in silico modeling to predict the structural differences between the spacer domain in ADAMTS17 and ADAMTS17A. We first modeled the structure of the canonical ADAMTS17 spacer based on a high-resolution crystal structure of the homologous ADAMTS13 spacer as template (Figure 3A).29 The spacerdomains of ADAMTS13 and ADAMTS17 are composed of a 10-stranded β-sheet that forms a “β-sandwich” fold. In the ADAMTS13 spacer domain, there are several small helical turns in the loop between strands 9 and 10, but this loop is shorter in ADAMTS17 and lacks these helices. Based on their respective sequences, we predicted other differences in the lengths of the loops connecting the β-sheets in ADAMTS13 and ADAMTS17. For example, in ADAMTS13, the loop that connects strands 6 and 7 makes contacts with the upstream cysteine-rich domain through an Asp-Leu-Glu sequence. This contact potentially enhances the rigidity of the overall structure of the C-terminal half of ADAMTS13. In ADAMTS17, this loop is shorter and has amino acid residues with smaller side chains. It is unclear whether these amino acid residues can engage in similar interactions with the cysteine-rich domain in ADAMTS17 and may thus result in greater interdomain flexibility in ADAMTS17 compared to ADAMTS13. Inclusion of exon 16 in the ADAMTS17A spacer domain provides an extra loop and a short α-helix between β-strands 1 and 2 of the “β-sandwich” fold. The extra loop introduces four cysteine residues into the spacer domain of ADAMTS17A, which are positioned to form two predicted disulfide bonds connecting Cys-718 and Cys-724 and Cys-733 and Cys-736, respectively (Figure 3B). To validate the predicted disulfide bond pattern, we expressed the recombinant ADAMTS17A spacer protein in HEK293 cells and analyzed trypsin digests of the purified native peptide, that is, nonreduced, by mass spectrometry (Figure 3C). This experimental design likely preserved the native disulfide bonding pattern and yielded tryptic peptides linked by an intact disulfide bond for mass spectrometry analysis. The formation of a disulfide bond reduces the mass of the corresponding peptide by 2.0158 Da and that mass difference was used to identify disulfide-linked peptides (Figure 3D,E). Nine peptides harboring the four cysteine residues were identified when the native protein was digested with trypsin without prior reduction or alkylation (Figure 3D). Disulfide bonds were identified between Cys-718 and Cys-724 and between Cys-733 and Cys-736, respectively, supporting the disulfide bond pattern predicted from the molecular model (Figure 3A). The disulfide bonds were further validated by LC-MS/MS analysis of tryptic peptides derived from recombinant ADAMTS17A spacer protein after prior alkylation of free cysteine residues with iodoacetamide, which results in a carbamidomethyl modification of free cysteine residues. Three different carbamidomethylated peptides were identified (Figure 3D, bottom). However, the overall abundance of these carbamidomethylated peptides was low, either due to the absence of free cysteine residues or due to reduced susceptibility of potentially free cysteine residues to alkylation. In addition, analysis of ADAMTS17A spacer protein under nonreducing conditions by polyacrylamide gel electrophoresis followed by Coomassie staining revealed no pronounced disulfide-mediated oligomerization, excluding the possibility of intermolecular disulfide bond formation (Figure 3C). Collectively, the molecular modeling and experimental mass spectrometry data support a spacer structure of ADAMTS17A in which the four cysteine residues form two intramolecular disulfide bonds. It is likely that these disulfide bonds stabilize or rigidify the structure of the finger-like spacer domain extension in ADAMTS17A.
3.4 |. Autoproteolysis is altered in ADAMTS17A
When we analyzed conditioned medium and cell lysate containing full-length ADAMTS17A or ADAMTS17AEA, we detected both proteins in the cell lysate and the conditioned medium with the α-Myc antibody (Figure 4). When we compared active ADAMTS17 with ADAMTS17A and with their respective proteolytically inactive forms, we found robust autoproteolysis for ADAMTS17, but not for ADAMTS17A, suggesting that the extension of the spacer domain by inclusion of exon 16 modulates the autoproteolytic activity of ADAMTS17. Based on our previous report, we detected multiple autoproteolytic cleavage sites in ADAMTS17 that are distributed throughout the protein, with one being located very close to the C-terminus, and others clustering in the N-terminal half, close to the active site of ADAMTS17.28 This suggests that ADAMTS17A is inactive or does not recognize itself as a substrate.
FIGURE 4.

Western blot analysis of full-length ADAMTS17A and ADAMTS17 and their respective active site mutants in medium (M) and cell lysate (L) probed with the mAB ADAMTS17 antibody (left panel, green) and the polyclonal α-Myc antibody (middle panel, red). Arrowheads indicate the autoproteolytic ADAMTS17 peptides detected with the mAB ADAMTS17 antibody, that were absent in ADAMTS17A. Asterisks indicate nonspecific bands detected with the polyclonal α-Myc antibody. Z, zymogen, M, mature enzyme
3.5 |. ADAMTS17A localizes to fibrillin-1 microfibrils in the ECM
We previously showed that exogenously added recombinant ADAMTS17 peptides localized to fibrillin-1 microfibrils in the ECM of HDFs.28 Since full-length wild-type ADAMTS protein cannot be purified due to autoproteolysis, we used a co-culture system of HDFs with HEK293 cells that stably express ADAMTS17, ADAMTS17EA, ADAMTS17A, or ADAMTS17AEA to analyze their localization relative to fibrillin-1 microfibrils (Figure 5).36,37 In co-culture, HEK293 cells and HDFs did not mix homogenously, but were arranged in patches of HEK293 cells, interspersed with streaks of HDFs. Fibrillin-1 microfibril deposition followed HDF cell arrangement and fibrillin microfibrils were rarely detected crossing over patches of HEK293 cells. ADAMTS17 produced by HEK cells co-stained with fibrillin-1 microfibrils assembled by HDFs when microfibrils were in close vicinity to the HEK293 cells, but not when farther away (Figure 5A, top). We observed more extended co-staining of ADAMTS17EA with fibrillin-1 microfibrils consistent with protection of the proteolytic inactive mutant form from autoproteolysis (Figure 5A, bottom). Similar to ADAMTS17, co-staining of ADAMTS17EA and fibrillin-1 was limited to fibrillin-1 microfibrils that crossed into areas of HEK293 cells. This suggested that localization of ADAMTS17 to fibrillin microfibrils may not be controlled simply by diffusion, but may require direct cell (HEK293) - fibrillin-microfibril or cell (HEK293) - cell (HDF) contact. ADAMTS17A and ADAMTS17AEA showed a very similar co-staining pattern with fibrillin-1 microfibrils (Figure 5B). In addition, we did not observe an appreciable change in the quantity of microfibrils deposited in the ECM when ADAMTS17 or ADAMTS17A was present. Controls with nontransfected HEK293 cells showed no staining with the ADAMTS17 mAB antibody (Figure 5C). As shown previously for ADAMTS17, ADAMTS17A did not result in apparent cleavage of fibrillin-1 or −2 when co-expressed in HEK293 cells (data not shown).28
FIGURE 5.

Co-staining of ADAMTS17 and ADAMTS17EA (A) and ADAMTS17A and ADAMTS17AEA (B) with fibrillin-1 microfibrils in HEK293/HDF co-cultures. Equal numbers of cells were cultured for 4 days and stained with antibodies against fibrillin-1 (red, left) and ADAMTS17 (green, middle). Nuclei were counterstained with DAPI (blue). Boxed areas in the merged images (right) indicate the location of the digitally magnified image shown as inset. Arrows show co-staining of ADAMTS17 with fibrillin-1 microfibrils. C, Control HEK293-HDF co-cultures show no ADAMTS17 signal in the green channel
4 |. DISCUSSION
In addition to the role of ADAMTS17 mutations in human WMS 4, polymorphisms in ADAMTS17 are associated with primary open angle glaucoma and reduced height in several dog breeds, providing compelling arguments for understanding the molecular biology, structure, and function of this protease.38,39 However, it is equally important to understand the role of tissue-specific ADAMTS17 isoforms, generated by posttranscriptional modifications, such as alternative splicing. Here, we describe two novel isoforms of ADAMTS17 that are generated by alternative splicing of the ADAMTS17 spacer domain. Compared to ADAMTS17, the isoform harboring the canonical spacer domain, alternative splicing of the ADAMTS17 spacer domain resulted in retention of the ADAMTS17B isoform inside the cell. Moreover, alternative splicing modulated (auto)catalytic properties in the ADAMTS17A isoform. To elucidate the biological functions of the ADAMTS17 protease, it will be paramount to identify the substrates for ADAMTS17 and determine if they differ between its isoforms.40 Such efforts are currently underway in the laboratory. In the meantime, we capitalized on the known autoproteolytic activity as a surrogate substrate to test the functional impact of the new ADAMTS17 isoforms.
Splice variants for other ADAMTS proteases, such as ADAMTS2, −6, −7, −9, and −13 have been described previously.4,15,16,41 However, functional consequences of alternative splicing were not analyzed in detail in most instances. Due to the involvement of ADAMTS4 and ADAMTS5 in osteoarthritis, consequences of alternatively spliced ADAMTS4 and ADAMTS5 isoforms have been studied.42–45 The canonical cleavage of aggrecan and versican, which contain well-defined ADAMTS cleavage sites, and the availability of validated reagents for monitoring cleavage events were exploited to study the functional significance of ADAMTS4 and ADAMTS5 splicing.46,47 Splicing of the ADAMTS4 spacer domain due through utilization of a cryptic splice site has been discovered in synovium from patients with osteoarthritis.42 The alternatively spliced ADAMTS4 variant is predicted to lack the entire spacer domain and the consecutive domains of the ancillary domain. The presence of the ADAMTS4 spacer domain resulted in decreased proteolytic cleavage of aggrecan and, through binding to fibronectin, inhibited cleavage of aggrecan.43 It was, therefore, suggested that removal of the spacer domain switches the ADAMTS4 activity from ECM formation to pathological ECM destruction.43,48,49 In contrast to ADAMTS4, the splice variants of ADAMTS17 did not remove the entire spacer domain, but resulted in smaller, yet, significant, in-frame sequence alterations. In a more recent study, removal of the spacer domain in ADAMTS4 markedly reduced versican cleavage, which suggests that the spacer domain is crucial for versican substrate recognition by ADAMTS4.50 The sequences that are required for versican cleavage by ADAMTS4 and ADAMTS5 were localized to distinct loops connecting individual β-sheets of the spacer domain. The ADAMTS17 isoforms that we identified are also predicted to have isoform-specific changes of the loop segments of the extended spacer domain and we found evidence for altered autoproteolysis. Therefore, we propose that the alternative splicing that yields the spacer domain resulting in the ADAMTS17A isoform preserves the stability of full-length ADAMTS17A for more effective long-lived proteolysis. In addition, ADAMTS17A may engage in novel intermolecular interactions, including with substrates that are not cleaved by canonical ADAMTS17. However, since no ADAMTS17 substrates are known, this hypothesis will need to be tested in the future. Alternatively, ADAMTS17 and ADAMTS17A may cleave each other.
Molecular modeling provided insights into potentially substantial structural changes resulting from the inclusion or exclusion of exons 16 and 17. Molecular modeling could be applied with reasonable confidence based on the availability of a experimentally determined high-resolution crystal structure for ADAMTS13.29 The modeling suggested that ADAMTS17A incorporates a novel finger-like extension not previously identified in other ADAMTS proteins. Indeed, detailed searches of protein and expressed sequence tag databases found no equivalent insertion in any species, nor other inserted sequences that were substantially homologous to the protein sequence encoded by exon 16, suggesting an entirely novel structure. Although a detailed structure of this insertion needs to be determined experimentally, molecular modeling predicted a disulfide bonding assignment for the four cysteine residues that was strongly supported by mass spectrometry.
In addition to possible alteration in catalytic activities due to ADAMTS17A alternative splicing, we found that a shorter spacer in ADAMTS17B prevented passage through the secretory pathway and resulted in the intracellular retention of ADAMTS17B. This level of regulation could result in enrichment of the proteolytic activity of ADAMTS17 in the secretory pathway or inside the cell. Intracellular roles for extracellular proteins, including ADAMTS proteases are emerging and we localized at least a fraction of endogenous ADAMTS17 to the centrosome with two ADAMTS17 antibodies (Hubmacher, Apte, unpublished).11,51,52 Individual loops of the spacer domains of ADAMTS1, 4, 5, and 13 were shown to be critical for ECM binding.29,53–56 We propose that ADAMTS17 localization in the ECM is achieved, at least in part, via the spacer domain. We previously found that the ancillary domain of ADAMTS17 binds the N-terminus of fibrillin-2 and localizes to fibrillin-1 microfibrils in the ECM of human dermal fibroblasts.28 However, a similar behavior was also observed for the N-terminal ADAMTS17 protease domain, indicating that ADAMTS17 may have at least two ECM binding sites. When we analyzed the localization of ADAMTS17 and ADAMTS17A, we found that they both localized to fibrillin-1 microfibrils, predominantly to the microfibrils that run adjacent to the HEK293 cells that express recombinant ADAMTS17 isoforms. This suggests that cell proximity may be required for the deposition of ADAMTS17 isoforms onto fibrillin-1 microfibrils. Since ADAMTS17 and ADAMTS17A did not localize to more distal fibrillin-1 microfibrils, secreted ADAMTS17 may not be able to freely diffuse in the ECM and be deposited more uniformly onto fibrillin microfibrils. The in vivo relevance of this pattern of ADAMTS17-fibrillin-1 microfibril association and the underlying mechanisms will need to be established in the future, for example by using co-culture systems that allow for the spatial separation of the involved cell types.
In summary, we report two novel splice variants that alter the sequence of the ADAMTS17 spacer and result in intracellular retention or modulation of the autoproteolytic properties of ADAMTS17, respectively. We propose that the ADAMTS17 isoform with the short spacer, ADAMTS17B, is generated to reroute ADAMTS17 or to prevent proteolytic events in the ECM. In contrast, ADAMTS17A, the isoform that contains the extended spacer is generated to protect against autoproteolysis or to alter substrate specificity of ADAMTS17 substrates. Therefore, the regulation of ADAMTS17A production could modulate the substrate spectrum of ADAMTS17, mediate binding to tissue-specific ECM components, or regulate the role of autoproteolytic peptides derived from ADAMTS17 or ADAMTS17A. However, the in vivo relevance of alternative splicing of ADAMTS17 needs to be established in future studies and the identification of potential isoform-specific ADAMTS17 substrates or cell surface and ECM binding partners may be achieved through application of advanced proteomics approaches.
ACKNOWLEDGMENTS
This study was supported by NIH grants R01AR070748 to DH and R01EY021151-01 to SSA, an award from the Marfan Foundation to DH, and by the Canadian NSERC (RGPIN-2016-06278) to DPR. The Orbitrap Elite instrument in the Cleveland Clinic Lerner Research Institute Proteomics core was purchased via an NIH shared instrument grant, 1S10RR031537-01. We thank Dr Suzie Comhair (Cleveland Clinic Lerner Research Institute) for providing primary bronchial smooth muscle cells.
Abbreviations:
- ADAMTS
a disintegrin and metalloprotease with thrombospondin type 1 motifs
- DMEM
Dulbecco’s Modified Eagle’s Medium
- ECM
extracellular matrix
- GON-1
abnormal gonad development-1
- PBS
phosphate-buffered saline
- PLAC
protease and lacunin domain
- TBS
Tris-buffered saline
- TSR
thrombospondin type 1 repeat
- WMS
Weill-Marchesani syndrome
REFERENCES
- 1.Apte SS. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem. 2009;284:31493–31497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mead TJ, Apte SS. ADAMTS proteins in human disorders. Matrix Biol. 2018;71–72:225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bekhouche M, Colige A. The procollagen N-proteinases ADAMTS2, 3 and 14 in pathophysiology. Matrix Biol. 2015;44–46:46–53. [DOI] [PubMed] [Google Scholar]
- 4.Colige A, Sieron AL, Li SW, et al. Human Ehlers-Danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am J Hum Genet. 1999;65:308–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeltsch M, Jha SK, Tvorogov D, et al. CCBE1 enhances lymphangiogenesis via A disintegrin and metalloprotease with thrombospondin motifs-3-mediated vascular endothelial growth factor-C activation. Circulation. 2014;129:1962–1971. [DOI] [PubMed] [Google Scholar]
- 6.Brouillard P, Dupont L, Helaers R, et al. Loss of ADAMTS3 activity causes Hennekam lymphangiectasia-lymphedema syndrome 3. Hum Mol Genet. 2017;26:4095–4104. [DOI] [PubMed] [Google Scholar]
- 7.Song RH, Tortorella MD, Malfait AM, et al. Aggrecan degradation in human articular cartilage explants is mediated by both ADAMTS-4 and ADAMTS-5. Arthritis Rheum. 2007;56:575–585. [DOI] [PubMed] [Google Scholar]
- 8.Majumdar MK, Askew R, Schelling S, et al. Double-knockout of ADAMTS-4 and ADAMTS-5 in mice results in physiologically normal animals and prevents the progression of osteoarthritis. Arthritis Rheum. 2007;56:3670–3674. [DOI] [PubMed] [Google Scholar]
- 9.Stanton H, Rogerson FM, East CJ, et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature. 2005;434:648–652. [DOI] [PubMed] [Google Scholar]
- 10.Dupuis LE, Nelson EL, Hozik B, et al. Adamts5(−/−) Mice Exhibit Altered Aggrecan Proteolytic Profiles That Correlate With Ascending Aortic Anomalies. Arterioscler. Thromb Vasc Biol 2019;39:2067–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nandadasa S, Kraft CM, Wang LW, et al. Secreted metalloproteases ADAMTS9 and ADAMTS20 have a non-canonical role in ciliary vesicle growth during ciliogenesis. Nat Commun. 2019;10:953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubail J, Apte SS. Insights on ADAMTS proteases and ADAMTS-like proteins from mammalian genetics. Matrix Biol. 2015;44–46:24–37. [DOI] [PubMed] [Google Scholar]
- 13.Takeda S ADAM and ADAMTS family proteins and snake venom metalloproteinases: a structural overview. Toxins (Basel). 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foulcer SJ, Nelson CM, Quintero MV, et al. Determinants of versican-V1 proteoglycan processing by the metalloproteinase ADAMTS5. J Biol Chem. 2014;289:27859–27873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bevitt DJ, Li Z, Lindrop JL, Barker MD, Clarke MP, McKie N. Analysis of full length ADAMTS6 transcript reveals alternative splicing and a role for the 5’ untranslated region in translational control. Gene. 2005;359:99–110. [DOI] [PubMed] [Google Scholar]
- 16.Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, Fujikawa K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem. 2001;276:41059–41063. [DOI] [PubMed] [Google Scholar]
- 17.Karoulias SZ, Taye N, Stanley S, Hubmacher D. The ADAMTS/Fibrillin connection: insights into the biological functions of ADAMTS10 and ADAMTS17 and their respective sister proteases. Biomolecules. 2020;10(4):596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan AO, Aldahmesh MA, Al-Ghadeer H, Mohamed JY, Alkuraya FS. Familial spherophakia with short stature caused by a novel homozygous ADAMTS17 mutation. Ophthalmic Genet. 2012;33:235–239. [DOI] [PubMed] [Google Scholar]
- 19.Morales J, Al-Sharif L, Khalil DS, et al. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am J Hum Genet. 2009;85:558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karoulias SZ, Beyens A, Balic Z, et al. A novel ADAMTS17 variant that causes Weill-Marchesani syndrome 4 alters fibrillin-1 and collagen type I deposition in the extracellular matrix. Matrix Biol. 2020;88:1–18. [DOI] [PubMed] [Google Scholar]
- 21.Evans DR, Green JS, Fahiminiya S, et al. A novel pathogenic missense ADAMTS17 variant that impairs secretion causes Weill-Marchesani Syndrome with variably dysmorphic hand features. Sci Rep. 2020;10:10827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hubmacher D, Apte SS. ADAMTS proteins as modulators of microfibril formation and function. Matrix Biol. 2015;47:34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kutz WE, Wang LW, Dagoneau N, et al. Functional analysis of an ADAMTS10 signal peptide mutation in Weill-Marchesani syndrome demonstrates a long-range effect on secretion of the full-length enzyme. Hum Mutat. 2008;29:1425–1434. [DOI] [PubMed] [Google Scholar]
- 24.Dagoneau N, Benoist-Lasselin C, Huber C, et al. ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am J Hum Genet. 2004;75:801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faivre L, Gorlin RJ, Wirtz MK, et al. In frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndrome. J Med Genet. 2003;40:34–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stanley S, Balic Z, Hubmacher D. Acromelic dysplasias: how rare musculoskeletal disorders reveal biological functions of extracellular matrix proteins. Ann N Y Acad Sci. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oichi T, Taniguchi Y, Soma K, et al. Adamts17 is involved in skeletogenesis through modulation of BMP-Smad1/5/8 pathway. Cell Mol Life Sci. 2019;76(23):4795–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hubmacher D, Schneider M, Berardinelli SJ, et al. Unusual life cycle and impact on microfibril assembly of ADAMTS17, a secreted metalloprotease mutated in genetic eye disease. Sci Rep. 2017;7:41871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akiyama M, Takeda S, Kokame K, Takagi J, Miyata T. Crystal structures of the noncatalytic domains of ADAMTS13 reveal multiple discontinuous exosites for von Willebrand factor. Proc Natl Acad Sci USA. 2009;106:19274–19279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akiyama M, Nakayama D, Takeda S, Kokame K, Takagi J, Miyata T. Crystal structure and enzymatic activity of an ADAMTS-13 mutant with the East Asian-specific P475S polymorphism. J Thromb Haemost. 2013;11:1399–1406. [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, Liang Y, Zhang Y. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure. 2011;19:1784–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tiedemann K, Batge B, Muller PK, Reinhardt DP. Interactions of fibrillin-1 with heparin/heparan sulfate, implications for microfibrillar assembly. J Biol Chem. 2001;276:36035–36042. [DOI] [PubMed] [Google Scholar]
- 36.Hubmacher D Cell-Based Interaction Analysis of ADAMTS Proteases and ADAMTS-Like Proteins with Fibrillin Microfibrils. Methods Mol Biol 2020;2043:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hubmacher D, Bergeron E, Fagotto-Kaufmann C, Sakai LY, Reinhardt DP. Early fibrillin-1 assembly monitored through a modifiable recombinant cell approach. Biomacromol. 2014;15:1456–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oliver JA, Forman OP, Pettitt L, Mellersh CS. Two independent mutations in ADAMTS17 are associated with primary open angle glaucoma in the basset hound and basset fauve de Bretagne breeds of dog. PLoS One. 2015;10:e0140436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeanes EC, Oliver JAC, Ricketts SL, Gould DJ, Mellersh CS. Glaucoma-causing ADAMTS17 mutations are also reproducibly associated with height in two domestic dog breeds: selection for short stature may have contributed to increased prevalence of glaucoma. Canine Genet Epidemiol. 2019;6:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Satz-Jacobowitz B, Hubmacher D. The quest for substrates and binding partners: A critical barrier for understanding the role of ADAMTS proteases in musculoskeletal development and disease. Dev Dyn. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bevitt DJ, Mohamed J, Catterall JB, et al. Expression of ADAMTS metalloproteinases in the retinal pigment epithelium derived cell line ARPE-19: transcriptional regulation by TNFalpha. Biochim Biophys Acta. 2003;1626:83–91. [DOI] [PubMed] [Google Scholar]
- 42.Wainwright SD, Bondeson J, Hughes CE. An alternative spliced transcript of ADAMTS4 is present in human synovium from OA patients. Matrix Biol. 2006;25:317–320. [DOI] [PubMed] [Google Scholar]
- 43.Hashimoto G, Shimoda M, Okada Y. ADAMTS4 (aggrecanase-1) interaction with the C-terminal domain of fibronectin inhibits proteolysis of aggrecan. J Biol Chem. 2004;279:32483–32491. [DOI] [PubMed] [Google Scholar]
- 44.Gao G, Westling J, Thompson VP, Howell TD, Gottschall PE, Sandy JD. Activation of the proteolytic activity of ADAMTS4 (aggrecanase-1) by C-terminal truncation. J Biol. Chem 2002;277:11034–11041. [DOI] [PubMed] [Google Scholar]
- 45.Gendron C, Kashiwagi M, Lim NH, et al. Proteolytic activities of human ADAMTS-5: comparative studies with ADAMTS-4. J Biol Chem. 2007;282:18294–18306. [DOI] [PubMed] [Google Scholar]
- 46.Foulcer SJ, Day AJ, Apte SS. Isolation and purification of versican and analysis of versican proteolysis. Methods Mol Biol. 2015;1229:587–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang K, Wu LD. Aggrecanase and aggrecan degradation in osteoarthritis: a review. J Int Med Res. 2008;36:1149–1160. [DOI] [PubMed] [Google Scholar]
- 48.Kashiwagi M, Enghild JJ, Gendron C, et al. Altered proteolytic activities of ADAMTS-4 expressed by C-terminal processing. J Biol Chem. 2004;279:10109–10119. [DOI] [PubMed] [Google Scholar]
- 49.Gao G, Plaas A, Thompson VP, Jin S, Zuo F, Sandy JD. ADAMTS4 (aggrecanase-1) activation on the cell surface involves C-terminal cleavage by glycosylphosphatidyl inositol-anchored membrane type 4-matrix metalloproteinase and binding of the activated proteinase to chondroitin sulfate and heparan sulfate on syndecan-1. J Biol Chem. 2004;279:10042–10051. [DOI] [PubMed] [Google Scholar]
- 50.Santamaria S, Yamamoto K, Teraz-Orosz A, et al. Exosites in hypervariable loops of ADAMTS spacer domains control substrate recognition and proteolysis. Sci Rep. 2019;9:10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Silva SV, Lima MA, Cella N, Jaeger RG, Freitas VM. ADAMTS-1 is found in the nuclei of normal and tumoral breast cells. PLoS One. 2016;11:e0165061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jobin PG, Butler GS, Overall CM. New intracellular activities of matrix metalloproteinases shine in the moonlight. Biochim Biophys Acta Mol Cell Res. 2017;1864:2043–2055. [DOI] [PubMed] [Google Scholar]
- 53.Kuno K, Matsushima K. ADAMTS-1 protein anchors at the extracellular matrix through the thrombospondin type I motifs and its spacing region. J Biol Chem. 1998;273:13912–13917. [DOI] [PubMed] [Google Scholar]
- 54.Pos W, Crawley JT, Fijnheer R, Voorberg J, Lane DA, Luken BM. An autoantibody epitope comprising residues R660, Y661, and Y665 in the ADAMTS13 spacer domain identifies a binding site for the A2 domain of VWF. Blood. 2010;115:1640–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flannery CR, Zeng W, Corcoran C, et al. Autocatalytic cleavage of ADAMTS-4 (Aggrecanase-1) reveals multiple glycosaminoglycan-binding sites. J Biol Chem. 2002;277:42775–42780. [DOI] [PubMed] [Google Scholar]
- 56.Troeberg L, Mulloy B, Ghosh P, Lee MH, Murphy G, Nagase H. Pentosan polysulfate increases affinity between ADAMTS-5 and TIMP-3 through formation of an electrostatically driven trimolecular complex. Biochem J. 2012;443:307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]