

Abstract
Emerging clonal complexity has brought into question the way in which we perceive and, in turn, treat disorders of the hematopoietic system. Former models of cell-intrinsic clonal dominance driven by acquisition of driver genes in a stereotypic sequence are often insufficient in explaining observations such as clonal hematopoiesis, and new paradigms are in order. Here, we review the evidence within the hematologic malignancy field and also borrow from perspectives rooted in evolutionary biology to reframe pathogenesis of hematologic disorders as dynamic processes involving complex interplays of genetic and nongenetic subclones and the tissue microenvironment in which they reside.
Significance:
Hematopoietic malignant and premalignant syndromes exhibit vast clonal diversity that is subject to selection imposed by the tissue microenvironment, as well as artificial selection by therapy. Tackling these disorders requires an appreciation of heterogeneity at both genetic and nongenetic levels, which can be borrowed from evolutionary biology principles. Models and drug development strategies that veer away from targeting solely dominant clones and, instead, embrace this complexity to outsmart it are required for long-term remission.
Introduction
Hematopoietic malignancies, such as myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML), are notoriously heterogeneous groups of diseases impacting myeloid lineages of the hematopoietic system within the bone marrow. Typically penetrating in the late years of life, these diseases are defined not only by uncontrolled, invasive proliferation but also uniquely by a block in the ability to differentiate, or effectively reconstitute the various lineages of the blood. In fact, this differentiation block can be the predominant disease feature, particularly in low-risk MDS in which 70% of patients lose their ability to form red cells, platelets, and/or an effective immune system and die from bone marrow failure rather than progression to secondary leukemia (1).
Importantly, the hematopoietic system comprises all cellular components of the blood and is, therefore, uniquely dynamic, diverse, and tightly responsive to the host's environment. Classically thought to differentiate unidirectionally and governed by strict hierarchical rules, it is now appreciated that hematopoietic stem and progenitor cells (HSPC) comprise a highly plastic continuum of cell states and populations, exhibiting more plasticity than previously thought and providing an elegant model for evolutionary adaptability (2,3,4). Hematopoiesis is maintained throughout life despite an onslaught of genetic and environmental insults; however, certain molecular footprints exist that may mark the early stages of its derailment.
Over the past decade, sequencing advances have led to seminal findings in both individuals with overt disease as well as healthy individuals exhibiting clonal expansion of hematopoietic cells but lacking symptoms typical of hematologic disorders. The latter phenomenon is now known as clonal hematopoiesis (CH) and has brought into question our conventional understanding of the biology underlying clonally complex diseases. Clonality in the bone marrow, defined by expansion of one or more genetically distinct cell populations, is now seen as inevitable with aging. Discovered initially due to observations of X-chromosome inactivation, CH has exposed the inevitability of somatic nongenetic and genetic changes within the bone marrow with aging that, in some cases, lead to a fitness advantage and consequent clonal expansion (5). Strikingly, mutations in classic oncogenes like TP53 are found in hematopoietic cells of seemingly healthy individuals (6, 7). Moreover, the most common genes mutated in CH are also implicated in animal models of leukemia—DNMT3A, TET2, and ASXL1—yet often lack a clear, consistent link to overall survival in humans (7). This new knowledge begs the reevaluation of traditional views of hematologic malignancy defined by “clonal dominance” or the predominance of a single clone that has acquired classic “driver” mutations in a stereotypical sequence and consequently appropriated the bone marrow. Even if clonal dominance is observed, increasing bodies of evidence support preexisting diversity in the form of both genetic and nongenetic variation that are often only brought to light in the face of substantial environmental perturbation, including selection imposed by treatment (8,9,10,11,12,13,14,15).
Glossary of terms
| Term | Definition |
|---|---|
| Attractor state | A particularly stable transcriptional state that attracts cells in less stable states toward it, that is, cells exhibiting violations of regulatory rules will tend to gravitate toward the nearest attractor state; a modern descriptor of a canalized state attributed to transcriptional networks |
| Canalization | The process of producing phenotypic constancy in the presence of varying conditions |
| Capacitor | Gene product involved in maintaining robustness to environmental stress |
| Chromothripsis | Multiple chromosomal rearrangements occurring within a single event either within a single chromosome or across multiple chromosomes; chromosome shattering |
| Epigenetic landscape | A metaphor describing the regulation of developmental trajectories by buffering mechanisms that dictate which phenotypes cells are likely to adopt; the shape of the landscape is derived in part due to the cell-intrinsic action of gene regulatory networks and is not equivalent with the modern term “epigenetics” |
| Evolvability | An organism's capacity to generate heritable phenotypic variation |
| Fitness | The propensity of an individual to exhibit reproductive success (if the individual is a cell, it is the number of daughter cells a parent cell is likely to produce); a quantitative metric for selection representing how well adapted an organism is to its environment |
| Gene regulatory network, or GRN | A collection of regulatory molecules that work in concert to determine expression of gene products, the transcriptional state, and, in turn, the final phenotype of a cell; a mechanistic descriptor for the buffering interactions within a cell that contribute to the final shape of Waddington's epigenetic landscape |
| Mutator phenotype | A phenotype exhibiting higher than normal mutation rates |
| Punctuated equilibria | An alternative hypothesis to gradualism, or slow and steady evolutionary change, in which periods of equilibria are disturbed by “rapid and episodic events of speciation” |
| Purifying selection | Selection resulting in reduction in frequency of individuals; negative selection |
| Robustness | The ability of a cell, tissue, or organism to maintain a constant phenotype despite various genotypic and environmental perturbations |
| Selection | A force that acts upon phenotype and results in preferential survival of individual organisms due to greater relative fitness within an environment; the key deterministic mechanism of evolution |
As such, the hematologic malignancy community is faced with somewhat of a conundrum: how to reconcile conventional wisdom of somatic mutation theory—or specific mutated genes “driving” disease in a direct, causal manner—with the now undeniable evidence that many of these mutations can occur without the expected consequences. We argue that context matters, and, while a mutation in an oncogene may very well have “driving” effects, those effects are subject to a slew of buffering mechanisms both within and surrounding a cell that favor a “wild-type” outcome and vary with existing genetic, nongenetic, and environmental variation. Moreover, underlying clonal heterogeneity should be assumed even in the absence of its detection, which is inherently limited by standard diagnostic sequencing strategies. Deciphering the highly dynamic and nonlinear nature of clonal evolution requires systems-based perspectives rooted in evolutionary biology principles. This review will investigate the complex, multifactorial nature of clonal competition in the bone marrow, as well as consider in silico approaches to aid in its investigation.
Deciphering Evolution: the Importance of Underlying Genetic Variation and Simultaneous Phenotypic Robustness
The existence of relatively consistent phenotypes in the face of vast genetic diversity was initially described in 1942 by C.H. Waddington in his published works on the developmental epigenetic landscape (16). Waddington introduced the idea of canalization, or the tendency of organisms to adopt homogeneous intermediate phenotypes with minimal variation despite considerable environmental and genotypic variation (Fig. 1). These intermediate phenotypes have since been coined “attractor states,” which are largely attributed to the existence of gene regulatory networks, and are discussed in-depth in the following sections. Waddington noted that wild-type animals typically seen in nature are “amazingly constant… [like] peas in a pod,” while individuals of the “mutant races” exhibit much higher-order phenotypic variation (ref. 16, p. 564). He attributed the constancy of the wild type to “buffering of the genotype against minor variations not only in the environment in which the animals developed but also in its genetic make-up” (ref. 16, p. 564). Individuals deviating from wild type have, accordingly, experienced either extreme genetic or environmental variation, such that the system of buffering mechanisms has deteriorated.
Figure 1.

Waddington's epigenetic landscape depicts the process of development as balls rolling down a hill that tend toward more stable phenotypes (represented by valleys or “canals”). Waddington's epigenetic landscape depicts cell fate (differentiation) trajectories as balls rolling down a rugged landscape. The likelihood of a cell adopting a given state is represented by the elevation or depression of the landscape, that is, a likely, stable cellular state (as determined by its gene expression profile) is depicted as a valley and unlikely states are represented by elevated hilltops. More recently, mathematical modeling of transcriptional dynamics has, in part, provided the mechanistic underpinnings of this phenomenon of canalization. For a cell to change its phenotype, the cell must transition from an original phenotype that is evolutionarily adaptive (and, therefore, very stable, e.g., attractor state 1) to a new stable phenotype (e.g., attractor state 2). This transformation is driven by the gene regulatory network (GRN), or the overarching architecture between all gene products that orchestrates gene expression profiles and, in turn, determines the final cellular “state” or phenotype. While a given cell can adopt different states along its differentiation trajectory, it can also undergo a state transition or exhibit plasticity between states at a given time point in its development.
A decade later, citing seminal experiments in Drosophila, Waddington and Paton illustrated that extreme environmental perturbation via temperature shock appeared to induce a change in wing morphology that the authors referred to as “crossveinless” (17). While, initially, the majority of these changes were plastic and limited to the somatic life span of the organism, the authors observed that, after five generations, phenotypes could be inherited, that is, persist in the progeny of flies despite removal of the stressor. In an elegant display of artificial selection, they showed that a complex, multicellular organism could “evolve” at a remarkable pace in the face of environmental stress, adapting with heritable phenotypic changes within only a few generations (17). The speed at which this process occurred and the heritable nature of the features suggest an interplay between genetic diversity and transient transcriptional changes in precipitating evolutionary change. The degree to which environmental pressures induce nongenetic changes that are eventually replaced with genetic changes, or simply expose a reservoir of existing rare genetic subpopulations, remains incompletely resolved.
As a corollary to Waddington's findings, Rutherford and Lindquist put forth Hsp90 as a capacitor, or gene product that plays a fundamental role in maintaining robustness to environmental stress. In an elegant series of Drosophila genetic models, they showed that mutating Hsp90 reveals preexisting variability (18) in a wide array of detectable traits across multiple tissues. They conclude that Hsp90 acts as an interface between the environment and fundamental signaling processes within a cell—specifically, that Hsp90 is a crucial environmental sensor that maintains homeostasis via buffering genetic variability (18). Rutherford and Lindquist parallel Waddington's findings in describing not only expression of cryptic variation but also its important consequence: When the source of the expressed trait is heritable and occurs in germline tissue, it can lead to Hsp90-independent expression across a lineage. This process, coined genetic assimilation, is the result of inheritance (vertical transmission from parents to offspring) of stress-induced phenotypic variation. Thus, transition from cryptic variation (lacking phenotypic consequence) to an expressed trait surrenders the organism to the mercy of natural selection, thereby losing its adaptive neutrality.
In short, genetic variation is not a mere byproduct of stochastic genotoxic insult, but an essential source of evolution and adaptability. Importantly, mutations depend upon both external environmental context and internal regulatory networks in their translation to phenotype. Applying these concepts to hematopoiesis will aid in our interpretation of phenomena like CH, mechanisms of transformation to hematologic malignancies, and the role of mutations in cancer. While germline context undoubtedly plays a role in shaping phenotypes, this review will focus on de novo somatic changes involved in cancer, as they represent a major factor in malignant transformation and source of variability in clonal selection. Germline impacts are discussed in detail elsewhere (19).
Applying Evolutionary Concepts to Clonality in the Bone Marrow
Because the median ages of AML and MDS diagnoses are 68 and 71, respectively, patients suffering from myeloid disorders have experienced a lifetime of environmental exposures, including progression of natural biological mechanisms associated with aging, as well as direct genotoxic exposures from chemotherapy and radiation (20). In somewhat of an oversimplification, the molecular events leading to the symptomatic consequences of hematologic malignancy comprise three essential, interdependent elements: mutations, nongenetic aberrations, and changes to the microenvironmental milieu (13, 21). While the relative contributions of these events may differ between patients, it has become clear that malignant transformation requires more than any individual mutation or even combination of mutations; transformation is, rather, a more insidious and global breakdown of the very mechanisms that protect hematopoietic integrity and ensure production of properly differentiated blood cells.
The hematopoietic system is, by design, beautifully structured and buffered to ensure maintenance of healthy hematopoiesis throughout most of life. For one, the hierarchical nature of the system ensures that the differentiated cells comprising the functioning immune system, red blood cells, and platelets have a limited life span (22,23,24). Therefore, a premeditated inverse relationship exists between differentiation/proliferation and self-renewal, such that the most rapidly dividing cells with the highest mutation rates will be cleared from the body within hours to weeks (with some exceptions). Long-term hematopoietic stem cells (HSC) at the apex of this hierarchy are largely quiescent, protected by the stem cell niche, and reserved for reconstituting the blood only in the presence of specific cues, leaving the more proliferative multipotent progenitors to do the majority of the heavy lifting (25).
The interdependency of hematopoietic cells with one another as well as their surrounding stroma (extrinsic factors), therefore, can be seen as one layer of a protective buffering mechanism. Internal epistatic networks within the HSCs themselves (intrinsic factors) form another layer. An increasing body of evidence has illustrated that each layer of buffering is slowly disrupted with aging (26). Over time, HSCs respond more aberrantly to mutation, heritable nongenetic, and microenvironmental changes, ultimately, being remodeled into their leukemic counterparts (26, 27). While this suggests that disease-relevant stem cells and stroma bear little resemblance to healthy bone marrow by the time of clinical presentation of myeloid neoplasms, it also presents the field with opportunity to approach our understanding of leukemogenesis in a new light.
Overall, striking congruences exist between evolutionary theory and hematopoietic malignancies, the original examples of cancer stem cells. One can intuit clonal competition as Darwinian evolution on a microscopic scale: Long-lived stem cells in the microcosm of the bone marrow acquire genetic and nongenetic diversity over time, adding heterogeneous subpopulations to the overall stem cell pool upon which selection can act, in a manner not unlike competition between organisms of the same species in natural selection. In fact, the fitness of clonal populations of cells, or their propensity to replicate, can be depicted as Sewall Wright's fitness landscape, which is a historic visualization illustrating the relationship between an organism's genotype and its reproductive success (Fig. 2). In these representations, cells move across the x, y-plane (or genotypic space) as they acquire mutations. Certain combinations of mutations may result in greater fitness, represented as an upward inflection on the z-axis, that is, a hilltop. Of note, this convention is the opposite of Waddington's epigenetic landscape, in which more favored states are depicted as valleys rather than hilltops. In a healthy microenvironment, the vast majority of mutations will not produce an immediate phenotype and remain at low frequencies precluding detection, but this shifts over time with environmental stress, such as aging, inflammation, chemotherapy, and targeted interventions. Ultimately, underlying genetic variation is exposed phenotypically, selected for, and, if occurring in a stem cell, goes on to persist in all daughter cells. Just as Drosophila can evolve a crossveinless wing phenotype in the presence of heat, stem cells can evolve phenotypes consistent with relapse in the presence of therapy (17, 18).
Figure 2.
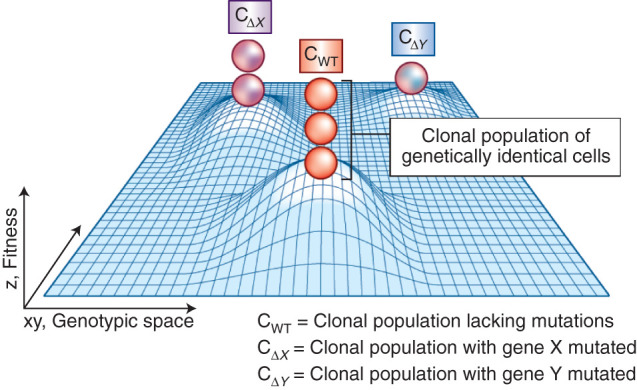
Clonal evolution can be visualized as a fitness landscape, in which individual somatic mutations are minor deviations in genotypic space toward a local fitness optimum. The fitness, or propensity of an individual clone to successfully divide into daughter cells, is depicted here in reduced dimension as a three-dimensional fitness landscape, first described by Sewall Wright in 1931 (113). A mutation causes an individual cell to travel across genotypic space (depicted as the x,y-axes), which can impact fitness (depicted as the z-axis). Certain combinations of mutations may result in greater fitness and subsequent clonal expansion, represented as an upward inflection on the z-axis, that is, a hilltop. An actively adapting population will climb uphill by a sequence of minute genetic changes until a local optimum is achieved. Clonal expansion may occur if evolution slows as these optimums are reached, that is, conditions stabilize. Of note, this convention is the opposite of Waddington's epigenetic landscape, in which more favored states are depicted as valleys rather than hilltops.
The learnings from this analogy are three-fold: (i) Stem cells or cells acquiring stem-like properties are the source of persistent genetic diversity; (ii) this underlying genetic variation should be assumed, is often cryptic (i.e., does not produce a phenotype), and contributes to the capacity for clonal adaptation; and (iii) understanding how perturbations of the system produce malignant transformation presents a unique opportunity to approach therapeutic management as not just targeting any individual mutation but rather shifting or resetting the entire fitness landscape.
Microenvironmental Context Governs Cell-intrinsic Mutations
Conventional genomic sequencing strategies frequently present with two fundamental limitations: (i) Studies have largely focused on either dominant clones or subclones that have sufficiently expanded in the bulk tumor, largely ignoring early and rare events, and (ii) the sole evaluation of genomic alterations in tumor cells does not take into account transcriptional states, as well as significant alterations to the surrounding microenvironment that are fueling clonal expansions (7, 28, 29). To gain a more complete understanding of the processes leading up to leukemogenesis, one must appreciate the impact of a given mutation as a function of the selective pressures surrounding it.
By the time of clinical presentation, tumors have substantially metamorphosed into malignant tissues that, at times, hardly resemble their wild-type precursors (30). While impacts of mutations at this stage are well studied, the early and very rare mutations that do not produce immediate expansion or symptomatic consequences are poorly understood (Fig. 3; ref. 31). However, sequencing advancements and the discovery of CH provide a window of opportunity during which we can study very early “premalignant” events, including the paradigm-shifting discovery that healthy individuals can harbor TP53 mutations in the bone marrow (7). Our understanding of why TP53 loss of function leads directly to malignant transformation in some individuals but exhibits relatively slow growth in others remains limited.
Figure 3.

Dynamic selective pressures imposed by the microenvironment with aging and disease pathogenesis result in differential expansion of clones. In early life, immune surveillance results in purifying selection and effective removal of (pre-)malignant clones, thereby preventing their expansion and, in turn, detection. Immune clearance of malignant cells evidently declines with age in the phenomenon known as immunosenescence, in part, providing an explanation for the greater incidences of myeloid malignancies in the elderly (114). For a number of reasons, including thymic involution and increasing ratios of regulatory T cells to cytotoxic T cells, (pre-)malignant cells expressing neoantigens that would otherwise be eliminated by immune surveillance are permitted to persist and expand in old age (114,115,116,117,118). Later in life, the increasing selective pressure of inflammaging can lead to clonal expansions and clonal hematopoiesis. Finally, in leukemia, malignant clones are selected for and completely consume the bone marrow, leading to symptomatic consequences. The upper range depicted in red represents the threshold at which the buffering by remaining healthy hematopoietic clones is no longer sufficient to achieve a canalized healthy phenotype and clinical symptoms emerge. The lower range depicted in dark peach represents the sequencing limit of detection. The green represents phenotypically wild-type clones, while the other colors represent (pre-)malignant clones with pathogenic mutation(s). In the aging microenvironment, the wild-type clones retain enough of a selective advantage to sustain hematopoiesis and prevent any clinical consequences; however, upon progression to leukemia, the bone marrow microenvironment has been remodeled to favor mutated clones over wild-type (green). The wild-type clones can no longer sustain healthy hematopoiesis, and the patient will present with symptoms of leukemia.
One possible explanation for this is the differential selection pressures imposed by the surrounding microenvironment, including both stromal cells as well as additional HSPCs competing within the same pool. In patients with bone marrow failure syndromes, such as Fanconi anemia, Diamond–Blackfan anemia, and 5q- syndrome (MDS), p53 is upregulated in response to defective DNA repair or ribosomal biogenesis (32,33,34). Evidently, erythroid progenitor cells are particularly sensitive to DNA repair and ribosomal defects, consequently activating apoptotic signaling via p53, reducing the pool of cells capable of erythropoiesis, and producing a macrocytic anemia (33, 34). In an effort to abate this process, cells that lose function of a single TP53 allele might “rescue” the proapoptotic phenotype, and, in turn, undergo positive selection. The bone marrow microenvironment, responding to the reduced pool of red blood cells and consequent insufficient oxygen delivery to tissues, may provide a minor fitness advantage to clones harboring heterozygous loss of TP53 (Fig. 4; ref. 34). In fact, TP53 loss has been shown to be a rescue mechanism for a variety of developmental diseases in which TP53 is pathogenically activated (35, 36).
Figure 4.

Fitness landscapes are themselves dynamic and can change in the context of a healthy versus aberrant microenvironment. In the healthy microenvironmental context (left), the nonmutated, wild-type clone exhibits the greatest fitness advantage as compared with TP53-mutated clones with mono- and biallelic loss-of-function mutations. The endogenous bone marrow microenvironment favors wild-type clones as an evolutionary mechanism to maintain intact hematopoiesis, a feature essential for viable life during reproductive years. In contrast, an anemic microenvironment (right) may sustain aberrant growth signals in an effort to boost erythropoiesis, perhaps providing a selective advantage to the more proliferative TP53-mutated clones. WT, wild-type.
In the bone marrow of a healthy young individual, on the other hand, the predominant selective pressure on a heterozygous TP53 clone may be negative, for example, acquisition of minor DNA damage might result in subsequent clearance by the immune system. Furthermore, given p53's role in maintenance of genomic integrity, its loss of function can result in a “mutator phenotype,” which generally reduces fitness of cells due to the bias in favor of mutations resulting in detrimental effects in a stable microenvironment (37). This scenario is somewhat speculative given the lack of data on extremely rare clones as well as accompanying microenvironmental data, but it nevertheless supports a fundamental theme that is frequently ignored by solely cell-intrinsic perspectives: One bone marrow does not fit all. Roughly estimating, if the total pool of HSCs is 50,000 in humans (a conservative estimate), the per base mutation rate per cell division is 10−8, and the coding length of TP53 is 1,180 bases, there is a 0.59 probability that a coding base will be mutated with each round of division in any one of the 50,000 stem cells [Eq. (A); ref. 31]. It is, therefore, reasonable to deduce that the likelihood of accruing a detrimental TP53 mutation within a given person's lifetime is far greater than the fraction of individuals who will go on to develop TP53-driven malignancies; therefore, either small such clones may simply be tolerated in the absence of further aberrations, or negative or purifying selection is likely to play a critical role in curbing somatic evolution and ridding the body of cells with mutated oncogenes or tumor suppressors throughout life (38, 39). While this calculation neglects precise quantitation of the number and probability of transitions or transversions at a given base producing functional consequence, as well as rates of cell division–independent mutagenesis (such as cytosine deamination), it serves as a reasonable approximation.
 |
Using publicly available human SNP data, Chu and Wei have illustrated that mutations in cancer-related genes undergo strong purifying selection when compared with other genes (40). From an evolutionary perspective, cells within healthy and stable microenvironments are more likely to be optimally adapted; consequently, the few mutations that do produce a functional impact (the vast majority of mutations will be selectively neutral) are overwhelmingly in favor of producing a detrimental impact on fitness given that the surrounding environment maintains relative stability and retention of normal tissue structure (37, 40). The phenomenon of cells harboring mutations faring worse than wild-type cells has also been illustrated in a competitive bone marrow transplantation model. Bondar and Medzhitov showed that irradiated transplanted HSCs were outcompeted by nonirradiated HSCs (41). Furthermore, IDH1 mutations have been shown to paradoxically reduce HSC self-renewal (37, 42). Evidently, cell competition is itself an evolutionary strategy for tissue fitness. In a Drosophila model, another group described the mechanistic underpinnings of cell competition, in which cells with lower division rates engage specific transcriptional programs that, ultimately, result in their programmed death, thereby, providing a basis for the systematic elimination of comparatively “less fit” cells (43). These findings have profound implications on interpretation of classic monogenic mouse models of hematopoietic malignancies. If all cells within the bone marrow harbor a mutation in a given cancer gene, the pool of wild-type cells to compete against the mutated cells is effectively nonexistent. Notably, the fitness landscape bears little resemblance to that of a healthy human for the majority of life.
In addition to the character and quantity of competing stem cells, increasing bodies of evidence support a crucial role of inflammation in sculpting the fitness landscape. The phenomenon known as inflammaging contributes to bone marrow clonal expansion, epigenetic aging signatures, and consequent myeloid bias and is reviewed in detail elsewhere (26,44,45,46). In the context of inflammation, cells that were once optimally adapted are no longer the fittest and clones with previously neutral or even mildly detrimental phenotypes can now be positively selected for. By this logic, it is perhaps not surprising that bone marrow failure syndromes that paradoxically present with hypocellularity of the bone marrow are risk factors for hematopoietic malignancies (47). Healthy individuals producing normal blood counts are unlikely to accommodate abnormal cues favoring sustained proliferation of aberrant clones, opting for maintenance of an evenly distributed HSC pool and elimination of clones exhibiting early signs of malignancy. Opposingly, anemic patients are likely to sustain a milieu with chronic growth signals that grants greater fitness advantages to specific clones. In an effort to replete cell counts lost to apoptosis, the TP53 clone that would have typically been cleared is permitted to persist and expand long enough that it may eventually acquire complete loss of function of TP53 and produce a secondary leukemia.
The profound significance of microenvironmental influence on (pre-)malignant syndromes was recognized in 1889, when Paget first described the seed and soil hypothesis (48). Very important work by Beatrice Mintz and Mina Bissell in 1975 and 1984, respectively, has illustrated that cancer is indeed as much a product of greater tissue architecture as it is one of cell-intrinsic mutations (49,50,51,52). Through elegant experiments involving injection of malignant mouse teratocarcinoma into blastocysts, Mintz and Illmensee were able to generate phenotypically wild-type mice that were effective mosaics of the tumor-derived cells and those of the host blastocyst (49). Strikingly, cells originating from the teratocarcinoma successfully recapitulated developmentally unrelated tissues that were phenotypically normal in spite of their malignant origins, which was also elaborated on by Bissell in a Rous sarcoma virus (RSV) model of malignancy (49). Echoing prior findings by Duran-Reynals, Dolberg and Bissell illustrated that RSV is neither tumorigenic nor teratogenic when injected into a 4-day-old chicken embryo despite elevated activity of the suspect v-Src oncogenic kinase (50, 53). Interestingly, they found that the inoculated embryonic cells exhibited delayed transformation when placed in culture, suggestive of some inhibitory quality of the embryonic microenvironment on tumorigenesis. In other words, the endogenous embryo was simply not the correct soil. They summarized that, despite v-Src kinase being necessary for RSV-mediated transformation, “there are one or several other developmentally related criteria that must be met before expression of the malignant phenotype is possible” (ref. 50, p. 556).
In 1985, Bissell took her RSV studies a step further and began to tease out the precise qualities of the soil that led to malignant transformation, beginning with the logical acknowledgment that the process of injection of RSV into the body of an animal created local wounding (51). Furthermore, the ubiquitous presence of nonmalignant hemorrhagic lesions characteristic of RSV infection throughout the body but rarity of sarcomas distal to the injection site led her to deduce that the wounding process itself was one of the aforementioned “developmentally related criteria” required for transformation. The authors illustrated that infliction of injury to the wing opposite the injection site was sufficient to cause formation of tumors that were indistinguishable from those at the original site within 8 to 9 days (51).
In short, the impact of a mutation is not fixed and often not predictable in the absence of understanding context; even the most malignant leukemic stem cell (LSC) does not exist in a vacuum and is, rather, part of a hierarchical multicellular tissue. Clonal fitness leading to cancer is as much a function of the higher-order ecosystem of the bone marrow as it is a disease of mutation (37). Instead of focusing on late-stage, largely “cell-intrinsic” expansion of clones harboring complete TP53 loss of function, greater focus should be placed on the mechanisms that ensure that this does not happen for most of life despite the constant onslaught of stochastic mutations and genotoxic exposures (27, 30, 37). In fact, we understand very little of the early processes governing how genetic variation eventually produces a malignant phenotype and why certain phenotypes are selected for or against. Taken together, it is perhaps not surprising that even the most sinister mutations can be compatible with normal hematopoiesis and that this property is by design.
The Role of the Gene Regulatory Network and Nongenetic Diversity in Normal hematopoiesis And leukemogenesis
Nongenetic factors, including transcription factors, DNA methylation, and histone modifications, play a key role in phenotypic diversification in normal hematopoiesis, as well as in MDS and AML (4, 54, 55). Essential features in the development and physiology of multicellular organisms, nongenetic phenomena are highly plastic and often tightly responsive to the environment, rendering them ideal mechanisms to be misappropriated by cancer. The aspects of nongenetic heterogeneity as an unintentional consequence of tumor evolution, as a buffer against shifting selective pressures, and as a reflection of hierarchical multicellular organization remain to be resolved (56). Adapting the paradigm of normal development, the role of nongenetic factors in maintaining higher-order tissue architecture provides an intuitive explanation for self-sustaining microenvironmental changes and differentiation hierarchies within tumors.
Herein lies a notable paradox in current paradigms: The production of diverse cellular phenotypes during normal development is almost universally attributed to nongenetic phenomena, independent of genomic alteration; yet, phenotypic diversification during tumorigenesis has overwhelmingly been attributed to genetic events, that is, mutations (57). Both processes ultimately require generation of a stable cellular phenotype by means of heritable and successive directional changes from parent to daughter cell, yet our understanding of the underlying mechanisms has been incongruous. Traditionally, neoplastic transformation has been viewed as a cell-intrinsic, linear process of acquiring driver mutations in a small number of “oncogenes” and “tumor suppressor genes.” Yet, mutations are components of a much greater system. A mutation altering gene regulatory or amino acid–coding sequence feeds into a gene regulatory network (GRN; ref. 57), which orchestrates the gene expression profile. This, in turn, determines the final cellular “state” or phenotype that the cell will adopt: either a stable expression state compatible with normal phenotype or one compatible with uncontrolled proliferation characteristic of malignancy. Put simply, just as external cues from the microenvironment impact the phenotype of a clone, so do internal elements of the GRN, such as the presence or absence of transcription factors or histone marks necessary for a mutated gene product to be transcribed or exert its function within the cell.
To understand the role of nongenetic heterogeneity in leukemia, we must first understand its evolutionary significance in maintenance of normal hematopoiesis. One group showed that transcriptional stochasticity previously viewed as inconsequential harbors subpopulations bearing distinct and meaningful biological properties (58). Using Gaussian mixture modeling, they inferred that stochastic transcriptional fluctuations can generate stable phenotypes. They observed that over extended culture, isolated Sca-1highest and Sca-1lowest HSPCs populations tended to relax toward central populations not only in terms of Sca-1 levels but also gene profiles and differentiation potential, supporting the notion of a noncommitted HSPC attractor state. Attractor states are a product of the GRN and represent highly stable expression states to which cells in more unstable states (that is, those violating regulatory interactions) spontaneously gravitate (59). Frequently depicted as “valleys” in Waddington's epigenetic landscape, they account for the existence of distinct cellular phenotypes, are a necessary manifestation of nonlinear dynamics, and have been shown by in silico experiments to be an inevitable consequence of increasing network complexity with evolution (see Fig. 1; ref. 60). More recent work by Wheat and colleagues directly illustrated that transcriptional dynamics of several identity-defining transcription factors in HSPCs exhibit infrequent and stochastic burst–like behavior by means of imaging single mRNA molecules (4). This suggests a model by which HSPCs reside in a “transcriptional space” rather than a fate-determined “transcriptionally primed” state with restricted directionality and limited fate potential. As such, hematopoietic cell fate transitions in early HSPCs may be fluid and reversible. By allowing HSPCs to continuously sample a number of stable expression states, homeostatic robustness of the HSC and early progenitor cell pool may be in fact aided by nongenetic heterogeneity (4). This work has elucidated properties of the hematopoietic system that leverage inevitable biophysical stochasticity as a fundamental tool enabling physiologic choices, suggesting that nongenetic heterogeneity is an essential feature of multicellular tissues rather than simply unwanted “noise” in the system or a byproduct of natural diversification with time. Similar to the role of genetic heterogeneity in evolution, nongenetic heterogeneity is integral to normal development and tissue homeostasis.
While phenomena supporting transcriptional plasticity in cancer have long been appreciated (such as epithelial-to-mesenchymal transition, dedifferentiation, and drug-induced tolerance), direct experimental evidence for the existence of heterogeneous and dynamic transcriptional states in cancer has been lacking until recently. Novel insights are beginning to shed light on the processes by which robust malignant cell states (e.g., treatment-resistant LSC phenotypes) are produced by noise-driven transcriptome dispersion (59). Recently, Shaffer and colleagues used bulk transcriptome sequencing of single-cell–derived isogenic colonies to document the existence of distinct and heritable transcriptomic states in melanoma cell lines (55). Through direct observation by means of single-molecule RNA fluorescence in situ hybridization and time-lapse microscopy, they also functionally linked heritable transcriptional states to treatment resistance.
Another group sought to derive how certain cells transition into specific transcriptional states. By means of in silico modeling, they illustrated that cancer cells are capable of entering therapy-resistant states of transient, coordinated high expression of several genes (61). Interestingly, entry into these high expression states is dependent upon transcriptional bursting, a well-described phenomenon of genes switching between on and off states and is independent of any specific gene or pathway. This suggests that the inherent stochasticity of GRNs is sufficient in explaining nongenetic heterogeneity and plasticity in cancer (reviewed in ref. 62).
Notably, mutations in gene products involved in DNA methylation, demethylation, and chromatin-modifying enzymes are highly frequent in MDS and AML (28, 29). The impact of these mutations on the availability of transcriptional states upon which selection can act remains largely unresolved. Recently, one group showed that loss of a histone acetyltransferase, Kat2a, enhances transcriptional noise and, in turn, depletes LSCs (63). It is intriguing to speculate that targeting transcriptional noise as a therapeutic strategy may destabilize leukemic transcriptional programs and cause differentiation and, therefore, depletion of LSCs; however, additional studies are needed.
Evolvability of the Bone Marrow: Punctuated Versus Continuous?
Overall, genetic and nongenetic heterogeneity work together to produce a remarkable diversity of cells within the bone marrow in disease states, as well as preceding stages. The inevitability of such diversity warrants discussion of the concept of evolvability, defined as “an organism's capacity to generate heritable phenotypic variation” (ref. 64, p. 8240).
The most widely accepted mechanism of accelerated evolvability in cancer is mutator phenotype. It is well described in colon and breast cancers with mutations in DNA mismatch repair genes and BRCA1/2 (65, 66). In addition, complete loss of function of TP53 is associated with complex karyotypes and copy number changes in AML and is a well-studied mechanism of leukemic transformation in both MDS and MPN (67,68,69,70). Intuitively, loss of DNA repair function or genomic integrity accelerates the rate of somatic mutation acquisition, and, in turn, resident cells will more rapidly diversify on the level of the genome. More recently, the phenomenon known as chromothripsis or “chromosome shattering,” in which isolated chromosomal regions undergo catastrophic rearrangements, has gained attention (71). Interestingly, two studies of pan-cancer whole-genome sequencing elucidated that chromothripsis is pervasive, arising in over 50% of all cancers, and frequently occurs as an early event in oncogenesis (71). Initially described in chronic lymphocytic leukemia and osteosarcoma, chromothripsis has been shown to occur in 6.6% of AML cases (72, 73). The gravity of this phenomenon is in the rate at which genomic change can occur: In a single instance, chromosome(s) can undergo profound rearrangements and somehow maintain cell viability. This bears uncanny resemblance to the theory of punctuated equilibria, or the periodic rapid bursts of evolution demonstrated by the fossil record (74). While there is a tendency to view evolution as gradual, history has illustrated that sweeping changes to the environment can engender mass extinctions and simultaneous rapid evolution of species (74). In this manner, chromothripsis can be viewed as an attractive mechanism by which cells can immediately adapt in the face of substantial selective pressures (e.g., therapy). However, whether chromothripsis directly promotes cancer evolution or is largely a consequence of telomere crisis remains to be resolved.
While mutator phenotypes are an intuitively appealing mechanism of acquired evolvability, it is important to note that they can reduce fitness and are not a requirement for malignant transformation (75). Importantly, healthy individuals exhibit linear mutation acquisition throughout life, which suggests that mutator phenotypes are naturally outcompeted or actively removed by the immune system in normal physiologic states (31). Because mutator phenotypes appear to be more of an exception than a rule in AML, nongenetic heterogeneity has gained traction as a pertinent source of evolvability in leukemogenesis. In fact, studies have linked acquisition of relapse characteristics with differences in chromatin structure (76). Furthermore, the shorter time frame in which heritable nongenetic fluctuations occur make them more attractive in explaining rapid plasticity, such as that observed in resistance to targeted therapies—time scales often unlikely to accommodate occurrence of novel mutations (though not precluding selection of preexistent ones).
Li and colleagues described the distinct kinetics of genetic and epigenetic heterogeneity via a single-cell multi-omics approach (13). They defined “epiallelic burden” (EPM) as a metric for changes in genomic DNA methylation sites at specific loci (“epialleles”) and compared EPM and tumor mutational burden (TMB) shifts in AML during relapse. They describe alternating patterns in epigenetic- or genetic-derived heterogeneity, that is, patients exhibiting high TMB tend to have low EPM and vice versa. Inferior outcomes were evident in patients with greater EPM burden, which could be traced to specific promoter regions, suggestive of a role of transcriptional regulation in disease progression. Interestingly, different treatments result in distinct epigenetic patterns as exhibited by differential patterns in de novo acquisition of epigenetic hypervariability (13).
While Li and colleagues demonstrated orthogonal evolution of TMB and EPM, Shlush and colleagues demonstrated orthogonal evolution at the level of hematopoietic cell populations contributing to relapse (10). Through both sequencing and functional studies, they showed that relapse can originate either from bona fide immunophenotypic stem cells or more committed non-stem cells that retain stem-like properties via transcriptional reprogramming. While these populations have disparate origins, they nevertheless evolve to bear considerable resemblance to one another. This illustration of distinct events leading to functional similarities—in this case, relapse phenotypes—supports the notion of vast preexisting heritable heterogeneity.
Some studies have taken the concept of evolvability to the next step, attempting to explain the mechanisms by which developmentally responsive epigenetic variability can produce long-lasting genetic variability (a putative model for genetic assimilation). An in silico study described a model by which phenotypic changes are initially epigenetically driven and subsequently replaced with incremental genetic mutations over time (77). This model is attractive due to epigenetic alterations compensating for the generally longer time frame by which genomic mutations are acquired and, in turn, allowed to persist. Another group described a biochemical mechanism by which epigenetic adaptation can be translated into genetic adaptation. Their model involves enrichment of G-quadruplexes in abnormally hypomethylated regions, which can act as mutagenic factors in driving tissue-specific mutational landscapes (78). In short, stable G-quadruplexes reside in regions with methylated CpGs and can stall DNA polymerase, introducing DNA breaks and concomitant gain or loss of genomic material (78). Although the prevalence of a direct causative relationship between epigenetic and genetic variation in leukemogenesis remains unclear, understanding and targeting the capacity of a clone to evolve may be a useful therapeutic or preventative strategy.
Targeting Cancer Evolution
Lessons from the Past: Convergent Patterns of Therapeutic Resistance and the Need for Adaptive Therapy Regimens
Over the past few decades, many therapeutic strategies have been attempted for MDS and AML, ranging from more broadly acting hypomethylating agents (HMA) to exquisitely specific small molecules targeting not only individual gene products but their mutated variants. Targeted FLT3, IDH1, and IDH2 inhibitors midostaurin, ivosidenib, and enasidenib, on one hand, have been shown to induce hematologic responses and prolong remission, but patient responses rarely extend past a single year and are often isolated to small subsets of patients with AML harboring specific molecular or cytogenetic characteristics (79, 80). Evidently, both intra- and interpatient heterogeneity complicate response rates, warranting in-depth discussion of patient eligibility and appropriate metrics for therapeutic efficacy (81). In addition to clinical endpoints, targeted therapy efficacy is established by molecular responses, or (cyto-)genetic evidence of reduction of the dominant clone; however, the preexistence of rare genetic clones and nongenetic subpopulations that contribute to relapse as well as the potential induction of novel pathogenic subpopulations with treatment are often overlooked.
In fact, IDH inhibitor resistance mechanisms are complex and frequent, including, but not limited to, isoform switching and cis dimer-interface mutations (82,83,84). In the case of isoform switching in IDH1-mutated leukemias treated with ivosidenib, rare IDH2-mutated clones emerge to sustain production of oncometabolites and consequently propel leukemogenesis (82). This remarkable adaptability speaks to the extent of preexisting heterogeneity and evolvability of hematologic malignancies. Currently, relapse in the clinic is largely dealt with ad hoc, with therapies often administered only after substantial clonal expansion enables detection in the bulk. In the case of IDH inhibitors, it seems sensible to closely monitor patients for isoform switching or acquisition of additional point mutations in IDH enzymes in the stem cell population and treat resistant clones early rather than waiting for them to expand. As more data emerges on the prevalence of isoform switching and other resistance mechanisms, various combination therapies should be explored, including potential oscillatory administration of ivosidenib and enasidenib to relieve the selective pressure of any single agent and prolong time to relapse (82).
Resistance mechanisms are also common and complex for lenalidomide, a therapy targeting the del(5q) clone frequently administered to patients with low-risk MDS. Lenalidomide produces initial hematologic responses in 70% of patients; however, approximately half of those patients will become resistant to therapy in less than 2 years (85). Because the mutations that confer resistance to lenalidomide therapy are similar with those causing transformation to secondary leukemia (namely TP53, NRAS, and KRAS mutations), strategies to prevent expansion of resistant subclones are urgently needed (67, 86, 87). Importantly, the consequences of altering the fitness environment by means of drug administration, including the possibility of selecting for increasingly malignant clones, must be considered in the design of therapeutic regimens. The particularly well-known prevalence of TP53 resistance mechanisms during lenalidomide therapy warrants investigation of oscillatory administration of lenalidomide and drugs effective against cells harboring TP53 mutations. For example, HMAs or low-dose chemotherapy might select against phenotypes with existing genomic instability. In fact, patients given hypomethylating agents shortly after lenalidomide therapy failure exhibited significantly longer survival rates than those treated with standard supportive therapy (88). As the repertoire of agents that selectively kill certain clones continues to expand, so should the ways and combinations with which we use these agents.
In an effort to epigenetically target hematologic malignancies, bromodomain and extra-terminal (BET) protein inhibitors have also been developed. BET inhibitors have high affinity for acetylated histones within chromatin and have been shown to repress c-Myc expression; however, resistance is frequent (89). The mechanisms of action and resistance are broader than initially established. For one, the emerging evidence supports much broader action of BET inhibitors encompassing a multitude of superenhancer regions rather than sole c-Myc repression (90,91,92). Furthermore, Guo and colleagues illustrated a role of genome-wide enhancer remodeling in acquisition of resistance (21, 91). In particular, they showed that enhancer remodeling results in compensatory expression of oncogenes, and that, importantly, this can be overcome with combination therapy. In a clever demonstration of synergistic lethality, repressing Cdk7 RNA polymerase II (RNAPII) activity with a CDK7 inhibitor in conjunction with a BET inhibitor effectively prevented compensatory RNAPII recruitment at a key enhancer–promoter loop (21).
As a comparison to agents targeting specific molecular pathways, the HMAs azacytidine and decitabine are more broadly acting, have been relatively successful in treating myeloid malignancies, and are now standard of care, with two thirds of patients with MDS achieving transfusion independence (93). This success is, in part, attributed to reduction in intrapatient genetic and nongenetic heterogeneity. While the precise contribution of each remains unclear, these agents are particularly efficacious in AML patients exhibiting adverse cytogenetics, suggestive of their role in induction of genomic instability as a mechanism for cell killing (94). Furthermore, identification of specific promoter regions involved in HMA response has been altogether unsuccessful, implicating global hypomethylation and collapse of nongenetic diversity as contributing mechanisms in the efficacy of these agents (95, 96).
Overall, the failures and successes of the past few decades support the necessity for adaptive therapies. In light of ever-increasing clonal complexity and nongenetic plasticity, the goal of developing “blockbuster” targeted monotherapies against specific mutated gene products has become increasingly distant. Despite initial therapeutic responses, the adaptivestrategies evolved by malignancies are numerous, leading to almost invariable resistance (8). Consideration of more nuanced concepts of clonal balance and relative fitness may be required to outsmart rapidly evolving myeloid malignancies. Herein lies an opportunity to reevaluate both drug development strategies as well as metrics for drug efficacy toward the goal of achieving long-term response rates.
Game Theory–Based Adaptive Therapy Regimens Model Dynamic Fitness Landscapes
Because appreciation of the true extent of subclonal heterogeneity is inherently restricted by current sequencing limits of detection and clinical diagnostic capabilities, in silico modeling of clonal evolution can be instrumental. To this end, various mathematical models of resistance have been put forth, inspired by seminal work on bacterial antibiotic resistance that dates back to 1943 (97). Birth rates of novel subclones, for instance, can be modeled by stochastic acquisition of mutations and death rates determined by drug administration and competition for nutrients (97).
Variations of the classic Luria-Delbrück model have been successful in depicting numerous applications of drug resistance; however, they often disregard the dynamic nature of fitness landscapes. That is, while the environment shapes the fitness landscape and selects for certain adaptations, these adaptations simultaneously alter the environment itself (98). This concept is most intuitive in situations in which clones are in direct competition with each other for microenvironmental resources. For instance, one clone's successful proliferation is likely to coincide with depletion of local resources and paracrine signals, which, in turn, can fundamentally alter the fitness landscape for competing clones. This principle warrants the use of game theory in modeling Darwinian clonal dynamics over more classic optimization-based theories that operate under the assumption of a more constant environment (98).
Originating in economics, game theory has been employed to model Darwinian competition between both individuals of the same species and those of contending species since John Maynard Smith and George Price first described the concept of evolutionary stable strategies in 1973 (99). More recently, these efforts have themselves adapted, shifting from habitat-scale ecologic environments to the microcosm of the tumor microenvironment (100, 101). For instance, Vincent and Gatenby developed the first mathematical framework reconciling greater tissue-level changes with gradual (sub-)cellular genotypic and phenotypic events. In their model, they describe clonal evolution of epithelial cancers in response to microenvironmental selective pressures, with the cancer adopting a strategy of optimized proliferation. Their results capture the adaptive landscapes in which (sub-)clonal populations compete as a result of random mutations and, importantly, describe the evolutionary potential of cells through mutual interactions with their environment (100).
Another group added an additional layer of complexity by incorporating the clinician as well as competing clonal dynamics in a predator–prey model (102). In this model, the physician plays in opposition to the malignant clones. While the clones are, again, optimizing their own proliferation, the physician oncologist strives to balance therapeutic efficacy with toxicity for the patient. Notably, only the clinician is capable of playing rationally and begins by playing first, exhibiting leader–follower or “Stackelberg” dynamics. These critical asymmetries do indeed reflect reality, in which scientists and clinicians have the capacity to use past knowledge of relapse patterns to guide rational therapeutic targeting, while a cancer can merely adapt to an environment that it is already experiencing. Although it is tempting to view evolution as trending intentionally toward some greater “optimum,” in reality, it possesses neither directionality nor greater stepwise plan (98). As demonstrated by Stankova, these subtleties can be modeled and used to our advantage in the clinic (102). Notably, they illustrated that standard-of-care repeated administration of monotherapies is equivalent to a “fixed” strategy played by the physician that allows for the cancer to progressively adapt. Furthermore, the act of waiting for substantial tumor progression prior to revision of therapy effectively relinquishes leadership to the malignancy, precipitating inevitable therapeutic relapse. This important work illustrates the necessity of adopting dynamic therapeutic strategies in achieving long-term remission.
The application of game theory in cancer evolution extends far beyond modeling therapies targeting specific clones. Designed to quantify stability of equilibria and dynamics resulting from perturbation, game theory provides a means to study the ways in which perturbing the environment impacts evolutionary strategies employed by cancers (103). As our understanding of the reciprocal interplay of malignant clones and their environments increases, so will our capacity to consider treatment strategies that focus not only on targeting malignant clones in a cell-intrinsic manner but rather shifting the entire fitness landscape in which they reside. Ideally, it would be possible to rejuvenate the bone marrow microenvironment to ensure longevity and robustness of the most benign HSCs that retain the capacity to differentiate and repopulate the diverse array of cells within the hematopoietic system. The microenvironment could hypothetically be recalibrated such that healthy HSCs exhibit optimum fitness, as is the case in early life during which phenotypically wild-type cells are favored by design (37).
The mechanisms by which we can target the microenvironment are as diverse as those employed by cancers to sustain their proliferation. By virtue of the reliance of LSCs on external cues and nutrients, the microenvironment represents a key vulnerability for leukemia. Examples of ways in which we can drug the microenvironment include targeting the inflammatory milieu of MDS and AML, in which key secreted factors such as IL1, IL6, IL8, and TGFβ (among many others) supply aberrant growth signals to LSCs (104,105,106,107). CXCR4 and its ligand CXCL12, for example, play an essential role in the survival of LSCs and their homing to the bone marrow (108, 109). LSCs are conveniently protected by the stem cell niche, leading to frequent chemoresistance (110). Accordingly, we stand to benefit from optimizing treatment paradigms that “prime” leukemic clones by means of removing them from the niche, thereby reducing their fitness advantages upon exposure to therapy. In fact, CXCR4 inhibition has exhibited synergy with chemotherapy preclinically and is currently being investigated in clinical trials (110, 111).
In addition to inflammation, metabolic adaptations of leukemic cells could be targeted strategically. For example, previous studies have demonstrated reprogramming of lipolysis in response to leukemic blasts (112). With normal aging, bone marrow adiposity increases from about 15% in younger years to up to 60% in older age, resculpting the overall fitness landscape and providing opportunity for leukemic clones to emerge. AML cells take advantage of surrounding adipocytes via activating lipolysis, liberating fatty acids for ATP production, and creating a milieu that supports sustained leukemogenesis (112). This dependency opens a window of vulnerability for therapeutic intervention. Pharmacologic reduction of available fatty acids could decrease the survival advantage of LSCs, thereby favoring more benign clones with lesser dependency on abundant ATP production. Perhaps with the emergence of autologous cellular therapies, one could even engineer a therapeutic defector cell to consume local nutrients and render the environment unfavorable to highly aggressive clones. In fact, using a mixed population game theory model, Archetti showed that defector cells reached an evolutionarily stable state of coexistence with benign clones that was robust to invasion by resistant clones (Fig. 5; ref. 103). This approach could be utilized to disrupt the reliance on intrinsic oncometabolites in the case of IDH-mutated leukemias as well.
Figure 5.

Addition of a defector clone (CD) results in decline of fitness of a malignant clone (CM) but stable coexistence with a benign clone (CB). With aging and various other insults, the leukemic microenvironment is transformed, no longer resembling the greater tissue architecture of healthy bone marrow. In the absence of therapy, this aberrant microenvironment may possess growth signals and disrupted tissue organization such that a malignant clone bears a fitness advantage (left). Upon administration of cellular therapy, a defector cell enters the fitness landscape, depleting microenvironmental nutrients critical for sustaining the highly proliferative malignant clone, and, thereby, altering the entire fitness landscape (right). The outcome of this therapy is a stable coexistence of defector cells and benign clones capable of sustaining hematopoiesis. Of note, this depiction differs slightly from previous figures and Wright's classic fitness landscape, as clones are more loosely defined as traveling across phenotypic space, which encompasses both genetic and nongenetic (including transcriptional and epigenetic) components. Within a given population of genetically identical (clonal) cells, there exists subpopulations of cells exhibiting varying transcriptional states that will contribute to clonal evolution and ultimately dictate phenotype.
Future Directions and Overall Takeaways
Malignant stem cells can be thought of as possessing an arsenal of deadly weapons: They are long-lived, are transcriptionally heterogeneous, and possess underlying cryptic genetic variability. If we are to continue with current treatment paradigms and neglect the extensive mechanisms underlying this evolvability, malignancies will always be a few steps ahead. Overall, genetic variability is an essential element of evolution, while nongenetic variability is essential for higher-order tissue organization. Therefore, we must embrace heterogeneity to outsmart it and view targeted therapies as a form of artificial selection that will expose underlying genetic diversity but not eliminate it.
The true extent of genetic, nongenetic, and microenvironmental diversity and their roles in initial leukemogenesis and therapeutic resistance remain incompletely explored. An improved understanding of related mechanisms will be instrumental in increasing predictive capabilities and development of adaptive treatment regimens, including for possible prevention of transformation or relapse.
Acknowledgments
The authors acknowledge Katie Vicari for her expertise in design of the figures presented in this review. E. Schwenger would like to thank Drs. Aviv Bergman and James DeGregori for their inspiring discussions.
Footnotes
Blood Cancer Discov 2021;2:201–15
Authors' Disclosures
U. Steidl reports grants and personal fees from Bayer Healthcare and Aileron Therapeutics; grants from GlaxoSmithKline; and personal fees from Celgene, Stelexis Therapeutics, Pieris Pharmaceuticals, Novartis, and Vor Biopharma outside the submitted work. U. Steidl is a scientific cofounder of and has equity interests in Stelexis Therapeutics. No disclosures were reported by the other author.
References
- 1.World Health Organization. WHO classification of tumours of haematopoietic and lymphoid tissues. Geneva (Switzerland): WHO; 2017. [Google Scholar]
- 2.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730–7. [DOI] [PubMed] [Google Scholar]
- 3.Laurenti E, Göttgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018;553:418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wheat JC, Sella Y, Willcockson M, Skoultchi AI, Bergman A, Singer RH, et al. Single-molecule imaging of transcription dynamics in somatic stem cells. Nature 2020;583:431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busque L, Mio R, Mattioli J, Brais E, Blais N, Lalonde Y, et al. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood 1996;88:59–65. [PubMed] [Google Scholar]
- 6.Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 2012;44:1179–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, Kao YR, Sun D, Todorova TI, Reynolds D, Narayanagari SR, et al. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat Med 2019;25:103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013;152:714–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017;547:104–8. [DOI] [PubMed] [Google Scholar]
- 11.Miles LA, Bowman RL, Merlinsky TR, Csete IS, Ooi AT, Durruthy-Durruthy R, et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020;587:477–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Galen P, Hovestadt V, Wadsworth MH, II, Hughes TK, Griffin GK, Battaglia S, et al. Single-cell RNA-Seq reveals AML hierarchies relevant to disease progression and immunity. Cell 2019;176:1265–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li S, Garrett-Bakelman FE, Chung SS, Sanders MA, Hricik T, Rapaport F, et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med 2016;22:792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams N, Lee J, Moore L, Baxter EJ, Hewinson J, Dawson KJ, et al. Phylogenetic reconstruction of myeloproliferative neoplasm reveals very early origins and lifelong evolution. BioRxiv 2020.11.09.374710 [Preprint]. 2020. Available from: 10.1101/2020.11.09.374710. [DOI] [Google Scholar]
- 15.Van Egeren D, Escabi J, Nguyen M, Liu S, Reilly CR, Patel S, et al. Reconstructing the lineage histories and differentiation trajectories of individual hematopoietic stem cells in JAK2-mutant myeloproliferative neoplasms. Blood 2020;135 Suppl 1:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waddington CH. Canalization of development and the inheritance of acquired characters. Nature 1942;150:563–5. [DOI] [PubMed] [Google Scholar]
- 17.Waddington CH. Genetic assimilation of an acquired character. Evolution 1953;7:118–26. [Google Scholar]
- 18.Rutherford SL, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature 1998;396:336–42. [DOI] [PubMed] [Google Scholar]
- 19.Godley LA, Shimamura A. Genetic predisposition to hematologic malignancies: management and surveillance. Blood 2017;130:424–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma X. Epidemiology of myelodysplastic syndromes. Am J Med 2012;125:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo L, Li J, Zeng H, Guzman AG, Li T, Lee M, et al. A combination strategy targeting enhancer plasticity exerts synergistic lethality against BETi-resistant leukemia cells. Nat Commun 2020;11:740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashby W. The determination of the length of life of transfused blood corpuscles in man. J Exp Med 1919;29:267–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tough DF, Sprent J. Life span of naive and memory t cells. Stem Cells 1995;13:242–9. [DOI] [PubMed] [Google Scholar]
- 24.Saverymuttu SH, Peters AM, Keshavarzian A, Reavy HJ, Lavender JP. The kinetics of 111Indium distribution following injection of 111Indium labelled autologous granulocytes in man. Br J Haematol 1985;61:675–85. [DOI] [PubMed] [Google Scholar]
- 25.Seita J, Weissman IL. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med 2010;2:640–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verovskaya EV, Dellorusso PV, Passegué E. Losing sense of self and surroundings: hematopoietic stem cell aging and leukemic transformation. Trends Mol Med 2019;25:494–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeGregori J. Evolved tumor suppression: why are we so good at not getting cancer? Cancer Res 2011;71:3739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas Research Network. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018;562:526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rozhok AI, DeGregori J. The evolution of lifespan and age-dependent cancer risk. Trends Cancer 2016;2:552–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee-Six H, Øbro NF, Shepherd MS, Grossmann S, Dawson K, Belmonte M, et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 2018;561:473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barlow JL, Drynan LF, Hewett DR, Holmes LR, Lorenzo-Abalde S, Lane AL, et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q-syndrome. Nat Med 2010;16:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boultwood J, Pellagatti A, Wainscoat JS. Haploinsufficiency of ribosomal proteins and p53 activation in anemia: Diamond-Blackfan anemia and the 5q- syndrome. Adv Biol Regul 2012;52:196–203. [DOI] [PubMed] [Google Scholar]
- 34.Ceccaldi R, Parmar K, Mouly E, Delord M, Kim JM, Regairaz M, et al. Bone marrow failure in fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 2012;11:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, Megerdichian C, et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 2011;117:2567–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pellagatti A, Marafioti T, Paterson JC, Barlow JL, Drynan LF, Giagounidis A, et al. Induction of p53 and up-regulation of the p53 pathway in the human 5q- syndrome. Blood 2010;115:2721–3. [DOI] [PubMed] [Google Scholar]
- 37.DeGregori J. Adaptive oncogenesis: a new understanding of how cancer evolves inside us. Cambridge (MA): Harvard University Press; 2018. [Google Scholar]
- 38.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNγ, and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001;410:1107–11. [DOI] [PubMed] [Google Scholar]
- 39.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012;482:400–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chu D, Wei L. Nonsynonymous, synonymous and nonsense mutations in human cancer-related genes undergo stronger purifying selections than expectation. BMC Cancer 2019;19:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bondar T, Medzhitov R. p53-Mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell 2010;6:309–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inoue S, Li WY, Tseng A, Beerman I, Elia AJ, Bendall SC, et al. Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer Cell 2016;30:337–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moreno E, Basler K, Morata G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature 2002;416:755–9. [DOI] [PubMed] [Google Scholar]
- 44.Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood 2017;130:1693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cook EK, Luo M, Rauh MJ. Clonal hematopoiesis and inflammation: partners in leukemogenesis and comorbidity. Exp Hematol 2020;83:85–94. [DOI] [PubMed] [Google Scholar]
- 46.King KY, Huang Y, Nakada D, Goodell MA. Environmental influences on clonal hematopoiesis. Exp Hematol 2020;83:66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bagby GC, Meyers G. Bone marrow failure as a risk factor for clonal evolution: prospects for leukemia prevention. Hematology Am Soc Hematol Educ Program 2007;2007:40–6. [DOI] [PubMed] [Google Scholar]
- 48.Paget S. The distribution of secondary growths in cancer of the breast. Lancet 1889;8:98–101. [PubMed] [Google Scholar]
- 49.Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci U S A 1975;72:3585–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dolberg DS, Bissell MJ. Inability of Rous sarcoma virus to cause sarcomas in the avian embryo. Nature 1984;309:552–6. [DOI] [PubMed] [Google Scholar]
- 51.Dolberg DS, Hollingsworth R, Hertle M, Bissell MJ. Wounding and its role in RSV-mediated tumor formation. Science 1985;230:676–8. [DOI] [PubMed] [Google Scholar]
- 52.Mintz B, Fleischman RA. Teratocarcinomas and other neoplasms as develop-mental defects in gene expression. Adv Cancer Res 1981;34:211–78. [DOI] [PubMed] [Google Scholar]
- 53.Milford JJ, Duran-Reynals F. Growth of a chicken sarcoma virus in the chick embryo in the absence of neoplasia. Cancer Res 1943;3:578–84. [Google Scholar]
- 54.Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017;546:431–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shaffer SM, Emert BL, Reyes Hueros RA, Cote C, Harmange G, Schaff DL, et al. Memory sequencing reveals heritable single-cell gene expression programs associated with distinct cellular behaviors. Cell 2020;182:947–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shaknovich R, De S, Michor F. Epigenetic diversity in hematopoietic neoplasms. Biochim Biophys Acta 2014;1846:477–84. [DOI] [PubMed] [Google Scholar]
- 57.Huang S. Tumor progression: chance and necessity in Darwinian and Lamarckian somatic (mutationless) evolution. Prog Biophys Mol Biol 2012;110:69–86. [DOI] [PubMed] [Google Scholar]
- 58.Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 2008;453:544–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang S. Reconciling non-genetic plasticity with somatic evolution in cancer. Trends Cancer 2021;7:309–22. [DOI] [PubMed] [Google Scholar]
- 60.Siegal ML, Bergman A. Waddington's canalization revisited: develop-mental stability and evolution. Proc Natl Acad Sci U S A 2002;99:10528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schuh L, Saint-Antoine M, Sanford EM, Emert BL, Singh A, Marr C, et al. Gene networks with transcriptional bursting recapitulate rare transient coordinated high expression states in cancer. Cell Syst 2020;10:363–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wheat JC Steidl U.. Gene expression at a single molecule level: implications for MDS and AML. Blood In press. [DOI] [PMC free article] [PubMed]
- 63.Domingues AF, Kulkarni R, Giotopoulos G, Gupta S, Vinnenberg L, Arede L, et al. Loss of KAT2A enhances transcriptional noise and depletes acute myeloid leukemia stem-like cells. Elife 2020;9:e51754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kirschner M, Gerhart J. Evolvability. Proc Natl Acad Sci U S A 1998;95:8420–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peltomäki P. Deficient DNA mismatch repair: a common etiologic factor for colon cancer. Hum Mol Genet 2001;10:735–40. [DOI] [PubMed] [Google Scholar]
- 66.Semmler L, Reiter-Brennan C, Klein A. BRCA1 and breast cancer: a review of the underlying mechanisms resulting in the tissue-specific tumorigenesis in mutation carriers. J Breast Cancer 2019;22:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.da Silva-Coelho P, Kroeze LI, Yoshida K, Koorenhof-Scheele TN, Knops R, van de Locht LT, et al. Clonal evolution in myelodysplastic syndromes. Nat Commun 2017;8:15099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rodriguez-Meira A, Buck G, Clark S-A, Povinelli BJ, Alcolea V, Louka E, et al. Unravelling intratumoral heterogeneity through high-sensitivity single-cell mutational analysis and parallel RNA sequencing. Mol Cell 2019;73:1292–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012;119:2114–21. [DOI] [PubMed] [Google Scholar]
- 70.Will B, Vogler TO, Narayanagari S, Bartholdy B, Todorova TI, da Silva Ferreira M, et al. Minimal PU.1 reduction induces a preleukemic state and promotes development of acute myeloid leukemia. Nat Med 2015;21:1172–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cortés-Ciriano I, Lee JJK, Xi R, Jain D, Jung YL, Yang L, et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet 2020;52:331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fontana MC, Marconi G, Feenstra JDM, Fonzi E, Papayannidis C, Ghelli Luserna di Rorá A, et al. Chromothripsis in acute myeloid leukemia: biological features and impact on survival. Leukemia 2018;32:1609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011;144:27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gould SJ, Eldredge N. Punctuated equilibria: the tempo and mode of evolution reconsidered. Paleobiology 1977;2:115–51. [Google Scholar]
- 75.Tomlinson IPM, Novelli MR, Bodmer WF. The mutation rate and cancer. Proc Natl Acad Sci U S A 1996;93:14800–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 2016;48:1193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nishikawa K, Kinjo AR. Mechanism of evolution by genetic assimilation: equivalence and independence of genetic mutation and epigenetic modulation in phenotypic expression. Biophys Rev 2018;10:667–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De S, Michor F. DNA secondary structures and epigenetic determinants of cancer genome evolution. Nat Struct Mol Biol 2011;18:950–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017;130:722–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017;377:454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Estey E, Levine RL, Löwenberg B. Current challenges in clinical development of ‘targeted therapies’: the case of acute myeloid leukemia. Blood 2015;125:2461–6. [DOI] [PubMed] [Google Scholar]
- 82.Harding JJ, Lowery MA, Shih AH, Schvartzman JM, Hou S, Famulare C, et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition. Cancer Discov 2018;8:1540–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Intlekofer AM, Shih AH, Wang Bo, Nazir A, Rustenburg AS, Albanese SK, et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018;559:125–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Quek L, David MD, Kennedy A, Metzner M, Amatangelo M, Shih A, et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat Med 2018;24:1167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Giagounidis AAN, Germing U, Haase S, Aul C. Lenalidomide: a brief review of its therapeutic potential in myelodysplastic syndromes. Ther Clin Risk Manag 2007;3:553–62. [PMC free article] [PubMed] [Google Scholar]
- 86.Jädersten M, Saft L, Smith A, Kulasekararaj A, Pomplun S, Göhring G, et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J Clin Oncol 2011;29:1971–9. [DOI] [PubMed] [Google Scholar]
- 87.Mossner M, Jann JC, Wittig J, Nolte F, Fey S, Nowak V, et al. Mutational hierarchies in myelodysplastic syndromes dynamically adapt and evolve upon therapy response and failure. Blood 2016;128:1246–9. [DOI] [PubMed] [Google Scholar]
- 88.Prebet T, Cluzeau T, Park S, Sekeres MA, Germing U, Ades L, et al. Outcome of patients treated for myelodysplastic syndromes with 5q deletion after failure of lenalidomide therapy. Oncotarget 2017;8:81926–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A 2011;108:16669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153:320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bell CC, Fennell KA, Chan YC, Rambow F, Yeung MM, Vassiliadis D, et al. Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nat Commun 2019;10:2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Marine JC, Dawson SJ, Dawson MA. Non-genetic mechanisms of therapeutic resistance in cancer. Nat Rev Cancer 2020;20:743–56. [DOI] [PubMed] [Google Scholar]
- 93.Kaminskas E, Farrell AT, Wang Y, Sridhara R, Pazdur R. FDA drug approval summary: azacitidine (5-azacytidine, VidazaTM) for injectable suspension. Oncologist 2005;10:176–82. [DOI] [PubMed] [Google Scholar]
- 94.Döhner H, Dolnik A, Tang L, Seymour JF, Minden MD, Stone RM, et al. Cytogenetics and gene mutations influence survival in older patients with acute myeloid leukemia treated with azacitidine or conventional care. Leukemia 2018;32:2546–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fandy TE, Herman JG, Kerns P, Jiemjit A, Sugar EA, Choi SH, et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood 2009;114:2764–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shen L, Kantarjian H, Guo Yi, Lin E, Shan J, Huang X, et al. DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J Clin Oncol 2010;28:605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Luria SE, Delbrück M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 1943;28:491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nowak MA, Sigmund K. Evolutionary dynamics of biological games. Science 2004;303:793–9. [DOI] [PubMed] [Google Scholar]
- 99.Smith JM, Price GR. The logic of animal conflict. Nature 1973;246:15–8. [Google Scholar]
- 100.Gatenby RA, Vincent TL. An evolutionary model of carcinogenesis. Cancer Res 2003;63:6212–20. [PubMed] [Google Scholar]
- 101.Zhang J, Cunningham JJ, Brown JS, Gatenby RA. Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nat Commun 2017;8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Staňková K, Brown JS, Dalton WS, Gatenby RA. Optimizing cancer treatment using game theory: a review. JAMA Oncol 2019;5:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Archetti M. Evolutionarily stable anti-cancer therapies by autologous cell defection. Evol Med Public Heal 2013;2013:161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Abdul-Aziz AM, Shafat MS, Mehta TK, Di Palma F, Lawes MJ, Rushworth SA, et al. MIF-induced stromal PKCβ/IL8 is essential in human acute myeloid leukemia. Cancer Res 2017;77:303–11. [DOI] [PubMed] [Google Scholar]
- 105.Schinke C, Giricz O, Li W, Shastri A, Gordon S, Barreyro L, et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood 2015;125:3144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Reynaud D, Pietras E, Barry-Holson K, Mir A, Binnewies M, Jeanne M, et al. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell 2011;20:661–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhou L, Nguyen AN, Sohal D, Ying Ma J, Pahanish P, Gundabolu K, et al. Inhibition of the TGF-β receptor I kinase promotes hematopoiesis in MDS. Blood 2008;112:3434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013;495:231–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Greenbaum A, Hsu Y-MS, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013;495:227–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cho BS, Zeng Z, Mu H, Wang Z, Konoplev S, McQueen T, et al. Antileukemia activity of the novel peptidic CXCR4 antagonist LY2510924 as monotherapy and in combination with chemotherapy. Blood 2015;126:222–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Boddu P, Borthakur G, Koneru M, Huang X, Naqvi K, Wierda W, et al. Initial report of a phase I study of LY2510924, idarubicin, and cytarabine in relapsed/refractory acute myeloid leukemia. Front Oncol 2018;8:369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shafat MS, Oellerich T, Mohr S, Robinson SD, Edwards DR, Marlein CR, et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017;129:1320–32. [DOI] [PubMed] [Google Scholar]
- 113.Wright S. Evolution in mendelian populations. Genetics 1931;16:97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med 2015;373:1136–52. [DOI] [PubMed] [Google Scholar]
- 115.Goronzy JJ, Fang F, Cavanagh MM, Qi Q, Weyand CM. Naive T cell maintenance and function in human aging. J Immunol 2015;194:4073–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Isidori A, Loscocco F, Ciciarello M, Corradi G, Lecciso M, Ocadlikova D, et al. Immunosenescence and immunotherapy in elderly acute myeloid leukemia patients: time for a biology-driven approach. Cancers 2018;10:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sauce D, Larsen M, Fastenackels S, Duperrier A, Keller M, Grubeck-Loebenstein B, et al. Evidence of premature immune aging in patients thymectomized during early childhood. J Clin Invest 2009;119:3070–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wertheimer AM, Bennett MS, Park B, Uhrlaub JL, Martinez C, Pulko V, et al. Aging and cytomegalovirus infection differentially and jointly affect distinct circulating T cell subsets in humans. J Immunol 2014;192:2143–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Westera L, van Hoeven V, Drylewicz J, Spierenburg G, van Velzen JF, de Boer RJ, et al. Lymphocyte maintenance during healthy aging requires no substantial alterations in cellular turnover. Aging Cell 2015;14:219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]