

Understanding the dependence of cancers and virus-infected and immune cells on nucleotides can tilt the balance to benefit therapy.
Abstract
Virus-infected cells and cancers share metabolic commonalities that stem from their insatiable need to replicate while evading the host immune system. These similarities include hijacking signaling mechanisms that induce metabolic rewiring in the host to up-regulate nucleotide metabolism and, in parallel, suppress the immune response. In both cancer and viral infections, the host immune cells and, specifically, lymphocytes augment nucleotide synthesis to support their own proliferation and effector functions. Consequently, established treatment modalities targeting nucleotide metabolism against cancers and virally infected cells may result in restricted immune response. Encouragingly, following the introduction of immunotherapy against cancers, multiple studies improved our understanding for improving antigen presentation to the immune system. We propose here that understanding the immune consequences of targeting nucleotide metabolism against cancers may be harnessed to optimize therapy against viral infections.
INTRODUCTION
All living cells require nucleotides as building blocks for deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) synthesis for replication, transcription, and translation of genetic information, enabling cellular biomass increase. To ensure the accuracy of these processes, nucleotide metabolism is tightly regulated at all levels to maintain constant pools of pyrimidines—cytosine, uracil, and thymine (C, U, and T, respectively) and purines—adenine and guanine (A and G, respectively). Cancer cells and virus-infected cells share a metabolic dependency on nucleotide synthesis to support their unrestrained proliferation (1, 2). From the disease standpoint, a favorable outcome of uncontrolled proliferation lies in inducing mutations that further promote disease virulence and evolvability and enable survival in continuously changing environments (3, 4). Indeed, different drugs targeting nucleotide metabolism in either cancer or viral diseases constitute a shared therapeutic strategy to restrict replication.
The benefit from inhibiting nucleotide synthesis goes beyond restraining proliferation. In cancer, drugs that disrupt the balance of nucleotide pools can generate mutations that affect antigen presentation and, consequently, the immune response against the disease (5, 6). In addition, inhibition of purine synthesis can directly alleviate immune suppression, as secreted purines directly bind inhibitory receptors on immune cells (7, 8). On the other hand, targeting nucleotide metabolism could negatively affect the response of the host immune system. An early and essential step in the adaptive immune response against cancers and virally infected cells involves the rapid proliferation of lymphocytes (9), which will be disrupted by nucleotide deficiency. Therefore, any therapeutic strategy that targets nucleotide metabolism may potentially exert secondary effects on immune cells.
Despite the general characteristics discussed above, it is important to note that different viruses can impose distinct metabolic alterations of nucleotide metabolism in the infected cells. Furthermore, in cancer, metabolic heterogeneity is found among cells, locations, and at different stages of the same tumor. Here, we focus on the shared commonalities in nucleotide metabolism between cancer and virally infected cells and on the cross-talk it generates with immune cells along disease courses and during therapy. We highlight the advantages this cross-talk provides for disease progression, as well as the vulnerabilities it introduces that may constitute targets for therapy.
NUCLEOTIDE METABOLISM IN CANCERS AND VIRALLY INFECTED CELLS
Reprogramming of nucleotide metabolism to increase synthesis is regulated in cancers and virus-infected cells by similar signaling and metabolic pathways. In cancers, mutations and genomic aberrations in pro-growth and biosynthetic pathways are selected for promoting nucleotide synthesis (10). Virus-infected cells use other strategies, such as protein-protein interactions, to turn on the same biosynthetic machinery within the host cell. In both diseases, inhibition of tumor suppressors and activation of oncogenes, together with changes in expression of metabolic enzymes, lead to a shared outcome of increased nucleotide levels.
Rewiring of nucleotide metabolism via regulation of cell signaling
Several major signaling pathways and transcription regulators are commonly altered in cancers and virus-infected cells to increase nucleotide synthesis. These include MYC, RAS, P53, and mammalian target of rapamycin (mTOR). As the metabolic rewiring induced by these pathways have been extensively reviewed elsewhere (11, 12), here we will only briefly describe the most relevant metabolic alterations that augment nucleotide metabolism (Fig. 1).
Fig. 1. Shared signaling and metabolic rewiring between virus-infected cells, cancers, and lymphocytes.
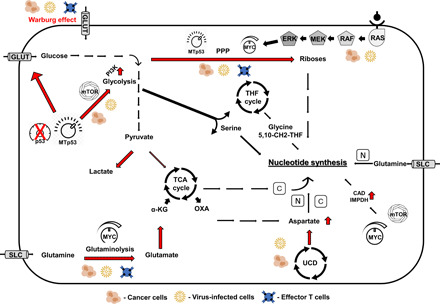
Virus-infected cells, cancers, and lymphocytes rewire cell metabolism to promote anabolism in general and that of nucleic acids specifically as building blocks for their enhanced proliferation. In addition to the “Warburg effect” that enhances glycolysis, the scheme depicts the metabolic reprogramming that is promoted by major signaling pathways such as p53, MYC, mitogen-activated protein kinase (MAPK), and mTOR to increase nucleotide synthesis (red arrows) for carbon (C) and nitrogen (N) utilization. UCD, urea cycle dysregulation; SLC, solute carrier; THF, tetrahydrofolate; TCA, tricarboxylic acid, MTp53, mutated p53; and OXA, oxaloacetate.
MYC is considered a “master regulator” of cell proliferation, growth, and metabolism (10, 13) and is overexpressed and/or activated by oncogenic or epigenetic events in most human cancers (14). MYC directly binds and enhances expression of bottleneck enzymes in nucleotide biosynthesis, including CAD (the trifunctional enzyme carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase), thymidylate synthase, and IMPDH (inosine monophosphate dehydrogenase) (15–18). MYC further supports nucleotide synthesis by augmenting glycolysis and glutaminolysis to supply carbon and nitrogen precursors and by promoting the alternative splicing isoform of pyruvate kinase, PKM2, over PKM1. PKM2 increases the availability of glycolytic intermediates for branching the anabolic pathways: the pentose phosphate pathway (PPP) and serine synthesis (19, 20), which generate precursors for nucleotide synthesis. In virus-infected cells, MYC can be activated by various strategies, such as protein-protein interaction, and by induction of its transcription. For example, adenovirus-encoded protein E4ORF1 translocates to the nucleus, where it binds MYC and enhances its binding to metabolic genes (21). In addition, hepatitis C virus (HCV) enhances MYC transcription by activating other signaling pathways such as AKT and β-catenin (22).
RAS oncogene promotes nucleotide synthesis in multiple ways, which include MYC activation (12, 23), and by activation of extracellular signal–regulated kinase, which phosphorylates and stimulates the purine synthetic enzyme phosphoribosylformylglycinamidine synthase (24) and the pyrimidine synthetic enzyme CAD (25). Viruses also use RAS signaling to promote proliferation. In human herpesvirus–infected cells, RAS activation is induced by interaction of viral glycoproteins and cellular receptors, as reviewed in (26). Similarly, the HCV generates replication complexes that are composed of virally-encoded proteins and Ras-GTPase–activating protein-binding protein 1 (27).
Cancer and virally infected cells further promote nucleotide synthesis by inhibiting signaling of tumor suppressors such as p53. In cancers, mutated p53, or its loss of function, reprograms cell metabolism to support growth, proliferation, and macromolecule synthesis. In DNA virus–infected cells, specific inactivating proteins or viral regulatory factors form complexes to inactivate p53 (28–30). In addition to its direct effects on metabolic pathways such as glycolysis and the salvage nucleotide pathway, p53 is among the plethora of genes, nutrients, and stress and growth signals that regulate the mTOR pathway, which is a critical regulator of mammalian metabolism (31). Loss of p53 activates mTOR complex 1 (mTORC1) to stimulate de novo pyrimidine and purine synthesis through activation of CAD enzyme and induction of one-carbon metabolism, which uses serine and glycine to generate one-carbon units for thymidine and purine synthesis (32–34). Similarly to MYC and mutated p53, mTORC1 stimulates glycolysis, and the PPP thus increases supply of precursors for nucleotide production (35). Activation of mTOR signaling has been demonstrated in multiple cancers (36) and following viral infections (37) and is mediated by various oncogenic alterations and virally induced regulatory proteins, respectively.
Rewiring of nucleotide synthesis via a direct regulation of metabolic enzymes
Mutations and genomic aberrations in metabolic enzymes are selected in cancer to promote proliferation and increase nucleotide synthesis. These include mutations that drive overexpression of enzymes that promote production of precursors for nucleotide synthesis, such as the glycolytic enzymes: hexokinase, phosphofructokinase and glyceraldehyde-3-phosphate dehydrogenase, and pyruvate kinase (38–41). In some cancers, such as melanoma and subtypes of breast and lung carcinomas, an amplification of a region on chromosome 1p increases the expression of phosphoglycerate dehydrogenase, the committed step in serine biosynthesis, leading to enhancement of the tetrahydrofolate cycle and purine synthesis (42, 43). In addition, amplifications of CAD enzyme that catalyzes the initiating step in pyrimidine synthesis and of several enzymes in the purinosome, the multienzyme complex that catalyzes purine synthesis, have been found in different cancers to directly increase nucleotide levels (44, 45). Similarly, some virus-infected cells induce nucleotide synthesis by directly encoding for metabolic enzymes. For example, the herpes simplex virus 1 (HSV1) encodes thymidine kinase, ribonucleotide reductase, dUTPase, and uracil-DNA glycosylase (46), all of which increase pyrimidine production.
Mechanisms used by cancers and viruses for increasing substrate levels necessary for nucleotide synthesis include not only the induction of anabolic enzymes but also interference with catabolic pathways. One example is the rewiring of the urea cycle (UC), the main pathway for disposal of excess nitrogen. Altered expression of UC enzymes is detected in many tumors (5). For example, overexpression of the UC enzyme CPS1, as seen in lung cancer, increases the availability of carbamoyl phosphate for pyrimidine synthesis (47). In addition, down-regulation of the UC enzyme argininosuccinate synthase 1 (ASS1), as seen in multiple cancers [e.g., osteosarcoma, melanoma, and mesothelioma (48, 49)], increases the availability of its substrate aspartate, thus facilitating pyrimidine synthesis and cancer proliferation (50). Aspartate is also essential for asparagine activation of mTOR-dependent nucleotide synthesis (51, 52). Likewise, down-regulation of ASS1 enhances viral genome replication and production of infectious HSV1 (53). Interestingly, such changes may also affect the immune response. For example, lymphocytic choriomeningitis virus has been demonstrated to repress the transcription of the UC enzymes OTC (Ornithine transcarbamylase) and ASS1 in the liver, consequently leading to decreased arginine and increased ornithine concentrations in the circulation, which suppress virus-specific cytotoxic T cell responses (54).
These studies highlight different mechanisms by which viruses and cancers rewire the host cellular and systemic metabolism to provide their insatiable need for nucleotides. Consequently, targeting nucleotide metabolism is an established treatment approach to restrain proliferation in both diseases.
TARGETING NUCLEOTIDE METABOLISM AGAINST CANCERS AND VIRAL INFECTIONS
Over the years, different therapeutic strategies have been used against cancers and virus-infected cells. Interestingly, while targeting nucleotide synthesis induces similar effects on cancers and virus-infected cells, attempting at hampering nucleotide metabolism by targeting metabolic master regulators can generate differential effects against each disease. For example, the mTOR inhibitors (Rapalogs), which are widely used against various cancers (55, 56), have variable effects against different viruses and have been shown to diminish cytomegalovirus and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) replication (57, 58) and, on the other hand, to facilitate influenza infection and polyomavirus replication (59–61). The variability in response of different viruses to mTOR inhibitors could result from the dependence of the virus on mTOR for replication as well as from the level of effect the inhibitor has on the antiviral immune response. In contrast, similar benefits have been demonstrated against both diseases when using nucleotide analogs and agents that directly inhibit the enzymes involved in nucleotide metabolism. Surprisingly, despite the shared mechanisms of action, there are no drug agents that are approved against both cancers and viral infections (tables S1 and S2).
Nucleotide analog treatment modality
One main strategy to halt cellular replication uses modified purine and pyrimidine nucleosides that terminate DNA or RNA polymerase activity. In these synthetic nucleosides, the deoxyribose moiety is replaced with nonfunctional modifications such as azide or hydrogen, which inhibit elongation of the DNA or RNA strands. Purine and pyrimidine analogs used in cancer treatment are summarized in table S1, together with their labeled indications and mechanisms of action. Most of these drugs are incorporated into the DNA and inhibit the activity of DNA polymerase (62). New generations of nucleosides are more sophisticated and display additional metabolic consequences in their cellular cytotoxicity. For example, in addition to blocking RNA synthesis, the adenosine analog 8-amino-adenosine causes an energy crisis and induces cell death by decreasing the intracellular concentrations of adenosine triphosphate (ATP) (table S1) (63). Nucleosides are efficient drugs; however, cancers can develop mechanisms that overcome the metabolic hurdles they impose. For example, gemcitabine is a pyrimidine prodrug analog of deoxycytidine that has been approved for the treatment of non–small cell lung cancer, pancreatic cancer, bladder cancer, and breast cancer (64). However, it was recently demonstrated that tumor-infiltrating activated macrophages synthesize and release a spectrum of pyrimidine nucleosides including deoxycytidine that are consumed by the cancer cells (65). These nucleosides directly compete with gemcitabine, hindering its efficiency as a chemotherapy.
Nucleoside and nucleotide analogs represent one of the largest classes of small-molecule antiviral drugs (summarized in table S2). The mechanisms of action of these drugs include lethal mutagenesis, specific or nonspecific chain termination, and inhibition of nucleotide biosynthesis (66, 67). Indeed, for the SARS-CoV-2, one of the trialed antiviral drugs used is remdesivir, a prodrug of a nucleotide analog that is intracellularly metabolized to an ATP analog, which inhibits the activity of viral RNA polymerases (68). Another example of a nucleoside analog used as viral therapy is acyclovir and its related drugs: valacyclovir, penciclovir, and famciclovir, used against infections with HSV-1, HSV-2 and varicella zoster virus. Ganciclovir and its related drug valganciclovir are additional nucleoside analogs used against members of the family Herpesviridae (69). The mechanism of action of these drugs requires phosphorylation by a virally encoded enzyme that induces chain termination only in infected cells. Ribavirin, a Food and Drug Administration (FDA)–approved guanosine analog, is standard for care against several viruses, including respiratory syncytial virus and HCV (70). Although its mechanism of action is a matter of debate, several possibilities have been proposed, including depletion of guanine nucleotides through inhibition of IMPDH (71). Intriguingly, ribavirin’s antiviral effect against HCV is associated with induction of transition mutations in the viral genome, resulting in the generation of noninfectious virions (72).
Inhibition of metabolic enzymes involved in nucleotide metabolism
Direct inhibition of enzymes involved in DNA and RNA synthesis is another known treatment strategy against both cancers and viral infections. In fact, one of the first FDA-approved molecules for cancer treatment was the purine inhibitor 6-mercaptopurine (6-MP). 6-MP inhibits the first enzyme of de novo purine synthesis, 5-phosphoribosyl-1-pyrophosphatase (73), and the purine salvage enzyme, hypoxanthine-guanine phosphoribosyltransferase (74). Merimepodib, which inhibits IMPDH, an enzyme catalyzing de novo synthesis of guanosine nucleotides, has been demonstrated to suppress replication of a variety of RNA viruses, including SARS-CoV-2 replication in vitro (75, 76).
Inhibitors of pyrimidine biosynthesis currently used for cancer treatment include 5-fluorouracil, a thymidylate synthase inhibitor, and methotrexate, which inhibits dihydrofolate reductase and causes a drop in cellular levels of thymidine (77, 78). Not surprisingly, efforts have been made to target CAD and dihydroorotate dehydrogenase (DHODH) pyrimidine synthetic proteins (79, 80). While potent CAD inhibitors are still lacking, inhibitors of DHODH, such as brequinar sodium, are currently being tested in clinical trials as cancer therapy for acute myeloid leukemia (81). DHODH inhibitors have also been tested in different models of viral infections (82). The repression in viral growth induced by these inhibitors was attributed to enhanced innate immune response in reaction to pyrimidine deprivation (83).
Targeting a specific nucleotide pathway for the synthesis of either purines or pyrimidines can generate a nucleotide pool imbalance by decreasing the levels of one pool relative to the other. Since the ratio between the two pools is tightly regulated, induced imbalance can subsequently cause genotoxic stress and increase mutagenesis (84, 85). While prompting mutations can increase fitness and survival of cancers and virally infected cells, high mutation number will ultimately generate more neoantigens that can improve the response of immune cells (86).
NUCLEOTIDE METABOLISM REGULATES THE HOST IMMUNE SYSTEM
The rationale and benefit from targeting nucleotide metabolism to inhibit replication of tumor cells or viruses during infection are clear. However, we should not overlook the potential for consequential deleterious effects on the immune response against both diseases, specifically that of T lymphocytes. T lymphocytes are the cellular arm of the adaptive immune system and can be divided into different subpopulations with distinct functions: CD8+ cytotoxic T cells kill cells that express foreign antigens including cells infected with virus and tumor cells. CD4+ T helper cells regulate the function of other immune cells, including CD8+ cytotoxic T cells. Upon activation, T cells induce anabolic metabolism to support rapid growth, proliferation, effector molecule production, and differentiation using the same signaling and metabolic pathways induced to support proliferation in cancer cells and virally infected cells (87). Signals received through the T cell receptor activate MYC and mTOR to induce transcription of multiple metabolic enzymes (88). In addition, wild-type P53 is down-regulated in lymphocytes by its regulator mouse double minute 2 (MDM2) (Fig. 2) (89). Consequently, glucose uptake and glycolysis are induced, with increased serine biosynthesis and increased metabolic flux through the PPP, producing five-carbon sugars for nucleotide synthesis (88). In parallel to a large increase in aerobic glycolysis, T cell activation induces a robust and highly synchronized program of mitochondrial biogenesis (90, 91), giving rise to one-carbon metabolism that uses serine to generate glycine and one-carbon units for de novo purine biosynthesis (91). Mitochondrial respiration further supports nucleotide synthesis and proliferation through production of aspartate, a precursor for CAD enzyme (Fig. 1) (92, 93). Thus, T cells engage in nucleotide metabolism to support their growth, proliferation, and redox balance using the same pathways “hijacked” by tumors and virally infected cells (Fig. 2). Not surprisingly, similar to the anticancer and antiviral therapies, drugs that inhibit nucleotide synthesis have long been used to treat inflammatory autoimmune diseases such as rheumatoid arthritis, Crohn’s disease, and psoriasis and to prevent host versus graft disease following organ transplantation (table S3).
Fig. 2. p53: Engaging a common pathway by different mechanisms.
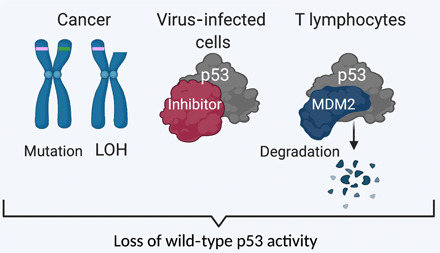
Nucleotide synthesis is induced in T cells, cancers, and virally infected cells through engagement of the same signaling pathways but by different mechanisms, as nicely illustrated in the case of p53. Cancer cells select for loss of wild-type p53 activity via mutations or loss of heterozygosity (LOH). Virus-infected cells inhibit p53 through protein-protein interaction. In T lymphocytes, MDM2 facilitates p53 degradation. This figure was generated using https://biorender.com/.
Inhibition of nucleotide synthesis as a treatment for cancer and viral infections is not all bad for T cells and, under certain circumstances, could even promote a protective immune response. Cancers and virally infected cells actively generate an immunosuppressive microenvironment by secreting purines, especially adenosine, into the extracellular space (8, 94). By engaging with adenosine receptors that are expressed on most immune cells, adenosine halts immune cell differentiation and maturation, induces the expression of checkpoint molecules, such as programmed cell death protein 1 and cytotoxic T lymphocyte–associated protein-4, and interferes with secretion of chemokines and cytokines (7). Accordingly, in tumors, drugs targeting the adenosine pathway were shown to convert an immunosuppressive microenvironment to a more immuno-permissive one and to reduce metastasis and resistance to therapy (95). Such drugs are currently undergoing their first clinical trials in humans, both as single agents and in combination with other immune therapies (96).
Following the introduction of immunotherapy against cancers, multiple studies advanced our knowledge for improving the immune response via regulating antigen presentation by the cancer cells (97). One such potential strategy to increase the presentation of more immunogenic antigens on the cell surface is by promoting the presentation of more hydrophobic neoantigens (98, 99). Since presented antigens are continuously translated from the cellular mRNAs, modulating nucleotide metabolism can regulate the antigens’ properties. Along these lines, we found that inducing a high pyrimidine-to-purine ratio in different cancers promotes the generation of a specific mutation signature, leading to the production of more hydrophobic, and therefore more immunogenic, neoantigens (5). Furthermore, as a proof of concept, we demonstrated that mizoribine, an inhibitor of IMPDH and consequently of purine synthesis, augmented pyrimidine-to-purine ratio and improved the response to immunotherapy in previously nonresponsive tumors (6). Of note, to avoid the potential immunosuppressive effect of mizoribine (6), it was given to cancer cells before treatment with immunotherapy. The resultant beneficial outcome exemplified by tumor growth restriction suggests the rationale that sequential drug regimens can optimize therapeutic consequences. We hence propose that better understanding of the secondary effects of drugs that target nucleotide synthesis on immune cells will minimize the inhibitory effect on T cells, enhance immunogenicity of infected or transformed cells, and thus improve existing therapies against viral infections and cancers (84, 85).
OUTLOOK
Nucleotide metabolism provides cancers and viruses with means to proliferate and evade the immune system; hence, it is a well-established therapeutic target against both diseases. Although the therapeutic strategy is shared between the two diseases, there are currently no drugs that are commonly used against both. These days, immunotherapy advances our knowledge regarding potential recruitment of the immune response as therapy against cancer. The similar cross-talks between cancers and virally-infected cells with the immune system via nucleotide metabolism offer an opportunity to take advantage of this shared vulnerability and modulate nucleotide metabolism in a way that would boost the immune response and benefit therapy. On the basis of recent insights, modulating nucleotide metabolism, for example, by generating an imbalance that favors pyrimidine synthesis, may potentially improve the immune response, may reduce tumor-induced immune suppression, and may increase genotoxic stress, consequently leading to cell death (Fig. 3).
Fig. 3. Modulating nucleotide metabolism to favor pyrimidine synthesis can increase the host immune response against viral attack and cancer.
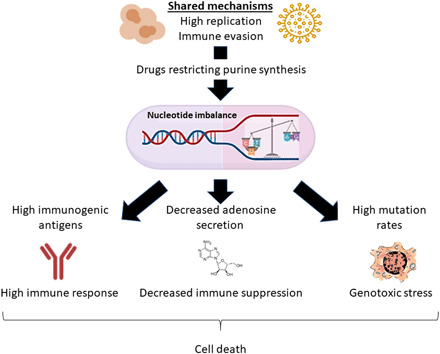
Inducing a high pyrimidine-to-purine ratio may promote the immune response against cancer and virus-infected cells by inducing genotoxic stress, decreasing immune suppression, and augmenting the antigens’ immunogenicity, all contributing to virally infected and cancer cells’ death.
Acknowledgments
We are grateful to N. Stern-Ginossar and A.-Ileana Zaromytidou for instrumental comments. Funding: We acknowledge and thank the Weizmann Institute of Science for providing financial and infrastructure support. A.E. was supported by research grants from the European Research Council (ERC818943) and from the Israel Science Foundation (860/18). A.E. received additional support from the Moross Integrated Cancer Center, Sagol Institute for Longevity Research, Adelis Foundation, Rising Tide Foundation, and Manya and Adolph Zarovinsky. H.R.C. was supported by a Research Scholar grant (RSG-16-111-01-MPC) from the American Cancer Society, NIH/NCI R01 CA215185, NIH/NIAMS R01 AR070245, the UCLA Jonsson Comprehensive Cancer Center, and the Eli and Edythe Broad Center for Regenerative Medicine Ablon Scholars Program. J.H.C. is a St. Baldrick’s Foundation Fellow and supported by their fellowship award. N.R.-H. was supported by grants from the Israel Science Foundation (1536/19) and the Israel Cancer Association (2019-0111). Author contributions: Y.A. generated Figs. 1 and 3 and wrote the cancer metabolism–related sections. J.H.C. generated the supplementary tables and wrote the virally infected cell metabolism sections. N.R.-H. wrote the immunometabolism-related section and generated Fig. 2 with BioRender. H.R.C., N.R.-H., and A.E. initiated and pursued the concepts described in the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/21/eabg6165/DC1
REFERENCES AND NOTES
- 1.Thaker S. K., Ch’ng J., Christofk H. R., Viral hijacking of cellular metabolism. BMC Biol. 17, 59 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goodwin C. M., Xu S., Munger J., Stealing the keys to the kitchen: Viral manipulation of the host cell metabolic network. Trends Microbiol. 23, 789–798 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Domingo E., de Avila A. I., Gallego I., Sheldon J., Perales C., Viral fitness: History and relevance for viral pathogenesis and antiviral interventions. Pathog. Dis. 77, ftz021 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Chen Y., Williams V., Filippova M., Filippov V., Duerksen-Hughes P., Viral carcinogenesis: Factors inducing DNA damage and virus integration. Cancers 6, 2155–2186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee J. S., Adler L., Karathia H., Carmel N., Rabinovich S., Auslander N., Keshet R., Stettner N., Silberman A., Agemy L., Helbling D., Eilam R., Sun Q., Brandis A., Malitsky S., Itkin M., Weiss H., Pinto S., Kalaora S., Levy R., Barnea E., Admon A., Dimmock D., Stern-Ginossar N., Scherz A., Nagamani S. C. S., Unda M., Wilson D. M. III, Elhasid R., Carracedo A., Samuels Y., Hannenhalli S., Ruppin E., Erez A., Urea cycle dysregulation generates clinically relevant genomic and biochemical signatures. Cell 174, 1559–1570.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keshet J. S. L. R., Adler L., Iraqi M., Ariav Y., Lim L. Q. J., Lerner S., Rabinovich S., Oren R., Katzir R., Tishler H. W., Stettner N., Goldman O., Landesman H., Galai S., Kuperman Y., Kuznetsov Y., Brandis A., Mehlman T., Malitsky S., Itkin M., Koehler S. E., Zhao Y., Talsania K., Shen T.-w., Peled N., Ulitsky I., Porgador A., Ruppin E., Erez A., Targeting purine synthesis in ASS1-expressing tumors enhances the response to immune checkpoint inhibitors. Nat. Cancer 1, 894–908 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Passos D. F., Bernardes V. M., da Silva J. L. G., Schetinger M. R. C., Leal D. B. R., Adenosine signaling and adenosine deaminase regulation of immune responses: Impact on the immunopathogenesis of HIV infection. Purinergic Signal 14, 309–320 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mastelic-Gavillet B., Navarro Rodrigo B., Décombaz L., Wang H., Ercolano G., Ahmed R., Lozano L. E., Ianaro A., Derré L., Valerio M., Tawadros T., Jichlinski P., Nguyen-Ngoc T., Speiser D. E., Verdeil G., Gestermann N., Dormond O., Kandalaft L., Coukos G., Jandus C., Ménétrier-Caux C., Caux C., Ho P. C., Romero P., Harari A., Vigano S., Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8+ T cells. J. Immunother. Cancer 7, 257 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quemeneur L., Gerland L.-M., Flacher M., Ffrench M., Revillard J.-P., Genestier L., Differential control of cell cycle, proliferation, and survival of primary T lymphocytes by purine and pyrimidine nucleotides. J. Immunol. 170, 4986–4995 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Dang C. V., MYC on the path to cancer. Cell 149, 22–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nascimento R., Costa H., Parkhouse R. M., Virus manipulation of cell cycle. Protoplasma 249, 519–528 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Villa E., Ali E. S., Sahu U., Ben-Sahra I., Cancer cells tune the signaling pathways to empower de novo synthesis of nucleotides. Cancers 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim J. W., Zeller K. I., Wang Y., Jegga A. G., Aronow B. J., O’Donnell K. A., Dang C. V., Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell. Biol. 24, 5923–5936 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gabay M., Li Y., Felsher D. W., MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 4, a014241 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeller K. I., Zhao X., Lee C. W. H., Chiu K. P., Yao F., Yustein J. T., Ooi H. S., Orlov Y. L., Shahab A., Yong H. C., Fu Y., Weng Z., Kuznetsov V. A., Sung W. K., Ruan Y., Dang C. V., Wei C. L., Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc. Natl. Acad. Sci. U.S.A. 103, 17834–17839 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mannava S., Grachtchouk V., Wheeler L. J., Im M., Zhuang D., Slavina E. G., Mathews C. K., Shewach D. S., Nikiforov M. A., Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 7, 2392–2400 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y. C., Li F., Handler J., Huang C. R. L., Xiang Y., Neretti N., Sedivy J. M., Zeller K. I., Dang C. V., Global regulation of nucleotide biosynthetic genes by c-Myc. PLOS ONE 3, e2722 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyd K. E., Farnham P. J., Myc versus USF: Discrimination at the cad gene is determined by core promoter elements. Mol. Cell. Biol. 17, 2529–2537 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.David C. J., Chen M., Assanah M., Canoll P., Manley J. L., HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463, 364–368 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vazquez A., Markert E. K., Oltvai Z. N., Serine biosynthesis with one carbon catabolism and the glycine cleavage system represents a novel pathway for ATP generation. PLOS ONE 6, e25881 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thai M., Graham N. A., Braas D., Nehil M., Komisopoulou E., Kurdistani S. K., McCormick F., Graeber T. G., Christofk H. R., Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab. 19, 694–701 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma H. C., Lin T. W., Li H., Iguchi-Ariga S. M. M., Ariga H., Chuang Y. L., Ou J. H., Lo S. Y., Hepatitis C virus ARFP/F protein interacts with cellular MM-1 protein and enhances the gene trans-activation activity of c-Myc. J. Biomed. Sci. 15, 417–425 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Santana-Codina N., Roeth A. A., Zhang Y., Yang A., Mashadova O., Asara J. M., Wang X., Bronson R. T., Lyssiotis C. A., Ying H., Kimmelman A. C., Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 9, 4945 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ali E. S., Sahu U., Villa E., O’Hara B. P., Gao P., Beaudet C., Wood A. W., Asara J. M., Ben-Sahra I., ERK2 phosphorylates PFAS to mediate posttranslational control of de novo purine synthesis. Mol. Cell 78, 1178–1191.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graves L. M., Guy H. I., Kozlowski P., Huang M., Lazarowski E., Pope R. M., Collins M. A., Dahlstrand E. N., Earp H. S. III, Evans D. R., Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature 403, 328–332 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Filippakis H., Spandidos D. A., Sourvinos G., Herpesviruses: Hijacking the Ras signaling pathway. Biochim. Biophys. Acta 1803, 777–785 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Yi Z., Pan T., Wu X., Song W., Wang S., Xu Y., Rice C. M., MacDonald M. R., Yuan Z., Hepatitis C virus co-opts Ras-GTPase–activating protein-binding protein 1 for its genome replication. J. Virol. 85, 6996–7004 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian W., Kashuba E., Magnusson K. P., Pokrovskaja K., Okan I., Klein G., Wiman K. G., Role of p53 mutation in polyomavirus-induced tumorigenesis. J. Gen. Virol. 78 ( Pt. 4), 893–903 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Fries K. L., Miller W. E., Raab-Traub N., Epstein-Barr virus latent membrane protein 1 blocks p53-mediated apoptosis through the induction of the A20 gene. J. Virol. 70, 8653–8659 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheffner M., Werness B. A., Huibregtse J. M., Levine A. J., Howley P. M., The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63, 1129–1136 (1990). [DOI] [PubMed] [Google Scholar]
- 31.Sabatini D. M., Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. U.S.A. 114, 11818–11825 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valvezan A. J., Turner M., Belaid A., Lam H. C., Miller S. K., Namara M. C. M., Baglini C., Housden B. E., Perrimon N., Kwiatkowski D. J., Asara J. M., Henske E. P., Manning B. D., mTORC1 couples nucleotide synthesis to nucleotide demand resulting in a targetable metabolic vulnerability. Cancer Cell 32, 624–638.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ben-Sahra I., Hoxhaj G., Ricoult S. J. H., Asara J. M., Manning B. D., mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ben-Sahra I., Howell J. J., Asara J. M., Manning B. D., Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339, 1323–1328 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duvel K., Yecies J. L., Menon S., Raman P., Lipovsky A. I., Souza A. L., Triantafellow E., Ma Q., Gorski R., Cleaver S., Heiden M. G. V., Keigan J. P. M., Finan P. M., Clish C. B., Murphy L. O., Manning B. D., Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu J., Pham C. G., Albanese S. K., Dong Y., Oyama T., Lee C. H., Rodrik-Outmezguine V., Yao Z., Han S., Chen D., Parton D. L., Chodera J. D., Rosen N., Cheng E. H., Hsieh J. J., Mechanistically distinct cancer-associated mTOR activation clusters predict sensitivity to rapamycin. J. Clin. Invest. 126, 3526–3540 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez-Sanchez I., Schafer X. L., Monaghan M., Munger J., The Human Cytomegalovirus UL38 protein drives mTOR-independent metabolic flux reprogramming by inhibiting TSC2. PLOS Pathog. 15, e1007569 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rempel A., Mathupala S. P., Griffin C. A., Hawkins A. L., Pedersen P. L., Glucose catabolism in cancer cells: Amplification of the gene encoding type II hexokinase. Cancer Res. 56, 2468–2471 (1996). [PubMed] [Google Scholar]
- 39.Webb B. A., Forouhar F., Szu F. E., Seetharaman J., Tong L., Barber D. L., Structures of human phosphofructokinase-1 and atomic basis of cancer-associated mutations. Nature 523, 111–114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu P., Zhong Y., Cao T., Sheng X., Huang H., A frequent somatic mutation in the 3′UTR of GAPDH facilitates the development of ovarian cancer by creating a miR‑125b binding site. Oncol. Rep. 44, 887–896 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu V. M., Howell A. J., Hosios A. M., Li Z., Israelsen W. J., Vander Heiden M. G., Cancer-associated mutations in human pyruvate kinase M2 impair enzyme activity. FEBS Lett. 594, 646–664 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Locasale J. W., Grassian A. R., Melman T., Lyssiotis C. A., Mattaini K. R., Bass A. J., Heffron G., Metallo C. M., Muranen T., Sharfi H., Sasaki A. T., Anastasiou D., Mullarky E., Vokes N. I., Sasaki M., Beroukhim R., Stephanopoulos G., Ligon A. H., Meyerson M., Richardson A. L., Chin L., Wagner G., Asara J. M., Brugge J. S., Cantley L. C., Vander Heiden M. G., Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43, 869–874 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang B., Zheng A., Hydbring P., Ambroise G., Ouchida A. T., Goiny M., Vakifahmetoglu-Norberg H., Norberg E., PHGDH defines a metabolic subtype in lung adenocarcinomas with poor prognosis. Cell Rep. 19, 2289–2303 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Chen S., Bigner S. H., Modrich P., High rate of CAD gene amplification in human cells deficient in MLH1 or MSH6. Proc. Natl. Acad. Sci. U.S.A. 98, 13802–13807 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia-Gil M., Camici M., Allegrini S., Pesi R., Petrotto E., Tozzi M., Emerging role of purine metabolizing enzymes in brain function and tumors. Int. J. Mol. Sci. 19, 3598 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boehmer P. E., Lehman I. R., Herpes simplex virus DNA replication. Annu. Rev. Biochem. 66, 347–384 (1997). [DOI] [PubMed] [Google Scholar]
- 47.Kim J., Hu Z., Cai L., Li K., Choi E., Faubert B., Bezwada D., Rodriguez-Canales J., Villalobos P., Lin Y. F., Ni M., Huffman K. E., Girard L., Byers L. A., Unsal-Kacmaz K., Peña C. G., Heymach J. V., Wauters E., Vansteenkiste J., Castrillon D. H., Chen B. P. C., Wistuba I., Lambrechts D., Xu J., Minna J. D., DeBerardinis R. J., CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546, 168–172 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Long Y., Tsai W. B., Wangpaichitr M., Tsukamoto T., Savaraj N., Feun L. G., Kuo M. T., Arginine deiminase resistance in melanoma cells is associated with metabolic reprogramming, glucose dependence, and glutamine addiction. Mol. Cancer Ther. 12, 2581–2590 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delage B., Fennell D. A., Nicholson L., McNeish I., Lemoine N. R., Crook T., Szlosarek P. W., Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int. J. Cancer 126, 2762–2772 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Rabinovich S., Adler L., Yizhak K., Sarver A., Silberman A., Agron S., Stettner N., Sun Q., Brandis A., Helbling D., Korman S., Itzkovitz S., Dimmock D., Ulitsky I., Nagamani S. C. S., Ruppin E., Erez A., Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 527, 379–383 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pathria G., Verma S., Yin J., Scott D. A., Ronai Z. A., MAPK signaling regulates c-MYC for melanoma cell adaptation to asparagine restriction. EMBO Rep. 22, e51436 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krall A. S., Mullen P. J., Surjono F., Momcilovic M., Schmid E. W., Halbrook C. J., Thambundit A., Mittelman S. D., Lyssiotis C. A., Shackelford D. B., Knott S. R. V., Christofk H. R., Asparagine couples mitochondrial respiration to ATF4 activity and tumor growth. Cell Metab., S1550-4131(21)00057-7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grady S. L., Purdy J. G., Rabinowitz J. D., Shenk T., Argininosuccinate synthetase 1 depletion produces a metabolic state conducive to herpes simplex virus 1 infection. Proc. Natl. Acad. Sci. U.S.A. 110, E5006–E5015 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lercher A., Bhattacharya A., Popa A. M., Caldera M., Schlapansky M. F., Baazim H., Agerer B., Gürtl B., Kosack L., Májek P., Brunner J. S., Vitko D., Pinter T., Genger J.-W., Orlova A., Pikor N., Reil D., Ozsvár-Kozma M., Kalinke U., Ludewig B., Moriggl R., Bennett K. L., Menche J., Cheng P. N., Schabbauer G., Trauner M., Klavins K., Bergthaler A., Type I interferon signaling disrupts the hepatic urea cycle and alters systemic metabolism to suppress T cell function. Immunity 51, 1074–1087.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.G. Hudes, M. Carducci, P. Tomczak, J. Dutcher, R. Figlin, A. Kapoor, E. Staroslawska, J. Sosman, D. M. Dermott, I. Bodrogi, Z. Kovacevic, V. Lesovoy, I. G. H. Schmidt-Wolf, O. Barbarash, E. Gokmen, T. O’Toole, S. Lustgarten, L. Moore, R. J. Motzer, Global ARCC Trial, Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. 356, 2271–2281 (2007). [DOI] [PubMed]
- 56.Chung J., Kuo C. J., Crabtree G. R., Blenis J., Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 69, 1227–1236 (1992). [DOI] [PubMed] [Google Scholar]
- 57.Moorman N. J., Shenk T., Rapamycin-resistant mTORC1 kinase activity is required for herpesvirus replication. J. Virol. 84, 5260–5269 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mullen P. J., Garcia G. Jr., Purkayastha A., Matulionis N., Schmid E. W., Momcilovic M., Sen C., Langerman J., Ramaiah A., Shackelford D. B., Damoiseaux R., French S. W., Plath K., Gomperts B. N., Arumugaswami V., Christofk H. R., SARS-CoV-2 infection rewires host cell metabolism and is potentially susceptible to mTORC1 inhibition. Nat. Commun. 12, 1876 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi G., Ozog S., Torbett B. E., Compton A. A., mTOR inhibitors lower an intrinsic barrier to virus infection mediated by IFITM3. Proc. Natl. Acad. Sci. U.S.A. 115, E10069–E10078 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alvarez Orellana J., Kwun H. J., Artusi S., Chang Y., Moore P. S., Sirolimus and other mechanistic target of rapamycin inhibitors directly activate latent pathogenic human polyomavirus replication. J Infect Dis , jiaa071 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang C. T., Hung C.-Y., Chen T.-C., Lin C.-Y., Lin Y.-C., Chang C.-S., He Y.-C., Huang Y.-L., Dutta A., Rapamycin adjuvant and exacerbation of severe influenza in an experimental mouse model. Sci. Rep. 7, 4136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berdis A. J., DNA polymerases as therapeutic targets. Biochemistry 47, 8253–8260 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gandhi V., Ayres M., Halgren R. G., Krett N. L., Newman R. A., Rosen S. T., 8-chloro-cAMP and 8-chloro-adenosine act by the same mechanism in multiple myeloma cells. Cancer Res. 61, 5474–5479 (2001). [PubMed] [Google Scholar]
- 64.Toschi L., Finocchiaro G., Bartolini S., Gioia V., Cappuzzo F., Role of gemcitabine in cancer therapy. Future Oncol. 1, 7–17 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Halbrook C. J., Pontious C., Kovalenko I., Lapienyte L., Dreyer S., Lee H.-J., Thurston G., Zhang Y., Lazarus J., Sajjakulnukit P., Hong H. S., Kremer D. M., Nelson B. S., Kemp S., Zhang L., Chang D., Biankin A., Shi J., Frankel T. L., Crawford H. C., Morton J. P., di Magliano M. P., Lyssiotis C. A., Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 29, 1390–1399.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eyer L., Nencka R., de Clercq E., Seley-Radtke K., Ruzek D., Nucleoside analogs as a rich source of antiviral agents active against arthropod-borne flaviviruses. Antivir. Chem. Chemother. 26, 2040206618761299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lu H., Drug treatment options for the 2019-new coronavirus (2019-nCoV). Biosci. Trends 14, 69–71 (2020). [DOI] [PubMed] [Google Scholar]
- 68.Warren T. K., Jordan R., Lo M. K., Ray A. S., Mackman R. L., Soloveva V., Siegel D., Perron M., Bannister R., Hui H. C., Larson N., Strickley R., Wells J., Stuthman K. S., van Tongeren S. A., Garza N. L., Donnelly G., Shurtleff A. C., Retterer C. J., Gharaibeh D., Zamani R., Kenny T., Eaton B. P., Grimes E., Welch L. S., Gomba L., Wilhelmsen C. L., Nichols D. K., Nuss J. E., Nagle E. R., Kugelman J. R., Palacios G., Doerffler E., Neville S., Carra E., Clarke M. O., Zhang L., Lew W., Ross B., Wang Q., Chun K., Wolfe L., Babusis D., Park Y., Stray K. M., Trancheva I., Feng J. Y., Barauskas O., Xu Y., Wong P., Braun M. R., Flint M., McMullan L. K., Chen S. S., Fearns R., Swaminathan S., Mayers D. L., Spiropoulou C. F., Lee W. A., Nichol S. T., Cihlar T., Bavari S., Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 531, 381–385 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Poole C. L., James S. H., Antiviral therapies for herpesviruses: Current agents and new directions. Clin. Ther. 40, 1282–1298 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feld J. J., Hoofnagle J. H., Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 436, 967–972 (2005). [DOI] [PubMed] [Google Scholar]
- 71.Keppeke G. D., Calise S. J., Chan E. K. L., Andrade L. E. C., Ribavirin induces widespread accumulation of IMP dehydrogenase into rods/rings structures in multiple major mouse organs. Antiviral Res. 162, 130–135 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Galli A., Mens H., Gottwein J. M., Gerstoft J., Bukh J., Antiviral effect of ribavirin against HCV associated with increased frequency of G-to-A and C-to-U transitions in infectious cell culture model. Sci. Rep. 8, 4619 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Elion G. B., The purine path to chemotherapy. Science 244, 41–47 (1989). [DOI] [PubMed] [Google Scholar]
- 74.Elgemeie G. H., Thioguanine, mercaptopurine: Their analogs and nucleosides as antimetabolites. Curr. Pharm. Des. 9, 2627–2642 (2003). [DOI] [PubMed] [Google Scholar]
- 75.Tong X., Smith J., Bukreyeva N., Koma T., Manning J. T., Kalkeri R., Kwong A. D., Paessler S., Merimepodib, an IMPDH inhibitor, suppresses replication of Zika virus and other emerging viral pathogens. Antiviral Res. 149, 34–40 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Natalya Bukreyeva E. K. M., Sattler R. A., Huang C., Paessler S., Zeldis J., The IMPDH inhibitor merimepodib suppresses SARS-CoV-2 replication in vitro. BioRxiv, 2020.04.07.028589 (2020). [Google Scholar]
- 77.Longley D. B., Harkin D. P., Johnston P. G., 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 3, 330–338 (2003). [DOI] [PubMed] [Google Scholar]
- 78.Burger K., Mühl B., Harasim T., Rohrmoser M., Malamoussi A., Orban M., Kellner M., Gruber-Eber A., Kremmer E., Hölzel M., Eick D., Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 285, 12416–12425 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Palmer A. M., Teriflunomide, an inhibitor of dihydroorotate dehydrogenase for the potential oral treatment of multiple sclerosis. Curr. Opin. Investig. Drugs 11, 1313–1323 (2010). [PubMed] [Google Scholar]
- 80.Paridaens R., Mouridsen H. T., Palshof T., Cocconi G., van Oosterom A., Rotmensz N., Sylvester R., Heuson J. C., Rozencweig M., N-(phosphonacetyl)-L-aspartate (PALA) in advanced breast cancer: A phase II trial of the EORTC breast cancer cooperative group. Eur. J. Cancer Clin. Oncol. 18, 67–70 (1982). [DOI] [PubMed] [Google Scholar]
- 81.Sykes D. B., The emergence of dihydroorotate dehydrogenase (DHODH) as a therapeutic target in acute myeloid leukemia. Expert Opin. Ther. Targets 22, 893–898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ortiz-Riano E., Ngo N., Devito S., Eggink D., Munger J., Shaw M. L., de la Torre J. C., Martinez-Sobrido L., Inhibition of arenavirus by A3, a pyrimidine biosynthesis inhibitor. J. Virol. 88, 878–889 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lucas-Hourani M., Dauzonne D., Jorda P., Cousin G., Lupan A., Helynck O., Caignard G., Janvier G., André-Leroux G., Khiar S., Escriou N., Desprès P., Jacob Y., Munier-Lehmann H., Tangy F., Vidalain P.-O., Inhibition of pyrimidine biosynthesis pathway suppresses viral growth through innate immunity. PLOS. Pathog. 9, e1003678 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang J., Dong H., Chionh Y. H., McBee M. E., Sirirungruang S., Cunningham R. P., Shi P. Y., Dedon P. C., The role of sequence context, nucleotide pool balance and stress in 2′-deoxynucleotide misincorporation in viral, bacterial and mammalian RNA. Nucleic Acids Res. 44, 8962–8975 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kohnken R., Kodigepalli K. M., Wu L., Regulation of deoxynucleotide metabolism in cancer: Novel mechanisms and therapeutic implications. Mol. Cancer 14, 176 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Samstein R. M., Lee C. H., Shoushtari A. N., Hellmann M. D., Shen R., Janjigian Y. Y., Barron D. A., Zehir A., Jordan E. J., Omuro A., Kaley T. J., Kendall S. M., Motzer R. J., Hakimi A. A., Voss M. H., Russo P., Rosenberg J., Iyer G., Bochner B. H., Bajorin D. F., al-Ahmadie H. A., Chaft J. E., Rudin C. M., Riely G. J., Baxi S., Ho A. L., Wong R. J., Pfister D. G., Wolchok J. D., Barker C. A., Gutin P. H., Brennan C. W., Tabar V., Mellinghoff I. K., DeAngelis L. M., Ariyan C. E., Lee N., Tap W. D., Gounder M. M., D’Angelo S. P., Saltz L., Stadler Z. K., Scher H. I., Baselga J., Razavi P., Klebanoff C. A., Yaeger R., Segal N. H., Ku G. Y., DeMatteo R. P., Ladanyi M., Rizvi N. A., Berger M. F., Riaz N., Solit D. B., Chan T. A., Morris L. G. T., Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 51, 202–206 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chapman N. M., Boothby M. R., Chi H., Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 20, 55–70 (2020). [DOI] [PubMed] [Google Scholar]
- 88.Wang R., Dillon C. P., Shi L. Z., Milasta S., Carter R., Finkelstein D., McCormick L. L., Fitzgerald P., Chi H., Munger J., Green D. R., The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Watanabe M., Moon K. D., Vacchio M. S., Hathcock K. S., Hodes R. J., Downmodulation of tumor suppressor p53 by T cell receptor signaling is critical for antigen-specific CD4+ T cell responses. Immunity 40, 681–691 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Akkaya B., Roesler A. S., Miozzo P., Theall B. P., al Souz J., Smelkinson M. G., Kabat J., Traba J., Sack M. N., Brzostowski J. A., Pena M., Dorward D. W., Pierce S. K., Akkaya M., Increased mitochondrial biogenesis and reactive oxygen species production accompany prolonged CD4+ T cell activation. J. Immunol. 201, 3294–3306 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ron-Harel N., Santos D., Ghergurovich J. M., Sage P. T., Reddy A., Lovitch S. B., Dephoure N., Satterstrom F. K., Sheffer M., Spinelli J. B., Gygi S., Rabinowitz J. D., Sharpe A. H., Haigis M. C., Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab. 24, 104–117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Birsoy K., Wang T., Chen W. W., Freinkman E., Abu-Remaileh M., Sabatini D. M., An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bailis W., Shyer J. A., Zhao J., Canaveras J. C. G., al Khazal F. J., Qu R., Steach H. R., Bielecki P., Khan O., Jackson R., Kluger Y., Maher L. J. III, Rabinowitz J., Craft J., Flavell R. A., Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 571, 403–407 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.He T., Brocca-Cofano E., Gillespie D. G., Xu C., Stock J. L., Ma D., Policicchio B. B., Raehtz K. D., Rinaldo C. R., Apetrei C., Jackson E. K., Macatangay B. J. C., Pandrea I., Critical role for the adenosine pathway in controlling simian immunodeficiency virus-related immune activation and inflammation in gut mucosal tissues. J. Virol. 89, 9616–9630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ohta A., A metabolic immune checkpoint: Adenosine in tumor microenvironment. Front. Immunol. 7, 109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vigano S., Alatzoglou D., Irving M., Ménétrier-Caux C., Caux C., Romero P., Coukos G., Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front. Immunol. 10, 925 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Esfahani K., Roudaia L., Buhlaiga N., del Rincon S., Papneja N., Miller W. H. Jr., A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 27, S87–S97 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chowell D., Krishna S., Becker P. D., Cocita C., Shu J., Tan X., Greenberg P. D., Klavinskis L. S., Blattman J. N., Anderson K. S., TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proc. Natl. Acad. Sci. U.S.A. 112, E1754–E1762 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boichard A., Pham T. V., Yeerna H., Goodman A., Tamayo P., Lippman S., Frampton G. M., Tsigelny I. F., Kurzrock R., APOBEC-related mutagenesis and neo-peptide hydrophobicity: Implications for response to immunotherapy. Onco. Targets. Ther. 8, 1550341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wisitpitthaya S., Zhao Y., Long M. J. C., Li M., Fletcher E. A., Blessing W. A., Weiss R. S., Aye Y., Cladribine and fludarabine nucleotides induce distinct hexamers defining a common mode of reversible rnr inhibition. ACS Chem. Biol. 11, 2021–2032 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Leist T. P., Weissert R., Cladribine: Mode of action and implications for treatment of multiple sclerosis. Clin. Neuropharmacol. 34, 28–35 (2011). [DOI] [PubMed] [Google Scholar]
- 102.Bonate P. L., Arthaud L., Cantrell W. R. Jr., Stephenson K., Secrist J. A. III, Weitman S., Discovery and development of clofarabine: A nucleoside analogue for treating cancer. Nat. Rev. Drug Discov. 5, 855–863 (2006). [DOI] [PubMed] [Google Scholar]
- 103.Aye Y., Stubbe J., Clofarabine 5′-di and -triphosphates inhibit human ribonucleotide reductase by altering the quaternary structure of its large subunit. Proc. Natl. Acad. Sci. U.S.A. 108, 9815–9820 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Holzer S., Rzechorzek N. J., Short I. R., Jenkyn-Bedford M., Pellegrini L., Kilkenny M. L., Structural basis for inhibition of human primase by arabinofuranosyl nucleoside analogues fludarabine and vidarabine. ACS Chem. Biol. 14, 1904–1912 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Huang P., Chubb S., Plunkett W., Termination of DNA synthesis by 9-beta-D-arabinofuranosyl-2-fluoroadenine. A mechanism for cytotoxicity. J. Biol. Chem. 265, 16617–16625 (1990). [PubMed] [Google Scholar]
- 106.Yang S. W., Huang P., Plunkett W., Becker F. F., Chan J. Y., Dual mode of inhibition of purified DNA ligase I from human cells by 9-beta-D-arabinofuranosyl-2-fluoroadenine triphosphate. J. Biol. Chem. 267, 2345–2349 (1992). [PubMed] [Google Scholar]
- 107.Huang R. S., Ratain M. J., Pharmacogenetics and pharmacogenomics of anticancer agents. CA Cancer J. Clin. 59, 42–55 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cooper T. M., Role of nelarabine in the treatment of T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Ther. Clin. Risk Manag. 3, 1135–1141 (2007). [PMC free article] [PubMed] [Google Scholar]
- 109.Maevis V., Mey U., Schmidt-Wolf G., Schmidt-Wolf I. G., Hairy cell leukemia: Short review, today’s recommendations and outlook. Blood Cancer J. 4, e184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yoon J. H., Choudhury J. R., Prakash L., Prakash S., Translesion synthesis DNA polymerases η, ι, and ν promote mutagenic replication through the anticancer nucleoside cytarabine. J. Biol. Chem. 294, 19048–19054 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Terwilliger T., Abdul-Hay M., Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 7, e577 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jabbour E., Kantarjian H., Chronic myeloid leukemia: 2018 update on diagnosis, therapy and monitoring. Am. J. Hematol. 93, 442–459 (2018). [DOI] [PubMed] [Google Scholar]
- 113.Tsume Y., Hilfinger J. M., Amidon G. L., Enhanced cancer cell growth inhibition by dipeptide prodrugs of floxuridine: Increased transporter affinity and metabolic stability. Mol. Pharm. 5, 717–727 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mojardín L., Botet J., Quintales L., Moreno S., Salas M., New insights into the RNA-based mechanism of action of the anticancer drug 5′-fluorouracil in eukaryotic cells. PLOS ONE 8, e78172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Aguado C., García-Paredes B., Sotelo M. J., Sastre J., Díaz-Rubio E., Should capecitabine replace 5-fluorouracil in the first-line treatment of metastatic colorectal cancer? World J. Gastroenterol. 20, 6092–6101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Moysan E., Bastiat G., Benoit J. P., Gemcitabine versus Modified Gemcitabine: A review of several promising chemical modifications. Mol. Pharm. 10, 430–444 (2013). [DOI] [PubMed] [Google Scholar]
- 117.Matsuoka K., Nakagawa F., Kobunai T., Takechi T., Trifluridine/tipiracil overcomes the resistance of human gastric 5-fluorouracil-refractory cells with high thymidylate synthase expression. Oncotarget 9, 13438–13450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Peeters M., Cervantes A., Moreno Vera S., Taieb J., Trifluridine/tipiracil: An emerging strategy for the management of gastrointestinal cancers. Future Oncol. 14, 1629–1645 (2018). [DOI] [PubMed] [Google Scholar]
- 119.Mikkelsen T. S., Thorn C. F., Yang J. J., Ulrich C. M., French D., Zaza G., Dunnenberger H. M., Marsh S., McLeod H., Giacomini K., Becker M. L., Gaedigk R., Leeder J. S., Kager L., Relling M. V., Evans W., Klein T. E., Altman R. B., PharmGKB summary: Methotrexate pathway. Pharmacogenet. Genomics 21, 679–686 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Smyth J. F., Ford H. T., Methotrexate in the chemotherapy of lung cancer. Cancer Treat. Rep. 65 ( Suppl. 1), 161–163 (1981). [PubMed] [Google Scholar]
- 121.Elion G. B., Mechanism of action and selectivity of acyclovir. Am. J. Med. 73, 7–13 (1982). [DOI] [PubMed] [Google Scholar]
- 122.Acosta E. P., Fletcher C. V., Valacyclovir. Ann. Pharmacother. 31, 185–191 (1997). [DOI] [PubMed] [Google Scholar]
- 123.De Clercq E., Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2, 704–720 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Matthews T., Boehme R., Antiviral activity and mechanism of action of ganciclovir. Rev. Infect. Dis. 10 ( Suppl. 3), S490–S494 (1988). [DOI] [PubMed] [Google Scholar]
- 125.Chen H., Beardsley G. P., Coen D. M., Mechanism of ganciclovir-induced chain termination revealed by resistant viral polymerase mutants with reduced exonuclease activity. Proc. Natl. Acad. Sci. U.S.A. 111, 17462–17467 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Patil A. J., Sharma A., Kenney M. C., Kuppermann B. D., Valganciclovir in the treatment of cytomegalovirus retinitis in HIV-infected patients. Clin. Ophthalmol. 4, 111–119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hodge R. A. V., Cheng Y.-C., The mode of action of penciclovir. Antiviral Chem. Chemother. 4, 13–24 (1993). [Google Scholar]
- 128.Crumpacker C., The pharmacological profile of famciclovir. Semin. Dermatol. 15, 14–26 (1996). [PubMed] [Google Scholar]
- 129.Nyström K., Waldenström J., Tang K.-W., Lagging M., Ribavirin: Pharmacology, multiple modes of action and possible future perspectives. Future Virol. 14, 153–160 (2019). [Google Scholar]
- 130.Delaney W. E. IV, Ray A. S., Yang H., Qi X., Xiong S., Zhu Y., Miller M. D., Intracellular metabolism and in vitro activity of tenofovir against hepatitis B virus. Antimicrob. Agents Chemother. 50, 2471–2477 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vajpayee M., Malhotra N., Antiviral drugs against herpes infections. Indian J. Pharmacol. 32, 330–338 (2000). [Google Scholar]
- 132.De Clercq E., Li G., Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 29, 695–747 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dong L., Hu S., Gao J., Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov. Ther. 14, 58–60 (2020). [DOI] [PubMed] [Google Scholar]
- 134.Andrei G., Topalis D., De Schutter T., Snoeck R., Insights into the mechanism of action of cidofovir and other acyclic nucleoside phosphonates against polyoma- and papillomaviruses and non-viral induced neoplasia. Antiviral Res. 114, 21–46 (2015). [DOI] [PubMed] [Google Scholar]
- 135.Coen D. M., Schaffer P. A., Antiherpesvirus drugs: A promising spectrum of new drugs and drug targets. Nat. Rev. Drug Discov. 2, 278–288 (2003). [DOI] [PubMed] [Google Scholar]
- 136.Anderson P. L., Rower J. E., Zidovudine and Lamivudine for HIV Infection. Clin Med Rev Ther 2, a2004 (2010). [PMC free article] [PubMed] [Google Scholar]
- 137.Kamiya N., The mechanisms of action of antivirals against hepatitis B virus infection. J. Antimicrob. Chemother. 51, 1085–1089 (2003). [DOI] [PubMed] [Google Scholar]
- 138.Bhatia H. K., Singh H., Grewal N., Natt N. K., Sofosbuvir: A novel treatment option for chronic hepatitis C infection. J Pharmacol Pharmacother 5, 278–284 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Athmanathan S., Garg P., Rao G., Ophthalmic antiviral chemotherapy : An overview. Indian J. Ophthalmol. 45, 203–210 (1997). [PubMed] [Google Scholar]
- 140.Turner L. D., Beckingsale P., Acyclovir-resistant herpetic keratitis in a solid-organ transplant recipient on systemic immunosuppression. Clin. Ophthalmol. 7, 229–232 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Amarapurkar D. N., Telbivudine: A new treatment for chronic hepatitis B. World J. Gastroenterol. 13, 6150–6155 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Furman P. A., Barry D. W., Spectrum of antiviral activity and mechanism of action of zidovudine. An overview. Am. J. Med. 85, 176–181 (1988). [PubMed] [Google Scholar]
- 143.Staatz C. E., Tett S. E., Pharmacology and toxicology of mycophenolate in organ transplant recipients: An update. Arch. Toxicol. 88, 1351–1389 (2014). [DOI] [PubMed] [Google Scholar]
- 144.Kawasaki Y., Mizoribine: A new approach in the treatment of renal disease. Clin. Dev. Immunol. 2009, 681482 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Maltzman J. S., Koretzky G. A., Azathioprine: Old drug, new actions. J. Clin. Invest. 111, 1122–1124 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Breedveld F. C., Dayer J. M., Leflunomide: Mode of action in the treatment of rheumatoid arthritis. Ann. Rheum. Dis. 59, 841–849 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Aly L., Hemmer B., Korn T., From leflunomide to teriflunomide: Drug development and immunosuppressive oral drugs in the treatment of multiple sclerosis. Curr. Neuropharmacol. 15, 874–891 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Palsson-McDermott E. M., O’Neill L. A. J., Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 30, 300–314 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Czarnecka-Operacz M., Sadowska-Przytocka A., The possibilities and principles of methotrexate treatment of psoriasis—The updated knowledge. Postepy Dermatol. Alergol. 31, 392–400 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cutolo M., Sulli A., Pizzorni C., Seriolo B., Straub R. H., Anti-inflammatory mechanisms of methotrexate in rheumatoid arthritis. Ann. Rheum. Dis. 60, 729–735 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/21/eabg6165/DC1