

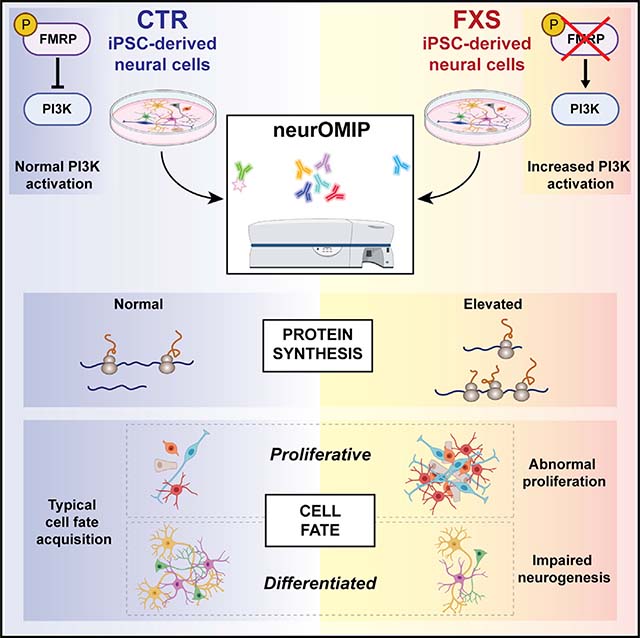
SUMMARY
Transcriptional silencing of the FMR1 gene in fragile X syndrome (FXS) leads to the loss of the RNA-binding protein FMRP. In addition to regulating mRNA translation and protein synthesis, emerging evidence suggests that FMRP acts to coordinate proliferation and differentiation during early neural development. However, whether loss of FMRP-mediated translational control is related to impaired cell fate specification in the developing human brain remains unknown. Here, we use human patient induced pluripotent stem cell (iPSC)-derived neural progenitor cells and organoids to model neurogenesis in FXS. We developed a high-throughput, in vitro assay that allows for the simultaneous quantification of protein synthesis and proliferation within defined neural subpopulations. We demonstrate that abnormal protein synthesis in FXS is coupled to altered cellular decisions to favor proliferative over neurogenic cell fates during early development. Furthermore, pharmacologic inhibition of elevated phosphoinositide 3-kinase (PI3K) signaling corrects both excess protein synthesis and cell proliferation in a subset of patient neural cells.
In brief
Raj et al. developed a multiparametric assay to measure cellular and molecular phenotypes during neurogenesis in fragile X syndrome iPSC-derived neural cells. Relative to controls, FXS patient cultures have more proliferative cells with increased protein synthesis. Defects in cell fate acquisition can be normalized by inhibiting overactive PI3K signaling.
Graphical Abstract
INTRODUCTION
The translation of mRNAs is a tightly regulated process that is essential to cellular development and function (Stiles and Jernigan, 2010). Dysregulated translation is a phenotype common to several neurodevelopmental disorders (Berg et al., 2015; Bhattacharya et al., 2012; Gkogkas et al., 2013; Hoeffer et al., 2012; Ricciardi et al., 2011; Wang et al., 2016), including fragile X syndrome (FXS), which is the leading genetic form of autism and inherited intellectual disability. FXS is primarily caused by the expansion of a microsatellite trinucleotide repeat in the promoter of the FMR1 gene, which results in transcriptional silencing and the subsequent loss of the RNA-binding protein FMRP (Ashley et al., 1993; Pieretti et al., 1991). Intellectual and behavioral impairments are characteristic clinical features of FXS, whereas dysregulated signaling and elevated neuronal protein synthesis are hallmark phenotypes reported in animal models (Richter et al., 2015). Some mRNA targets of FMRP are known to mediate cell proliferation and neurogenesis (Luo et al., 2010), and FMRP deficiency in murine neural stem cells disrupts translation and neuronal differentiation (Liu et al., 2018; Saffary and Xie, 2011). Recent studies have also shown that genes related to protein synthesis, neural development, and migration are aberrantly expressed in FXS-patient-derived neural progenitor cells (NPCs) (Boland et al., 2017; Halevy et al., 2015; Sunamura et al., 2018; Utami et al., 2020). Abnormal signaling via phosphoinositide 3-kinase (PI3K), extracellular signal-regulated kinase-1/2 (ERK1/2), and p70 ribosomal S6 kinase (S6K1) pathways have been reported in FXS, and genetic or pharmacological reduction of PI3K, ERK1/2, and S6K1 have all been shown to ameliorate defects in translation in rodent models of FXS (Asiminas et al., 2019; Bhattacharya et al., 2012, 2016; Gross et al., 2015b, 2019; Gross and Bassell, 2012; Osterweil et al., 2010; Utami et al., 2020). However, whether dysregulated signaling and loss of FMRP-mediated translational control are linked to aberrant neurogenesis during human neural development remains unknown.
Since the discovery of the causal mutation in FMR1, enormous strides have been made in understanding FXS, with the ultimate goal of developing effective therapeutics. Animal models have provided invaluable insight into the normal cellular and molecular functions of FMRP, in particular, by illustrating the consequences of its absence. However, an effective treatment for the disorder is still lacking. Despite successful preclinical studies in animal models, most clinical trials in FXS failed to meet their defined primary endpoints (Berry-Kravis et al., 2018; Erickson et al., 2017; Gross et al., 2015a). Although this could be attributed to several factors, the importance of validating the efficacy of drugs in human-patient-derived, disease-relevant cell types that exhibit FXS-associated molecular phenotypes is becoming increasingly apparent. Here, we developed an induced pluripotent stem cell (iPSC) model to study FMRP-mediated regulation of protein synthesis and signaling during human neurogenesis.
The ability to characterize how loss of FMRP may affect multiple molecular and cellular phenotypes across stages of neural development is hampered by a lack of suitable methods. To circumvent these limitations, we developed a flow-cytometry-based assay (neurOMIP – neuronal optimized multicolor immunophenotyping panel) that allows for the simultaneous measurement of protein synthesis and markers of proliferation and differentiation in defined neural subtypes. We observe that FXS patient cells exhibit abnormally elevated global protein synthesis and altered proliferation during neurogenesis. Cell fate decisions are altered to favor proliferative cell types in FXS, and protein synthesis defects are more profound in these cells. Furthermore, overactive PI3K signaling underlies these defects at a critical early developmental time point. This study provides biological insight into the developmental function of FMRP to balance PI3K signaling via the p110β catalytic subunit, which in turn, regulates protein synthesis and neuronal differentiation. We identified cell-type-specific defects in protein synthesis in a human iPSC-derived neural model of FXS and demonstrated that elevated translation is mechanistically linked to altered neurogenesis via elevated PI3K activity.
RESULTS
Generation of a cellular model of neurodevelopment in FXS
To model FXS and study the consequence of FMRP deficiency in the context of early neural development, we generated clonal iPSC lines from several male control and FXS patient dermal fibroblasts (Table S1). All iPSC lines used in this study were validated using RT-PCR and immunofluorescence for markers of pluripotency and were confirmed to be karyotypically normal (Figures S1A and S1B). We ensured that all lines used in this study could be differentiated using established protocols into NPCs, neurons, and organoids that expressed cell-type-specific markers (Figure S1C). We further confirmed that FMR1 mRNA and FMRP were absent in all FXS patient iPSC lines, as well as in NPCs, neurons, and organoids differentiated from those lines (Figures S1D and S1E). We observed no evidence of variable or low FMRP expression in any of the FXS patient cell lines used in this study.
A major concern in the field of human PSC research is the challenges of reproducibility and heterogeneity within and across patient cultures. To address this, we performed all experiments across multiple individual control and FXS patient lines. Seven FXS patient lines and five control lines were generated for this study, and in addition, we obtained two isogenic control-disease pairs (Table S2). All experiments described in this study were performed using at least three FXS patient and three control lines. Additionally, we were able to reproduce key findings from FXS iPSCs in patient postmortem brain tissue (Table S2), further validating the utility of our iPSC model to evaluate FXS-associated molecular phenotypes.
Global protein synthesis is elevated and normalized by PI3K inhibition in a human neural model of FXS
A fundamental role of FMRP is to bind to specific mRNAs and regulate their translation, typically by acting as a translational repressor. Although global protein synthesis is known to be elevated in animal models of FXS, as well as in human patient lymphoblastoid cells and fibroblasts (Gross and Bassell, 2012; Jacquemont et al., 2018), these defects have not been extensively analyzed in disease-relevant human neural cells. Here, we used two independent well-established assays—bio-orthogonal noncanonical amino acid tagging (BONCAT) (Dieterich et al., 2006) and surface sensing of translation (SUnSET) (Schmidt et al., 2009)—to demonstrate that de novo global protein synthesis was significantly elevated in FXS-patient-derived NPCs compared with that of controls (Figures 1A and 1B). Total protein synthesis in an engineered FMR1 knockout (KO) line (ISO KO) was also increased relative to its isogenic control (ISO CT) (Figures S2A and S2B), suggesting that this defect is a direct consequence of the loss of FMRP. To assess alterations in protein synthesis at a single-cell level, we adapted these assays to measure puromycin and azidohomoalanine (AHA) incorporation in a homogeneous population of human NPCs using flow cytometry. Consistent with our earlier results, FMRP-deficient human NPCs exhibited significantly increased protein synthesis compared with that of control cells in both methods (Figures 1C, 1D, and S2D–S2F). We further treated control and FXS NPCs with fibroblast growth factor (FGF) for 30 min and measured phosphorylation of S6K1 as a readout of translation. Although control NPCs exhibited increased phosphorylation of S6K1, FXS patient NPCs showed no response to FGF stimulation (Figure S2C), indicating that FMRP deficiency in human NPCs leads to defects in both global and stimulus-induced translation.
Figure 1. Elevated global protein synthesis in FXS patient-derived NPCs is corrected with inhibition of PI3K signaling.

(A) Representative immunoblot and densitometry analysis showing increased AHA incorporation in FXS NPCs (unpaired t test, **p < 0.01, n = 3 CTR, 3 FXS).
(B) Representative immunoblot and densitometry analysis showing increased puromycin incorporation in FXS NPCs (unpaired t test, **p < 0.01, n = 3 CTR, 3 FXS).
(C) Flow cytometry analysis of AHA incorporation shows increased global protein synthesis in FXS patient NPCs (Mann-Whitney test, **p < 0.01, n = 3 CTR, 3 FXS).
(D) Flow cytometry analysis of puromycin incorporation shows increased global protein synthesis in FXS NPCs (Mann-Whitney test, **p < 0.01, n = 3 CTR, 3 FXS).
(E) Representative immunoblots for p110β and GAPDH in CTR and FXS NPCs and postmortem frontal cortex tissue.
(F) Densitometry analysis of p110β normalized to GAPDH (Mann-Whitney test, *p < 0.05, n = 4 CTR, 4 FXS NPCs; unpaired t test, **p < 0.01, n = 3 CTR, 3 FXS postmortem FC).
(G) Treatment with either p110β inhibitor (TGX) or S6K1 inhibitor (PF) normalizes elevated protein synthesis in FXS patient NPCs (red bars) relative to control NPCs (gray bars) as measured by flow cytometry analysis of AHA incorporation (two-way ANOVA, Tukey’s test, ***adjusted p = 0.0004; n = 3 CTR, 3 FXS NPCs, across two experiments). Each data point represents an average of 2–3 experimental replicates. All data are shown as means ± SEM. MFI, median fluorescent intensity.
Given that FXS-patient-derived NPCs recapitulate the global translational dysregulation seen in animal models, we asked whether hallmark signaling defects upstream of protein synthesis were also present in patient-derived neural cells. We previously identified p110β as a driver of increased PI3K signaling and global protein synthesis at synapses in the adult brains of Fmr1 KO mice; however, whether PI3K is elevated during early human neurogenesis in FXS is not known. Here, we show that expression of p110β was significantly increased in FXS patient NPCs compared with that of control NPCs (Figures 1E and 1F), and importantly, p110β expression was increased in postmortem frontal cortex tissue from FXS patients compared with that of controls (Figures 1E and 1F). We then tested whether inhibition of the PI3K signaling pathway would normalize elevated protein synthesis in FXS-patient-derived NPCs. Indeed, an acute treatment with either a p110β subunit-specific inhibitor (TGX-221, 1 μM) or an S6K1 inhibitor (PF-4708671, 10 μM) attenuated the protein-synthesis defect in FXS patient NPCs (Figure 1G). Taken together, these results support a model of disrupted PI3K signaling underlying aberrant translation in human FXS neural cells.
Multiparametric analysis reveals altered cell fate commitment and cell-type-specific translational dysregulation in FXS patient neural cells
A key question we aimed to address in this study was whether cells at different stages of neurogenesis would be differentially vulnerable to the effect of the loss of FMRP-mediated translational control. To that end, we developed a multiparametric flow-cytometry-based assay, neurOMIP, which enabled us to simultaneously measure multiple molecular phenotypes within cellular subtypes defined by validated neural lineage and differentiation markers (Figures 2A, S3I, and S3J; Method details). We optimized a panel of fluorophore-conjugated antibodies that, either individually or in combination with each other, served as identifiers of specific cell types (Table S3). Additionally, we included fluorophore-conjugated antibodies to Ki67 and puromycin to allow for the quantification of proliferation and protein synthesis, respectively (Table S3).
Figure 2. Multiparametric analysis reveals elevated protein synthesis coupled to altered proliferation and neuronal cell fate decisions in FXS patient-derived cells.

(A) Schematic outlining the neuronal optimized multicolor immunophenotyping panel (neurOMIP) assay.
(B) Increased translation within NESTIN+/Ki67+ actively proliferating cells and higher abundance of proliferative cells in FXS cultures compared with that of controls. NESTIN+/SOX2+ cells (top left) and NESTIN+/DCX+ cells (bottom left) showed significantly increased protein synthesis in FXS cultures compared with that of controls. NESTIN+/SOX2+ (top right) and NESTIN+/DCX+ (bottom right) cells both showed increased abundance in FXS cultures.
(C) No significant difference in abundance or protein synthesis in NESTIN-/Ki67- cells between control and FXS cultures. Ki67-/DCX+/MAP2+ cells (top left) and Ki67-/GFAP-/MAP2+ cells (bottom left) showed similar levels of protein synthesis across control and FXS cultures. Ki67-/DCX+/MAP2+ cells (top right) and Ki67-/GFAP-/MAP2+ cells (bottom right) were reduced in FXS cultures. Each subpopulation was analyzed using Mann-Whitney tests, n = 4 CTR, 6 FXS, *p < 0.05, **p < 0.01). Data are shown as means ± SEM.
(D) Summary of abundance of proliferative and non-proliferative populations in CTR versus FXS patient-derived cultures.
We reasoned that differentiating NPCs would provide a unique in vitro system to model multiple stages of neural lineage progression, allowing us to measure protein synthesis in several cell types within the same culture. We initiated differentiation in control and FXS NPCs at day 6 and performed neurOMIP analysis at 4 days after induction, in a heterogenous cell population at various stages of proliferation and differentiation into immature neurons. We observed a significant increase in puromycin incorporation in FXS cultures, relative to controls, which was present only in a subset of cell populations (Figures 2B–2D). Those cells expressed high levels of the proliferative protein Ki67, together with additional markers specific to early stages of neurogenesis, such as NESTIN, SOX2, GFAP (neural stem cells and radial glia), and TBR2 and DCX (intermediate progenitors and neuroblasts) (Figures 2B and S3A–S3D; Table S3). In contrast, there was no significant difference in puromycin incorporation between control and FXS patient cultures in more lineage-committed, non-proliferating populations that no longer expressed NESTIN and Ki67 (Figures 2C and S3E–S3H), suggesting that the protein-synthesis defect in FMRP-deficient cells is more profound in early, proliferative cell types (Figure 2D).
These results led us to ask whether the cell-type specificity of translational defects may be reflective of an altered differentiation trajectory in FXS, such that populations of cells with high-protein synthesis were overrepresented in FXS cultures. Indeed, quantification of the relative abundance of cell types revealed that FXS patient cultures were composed of more proliferating cells compared with that of controls, including neural progenitor cells (Ki67+NESTIN+SOX2+), immature neuroblasts (Ki67+DCX+MAP2−) (Figures 2B and 2D) as well as several other proliferative cell types (Figures S3A–S3D). In contrast, non-proliferating neural cells that have acquired MAP2 expression (Ki67−DCX+MAP2+) as well as more mature MAP2+ cells (DCX−GFAP−MAP2+) were less abundant in FXS compared with that of controls (Figures 2C and 2D). Interestingly, although there are more actively proliferating Ki67+NESTIN+GFAP+ cells in FXS cultures, there are fewer Ki67−NESTIN−GFAP+ cells in FXS compared with that of controls (Figures 2B–2D and S3E–S3H). Flow cytometry analysis also revealed that FMRP-deficient isogenic NPCs as well as FXS patient NPCs both had more NESTIN+ Ki67+ cells compared with that of controls (Figures S4A and S4B). Thus, there appears to be a higher abundance of proliferative cell types and fewer non-proliferative cells in FXS cultures, and the effect of the loss of FMRP-mediated translational control is more profound in the proliferative cell populations.
Aberrant proliferation and cell cycle alterations in FXS patient neural cells
The striking shift toward proliferative cell fates suggests a role for FMRP in regulating global cellular proliferation in human neural cells. To further assess that relationship using an independent approach, we performed quantitative immunofluorescence in homogeneous NPC cultures using Ki67 to identify all actively proliferating cells, and phosphohistone H3 (pHH3) as a marker of cells in the late G2/M phase of the cell cycle. FXS patient NPCs had a higher mitotic index (Ki67+pHH3+ double-positive cells/total cells) compared with that of controls as well as greater overall proliferation (Ki67+ cells/total cells) (Figures 3A–3C). To determine whether that was a consequence of alterations in the cell cycle, we first used a colorimetric ELISA assay to measure incorporation of the thymidine analog 5-bromo-2′-deoxyuridine (BrdU) into cellular DNA following a 16-h labeling period. FXS patient NPCs had increased BrdU incorporation compared with that of control NPCs, and that result was also consistent in isogenic NPCs (Figure 3D). Cell cycle analysis of DNA content and 5-ethynyl-2′-deoxyuridine (EdU) incorporation using flow cytometry after a 30-min EdU pulse in asynchronous NPCs revealed that FXS patient NPCs had fewer cells in the G1 phase and more cells in the S phase compared with that of controls (Figures 3E and 3F). That decrease of patient NPCs in G0/G1 further supports a model of reduced fate commitment and increased proliferation in FXS (Lange et al., 2009; Pauklin and Vallier, 2013). As further evidence, we also generated three-dimensional (3D) cortical organoids from three independent control and FXS patient iPSCs and found that 28-day-old (D28) FXS patient organoids had a higher percentage of Ki67+SOX2+ proliferative cells compared with that of controls (Figures 3G and 3H).
Figure 3. Abnormal proliferation and altered cell-cycle distribution in FXS patient NPCs.

(A) Representative images of CTR and FXS NPCs stained for Ki67, pHH3, and DAPI. Scale bar, 50 μm.
(B and C) High-content image analysis showed FXS patient NPCs have higher mitotic activity as evidenced by more pH3 and Ki67-positive cells normalized to total cells (Mann-Whitney test, **p < 0.01, ***p < 0.001, n = 4 CTR, 4 FXS NPCs, ~3,500 cells imaged per line).
(D and E) Colorimetric ELISA shows greater BrdU incorporation in FXS patient NPCs and a FXS isogenic NPC line compared with controls (Mann-Whitney test, **p < 0.01, n = 3 CTR, 3 FXS NPCs, each data point represents an average of two replicates; unpaired t test, *p < 0.05, n = 3 experiments in isogenic control-disease NPC pair. (E) Representative bivariate dot plot for DNA content (FxCycle) and DNA synthesis (EdU). Gates for cells in G0/G1, S, and G2/M phases are indicated.
(F) Cell cycle distribution of CTR and FXS NPCs after a 30-min EdU pulse demonstrates significant reduction of cells in G0/G1 and an increase of cells in the S phase. (Two-way ANOVA corrected for multiple comparisons using Holm-Sidak method, *adjusted p < 0.05, **adjusted p < 0.01, n = 3 CTR, 3 FXS NPCs.)
(G) Representative images of CTR and FXS 3D organoids stained for proliferative markers Ki67 and SOX2. Scale bar, 50 μm.
(H) Quantification of the proportion of Ki67+SOX2+ cells in CTR and FXS organoids at day 28 shows increased actively proliferating cells in FXS (ANOVA, ***p < 0.001, ANOVA, n = 3 CTR, 3 FXS).
We further performed transcriptomic analysis of control and FXS patient iPSC-derived cortical organoids. Differential gene expression analysis revealed a total of 218 differentially expressed genes (DEGs) (Figure S4C), and Gene Ontology (GO) analysis of DEGs provided additional support for our results, showing altered cell-fate commitment and differentiation in FXS (Figure S4D). Significantly downregulated genes in FXS were enriched for GO terms relating to neuronal fate specification, migration, differentiation, and maturation, whereas significantly upregulated genes were enriched for GO terms related to proliferation (Figure S4D).
Inhibition of PI3K signaling corrects aberrant proliferation and selectively normalizes altered neuronal-differentiation trajectories in FXS
With the observation of abnormal cellular proliferation coupled to cell-type-specific translational dysregulation in human FXS patient-derived cells, we hypothesized that overactive PI3K activity due to loss of FMRP may link defects in protein synthesis and proliferation in FXS. The increased population of actively proliferating cells in FXS NPCs (Figures S4A and S4B) also had elevated levels of global protein synthesis relative to that of the controls (Figure 4A). Given that acute reduction of overactive PI3K signaling ameliorated defects in global protein synthesis (Figure 1G), we asked whether inhibition of the PI3K pathway would also correct defects in proliferation. We treated control and FXS NPCs with a p110β inhibitor (TGX-221, 1 μM) and an S6K1 inhibitor (PF-4708671, 10 μM) for 48 h (D4–D6) and subsequently quantified expression of Ki67 and pHH3 by immunofluorescence (Figures 4B–4D). Both drug treatments normalized the population of total Ki67+ proliferative cells (Figure 4C) as well as mitotic pHH3+Ki67+ cells (Figure 4D) in FXS NPCs relative to that of control NPCs, suggesting that increased PI3K in FXS is a key driver of downstream defects in both protein synthesis and cell proliferation in early neural progenitor cell populations.
Figure 4. Overactive PI3K signaling links translational dysregulation and aberrant proliferation in FXS-patient-derived cells.

(A) Representative overlaid dot plots of one control (gray) and one FXS NPC line (red) showing puromycin and Ki67 signals. FXS NPCs have more Ki67+ cells and increased protein synthesis (Mann-Whitney test, *p < 0.05, n = 3 CTR, 3 FXS lines; each data point represents an average of two experimental replicates).
(B) Representative images of NPCs stained for Ki67, pHH3 and DAPI after treatment with p110β inhibitor (TGX) and S6K1 inhibitors (PF). Scale bar, 50 μm.
(C and D) Treatment with either inhibitor normalizes aberrant proliferation in FXS NPCs (n = 4 CTR, 4 FXS NPCs; two-way ANOVA, Tukey’s test, *adjusted p < 0.05, **adjusted p < 0.01, ***adjusted p < 0.001, ****adjusted p < 0.0001).
(E) Chronic (12-day) treatment using TGX normalizes increased abundance and corrects elevated protein synthesis in proliferating cell populations in FXS cultures. TGX does not significantly alter abundance of non-proliferating cell populations, although they trended toward increased abundance. (n = 4 CTR, 4 FXS NPCs; two-way ANOVA, Tukey’s test, *adjusted p < 0.05, **adjusted p < 0.01, ***adjusted p < 0.001). All data are shown as means ± SEM.
We then examined whether the altered differentiation trajectory that we observed in FXS (Figures 2B–2D) is a direct consequence of aberrant proliferation because of elevated PI3K signaling. To assess that, we followed the 48-h treatment paradigm that was effective in correcting increased proliferation in FXS NPCs (Figures 4B–4D), but instead of harvesting cells at day 6, we initiated differentiation and performed neurOMIP analyses at day 4 after induction (Figure S4E). Consistent with the immunofluorescence data (Figures 4B–4D), this acute treatment reduced both Ki67 and puromycin signals in proliferative cell populations (Figures S4F and S4G). However, there was no significant effect on the altered differentiation profile in FXS cultures (Figure S4H). We then employed a chronic treatment strategy in which, cells were treated with TGX-221 for 12 days before performing neurOMIP analysis (Figures 4E and S4I). Intriguingly, chronic PI3K inhibition appears to normalize cell fate selectively in some, but not all, early proliferative cell types. We observed that TGX-221 treatment normalized the increased abundance of Ki67+NESTIN+SOX2+ and Ki67+DCX+MAP2− cells in FXS cultures to the level of the controls (Figure 4E). Although not significant, the proportion of Ki67+NESTIN+GFAP+ cells in TGXtreated FXS patient cultures was also reduced (Figure S4J). Although PI3K inhibition did not significantly affect the abundance of non-proliferating cell populations, elevated puromycin incorporation and Ki67 levels were normalized across most cell populations (Figures 4E, S4K, and S4L). Taken together, these findings indicate that elevated PI3K activity via the p110β subunit drives defects in protein synthesis that are coupled to the timing of cell-fate decisions in proliferative cells.
DISCUSSION
FMRP is an RNA-binding protein that regulates the translation of a subset of mRNAs. Several lines of evidence from animal models suggest that the loss of FMRP leads to “runaway” protein synthesis at synapses in the adult brain, which consequently gives rise to the characteristic behavioral and cognitive impairments seen in the disorder (Richter et al., 2015). Although a primary focus has been to investigate the consequences of loss of function of FMRP in post-mitotic neurons, it is becoming increasingly apparent that FMRP has several key roles at earlier time points during brain development. To better understand the role of FMRP to regulate protein synthesis during early developmental transitions, and, in particular, how that translational control may affect neurogenesis, we developed a flow-cytometry-based assay (neurOMIP) to measure protein synthesis and active proliferation within distinct neuronal subtypes. Our study provides mechanistic insight into how FMRP can control p110β to regulate both protein synthesis and cell fate decisions during early brain development. This demonstrates conservation of a mechanism that functions in a very different context at mature synapses in the adult brain to regulate protein synthesis affecting dendritic spine morphology and synaptic strength.
Our results suggest that neurogenic cell-fate commitment decisions are compromised in FXS, and cells that remain in a prolonged proliferative state exhibit more profound defects in translation. These findings have important implications for understanding the role of FMRP in typical brain development. Ribosome biogenesis and protein synthesis are thought to decline as cells become more lineage-restricted and progress through differentiation in the developing forebrain (Chau et al., 2018). Aberrant protein synthesis during early neurogenesis could thus reasonably underlie an altered neurogenic program in FXS. Furthermore, the FMRP-regulated translatome in the human brain likely varies significantly across cell types and stages of neural lineage progression, and thus, loss of FMRP in the developing human brain could result in dysregulated translation of specific mRNA targets that drive neurogenesis and lineage transitions. This hypothesis is supported by a recent study that describes “transcriptional priming,” wherein progenitor cells co-express mRNAs encoding several neuronal subtypes and rely on translational regulators to rapidly determine neuronal specification (Zahr et al., 2018).
A key finding of this study, made possible by the single-cell neurOMIP approach, is that protein synthesis defects in FMRP-deficient cells are more profound in actively proliferating cells compared with that of terminally differentiated neurons. Although this contradicts previous studies that have shown elevated protein synthesis in postmitotic Fmr1 KO mouse neurons, there are key considerations that may account for the differences we see in our results. First, we conducted these experiments in early, differentiating cultures that contain very few mature neurons. The utility of neurOMIP to identify and quantify protein synthesis within specific neural subtypes and to assess altered neurogenesis is highlighted when applied to a heterogeneous culture containing progenitors as well as terminally differentiated cells. Here, we distinguish between immature neurons and more-terminally differentiated neurons based on the expression of MAP2; however, even those MAP2+ neurons are likely still relatively immature (Marchetto et al., 2017). It is possible that a global translational defect in FXS patient iPSC-derived neurons may eventually emerge in neurons with more numerous and established synaptic connections. Furthermore, we note that although several studies have shown a moderate elevation in global basal protein synthesis in FXS, those studies were conducted on slices from rodent brains (Bear et al., 2004; Hou et al., 2006; Huber et al., 2002; Santini et al., 2017). Similar results may be less pronounced in dissociated cells that are cultured for only a few weeks. Nonetheless, a direct consequence of loss of FMRP in postmitotic neurons is not precluded by our results but, rather, a distinct functional effect of translational control and its interplay with signaling cascades on stem and progenitor populations is indicated.
Our approach to use human patient iPSC-derived neural cells combined with the neurOMIP assay allowed us to generate a unique picture of the dynamic translational landscape through human neurogenesis in the context of FMRP deficiency. Importantly, results of our RNA sequencing (RNA-seq) analysis (Figure S4) were complementary to our findings using the neurOMIP assay. Our results showing elevated p110β expression in multiple human FXS NPCs and 3D organoids are an important validation of previous work in the mouse model. Notably, we also observed an increase in p110β expression in human FXS postmortem brains, further supporting the translational relevance and utility of the patient-derived iPSC model to model FXS. Although previous studies have shown that p110β-inhibition normalizes synaptic defects in the postmitotic neurons in the Fmr1 KO mouse, our work demonstrates an effect of correcting aberrant early fate bias in human FXS patient cells. Thus, the potential therapeutic value of manipulating the PI3K pathway in patients may greatly depend on the developmental time point of intervention. We and others have observed that signaling via PI3K, ERK1/2 and other key signal transduction pathways all appear to be vulnerable to the loss of FMRP in the adult brain (Bhattacharya et al., 2016; Gross and Bassell, 2012; Gross et al., 2010; Osterweil et al., 2010). These pathways have overlapping functions and targets with a broad effect in dividing and post-mitotic cells across development. Future studies should consider the role of FMRP as a key integrator of these signaling pathways to precisely regulate protein synthesis and modulate the switch between proliferative and neurogenic programs in the human brain. Here, we uncover a role for FMRP-mediated regulation of PI3K during early neural development to drive excess protein synthesis and cell proliferation. Importantly, defects in signaling and neurogenesis have been linked to several autism-related disorders (Courchesne et al., 2019; Marchetto et al., 2017; Richter et al., 2019; Sundberg et al., 2018). Thus, identifying similar critical windows of intervention to normalize global imbalances in signal transduction cascades upstream of protein synthesis and fate commitment may be an effective strategy to rescue impairments across several neurodevelopmental disorders.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gary Bassell (gbassel@emory.edu).
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
The sequencing data generated in this study can be found at the Gene Expression Omnibus (GEO) under the accession number GSE146339.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human fibroblast and iPSC cultures
We generated the five control and seven FXS patient iPSC lines used in this study from dermal fibroblasts. All dermal fibroblasts from healthy male controls and male FXS patients were obtained with patient consent and under institutional approval from multiple repositories/sources as listed in Table S1. Normal karyotypes were verified in control and FXS iPSC lines used in this study (Figure S1). NPCs, neurons and cortical organoids were generated from these iPSC lines as listed in Table S2. FMR1 mRNA expression measured by specific taqman assays in iPSCs, NPCs and neurons shows loss of expression of FMR1 in all patient lines. Absence of expression of FMRP protein in iPSCs, NPCs, neurons and organoids was confirmed by western blot (Figure S1). Immunofluorescence further confirmed lack of FMRP expression across FXS patient lines. A pair of isogenic hESC lines was obtained from Dr. Marius Wernig in which exon 1 of the FMR1 gene in a control line (ISO CT) was excised to generate an FMRP null line (ISO KO) (Zhang et al., 2018). A second pair of isogenic lines was provided by Dr. Steve Warren, in which the expanded CGG repeat in an FXS patient line was collapsed to generate a control line (Xie et al., 2016).
Human postmortem tissue
Frozen postmortem human frontal cortex tissue from 3 control males and 3 full-mutation male patients were obtained from the FXS/FXTAS Brain Repository at the University of California at Davis in Sacramento, CA, under approved IRB protocols as listed in Table S1 (University of California, Davis).
METHOD DETAILS
Derivation of iPSC lines from control and FXS patient fibroblasts
Although all iPSC lines were not generated at the same time, disease-control pairs from the same source repository were established and reprogrammed simultaneously. All fibroblasts were reprogrammed using the CytoTune-iPS 2.0 Sendai virus reprogramming kit (ThermoFisher), following the manufacturer protocol. Briefly, fibroblasts were transduced with a Sendai virus cocktail containing four transcription factors (hOct3/4, hSOX2, hKlf4, hc-Myc) and maintained in Fibroblast medium (DMEM, GlutaMAX, FBS; ThermoFisher) for one week. Transduced fibroblasts were replated onto Vitronectin coated dishes in Essential-8 media (ThermoFisher) and individual iPSC colonies began to emerge within two weeks. We manually isolated clonal iPSC colonies from each culture and expanded two clones per iPSC line for characterization.
Maintenance of feeder-free iPSCs
iPSCs were cultured on Matrigel (Corning) coated dishes and maintained in complete mTeSR medium (Stemcell technologies). iPSCs were passaged every 6 days using ReLeSR (Stemcell technologies) following manufacturer’s protocol. For immunofluorescence characterization, iPSCs were passaged onto Matrigel coated coverslips and fixed after 24 hours. For cryostorage of iPSCs, cells were treated with ReLeSR, spun down and resuspended in cryoSTOR-10 freezing media (Stemcell technologies) and samples were stored in liquid nitrogen.
Generation of iPSC-derived NPCs
NPCs were generated using two different methods based on dual-SMAD inhibition. The EB-method used was similar to previously published methods, with minor modifications (Chailangkarn et al., 2016; Marchetto et al., 2010). Briefly, iPSCs were dissociated into single cells and cultured in suspension as EBs in neural induction media (DMEM/F-12, GlutaMAX, HEPES, N2; ThermoFisher) with dual-SMAD inhibition factors SB431542 (10uM; Stemgent) and DMH1 (5uM; Tocris) for 1 week. EBs were plated on matrigel coated dishes in neural progenitor medium (DMEM/F-12, GlutaMAX, HEPES, N2, B27; ThermoFisher) supplemented with bFGF (20 ng/ml; Peprotech). Within one week, neural rosettes formed and were manually isolated, spun down, and replated without dissociation on matrigel coated dishes. After another week, manual rosette selection was repeated, and rosettes were dissociated to single cells using Accutase and plated on matrigel dishes to generate a pure population of neural precursor cells (NPCs). NPCs were cultured in FGF supplemented NPC medium (DMEM/F-12, GlutaMAX, HEPES, N2, B27; ThermoFisher) and passaged every 6 days (Chailangkarn et al., 2016; Marchetto et al., 2017). NPCs generated using this method were used to generate data for Figures 1A–1D. The monolayer method of NPC generation was used for all other data presented and followed the manufacturers recommended protocol. Briefly, iPSCs were dissociated to single cells and plated at a high density and maintained in STEMdiff SMADi Neural Induction Medium (Stemcell technologies) for up to 9 days. Cells were passaged at ~80% confluence twice in STEMdiff Neural Induction Medium (Stemcell technologies), before passaging into Complete STEMdiff Neural Progenitor Medium (Stemcell technologies). NPCs were passaged every six days and maintained in STEMdiff Neural Progenitor Medium (Stemcell technologies) for at least another two passages. NPCs were passed into homemade NPC medium (DMEM/F-12, GlutaMAX, HEPES, N2, B27; ThermoFisher) supplemented with bFGF (20 ng/ml; Peprotech) at least one week prior to being used in experiments. NPCs generated using both methods can be expanded, frozen down, and thawed out successfully.
Generation of neurons from NPCs
In order to generate neurons, NPCs were plated at a density of 15,000 cells/cm2 onto poly-ornithine (25 ug/ml; Sigma) and laminin (3.3 ug/ml; ThermoFisher) coated dishes into NPC medium (DMEM/F-12, GlutaMAX, HEPES, N2, B27; ThermoFisher) without FGF and supplemented with ROCK inhibitor Y-27632 (10uM; Stemgent). After 24 hours, media was changed to neuronal media (Neurobasal/B27/GlutaMAX; ThermoFisher) supplemented with DAPT (10uM; Tocris), BDNF (10ng/ml; Peprotech) and GDNF (10ng/ml; Peprotech). Media changes were performed every other day for the first week, and every 3–4 days subsequently. Neurons were harvested according to experimental requirements. For neurons maintained in culture beyond 7 days, DAPT was removed on day 7 and BDNF and GDNF were removed on day 14.
Generation of iPSC-derived organoids
3D organoids were generated following published protocols (Qian et al., 2018). Briefly, iPSCs colonies were detached from Matrigel coated plates with collagenase (1mg/ml; Invitrogen) treatment for 1hr and suspended in EB medium, comprising of FGF-2-free hESC medium supplemented with Dorsomorphin (2 μM; Tocris) and A-83 (2 μM; Tocris), in non-treated polystyrene plates for 4 days with a daily medium change. On days 5–6, half of the medium was replaced with induction medium consisting of DMEM/F-12, N2 Supplement (ThermoFisher), 10 μg/ml Heparin (Sigma), NEAA (1X), GlutaMAX (1X), WNT-3A (4 ng/ml; R&D Systems), CHIR99021 (1 μM; Cellagentech), and SB-431542 (1 μM; Cellagentech). On day 7, organoids were embedded in Matrigel (Corning) and continued to grow in induction medium for 6 more days. On day 14, embedded organoids were mechanically dissociated from Matrigel by pipetting up and down onto the plate with a 5ml pipette tip. Typically, 10 – 20 organoids were transferred to each well of a 12-well spinning bioreactor (SpinΩ) containing differentiation medium, consisting of DMEM:F12, N2 (1X), B27 (1X; Invitrogen), 2-Mercaptoenthanol (100μM), NEAA (1X), Insulin (2.5 μg/ml; Sigma). Media was changed every other day until day 28 when organoids were collected for immunofluorescence analysis or RNA.
RNA extraction and quantitative reverse transcription with PCR (RT-PCR)
RNA was extracted using the Quick RNA kit (Zymo Research) with a combined on-column DNase I digestion step. Adherent cells were directly lysed in the culture dish and RNA extraction proceeded per the manufacturer’s protocol. cDNA was obtained via RT-PCR using the High Capacity cDNA Reverse Transcription Kit (ThermoFisher). To quantify relative mRNA expression for FMR1 qPCR was performed for each sample using specific Taqman gene expression assays (ThermoFisher) on a Quantstudio 6 Flex system (Applied Biosystems). Relative mRNA expression was normalized to geometric mean Ct values of RPLP0.
RNA-seq analysis
Total RNA was extracted from organoids using TRIzol Reagent (ThermoFisher), treated with DNase I, and cleaned with RNA Clean & Concentrator −5 (Zymo Research). In day 28 organoids, bulk mRNA-seq libraries were generated from triplicated samples per condition by Novogene. An Agilent 2100 BioAnalyzer and a DNA1000 kit (Agilent) were used to quantify amplified cDNA and to control the quality of the libraries. Illumina HiSeq4000 was used to perform 150-cycle pair-end sequencing. Image processing and sequence extraction were performed using the standard Illumina pipeline. Paired-end reads were first aligned to human genome assembly and transcriptome annotations (hg19) by Tophat v2.1.1 (Kim et al., 2013) with default settings. Transcript abundances were quantified by Stringtie v1.3.4 (Pertea et al., 2015). Pairwise comparison between FXS and control organoids were performed to detect differentially expressed genes (DEGs) using Cuffdiff v2.2.1 (Trapnell et al., 2013). DEGs are defined as those with q-value less than 0.05. Gene ontology (GO) analyses on biological processes were performed using GOrilla (Eden et al., 2009).
Lysate preparation
Cells were directly lysed in ice-cold lysis buffer (50mM Tris–HCl, pH 7.4, 300mM NaCl, 1% Triton X-100, 5mM EDTA, 2% SDS) with 1x HALT protease and phosphatase inhibitors (Pierce). Lysates were sonicated at 30% amplitude for 3 cycles of 5 s on/5 s off and cleared by centrifugation at 13500 RPM for 15 mins at 4°C. Protein quantification was done using a BCA assay (Pierce) and protein concentration was normalized across samples.
Immunoblotting
For western blots, protein lysates were prepared in 4X loading buffer, heat-denatured at 90°C for 5 minutes and resolved on 10% BisTris gels (Thermo Fisher) or 4%–20% TGX gels (Bio-Rad). Gels were transferred to 0.44 μm Nitrocellulose membranes and blocked in Odyssey blocking buffer (LI-COR) for 1 hour. Membranes were incubated overnight at 4°C with primary antibodies diluted in blocking buffer. GAPDH was used as an endogenous loading control. The following day, secondary antibodies were diluted in blocking buffer and applied to membrane for 1 hour at R.T. Blots were visualized using ChemiDoc MP imaging system (BioRad) and band/lane intensities were quantified using Image Studio (LI-COR)
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 10 minutes at R.T., washed 3x with 1x PBS and permeabilized for 10 min in 0.1% Triton-X/PBS. Cells were then blocked in 5% normal donkey serum (Jackson Labs) for 1 hour at R.T. and incubated with primary antibodies overnight at 4°C. The following day cells were washed and incubated with secondary antibodies for 1 hour. Organoids were embedded in OCT and 12um sections were cut on a cryostat. For staining, sections were washed in 1x PBS and permeabilized for 10 min in 0.1% Triton-X/PBS. Sections were blocked in 10% normal donkey serum (Jackson Labs) for 1 hour at R.T. and incubated with primary antibodies overnight at 4°C. The following day sections were washed, and secondary antibodies were applied for 2 hours at R.T. Sections were mounted using Prolong Gold.
AHA labeling and click reaction for BONCAT and flow cytometry
Cells were incubated in methionine-free DMEM (ThermoFisher) supplemented with B27 for 1 hour to deplete available methionine. Azidohomoalanine (AHA, ThermoFisher) was added at a final concentration of 1mM to cells. For BONCAT, cells were incubated with AHA for two hours, washed with 1x PBS and lysed in Tris-HCl/SDS lysis buffer. Cells were scraped into 1.5ml tubes and incubated on ice for 10 minutes before sonication at 30% amplitude for 3 cycles of 5 s on/5 s off. Protein concentration was measured using BCA assay (Pierce) and each sample was made up with equal amount of protein up to a volume of 60ul. Click reaction was performed according to manufacturer’s protocol (Protein Reaction Buffer kit, ThermoFisher) to conjugate Biotin-alkyne (ThermoFisher) to incorporated AHA via copper-catalyzed click chemistry. Excess biotin was removed by methanol precipitation and pellets were resolubilized using 8M Urea buffer. Newly synthesized proteins were immunoprecipitated by incubating samples with Streptavidin Dynabeads (ThermoFisher) and rotating for 2 hours at R.T. Beads were resuspended in 4X laemmli buffer and western blots were performed as described earlier. BONCAT signal was measured using an anti-Streptavidin antibody (Li-Cor). For flow cytometry experiments, cells were incubated in AHA media for 45 minutes and then rinsed 2X in warm media. Adherent cells were lifted with Accutase, washed in ice-cold 1x PBS and then resuspended in 1x PBS. A Live/Dead distinguishing marker was added to cells in PBS (Live/Dead-Aqua, 1:1000, ThermoFisher) for 15 mins in the dark at R.T. Cells were fixed in 4% PFA for 15 mins at R.T., and permeabilized in 0.25% Triton X-100/PBS for 15 mins. Cells were washed in 3% BSA/PBS and then the click reaction was performed according to manufacturer’s protocol (Cell reaction buffer, ThermoFisher) to label newly synthesized proteins. Cells were washed and analyzed on a BD LSR II flow cytometer.
Puromycin labeling
Cells were washed 2X in PBS and then incubated with Puromycin (10ug/ml, Sigma) for 30 mins at 37°C, following which cells were rinsed with 1x PBS. Biological negative controls of “no puromycin” and “puromycin + anisomycin” were included to ensure that the signal was puromcyin- and protein synthesis-dependent. Cells were lifted using Accutase and resuspended in ice-cold PBS. For western blotting, cells were spun down and resuspended in lysis buffer and lysate preparation and immunoblotting was carried out as described previously. Newly synthesized proteins were visualized on a gel using anti-Puromycin antibody (1:1000, Millipore). For flow cytometry analysis cells were resuspended in ice-cold PBS and Live/Dead marker (1:1000, Live/Dead Aqua or Live/Dead Near-IR, ThermoFisher) was added for 15 mins in the dark at R.T. Cells were resuspended in TFP Fix/Perm buffer (BD Biosciences) for 15 mins in the dark at R.T. and then washed 2X in TFP Perm/Wash buffer (BD Biosciences). For analysis of global protein synthesis in all cells, anti-Puromycin-Alexa 488 was added to cells for 1 hour in the dark at R.T., and cells were subsequently washed 1X in Perm/Wash buffer and 2X in 2% FBS/PBS and then analyzed on a BD LSR II flow cytometer.
Flow cytometry
Flow cytometry was conducted on a BD LSR II (BD Biosciences) and data were analyzed using FlowJO (Tree Star). Cell suspensions were run through the cytometry on the lowest speed setting and voltages were maintained across experiments. For each experiment, compensation controls and FMO controls were included in addition to biological controls. Compensation was calculated using FACS Diva at the time of sample collection and all samples were run in the same sitting. Protein synthesis and proliferation were measured based on Mean Fluorescence Intensity (MFI) of Puromycin and Ki67 signals, respectively, and percentages of cells positive for each set of markers were calculated based on predetermined gating strategy.
Neuronal optimized multicolor immunophenotyping (neurOMIP)
For cell-type specific profiling of molecular phenotypes we developed optimized panels of antibodies to identify neuronal subpopulations and measure proliferation and protein synthesis. Cells were labeled using puromycin as described and dead cells were identified using Live/Dead markers. Antibodies for each panel were selected after careful evaluation of their ability to label specific subpopulations. All antibodies were titered for optimal staining and minimal spillover into neighboring detectors. To account for any spillover, single-stained compensation controls were included for each color used in the panel, and compensation was calculated and accounted for using BD FACS Diva software. During panel development, antibody titrations, compensation assessments, and gating were all measured using consistent number of cells (1×106 cells/test) resuspended in 200ul of FACS buffer. Cytometer settings and gating strategy were designed to yield the least amount of background and maximal signal. For each antibody within the panel, a fluorescence-minus-one (FMO) sample was included (Figures S3J and S3I) to discriminate between positive and negative events and establish gates for each color. The gate for identifying positive events for puromycin signal was set based on the “no puromycin” negative control sample (Figure S3I).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed using GraphPad Prism. All pooled data are presented as mean ± standard error of the mean (SEM). Details regarding number of technical and biological replicates are provided in the figure legends, along with the specific statistical analysis test used. Unless otherwise indicated, unpaired Mann-Whitney test was performed for all non-parametric data. ANOVA analyses were used for datasets with more than two groups. Kruskal-Wallis analysis of variance, one-way ANOVA followed by Dunn’s post hoc or Dunnett’s post hoc analysis, or two-way ANOVA followed by post hoc Holm-Sidak’s test or Tukey’s test (GraphPad Prism 8). Differences were considered statistically significant at *(p < 0.05), **(p < 0.01), ***(p < 0.001), and ****(p < 0.0001).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Oct4A (clone C30A3) | Cell Signaling Technology | Cat#: 2840; RRID: AB_2167691 |
| Mouse monoclonal anti-SSEA4 (clone MC813) | Cell Signaling Technology | Cat#: 4755; RRID: AB_1264259 |
| Goat polyclonal anti-Nanog | R&D Systems | Cat#: AF1997; RRID: AB_355097 |
| Rabbit polyclonal anti-SOX2 | ThermoFisher | Cat#: PA1–16968; RRID: AB_2195781 |
| Mouse monoclonal anti-TRA-1–60 (clone TRA-1–60) | Millipore | Cat#: MAB4360; RRID:AB_2119183 |
| Mouse monoclonal anti-TRA-1–81 (clone TRA-1–81) | Millipore | Cat#: MAB4381; RRID:AB_177638 |
| Mouse monoclonal anti-Nestin (clone 10C2) | Abcam | Cat#: ab22035; RRID: AB_446723 |
| Rabbit monoclonal anti-Ki-67 (clone 12H15 L5) | ThermoFisher | Cat#: 701198; RRID: AB_2532426 |
| Mouse monoclonal anti-Ki-67 (clone B56) | BD Biosciences | Cat#: 550609; RRID: AB_393778 |
| Rabbit polyclonal anti-Phospho-Histone H3 (Ser10) | Cell Signaling Technology | Cat#: 9701; RRID: AB_331535 |
| Guinea Pig polyclonal anti-MAP2 | Synaptic Systems | Cat#: 188004; RRID: AB_2138181 |
| Rabbit polyclonal anti-VGLUT1 | Synaptic Systems | Cat#: 135302; RRID: AB_2138181 |
| Mouse monoclonal anti-Puromycin (clone 12D10) | Millipore | Cat#: MABE343; RRID: AB_2566826 |
| Rabbit polyclonal anti-PI3K p110β | Millipore | Cat#: 09–482; RRID: AB_1977425 |
| Rabbit monoclonal anti-GAPDH (clone D16H11) | Cell Signaling Technology | Cat#: 5174; RRID: AB_10622025 |
| Mouse monoclonal anti-GAPDH (clone D4C6R) | Cell Signaling Technology | Cat#: 97166; RRID: AB_2756824 |
| PE-Cy7 Mouse anti-Ki-67 (clone B56) | BD Biosciences | Cat#: 561283; RRID: AB_10716060 |
| V450 Mouse anti-Nestin (clone 25) | BD Biosciences | Cat#: 561551; RRID: AB_10717122 |
| PerCP-Cy5.5 Mouse anti-Sox2 (clone O30–678) | BD Biosciences | Cat#: 561506; RRID: AB_10646039 |
| PE Mouse anti-Doublecortin (clone 30) | BD Biosciences | Cat#: 561505; RRID: AB_10643766 |
| Alexa Fluor® 647 Mouse anti-MAP2B (clone 18) | BD Biosciences | Cat#: 560382; RRID: AB_1645423 |
| Alexa Fluor® 700 Mouse anti-GFAP (clone SPM248) | Novus | Cat#: NBP2–34401AF700 |
| Alexa Fluor® 488 Mouse anti-puromycin (clone 12D10) | Millipore | Cat#: MABE343-AF488; RRID: AB_2736875 |
| Mouse monoclonal anti-FMRP (clone 5C2) | Biolegend | Cat#: 834701; RRID: AB_2564993 |
| Rabbit polyclonal anti-FMRP | Cell Signaling Technology | Cat#: 4317; RRID: AB_1903978 |
| Biological samples | ||
| Human postmortem frontal cortex tissue (see Table S1) | FXS/FXTAS Brain Repository (UC Davis) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| TGX-221, p110β-inhibitor | Selleck Chemicals | Cat#: S1169; CAS: 663619–89–4 |
| PF-4708671, S6K1 inhibitor | Tocris | Cat#: 4032; CAS: 1255517–76–0 |
| L-Azidohomoalanine (AHA) | Click Chemistry Tools | Cat#: 1066 |
| Puromycin dihydrochloride | Sigma | Cat#: P8833 |
| Critical commercial assays | ||
| Pierce BCA Protein Assay | ThermoFisher | Cat#: 23225 |
| Click-iT Cell Reaction Buffer Kit | ThermoFisher | Cat#: C10269 |
| Click-iT Protein Reaction Buffer Kit | ThermoFisher | Cat#: C10276 |
| Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit | ThermoFisher | Cat#: C10634 |
| Cell Proliferation ELISA, BrdU (colorimetric) | Roche | Cat#: 11647229001 |
| Deposited data | ||
| Raw and analyzed RNaseq data | This study | GEO: GSE146339 |
| Experimental Models: Cell Lines | ||
| Induced pluripotent stem cells | This study | N/A |
| Oligonucleotides | ||
| FMR1 Taqman probe | ThermoFisher | Hs00924547_m1 |
| RPLP0 Taqman probe | ThermoFisher | 4310879E |
| Software and algorithms | ||
| ImageJ | ImageJ | https://imagej.nih.gov/ij/index.html |
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com:443/ |
| Image Lab | Bio-Rad | https://www.bio-rad.com/ |
| FlowJo v9 | Becton Dickinson | https://www.flowjo.com |
Highlights.
Global protein synthesis and PI3K signaling are elevated in FXS patient NPCs
neurOMIP provides cell-type-specific readouts of translation and proliferation
Translational defect in FXS is more profound during early neurogenesis
PI3K inhibition selectively corrects cell fate and protein synthesis defects
ACKNOWLEDGMENTS
We are extremely grateful to the patients and their families for participating in this study. We thank Christina Gross and Joel Richter for helpful discussions, Joseph Cubells (Emory University) for providing access to patient samples and clinical information, and Alysson Muotri (UCSD), Cleber Trujillo (UCSD), and Eric Wang (University of Florida) for technical support. This work was largely supported by a NICHD Fragile X Center project grant 1U54HD082013–01 (G.J.B) and, more recently, by 1P50HD104458 (G.J.B). This research project was supported in part by the Emory University Integrated Cellular Imaging Microscopy Core. C.M.H was supported by a National Institute of Neurological Disorders and Stroke award (K08-NS087121). J.M.T was supported by the RNA Bioscience Initiative at the University of Colorado Anschutz Medical Campus and the Webb-Waring Early Career Investigator Award from the Boettcher Foundation (AWD-182937).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108991.
REFERENCES
- Ashley CT Jr., Wilkinson KD, Reines D, and Warren ST (1993). FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262, 563–566. [DOI] [PubMed] [Google Scholar]
- Asiminas A, Jackson AD, Louros SR, Till SM, Spano T, Dando O, Bear MF, Chattarji S, Hardingham GE, Osterweil EK, et al. (2019). Sustained correction of associative learning deficits after brief, early treatment in a rat model of Fragile X Syndrome. Sci. Transl. Med 11, eaao0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, and Warren ST (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci 27, 370–377. [DOI] [PubMed] [Google Scholar]
- Berg JM, Lee C, Chen L, Galvan L, Cepeda C, Chen JY, Peñagarikano O, Stein JL, Li A, Oguro-Ando A, et al. (2015). JAKMIP1, a novel regulator of neuronal translation, modulates synaptic function and autistic-like behaviors in mouse. Neuron 88, 1173–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis EM, Lindemann L, Jønch AE, Apostol G, Bear MF, Carpenter RL, Crawley JN, Curie A, Des Portes V, Hossain F, et al. (2018). Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Nat. Rev. Drug Discov 17, 280–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, and Klann E (2012). Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 76, 325–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Mamcarz M, Mullins C, Choudhury A, Boyle RG, Smith DG, Walker DW, and Klann E (2016). Targeting translation control with p70 S6 kinase 1 inhibitors to reverse phenotypes in fragile X syndrome mice. Neuropsychopharmacology 41, 1991–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland MJ, Nazor KL, Tran HT, Szücs A, Lynch CL, Paredes R, Tassone F, Sanna PP, Hagerman RJ, and Loring JF (2017). Molecular analyses of neurogenic defects in a human pluripotent stem cell model of fragile X syndrome. Brain 140, 582–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chailangkarn T, Trujillo CA, Freitas BC, Hrvoj-Mihic B, Herai RH, Yu DX, Brown TT, Marchetto MC, Bardy C, McHenry L, et al. (2016). A human neurodevelopmental model for Williams syndrome. Nature 536, 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau KF, Shannon ML, Fame RM, Fonseca E, Mullan H, Johnson MB, Sendamarai AK, Springel MW, Laurent B, and Lehtinen MK (2018). Downregulation of ribosome biogenesis during early forebrain development. eLife 7, e36998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Pramparo T, Gazestani VH, Lombardo MV, Pierce K, and Lewis NE (2019). The ASD living biology: from cell proliferation to clinical phenotype. Mol. Psychiatry 24, 88–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich DC, Link AJ, Graumann J, Tirrell DA, and Schuman EM (2006). Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl. Acad. Sci. USA 103, 9482–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson CA, Davenport MH, Schaefer TL, Wink LK, Pedapati EV, Sweeney JA, Fitzpatrick SE, Brown WT, Budimirovic D, Hagerman RJ, et al. (2017). Fragile X targeted pharmacotherapy: lessons learned and future directions. J. Neurodev. Disord 9, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, et al. (2013). Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, and Bassell GJ (2012). Excess protein synthesis in FXS patient lymphoblastoid cells can be rescued with a p110β-selective inhibitor. Mol. Med 18, 336–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Nakamoto M, Yao X, Chan CB, Yim SY, Ye K, Warren ST, and Bassell GJ (2010). Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J. Neurosci 30, 10624–10638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Hoffmann A, Bassell GJ, and Berry-Kravis EM (2015a). Therapeutic strategies in fragile X syndrome: from bench to bedside and back. Neurotherapeutics 12, 584–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Raj N, Molinaro G, Allen AG, Whyte AJ, Gibson JR, Huber KM, Gourley SL, and Bassell GJ (2015b). Selective role of the catalytic PI3K subunit p110β in impaired higher order cognition in fragile X syndrome. Cell Rep 11, 681–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Banerjee A, Tiwari D, Longo F, White AR, Allen AG, Schroeder-Carter LM, Krzeski JC, Elsayed NA, Puckett R, et al. (2019). Isoform-selective phosphoinositide 3-kinase inhibition ameliorates a broad range of fragile X syndrome-associated deficits in a mouse model. Neuropsychopharmacology 44, 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halevy T, Czech C, and Benvenisty N (2015). Molecular mechanisms regulating the defects in fragile X syndrome neurons derived from human pluripotent stem cells. Stem Cell Reports 4, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Sanchez E, Hagerman RJ, Mu Y, Nguyen DV, Wong H, Whelan AM, Zukin RS, Klann E, and Tassone F (2012). Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav 11, 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, and Klann E (2006). Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51, 441–454. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, and Bear MF (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 99, 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Pacini L, Jønch AE, Cencelli G, Rozenberg I, He Y, D’Andrea L, Pedini G, Eldeeb M, Willemsen R, et al. (2018). Protein synthesis levels are increased in a subset of individuals with fragile X syndrome. Hum. Mol. Genet 27, 3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C, Huttner WB, and Calegari F (2009). Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 5, 320–331. [DOI] [PubMed] [Google Scholar]
- Liu B, Li Y, Stackpole EE, Novak A, Gao Y, Zhao Y, Zhao X, and Richter JD (2018). Regulatory discrimination of mRNAs by FMRP controls mouse adult neural stem cell differentiation. Proc. Natl. Acad. Sci. USA 115, E11397–E11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Shan G, Guo W, Smrt RD, Johnson EB, Li X, Pfeiffer RL, Szulwach KE, Duan R, Barkho BZ, et al. (2010). Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet 6, e1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, and Muotri AR (2010). A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MC, Belinson H, Tian Y, Freitas BC, Fu C, Vadodaria K, Beltrao-Braga P, Trujillo CA, Mendes APD, Padmanabhan K, et al. (2017). Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol. Psychiatry 22, 820–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, and Bear MF (2010). Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci 30, 15616–15627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauklin S, and Vallier L (2013). The cell-cycle state of stem cells determines cell fate propensity. Cell 155, 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, and Nelson DL (1991). Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822. [DOI] [PubMed] [Google Scholar]
- Qian X, Jacob F, Song MM, Nguyen HN, Song H, and Ming GL (2018). Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nat. Protoc 13, 565–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciardi S, Boggio EM, Grosso S, Lonetti G, Forlani G, Stefanelli G, Calcagno E, Morello N, Landsberger N, Biffo S, et al. (2011). Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum. Mol. Genet 20, 1182–1196. [DOI] [PubMed] [Google Scholar]
- Richter JD, Bassell GJ, and Klann E (2015). Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat. Rev. Neurosci 16, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M, Murtaza N, Scharrenberg R, White SH, Johanns O, Walker S, Yuen RKC, Schwanke B, Bedürftig B, Henis M, et al. (2019). Altered TAOK2 activity causes autism-related neurodevelopmental and cognitive abnormalities through RhoA signaling. Mol. Psychiatry 24, 1329–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffary R, and Xie Z (2011). FMRP regulates the transition from radial glial cells to intermediate progenitor cells during neocortical development. J. Neurosci 31, 1427–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Huynh TN, Longo F, Koo SY, Mojica E, D’Andrea L, Bagni C, and Klann E (2017). Reducing eIF4E-eIF4G interactions restores the balance between protein synthesis and actin dynamics in fragile X syndrome model mice. Sci. Signal. 10, eaan0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, and Pierre P (2009). SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275–277. [DOI] [PubMed] [Google Scholar]
- Stiles J, and Jernigan TL (2010). The basics of brain development. Neuropsychol. Rev 20, 327–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunamura N, Iwashita S, Enomoto K, Kadoshima T, and Isono F (2018). Loss of the fragile X mental retardation protein causes aberrant differentiation in human neural progenitor cells. Sci. Rep 8, 11585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg M, Tochitsky I, Buchholz DE, Winden K, Kujala V, Kapur K, Cataltepe D, Turner D, Han MJ, Woolf CJ, et al. (2018). Purkinje cells derived from TSC patients display hypoexcitability and synaptic deficits associated with reduced FMRP levels and reversed by rapamycin. Mol. Psychiatry 23, 2167–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utami KH, Yusof NABM, Kwa JE, Peteri UK, Castrén ML, and Pouladi MA (2020). Elevated de novo protein synthesis in FMRP-deficient human neurons and its correction by metformin treatment. Mol. Autism 11, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Taliaferro JM, Lee JA, Sudhakaran IP, Rossoll W, Gross C, Moss KR, and Bassell GJ (2016). Dysregulation of mRNA localization and translation in genetic disease. J. Neurosci 36, 11418–11426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie N, Gong H, Suhl JA, Chopra P, Wang T, and Warren ST (2016). Reactivation of FMR1 by CRISPR/Cas9-mediated deletion of the expanded CGG-repeat of the fragile X chromosome. PLoS ONE 11, e0165499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr SK, Yang G, Kazan H, Borrett MJ, Yuzwa SA, Voronova A, Kaplan DR, and Miller FD (2018). A translational repression complex in developing mammalian neural stem cells that regulates neuronal specification. Neuron 97, 520–537.e6. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Marro SG, Zhang Y, Arendt KL, Patzke C, Zhou B, Fair T, Yang N, Südhof TC, Wernig M, and Chen L (2018). The fragile X mutation impairs homeostatic plasticity in human neurons by blocking synaptic retinoic acid signaling. Sci. Transl. Med 10, eaar4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data generated in this study can be found at the Gene Expression Omnibus (GEO) under the accession number GSE146339.