

Abstract
BACKGROUND AND PURPOSE: Three families with adult-onset autosomal dominant leukodystrophy (ADLD) presenting autonomic dysfunction as the first symptom are reported. We describe detailed MR appearances of the brain in 2 new families and neuropathology in 2 patients and compare the findings with those in other adult-onset leukodystrophies.
METHODS: Twenty subjects (12 women and 8 men; age range, 29–70 years) from 2 unrelated families with ADLD were examined with MR. Six subjects were asymptomatic. Fourteen had autonomic dysfunction. Eleven of them also had pyramidal signs and ataxia. The brains of 2 autopsied patients were examined histopathologically.
RESULTS: Two subjects manifested no neurologic symptoms, signs, or MR pathology. Eighteen subjects displayed radiologic abnormalities ranging from subtle T2 high-signal-intensity changes in the upper corticospinal tract to extensive confluent white matter changes, predominantly in a frontoparietal distribution, along the corticospinal tracts down to the medulla oblongata and in the upper and middle cerebellar peduncles. Periventricular white matter was spared or less affected than the adjacent white matter. Histopathology revealed marked loss of cerebral and cerebellar myelin without signs of inflammation. Oligodendrocytes were relatively spared, the number of axons not markedly decreased, and reactive gliosis was modest. The number of Purkinje cells in the cerebellum was reduced.
CONCLUSIONS: Two families with adult-onset ADLD with the disease entity originally reported by Eldridge et al. (N Engl J Med 1984;311:948–53) were described. We propose naming the disease “adult-onset ADLD with autonomic symptoms.” The characteristic radiologic findings, combined with the clinical symptoms and mode of inheritance, enable the diagnosis.
Leukodystrophy is a progressive disease of myelin in which a genetically determined metabolic defect results in confluent destruction, or failed development, of central white matter.1 Most leukodystrophies are autosomal recessive or X-linked recessive with onset in early childhood. Dominantly inherited leukodystrophies with onset in adulthood are rare.
Adult-onset autosomal dominant leukodystrophy (ADLD; OMIM accession number 169500)2 was first reported in an American-Irish kindred in 19843 with some follow-up studies.4–6 Autonomic dysfunction involved the urinary bladder, bowel regulation, and orthostatic hypotension. Autonomic symptoms frequently preceded cerebellar and pyramidal dysfunction. The disease was initially misinterpreted as chronic progressive multiple sclerosis.3 CT and MR signs of leukodystrophy were most prominent in the frontoparietal and cerebellar white matter. In advanced disease, the occipital and, to a lesser extent, temporal lobes were also affected.4 Neuropathology has been reported in 3 patients.3,5, 6 Preservation of oligodendroglia and relative absence of astrogliosis in the demyelinated areas of the brain were regarded as unique features.6 The gene that causes the disease was located on chromosome 5q31.6 Only 2 additional families with what seems to be this disorder have been reported.7, 8 Other examples of adult-onset ADLD are also rare, displaying clinical and radiologic heterogeneity.9–16
We describe the MR appearances of the brain in detail in the patients and their relatives at risk in 2 families with the same type of symptoms and signs as in the kindred reported by Eldridge et al.3 We also present histopathologic findings in the brains of 2 deceased patients. Our findings are compared with those in other adult-onset leukodystrophies.
Materials and Methods
Subjects
The families were identified on the basis of clinical presentation with adult-onset autonomic symptoms preceding or occurring together with motor symptoms and imbalance, white matter changes on MR examination, and a family history compatible with autosomal dominant inheritance. Asymptomatic relatives were included because of their risk of being carriers. Twenty subjects (median age, 55 years; age range, 29–70 years) participated in the study. Eighteen subjects originated from 3 generations in family 1 (11 women and 7 men) and 2 from 2 generations in family 2 (1 woman and 1 man). The study was approved by the local medical research ethics committee. All subjects gave informed consent to enter the study.
Six subjects, all from family 1 (29–59 years of age), were asymptomatic and displayed no clinical signs of the disorder. Fourteen patients had the onset of symptoms, most frequently micturition urgency, in the fifth or sixth decade. Obstipation, impotence, and asymptomatic postural hypotension occurred as well. In family 2, postural hypotension was symptomatic. Three patients had autonomic symptoms only and no motor symptoms or ataxia. The remaining 11 of 14 patients had pyramidal signs and ataxia as well.
Clinical signs of peripheral neuropathy were absent. Peripheral nerve conduction studies (n = 10) were normal. CSF (n = 7) showed normal cell count, normal IgG-index, and absence of oligoclonal bands. In one patient, the CSF albumin/serum albumin ratio was increased, which suggests blood-brain barrier leakage.
MR Examinations
MR imaging of the brain was performed on 9 patients in local hospitals. Eleven subjects were examined at our hospital. This caused some diversity in sequences. The field strength varied from 0.5 to 1.5T. All examinations included T1-weighted sagittal and T2-weighted axial spin-echo (SE) images completed with other image series. In 16 of 20 examinations, a fluid-attenuated inversion recovery (FLAIR) sequence (TR [repetition time], 6000–11000 ms) was used. At our hospital, a standard protocol at 1.5T was used, including T1-weighted sagittal SE images (TR/echo time [TE], 509/14; section thickness, 5.0 mm; gap, 1.0 mm), T2-weighted axial and coronal turbo spin-echo (TSE) images (TR axial-coronal/TE, 6087–8116/100; section thickness, 4 mm; gap, 0.4 mm), and axial FLAIR images (TR inversion time [TI]/TE, 10,000/140/2500; section thickness, 4.0 mm; gap, 1.0 mm). The field of view was 230 mm, and the matrix was 177–207 × 256. Intravenous contrast agent was administered in 5 subjects.
All the images were reviewed by an experienced neuroradiologist blinded to the clinical data. The existence, localization, and pattern of signal-intensity alterations in the brain were recorded. The sizes of the CSF spaces were graded by using the copies of a standard image series used in a study of a neurologically healthy population.17 The measurements of the brain stem were compared with those in a healthy population.18
Postmortem MR Imaging and Neuropathology
A female patient died at age 56 years of lung embolus. She had a 3-year history of gait difficulties. She also had micturition urgency, ataxia, and pyramidal signs. A male patient died of pneumonia at 69 years of age, 17 years after the onset of neurologic symptoms. Besides impotence, urinary bladder dysfunction, and obstipation, he had ataxia, pseudobulbar palsy, and paralysis of the legs.
Both patients were autopsied, and the brains were fixed in 4% buffered formaldehyde. The male brain in toto and a parietal section and a piece of the cerebellum from the female brain were sent for postmortem MR and histopathologic examination. Postmortem MR was performed at 0.5T by using sagittal T1-weighted SE images (TR/TE, 509/14) and axial T2-weighted SE (TR/TE 2334/90) and FLAIR images (TR/TI/TE 6000/100/2000 and 11000/140/2725 ms). The interval between the in vivo MR study and the death/formalin fixation was 4 months for the female patient and 3.5 years for the male patient. The lesion extensiveness in the MR studies, in vivo and postmortem, was compared with that in pathologic studies. Samples for histologic specimens were taken from the areas with normal and abnormal appearances.
The samples, including whole-brain coronal sections through the male brain, were embedded in paraffin. Sections were stained with hematoxylin-eosin for general structures, Luxol fast blue-cresyl violet (LFB-CV) for myelin, and oil-Red O for lipids. Astrocytes were demonstrated immunohistochemically with rabbit polyclonal antiserum to glial fibrillar acidic protein (GFAP) and mouse monoclonal (mmc) antibody to vimentin (DakoCytomation, Glostrup, Denmark). The neurons and their processes were identified with mmc antibody to neurofilament (Euro/DPC, Gwynedd, United Kingdom) and phagocytic cells with mmc antibody to CD 68 (Dako). The bound primary antibodies were visualized by using peroxidase labeled secondary antibodies (Vector Laboratories, Burlingame, Calif), diaminobenzidine as the chromogen, and hematoxylin as the counterstain.
Results
MR Signal Intensity
Subjects without Signal-Intensity Abnormalities (n = 2).
Two subjects (57 and 59 years of age) had no signal-intensity abnormalities on MR imaging. Both lacked symptoms and clinical signs of the disease.
Asymptomatic Clinically Normal Subjects with Signal-Intensity Abnormalities (n = 4).
Two subjects (29 and 34 years of age) displayed a subtle bilateral high-T2-signal-intensity lesion in the white matter under the motor cortex and in the posterior limb of the internal capsule that was revealed only by FLAIR. In one of them, the latter finding was uncertain.
Two other subjects (34 and 37 years of age) showed high T2 signal intensity all along the corticospinal tracts—ie, from the subcortical white matter in the motor region via the posterior limb of the internal capsule, cerebral peduncles, and pontine nuclei into the anterior medulla oblongata down to the level of the foramen magnum (Fig 1). They also manifested high-signal-intensity changes in the upper cerebellar peduncles. One of them had high signal intensity in the middle cerebellar peduncles and in large white matter areas above the ventricle level in the frontal lobes and at the atria in the parietal lobes (Fig. 2). The periventricular area was less affected than the adjacent white matter. This 8-mm-thick area showed lines and spots of the same signal intensity as the adjacent affected white matter, but the areas between had a less pathologic signal intensity. This subject also had increased T2 signal intensities in the deep and posterior parts of the corpus callosum, whereas the other subject had probable callosal changes.
Fig 1.

T2 signal-intensity alterations in a 37-year-old asymptomatic family member. Subtle high-signal-intensity lesions in the corticospinal tracts under the motor cortex (A), in the posterior limb of the internal capsule (B, arrow), mesencephalon (C, arrow), pons (D, arrow), and medulla oblongata (E, arrow). Only a tiny signal-intensity abnormality is seen in the right corticospinal tract at the level of the pons. Note the lesions in the upper cerebellar peduncles (C, arrowhead). Fluid-attenuated inversion recovery (FLAIR), 10,000/140/2500.
Fig 2.

More extensive signal-intensity alterations in a 34-year-old asymptomatic subject.
A, Frontoparietal changes are less severe in the periventricular region.
B, Increased signal intensity in the internal capsules, in the anterior and posterior corpus callosum and in the vicinity of the atria (arrow).
C, Changes in the middle cerebellar peduncles and in the pontine nuclei. Turbo spin-echo (TSE), 5000/90.
Symptomatic Subjects with Signal-Intensity Abnormalities (n = 14).
Of the 14 patients (47–70 years of age) 12 were from family 1 and 2 were from family 2. In general, the severity of symptoms correlated with age and symptom duration. All patients had a high T2 signal intensity in the whole length of the corticospinal tracts, in the upper and middle cerebellar peduncles, lobar cerebral white matter, and corpus callosum. The changes in the cerebral and cerebellar white matter showed a low signal intensity on T1-weighted images. Almost all lesions were symmetrical.
In these 14 patients, the frontoparietal white matter was affected. In 2 patients, only the uppermost frontal white matter and a small part of the parietal white matter were involved, and the periventricular area was intact (Fig 3). In the other patients, most of the frontal and parietal lobes were pathologic and the process continued along the occipital horns. The posterior surroundings of the temporal horns were involved in 7 patients (Fig 4). The periventricular area around the frontal horns, cellae mediae, and the atria was less affected. The thickness of this area was 8 to 10 mm around the frontal horns. The coronal sections most clearly demonstrated the more and less affected layers around the ventricles (Figs 3 and 4). In addition to the circular layers of very high signal intensity—which could be seen as longitudinal lines in some axial images—spotlike, diffuse, and even radiating periventricular changes were found. The signal intensity was lower between these changes, but even higher than in normal white matter (Fig 4). The lobar white matter changes were mainly homogeneous and of a very high signal intensity. U fibers were mainly spared. In the patients with the most severe changes, the U fibers could not always be recognized in the uppermost areas. In 7 patients, there were signal-intensity abnormalities in or near the optic radiation. Two patients had signal-intensity changes in the anterior limb of the internal capsule.
Fig 3.

A 48-year-old woman with a history of urinary bladder dysfunction for the past 2 years. Frontoparietal white matter alteration does not involve the periventricular area (A and B). Increased signal intensity in the corticospinal tracts in the internal capsules (B, arrow).
A, Fluid-attenuated inversion recovery (FLAIR), 10,000/140/2500.
B, Turbo spin-echo (TSE), 8116/100.
Fig 4.

A 55-year-old man with a history of impotence, urinary bladder dysfunction, and obstipation for the past 6 years. Most of the frontoparietal and occipital white matter is affected (A–D and G). The periventricular white matter looks less pathologic (B, C, G). The circular periventricular layers are best seen in a coronal plane (G). The leukodystrophic area continues in the temporal lobes (D, arrow). The corticospinal tracts show a high signal intensity in the internal capsules (C), mesencephalon (D), pons (E), and medulla oblongata (F). The upper (arrowhead) and middle cerebellar peduncles are affected (D and E). High signal intensities are seen in the corpus callosum (B). Its posterior part (B) is very thin.
A–F, Fluid-attenuated inversion recovery (FLAIR), 10,000/140/2500.
G, Turbo spin-echo (TSE), 8116/100.
The signal-intensity changes revealed in the corpus callosum of all symptomatic subjects were situated in the deep and the posterior parts (Fig 4). The cerebellar signal-intensity abnormalities were symmetrical and well circumscribed in the middle cerebellar peduncles including the area adjacent to the fourth ventricle (Fig 4). The upper cerebellar peduncles were also involved in all symptomatic subjects.
There was no pathologic enhancement in the brains of the 5 subjects who received intravenous contrast medium. One of them had no symptoms or radiologic signs of the disease, whereas the other 4 had extensive white matter changes.
Brain Atrophy
The measurements of the brain stem revealed some size reduction in 16 of the 18 subjects with signal-intensity changes. The most-common reduced measure was the coronal diameter of the medulla oblongata (15 subjects), which lay outside the normal range even in one subject without signal-intensity changes in the brain stem. In the visual evaluation, slight thinning of the anterior medulla oblongata could be seen. The other common finding was atrophy of the corpus callosum. It was thinned in all but 2 symptomatic subjects. In 6 patients, the callosal atrophy was severe. Other signs of substance loss were not as constant. Of the 14 symptomatic subjects, 11 presented some indications of cerebral atrophy. Atrophy was mild in all but 2 subjects and without any predilection site. Six patients and one of the 2 asymptomatic subjects without any signal-intensity changes had mild cerebellar substance loss.
MR Findings of the Autopsied Patients
The in vivo and postmortem studies of the female patient showed identical changes: a lower T2 signal-intensity increase in the periventricular than in the lobar parietal white matter and a high signal intensity in the middle cerebellar peduncle.
The in vivo study of the male patient showed the typical pattern with extensive white matter changes involving all cerebral lobes and the corticospinal tracts. The corpus callosum and the upper and middle cerebellar peduncles were affected. In the postmortem examination, the pathologic areas in the temporal lobes, brain stem, and cerebellar peduncles were somewhat smaller. We concluded that radiologic brain involvement probably had not progressed despite clinical deterioration during the time elapsed from the in vivo study.
Gross Pathology
The discolored areas in the cerebral white matter (Fig 5A) and the middle cerebellar peduncles corresponded relatively well with the MR changes, but the periventricular less-affected areas were not discolored in macroscopic inspection, whereas the temporal changes were somewhat larger than on any MR examination. No definite discoloring was seen in the corpus callosum, mesencephalon, and cerebellum, but the corticospinal tracts were discolored in the pons and medulla oblongata.
Fig 5.
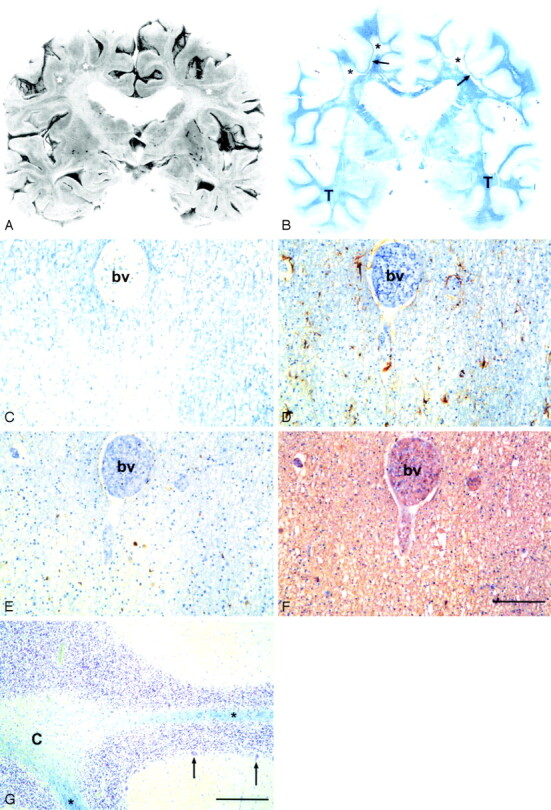
A, Coronal formalin-fixed section at the level of the basal ganglia of a 69-year-old man. White matter is reduced and there are irregularly shaped miscolored areas of demyelination (asterisk).
B, Whole-brain microscopic section from the same level. Widespread diffuse loss of myelin (pallor of the blue color) in the centrum semiovale bilaterally (asterisk, arrows) with sparing of the U fibers (arrows). The myelin is better preserved in the temporal lobes (T). The Sylvian fissures are widened and the lateral ventricles moderately dilated, which indicates mild brain atrophy.
C–F, Consecutive sections from the parietal white matter of a 56-year-old woman. C, Diffuse loss of myelin. Note the less severe pallor around the blood vessel (bv), which indicates that the myelin loss is not perivascular as in multiple sclerosis. Luxol fast blue-cresyl violet (LFB-CV) staining. D, There is only minor astrogliosis in the demyelinated area. The astrocytes (brown cells) are rather plump with irregular processes. Anti-glial fibrillar acidic protein (GFAP) + hematoxylin counterstain. E, No lymphocyte infiltrates are present, but scattered macrophages (brown cells) are seen in the demyelinated area. Anti-CD68 + hematoxylin counterstain. The number of oligodendroglial nuclei (blue) in panels D and E appears the same irrespective of the intensity of the myelin stain (C). F, The density of neurofilament positive axons did not appear to be affected by demyelination. Anti-neurofilament + hematoxylin counterstain. Scale bar, 100 μm.
G, Cerebellum of the 69-year-old man. Severe diffuse loss of myelin in the cerebellar white matter, more marked centrally (C) than in the folia (asterisk). The number of Purkinje cells (2 preserved cells marked with arrows) is greatly decreased, but there is only slightly increased number of reactive Bergmann astrocytes. LFB-CV. Scale bar, 100 μm.
Histopathology
Cerebrum.
In both patients, the cortex showed no significant pathology. There were irregular areas of abnormal white matter with pallor in LFB-CV staining, but the subcortical areas, including the U fibers, and the periphery of gyri were spared (Fig 5B). Variably severely affected areas could be seen around the lateral ventricles. The reduction of myelin was most prominent in the frontal, parietal, and occipital lobes but also occurred in the temporal lobes and corpus callosum.
Myelin appeared rarefied and vacuolated with better preservation around blood vessels (Fig 5C). No significant loss of oligodendrocytes was observed in the demyelinated regions. Despite the prominent reduction of myelin, there was only minimal reactive astrogliosis. GFAP staining displayed some astrocytes with abnormal thickening of processes (Fig 5D). There was no increase of lymphocytes or phagocytic cells (Fig 5E). The density of axons did not appear to be affected by demyelination (Fig 5F). There was a very discrete presence of lipid laden macrophages and fat droplets. No Rosenthal fibers or neuroaxonal spheroids were observed.
Cerebellum.
The white matter of both patients was rarefied in the same way as in the cerebrum with vacuolated myelin in both the cerebellar peduncles and the hemispheres (Fig 5G). The number of Purkinje cells was reduced and that of Bergmann astroglial cells slightly increased. The cerebellar hemispheres, however, had a normal MR appearance. There was no increase of phagocytic cells (microglia/macrophages) or lymphocytes.
Discussion
Three families with adult-onset ADLD with the first symptoms from the autonomic nervous system have been reported elsewhere.3–8 The disorder has been extensively described in English only in the American-Irish kindred.3–6 Our report presents 2 new families with the same clinical symptoms and findings as reported by Eldridge et al.3 Their 9 patients had the onset of symptoms in the fourth and fifth decades and our 14 patients manifested the onset in the fifth or sixth decade. Autonomic symptoms typically preceded motor symptoms and imbalance. Peripheral neuropathy was absent. Histopathologic examinations in our 2 autopsied patients showed loss of myelination in the cerebrum and cerebellum without signs of inflammation, preservation of oligodendroglia, and modest reactive gliosis. These findings are in line with the observations in the 3 previously reported patients,3,5, 6 which further indicates that our patients and the patients in the American-Irish family have the same disease.
Fourteen subjects in the American-Irish family were studied by MR imaging.4 The investigators found abnormalities in 5 patients (56–60 years of age) and in one asymptomatic family member. Later, additional patients in this kindred were examined with similar findings but without giving the number of the examinations or exact description of the abnormalities.6 The signal-intensity changes were most prominent in the frontoparietal and cerebellar—in fact, in the cerebellar peduncles—white matter. In advanced disease, abnormalities were also seen in the occipital and, to a lesser extent, temporal lobes.4 We confirmed these findings in our 14 patients (47–70 years of age). In addition, we observed that the whole lengths of the corticospinal tracts and the corpus callosum were affected in all symptomatic subjects. Sparing or less-severe alteration of the periventricular than lobar cerebral white matter was a typical finding. Although not discussed in the previous reports in English, the figures in the articles (all are axial sections) reveal the same characteristic appearance seen in our patients.4, 6, 8 Our coronal images revealed circular layers of higher and lower signal intensity in the affected periventricular area.
We detected signal-intensity changes in 4 asymptomatic and clinically unaffected subjects (29–37 years of age). These ranged from subtle changes in the uppermost corticospinal tract to a more extensive involvement than in some symptomatic subjects. Figures of the asymptomatic patient of Schwankhaus et al4 show abnormalities in the cerebellar peduncles and frontoparietal white matter but not periventricularly. Our observation of white matter abnormalities in the uppermost corticospinal tract underlying the motor cortex may represent the earliest radiologic manifestation of the disease or incomplete penetrance of the gene causing the disease. Long-term clinical follow-up will determine whether that MR abnormality represents the radiologic onset of the disease. It is possible, that the abnormality begins there and then spreads downward along the corticospinal tracts. The corpus callosum and cerebellar peduncles are involved quite early. Extensive spreading in the cerebral white matter may be the latest process. This theory needs confirmation in follow-up studies.
There is no radiologic explanation for the patients’ early autonomic symptoms. The cause of the urinary bladder dysfunction is unclear but was suggested to be a manifestation of frontal lobe disease.5 A distal lesion of sympathetic noradrenergic neurons has been suggested, and autonomic neuropathy was attended by severe adrenal medullary dysfunction.19 The involvement of the corticospinal tracts and cerebellum, seen radiologically and pathologically, explains the clinical pyramidal signs and ataxia. The extent of the changes in asymptomatic subjects is astonishing. Even most of the patients with very large pathologic areas manifested relatively mild symptoms. One explanation may be the lack of inflammation and preservation of neurons. Atrophy was not a striking feature in this disease, except in the corpus callosum. Brain stem atrophy was pathognomonic but most often became manifest only in measurements.
There was a relatively close match between grossly visible lesions in neuropathologic inspection and those revealed by MR imaging in our patients and in one patient described elsewhere.6 Microscopically, the disease extended beyond the MR lesions and grossly visible lesions, especially in the cerebellum.
Comparison with Other Adult-Onset ADLDs
In an Italian kindred with adult-onset ADLD, the clinical picture was similar but not identical.9, 10 It was characterized by onset in the fifth decade and progressive pyramidal and pseudobulbar signs and, in most patients, urinary incontinence. Their patients did not develop ataxia. Peripheral neuropathy was absent, as in our patients.20 Seventeen subjects (8 symptomatic and 9 asymptomatic) underwent MR examination. All the symptomatic subjects (44–63 years of age) and one asymptomatic (36 years of age) showed abnormalities appearing initially in the parietal white matter and then extending to the frontal and occipital regions.9, 10 The temporal lobes were less affected. The internal capsule (posterior limb) and corpus callosum were affected later, as was the corticospinal tract in the brain stem. In one article,9 lesions in the middle cerebellar peduncles were mentioned in advanced cases. In their asymptomatic subject, periventricular white matter was spared.9, 10 In their patient figures, the periventricular white matter was less or as much affected as the surrounding white matter. The latter finding is different from ours. Optic radiations were mostly spared, as was the case with our patients. Supratentorial atrophy was typical in their patients, but less pronounced in ours. The clinical symptoms and radiologic abnormalities developed in different order than in our patients. This may be another disease or a variant of the disease in our patients.
A Japanese kindred with ADLD has been reported with onset of pyramidal symptoms in the fourth decade and later urinary incontinence. MR imaging (3 patients), revealed diffuse, symmetrical areas of abnormal signal intensity in the white matter of the cerebrum, cerebellum, and brain stem.11 Frontal periventricular area can be seen in one of their figures and is as bright as the surrounding white matter. The optic radiations were spared. We conclude that this disease is not identical with the disease in our patients.
In another Japanese family with adult-onset ADLD, the patients presented with ataxia followed by pyramidal signs, but they had no autonomic symptoms.12 MR imaging (one patient) revealed brain atrophy and diffuse high signal intensity in the cerebral white matter and brain stem, located differently than in our patients. The authors mentioned that the neuropathology resembled that reported by Eldridge et al.,3 but it was not identical with their findings3, 5, 6 or ours. We conclude that this disease is different from the disease in our patients.
We reviewed the literature of other specified adult-onset autosomal dominant leukodystrophies/leukoencephalopathies (ie, Alexander disease,13–15 ADLD with neuroaxonal spheroids,16, 21, 22 and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy23–25). The MR alterations in these diseases are different from those of our patients.
Comparison with Other Adult-Onset Leukodystrophies
Leukodystrophies not inherited as an autosomal dominant trait usually present in childhood, but there are cases with adult onset. MR changes in adult-onset Krabbe disease occur in the corticospinal tracts from the upper part down to the cerebral peduncles or pons.26–29 This is the same area as in 2 of our asymptomatic subjects. Large cerebral white matter involvement has not been described in adult-onset Krabbe disease, and changes in the cerebellar peduncles have been described unilaterally in only one patient.30 Adult-onset Krabbe disease may otherwise be distinguished by differences in the mode of inheritance, clinical presentation, and biochemical test. Other diseases included autosomal recessive metachromatic leukodystrophy,31–34 X-linked recessive adrenoleukodystrophy/adrenomyeloneuropathy,35 X-linked recessive Pelizaeus-Merzbacher disease,36–39 and autosomal recessive leukoencephalopathy with vanishing white matter.40–43 The MR characteristics in these diseases are clearly different from those in our patients.
Specification of the Disease
Our clinical, radiologic, and histopathologic findings are in agreement with those first reported on the adult-onset ADLD by Eldridge et al.3 We have given a more detailed radiologic description of the disease and have not found any other disease with the exact same findings. White matter diseases with autonomic symptoms as the first manifestation are very rare, and we propose naming the disease adult-onset ADLD with autonomic symptoms.
Conclusion
Adult-onset ADLD with autonomic symptoms is a distinct disease entity. The MR lesion pattern is characteristic. The major findings were the affection of the corticospinal tracts and the presence of symmetrical extensive white matter changes predominantly in the frontal and parietal lobes and cerebellar peduncles. The periventricular white matter was spared or less affected. The corpus callosum was affected. Asymptomatic family members may show the same changes. Upon histopathologic examination, oligodendrocytes were found to be preserved in the demyelinated regions, and there were no major signs of gliosis and no signs of inflammation.
The radiologic findings per se are highly suggestive of the diagnosis. They, in combination with the typical clinical symptoms and mode of inheritance, enable the diagnosis.
Footnotes
Supported by grants from the Ländell Foundation, Selander Foundation, Hedberg Foundation for Medical Research, Swedish Medical Research Council, and the Swedish Association of Persons with Neurological Disabilities.
A.M. and L.H. contributed equally to this study
Presented in part at the 17th Symposium Neuroradiologicum, Paris, France, August 18–24, 2002.
References
- 1.Berger J, Moser HW, Forss-Petter S. Leukodystrophies: recent developments in genetics, molecular biology, pathogenesis and treatment. Curr Opin Neurol 2001;14:305–12 [DOI] [PubMed] [Google Scholar]
- 2.McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University and National Center for Biotechnology Information, National Library of Medicine. Online Mendelian inheritance in man, OMIM.2000. . Available at http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM/.
- 3.Eldridge R, Anayiotos CP, Schlesinger S, et al. Hereditary adult-onset leukodystrophy simulating chronic progressive multiple sclerosis. N Engl J Med 1984;311:948–53 [DOI] [PubMed] [Google Scholar]
- 4.Schwankhaus JD, Patronas N, Dorwart R, et al. Computed tomography and magnetic resonance imaging in adult-onset leukodystrophy. Arch Neurol 1988;45:1004–08 [DOI] [PubMed] [Google Scholar]
- 5.Schwankhaus JD, Katz DA, Eldridge R, et al. Clinical and pathological features of an autosomal dominant, adult-onset leukodystrophy simulating chronic progressive multiple sclerosis. Arch Neurol 1994;51:757–66 [DOI] [PubMed] [Google Scholar]
- 6.Coffeen CM, McKenna CE, Koeppen AH, et al. Genetic localisation of an autosomal dominant leukodystrophy mimicking chronic progressive multiple sclerosis to chromosome 5q31. Hum Mol Genet 2000;9:787–93 [DOI] [PubMed] [Google Scholar]
- 7.Laxova R, Hogan K, Haun J. A new autosomal dominant adult onset leukodystrophy. Am J Hum Genet 1985;37:A65 [Google Scholar]
- 8.Asahara H, Yoshimura T, Sada S, et al. A Japanese family with probably autosomal dominant adult onset leukodystrophy [in Japanese]. Rinsho Shinkeigaku 1996;36:968–72 [PubMed] [Google Scholar]
- 9.Quattrocolo G, Leombruni S, Vaula G, et al. Autosomal dominant late-onset leukoencephalopathy: clinical report of a new Italian family. Eur Neurol 1997;37:53–61 [DOI] [PubMed] [Google Scholar]
- 10.Bergui M, Bradac GB, Leombruni S, et al. MRI and CT in an autosomal-dominant, adult-onset leukodystrophy. Neuroradiology 1997;39:423–26 [DOI] [PubMed] [Google Scholar]
- 11.Abe K, Ikeda M, Watase K, et al. A kindred of hereditary adult-onset leukodystrophy with sparing of the optic radiations. Neuroradiology 1993;35:281–83 [DOI] [PubMed] [Google Scholar]
- 12.Tagawa A, Ono S, Inoue K, et al. A new familial adult-onset leukodystrophy manifesting as cerebellar ataxia and dementia. J Neurol Sci 2001;183:47–55 [DOI] [PubMed] [Google Scholar]
- 13.Okamoto Y, Mitsuyama H, Jonosono M, et al. Autosomal dominant palatal myoclonus and spinal cord atrophy. J Neurol Sci 2002;195:71–76 [DOI] [PubMed] [Google Scholar]
- 14.Stumpf E, Masson H, Duquette A, et al. Adult Alexander disease with autosomal dominant transmission: a distinct entity caused by mutation in the glial fibrillary acid protein gene. Arch Neurol 2003;60:1307–12 [DOI] [PubMed] [Google Scholar]
- 15.Thyagarajan D, Chataway T, Li R, et al. Dominantly-inherited adult-onset leukodystrophy with palatal tremor caused by a mutation in the glial fibrillary acidic protein gene. Mov Disord 2004;19:1244–48 [DOI] [PubMed] [Google Scholar]
- 16.van der Knaap MS, Naidu S, Kleinschmidt-Demasters BK, et al. Autosomal dominant diffuse leukoencephalopathy with neuroaxonal spheroids. Neurology 2000;54:463–68 [DOI] [PubMed] [Google Scholar]
- 17.Salonen O, Autti T, Raininko R, et al. MRI of the brain in neurologically healthy middle-aged and elderly individuals. Neuroradiology 1997;39:537–45 [DOI] [PubMed] [Google Scholar]
- 18.Raininko R, Autti T, Vanhanen SL, et al. The normal brain stem from infancy to old age: a morphometric MRI study. Neuroradiology 1994;36:364–68 [DOI] [PubMed] [Google Scholar]
- 19.Brown RT, Polinsky RJ, Schwankhaus J, et al. Adrenergic dysfunction in hereditary adult-onset leukodystrophy. Neurology 1987;37:1421–24 [DOI] [PubMed] [Google Scholar]
- 20.Leombruni S, Vaula G, Coletti Moja M, et al. Neurophysiological study in an Italian family with autosomal dominant late-onset leukodystrophy. Electromyogr Clin Neurophysiol 1998;38:131–35 [PubMed] [Google Scholar]
- 21.Axelsson R, Röyttä M, Sourander P, et al. Hereditary diffuse leucoencephalopathy with spheroids. Acta Psychiatr Scand 1984;69(suppl 314):7–65 [PubMed] [Google Scholar]
- 22.Marotti JD, Tobias S, Fratkin JD, et al. Adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: report of a family, historical perspective, and review of the literature. Acta Neuropathol 2004;107:481–88 [DOI] [PubMed] [Google Scholar]
- 23.Kalimo H, Ruchoux MM, Viitanen M, et al. CADASIL: a common form of hereditary arteriopathy causing brain infarcts and dementia. Brain Pathol 2002;12:371–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Auer DP, Pütz B, Gössl C, et al. Differential lesion patterns in CADASIL and sporadic subcortical arteriosclerotic encephalopathy: MR imaging study with statistical parametric group comparison. Radiology 2001;218:443–51 [DOI] [PubMed] [Google Scholar]
- 25.O’Sullivan M, Jarosz JM, Martin RJ, et al. MRI hyperintensities of the temporal lobe and external capsule in patients with CADASIL. Neurology 2001;56:628–34 [DOI] [PubMed] [Google Scholar]
- 26.Satoh J-I, Tokumoto H, Kurohara K, et al. Adult-onset Krabbe disease with homozygous T1853C mutation in the galactocerebrosidase gene. Neurology 1997;49:1392–99 [DOI] [PubMed] [Google Scholar]
- 27.Turazzini M, Beltramello A, Bassi R, et al. Adult onset Krabbe′s leukodystrophy: a report of 2 cases. Acta Neurol Scand 1997;96:413–15 [DOI] [PubMed] [Google Scholar]
- 28.De Stefano N, Dotti MT, Mortilla M, et al. Evidence of diffuse brain pathology and unspecific genetic characterization in a patient with an atypical form of adult-onset Krabbe disease. J Neurol 2000;247:226–28 [DOI] [PubMed] [Google Scholar]
- 29.Farina L, Bizzi A, Finocchiaro G, et al. MR imaging and proton MR spectroscopy in adult Krabbe disease. AJNR Am J Neuroradiol 2000;21:1478–82 [PMC free article] [PubMed] [Google Scholar]
- 30.Jardim LB, Giugliani R, Pires RF, et al. Protracted course of Krabbe disease in an adult patient bearing a novel mutation. Arch Neurol 1999;56:1014–17 [DOI] [PubMed] [Google Scholar]
- 31.Waltz G, Harik SI, Kaufman B. Adult metachromatic leukodystrophy. Arch Neurol 1987;44:225–27 [DOI] [PubMed] [Google Scholar]
- 32.Reider-Grosswasser I, Bornstein N. CT and MRI in late onset metachromatic leukodystrophy. Acta Neurol Scand 1987;75:64–69 [DOI] [PubMed] [Google Scholar]
- 33.Shapiro EG, Lockman LA, Knopman D, et al. Characteristics of the dementia in late-onset metachromatic leukodystrophy. Neurology 1994;44:662–65 [DOI] [PubMed] [Google Scholar]
- 34.Fukutani Y, Noriki Y, Sasaki K, et al. Adult-type metachromatic leukodystrophy with a compound heterozygote mutation showing character change and dementia. Psychiatry Clin Neurosci 1999;53:425–28 [DOI] [PubMed] [Google Scholar]
- 35.Kumar AJ, Köhler W, Kruse B, et al. MR findings in adult-onset adrenoleukodystrophy. AJNR Am J Neuroradiol 1995;16:1227–37 [PMC free article] [PubMed] [Google Scholar]
- 36.Koeppen AH, Robitaille Y. Pelizaeus-Merzbacher disease. J Neuropathol Exp Neurol 2002;61:747–59 [DOI] [PubMed] [Google Scholar]
- 37.Cailloux F, Gauthier-Barichard F, Mimault C, et al. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Eur J Hum Genet 2000;8:837–45 [DOI] [PubMed] [Google Scholar]
- 38.Sasaki A, Miyanaga K, Ototsuji M, et al. Two autopsy cases with Pelizaeus-Merzbacher disease phenotype of adult onset, without mutation of proteolipid protein gene. Acta Neuropathol 2000;99:7–13 [DOI] [PubMed] [Google Scholar]
- 39.Nance MA, Boyadjiev S, Pratt VM, et al. Adult-onset neurodegenerative disorder due to proteolipid protein gene mutation in the mother of a man with Pelizaeus-Merzbacher disease. Neurology 1996;47:1333–35 [DOI] [PubMed] [Google Scholar]
- 40.Prass K, Brück W, Schröder NWJ, et al. Adult-onset leukoencephalopathy with vanishing white matter presenting with dementia. Ann Neurol 2001;50:665–68 [DOI] [PubMed] [Google Scholar]
- 41.Biancheri R, Rossi A, Di Rocco M, et al. Leukoencephalopathy with vanishing white matter: an adult onset case. Neurology 2003;61:1818–19 [DOI] [PubMed] [Google Scholar]
- 42.Gallo A, Rocca MA, Falini A, et al. Multiparametric MRI in a patient with adult-onset leukoencephalopathy with vanishing white matter. Neurology 2004;62:323–26 [DOI] [PubMed] [Google Scholar]
- 43.Ohtake H, Shimohata T, Terajima K, et al. Adult-onset leukoencephalopathy with vanishing white matter with a missense mutation in EIF2B5. Neurology 2004;62:1601–03 [DOI] [PubMed] [Google Scholar]