

Abstract
BACKGROUND AND PURPOSE: The goal of thrombolytic therapy in patients with acute ischemic stroke is early recanalization, but this may result in delayed reperfusion injury. The purpose of this study was to evaluate the neuroprotective effect of agmatine in a transient ischemic cat model by using MR perfusion imaging and histopathologic analyses.
METHOD: One-hour temporary occlusion of the left middle cerebral artery of cats was performed in the control ischemia group (n = 10), and 100 mg/kg of agmatine was intravenously injected immediately after recanalization in the agmatine-treated group (n = 15). MR imaging was performed at 1, 24, and 48 hours after recanalization, and the perfusion patterns were investigated. Terminal–deoxynucleotidyl transferase mediated nick and end-labeling (TUNEL) and hematoxylin-eosin (H&E) stainings were performed at the corresponding sections.
RESULTS: In the control ischemia group, the number of TUNEL-positive cells was significantly increased in the areas with reperfusion hyperemia (P < .05). In the agmatine-treated group, no significant increase in the number of TUNEL-positive cells was noted in the areas of reperfusion hyperemia. The difference in the number of TUNEL-positive cells between the control ischemia and agmatine-treated group in the areas of reperfusion hyperemia was significant (P < .05). The total number of TUNEL-positive cells and the area of severe ischemic neuronal damage on H&E stain were also significantly attenuated in the agmatine-treated cats compared with the control ischemia cats (P < .05).
CONCLUSION: Our results suggest that agmatine has neuroprotective effects against reperfusion injury and ischemia.
Recently, the main emphasis in the management of hyperacute ischemic stroke has been placed in the recanalization of the occluded artery. Aggressive therapeutic trials have studied the effectiveness of intravenous, intra-arterial, mechanical, or combined thrombolysis in the treatment of hyperacute cerebral infarcts.1–4
In the case of recanalization in animals, reperfusion hyperemia usually occurs and is detected by perfusion-weighted imaging (PWI) or positron-emission tomography (PET). However, it is unclear whether this reperfusion hyperemia is beneficial or detrimental to the reperfused tissue. In rats, restoration of cerebral perfusion pressure after a period of ischemia consistently resulted in a marked and variably prolonged hyperperfusion, often followed by a phase of secondary hypoperfusion, which is generally associated with a poor tissue prognosis.5–8 In ketamine-anesthetized cats, the degree of postischemic hyperperfusion was strongly associated with the leakage of Evans blue and intracerebral petechial hemorrhages.9 However, in baboons, transient ischemia (20 minutes) followed by reperfusion hyperemia was not associated with alterations on CT and showed only minimal random neuronal injury.10 Thus, reperfusion hyperemia can save the ischemic tissue by rapid restoration of the cerebral blood flow, but delayed reperfusion injury may occur because of the presence of oxidants or free radical damage.11 In this regard, an effective neuroprotective agent that can prevent such potential harm after recanalization of the occluded vessel can become a valuable armamentarium for brain salvage.
Agmatine is a primary amine formed by the decarboxylation of L-arginine and is an endogenous clonidine-displacing substance, synthesized in the mammalian brain.12 It is known to be neuroprotective against glutamate-induced necrotic neuronal cell death in vitro and also acts as a competitive nitric oxide synthase (NOS) inhibitor, in which nitric oxide (NO) has been known to cause either apoptosis or necrosis of cells.13–16
The purpose of this study was to evaluate the neuroprotective effect of agmatine in a transient ischemic cat model by using MR perfusion imaging and histopathologic analyses of cellular outcomes.
Materials and Methods
Animal Preparation
The animal experiments were performed in accordance with a protocol approved by the Committee for the Care and Use of Laboratory Animals. Part I of the study consisted of the control ischemia group (n = 10). In this part of the study, the control ischemia reperfusion model was made in cats, and its effects were evaluated by using MR perfusion images and histopathologic specimens. The results of the control ischemia group have been published previously.17 In part II of the study (n = 15), the animals underwent the identical procedure except that agmatine was added to evaluate its effect in the reperfusion model.
In brief, cats of either sex weighing 3.5–4.0 kg were anesthetized by using 5 mg/kg of ketamine chloride and 2 mg/kg xylazine hydrochloride. Rectal temperature was maintained at 37°C by a heating pad during the operation. Oxygen saturation was maintained at >92%. The left brachial vein and the right femoral artery were cannulated for infusion of the fluid and physiologic monitoring. The left middle cerebral artery (MCA) was exposed via a transorbital approach and occluded by a microvascular clamp for a vessel size of 0.4–1.0 mm (Acland clamp, S&T Microlab AG, Rheinfall, Switzerland). One hour after vascular clamping, the clips were released, and the craniotomy site was sealed with bone wax. We injected 100 mg/kg of agmatine (Sigma Chemical Co, St. Louis, Mo) mixed in 0.9% saline solution intravenously immediately after reperfusion for the agmatine-treated group of cats. In 2 additional normal control cats, only enucleation-sphenoid craniotomy was performed, without the induction of transient cerebral ischemia.
Image Acquisition and Data Analysis
The animals were transferred to the MR imaging scanner (1.5T, Intera, Philips Medical Systems, Best, the Netherlands). The first MR imaging was performed 1 hour after reperfusion followed by serial MR imaging 24 and 48 hours after the first imaging session. We obtained coronal section images perpendicular to a theoretic line drawn from the anterior to the posterior commissure and transecting the bilateral striatum, which corresponded to the histologic sections. PWI (gradient-echo echo-planar imaging; TR/TE, 1500/40 msec; 128 × 128 matrix) was performed after injection of a double dose of gadolinium dimeglumine (Magnevist, Schering AG, Berlin, Germany). Overall, 40 phase images were obtained for each section and processed by commercially available postprocessing software (EasyVision, Philips Medical Systems), and the relative cerebral blood volume (rCBV) was calculated.
To determine the evolution of the ischemic brain tissue in accordance with the PWI findings, we investigated 5 different brain regions in each cat. The 5 brain regions were the superior/inferior frontal gyri, superior/inferior temporal gyri, and the striatum. The rCBV values were measured in each region, and the results were divided into 4 groups according to the appearance of perfusion changes as follows; normal perfusion (N), initial reperfusion hyperemia with late continuous hyperperfusion or late normal perfusion (I), initial reperfusion hyperemia and gradual depletion of rCBV (II), and persistent hypoperfusion throughout the experiment (III). The 1-hour and the 48-hour PWI were used as standards for determining early and late reperfusion patterns. The 24-hour PWI was used as an adjunct in determining the trend in the perfusion pattern changes. Increased or decreased rCBV of 20% compared with the contralateral normal side was regarded as hyper- or hypoperfusion.
Pathologic Specimens
The sagittal localizer images from which the coronal PWI had been acquired were used as reference for sectioning of the pathologic specimen. Terminal–deoxynucleotidyl transferase mediated nick and end-labeling (TUNEL)-stained, paraffin-embedded sections were deparaffinized by washing them twice with xylene for 5 minutes. The sections were then washed sequentially in 100%, 95%, and 75% ethanol before being incubated with 20 mg/mL of proteinase K (Sigma Chemical Co) for 5 minutes to strip the nuclear proteins. TUNEL was accomplished by using the In Situ Cell Death Detection Kit (Roche, Penzberg, Germany). After immersion in an equilibration buffer for 10 minutes, the sections were incubated with terminal–deoxynucleotidyl transferase (TdT) and deoxyuridine triphosphate (dUTP)-digoxigenin in a humidified chamber at 37°C for 1 hour and then incubated in a stop/wash buffer at 37°C for 30 minutes to stop the reaction. The sections were then washed with phosphate-buffered saline once before incubation in an antidigoxigenin–peroxidase solution for 30 minutes. They were colorized with diaminobenzidine-H2O2 solution (Sigma Chemical Co, 0.2 mg/mL tetrachloride and 0.005% H2O2 in 50 mmol/L Tris-HCl buffer) and then counterstained with methyl green or hematoxylin. The control sections were treated similarly but were incubated in the absence of TdT enzyme, dUTP-digoxigenin, or antidigoxigenin antibody.
From the multiple pathologic slides that were produced from this process, the slide that corresponded best with the MR imaging anatomic landmarks was selected. TUNEL-positive cells were counted in a blind fashion from 10 rectangular areas measuring 150 000 μm2 and averaged in each anatomic region as described previously by using high-powered-field (×200) light microscopy. Hematoxylin and eosin (H&E) staining was performed at the corresponding sections, and the area of severe ischemic neuronal damage characterized by moderate-to-severe neuronal shrinkage, increased eosinophilic neurons, nuclear basophilia, and nuclear pyknosis was assessed by using a computer assisted image analysis system (MCID, Imaging Research Inc., St. Catherines, ON, Canada). The area of severe ischemic neuronal damage was expressed as a percentage of the total area of the ipsilateral neocortex, striatum, and hemisphere and corrected for the presence of edema.18
Statistical Analysis
Statistical analysis was performed by using 1-way analysis of variance (ANOVA) with the Scheff é test to compare the differences among the reperfusion types. Independent t and Kolmogorov-Smirnov tests were performed to compare the differences between the control ischemia and agmatine-treated groups.
Results
Physiologic parameters, including body temperature, blood pressure, and arterial oxygen saturation levels, of all cats were within normal ranges and remained stable throughout the operation and imaging procedures.
Reperfusion Pattern After Temporary Ischemia
Early reperfusion hyperemia (type I or II) was noted in all cases of control ischemia and agmatine-treated cats. The most common reperfusion pattern was early hyperperfusion–late normo-/hyperperfusion (type I, 43%, n = 54/125). This pattern of reperfusion was distributed evenly in the hemisphere (Table).
Incidence of reperfusion types in the respective brain regions
| SF | IF | ST | IT | Striatum | Total (n=125) | |
|---|---|---|---|---|---|---|
| I | 9 (5/4) | 12 (6/6) | 10 (4/6) | 14 (4/10) | 9 (4/5) | 54 (23/31) |
| II | 2 (0/2) | 4 (1/3) | 6 (4/2) | 2 (1/1) | 4 (1/3) | 18 (7/11) |
| III | 3 (0/3) | 1 (0/1) | 4 (1/3) | 5 (3/2) | 9 (4/5) | 22 (8/14) |
| N | 11 (5/6) | 8 (3/5) | 5 (1/4) | 4 (2/2) | 3 (1/2) | 31 (12/19) |
Note:—SF indicates superior frontal lobe; IF, inferior frontal lobe; ST, superior temporal lobe; IT, inferior temporal lobe; I, initial reperfusion hyperemia with late continuous hyperperfusion or late normal perfusion; II, initial reperfusion hyperemia and gradual depletion of rCBV; III, persistent hypoperfusion; N, normal perfusion. Numbers in parentheses are related as follows: (Control ischemia group/agmatine-treated group).
Early hyperperfusion–late hypoperfusion pattern (type II) was the least commonly observed pattern of reperfusion (14%, n = 18/125). This pattern was most often observed in the superior temporal gyrus (33%, n = 6/18) but was also observed in other MCA territories.
Early hypoperfusion–late hypoperfusion pattern (type III) was noted in 18% (n = 22/125) of the sectors. Nine (41%) of the 22 sectors showing this pattern of reperfusion were located in the striatum.
Normal perfusion pattern (type N) was observed in 25% (n = 31/125) of the sectors, mostly in the frontal gyri (61%, n = 19/31).
Histopathologic Findings
TUNEL-Positive Cell Count
The mean number of total TUNEL-positive cells was higher in the control ischemia cats compared with the agmatine-treated cats (35.7 ± 15.8 versus 17.4 ± 34.8, P < .05).
In the control-ischemia cats, a significant difference in the number of TUNEL-positive cells was demonstrated between the reperfusion types (1-way ANOVA test, P < .05). The number of TUNEL-positive cells observed in the N-type reperfusion was 16.8 ± 5.2. However, significantly increased TUNEL-positive cell counts were demonstrated in the areas of type I and II reperfusions, with counts of 39.5 ± 13.4 and 43.6 ± 16.8, respectively. In the areas of type III reperfusion, TUNEL-positivity was lower than the other types of reperfusion hyperemia, with cell counts of 23.3 ± 7.0 (Scheff é test: N versus I, P < .05; N versus II, P < .05; II versus III, P < .05).
In the agmatine-treated cats, no statistically significant difference between the reperfusion types and the number of TUNEL-positive cells was demonstrated (1-way ANOVA). The number of TUNEL-positive cells observed in the N-type reperfusion was 14.1 ± 29.3. In the areas with reperfusion hyperemia types I and II, the TUNEL-positive cell counts were 18.4 ± 39.5 and 13.9 ± 30.2, respectively. In the area of type III reperfusion, TUNEL positivity was slightly higher than the other types of reperfusion, with the cell count being 23.0 ± 37.5 (Fig 1).
Fig 1.
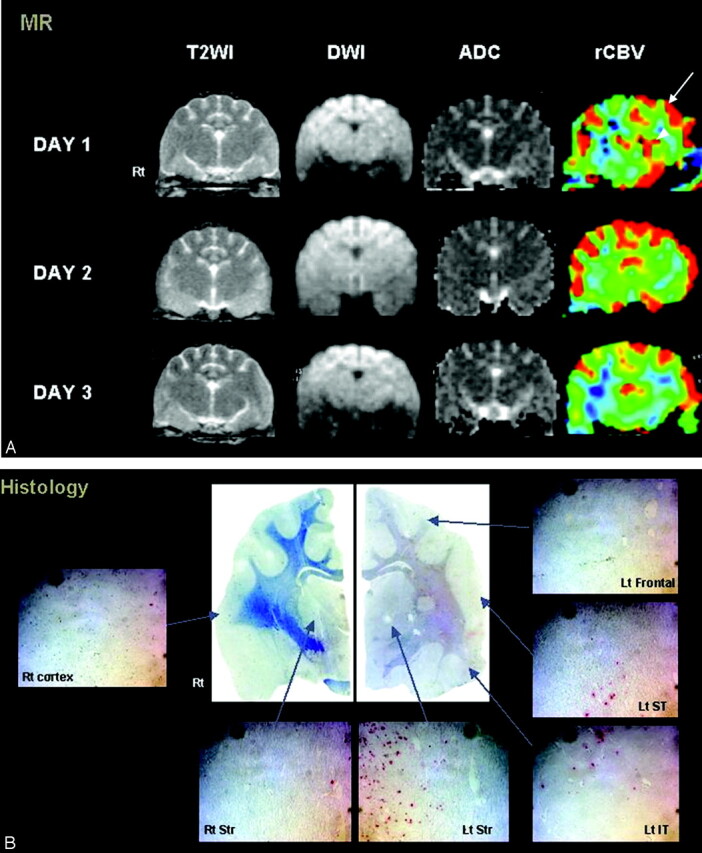
Agmatine-treated cat with type I and II reperfusion.
A, Persistent hyperperfusion is noted in the left frontal lobe (type I reperfusion, arrow). Initial hyperemia in the left striatum shows hypoperfusion on the last follow-up image (type II reperfusion, arrowhead). Notice the lack of signal-intensity changes on T2-weighted (T2WI) or diffusion-weighted images (DWI.). (B) A few red-stained TUNEL-positive cells are seen in all regions of the left hemisphere. ADC indicates apparent diffusion coefficient.
In terms of the difference of TUNEL-positive cells between the control ischemia and agmatine-treated groups in the respective reperfusion types, the type I and II reperfusions in the agmatine-treated group showed a significantly lower cell count compared with the corresponding control ischemia group reperfusion types (independent t test and Kolmogorov-Smirnov test, P < .05). Types N and III reperfusions did not show a significant difference in the number of TUNEL-positive cells between the control ischemia and agmatine treated groups.
In the 2 normal control cats, no abnormal findings on MR imaging were detected, and most parts of brain showed TUNEL negativity, except for 1 cat that showed a few scattered TUNEL-positive cells (<10) in the temporal lobe.
H&E Stain
The area of ischemic neuronal damage on H&E stain, indicated by the cells showing severe neuronal shrinkage, increased nuclear basophilia, and pyknosis, measured 25.6 ± 10.9% of the ipsilateral hemisphere in the control-ischemia group. The area of ischemic neuronal damage was significantly attenuated in the agmatine-treated group, measuring 16.7 ± 4.9% (P < .05).
Discussion
The results of this study have shown the neuroprotective effects of agmatine against reperfusion injury and ischemia in a transient ischemic cat model.
Histologically, the number of TUNEL-positive cells showed significant attenuation in the areas of reperfusion hyperemia with the administration of agmatine. In addition to the significant decrease of the total number of TUNEL-positive cells, the decrease of the area of ischemic neuronal damage on H&E stain in the agmatine-treated group also reflects the neuroprotective effect of agmatine.
Experimental studies have shown that restoration of cerebral blood flow after a period of ischemia consistently resulted in a marked and prolonged hyperperfusion. This hyperperfusion phase was often followed by a phase of secondary hypoperfusion generally associated with a poor tissue prognosis.5–8 By performing MR perfusion imaging for the evaluation of reperfusion patterns in this study, we were able to assess the various patterns of reperfusion with relatively high spatial resolution. Areas of reperfusion hyperemia—type I (early hyperperfusion–late normo-/hyperperfusion) and type II (early hyperperfusion–late hypoperfusion)—showed significant increases in the TUNEL-positive cell count in the control ischemia group, compared with the regions of normal reperfusion (type N), indicating the induction of cellular injury by reperfusion hyperemia. On the other hand, the TUNEL-positive cell count did not show any significant difference between the areas of type I or II hyperemia and normal reperfusion in the agmatine-treated group. Also, direct comparison of the TUNEL-positive cell count between the control ischemia and agmatine-treated groups in the type I and II hyperemic areas showed significant attenuation of the TUNEL-positive cells by agmatine.
The major causes of secondary brain damage after transient ischemic stroke and reperfusion are a combination of processes, including damage by excitotoxic amino acids, oxygen free radicals, apoptosis, and inflammatory reactions.19 Glutamate is a major excitatory neurotransmitter, and its toxicity has been ascribed to excessive excitatory neurotransmission. Ischemia causes massive efflux of glutamate into the extracellular space of the brain, raising its concentration to the toxic level. This induces intracellular Ca2+ increase, which then activates enzymes that cleave to structural or regulatory proteins and the membrane lipids, eventually leading to cell destruction.20,21 Structural damage during reperfusion is also thought to be a consequence of the excessive generation of oxygen free radicals followed by lipid peroxidation. The changes in the conformation of the free fatty acids resulting from lipid peroxidation alters the permeability and fluidity of the membrane and compromises the functions of receptors, ion channels, and other proteins.22 Peroxynitrite, formed by the reaction of O2 with NO (produced by neuronal constitutive [nNOS or iNOS]), is implicated as the lipid peroxidation–initiating radical species during reperfusion.23,24
Inflammatory reaction is also implicated as having a role in reperfusion injury. The recruitment of neutrophils to the area of ischemia, the first step to inflammation, involves the coordinated appearance of multiple adhesion molecules.19,25 Recently, apoptosis has gained much attention as a mechanism for secondary neuronal death after transient ischemia. By demonstrating the nucleosomal ladders of DNA fragments in transient ischemia and reperfusion models, previous reports have shown that cell death during ischemia is mainly necrotic, whereas damage induced by reperfusion caused additional cell death principally through apoptosis.26–28
The mechanism of neuroprotective effects of agmatine, as shown in this study, can be attributed to its multimolecular biologic actions.29 First, agmatine is known to inhibit the (NMDA) subtype of glutamatergic receptors, thus impeding the intracellular accumulation of Ca2+.13,30,31 Second, agmatine can inhibit all isoforms of NOS, with the highest reported activity as an irreversible inactivator of neuronal NOS, leading to reduced production of the neuromodulator NO.14,16,32 Third, agmatine interferes with the intracellular signaling pathways by inhibiting ADP–ribosylation of proteins, a process implicated in neuronal injury after cerebral ischemia in rats.33 These actions of agmatine can be postulated to counteract both the necrotic and apoptotic processes of cell death in this transient ischemia model.
Even though TUNEL staining has been accepted as a method for histologic assay for detection of apoptosis, it cannot always distinguish apoptotic and necrotic cells and sometimes falsely identifies cells in the process of DNA repair or cells in the process of active gene transcription.34,35 Thus, despite careful characterization of TUNEL staining to avoid inclusion of cells that demonstrated diffuse light staining, which could be observed with necrosis, the significant decrease in the total TUNEL-positive cell count in the agmatine-treated group in our study may include necrotic cells.36
Areas of type III reperfusion showing consistent hypoperfusion after recanalization failed to show a decrease in the number of TUNEL-positive cells after agmatine treatment in comparison with the control ischemia group. This type of reperfusion may represent areas of nonrecanalization due to persistent proximal occlusion or the “no-reflow phenomenon,” in which the neuroprotective agent was not able to reach the ischemic tissue in effective concentrations. This no-reflow phenomenon is known to be caused by a multiplicity of factors such as increased blood viscosity, intravascular coagulation, and microvascular occlusion by leukocytes.37
The number of TUNEL-positive cells in the agmatine-treated groups showed a large variation. The main reason seems to be that in a small number of cats, agmatine appeared to be ineffective in neuroprotection. The TUNEL-positive cell counts were very high in these cats despite agmatine infusion. The reason for this is unclear, but we suspect that this may also be due to the no-reflow phenomenon in some instances, relatively more severe damage during the 1-hour occlusion—perhaps due to poor collateral circulation, or unknown mechanisms of agmatine resistance in some cats.
Previous studies on the effects of exogenously applied agmatine have also shown its biologic activities in the brain. Agmatine decreased the extent of neuronal loss and improved the motor function after ischemic and traumatic spinal cord injury in rats.29,38 The weight deficit of the ischemic hemisphere was attenuated in hypoxic ischemic models in rat pups by using agmatine doses of 50 and 100 mg/kg.39 The neuroprotective effect of agmatine against delayed neuronal cell death was also shown in the gerbil hippocampus after global forebrain ischemia.40
The timing of intravenous administration of agmatine has been selected to coincide with the onset of reperfusion in this study. Many previous animal studies have shown the effectiveness of agmatine when administered with the onset of reperfusion.29,38–40 This protocol is practical because it simulates the actual clinical setting of hyperacute ischemic stroke and recanalization therapy. However, a recent study has shown the neuroprotective effects of agmatine when administered before or during occlusion and even 2 hours postrecanalization, by using in vivo stroke models.16 Future studies with administration protocols reflecting such a time window of effects may provide further evidence for the use of agmatine as a preventive treatment in the high-risk group or as an adjunctive therapeutic option in the extended time window.
The use of ketamine as an anesthetic in our study is a limitation. Ketamine interacts with the phencyclidine binding site, which is located in the NMDA receptor–associated ion channel. It inhibits the influx of Na+ and Ca2+ ions through this channel, thus noncompetitively antagonizing the actions of NMDA agonists and acting as a neuroprotective agent.41,42 Some animal studies have used inhalation anesthetics such as halothane or isoflurane in conjunction with ketamine to reduce the neuroprotective effects.43,44 However, the design of our study did not permit the use of inhalation agents because of the lack of a MR imaging–compatible inhalation anesthetic machinery. Our study used MR imaging to analyze the temporal changes of reperfusion and correlated its outcome with histologic findings. The advantage of MR imaging in terms of spatial resolution is critical in assessing the variable patterns of reperfusion within the brain of a small animal. Also, we believe that the use of ketamine did not significantly influence the outcome because identical anesthetic conditions were applied to both the control ischemia and agmatine-treated groups.
In conclusion, the neuroprotective effects of agmatine against reperfusion injury and ischemia have been demonstrated in a transient ischemic cat model designed to simulate the clinical situation of hyperacute ischemic stroke and its treatment by recanalization. Further validation of agmatine as a neuroprotective agent in terms of the drug administration protocol, combined reperfusion therapy, and prolonged ischemia is warranted.
Footnotes
Supported by a grant from the Korea Health 21 R&D Project, (02-PJ1-PG3–21301–0013), Ministry of Health and Welfare, Korea.
References
- 1.The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med 1995;333:1581–87 [DOI] [PubMed] [Google Scholar]
- 2.Furlan A, Higashida R, Wechsler L, et al. Intra-arterial prourokinase for acute ischemic stroke. The PROACT II study: a randomized controlled trial. Prolyse in Acute Cerebral Thromboembolism. JAMA 1999;282:2003–11 [DOI] [PubMed] [Google Scholar]
- 3.The Interventional Management of Stroke Study. Combined intravenous and intra-arterial recanalization for acute ischemic stroke. Stroke 2004;35:904–11 [DOI] [PubMed] [Google Scholar]
- 4.Zaidat OO, Suarez JI, Sunshine JL, et al. Thrombolytic therapy of acute ischemic stroke: correlation of angiographic recanalization with clinical outcome. AJNR Am J Neuroradiol 2005;26:880–84 [PMC free article] [PubMed] [Google Scholar]
- 5.Traupe H, Kruse E, Heiss WD. Reperfusion of focal ischemia of varying duration: postischemic hyper- and hypo-perfusion. Stroke 1982;13:615–22 [DOI] [PubMed] [Google Scholar]
- 6.Gourley JK, Heistad DD. Characteristics of reactive hyperemia in the cerebral circulation. Am J Physiol 1984;246:H52–58 [DOI] [PubMed] [Google Scholar]
- 7.Todd NV, Picozzi P, Crockard HA, et al. Reperfusion after cerebral ischemia: influence of duration of ischemia. Stroke 1986;17:460–66 [DOI] [PubMed] [Google Scholar]
- 8.Tasdemiroglu E, Macfarlane R, Wei EP, et al. Pial vessel caliber and cerebral blood flow become dissociated during ischemia-reperfusion in cats. Am J Physiol 1992;263:H533–36 [DOI] [PubMed] [Google Scholar]
- 9.Tamura A, Asano T, Sano K. Correlation between rCBF and histological changes following temporary middle cerebral artery occlusion. Stroke 1980;11:487–93 [DOI] [PubMed] [Google Scholar]
- 10.Yonas H, Gur D, Claassen D, et al. Stable xenon-enhanced CT measurement of cerebral blood flow in reversible focal ischemia in baboons. J Neurosurg 1990;73:266–73 [DOI] [PubMed] [Google Scholar]
- 11.Marchal G, Young AR, Baron JC. Early postischemic hyperperfusion: pathophysiologic insights from positron emission tomography. J Cereb Blood Flow Metab 1999;19:467–82 [DOI] [PubMed] [Google Scholar]
- 12.Li G, Regunathan S, Barrow CJ, et al. Agmatine: an endogenous clonidine-displacing substance in the brain. Science 1994;263:966–69 [DOI] [PubMed] [Google Scholar]
- 13.Yang X, Reis D. Agmatine selectively blocks the N-methyl-D-aspartate subclass of glutamate receptor channels in rat hippocampal neurons. J Pharmacol Exp Ther 1999;288:544–49 [PubMed] [Google Scholar]
- 14.Galea E, Regunathan S, Eliopoulos V, et al. Inhibition of mammalian nitric oxide synthases by agmatine: an endogenous polyamine formed by decarboxylation of arginine. Biochem J 1996;316 (pt 1):247–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abe K, Abe Y, Saito H. Agmatine suppresses nitric oxide production in microglia. Brain Res 2000;872:141–48 [DOI] [PubMed] [Google Scholar]
- 16.Kim JH, Yenari MA, Giffard RG, et al. Agmatine reduces infarct area in a mouse model of transient focal cerebral ischemia and protects cultured neurons from ischemia-like injury. Exp Neurol 2004;189:122–30 [DOI] [PubMed] [Google Scholar]
- 17.Lee SK, Kim DI, Kim SY, et al. Reperfusion cellular injury in an animal model of transient ischemia. AJNR Am J Neuroradiol 2004;25:1342–47 [PMC free article] [PubMed] [Google Scholar]
- 18.Dereski MO, Chopp M, Knight RA, et al. The heterogeneous temporal evolution of focal ischemic neuronal damage in the rat. Acta Neuropathol (Berl) 1993;85:327–33 [DOI] [PubMed] [Google Scholar]
- 19.Jean WC, Spellman SR, Nussbaum ES, et al. Reperfusion injury after focal cerebral ischemia: the role of inflammation and the therapeutic horizon. Neurosurgery 1998;43:1382–97 [DOI] [PubMed] [Google Scholar]
- 20.Kirino T. Delayed neuronal death. Neuropathology 2000;20(suppl):S95–97 [DOI] [PubMed] [Google Scholar]
- 21.Benveniste H, Drejer J, Schousboe A, et al. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem 1984;43:1369–74 [DOI] [PubMed] [Google Scholar]
- 22.White BC, Sullivan JM, DeGracia DJ, et al. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci 2000;179:1–33 [DOI] [PubMed] [Google Scholar]
- 23.Beckman JS. The double-edged role of nitric oxide in brain function and superoxide-mediated injury. J Dev Physiol 1991;15:53–59 [PubMed] [Google Scholar]
- 24.Samdani AF, Dawson TM, Dawson VL. Nitric oxide synthase in models of focal ischemia. Stroke 1997;28:1283–88 [DOI] [PubMed] [Google Scholar]
- 25.Bowes MP, Rothlein R, Fagan SC, et al. Monoclonal antibodies preventing leukocyte activation reduce experimental neurologic injury and enhance efficacy of thrombolytic therapy. Neurology 1995;45:815–19 [DOI] [PubMed] [Google Scholar]
- 26.Okubo S, Tanabe Y, Takeda K, et al. Ischemic preconditioning and morphine attenuate myocardial apoptosis and infarction following ischemia/reperfusion in rabbits: role of delta-opioid receptor. Am J Physiol Heart Circ Physiol 2004. :287:H1786–91. Epub 2004 Jul 1 [DOI] [PubMed] [Google Scholar]
- 27.Gottlieb RA, Burleson KO, Kloner RA, et al. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest 1994;94:1621–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res 1996;79:949–56 [DOI] [PubMed] [Google Scholar]
- 29.Gilad GM, Gilad VH. Accelerated functional recovery and neuroprotection by agmatine after spinal cord ischemia in rats. Neurosci Lett 2000;296:97–100 [DOI] [PubMed] [Google Scholar]
- 30.Olmos G, DeGregorio-Rocasolano N, Paz Regalado M, et al. Protection by imidazol(ine) drugs and agmatine of glutamate-induced neurotoxicity in cultured cerebellar granule cells through blockade of NMDA receptor. Br J Pharmacol 1999;127:1317–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu MY, Piletz JE, Halaris A, et al. Effect of agmatine against cell death induced by NMDA and glutamate in neurons and PC12 cells. Cell Mol Neurobiol 2003;23:865–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Demady DR, Jianmongkol S, Vuletich JL, et al. Agmatine enhances the NADPH oxidase activity of neuronal NO synthase and leads to oxidative inactivation of the enzyme. Mol Pharmacol 2001;59:24–29 [DOI] [PubMed] [Google Scholar]
- 33.Takahashi K, Greenberg JH, Jackson P, et al. Neuroprotective effects of inhibiting poly(ADP-ribose) synthetase on focal cerebral ischemia in rats. J Cereb Blood Flow Metab 1997;17:1137–42 [DOI] [PubMed] [Google Scholar]
- 34.Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, et al. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology 1995;21:1465–68 [DOI] [PubMed] [Google Scholar]
- 35.Kockx MM, Muhring J, Knaapen MW, et al. RNA synthesis and splicing interferes with DNA in situ end labeling techniques used to detect apoptosis. Am J Pathol 1998;152:885–88 [PMC free article] [PubMed] [Google Scholar]
- 36.Qureshi AI, Suri MF, Ostrow PT, et al. Apoptosis as a form of cell death in intracerebral hemorrhage. Neurosurgery 2003;52:1041–48 [PubMed] [Google Scholar]
- 37.Hossmann KA. Ischemia-mediated neuronal injury. Resuscitation 1993;26:225–35 [DOI] [PubMed] [Google Scholar]
- 38.Yu CG, Marcillo AE, Fairbanks CA, et al. Agmatine improves locomotor function and reduces tissue damage following spinal cord injury. Neuroreport 2000;11:3203–07 [DOI] [PubMed] [Google Scholar]
- 39.Feng Y, Piletz JE, Leblanc MH. Agmatine suppresses nitric oxide production and attenuates hypoxic-ischemic brain injury in neonatal rats. Pediatr Res 2002;52:606–11 [DOI] [PubMed] [Google Scholar]
- 40.Gilad GM, Salame K, Rabey JM, et al. Agmatine treatment is neuroprotective in rodent brain injury models. Life Sci 1996;58:PL41–46 [DOI] [PubMed] [Google Scholar]
- 41.Anis NA, Berry SC, Burton NR, et al. The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br J Pharmacol 1983;79:565–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Church J, Zeman S, Lodge D. The neuroprotective action of ketamine and MK-801 after transient cerebral ischemia in rats. Anesthesiology 1988;69:702–09 [DOI] [PubMed] [Google Scholar]
- 43.Lees GJ. Halothane anaesthesia reverses the neuroprotective effect of ketamine against ibotenic acid toxicity in the rat hippocampus. Brain Res 1989;502:280–86 [DOI] [PubMed] [Google Scholar]
- 44.Warner DS, Ludwig PS, Pearlstein R, et al Halothane reduces focal ischemic injury in the rat when brain temperature is controlled. Anesthesiology 1995;82:1237–45, discussion 1227A [DOI] [PubMed] [Google Scholar]