

Abstract
Background
Men with early-onset prostate cancer are at increased risk for cancer-related mortality, yet the prevalence and spectrum of molecular alterations in this patient population is unknown. Here, we analyze comprehensive genomic profiling data to characterize the molecular drivers of early-onset prostate cancer in patients with clinically advanced and metastatic disease.
Methods
Next-generation sequencing was ordered as a part of routine clinical care for 10,189 patients with prostate cancer between 02/2013 and 03/2020 using commercially available comprehensive genomic profiling.
Results
Deidentified genomic data for 10,189 unique patients with prostate cancer were obtained (median age = 66 y, range = 34–90 y). 439 patients were ≤50 y (4.3%), 1928 patients were between ages of 51 and 59 y (18.9%), and 7822 patients were ≥60 y (76.8%). Of metastatic biopsy sites, lymph node, liver, and bone were the most common in all groups, accounting for 60.2% of all specimens. Overall, 97.4% of patients harbored pathologic genomic alterations. The most commonly altered genes were TP53, TMPRSS2–ERG, PTEN, AR, MYC, MLL2, RAD21, BRCA2, APC, SPOP, PIK3CA, RB1, MLL3, CDK12, ATM, and CTNNB1. Patients ≤50 y harbored significantly more TMPRSS2–ERG fusions than patients ≥60 y, while AR copy number alterations as well as SPOP and ASXL1 mutations were significantly less frequent.
Conclusions
Clinically advanced and metastatic early-onset prostate cancer is a distinct clinical subgroup with characteristic genomic alterations including increased frequency of TMPRSS2–ERG fusions and fewer AR, SPOP, and ASXL1 alterations.
Subject terms: Prostate cancer, Cancer genetics
Introduction
Prostate cancer (PC) is the leading cancer diagnosis among men in the United States with an estimated 191,930 new cases expected in 2020 and remains the second most common cause of cancer death for men in the United States, responsible for an estimated 33,330 deaths in 2020 [1]. While patients with local and regional disease are treated with curative intent and carry an excellent 5-year overall survival of nearly 100%, a significant portion of patients suffer from more aggressive disease with poor outcomes despite advances in systemic therapy [2, 3]. These data have led to the realization that PC is a heterogeneous disease that includes both indolent disease in the elderly and aggressive phenotypes such as clinically advanced early-onset PC [4]. The clinical and molecular features of early-onset PC are not well described on a large scale.
Patients with clinically advanced early-onset PC represent an increasing proportion of men diagnosed each year [4]. From 2004 to 2013 the proportion of patients ≤75 y presenting with metastatic PC rose from 2.7 to 4.0%, and during the same period the proportion of patients presenting with intermediate- and high-grade disease rose significantly, from 46.3 to 56.4% (p < 0.01) [5]. Furthermore, for men with early-onset PC, high Gleason grade or locally advanced cancer at diagnosis carries a particularly poor prognosis [6]. These patterns highlight a clinically advanced early-onset PC phenotype of increasing clinical importance.
While the incidence is low, clinically advanced early-onset PC represents a clinical subgroup of PC patients that are challenging to manage. There are limited studies describing this disease phenotype and management relies heavily on data from older patients. Early-onset PC patients have a lower prevalence of cancer risk factors, and their tumors may be enriched for genetic alterations that specifically increase susceptibility to early-onset disease [7]. Understanding the genomic alterations specific to this population offers the potential to better risk-stratify patients and identify those who may benefit from early systemic interventions [8–10]. Improved characterization is needed to define the optimal treatment strategy and identify novel interventional approaches to care for these young cancer patients. To improve our understanding of the biology and our ability to deliver optimally managed care, herein we detail the landscape of molecular alterations that drive clinically advanced early-onset PC.
Methods
Design
We analyzed patient age at time of tissue specimen collection and disease spread together with comprehensive genomic profiling data. Patients were stratified by age and tissue site, and the frequency of molecular alterations including point mutations, copy number alterations, gene fusions, and genomic instability markers were characterized across groups.
Patients data
Approval for this study, including a waiver of informed consent and a HIPAA waiver of authorization, was obtained from the Western Institutional Review Board (Protocol No. 20152817). Deidentified genomics data for 10,189 men with metastatic and clinically advanced PC were obtained from Foundation Medicine, Inc. (Cambridge, MA). Testing was ordered as part of routine clinical care between February 2013 to March 2020. Pathologic classification of tumor specimens sent for testing included the following: prostate acinar adenocarcinoma (n = 9808), prostate undifferentiated carcinoma (n = 226), prostate ductal adenocarcinoma (n = 148), prostate carcinosarcoma (n = 5), and prostate basal cell carcinoma (n = 2).
Sample collection
Both fresh biopsy and archival tissue samples were analyzed. Patient age at specimen collection and the anatomic location of the biopsied tissue was obtained from accompanying sample records. For patients with multiple specimens, the first sample passing sequencing quality metrics was chosen. Pathology review of tissue was performed by board-certified pathologists to confirm tumor content and diagnosis.
Tumor sequencing
Comprehensive genomic profiling was performed by Foundation Medicine, Inc. as previously described [11]. Tumor mutational burden (TMB), microsatellite instability (MSI), and patient ancestry were determined as previously described [12–14]. TMB cutoffs were defined as: low TMB (<6.0 Mutations/Mb), intermediate TMB (6–19 Mutations/Mb), and high TMB (≥20 Mutations/Mb).
Results
Patient characteristics
Tissue samples from 10,189 unique patients were submitted for comprehensive genomic profiling as part of routine clinical care for metastatic and clinically advanced PC between 02/2013 and 03/2020. Archived tissue was allowed with samples collected between 09/1998 and 03/2020.
Median age at the time of biopsy was 66 y (range 34–90 y) (Fig. 1A and eTable 1). The majority of patients were from European ancestry (n = 6435), while African ancestry (n = 983) and other/unknown (n = 2771) were less frequent. In comparison to patients with European ancestry, patients with African ancestry were significantly younger, with a median age of 64 y (range 35–90 y) compared to a median age of 67 y (range 34–90 y) for patients of European ancestry (p < 0.0001, eFig. 1). Samples from patients for genomic profiling included tissue biopsies from both primary prostate (n = 5386, median age = 64 y) and metastatic sites (n = 4092, median age = 69 y) (eTable 2). Locally invasive sites (n = 711) were excluded from primary versus metastatic analyses.
Fig. 1. Clinical characteristics of 10,189 men with prostate cancer.
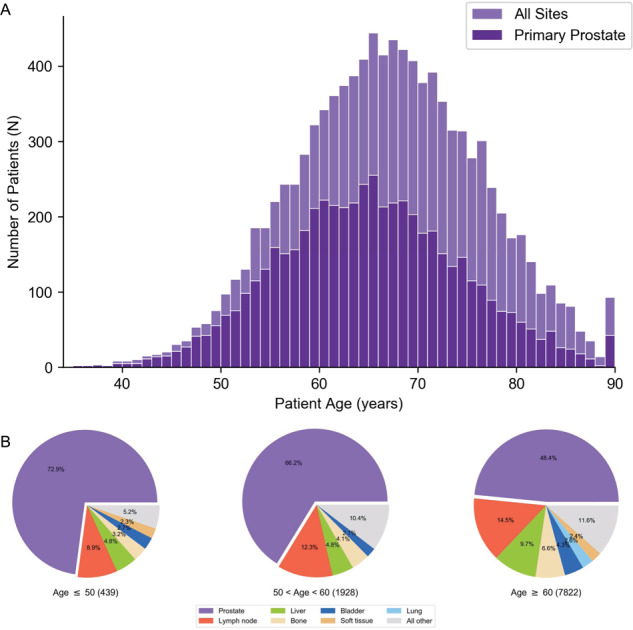
A Histogram of patient age at the time of sample collection for all sites superimposed with only primary prostate biopsies. B Distribution of tumor biopsy sites observed in patients with early- (≤50 y), intermediate- (51–59 y), and typical- (≥60 y) onset prostate cancer. All other includes sites with <2.0% representation.
Patients were stratified into early (≤50 y), intermediate (51–59 y), and typical (≥60 y) age cohorts to compare clinical characteristics and molecular drivers of disease. Overall, 439 patients had early-onset disease with a diagnosis prior to 51 y, 1928 patients were between 51 and 59 y, and 7822 patients were ≥60 y (eTable 1). Genomic profiling for patients with early-onset disease was more likely to be sent from primary prostate tissue than metastatic sites when compared to patients in the intermediate or typical age cohorts 73% vs. 66% vs. 48% respectively (Fig. 1B). Lymph node, liver, and bone were the three most common metastatic sites sent for testing, accounting for 60.2% of all metastatic site specimens, although a broad diversity in the location of metastatic disease sites were reported by the ordering provider (eTable 2).
Genomic alterations in prostate cancer
Overall 97.4% of patients harbored a pathologic mutation with an average of 4.1 mutations per tumor. Frequent genomic alterations were observed in TP53 (40.2%), TMPRSS2–ERG (30.1%), PTEN (29.7%), AR (17.9%), MYC (10.7%), MLL2 (10.5%), RAD21 (10.4%), BRCA2 (8.6%), APC (8.3%), SPOP (7.3%), PIK3CA (6.6%), RB1 (6.4%), MLL3 (5.7%), CDK12 (5.6%), ATM (5.2%), and CTNNB1 (5.2%) (eTable 3). Other pathways of interest with alterations in genes that were less frequent than 5% include: FGF19 (3.2%), FGF3 (3.1%), FGF4 (2.9%), FGFR1 (2.0%); AKT1 (2.3%), PIK3R1 (2.2%), PIK3CB (2.0%), PIK3C2B (1.9%); BRAF (2.1%), and KRAS (1.9%).
By alteration type, the most common short variants were in TP53 (37.8%), MLL2 (10.4%), PTEN (7.8%), APC (7.5%), and SPOP (7.3%) (eTable 4). Copy number alterations occurred most frequently in PTEN (22.0%), AR (13.0%), MYC (10.7%), and RAD21 (10.3%) (eTable 5). Notable gene rearrangements occurred with TMPRSS2 (32.3%) and BRAF (1.3%) (eTable 6). TMPRSS2 rearrangements most commonly occurred with ERG as a fusion partner (93.1%) although rare fusions with ETV1/4/5 were also observed (1.9%, 1.9%, and 0.8%, respectively). Additionally, TMPRSS2 fusions and SPOP alterations showed significant mutual exclusivity; only 20 out of 3065 (0.7%) patients with a TMPRSS2–ERG rearrangement have an SPOP alteration, and 20 out of 743 (2.7%) samples with an SPOP alteration have a TMPRSS2–ERG rearrangement (p < 0.0001).
Age-associated genomic alterations
Median age for patients harboring a given genomic alteration ranged from 63 to 69 y for primary prostate samples (Fig. 2A) and 67–73 y for metastatic samples (Fig. 2B). An increased number of frequently altered genes (>2%) were observed in metastatic disease (47 genes) compared to primary prostate lesions (24 genes). Alterations in gene copy number, single nucleotide variations, and gene fusions were broadly observed across all ages.
Fig. 2. Trends in patient age for patients harboring a known oncogenic alterations.

Genomic alterations with at least 2% frequency are shown for (A) primary and (B) metastatic prostate cancer. The white point indicates median age, the black box represents the interquartile range, and the violin shows age distribution for all patients with the indicated alteration, colored by alteration class. Median age of all patients is indicated with the black line. The total number of patients with each alteration is shown along the x-axis. Gray all patients, Green short variant, Red copy number, Blue fusion.
Overall, alterations increased with age with an average mutation rate of 3.4 mutations/tumor (median 3, range 0–17) in the early-onset cohort compared to 3.5 (median 3, range 0–41) in the intermediate and 4.3 (median 4, range 0–60) in the typical age cohort, a finding partially driven by the increased rate of metastasis in the typical aged cohort (eTable 7). Patients ≥60 y harbored significantly more alterations in AR (FDR = 4.0 × 10−12), SPOP (FDR = 5.2 × 10–4), and ASXL1 (FDR = 0.03) compared to patients ≤50 y (Fig. 3A, eTable 3). Specifically, for short variants, the change in frequency of these alterations was 3.0 vs. 8.2% for SPOP, 1.6 vs. 6.7% for AR, and 1.1 vs. 3.9% for ASXL1 when comparing patients ≤50 y to those ≥60 y (eTable 4). There were no significant differences in the frequency of TP53, PTEN, MYC, MLL2, RAD21, BRCA2, APC, PIK3CA, RB1, and CDK12 alterations between the three age cohorts. While most alterations in individual genes showed a tendency to increase in frequency with age, TMPRSS2 fusions were significantly more common in patients ≤50 y compared to patients ≥60 y, and the alteration rate was inversely associated with age. For patients ≤50 y, 38.7% had TMPRSS2 fusion compared to 34.1% in patients 51–59 y and 31.5% for patients ≥60 y (FDR = 0.03, eTable 6). The observed age-related variation in AR alterations among all samples was primarily driven by differences in alteration frequency observed in metastatic disease as no difference in AR alteration frequency was observed between age cohorts for primary prostate biopsies (Fig. 3B vs. Fig. 3C).
Fig. 3. Frequency of known oncogenic alterations in patients with early- and typical-onset prostate cancer.

Comparison of the frequency of genomic alterations in the indicated gene between early- (≤50 y) and typical- (≥60 y) onset groups for (A) all patients, (B) only primary, and (C) only metastatic cases. Colored markers indicate an FDR-corrected p value < 0.05.
Oncogenic alterations were further assessed by grouping genes into 11 clinically relevant pathways (eTable 8) [15]. Mutation frequency in each pathway tended to increase with age (Fig. 4A), and this trend was observed in both primary prostate biopsies and distant metastatic site biopsies (Fig. 4B, C). As in the individual gene analysis, alterations in TMPRSS2 were significantly more likely to occur in patients with early-onset PC compared to typical aged patients (Fig. 4A). Unlike other pathways, a wide age-related variation in alteration frequency for TMPRSS2 was observed across the entire dataset, as well as in both primary prostate biopsies and metastatic disease site biopsies (Fig. 4B, C). Similar alteration frequencies in TMPRSS2 were observed regardless of biopsy site (primary vs. metastatic). Alterations in androgen receptor axis signaling, SPOP, and mismatch repair proteins were more likely to occur in patients with increased age. In contrast to TMPRSS2, the increase in AR pathway alterations is largely driven by alterations in metastatic disease, and/or exposure to systemic therapies, as no age-related difference was observed in biopsies from primary prostate tumors (Fig. 4B, C).
Fig. 4. Pathway alterations in early-onset prostate cancer.

Frequency of an alteration in select genes or pathways related to prostate cancer for (A) all patients, (B) primary disease, or (C) metastatic disease. FDR-corrected two-sided Fisher’s exact test used in all cases to compare early and typical groups: ns not significant; *p < 0.05; **p < 0.01. Gene pathways are listed in eTable 8.
No significant age-associated differences were observed in the PI3K—(PTEN, PIK3CA, AKT, and mTOR), WNT—(CTNNB1, APC, RNF43, and AXIN1), and MAPK—(RAS, RAF, and MEK) signaling pathways. Additionally, no age-related variation was observed in DNA repair pathway genes including BRCA1/2 as well as other genes with a direct or indirect role in homologous recombination that may confer sensitivity to PARP inhibitor therapy or platinum agents for metastatic PC (Fig. 4A) [16–18].
TMB and MSI
MSI status and TMB were assessable for 9677 cases (95.0%) and 8137 cases (79.9%), respectively. Overall 2.6% of patients with assessable MSI status were found to have MSI-H tumors with a higher rate in metastatic disease (3.3%) compared to primary site (1.8%). Similarly, 3.1% of patients had a TMB high phenotype with a frequency of 3.7% and 2.2% observed in metastatic and primary sites, respectively (eTable 7). Of the 249 patients with high TMB, 191 (77%) were also MSI-H, and conversely, 191 of 249 MSI-H patients (77%) were also TMB high, showing marked, but incomplete overlap of these two instability biomarkers. In localized (Fig. 5A, B) and metastatic (Fig. 5C, D) patients, increasing TMB and MSI status were significantly associated with increasing patient age. Independent of tumor site, the average age of patients with TMB high or MSI-H was significantly greater (eFig. 2), and the frequency of instability biomarkers in the typical-onset age group was significantly higher (eFig. 3), consistent with prior studies that have shown increased genomic instability in later-onset disease [12, 19].
Fig. 5. Age-related variation of MSI and TMB status.

Samples from primary (A, B) and metastatic (C, D) prostate cancer are shown. The white point indicates median age, black box represents the interquartile range, and the violin shows age distribution for all patients of the indicated phenotype. Total number of patients shown along the x-axis. MSS microsatellite stable, MSI-H microsatellite-instable, TMB tumor mutational burden, low: TMB < 6, intermediate: 6 ≤ TMB < 20, high: TMB ≥ 20. Two-sided Student’s t test used to compare indicated groups: ns not significant; *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001.
Discussion
Patients with early-onset, clinically advanced and metastatic PC are challenging to manage due to the paucity of literature characterizing their disease, the requirement for multiple lines of therapy, and the absence of data guiding optimal management. Herein, we reported genomic alterations that drive disease in this clinical subgroup. In our analyses of genomics data collected during routine clinical care, we compared three cohorts by age and identified early-onset PC as a molecular subtype of disease driven more frequently by TMPRSS2 fusions and less commonly by alterations in AR, SPOP, and ASXL1. Secondary findings include an increase in AR alterations with age, a finding partially attributable to metastasis and systemic therapy exposure. MSI and increased TMB were both associated with increasing age.
Strengths of this study include a large, comprehensive dataset encompassing real-world patient genomics. Limitations include restricted access to treatment data, over representation of patients with European ancestry, and a retrospective cohort study design. The set of genes sequenced in this study have known or likely clinical/prognostic significance, however, an unbiased approach to sequence less well-known genes and germline variants underlying familial PC may provide additional insights [20]. The data presented for young patients accurately reflects that which is observed in clinical practice, where despite a low incidence, early-onset disease is frequently encountered as these patients may survive for years and often require multiple lines of systemic therapy. Clinical management of these patients is challenged by a lack of literature describing early-onset PC, with health care providers often relying on data from older populations of PC patients to determine how to manage this disease. While this study is reflective of real-world molecular alterations observed by physicians in the clinic, conclusions regarding alteration frequency before or after treatment require further investigation.
Consistent with clinical practice, specimens in this study would commonly be sent for genomic testing when a patient presents with metastatic disease or when a patient progresses on a line of systemic therapy. Tissue diagnosis prior to initiation of systemic therapy is a cornerstone of oncology practice, and neoadjuvant therapy is not currently recommended in PC. Therefore, prostate biopsies, including samples from radical prostatectomy, were likely collected prior to exposure to systemic therapies including androgen deprivation therapy, and represent hormone sensitive PC. Metastatic sites include both hormone sensitive and castration resistant disease states, and are more likely to be tested after progression on systemic therapy, reflecting alterations that arise with disease evolution. Limited access to patient level clinical data prevented further subclassification of patients. Data to support this include the increased frequency of AR mutations with age and in metastatic samples, a finding known to be associated with exposure to systemic therapies including androgen deprivation therapy [21–23]. The increased TMPRSS2 alteration frequency in younger patients is notable in that it is present regardless of biopsy site, and therefore unlikely attributed to prior systemic therapy exposure or metastasis.
Baseline demographics in this study may not be representative of the national/global diversity of patients with PC, and there is evidence reported in the literature of distinct driver mutations within different ancestries [1, 14, 24]. Molecular differences in PC are reported in men of East Asians and African decent compared to men of European decent, and PC carries an increased risk of morbidity and mortality in African Americans [25]. Together, these studies highlight the need for further investigations into the environmental and social determinants contributing to differences in outcomes by race, including in patients with early-onset PC.
These data bolster findings from a whole genome sequencing study of 11 patients with early-onset PC (median age 47 y) who had frequent TMPRSS2–ERG fusions compared to patients with typical-onset PC (median age 65 y) [26, 27]. The TMPRSS2–ERG fusion is an early event in PC pathogenesis, and there is increasing evidence that TMPRSS2–ERG fusions represent a unique molecular subtype of PC [16, 28, 29]. Consistent with data reported here, the TMPRSS2–ERG fusion is seen in both primary and metastatic PC sites; however, the clinical significance of the rearrangement, and of specific fusion constructs, is less clear [30, 31].
The prognostic significance of TMPRSS2 fusions, and their suitability as therapeutic targets, in advanced PC has been described by multiple studies and reviewed recently [32–36]. The data presented here adds to this body of work with increasing statistical power to define a distinct clinical subgroup of young patients with aggressive disease driven more frequently by TMPRSS2–ERG alterations. It also points to the need for further studies on the prognostic value of TMPRSS2–ERG alterations in this population, the possibility of early PC-related mortality due to early-onset disease, and optimal sequencing of systemic therapies in this distinct molecular subtype.
As evidenced here, significant molecular and clinical heterogeneity exists in PC with respect to age in addition to environmental exposures, family history, and germline predispositions [37, 38]. In an era of precision medicine, stratifying PC patients into specific clinical and molecular subgroups is of increasing importance as it provides insights into disease course and permits tailored clinical management [16]. TMPRSS2–ERG rearrangements have been shown to be mutually exclusive with SPOP mutations, suggesting divergent driver events leading to PC tumorigenesis [39], as we have verified. SPOP mutations, CDK12 loss, and homologous recombination repair pathway alterations are associated with clinical outcomes allow molecularly guided systemic therapies in PC [17, 18, 40–42]. Further clinical and molecular subclassification of PC, including at the gene and pathway level, may aid in identifying additional alterations to guide personalized therapy.
This study demonstrates the genomic heterogeneity of PC across the temporal spectrum of the disease, and describes the genomic landscape in the largest collection of early-onset cases reported. Patients with early-onset PC represent a clinical subgroup with disease driven by an increased frequency of TMPRSS2–ERG fusions and fewer AR, SPOP, and ASXL1 alterations. Prospective controlled trials, specifically focused on early-onset PC as a distinct clinical and molecular entity, are warranted to optimize the clinical management of patients with this disease.
Supplementary information
Funding
This work was supported by grants from the National Cancer Institute: F30CA250248, P50CA180995, and by the Prostate Cancer Foundation: 2017CHAL2044.
Code availability
Scripts used to analyze and present the data in this paper are available upon request (Python 2.7.16).
Compliance with ethical standards
Conflict of interest
EME, GMF, and JSR are employees of Foundation Medicine, Inc. No other potential conflicts of interest were reported by the authors.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Zachary R. Chalmers, Michael C. Burns
These authors jointly supervised this work: Maha H. A. Hussain, Sarki A. Abdulkadir
Contributor Information
Maha H. A. Hussain, Email: maha.hussain@northwestern.edu
Sarki A. Abdulkadir, Email: sarki.abdulkadir@northwestern.edu
Supplementary information
The online version of this article (10.1038/s41391-020-00314-z) contains supplementary material, which is available to authorized users.
References
- 1.Henley SJ, Ward EM, Scott S, Ma J, Anderson RN, Firth AU, et al. Annual report to the nation on the status of cancer, part I: National cancer statistics. Cancer. 2020;126:2225–49. doi: 10.1002/cncr.32802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Etzioni R, Tsodikov A, Mariotto A, Szabo A, Falcon S, Wegelin J, et al. Quantifying the role of PSA screening in the US prostate cancer mortality decline. Cancer Causes Control. 2008;19:175–81. doi: 10.1007/s10552-007-9083-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collin SM, Martin RM, Metcalfe C, Gunnell D, Albertsen PC, Neal D, et al. Prostate-cancer mortality in the USA and UK in 1975–2004: an ecological study. Lancet Oncol. 2008;9:445–52. doi: 10.1016/S1470-2045(08)70104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salinas CA, Tsodikov A, Ishak-Howard M, Cooney KA. Prostate cancer in young men: an important clinical entity. Nat Rev Urol. 2014;11:317–23. doi: 10.1038/nrurol.2014.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu JC, Nguyen P, Mao J, Halpern J, Shoag J, Wright JD, et al. Increase in prostate cancer distant metastases at diagnosis in the United States. JAMA Oncol. 2017;3:705–7. doi: 10.1001/jamaoncol.2016.5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin DW, Porter M, Montgomery B. Treatment and survival outcomes in young men diagnosed with prostate cancer: a population-based cohort study. Cancer. 2009;115:2863–71. doi: 10.1002/cncr.24324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lange EM, Salinas CA, Zuhlke KA, Ray AM, Wang Y, Lu Y, et al. Early onset prostate cancer has a significant genetic component. Prostate. 2012;72:147–56. doi: 10.1002/pros.21414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson D, Van Allen EM, Wu Y-M, Schultz N, Lonigro RJ, Mosquera J-M, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hussain M, Daignault-Newton S, Twardowski PW, Albany C, Stein MN, Kunju LP, et al. Targeting androgen receptor and DNA repair in metastatic castration-resistant prostate cancer: results from NCI 9012. J Clin Oncol. 2017;36:991–9. doi: 10.1200/JCO.2017.75.7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thorstenson A, Garmo H, Adolfsson J, Bratt O. Cancer specific mortality in men diagnosed with prostate cancer before age 50 years: a nationwide population based study. J Urol. 2017;197:61–6. doi: 10.1016/j.juro.2016.06.080. [DOI] [PubMed] [Google Scholar]
- 11.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotech. 2013;31:1023–31. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trabucco SE, Gowen K, Maund SL, Sanford E, Fabrizio DA, Hall MJ, et al. A novel next-generation sequencing approach to detecting microsatellite instability and pan-tumor characterization of 1000 microsatellite instability–high cases in 67,000 patient samples. J Mol Diagn. 2019;21:1053–66. doi: 10.1016/j.jmoldx.2019.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koga Y, Song H, Chalmers ZR, Newberg J, Kim E, Carrot-Zhang J, et al. Genomic profiling of prostate cancers from men with African and European ancestry. Clin Cancer Res. 2020;26:4651–60. doi: 10.1158/1078-0432.CCR-19-4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stopsack KH, Nandakumar S, Wibmer AG, Haywood S, Weg ES, Barnett ES, et al. Oncogenic genomic alterations, clinical phenotypes, and outcomes in metastatic castration-sensitive prostate cancer. Clin Cancer Res. 2020;26:3230–8. doi: 10.1158/1078-0432.CCR-20-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abeshouse A, Ahn J, Akbani R, Ally A, Amin S, Andry CD, et al. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–25. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, et al. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the phase II TRITON2 study. Clin Cancer Res. 2020;26:2487–96. doi: 10.1158/1078-0432.CCR-20-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382:2091–102. doi: 10.1056/NEJMoa1911440. [DOI] [PubMed] [Google Scholar]
- 19.Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598–608. doi: 10.1158/1535-7163.MCT-17-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maertens A, Tran VP, Maertens M, Kleensang A, Luechtefeld TH, Hartung T, et al. Functionally enigmatic genes in cancer: using TCGA data to map the limitations of annotations. Sci Rep. 2020;10:4106. doi: 10.1038/s41598-020-60456-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lallous N, Volik SV, Awrey S, Leblanc E, Tse R, Murillo J, et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. 2016;17:10. doi: 10.1186/s13059-015-0864-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat Commun. 2016;7:13668. doi: 10.1038/ncomms13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chung JH, Dewal N, Sokol E, Mathew P, Whitehead R, Millis SZ, et al. Prospective comprehensive genomic profiling of primary and metastatic prostate tumors. JCO Precis Oncol. 2019;3:1–23. doi: 10.1200/PO.18.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J, Xu C, Lee HJ, Ren S, Zi X, Zhang Z, et al. A genomic and epigenomic atlas of prostate cancer in Asian populations. Nature. 2020;580:93–9. doi: 10.1038/s41586-020-2135-x. [DOI] [PubMed] [Google Scholar]
- 25.Khani F, Mosquera JM, Park K, Blattner M, O’Reilly C, MacDonald TY, et al. Evidence for molecular differences in prostate cancer between African American and Caucasian men. Clin Cancer Res. 2014;20:4925–34. [DOI] [PMC free article] [PubMed]
- 26.Weischenfeldt J, Simon R, Feuerbach L, Schlangen K, Weichenhan D, Minner S, et al. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell. 2013;23:159–70. doi: 10.1016/j.ccr.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 27.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–20. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mosquera J-M, Mehra R, Regan MM, Perner S, Genega EM, Bueti G, et al. Prevalence of TMPRSS2-ERG fusion prostate cancer among men undergoing prostate biopsy in the United States. Clin Cancer Res. 2009;15:4706–11. doi: 10.1158/1078-0432.CCR-08-2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun X-W, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 30.Perner S, Demichelis F, Beroukhim R, Schmidt FH, Mosquera J-M, Setlur S, et al. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006;66:8337–41. doi: 10.1158/0008-5472.CAN-06-1482. [DOI] [PubMed] [Google Scholar]
- 31.Mehra R, Tomlins SA, Yu J, Cao X, Wang L, Menon A, et al. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res. 2008;68:3584–90. doi: 10.1158/0008-5472.CAN-07-6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark JP, Cooper CS. ETS gene fusions in prostate cancer. Nat Rev Urol. 2009;6:429–39. doi: 10.1038/nrurol.2009.127. [DOI] [PubMed] [Google Scholar]
- 33.Rubin MA, Maher CA, Chinnaiyan AM. Common gene rearrangements in prostate cancer. J Clin Oncol. 2011;29:3659–68. doi: 10.1200/JCO.2011.35.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Demichelis F, Rubin MA. TMPRSS2-ETS fusion prostate cancer: biological and clinical implications. J Clin Pathol. 2007;60:1185–6. doi: 10.1136/jcp.2007.046557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.FitzGerald LM, Agalliu I, Johnson K, Miller MA, Kwon EM, Hurtado-Coll A, et al. Association of TMPRSS2-ERG gene fusion with clinical characteristics and outcomes: results from a population-based study of prostate cancer. BMC Cancer. 2008;8:230. doi: 10.1186/1471-2407-8-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, Flohr P, et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene. 2008;27:253–63. doi: 10.1038/sj.onc.1210640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weischenfeldt J, Korbel JO. Genomes of early onset prostate cancer. Curr Opin Urol. 2017;27:481–7. doi: 10.1097/MOU.0000000000000422. [DOI] [PubMed] [Google Scholar]
- 38.Bostwick DG, Burke HB, Djakiew D, Euling S, Ho S-m, Landolph J, et al. Human prostate cancer risk factors. Cancer. 2004;101:2371–490. doi: 10.1002/cncr.20408. [DOI] [PubMed] [Google Scholar]
- 39.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat J-P, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–9. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antonarakis ES, Isaacsson Velho P, Fu W, Wang H, Agarwal N, Santos VS, et al. CDK12-altered prostate cancer: clinical features and therapeutic outcomes to standard systemic therapies, Poly (ADP-ribose) polymerase inhibitors, and PD-1 inhibitors. JCO Precis Oncol. 2020;4:370–81. doi: 10.1200/PO.19.00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swami U, Velho PI, Nussenzveig R, Chipman J, Santos VS, Erickson S, et al. Association of SPOP mutations with outcomes in men with de novo metastatic castration-sensitive prostate cancer. Eur Urol. 2020;78:652–6. [DOI] [PMC free article] [PubMed]
- 42.Boysen G, Rodrigues DN, Rescigno P, Seed G, Dolling D, Riisnaes R, et al. SPOP-mutated/CHD1-deleted lethal prostate cancer and abiraterone sensitivity. Clin Cancer Res. 2018;24:5585–93. doi: 10.1158/1078-0432.CCR-18-0937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Scripts used to analyze and present the data in this paper are available upon request (Python 2.7.16).