

Abstract
BACKGROUND AND PURPOSE: Radiologic markers in multicenter trials are often confounded by different instrumentation used. Our goal was to estimate the variance of the global concentration of the neuronal cell marker N-acetylaspartate (NAA) among research centers using MR imaging scanners of different models, from different manufacturers, and of different magnetic field strength.
MATERIALS AND METHODS: Absolute millimolar amounts of whole-brain NAA (WBNAA) were quantified with nonlocalizing proton MR spectroscopy in the brains of 101 healthy subjects (53 women, 48 men) aged 16–59 years (mean, 34.2 years). Twenty-three were scanned at 1 institute in a 1.5T Siemens Vision; 31 from another institute were studied with a 1.5T Siemens SP63; 36 were scanned at a third institute (24 with a 1.5T Vision, 12 with a 3T Siemens Trio); and 11 were obtained at a fourth institute using a 4T GE Signa 5.x. The NAA amounts were quantified with phantom-replacement and divided by the brain volume, segmented from MR imaging, to yield the concentration, a metric independent of brain size suitable for cross-sectional comparison.
RESULTS: The average WBNAA concentration among institutions was 12.2 ± 1.2 mmol/L. The subjects’ WBNAA distributions did not differ significantly (p > .237) among the 4 centers, regardless of scanner manufacturer, model, or field strength and irrespective of whether adjustments were made for age or sex.
CONCLUSION: Absolute quantification against a standard makes the WBNAA concentration insensitive to the MR hardware used to acquire it. This important attribute renders it a robust surrogate marker for multicenter neurologic trials.
Multicenter phase II clinical trials for neurologic disorders frequently rely on surrogate markers to provide outcome measures sooner and with many fewer patients than clinical assessments.1–4 Such surrogates are often clinical MR imaging metrics, because of their sensitivity to disease-related abnormalities and their changes over time.1, 5 Still, despite its sensitivity, MR imaging is not specific to the nature or true extent of brain tissue damage.1, 6 Therefore, quantitative MR techniques (such as volumetry, magnetization transfer [MT], diffusion-weighted functional imaging, and proton MR spectroscopy [1H-MR spectroscopy]) are sometimes also used.7, 8 Although they could all potentially complement MR imaging,5, 9 their interscanner variability, which is critical to avoid confounds in the data-consolidating stage, has not yet been fully established.10
One of the most prominent candidates for a neurologic surrogate marker is the amino acid derivative N-acetylaspartate (NAA), the second most abundant amino acid in the mammalian brain.11–14 Almost exclusive to neurons and their processes,15–17 its level is considered to reflect their health and attenuation.13 Because NAA can be quantified noninvasively with 1H-MR spectroscopy,12 this “marker + technique” duo is ideally suited to probe the underlying biochemistry of neurologic disorders,13 both as structural abnormalities and as MR-occult pathologies.12
Unfortunately, brain 1H-MR spectroscopy so far employs either small (1–8 cm3, single voxel) or large (≤100 cm3) 2D volumes-of-interest (VOI) placed away from the skull to avoid lipid contamination, missing most of the cortex.18 These limited sizes account for less than 10% of the brain volume, at best.19–21 Because many neurologic disorders are diffuse, missing 90%–99% of the brain implies that the overall extent of neuronal damage is represented by NAA decline in the VOI.22 Interpretation of 1H-MR spectroscopy is further complicated by longitudinal VOI repositioning errors and the use of MR imaging scanners of various makes, model, and field strengths.
Although the repositioning and partial coverage limitations have recently been overcome with nonlocalizing 1H-MR spectroscopy quantification of the whole-brain NAA (WBNAA) concentration, 23–25 its sensitivity to instrumentation change has not yet been established. This issue is examined in the present article by comparing the distribution of WBNAA from several research centers and using various imagers by 2 manufacturers operating at different magnetic field strengths.
Materials and Methods
Human Subjects and MR Scanners
Because healthy persons are presumed, by definition, to be as similar as biologically possible, separate cohorts can be conveniently studied at different institutions, as opposed to transporting (the same) patient group around. Therefore, 101 healthy subjects (53 women, 48 men) aged 16–59 years (mean, 34.2 years) were recruited as follows: 23 subjects aged 31.0 ± 1.5 years (mean ± SD) were studied at the first institution using a 1.5T Siemens Vision (Siemens, Erlangen Germany). Thirty-one subjects aged 33.0 ± 9.5 years were studied at the second institution using a 1.5T Siemens SP63. Thirty-six subjects aged 34.0 ± 8.9 years were studied at the third institution, as follows: 24 in a 1.5T Vision scanner and 12 in a 3T Siemens Trio. Finally, 11 subjects aged 43 ± 15 years were imaged at the fourth institution in a 4T Signa 5.x (GE Medical Systems, Milwaukee, Wis). All gave written informed consent, and the study was approved by the respective Institutional Review Boards.
MR Imaging—Brain volume, VB, Segmentation
Each subject’s brain parenchymal volume, VB, was obtained from high-resolution T1-weighted sagittal magnetization-prepared rapid gradient-echo (TE/TR/TI, 7.0/14.7/300 ms; 128 1.5-mm sections; matrix, 256 × 256; FOV, 210 × 210-mm2). The images were segmented into tissue and CSF, as described previously,26–28 with only the parenchyma accounted for in VB.
MR Spectroscopy—WBNAA quantification
The amount of WBNAA, QNAA, was measured in each institution by using their imager in its standard neuro-MR configuration with a transmit-receive head coil. In each case, shimming yielded consistent 12 ± 3, 20 ± 5, and 26 ± 7 Hz whole-head water linewidths at 1.5-, 3-, and 4T, respectively, followed by the same nonlocalizing TE/TI/TR = 0/970/10,000 ms WBNAA 1H-MR spectroscopy sequence.23 The sequence nulls the NAA signal intensity every second shot with inversion-recovery, whereas the lipids’ signals, with their short T1, ∼220 ms, are always thermal (at either the TR of 10 seconds, or the TI of 970 ms). Therefore, an add-subtract scheme destructively interferes with their signal intensity, 23 as shown in Fig 1. Absolute quantification was done against a reference 3-L sphere of 1.5 × 10−2 mol of NAA in water. Subject and reference NAA peaks, SS and SR, were integrated from the resultant spectrum, as shown in Fig 1, and QNAA was obtained as23, 29:
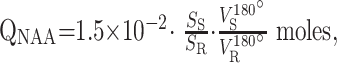 |
1) |
where VR180° and VS180° are the transmitter voltages into 50 Ω for nonselective 1-ms 180° inversion pulses on the reference and subject, respectively, reflecting relative coil loading. Note that slight changes were made to the sequence, increase of the chemical shift selection water suppression bandwidth, and decrease in the 13̅31̅ interpulse delays,23 proportional to the magnetic field strength.
Fig 1.
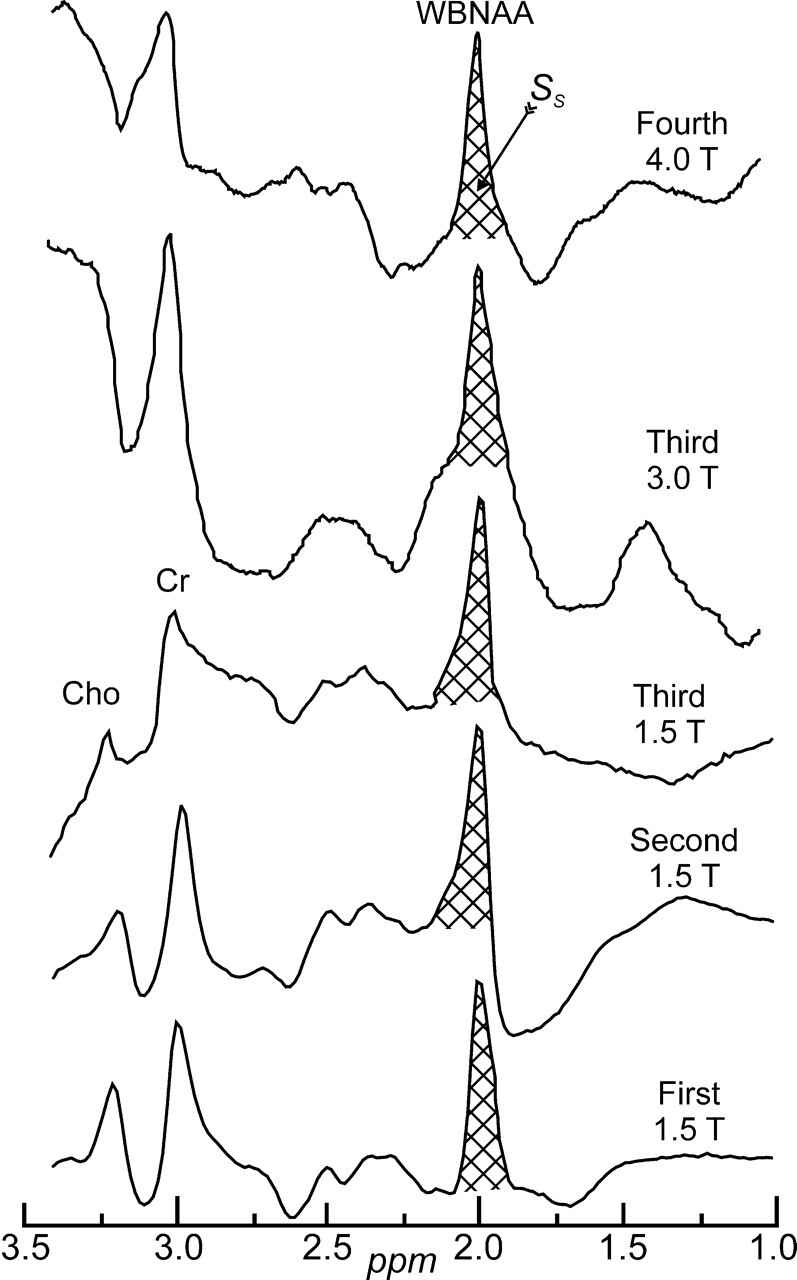
Sample whole-head 1H-MR spectroscopy spectra from each of the MR imaging instruments used, with institution and field strength indicated. Although all the proton metabolites can be seen in the spectrum, note that because it was acquired in a whole-head, nonlocalized fashion, only the NAA peak is implicitly localized to the brain. The area of the NAA peak of the subject SS, was integrated in each subject as shown and converted into absolute millimoles, QNAA, by comparing with the NAA peak area, SR, in a reference phantom of known concentration as described by Eq. 1. Note the excellent signal-to-noise ratio of this short acquisition and the nearly total elimination of the skull’s lipid signals from obscuring the spectrum.
To account for natural variations in human head sizes, each subject’s QNAA was divided by their VB to yield the concentration
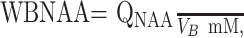 |
2) |
which is independent of brain size and therefore suitable for cross-sectional comparison. The intrasubject and intersubject variability of this metric was shown previously to be better than ±7%.18, 23
Statistics
Analysis of variance (ANOVA) was used to compare sites and field strengths with respect to WBNAA. In particular, the Tukey honestly significant difference (HSD) procedure (to correct for multiple comparisons) was used in the ANOVA framework to make all pairwise comparisons among sites and field strengths with respect to WBNAA while maintaining the experiment-wise type I error rate for the set of comparisons at or below the 5% level. The ANOVA used WBNAA as the dependent variable and the model included subject age as a numeric factor and subject sex and either site or field strength as classification factors.
Results
The distribution of WBNAA values from each of the 5 scanners at the 4 centers are shown in Fig 2. There were no significant differences between them in terms of either the raw, unadjusted SDs or the SDs after adjustment for variation attributable to age or sex (p > .237), as compiled in the Table. Furthermore, there were no significant differences between the mean WBNAA concentrations among the different instruments or research centers. Therefore, the invariant WBNAA concentration of healthy subjects is 12.2 ± 1.2 mmol/L.
Fig 2.

Box plot showing the 25%, 50% (median), and 75% quartiles (box) and ±95% (whiskers), of the distributions of the subjects’ WBNAA concentrations in each of the 4 institutions, 5 scanners, 3 field strengths, and 2 manufacturers used in this study. Note that the differences among the distributions (means and SDs) are statistically insignificant, independent of the (healthy) subjects’ age or sex, indicating that the methodology is robust to instrumentation differences.
Estimated mean ± SD NAA concentrations at each institution.
| Institution | Subjects | Scanner Field, Manufacturer, Model | Mean WBNAA (mmol/L) | SD (Unadjusted) (mmol/L) | SD (adjusted) |
|---|---|---|---|---|---|
| First | n = 23 | 1.5 T, Siemens, Vision | 11.7 | 1.3 | 1.3 |
| Second | n = 31 | 1.5 T, Siemens, SP63 | 12.3 | 1.1 | 1.1 |
| Third | n = 24 | 1.5 T, Siemens, Vision | 12.6 | 1.2 | 1.2 |
| n = 12 | 3.0 T, Siemens, Trio | 11.7 | 1.1 | 1.1 | |
| Fourth | n = 11 | 4.0 T, GE, Signa 5.x | 12.8 | 1.5 | 1.5 |
| Total | n = 101 | 12.2* | 1.2* | 1.2 |
Note:—WBNAA indicates whole-brain N-acetylaspartate. The SDs are presented as both unadjusted and adjusted with respect to confounding effects from age and gender (differences were in second decimal digit or smaller).
Average of all subjects at all institutions.
Tukey HSD indicated that there were no significant differences (p > .27) between any pair of sites with respect to mean level of WBNAA. Furthermore, the same test has shown no significant difference between any 2 field strengths (HSD adjusted p > .12).
Discussion
Because of their exquisite sensitivity to brain abnormalities, MR-derived metrics are increasingly used as surrogates for efficacy in clinical trials of treatments for neurologic disorders.1, 2, 5, 7–9, 30 The problem, however, is that current clinical MR metrics do not fulfill some of the prerequisites needed to define a valid paraclinical surrogate.3, 4 Key among these are that it be 1) simple to implement, 2) reproducible, 3) clinically meaningful, 4) pathologically specific, 5) sensitive to disease changes over time, and 6) of reasonably low labor intensity. The goals of this study were to determine the influence of center and hardware on WBNAA, an issue critical to the consideration of this technique for inclusion in multicenter clinical trials.
Prerequisite 1), above, is met by the WBNAA technique, as demonstrated by implementation in this study, across the multiple centers and scanners of different vintage, manufacturer, and magnetic field strength. These implementations also reflect, in a more subtle way, that the conjugate requirement 6), that of low labor intensity, is also met; ie, WBNAA acquisition and postprocessing are no more time-consuming than other quantitative MR imaging metrics. These 2 are “baseline” conditions for any technique to be considered for use in any multicenter trial.
Reproducibility, requirement 2) above, is also satisfied, as shown by Fig 2, as well as the statistical analyses in the Results, summarized in Table 1. Specifically, neither the 12.2 mmol/L average mean of the WBNAA distributions, nor its ± 1.2 mmol/L average SD, differs among imagers or centers. In other words, the variability of WBNAA across multiple imagers, ±10%, is similar to that of any single instrument, assessed previously at ±6%–7% in healthy control subjects.18, 23 This finding is of particular importance if WBNAA is to be considered an adjunctive treatment efficacy measure for 2 main reasons: first, the scanners used represent many clinical research centers worldwide; second, most trials have treatment and control arms. Therefore, knowledge of the intrainstrument and interinstrument variability in the control subjects will assist in determining the subject population required to detect a given expected treatment effect in the patients.
Previous studies have shown that the scan-rescan variability is a serious issue for quantities derived from conventional31, 32 and MT MR imaging.33 This has led to the formulation of rules standardizing acquisition across multiple centers, which suggested that upgrades or changes to scanners be limited during trials.9, 31 This is a difficult rule to follow considering 1) the 2–3-year course of most clinical neurology trials versus the 5-year life span of modern MR scanners and 2) the manufacturers’ annual software upgrade cycles, some mandatory for regulatory compliance and others to keep the system current for its prime mission of clinical diagnostic MR imaging. The use of a calibration reference for the WBNAA measurement shows its potential to preempt both concerns.
The WBNAA method, however, is not without its limitations that need to be considered as part of a study design. Its main weakness is the total loss of localization. As a result, it is insensitive to changes smaller than 5%–6% (ie, oblivious to focal disorders [eg, stroke] and unable to discern between several foci of great loss and diffuse, overall smaller NAA decline). It also misses the NAA signal intensity from regions that suffer extreme susceptibility-driven field inhomogeneities (eg, the inferior frontal lobes, temporal lobes, and around the auditory canals). This loss, however, has been quantified previously at less than 10%.25
In conclusion, this study indicates that the use of different MR scanners should not be considered a source of confounding variability for use of WBNAA in multicenter studies of neurologic disorders. Using it, therefore, allows the study designers to focus on the much more relevant criterion of patient accrual capability rather than hardware compatibility. The premise that this approach allows an overall assessment of neuroaxonal viability makes WBNAA an attractive surrogate marker for large-scale studies of treatment outcomes, especially for conditions characterized by diffuse or widespread brain damage, (eg, multiple sclerosis, AIDS, traumatic brain injury, Alzheimer disease, normal aging).
Footnotes
This work was supported by National Institutes of Health grants EB01015, NS0050520, and NS39135.
References
- 1.Miller DH, Grossman RI, Reingold SC, et al. The role of magnetic resonance techniques in understanding and managing multiple sclerosis. Brain 1998;121:3–24 [DOI] [PubMed] [Google Scholar]
- 2.Arnold DL, Matthews PM. MRI in the diagnosis and management of multiple sclerosis. Neurology 2002;58:S23–31 [DOI] [PubMed] [Google Scholar]
- 3.Barkhof F, Held U, Simon JH, et al. Predicting gadolinium enhancement status in MS patients eligible for randomized clinical trials. Neurology 2005;65:1447–54 [DOI] [PubMed] [Google Scholar]
- 4.Sormani MP, Bruzzi P, Comi G, et al. The distribution of the magnetic resonance imaging response to glatiramer acetate in multiple sclerosis. Mult Scler 2005;11:447–49 [DOI] [PubMed] [Google Scholar]
- 5.Sormani MP, Bruzzi P, Comi G, et al. MRI metrics as surrogate markers for clinical relapse rate in relapsing-remitting MS patients. Neurology 2002;58:417–21 [DOI] [PubMed] [Google Scholar]
- 6.Filippi M, Horsfield MA, Tofts PS, et al. Quantitative assessment of MRI lesion load in monitoring the evolution of multiple sclerosis. Brain 1995;118:1601–12 [DOI] [PubMed] [Google Scholar]
- 7.Rovaris M, Filippi M. Magnetic resonance techniques to monitor disease evolution and treatment trial outcomes in multiple sclerosis. Curr Opin Neurol 1999;12:337–44 [DOI] [PubMed] [Google Scholar]
- 8.Filippi M. In-vivo tissue characterization of multiple sclerosis and other white matter diseases using magnetic resonance based techniques. J Neurol 2001;248:1019–29 [DOI] [PubMed] [Google Scholar]
- 9.Filippi M, Dousset V, McFarland HF, et al. Role of magnetic resonance imaging in the diagnosis and monitoring of multiple sclerosis: consensus report of the White Matter Study Group. J Magn Reson Imaging 2002;15:499–504 [DOI] [PubMed] [Google Scholar]
- 10.Sormani MP, Iannucci G, Rocca MA, et al. Reproducibility of magnetization transfer ratio histogram-derived measures of the brain in healthy volunteers. AJNR Am J Neuroradiol 2000;21:133–36 [PMC free article] [PubMed] [Google Scholar]
- 11.Birken DL, Oldendorf WH. N-acetyl-L-aspartic acid: a literature review of a compound prominent in 1H-NMR spectroscopic studies of brain. Neurosci Biobehav Rev 1989;13:23–31 [DOI] [PubMed] [Google Scholar]
- 12.Danielsen EA, Ross B. Magnetic Resonance Spectroscopy Diagnosis of Neurological Diseases. New York: Marcel Dekker, Inc;1999. .
- 13.Arnold DL, de Stefano N, Matthews PM, et al. N-Acetylaspartate: usefulness as an indicator of viable neuronal tissue Ann Neurol 2001;50:823; discussion 824–25 [DOI] [PubMed] [Google Scholar]
- 14.Baslow MH. Functions of N-acetyl-L-aspartate and N-acetyl-l-aspartylglutamate in the vertebrate brain: role in glial cell-specific signaling. J Neurochem 2000;75:453–59 [DOI] [PubMed] [Google Scholar]
- 15.Simmons ML, Frondoza CG, Coyle JT. Immunocytochemical localization of N-acetyl-aspartate with monoclonal antibodies Neuroscience 1991;45:37–45 [DOI] [PubMed] [Google Scholar]
- 16.Moffett JR, Namboodiri MA, Cangro CB, et al. Immunohistochemical localization of N-acetylaspartate in rat brain. Neuroreport 1991;2:131–34 [DOI] [PubMed] [Google Scholar]
- 17.Jenkins BG, Klivenyi P, Kustermann E, et al. Nonlinear decrease over time in N-acetyl aspartate levels in the absence of neuronal loss and increases in glutamine and glucose in transgenic Huntington’s disease mice. J Neurochem 2000;74:2108–19 [DOI] [PubMed] [Google Scholar]
- 18.Inglese M, Ge Y, Filippi M, et al. Indirect evidence for early widespread gray matter involvement in relapsing-remitting multiple sclerosis. Neuroimage 2004;21:1825–29 [DOI] [PubMed] [Google Scholar]
- 19.Brooks WM, Friedman SD, Stidley CA. Reproducibility of 1H-MRS in vivo. Magn Reson Med 1999;41:193–97 [DOI] [PubMed] [Google Scholar]
- 20.Marshall I, Wardlaw J, Cannon J, et al. Reproducibility of metabolite peak areas in 1H MRS of brain. Magn Reson Imaging 1996;14:281–92 [DOI] [PubMed] [Google Scholar]
- 21.Bokde AL, Teipel SJ, Zebuhr Y, et al. A new rapid landmark-based regional MRI segmentation method of the brain. J Neurol Sci 2002;194:35–40 [DOI] [PubMed] [Google Scholar]
- 22.De Stefano N, Narayanan S, Francis SJ, et al. Diffuse axonal and tissue injury in patients with multiple sclerosis with low cerebral lesion load and no disability. Arch Neurol 2002;59:1565–71 [DOI] [PubMed] [Google Scholar]
- 23.Gonen O, Viswanathan AK, Catalaa I, et al. Total brain N-acetylaspartate concentration in normal, age-grouped females: quantitation with non-echo proton NMR spectroscopy. Magn Reson Med 1998;40:684–89 [DOI] [PubMed] [Google Scholar]
- 24.Gonen O, Catalaa I, Babb JS, et al. Total brain N-acetylaspartate: a new measure of disease load in MS. Neurology 2000;54:15–19 [DOI] [PubMed] [Google Scholar]
- 25.Gonen O, Grossman RI. The accuracy of whole brain N-acetylaspartate quantification. Magn Reson Imaging 2000;18:1255–58 [DOI] [PubMed] [Google Scholar]
- 26.Udupa JK, Odhner D, Samarasekera S. 3DVIEWNIX: an open, transportable, multdimensional, multimodality, multiparametric imging software system. Proc SPIE 1994;2164:58–73 [Google Scholar]
- 27.De Santi S, de Leon MJ, Rusinek H, et al. Hippocampal formation glucose metabolism and volume losses in MCI and AD Neurobiol Aging 2001;22:529–39 [DOI] [PubMed] [Google Scholar]
- 28.Smith SM, De Stefano N, Jenkinson M, et al. Normalized accurate measurement of longitudinal brain change. J Comput Assist Tomogr 2001;25:466–75 [DOI] [PubMed] [Google Scholar]
- 29.Soher BJ, van Zijl PC, Duyn JH, et al. Quantitative proton MR spectroscopic imaging of the human brain Magn Reson Med 1996;35:356–63 [DOI] [PubMed] [Google Scholar]
- 30.Filippi M, Horsfield MA, Ader HJ, et al. Guidelines for using quantitative measures of brain magnetic resonance imaging abnormalities in monitoring the treatment of multiple sclerosis. Ann Neurol 1998;43:499–506 [DOI] [PubMed] [Google Scholar]
- 31.Filippi M, van Waesberghe JH, Horsfield MA, et al. Interscanner variation in brain MRI lesion load measurements in MS: implications for clinical trials. Neurology 1997;49:371–77 [DOI] [PubMed] [Google Scholar]
- 32.Molyneux PD, Brex PA, Fogg C, et al. The precision of T1 hypointense lesion volume quantification in multiple sclerosis treatment trials: a multicenter study. Mult Scler 2000;6:237–40 [DOI] [PubMed] [Google Scholar]
- 33.Filippi M, Inglese M, Rovaris M, et al. Magnetization transfer imaging to monitor the evolution of MS: a 1-year follow-up study. Neurology 2000;55:940–4 [DOI] [PubMed] [Google Scholar]