

Abstract
SUMMARY: Proton MR spectroscopy (1H-MR spectroscopy) is a quantitative MR imaging technique often used to complement the sensitivity of conventional MR imaging with specific metabolic information. A key metabolite is the amino acid derivative N-acetylaspartate (NAA), which is almost exclusive to neurons and their processes and is, therefore, an accepted marker of their health and attenuation. Unfortunately, most 1H-MR spectroscopy studies only account for small 1- to 200-cm volumes of interest (VOI), representing less than 20% of the total brain volume. These VOIs have at least 5 additional restrictions: 1) To avoid contamination from subcutaneous and bone marrow lipids, they must be placed away from the skull, thereby missing most of the cortex. 2) They must be image-guided onto MR imaging–visible pathology, subjecting them to the implicit assumption that metabolic changes occur only there. 3) They encounter misregistration errors in serial studies. 4) The time needed to accumulate sufficient signal-intensity quality is often restrictive, and 5) they incur (unknown) T1- and T2-weighting. All these issues are avoided (at the cost of specific localization) by measuring the nonlocalized average NAA concentration over the entire brain. Indeed, whole-brain NAA quantification has been applied to several diffuse neurodegenerative diseases (where specific localization is less important than the total load of the pathology), and the results are presented in this review.
Although conventional MR imaging is exquisitely sensitive to morphologic soft-tissue changes, it is limited in its pathologic specificity and sensitivity to microscopic injury in normal-appearing brain tissue.1-3 Consequently, proton MR spectroscopy (1H-MR spectroscopy) is often used to provide specific biochemical information via the levels of several detectable neurometabolites and mobile lipids.4,5 Of these, the amino acid derivative N-acetylaspartate (NAA), first described by Tallan et al in 1956,6 is almost exclusive to neurons and their processes (though trace amounts are also found in liver, kidney, muscle, and oligodendrocyte progenitor cells). Consequently, NAA is considered a marker for neuronal health and attenuation.7-9 Indeed, with the exception of Canavan disease,10 its concentration has been reported to decline in all central nervous system (CNS) disorders.11,12 In addition to MR spectroscopy evidence, NAA loss has also been described in transected rat optic nerve axons13 and postmortem biopsies of multiple sclerosis (MS) lesions.14
NAA is synthesized in the brain through the acetylization by acetyl coenzyme A of free aspartate by the enzyme L-aspartate N-acetyltransferase, as shown in Fig 1,15 and catabolized by the enzyme aspartoacylase. Yet, despite more than 50 years of research, its role in the brain remains controversial, and hypotheses include (but are not limited to) the following: 1) an osmolite to remove water from neurons,16 2) an acetate contributor in myelin sheath synthesis,17 3) a mitochondrial energy source,18 4) a precursor for N-acetylaspartyl glutamate, and 5) a ligand for certain metabotropic glutamate receptors.19
Fig 1.

Schematic description of the structures and acetylization of aspartate into NAA in the mitochondria (top). The 3 protons of the methyl group of NAA provide the most prominent peak on the neurometabolite proton spectrum (bottom).
Because it is the second most abundant amino acid in the human CNS after glutamate (NAA comprises up to 0.1% wet weight of the brain 11,20-23), its singlet peak at 2.02 ppm, shown in Fig 1, is the most intense in 1H-MR spectroscopy of a healthy brain. Unfortunately, most of the many 1H-MR spectroscopy studies to date used either small (1–8 cm) single voxels or larger (up to ∼200 cm) 2D volumes of interest (VOIs), as shown in Fig 2.24-26 These VOIs must be placed away from the skull to avoid contamination from subcutaneous lipid and bone marrow signals, thereby missing most of the cortex.27 Due to their size, such VOIs also must be image-guided to MR imaging–visible pathologies, subjecting them to the implicit assumption that metabolic abnormalities occur only there. Furthermore, because many CNS disorders are diffuse, missing 80%–99% of the brain renders estimates of the true extent of their load susceptible to extrapolation errors.28
Fig 2.

Top: Schematic comparison of relative VOI size and coverage between single-voxel, 2D or 3D multivoxel, and WBNAA in the human brain. Note that the single- and multivoxel VOIs cover only small fractions of the brain volume and must be kept away from the skull, missing most of the cortex, whereas WBNAA accounts for the entire brain. Bottom: The consequence of a 1-mm (1 guiding-image pixel) placemat error in each direction, x, y, and z, on a 1-cm voxel in a serial study. This 10% error leads to only 70% of the VOI common to both measurements. Note that though this error is technically unavoidable in localized spectroscopy, it is not an issue for WBNAA, as shown in the top portion of the lower panel.
Localized 1H-MR spectroscopy is also confounded by 3 additional factors: First, it is susceptible to VOI repositioning errors in serial studies. For example, in a 1.5-cm VOI, common in single-voxel experiments, misregistration of just 1 mm (1 guiding MR imaging pixel) in each plane leads to <80% common volume (worse for smaller voxels) as shown in Fig 2. Second, it requires long acquisitions, ∼7 minutes for 3- to 8-cm and 20–30 minutes for 1-cm voxels to achieve sufficient signal intensity-to-noise ratio (SNR). Finally, an assumption must be made that the relaxation times, T1 and T2, are either known or the same in patients and controls in all regions studied.29
These concerns can be addressed by a nonecho, not T1-weighted, nonlocalizing 1H-MR spectroscopy sequence acquiring the 1H-MR spectroscopy signal intensity from the entire head, as shown in Fig 2.30 Because NAA is restricted to neuronal cells, its signal intensity is implicitly localized to the brain, and the lack of explicit localization removes issues of VOI guidance, serial misregistration, or SNR. These factors make the technique attractive to quantify global neuroaxonal injury and to monitor neuroprotection, either in response to the treatments in clinical routine or in response to experimental treatments in clinical trials of neurologic and neuropsychiatric diseases. This article reviews the method, its implementation, and the findings from several diffuse neurologic disorders.
The Method
The Whole-Brain NAA 1H-MR Spectroscopy Sequence
The amount of whole-brain NAA (WBNAA), QNAA, can be obtained by using the nonlocalizing 1H-MR spectroscopy sequence (TE/TI/TR = 0/940/10,000 ms) shown in Fig 3A.30 Unlike the previous lipid-nulling inversion-recovery approaches used in 1H-MR spectroscopy near the skull, this sequence relies on the much shorter, T1 ≈ 220 ms, of the lipids compared with the T1 ≈ 1.4 seconds of the NAA to null the latter every second acquisition.30 The long ln2 × T1 nulling delay of the NAA, which is greater than 4 × T1 of the lipids, ensures that the latter lipids are at thermal equilibrium in each acquisition, as shown in Fig 3B, whereas the NAA is thermal only in odd and null in every even acquisition.
Fig 3.

Top: A, Schematic representation of the 2-step WBNAA sequence. It comprises an alternating inversion pulse (applied every odd acquisition) followed by a TI designed to null the NAA signal. It ends with a chemical-shift-selective (CHESS) and a 13̄31̄ water-suppression pulse (4-ms interpulse delay at 1.5T). The latter also serves as the 90° readout pulse. Acquisition commences immediately (TE = 0), and every even acquisition is subtracted from every odd one. Because the TR is long, 10 seconds, no T1- or T2-weighting is incurred. Note that all the radio-frequency (RF) pulses are (spatially) nonselective and the gradient “blips” are just crushers. Center: B, The result of a 90° 13̄31̄ on a human head, demonstrating the problem of the immense metabolite-obscuring lipid signal from the bone marrow and adipose tissue. Bottom: C, The resulting spectrum from the WBNAA sequence on the same head as in B, demonstrating almost complete lipid suppression. Note that the NAA is implicitly localized to the brain, whereas with the other metabolites whose peaks are distinct and nonlocalized, it is impossible to ascertain where their signals come from. Also note the excellent SNR from this 2.5-minute acquisition.
Subtracting even from odd acquisitions, therefore, leads to destructive interference of the lipids but not the signals of the long T1 metabolites (Fig 3C). Because NAA is the only one of these exclusive to neurons, it is implicitly localized to the brain. All the others (eg, creatine, choline, myo-inositol, glutamate, and so forth) are in all tissues, and their brain contribution cannot be estimated.
Quantification is against a reference 3-L sphere of 1.5 × 10−2 mol NAA in water. Subject and phantom NAA peaks, SS and SR, are integrated, as shown in Figs 1 and 3C, and QNAA obtained as,
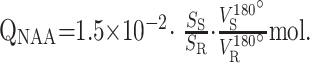 |
1) |
VR180° and VS180° are the transmitter voltages for nonselective 1-ms 180° inversion pulses on the reference and subject, reflecting relative coil loading and, by reciprocity sensitivity.
Normalization of WBNAA concentration
To normalize for differences in head size among subjects, we divided QNAA by total brain parenchyma volume, VB, to yield the WBNAA concentration.
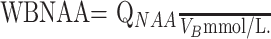 |
2) |
This is a specific brain size independent metric, and its intersubject variability in controls was shown to be better than ±6%.30 VB can be obtained with any validated automated or semiautomated brain-segmentation software, and several different ones have been used successfully (eg, 3DVIEWNIX,31 MIDAS,32 SPM,33 SIENA,34,35 and SIENAX.36).
Accuracy
Due to severe magnetic field inhomogeneities from air-tissue susceptibility differences at such interfaces (eg, at the air-filled sinuses and near the auditory canals), some of the NAA signal intensity may have been broadened or shifted outside the integration window shown in Fig 3C. The extent of the signal intensity (lost) outside this integration window was estimated by using high-spatial-resolution chemical-shift imaging of the water of the brain. Due to the high SNR of water, it is easy to measure how much of their signal intensity lies outside this interval. The measurement has shown that the brain water losses from susceptibility effects are <10%, suggesting that by analogy, the integrated SS in Fig 3C captures more than 90% of the NAA signal intensity of the brain.37
Intra- and Interindividual Precision
The individual variations reported in the first WBNAA study were small, ±5% intraindividual and 6% interindividual in a group of 5 healthy women.30 The interindividual variations in much larger cohorts of several score of healthy subjects, each, reported since then, have been consistent with coefficients of variations in the 6%–8% range.27,38,39
Multisite, Instrument, and Field Precision
In multicenter trials, the different instrumentation that each site may use could potentially confound the results at the final data-consolidation stage. To test the effectiveness of the absolute quantification strategy used by equation 1, we compared the WBNAA concentrations in healthy subjects across 6 MR imaging instruments from 2 manufacturers (GE Healthcare, Milwaukee, Wis; Siemens, Erlangen, Germany) operating at 3 different magnetic field strengths (1.5, 3, and 4T) at 5 different sites. Overall more than 120 subjects were enrolled, and the results are summarized in Fig 4.39
Fig 4.

Box plot showing the 25%, 50% (median), and 75% quartiles (box) and ±95% (whiskers) of the distributions of the subjects’ WBNAA concentrations in each of the 5 institutions, 6 scanners, 3 field strengths, and 2 manufacturers used in this study. Note that the differences among the distributions, 12.2 ± 1.2 mmol/L (means and SDs), for more than 120 healthy individuals are statistically insignificant, independent of these subjects’ ages or sex, indicating that the methodology is robust to instrumentation differences. HSR indicates Hospital San Rafael (Italy); FCCC, Fox Chase Cancer Center (USA); NYU, New York University (USA); Basel, Basel (Switzerland); U.PENN, University of Pennsylvania (USA).
These results demonstrate that the WBNAA distribution is statistically indistinguishable (P > .237) regardless of site, manufacturer, field strength, or scanner model.39 They indicate that the absolute global WBNAA concentration is 12.2 ± 1.2 mmol/L. There were no age or sex WBNAA differences found in this cohort, comprising approximately an equal number of men and women, ranging in age from late teens to late 50s. This indicates that absolute quantification against a phantom in each instrument combined with the simplicity of the sequence render the technique hardware-insensitive and, consequently, a robust marker in multicenter trials.39
Longitudinal WBNAA in Healthy Adults
Longitudinal WBNAA levels in healthy adults have been reported during a several-day period30 and, more recently, annually in 14 subjects during periods of 2–3 years, common in most treatment clinical trials. No significant change (adjusted for both sex and age) was found either inter- or intrapersonally during the entire duration of the study in the 25- to 50-years-of-age group studied.40 Little data, however, have been published chronicling the levels or temporal changes in elderly controls as a result of the normal aging processes.41 Although the normal rate of change in parenchymal volume has been reported to be approximately 0.25% per year during the life span of most healthy humans,42 WBNAA is hypothesized to remain relatively stable even in later years. Because it is normalized to the brain volume (equation 2), it should be relatively insensitive to this normal atrophy, reflecting only the health of neurons in the remaining tissue.
WBNAA in MS
MS is the most common autoimmune, demyelinating, neurodegenerative disease in young adults. It strikes nearly 2.5 million people worldwide at a cost of approximately $23 billion for treatment and management in the United States alone (http://www.nationalmssociety.org). It is of unknown etiology, though 2 of every 3 new patients are women in their late 20s. Its prevalence among men or of all people of African or Asian descent is less than half that of women.43
Most (85%–90%) new patients with MS follow a relapsing remitting (RR) course characterized by relapses of variable severity followed by remissions of varying duration.43 Nearly 20% will remain clinically stable for at least 2 decades, a phenotype referred to as benign MS.44 Within 25 years, however, most untreated patients with RRMS will evolve into a secondary-progressive (SP) phase of a steady increase in chronic disability. Some 10%–15% of new patients with MS follow a primary-progressive (PP) course, characterized by late onset and steady increase in disability.45
Although still without cure, 6 drugs are currently approved by the US Food and Drug Administration as disease-modifying agents that alter the natural history of MS. These include the following: intramuscular beta-interferon-la (Avonex), subcutaneous beta-interferon-1a (Rebif), subcutaneous beta-interferon-1b (Betaseron), glatiramer acetate (Copaxone), and natalizumab (Tysabri), a laboratory-produced monoclonal antibody given by intravenous infusion. For SPMS, the most convincing data favors mitoxantrone (Novantrone) as most likely to retard progression and delay disability.46-48
Extensive 1H-MR spectroscopy studies to characterize the metabolic profiles of MS lesions as well as the normal-appearing white and gray matter (NAGM) revealed loss of NAA in all tissue types, confirming the diffuse nature of the pathologic processes underlying the disease.49 This was confirmed by the first WBNAA study, which demonstrated age-dependent deficits in patients compared with matched controls in both RR-50 and PPMS.51 This dependence is probably due to the similar age of onset, transforming patients’ “age” to “disease-duration.”
Although lesions and atrophy, shown in Fig 5, the hallmarks of MS, represent end points of its complex pathologic processes, lesions do not identify these processes. Comparing WBNAA levels with atrophy (reflected by the fraction of parenchymal brain volume) as functions of disease duration in a cohort of 42 patients with RRMS has found the former to decrease nearly 3.5 times faster than the latter.52 This suggests that neuronal cell injury precedes atrophy and that that degenerating axons may leave behind their empty myelin sheaths, suggesting that WBNAA is a more sensitive indicator of disease progression than either lesion load or atrophy.52
Fig 5.

Top: Side-by-side axial T1 MR image from two 29-year-old-female patients with MS, both with a disease duration of 6 years and a very mild disability score (EDSS) of 1.5. Note that one exhibits noticeably more brain atrophy, reflected by enlarged subarachnoid spaces (1), ventricles (2), sulci (3) as well as T1-lesions (“black holes”) (4). These 2 images demonstrate the great heterogeneity in the MS course and pathogenesis. Bottom: The WBNAA spectrum from each of these women, normalized for brain tissue volume. Note that the one on the left is significantly lower than the one on the right, demonstrating that not only does she lose tissue faster but that the quality of the remaining tissue is worse (ie, neuronal dysfunction and damage precedes atrophy). mI indicates myo-Inositol; Cho, choline; Cr, creatine; glx, glutamine/glutamate; acq., acquisition.
For more than a century, MS was considered a white matter (WM) disease. Although evidence that gray matter (GM) is also affected has been available for more than 30 years, only recently has postmortem pathology shown many cortical and subcortical lesions that are usually missed by T2-weighted MR imaging.53 In addition, quantitative MR imaging techniques (eg, magnetization transfer and diffusion tensor imaging) revealed abnormal physical properties of otherwise NAGM. Because the WM:GM volume fractions of the brain are approximately 40:60 and the concentration of NAA in the former is approximately two thirds of the latter, WBNAA deficits exceeding 20% must reflect GM involvement. Indeed, of 71 patients with RRMS, 13 had 40% less WBNAA than controls, a loss that cannot be explained in terms of WM involvement alone, leading to a conclusion of extensive GM involvement, even in a relatively early RR stage of the disease.27
The paucity of reliable clinical prognostic metrics in MS confounds treatment choices, making laboratory tools to predict and monitor an individual's disease course crucial. To investigate the potential role of WBNAA, we quantified its rate of decline in 49 patients with RRMS by dividing their difference from the average of controls by their disease duration.54 We found 3 distinct subgroups of cross-sectional decline rates: ∼15% of the patients were considered “stable” (annual loss ∼0%), 65% were considered “moderate” (annual loss of 2.8%), and 20% were “rapid” with an annual loss of 28%.54 The average clinical Expanded Disability Status Scale (EDSS) score in each one of these subgroups was the same, 2.0, representing a very slight impairment in all. These results were recently replicated in a different 20-patient cohort,55 indicating that they are probably characteristics of MS corresponding to its known epidemiology, described, for example, by Weinshenker, 56 as “benign,” “intermediate,” and “acute,” rather than a particular recruitment pattern. Although they need to be confirmed in a longitudinal study, these findings suggest that WBNAA might be a sensitive gauge and predictor of clinical course and might help select and monitor treatment.
To ascertain how early neuroaxonal pathology does occur in MS, Filippi et al38 obtained the WBNAA levels in patients experiencing a clinically isolated syndrome suggestive of MS (CIS), the first presentation of the disease. Their WBNAA was significantly lower (mean, 20%) than that of their controls but not different from patients with or without enhancing lesions at baseline MR imaging or between patients with and without lesion dissemination in time,38 nor did the WBNAA correlate with lesion volumes. WBNAA scans were repeated at 1 year, showing an average 7% decrease in the patients with CIS, to assess whether such axonal damage is transient or persistent.57 These preliminary findings suggest that widespread axonal pathology may occur even at the earliest stage of MS, independent of MR imaging–visible inflammation and too extensive to be reversible. These results also suggest that axonal pathology may not always be the end stage of repeated inflammatory events, arguing strongly for early neuroprotective intervention.
WBNAA in Other Neurologic Disorders
Alzheimer Disease and Mild Cognitive Impairment
Alzheimer Disease (AD) is the leading cause of dementia affecting 5 million Americans (http://www.alz.org). It is often preceded by amnestic mild cognitive impairment (MCI) characterized by relative memory performance below typical age-related declines. Detecting the transition from MCI to AD has become increasingly important, with the prospect of disease-modifying therapies that may be most beneficial at the earliest stages. In analogy with CIS, where a WBNAA decrease has been shown at the earliest clinical symptoms, a study of 28 patients with AD, 27 with MCI, and 25 elderly controls found significant WBNAA decrease for both AD and MCI versus the controls, but not between them.41 The decrease in patients with AD also correlated moderately with their Mini-Mental State Examination score (R = 0.39, P < .001). These metabolic findings in MCI, the intermediate stage between healthy elderly and AD, may indicate that neuronal damage is already evident and widespread before the onset of clinical dementia.
Human Immunodeficiency Virus–Related Dementia
CNS involvement is a common complication of acquired immunodeficiency syndrome (AIDS), either as a direct effect of the virus or as the result of a secondary infection or toxicity. Cortical and WM lesions can be detected on conventional MR imaging in patients with AIDS, but these are not specific to the pathologic substrate. Because the disease is diffuse, Patel et al58 studied the relationship between global neuronal integrity, reflected by WBNAA and neuropsychological function in 15 patients infected with human immunodeficiency virus (HIV). Their WBNAA levels were lower (−9%, P = .03) and moderately correlated (R = −0.44, P = .05) with the processing speed subtest score and the AIDS dementia complex stage score. This suggests that WBNAA, reflecting both focal and diffuse damage, may be a better correlate of cognitive functions controlled by multicircuit networks.
Traumatic Brain Injury
More than 1.5 million Americans annually have traumatic brain injury (TBI), the leading cause of death in children and young adults. The “moderate” and “severe” forms of TBI together account for <10% of the total, and >85% of traumas are classified as “mild” (mTBI, Glasgow Comma Scale scores of 13–15). The current estimate that only 10% of patients with mTBI have persisting neurocognitive deficits, however, is considered an underestimation. Diffuse axonal injury (DAI) in the lobar WM, corpus callosum, and dorsolateral upper brain stem has been reported in a significant number of patients following severe, moderate, and even mTBI.59 Typically, DAI is microscopic and conventional MR imaging–occult. Because 1H-MR spectroscopy can detect such abnormalities in normal-appearing brain, WBNAA has been applied to 20 patients with mTBI.60 They exhibited an average of 12% WBNAA deficit (P < .05) that increased with age compared with that in healthy controls. Combined with −1.09% annual GM atrophy, these findings may provide noninvasive insight into the nature of mTBI progression and potentially identify individuals “at risk” for its neurologic and neuropsychological sequelae.
Glial Brain Tumors
High-grade (World Health Organization grades III and IV) gliomas portend a poor prognosis of an average postdiagnosis life expectancy of <2 years.61 They are highly vascular and have a tendency for infiltration with extensive areas of necrosis, hypoxia, and a breakdown of the blood-brain barrier. They almost always recur even after complete resection.62 One possible reason for the recurrence is cell invasion into the surrounding tissue, which is difficult to image with current radiologic methods. Indeed, glial tumors often infiltrate well beyond the T2 signal-intensity abnormalities on conventional MR imaging, complicating treatment planning and removal.63
WBNAA was measured in 17 newly diagnosed untreated patients with glioma and compared with age-matched controls to test the hypothesis that tumor cell invasion can be tracked via neuronal cell damage in its path. Patients’ WBNAA was significantly (average, 26%) lower both pre- and postsurgery, but the pre- versus postdifferences were insignificant.64 These tumors (on average <3% of brain volume) alone could not explain such NAA deficits, suggesting extensive diffuse tumor cell infiltration into the normal-appearing brain.64 This hypothesis is supported by the correlation found between tumor grade (but not volume) and WBNAA deficit, suggesting extensive tumor cell proliferation throughout the brain for higher grades. This conclusion is supported by another study reporting a strong inverse correlation (R = −0.88, P = .0008) between WBNAA and the mean diffusivity in “tumor-free” contralateral brain in these patients.65
Meningiomas
Meningiomas are the most common benign brain tumors.62 Although few are malignant, most are well circumscribed with very little infiltration into the adjacent brain. Although most small ones are asymptomatic and are often incidental findings, larger meningiomas can cause focal seizures and spastic weakness as a result of compression, depending on their size and location.66 Diagnosis is straightforward with MR imaging or CT, and surgery is often the best treatment.
In a case study of 2 presurgical meningiomas, the patients presented with neurologic symptoms and WBNAA concentrations significantly higher than those of matched controls when excluding the mass from their brain volume.67 Correction to “premass” brain volume left the WBNAA levels indistinguishable from those of the controls. Postoperative follow-up showed no physical or cognitive disability, suggesting that the clinical symptoms were due to brain compression (ie, transient) and not reflecting (permanent) neuronal damage.67
Radiation Therapy
Because of a high correlation between small cell lung cancer and brain tumors, prophylactic cranial irradiation (PCI) is often offered even in the absence of overt brain pathology.68 Radiation is inherently cytotoxic and runs the risk of ancillary brain damage confounding treatment cost-benefit analyses. WBNAA was measured in a cohort of 8 patients with small cell lung cancer immediately before and right after PCI, to quantify the overall brain damage caused by PCI. A significant WBNAA loss of approximately 10% was observed after 2 weeks of fractionated therapy, but there was no change in cognitive abilities as measured by the Mini-Mental State Examination.69 This suggests that WBNAA may be a more sensitive objective biomarker of the neurologic damage imparted by these 3000- to 5000-cGy dose levels.
Conclusions
WBNAA has the potential to quantify neurodegeneration and neuroaxonal dysfunction in several disorders. Due to its lack of localization, this technique is particularly attractive for diseases characterized by widespread or diffuse pathology. However, as shown in the brain tumor studies, it can also provide significant insights into the pathophysiology of seemingly “focal” diseases. Not only is WBNAA spectroscopy able to detect early neuroaxonal changes providing a potential prognostic indication of clinical outcome, but it can also yield a specific surrogate marker for monitoring neuroprotection in response to current and experimental treatments.
Acknowledgments
The authors are grateful to Drs. Achim Gass and Lutz Achtnichts of the Neurology Clinic of the University of Basel for making their unpublished WBNAA data available for this review.
Footnotes
This work was supported by National Institutes of Health Grants EB01015, NS050520, NS29029, NS39135, and NS051623.
References
- 1.McFarland H. The lesion in multiple sclerosis: clinical, pathological, and magnetic resonance imaging considerations. J Neurol Neurosurg Psychiatry 1998;64S1:S26–S30 [PubMed] [Google Scholar]
- 2.Ferguson B, Matyszak MK, Esiri MM, et al. Axonal damage in acute multiple sclerosis lesions. Brain 1997;120:393–99 [DOI] [PubMed] [Google Scholar]
- 3.Bitsch A, Bruhn H, Vougioukas V, et al. Inflammatory CNS demyelination: histopathologic correlation with in vivo quantitative proton MR spectroscopy. AJNR Am J Neuroradiol 1999;20:1619–27 [PMC free article] [PubMed] [Google Scholar]
- 4.Miller BL. A review of chemical issues in 1H NMR spectroscopy: N-acetyl-L-aspartate, creatine and choline. NMR Biomed 1991;4:47–52 [DOI] [PubMed] [Google Scholar]
- 5.Lin A, Ross BD, Harris K, et al. Efficacy of proton magnetic resonance spectroscopy in neurological diagnosis and neurotherapeutic decision making. NeuroRx 12005;2:197–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tallan HH, Moore S, Stein WH. N-acetyl-L-aspartic acid in brain. J Biol Chem 1956;219:257–64 [PubMed] [Google Scholar]
- 7.Simmons ML, Frondoza CG, Coyle JT. Immunocytochemical localization of N-acetyl-aspartate with monoclonal antibodies. Neuroscience 1991;45:37–45 [DOI] [PubMed] [Google Scholar]
- 8.Jenkins BG, Klivenyi P, Kustermann E, et al. Nonlinear decrease over time in N-acetyl aspartate levels in the absence of neuronal loss and increases in glutamine and glucose in transgenic Huntington's disease mice. J Neurochem 2000;74:2108–19 [DOI] [PubMed] [Google Scholar]
- 9.Moffett JR, Namboodiri MA, Cangro CB, et al. Immunohistochemical localization of N-acetylaspartate in rat brain. Neuroreport 1991;2:131–34 [DOI] [PubMed] [Google Scholar]
- 10.Baslow MH, Resnik TR. Canavan disease: analysis of the nature of the metabolic lesions responsible for development of the observed clinical symptoms. J Mol Neurosci 1997;9:109–25 [DOI] [PubMed] [Google Scholar]
- 11.Danielsen EA, Ross B. Magnetic Resonance Spectroscopy Diagnosis of Neurological Diseases. New York: Marcel Dekker;1999. .
- 12.Ross BD, Bluml S. Magnetic resonances of the human brain. Anat Rec 2001;265:54–84 [DOI] [PubMed] [Google Scholar]
- 13.Bjartmar C, Battistuta J, Terada N, et al. N-acetylaspartate is an axon-specific marker of mature white matter in vivo: a biochemical and immunohistochemical study on the rat optic nerve. Ann Neurol 2002;51:51–58 [DOI] [PubMed] [Google Scholar]
- 14.Bjartmar C, Kidd G, Mork S, et al. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol 2000;48:893–901 [PubMed] [Google Scholar]
- 15.Goldstein FB. Biosynthesis of N-acetyl-L-aspartic acid. Biochim Biophys Acta 1959;33:583–84 [DOI] [PubMed] [Google Scholar]
- 16.Baslow MH, Guilfoyle DN. Effect of N-acetylaspartic acid on the diffusion coefficient of water: a proton magnetic resonance phantom method for measurement of osmolyte-obligated water. Anal Biochem 2002;311:133–38 [DOI] [PubMed] [Google Scholar]
- 17.Chakraborty G, Mekala P, Yahya D, et al. Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipid synthesis: evidence for myelin-associated aspartoacylase. J Neurochem 2001;78:736–45 [DOI] [PubMed] [Google Scholar]
- 18.Madhavarao CN, Chinopoulos C, Chandrasekaran K, et al. Characterization of the N-acetylaspartate biosynthetic enzyme from rat brain. J Neurochem 2003;86:824–35 [DOI] [PubMed] [Google Scholar]
- 19.Yan HD, Ishihara K, Serikawa T, et al. Activation by N-acetyl-L-aspartate of acutely dissociated hippocampal neurons in rats via metabotropic glutamate receptors. Epilepsia 2003;44:1153–59 [DOI] [PubMed] [Google Scholar]
- 20.Kreis R, Ernst T, Ross BD. Absolute quantitation of water and metabolites in the human brain. II. metabolite concentrations. J Magn Reson Series B 1993;102:9–19 [Google Scholar]
- 21.Birken DL, Oldendorf WH. N-acetyl-L-aspartic acid: a literature review of a compound prominent in 1H-NMR spectroscopic studies of brain. Neurosci Biobehav Rev 1989;13:23–31 [DOI] [PubMed] [Google Scholar]
- 22.Arnold DL, de Stefano N, Matthews PM, et al. N-acetylaspartate: usefulness as an indicator of viable neuronal tissue. Ann Neurol 2001;50:823–25 [DOI] [PubMed] [Google Scholar]
- 23.Baslow MH. Functions of N-acetyl-L-aspartate and N-acetyl-L-aspartylglutamate in the vertebrate brain: role in glial cell-specific signaling. J Neurochem 2000;75:453–59 [DOI] [PubMed] [Google Scholar]
- 24.Brooks WM, Friedman SD, Stidley CA. Reproducibility of 1H-MRS in vivo. Magn Reson Med 1999;41:193–97 [DOI] [PubMed] [Google Scholar]
- 25.Marshall I, Wardlaw J, Cannon J, et al. Reproducibility of metabolite peak areas in 1H MRS of brain. Magn Reson Imaging 1996;14:281–92 [DOI] [PubMed] [Google Scholar]
- 26.Bokde AL, Teipel SJ, Zebuhr Y, et al. A new rapid landmark-based regional MRI segmentation method of the brain. J Neurol Sci 2002;194:35–40 [DOI] [PubMed] [Google Scholar]
- 27.Inglese M, Ge Y, Filippi M, et al. Indirect evidence for early widespread gray matter involvement in relapsing-remitting multiple sclerosis. Neuroimage 2004;21:1825–29 [DOI] [PubMed] [Google Scholar]
- 28.De Stefano N, Narayanan S, Francis SJ, et al. Diffuse axonal and tissue injury in patients with multiple sclerosis with low cerebral lesion load and no disability. Arch Neurol 2002;59:1565–71 [DOI] [PubMed] [Google Scholar]
- 29.Zaaraoui W, Fleysher L, Fleysher R, et al. Human brain-structure resolved T(2) relaxation times of proton metabolites at 3 Tesla. Magn Reson Med 2007;57:983–89 [DOI] [PubMed] [Google Scholar]
- 30.Gonen O, Viswanathan AK, Catalaa I, et al. Total brain N-acetylaspartate concentration in normal, age-grouped females: quantitation with non-echo proton NMR spectroscopy. Magn Reson Med 1998;40:684–89 [DOI] [PubMed] [Google Scholar]
- 31.Udupa JK, Odhner D, Samarasekera S. 3DVIEWNIX: an open, transportable, multdimensional, multimodality, multiparametric imaging software system. Proc SPIE 1994;2164:58–73 [Google Scholar]
- 32.De Santi S, de Leon MJ, Rusinek H, et al. Hippocampal formation glucose metabolism and volume losses in MCI and AD. Neurobiol Aging 2001;22:529–39 [DOI] [PubMed] [Google Scholar]
- 33.Ashburner J, Hutton C, Frackowiak R, et al. Identifying global anatomical differences: deformation-based morphometry. Hum Brain Mapp 1998;6:348–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith SM, De Stefano N, Jenkinson M, et al. Normalized accurate measurement of longitudinal brain change. J Comput Assist Tomogr 2001;25:466–75 [DOI] [PubMed] [Google Scholar]
- 35.Smith SM, Zhang Y, Jenkinson M, et al. Accurate, robust, and automated longitudinal and cross-sectional brain change analysis. Neuroimage 2002;17:479–89 [DOI] [PubMed] [Google Scholar]
- 36.Smith SM, Rao A, De Stefano N, et al. Longitudinal and cross-sectional analysis of atrophy in Alzheimer's disease: cross-validation of BSI, SIENA and SIENAX. Neuroimage 2007;36:1200–06. Epub 2007 Apr 27 [DOI] [PubMed] [Google Scholar]
- 37.Gonen O, Grossman RI. The accuracy of whole brain N-acetylaspartate quantification. Magn Reson Imaging 2000;18:1255–58 [DOI] [PubMed] [Google Scholar]
- 38.Filippi M, Bozzali M, Rovaris M, et al. Evidence for widespread axonal damage at the earliest clinical stage of multiple sclerosis. Brain 2003;126:433–37 [DOI] [PubMed] [Google Scholar]
- 39.Benedetti B, Rigotti DJ, Liu S, et al. Reproducibility of the whole-brain N-acetylaspartate level across institutions, MR scanners, and field strengths. AJNR Am J Neuroradiol 2007;28:72–75 [PMC free article] [PubMed] [Google Scholar]
- 40.Rigotti DJ, Inglese M, Babb JS, et al. Serial whole-brain N-acetylaspartate concentration in healthy young adults. AJNR Am J Neuroradiol. 2007;28:1650–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Falini A, Bozzali M, Magnani G, et al. A whole brain MR spectroscopy study from patients with Alzheimer's disease and mild cognitive impairment. Neuroimage 2005;26:1159–63 [DOI] [PubMed] [Google Scholar]
- 42.Scahill RI, Frost C, Jenkins R, et al. A longitudinal study of brain volume changes in normal aging using serial registered magnetic resonance imaging. Arch Neurol 2003;60:989–94 [DOI] [PubMed] [Google Scholar]
- 43.Compston A, Coles A. Multiple sclerosis. Lancet 2002;359:1221–31 [DOI] [PubMed] [Google Scholar]
- 44.Sayao AL, Devonshire V, Tremlett H. Longitudinal follow-up of “benign” multiple sclerosis at 20 years. Neurology 2007;68:496–500 [DOI] [PubMed] [Google Scholar]
- 45.Montalban X. Primary progressive multiple sclerosis. Curr Opin Neurol 2005;18:261–66 [DOI] [PubMed] [Google Scholar]
- 46.Cree B. Emerging monoclonal antibody therapies for multiple sclerosis. Neurologist 2006;12:171–78 [DOI] [PubMed] [Google Scholar]
- 47.Hartung HP, Bar-Or A, Zoukos Y. What do we know about the mechanism of action of disease-modifying treatments in MS? J Neurol 2004;251 (suppl 5):v12–v29 [DOI] [PubMed] [Google Scholar]
- 48.Fox EJ. Management of worsening multiple sclerosis with mitoxantrone: a review. Clin Ther 2006;28:461–74 [DOI] [PubMed] [Google Scholar]
- 49.Miller DH, Thompson AJ, Filippi M. Magnetic resonance studies of abnormalities in the normal-appearing white matter and gray matter in multiple sclerosis. J Neurol 2003;250:1407–19 [DOI] [PubMed] [Google Scholar]
- 50.Gonen O, Catalaa I, Babb JS, et al. Total brain N-acetylaspartate: a new measure of disease load in MS. Neurology 2000;54:15–19 [DOI] [PubMed] [Google Scholar]
- 51.Rovaris M, Gallo A, Falini A, et al. Axonal injury and overall tissue loss are not related in primary progressive multiple sclerosis. Arch Neurol 2005;62:898–902 [DOI] [PubMed] [Google Scholar]
- 52.Ge Y, Gonen O, Inglese M, et al. Neuronal cell injury precedes brain atrophy in multiple sclerosis. Neurology 2004;62:624–27 [DOI] [PubMed] [Google Scholar]
- 53.Lumsden C. The clinical immunology of multiple sclerosis. In: BG Vinken PJ, ed. Handbook of Clinical Neurology. Vol.9 . North-Holland, Amsterdam: Elsevier;1970. :217–309 [Google Scholar]
- 54.Gonen O, Moriarty DM, Li BS, et al. Relapsing-remitting multiple sclerosis and whole-brain N-acetylaspartate measurement: evidence for different clinical cohorts initial observations. Radiology 2002;225:261–68 [DOI] [PubMed] [Google Scholar]
- 55.Gonen O, Oberndorfer TA, Inglese M, et al. Reproducibility of 3 whole-brain N-acetylaspartate decline cohorts in relapsing-remitting multiple sclerosis. AJNR Am J Neuroradiol 2007;28:267–71 [PMC free article] [PubMed] [Google Scholar]
- 56.Weinshenker BG. Natural history of multiple-sclerosis. Ann Neurol 1994;36:S6–S11 [DOI] [PubMed] [Google Scholar]
- 57.Rovaris M, Gambini A, Gallo A, et al. Axonal injury in early multiple sclerosis is irreversible and independent of the short-term disease evolution. Neurology 2005;65:1626–30 [DOI] [PubMed] [Google Scholar]
- 58.Patel SH, Inglese M, Glosser G, et al. Whole-brain N-acetylaspartate level and cognitive performance in HIV infection. AJNR Am J Neuroradiol 2003;24:1587–91 [PMC free article] [PubMed] [Google Scholar]
- 59.Medana IM, Esiri MM. Axonal damage: a key predictor of outcome in human CNS diseases. Brain 2003;126:515–30 [DOI] [PubMed] [Google Scholar]
- 60.Cohen BA, Inglese M, Rusinek H, et al. Proton MR spectroscopy and MRI-volumetry in mild traumatic brain injury. AJNR Am J Neuroradiol 2007;28:907–13 [PMC free article] [PubMed] [Google Scholar]
- 61.DeAngelis LM. Brain tumors. N Engl J Med 2001;344:114–23 [DOI] [PubMed] [Google Scholar]
- 62.Surawicz TS, McCarthy BJ, Kupelian V, et al. Descriptive epidemiology of primary brain and CNS tumors: results from the Central Brain Tumor Registry of the United States, 1990–1994. Neuro Oncol 1999;1:14–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lunsford LD, Martinez AJ, Latchaw RE. Magnetic resonance imaging does not define tumor boundaries. Acta Radiol Suppl 1986;369:154–56 [PubMed] [Google Scholar]
- 64.Cohen BA, Knopp EA, Rusinek H, et al. Assessing global invasion of newly diagnosed glial tumors with whole-brain proton MR spectroscopy. AJNR Am J Neuroradiol 2005;26:2170–77 [PMC free article] [PubMed] [Google Scholar]
- 65.Inglese M, Brown S, Johnson G, et al. Whole-brain N-acetylaspartate spectroscopy and diffusion tensor imaging in patients with newly diagnosed gliomas: a preliminary study. AJNR Am J Neuroradiol 2006;27:2137–40 [PMC free article] [PubMed] [Google Scholar]
- 66.Curnes JT. MR imaging of peripheral intracranial neoplasms: extraaxial vs intraaxial masses. J Comput Assist Tomogr 1987;11:932–37 [DOI] [PubMed] [Google Scholar]
- 67.Cohen BA, Knopp EA, Rusinek H, et al. Brain compression without global neuronal loss in meningiomas: whole-brain proton MR spectroscopy report of 2 cases. AJNR Am J Neuroradiol 2005;26:2178–82 [PMC free article] [PubMed] [Google Scholar]
- 68.Aupérin A, Arriagada R, PIgnon JP, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. Prophylactic Cranial Irradiation Overview Collaborative Group. N Engl J Med 1999;341:476–84 [DOI] [PubMed] [Google Scholar]
- 69.Movsas B, Li BS, Babb JS, et al. Quantifying radiation therapy-induced brain injury with whole-brain proton MR spectroscopy: initial observations. Radiology 2001;221:327–31 [DOI] [PubMed] [Google Scholar]