

Abstract
Alzheimer's Disease (AD), the leading cause of senile dementia, is a progressive neurodegenerative disorder affecting millions of people worldwide and exerting tremendous socioeconomic burden on all societies. Although definitive diagnosis of AD is often made in the presence of clinical manifestations in late stages, it is now universally believed that AD is a continuum of disease commencing from the preclinical stage with typical neuropathological alterations appearing decades prior to its first symptom, to the prodromal stage with slight symptoms of amnesia (amnestic mild cognitive impairment, aMCI), and then to the terminal stage with extensive loss of basic cognitive functions, i.e., AD-dementia. Positron emission tomography (PET) radiotracers have been developed in a search to meet the increasing clinical need of early detection and treatment monitoring for AD, with reference to the pathophysiological targets in Alzheimer's brain. These include the pathological aggregations of misfolded proteins such as β-amyloid (Aβ) plagues and neurofibrillary tangles (NFTs), impaired neurotransmitter system, neuroinflammation, as well as deficient synaptic vesicles and glucose utilization. In this article we survey the various PET radiotracers available for AD imaging and discuss their clinical applications especially in terms of early detection and cognitive relevance.
Keywords: PET, neuroimaging, Alzheimer's disease, radiotracer, clinical research
Introduction
Alzheimer's Disease (AD), the leading cause of senile dementia, is a progressive neurodegenerative disorder affecting millions of people worldwide and exerting tremendous socioeconomic burden on all societies (Goedert and Spillantini, 2006; Querfurth and LaFerla, 2010). AD is neuropathologically characterized by deposition of senile plaques and neurofibrillary tangles in the brain tissue. Excessive aggregation of misfolded β-amyloid (Aβ) and hyperphosphorylated tau proteins leads to cytotoxicity and disruption of cytoarchitecture, and subsequent neuronal death and brain function decline. Neuroinflammation activation, cholinergic deficit, impaired glucose utilization and synaptic dysfunction are also outstanding characteristics of AD. Functional neuroimaging using positron emission tomography (PET) is able to reveal these in vivo pathological/pathophysiological alterations. With increasing viewpoint of AD as a continuum from asymptomatic preclinical stage, to prodromal stage with mild cognitive impairment (MCI), and finally to the advanced stage of dementia, integrated early diagnostic and differentiation paradigms with the help of PET imaging has been well-acknowledged in multiple diagnostic criteria (Dubois et al., 2007, 2014; Jack et al., 2011). In this review article, we describe the various PET radiotracers available for AD imaging and discuss their clinical applications especially in terms of early detection and cognitive relevance. Literature evidence on the predictive ability of PET imaging with various PET tracers for prodromal stage conversion and monitoring of disease progression will also be reviewed. Finally, comments on emerging biomarkers and their prospects in early detection of AD will be provided.
PET Tracers for Imaging Aβ
Overview
The disturbance of homeostasis between the accumulation of neurotoxic Aβ peptides and its clearance in the brain is believed to be the core event in AD etiology (Hardy and Allsop, 1991; Hardy and Higgins, 1992). The Aβ peptides are derived from the breakdown of amyloid precursor protein (APP) through cleavage by β- and γ-secretase. The soluble oligomers, believed to be the perpetrator of cytotoxicity, are aggregated by longer species of the Aβ peptides such as Aβ40 and Aβ42 released into the extracellular space. The accumulation of Aβ peptides, from neurotoxic oligomers to further aggregated insoluble β-sheet fibrils and dense fibrillary plaques, is believed to underlie subsequent neurofibrillary tangle formation and neuronal loss, which precede the onset of clinical symptoms by more than 10–15 years (Hardy and Gwinn-Hardy, 1998; Hardy et al., 1998). However, confirmation of AD neuropathology has long relied on immunohistochemical staining of Aβ aggregates in postmortem autopsy tissues. Since the early 2000s, the availability of antemortem in vivo PET imaging with Aβ radiotracers has greatly advanced our knowledge on the time course and correlation of Aβ aggregation, AD progression, and cognitive decline, and revolutionized AD diagnosis.
Tracer Development
The search for Aβ imaging tracers dated back to the mid-1990s, culminated in the first in vivo imaging of Aβ in an AD patient in 2002 with the 11C-labeled Pittsburgh compound B ([11C]PIB, Figure 1), which is derived from the Aβ staining agent thioflavin-T (Klunk et al., 2004). [11C]PIB has a relative high selectivity for Aβ of all forms from soluble oligomers to insoluble fibrils and plaques over other pathologic proteins such as tau and α-synuclein. Both visual inspection and quantitative analysis demonstrated higher cortical retention in AD patients than in cognitively intact subjects, especially in the orbitofrontal cortex, inferior parietal cortex, posterior cingulate cortex and precuneus, which resembled the pattern found in immunohistochemical studies (Rowe and Villemagne, 2011). To date, [11C]PIB is still the best and most widely used Aβ PET tracer and regarded as the gold standard.
Figure 1.

Structures of representative PET radiotracers for Aβ imaging.
The short radioactive half-life of ~20 min for the carbon-11 nuclide limits the use of [11C]PIB to institutions with on-site cyclotrons, thus inspiring the development of 18F-labeled Aβ radiotracers. Three of such tracers with favorable binding and imaging properties, [18F]florbetapir ([18F]AV-45), [18F]florbetaben ([18F]AV-1, [18F]BAY-94-9172), and [18F]flutemetamol ([18F]GE-067) (Figure 1), have since been approved by the United States Food and Drug Administration (FDA) for clinical diagnosis and differential diagnosis of AD (Barthel et al., 2011; Lister-James et al., 2011; Curtis et al., 2015). [18F]NAV4694 ([18F]AZD4694), another promising 18F-labeled tracer with rapid pharmacokinetics and lower non-specific binding in the cerebral white matter, is now in clinical trials (Cselenyi et al., 2012; Therriault et al., 2021). These tracers, with longer radioactive half-life of ~110 min, are suitable for long-distance distribution and thus can be more widely used in the clinics. They are presumed to yield less noisier images and therefore more precise quantitation of minute cerebral Aβ accumulation in the early stage of the disease, due to more abundant radioactivity counts in the later period of scan. Notably, imaging protocols provided for each tracer are quite different from one another in terms of scanning window as well as visual interpretation and quantitative analysis of the images (Mallik et al., 2017).
Imaging Research Findings and Clinical Relevance
In general, cortical Aβ retention detected by PET imaging is in good correlation with immunohistochemical staining of amyloid plagues at autopsy or biopsy brain samples (Clark et al., 2012; Rinne et al., 2012; Curtis et al., 2015; Sabri et al., 2015a). This qualifies the application of Aβ PET imaging as a non-invasive tool for in vivo detection of cortical Aβ deposition in the living brain. In the clinical daily-routine context, Aβ PET imaging provides plenty valuable information for the purpose of differential diagnosis between AD dementia and dementia disorders associated with non-Aβ pathologies such as frontotemporal lobe dementia (FTLD), which is sometimes indistinguishable from AD by neuropsychological assessments and conventional structural imaging modalities only (Rowe et al., 2008).
On the other hand, it has been emphasized that an amyloid-positive PET merely reflects the existence of amyloid neuropathology in vivo and does not necessarily guarantee a diagnosis of AD, concerning the fact that many otherwise cognitively normal subjects (even young healthy volunteers) have been found to be amyloid-positive judging from PET results, and that cognitive decline is more likely due to factors other than amyloid pathology in dementia with Lewy bodies (DLB) or in some co-morbid situations (Johnson et al., 2007; Ossenkoppele et al., 2015; Petrou et al., 2015). It has also been debated whether a negative amyloid scan could exclude the possibility of AD (Jack et al., 2016), since evidence of neurodegeneration in the absence of amyloid pathology challenges the proposed AD progression scheme (Sperling et al., 2011).
The ability of PET imaging with Aβ tracers to predict conversion from MCI due to AD, the prodromal stage of the disease defined by different diagnostic guidelines (McKhann et al., 2011; Dubois et al., 2014), to AD dementia has also been extensively investigated. Roughly 70% of amyloid-positive MCI patients converts to AD dementia within 3 years (Okello et al., 2009a). As compared to [18F]FDG PET, amyloid PET has higher sensitivity but relatively lower specificity (Teipel et al., 2015), which is consistent with the finding that amyloid accumulation commences at least a decade before the worsening of synaptic activity and brain function to a clinically significant level where amyloid deposition reaches a plateau (Jack and Holtzman, 2013; Jack et al., 2013a). Correlation of Aβ retention to cognitive performance has been shown to be greater in MCI and cognitively intact subjects than in AD dementia patients (Pike et al., 2007, 2011; Villemagne et al., 2008), which could also be explained by the hypothesized plateau model. Therefore, it is not surprising that regional hypometabolism and amyloid deposition in the temporoparietal regions are closely associated with each other whereas those in the frontal lobe, a region affected only in advanced AD, are not (Edison et al., 2007; Cohen et al., 2009).
The significance of Aβ PET in subjects without objective evidence of cognitive decline is emerging most recently. Large-scale comprehensive studies have shown that Aβ positivity is linked to high progression risk in subjective cognitive decline (SCD) and is associated with age and family history other than sex, education, marital or retirement status and self-reported lifestyle factors (Papp et al., 2020; Sperling et al., 2020). Longitudinal follow-up studies suggest that the duration of Aβ existence might be more important than age or binary result in affecting both deteriorating rate and the final status of cognition through elevated entorhinal tau burden (Hanseeuw et al., 2019; Koscik et al., 2020). Even if Aβ burden detected by PET is below threshold, its value is still positively correlated to the subject's future risk of cognitive decline (Guo et al., 2020).
The Centiloid Scaling Project
As the utilization of amyloid PET in clinical trials and research expands and multiple tracers are available for such imaging applications, the urgent need for inter-tracer standardization and for multi-center collaboration and longitudinal comparison drove the launch of the centiloid scaling project (Klunk et al., 2015). According to the concept of the project, one institute can follow a multi-step regime to create a scaling from 0 (young healthy controls) to 100 (typical AD patients) using its own amyloid PET data (Rowe et al., 2016). In this way a universal cutoff value could then be directly or indirectly applied in multi-center imaging and/or longitudinal studies to allow for inter-site/inter-tracer comparisons. The study group of the centiloid project has now made progress in the derivation and verification of converting formula, enabling the translation of non-[11C]PIB Aβ PET semi-quantitative values to standardized [11C]PIB counterparts (Rowe et al., 2017; Battle et al., 2018; Bourgeat et al., 2018; Navitsky et al., 2018). The authenticity of the centiloid approach has been confirmed neuropathologically (Amadoru et al., 2020).
Discussion
The amyloid cascade theory has been the predominant theory of AD etiology and drove the development of anti-Aβ therapeutics in the last 3 decades. The availability of Aβ PET imaging tracers and its applications in AD imaging have indicated that amyloid pathology may be a high risk factor for future cognitive decline. However, increasing evidence also indicates that cortical amyloid is not specific for the presence of cognitive symptoms, thus affecting the positive predictive value of Aβ PET imaging. Among populations without dementia, the prevalence of cerebral amyloid pathology as determined by Aβ PET imaging or cerebral spinal fluid (CSF) Aβ measurement is associated with age (Jansen et al., 2015), e.g., 33% of healthy elderly individuals have significant levels of Aβ deposition without apparent clinical symptoms (Rowe et al., 2010). Therefore, Aβ deposition alone cannot explain AD pathogenesis and progression. Repeated failures of clinical trials for many anti-Aβ drug candidates have dampened the hope for their efficacy as disease-modifying therapeutics. Nonetheless, it should be kept in mind that Aβ PET imaging will still remain the gold standard to investigate disease mechanisms as it provides information regarding the topography of Aβ lesions. Although there have been no successful anti-Aβ drugs up to date, Aβ PET imaging has provided useful outcome measures for anti-Aβ therapeutics in clinical trials (Salloway et al., 2014; Honig et al., 2018; Wessels et al., 2020).
PET Tracers for Imaging TAU Tangles
Overview
In addition to the β-amyloid peptides, microtubule-associated protein tau (MAPT), or simply tau protein, together with its misfolded products, has been more thoroughly studied in recent years to explore its relationship with AD. Similar to the case of senile plaques formed by Aβ, the formation of neurofibrillary tangles (NFTs) by paired helical filaments (PHFs) is also a neuropathological hallmark of AD (Braak and Braak, 1997). PHFs are aggregated by misfolded hyperphosphorylated tau protein whose binding affinity with the microtubules is weakened, causing neuronal cytoarchitecture breakdown and dysfunction (Hoover et al., 2010; Spillantini and Goedert, 2013).
With the repeated failures of anti-Aβ therapeutics in large scale clinical trials, the focus was shifted from Aβ to tau on the development of AD therapeutics and imaging agents (Giacobini and Gold, 2013). However imaging tau in vivo is more challenging than imaging Aβ. Tau protein has six unique isoforms characterized by the number of repeats of its microtubular binding domains, and multiple secondary/tertiary structures differentiated by the shape of the filaments (Spillantini and Goedert, 2013). In addition to its much lower abundance compared to Aβ peptides in the brain, MAPT's intraneuronal property demands the qualified tracer to cross neuron cell membrane in addition to the blood brain barrier. These factors collectively hamper the screening and identification of sensitive and specific compounds. Nevertheless, substantial progress has been made in overcoming these inherent obstacles, and preliminary studies have shown encouraging results worthy of the efforts (Hall et al., 2017; Leuzy et al., 2019).
Tracer Development
PET imaging of fibrillary tau traced back to about the same time for amyloid, with [18F]FDDNP arguably as the earliest tracer (Agdeppa et al., 2001). Indeed, this tracer labels both amyloid plaques and fibrillary tau tangles in vivo, but this property is also its biggest disadvantage, as it has comparable affinity for both amyloid and tau proteins, i.e., a lack of selectivity for either target. The first selective tau tracer [18F]THK523 was developed by Tohoku University of Japan in 2005 (Okamura et al., 2005). Later structural modifications led to the development of other tracers in the THK family: [18F]THK5105, [18F]THK5117, [18F]THK5317, and [18F]THK5351, with improved binding and in vivo pharmacokinetic properties (Okamura et al., 2013; Chiotis et al., 2016; Betthauser et al., 2017). However, tracers in this family were later found to have notable off-target binding to monoamine oxidase-B (MAO-B), which greatly limited their utility in imaging of tauopathies including AD (Ng et al., 2017).
[18F]Flortaucipir ([18F]AV1451, or formerly [18F]T807) is currently the most applied and the only FDA-approved tau radiotracer (Figure 2) (https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trial-snapshot-tauvid). It has a 25-fold higher affinity for tau than Aβ, as well as favorable kinetics for both uptake and washout in the brain without radioactive metabolites penetrating the blood-brain-barrier (Xia et al., 2013). [18F]flortaucipir has higher affinity to PHFs over straight filaments (SF), and to combined 3-repeat (3R) and 4-repeat (4R) isoforms over 3R or 4R isoforms alone, making it more suitable for imaging AD pathology than non-AD tauopathies such as progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) (Lowe et al., 2016). It is noted that suspected minor off-target binding to MAO-A in the basal ganglia and substantia nigra would limit the application of [18F]flortaucipir in imaging Parkinsonian tauopathies (Ono et al., 2017).
Figure 2.

Structures of representative PET radiotracers for tau imaging.
[11C]PBB3 is another selective tau tracer that has also been thoroughly studied in various tauopathies including AD (Figure 2). [11C]PBB3 binds to both 3R and 4R tau isoforms and its affinity for tau is over 40 times higher than that for amyloid, making it suitable for imaging of various tauopathies in vivo (Kimura et al., 2015). Minor structural modifications then afforded [18F]APN-1607 (PM-PBB3) and [18F]AM-PBB3, the next generation members of the PBB3 family. [18F]APN-1607, (Figure 2) has recently been reported to possess more favorable pharmacokinetics and provide higher gray-matter/white-matter contrast (Hsu et al., 2020; Su et al., 2020; Tagai et al., 2021).
The clinical development of several newer, second generation selective tau tracers are ongoing, including [18F]PI2620, [18F]MK6240, [18F]GTP1, [18F]RO-948 (RO6958948), [18F]JNJ-311 (JNJ64349311), and [18F]JNJ-067 (JNJ-64326067) (Figure 2). Designers of these new generation tracers are focusing on improving in vivo characteristics such as higher selectivity, faster brain penetration/washout, and less off-target binding. Preliminary studies for these newer tracers have shown promising results (Declercq et al., 2017; Kuwabara et al., 2018; Guehl et al., 2019; Rombouts et al., 2019; Teng et al., 2019; Schmidt et al., 2020).
Off-target binding has been a universal concern for tau tracers since the early [18F]FDDNP was found to be non-selective to both amyloid and tau (Thompson et al., 2009). In PET imaging of neurodegeneration, selectivity of tau tracers over other pathological proteins such as Aβ, α-synuclein, and TDP-43 would always require validation. Similarity of the secondary/tertiary structures of the binding sites of these proteins makes it difficult to find a truly selective and specific probe, not to mention the complexity introduced by potential comorbidity of the neurodegenerative disorders. In addition, off-target binding in the central nervous system (CNS) has been widely examined across the tau tracers. It is now known that MAO-A and MAO-B are the most frequent off-targets whose secreting neurons are highly overlapped with Parkinsonism-related brain regions (Lowe et al., 2016; Bischof et al., 2017; Okamura et al., 2018). Choroid plexus is frequently found to be a tissue with apparent off-target binding, the mechanism of which is yet unclear (Ikonomovic et al., 2016). It is postulated that melanin, neuromelanin, mineralized structures and hemorrhagic lesions can also cause off-target binding of tau tracers in various locations. There is also debate that the suspected “off-target” binding may be reflecting true tau-binding, or binding to some specific targets yet to be identified (Ikonomovic et al., 2016; Passamonti et al., 2017). These characteristics pose substantial difficulties to clinical differentiation and potential post-treatment evaluation (Passamonti et al., 2017).
Recent cryo-electron microscopic structure discoveries of AD tau filaments may provide new insights for the binding interactions between various tau tracers and tangles, and spur refinements on the design of novel, subtype-selective tau tracers (Fitzpatrick et al., 2017).
Imaging Research Findings and Clinical Relevance
According to the amyloid cascade hypothesis, neurodegeneration characterized by misfolded tau tangle aggregation is the downstream event secondary to major amyloid deposition (Jack et al., 2013b). In contrast to the globally elevated pattern seen in brain amyloid PET of AD dementia patients, the spatiotemporal distribution of tau tracers follows a typical neuropathological sequence of spreading (Braak's stage), as have been demonstrated in both cross-sectional and longitudinal studies in a wide-range of subjects from advanced AD dementia patients to cognitively intact elderly controls (Rabinovici and Jagust, 2009; Johnson et al., 2016; Wooten et al., 2017; Villemagne et al., 2018). This quantitative and sequential connection indicates that this in vivo tangle spreading trajectory may reflect not merely neuronal dysfunction but also disease progression. In parallel to postmortem immunohistochemical findings, in vivo tau deposition varies amongst the brain regions, starting from the medial temporal lobe, i.e., hippocampus and entorhinal cortex (Braak stage I–II), to the adjacent neocortices (Braak stage III–IV), and finally to the entire brain (Braak stage V–VI) (Cho et al., 2019; Leuzy et al., 2019; Baek et al., 2020; Fleisher et al., 2020). As expected, clinical manifestation of AD is closely related to tau retention in the responsible brain regions. For instance, while temporal lobe deposition correlates well with memory performances, frontal lobe retention strongly correlates with execution and global cognition (Ossenkoppele et al., 2016; Bejanin et al., 2017; Shimada et al., 2017).
According to current experience with all the available tau tracers, AD is not likely to be the diagnosis of a tau-negative individual, while non-tauopathies can be basically excluded from the cause of cognitive decline for a tau-positive individual (Bischof et al., 2017). Non-AD tauopathies such as FTLD, PSP and CBD, however, manifest different distribution patterns and trajectory from AD involving different sub-regions in the brain stem and basal ganglia as well as cortical regions, which could be of additional differential diagnostic value in the clinical context (Bischof et al., 2017; Leuzy et al., 2019). Recently, tau deposition in cognitively intact healthy elderly subjects has also been revealed by PET and confirmed by autopsy results. This phenomenon is coined primary age-related tauopathy (PART) and believed to produce only minute, if any, clinical symptoms (Crary et al., 2014). In the absence of amyloid, PART alone is believed to be insufficient to develop memory decline (Harrison et al., 2019). Its relationship to suspected non-amyloid pathology (SNAP) has also been proposed, which presumably underlies the preclinical abnormalities during the development of AD (Jack, 2014).
Hypometabolism in brain regions that are commonly affected in advanced AD, including posterior cingulate cortex, precuneus and temporoparietal association cortex, represents synaptic dysfunction of the neurons in the course of AD (see below section on FDG imaging of glucose metabolism). Cortical tau tracer deposition can be seen in the same areas, establishing a typical topographic “yin-yang” offset between the two biomarkers. Further, correlation between these two imaging findings grows stronger as tau burden increases (Figure 3) (Whitwell et al., 2018; Lu et al., 2020). These unique phenomena demonstrate outstanding pathophysiological coherency of excessive MAPT retention and subsequent destruction of cytoarchitecture, resulting in synaptic metabolic deficit. On the other hand, multi-modality studies also reveal that the correspondence between frontal hypometabolism and medial temporal tau retention is present independent of amyloid deposition, which can be interpreted as aging-related.
Figure 3.

Representative images of [11C]PIB, [18F]APN1607, and [18F]FDG PET and MRI of an AD patient, an MCI patient and a high-risk asymptomatic subject. Aβ was positive for the AD patient and equivocal for the MCI patient and the high-risk asymptomatic subject. Typical Braak Stage V–VI tau deposition was seen in the AD brain, whereas no tau deposition was seen in the brain of the MCI patient or the high-risk asymptomatic subject. Typical AD-like glucose-hypometabolism was seen in AD brain including posterior cingulate cortex, parietal, temporal, and pre-frontal cortices, whereas most of these regions are relatively spared in the brain of MCI patient and high-risk asymptomatic subject. Images provided by PET Center, Huashan Hospital, Fudan University.
The relationship between tau aggregation and cortical atrophy as demonstrated by structural MRI is similar to that between tau aggregation and hypometabolism. However the former was found to be weaker than the later, especially in conditions other than mild AD (Sepulcre et al., 2016; Iaccarino et al., 2018). In addition to local correlation, tau deposition in the parietal lobe and precuneus can also be correlated to medial temporal lobe atrophy (Shimada et al., 2017). These observations seem plausible due to the fact that substantial structural alterations often relate to the later stage of the disease.
In general, CSF phosphorylated tau (p-tau, representing misfolded tangle formation) and total tau (t-tau, representing neurodegeneration) levels parallel in vivo tau tracer binding (Mattsson et al., 2018; Leuzy et al., 2019; Okafor et al., 2020). However, discordant results have also been found in small subgroups, possibly due to varying detectability around the CSF threshold and uneven rate of tau tangle formation in different disease stages (Murray et al., 2015; Thal et al., 2015). Although it is believed that elevated CSF tau levels precede tau imaging manifestation, tau PET has the advantage of visualization and topographic quantitation capacity, as well as relative non-invasiveness (Wolters et al., 2020).
Discussion
The research and development of tau PET tracers has gained undeniable progress in the last few years. Although some issues remain, especially isoform selectivity and other potential off-target binding, the applications of these tracers in AD imaging have provided, and are expected to continue to provide valuable information on the time course and topography of tau tangles in AD, and the correlation with cognitive dysfunction. Longitudinal and cross-sectional multi-target and multi-modality studies are encouraged to further elucidate the role of tau in the course of AD, as well as its interaction and relationship with Aβ deposition, synaptic dysfunction, brain atrophy, and other pathological biomarkers. Another important application of tau imaging is its utility for patient selection and endpoint measurements in phase 2 and phase 3 clinical trials of disease-modifying anti-tau treatments that have been gaining increasing impetus (Giacobini and Gold, 2013).
Coming from different compound families, the currently available tau tracers have different affinities for the various MAPT isoforms or tangle structures, hence distinctive topographic binding patterns in the same tauopathy. Selectivity to isoforms and structures could be a future direction to design new probes as 3R-specific or 4R-specific. We could therefore foresee a future tau imaging landscape where different tracers “rule” their own pieces of territory (i.e., specific tauopathy characterized by specific isoform or structure) in case that the correlation of tau imaging results similar to the “Aβ centiloid” is not achievable.
PET Tracer for Imaging Glucose Metabolism: [18F]FDG
Overview
As a radionuclide-labeled analog of glucose, the major metabolic substrate of neurons, [18F]fluorodeoxyglucose ([18F]FDG, or FDG) has long been used in the investigation of CNS disorders to reflect neuronal degeneration and injury. In fact the first report of this most widely used tracer was a brain imaging study (Phelps et al., 1979), although now the majority of its clinical application is for oncological purposes. The characteristic AD pathology (McGeer et al., 1986) and neurodegenerative changes (Mielke et al., 1996; Scholl et al., 2011) are associated with cortical hypometabolism demonstrated in pre-mortem FDG PET imaging. Clinical application of FDG PET in AD lies mainly in differential diagnosis from other causes of dementia, as well as treatment effect evaluation of disease-modifying or progression-slowing therapies. Clinical value of FDG PET has also been investigated for its predictive ability for conversion to AD dementia of high-risk subjects assumed to be in the prodromal or asymptomatic stage.
AD Dementia Patients
The characteristic manifestation of FDG PET in AD dementia is hypometabolism in the posterior part of cerebrum including the posterior cingulate cortex (PCC), precuneus (PrC), and parietotemporal association cortices such as the angular gyrus. These regions are the most discriminating components of AD-specific cerebral hypometabolic pattern as well as strong indicators of disease severity and progression. It is of significant differential value that PCC and PrC are substantially spared in other minor causes of senile dementia including frontotemporal lobe dementia (FTLD), dementia with Lewy bodies (DLB), Parkinson's Disease dementia (PDD), and vascular dementia (VaD). The hippocampus and entorhinal cortex are involved in the earliest stages of AD according to neuropathological findings based on NFT formation (Braak and Braak, 1997), however glucose metabolism in these areas cannot be readily distinguished between AD patients and normal controls. This is largely due to the frequent presence of medial temporal hypometabolism even in normal aging, and the partial volume effect (PVE) correlated with regional cortical atrophy. Frontal lobe hypometabolism is often associated with advancement, executive dysfunction, or atypical behavior in AD cases, although it could also be observed in normal aging. Occipital lobe hypometabolism is related to the posterior cortical atrophy (PCA) subtype of AD, but care should be taken when differentiating dementia types because this is also a prominent sign of DLB.
MCI Patients
Early differentiation of MCI due to AD is crucial since early therapeutic intervention is indicated to be beneficial at least in slowing disease progression (Vellas et al., 2007; Molinuevo et al., 2011). It should be noted, though, that the etiology of MCI is heterogeneous, meaning that approximately half of the patients convert to dementia other than AD, or do not convert at all (Rowe et al., 2010; Bennett et al., 2012; Frisoni et al., 2017). Therefore, whether FDG PET has discriminative and predictive ability for MCI patients is of great clinical relevance.
Hypometabolism revealed by FDG PET in typical AD-affected brain regions including the inferior parietal lobe, precuneus and posterior cingulate cortex is present early in prodromal AD, namely MCI stage. In a meta-analysis comparing the accuracy of 3 different imaging modalities, FDG PET (sensitivity = 88.8%, specificity = 84.9%) exhibits higher sensitivity and higher specificity than cerebral blood flow SPECT (sensitivity = 83.8%, specificity = 70.4%) and structural MRI (sensitivity = 72.8%, specificity = 81%) in terms of predicting short-term conversion to AD dementia (Yuan et al., 2009). Interestingly, FDG PET performs better in excluding non-converters than [11C]PIB Aβ PET (specificity: 74.0 vs. 56.2%), while its sensitivity is lower (sensitivity: 78.7 vs. 93.5%) (Zhang et al., 2012). This could be explained by the compensatory mechanism of preserved cerebral synaptic function against amyloid burden. Aside from the advantage of accurate short-term predictive ability, FDG PET can also exclude other potential etiology underlying MCI such as FTLD and DLB, in contrast to other biomarkers including CSF Aβ1−42 and t-tau/p-tau assays (Arbizu et al., 2018).
The value of FDG PET in MCI has been acknowledged in existing diagnostic criteria (Albert et al., 2011; Dubois et al., 2014). Meanwhile, future researches in this field are encouraged to overcome the various current limitations in terms of methodology normalization, gold standard verification, and effectiveness/economy evaluation (Arbizu et al., 2018).
High-Risk Asymptomatic Subjects
People with subjective cognitive decline (SCD) rather than objective evidence of cognitive impairment, subjects burdening amyloidosis, and family members of AD patients expressing PSEN1/2 or APP mutation were reported to have higher tendencies to develop to AD dementia (Bateman et al., 2011; Villemagne et al., 2011; Wolfsgruber et al., 2016). It is hypothesized that neurodegenerative alterations could have be silently undergoing in these subjects.
Various confounding factors lead to heterogeneity of the underlying etiology for SCD, leaving controversy in the regional metabolic manifestation of typical AD-affected areas in this subgroup (Scheef et al., 2012; Brugnolo et al., 2014; Van Der Gucht et al., 2015). Although amyloid-positive asymptomatic subjects, as defined by CSF Aβ1−42 or amyloid PET, will have higher risk of converting to AD in a life-long period, the utility of FDG PET reflecting neurodegeneration in this population is still not comparable to that in MCI patients, due to its poor performance in prediction of short-term conversion (Villemagne et al., 2011). Glucose metabolic abnormalities prior to the onset of clinical symptoms were observed in asymptomatic carriers of mutated APP and PSEN1/2 genes (Mosconi et al., 2008; Benzinger et al., 2013), who theoretically would suffer from dementia eventually. Nonetheless, the relationship between cerebral hypometabolism and time-to-conversion cannot be easily concluded in the absence of well-designed longitudinal studies.
In view of the above-mentioned facts, it is not recommended to clinically apply FDG PET to asymptomatic subjects with only one risk factor for diagnostic or prognostic purposes. Further investigation is needed to verify whether FDG PET is useful for individuals with multiple risk factors (Mosconi et al., 2008; Vannini et al., 2017).
Treatment Monitoring
FDG PET has been used as an imaging biomarker for outcome assessment in multiple clinical trials of AD therapeutics (Hoyer, 2002; Landau et al., 2011; Herholz, 2012). Global as well as sub-global or regional FDG uptake alterations following medication or surgical treatment has been observed in multiple clinical research studies (Nordberg et al., 1992; Heiss et al., 1994; Mega et al., 2001; Potkin et al., 2001; Tune et al., 2003; Schmidt et al., 2008; Tzimopoulou et al., 2010; Craft et al., 2012; Smith et al., 2012).
Discussion
FDG PET has been most widely used in dementia research and as an important adjunct imaging tool in the diagnosis of AD. On the whole, FDG PET enables early diagnosis of AD and thus early therapeutic intervention, as well as treatment strategy optimization in a large proportion of cases (Laforce et al., 2010; Elias et al., 2014). Delaying of disease progression and prevention of life quality deterioration as instructed by FDG PET can help lessen the overall healthcare expenditure (Banerjee and Wittenberg, 2009; Getsios et al., 2012).
Although quite a few critical issues in the utility of FDG PET for AD diagnosis have already been resolved, there are still many others that require verification with larger cohorts and better-designed trials (Garibotto et al., 2017).
PET Tracers for Imaging Neuroinflammation
Overview
Increasing evidence has helped with the formation of neuroinflammation hypothesis of AD etiology (McGeer and McGeer, 2010; Morales et al., 2014). Similar to the cases in other systems of the human body, it is now believed that the impact on CNS under different phases of the inflammatory process could be different. While acute inflammation could be protective to the brain against the harmful effects of invading pathogens and traumatic injuries, consistent stimulation by inflammatory factors could, on the other hand, be detrimental to the neurons and eventually induce neurodegeneration and loss of cognitive functions (Wyss-Coray and Mucke, 2002; Mrak and Griffin, 2005). It is also suggested that the neuroinflammation underlying AD starts from the earliest stages without any obvious clinical symptoms, and lasts to the end stage of the disease (Vehmas et al., 2003; Hoozemans et al., 2005). Therefore it is hypothesized that the clinical onset of AD dementia could be partly prevented or postponed by anti-inflammation interventions, which is based on results from retrospective observation studies and prospective trials (McGeer et al., 1996; Hoozemans et al., 2011). Multiple pathophysiological factors can trigger the process of immunoactivation in the CNS, in which complements, cytokines, growth factors, reactive oxygen species, microglia and astrocytes participate (Barger and Harmon, 1997; Akiyama et al., 2000). PET radiotracers have been developed to image these neuroinflammatory targets in vivo (Zimmer et al., 2014; Varley et al., 2015).
Tracer Development
The 18-kDa translocator protein (TSPO) is a mitochondrial protein. Under normal circumstances, it is low-expressed in specific brain regions in the microglia, the monocyte-macrophage-dendritic cell family member majorly active in the CNS and comprising ~15% of the non-neuronal cells within. TSPO is found to participate in amino acid and cholesterol transportation, and to serve as a “switch” in activating microglia from the resting state in response to various stimuli including infection and traumatic injury (Streit et al., 2004). Pro-inflammatory cytokines and neurotoxic substances are released by significantly proliferated microglia after the induction of soluble or fibrillary Aβ and subsequent TSPO regulation, which could be related to the fact that amyloid plaques are found to be colocalized with activated microglia in vivo in the brain where massive neuronal destruction and brain atrophy occur (McGeer et al., 1988; Venneti et al., 2009; Schilling and Eder, 2011). These characteristics made TSPO the predominant target for imaging in vivo inflammatory process in AD.
[11C]PK11195 was the first successfully developed TSPO tracer for human PET imaging (Figure 4) (Cagnin et al., 2001), but it remains controversial whether its binding is correlated with amyloid deposition, probably due to its low specific binding signal and hence low sensitivity to detect small changes in TSPO under disease conditions (Edison et al., 2008; Wiley et al., 2009; Yokokura et al., 2011). To overcome the shortcomings of [11C]PK11195, a series of 2nd generation TSPO tracers were developed and evaluated, including [11C]PBR28, [11C]DAA1106, [18F]DPA713, [18F]DPA714, [18F]FEPPA, [18F]FEMPA, and [18F]FEDAA1106 (Figure 4; Varley et al., 2015; Calsolaro and Edison, 2016; Edison et al., 2018). Though the majority of these radioligands are more sensitive than [11C]PK11195, subsequent studies found that their brain uptake are regulated by the TSPO gene rs6971 polymorphism, thus requiring phenotyping of individual subjects to match imaging results to their TSPO affinity status (Owen et al., 2012). More recently, 3rd generation, putative “phenotype-insensitive” TSPO probes such as [11C]ER176 (Figure 4) are being developed and evaluated (Wadsworth et al., 2012; Ikawa et al., 2017).
Figure 4.

Structures of representative PET radiotracers for TSPO imaging.
Similar to the paradigm of TSPO imaging of microglial activation, MAO-B is found to be elevated in reactive astrocytes and chosen as the target for in vivo imaging of neuroinflammation. The selective MAO-B tracer [11C]deuterium-L-deprenyl ([11C]DED) has been applied to studies of neurodegenerative disorders including AD (Carter et al., 2012). Type-2 imidazoline receptor, a newly discovered target for imaging astrocyte, is now being evaluated for its potential in differentiating AD and control using [11C]BU99008 under ongoing trial (Wilson et al., 2019).
Imaging Research Findings and Clinical Relevance
In vivo TSPO imaging is generally able to discriminate between AD dementia patients and normal control subjects (Cagnin et al., 2001; Yasuno et al., 2008; Suridjan et al., 2015; Varrone et al., 2015; Hamelin et al., 2016; Kreisl et al., 2016a). The majority of the multi-modality studies report that regional TSPO binding correlates to [11C]PIB retention positively, and FDG uptake as well as cortical volume negatively (Edison et al., 2008; Yokokura et al., 2011; Kreisl et al., 2013, 2016b) (Figure 5). In most cases the extent of TSPO binding is associated with not only baseline cognitive performance, but also its deterioration over time (Edison et al., 2008; Okello et al., 2009b; Yokokura et al., 2011; Kreisl et al., 2013, 2016a). Elevated TSPO binding in advanced AD dementia patients is seen in various cortical regions following an anticipated distribution pattern (Cagnin et al., 2001; Hamelin et al., 2016). Apart from global discrimination, regional TSPO binding is correlated to attenuated corresponding brain function. One study uncovered an inverse correlation between visuospatial function and [18F]FEPPA binding in the parietal and posterior internal capsule, as well as correlation between language ability and binding in the latter region (Suridjan et al., 2015). Interestingly, early-onset AD (EOAD) is reported to be associated with higher TSPO binding than late-onset AD (LOAD), especially in the frontal and parietal cortices, suggesting greater microglial activation in the former condition (Kreisl et al., 2013). In addition, a longitudinal study revealed that TSPO binding in MCI converters to AD dementia is drastically different from that of non-converters (Kreisl et al., 2016a). These results are concordant with previous finding that chronic microglia participation may be associated to the brain in AD progression (McGeer and McGeer, 2010; Morales et al., 2014).
Figure 5.
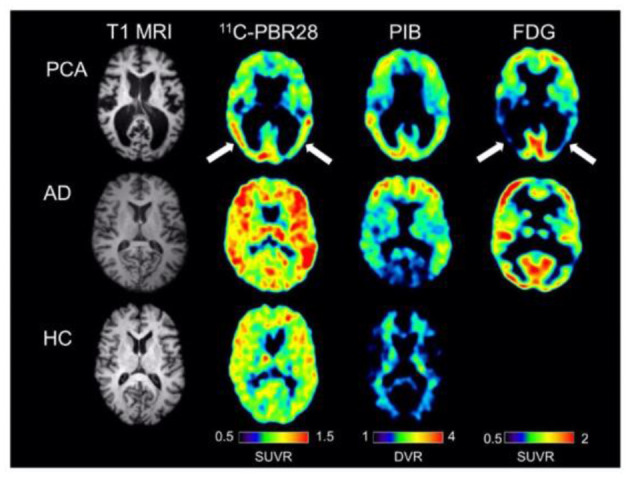
Single subject images from a patient with posterior cortical atrophy (top row), a patient with amnestic Alzheimer's disease (center row), and a healthy control subject (bottom row). The posterior cortical atrophy subject showed focal occipito-temporal [11C]PBR28 binding, with FDG hypometabolism in the same region (arrows). While the posterior cortical atrophy subjects showed occipito-temporal PIB binding, PIB binding was also found in frontal cortex. The subject with amnestic Alzheimer's disease showed more diffuse [11C]PBR28 binding, with occipital sparing on PIB and classic bilateral temporo-parietal hypometabolism on FDG imaging. The control subject showed low amounts of diffuse [11C]PBR28 binding and absence of cortical PIB [11C]PIB binding. Courtesy from Kreisl et al. (2016b).
However, conflicting results are also present in various aspects. Absence of the expected correlation between TSPO binding and cortical amyloid retention or neuronal metabolism has been repeatedly reported (Okello et al., 2009b; Wiley et al., 2009; Schuitemaker et al., 2013). There are also studies showing the inability of TSPO tracers to discriminate healthy subjects from patients with MCI or even advanced dementia (Okello et al., 2009b; Wiley et al., 2009; Schuitemaker et al., 2013; Golla et al., 2015). Likewise, significant association between TSPO binding and memory performance or disease severity is not always found (Yasuno et al., 2008; Schuitemaker et al., 2013). Single-nuclide polymorphism of rs6971 has been discovered to regulate TSPO binding in vivo and may partly contribute to the conflicting findings so far, since binding adjusted to individual phenotyping shows increased discriminating accuracy (Suridjan et al., 2015). In addition to doubts about the sensitivity of current TSPO tracers to detect subtle alterations in the prodromal stages of AD, there are data relating higher binding of [18F]DPA714 to slower cognitive decline, suggesting a neuroprotective role of microglial activation perhaps in the early phase of the disease (Hamelin et al., 2016).
According to a collective study with both [11C]DED and [11C]PIB, astrocytosis is most profound in amyloid-positive MCI subjects, followed by advanced AD patients as well as amyloid-negative MCI subjects and healthy controls (Carter et al., 2012; Rodriguez-Vieitez et al., 2016). This PET finding suggests that astrocytic reaction diminishes after prodromal AD converts to dementia, and is supported by postmortem autoradiographic study showing highest binding of MAO-B radioligand in the earliest Braak stages (Gulyas et al., 2011).
Discussion
It is universally acknowledged that neuroinflammation plays an active part in the course of AD. PET imaging of neuroinflammation targets have, in part, confirmed the correlation between Aβ deposition and elevated microglia/astrocyte activation/neuroinflammation. Nonetheless, in light of the complexity of both the process and components, questions such as “Is neuroinflammation beneficial at the beginning and harmful thereafter?” and “Is TSPO imaging and MAO-B imaging reflecting true activation of microglia and astrocytes?” remain to be answered. In order to obtain a clearer picture of the various aspects of inflammation in the CNS, further exploration is needed with some of the clues now at hand. First, microglia can be polarized to either M1, releasing neurotoxic substances, or to M2, releasing neuroprotective cytokines (Mosser and Edwards, 2008). Highly selective radioligands discriminating the two opposing activations will help answer these questions. Further, longitudinal studies, ideally tracking from the very early stage to the end stage of the disease across subjects, will be more informative than cross-sectional studies in mapping the time course and topography of microglia activation in disease progression. In addition, genotype-insensitive TSPO tracers under development require verification and validation in large-sample cohorts (Wadsworth et al., 2012; Zanotti-Fregonara et al., 2014; Ikawa et al., 2017). Finally, multi-modality imaging approaches will be helpful in revealing the interactions between neuroinflammation and various pathologic/pathophysiological components in AD.
PET Tracers for Imaging Targets in the Cholinergic System
Overview
The basal/rostral forebrain cholinergic pathways are believed to play an important role in a variety of neuropsychological functions including attention, consciousness and memory processing (Perry et al., 1999). Cholinergic replacement countering the loss of cholinergic neurotransmission in neurodegeneration is the theoretical basis of AD-treatment strategies using clinically approved medications (Bartus et al., 1982; Schliebs and Arendt, 2011). Cholinergic depletion and the resulting deficit in neuronal compensatory plasticity are confirmed in autopsy studies of AD patients (Bierer et al., 1995; Craig et al., 2011). PET imaging tracers targeting various aspects of cholinergic neurotransmission and metabolism can help us to better understand the role of cholinergic neuropathy and its interaction with other pathologic/pathophysiological components in the course of AD.
Tracer Development
Acetylcholinesterase (AChE) is the major target of AD medications, most of which are acetylcholinesterase inhibitors (AChEIs). These therapeutic agents, including galantamine, rivastigmine, tacrine and donepezil, block AChE to inhibit hydrolysis of ACh, thus increasing ACh level in the synaptic cleft. N-[11C]methyl-4-piperidinyl propionate ([11C]PMP) and N-[11C]methyl-4-piperidyl acetate ([11C]MP4A) are two selective substrates for AChE that have been successfully brought to in vivo human imaging research (Figure 6). Another presynaptic cholinergic PET tracer [18F]FEOBV, selective to the vesicle ACh transporter (VAChT), is reported to have been assessed in humans (Aghourian et al., 2017). Postsynaptic acetylcholine receptors can be classified into nicotinic acetylcholine receptors (nAChRs) and muscarinic acetylcholine receptors (mAChRs). While most of the previous human PET studies used α4β2 or non-selective nAChR tracers including [11C]nicotine, 2-[18F]F-A-85380 ([18F]2-FA), [18F]AZAN, and [18F]flubatine (also known as [18F]NCFHEB) (Figure 6) (Nordberg et al., 1990; Sabri et al., 2008, 2015b; Wong et al., 2013), successful development of selective tracers for new targets such α7 nAChR and M1 and M4 mAChR has been reported recently. Recent imaging evaluations in humans indicated that [18F]ASEM, [11C]LSN3172176, and [11C]MK-6884 (Figure 6) have appropriate kinetics and imaging properties and are promising tracers for their respective targets α7 nAChR and M1 and M4 mAChR (Hillmer et al., 2017; Wong et al., 2018; Masdeu et al., 2020; Naganawa et al., 2020a; Tong et al., 2020). [18F]ASEM and [11C]MK-6884 have been used in preliminary studies of α7 nAChR and M4 mAChR in AD (see below), while evaluation of M1 mAChR in AD is ongoing with [11C]LSN3172176 in our laboratories.
Figure 6.

Structures of representative PET radiotracers for cholinergic targets.
Imaging Research Findings and Clinical Relevance
AChE imaging using [11C]PMP and [11C]MP4A showed reduced cortical binding in AD patients than in healthy controls, especially in regions innervated with cholinergic projections (Iyo et al., 1997; Kuhl et al., 1999). AChE binding is lower in AD patients than in healthy controls, in parallel with VAChT reduction, and is further decreased by treatment with AChE inhibitors donepezil, rivastigmine and galantamine (Kuhl et al., 1999, 2000; Shinotoh et al., 2001; Kaasinen et al., 2002; Kadir et al., 2008). However this induced inhibition is absent for nAChR binding, suggesting that allosteric modulation effect maintains ACh signaling by up-regulating nAChR but not AChE (Maelicke et al., 2001). The degree of treatment-induced decline in AChE binding is more prominent in the frontal cortex than in the temporoparietal cortices, and correlated to improvement in frontal lobe functions such as execution and attention rather than episodic memory. Correlation between AChE hydrolysis and hypometabolism in the posterior cingulate cortex is also absent, while a connection between higher hydrolysis rate and APOE4 positivity exists (Kuhl et al., 1999; Eggers et al., 2006).
The binding of novel VAChT tracer [18F]FEOBV is found to be lower in AD than in healthy controls in a recent multi-tracer study involving [18F]FDG and [18F]NAV4694 as well (Aghourian et al., 2017). [18F]FEOBV PET is shown to have higher sensitivity than [18F]FDG in discriminating AD from healthy controls (Figure 7). There is also a positive correlation of [18F]FEOBV binding to minimal mental state examination (MMSE) and Montreal cognitive assessment (MoCA). A more recent study attributed basal forebrain degeneration in AD to the loss of cortico-amygdalar cholinergic input, as demonstrated by [18F]FEOBV binding decline (Schmitz et al., 2018).
Figure 7.
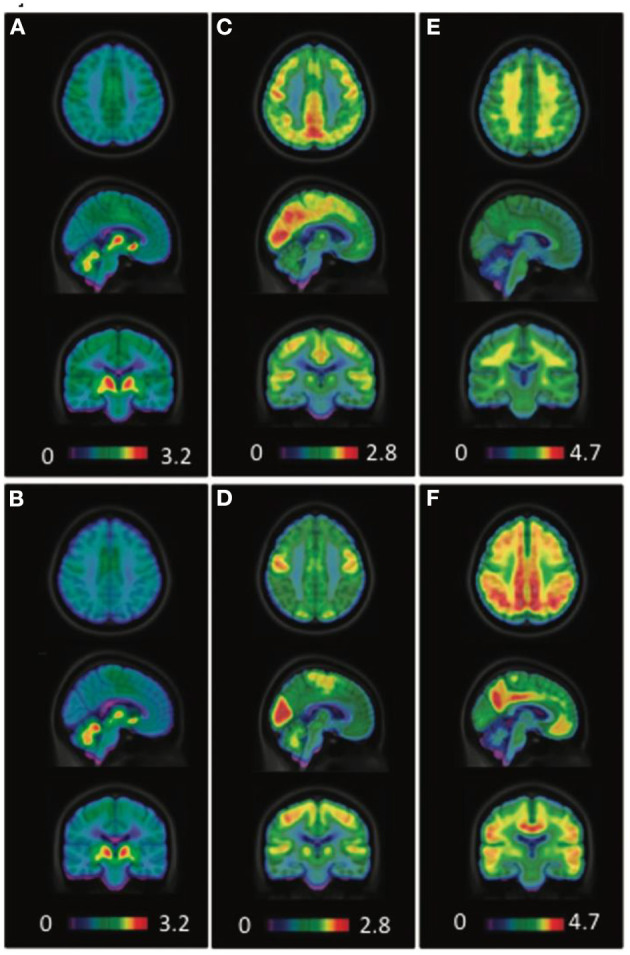
Averaged [18F]FEOBV VAChT PET (A,B), [18F]FDG PET (C,D), and [18F]NAV4694 Aβ PET (E,F) images of healthy controls (A,C,E) and AD patients (B,D,F). PET images in control subjects revealed the greatest [18F]FEOBV uptake in brain areas known to be the most innervated by the cholinergic systems, including the striatum, thalamus, cerebral cortex as a whole, hippocampal area and cerebellum (A). In AD patients, PET imaging revealed a significant reduction of [18F]FEOBV uptake in comparison with control subjects (B). Distinct cerebral hypometabolism (D) and amyloid deposition (F) were seen in the AD patients, as compared to control subjects (C,E). Courtesy from Aghourian et al. (2017).
Nicotinic AChR binding reduction is not only lower in AD and MCI subjects than in cognitively-intact volunteers, but also in MCI converters to AD dementia than in non-converters (Nordberg et al., 1990, 1995; Kendziorra et al., 2011). Binding quantitation of the AD-affected brain regions is significantly responsible for their coupling cognitive functions (Ellis et al., 2008; Sabri et al., 2008; Kendziorra et al., 2011; Okada et al., 2013). Frontal [11C]PIB retention is found to be inversely correlated with [18F]2-FA binding in the medial frontal cortex and nucleus basalis magnocellularis of AD patients (Maas et al., 2000). PET imaging with [18F]flubatine, a new-generation α4β2 tracer with more favorable kinetics, reveals receptor deficiency within the basal forebrain-cortical and septo-hippocampal cholinergic projections (Sabri et al., 2018). Episodic memory, working memory and executive functions are well-correlated with impairment of α4β2 nAChR in the corresponding cortices. Initial imaging studies with the α7 nAChR selective radioligand [18F]ASEM in healthy controls and MCI subjects indicated elevation of this receptor subtype with healthy aging, and in MCI, in a direction opposite of that for α4β2 nAChR in AD (Coughlin et al., 2018, 2019). A preliminary imaging study of M4 mAChR in AD revealed reduced uptake of [11C]MK-6884 primarily in the parietotemporal cortex in a pattern consistent with clinical symptom presentations and FDG hypometabolism (Masdeu et al., 2020).
Discussion
PET imaging studies with tracers for both pre- and post-synaptic cholinergic targets have confirmed the abnormalities of cholinergic transmission in vivo, in AD, as manifested in altered levels of AChE, VAChT, and α4β2 and α7 nAChR's, and their correlations with Aβ deposition and cognitive functions. The α7 nAChR is a target gaining increasing focus in recent years, due to its newly discovered participation in preventing amyloid toxicity and tau hyper-phosphorylation, in increasing synaptic strength and stability, and in modulating neuroinflammation by acting on non-neuronal cells (Wang et al., 2003; Conejero-Goldberg et al., 2008; Halff et al., 2014; de Oliveira et al., 2016; Maurer and Williams, 2017; Gamage et al., 2020). Renewed interests have also been seen in the mAChR, especially the M1 and M4 subtypes, for therapeutic development for AD (Levey, 1996; Schliebs and Arendt, 2011; Melancon et al., 2013). Investigation of these additional targets with recently available PET tracers will lead to a more thorough understanding of cholinergic involvement in AD etiology and progression.
PET Tracers for Imaging Synaptic Density
Overview
The total number of synapses in the neocortices is approximately 164 × 1012 (Tang et al., 2001). Synapses are critical for neurotransmission in neuron-neuron interaction, the deficiency of which would result in neuronal dysfunction and consequent occurrence of neuropsychiatric symptoms including amnesia, apathy and executive dysfunction. Loss of synapses has long been regarded as a pathologic hallmark of AD (Scheff et al., 1990; Terry et al., 1991; Scheff and Price, 2006) and correlates strongly with cognitive impairment (Hamos et al., 1989; DeKosky and Scheff, 1990; Terry et al., 1991; DeKosky et al., 1996; Robinson et al., 2014). Synaptic density reductions are seen in the neocortex and limbic system in MCI, a prodromal form of AD and other dementias (Masliah et al., 1994, 2001; Pham et al., 2010). Accumulation of toxic Aβ oligomers is believed to lead to the loss of synapses and presynaptic proteins in MCI patients (Pham et al., 2010; Wei et al., 2010; Beeri et al., 2012; Robinson et al., 2014). More recent research also suggests an emerging role for tau mediated toxicity at the synapses (Pooler et al., 2014; Wang and Mandelkow, 2016). Thus PET imaging of the synapses provides a tool for direct visualization of synaptic density loss along the AD pathogenesis and progression pathway.
Tracer Development
Synaptic vesicle glycoprotein 2 (SV2) is one of the synaptic proteins highly expressed on the presynaptic membrane. Among the 3 forms of SV2, only SV2A is extensively expressed in glutamatergic and GABAergic neurons throughout the CNS (Dong et al., 2006). Traditionally, synapses can only be observed by electron microscopic and immunohistochemical examinations in biopsy and autopsy tissue samples. Recently a series of novel PET radiotracers selectively targeting SV2A have been developed (Figure 8) (Mercier et al., 2014; Estrada et al., 2016; Nabulsi et al., 2016; Becker et al., 2017; Li et al., 2019; Cai et al., 2020a), enabling non-invasive observation and quantification of synaptic density in humans (Finnema et al., 2016; Bahri et al., 2017; Cai et al., 2020b; Naganawa et al., 2020b).
Figure 8.

Structures of representative PET radiotracers for SV2A.
Imaging Research Findings and Clinical Relevance
A recent human SV2A PET study using [11C]UCB-J in cohorts of MCI/AD patients and age-matched cognitively intact elderly subjects revealed significant reduction of synaptic density in the hippocampus and entorhinal cortex (44 and 27%, respectively) (Chen et al., 2018). Moreover, statistically significant correlations were found between hippocampal synaptic density and episodic memory (Logical Memory II and Rey Auditory Verbal Learning Test, R = 0.56, P = 0.01) and global function (Clinical Dementia Rating Sum of Boxes, R = −0.61, P = 0.003) (Chen et al., 2018). These findings suggest that [11C]UCB-J can capture the early pathological alterations in the hippocampus and entorhinal cortices, brain structures not readily analyzed in FDG PET compared to neocortices due to attenuated sensitivity introduced by size-related partial volume effect despite their early involvement in AD's Braak Staging (Braak and Braak, 1997).
The loss of synaptic density in the hippocampus of AD patients was replicated in a study using another SV2A radiotracer [18F]UCB-H, in which a 11 ~ 18% reductions of synaptic density in the basal forebrain and anterior/dorsomedial thalamus were found to be correlated with cognitive alterations (Bastin et al., 2020; Mecca et al., 2020). In a more recent [11C]UCB-J study with a larger cohort of 34 AD and MCI patients, the significant loss of synaptic density measured by PET in the hippocampus and entorhinal cortex followed by parahippocampal cortex, amygdala, lateral temporal cortex, pre-frontal cortex, posterior cingulate cortex/precuneus, lateral parietal cortex, and pericentral cortex was found to be generally correlated with atrophy measured by structural MRI which is in line with autopsy reports, and was more significantly correlated with CDR-SB (r = −0.54, P = 0.00003) and episodic memory (r = 0.56, P = 0.00001) than in the smaller sample reported earlier (de Wilde et al., 2016; Chen et al., 2018; Mecca et al., 2020). The relationship between cognitive performances and medial temporal (especially hippocampus and parahippocampal gyrus) synaptic density loss in aMCI was strengthened by a mutually correlated regional tau accumulation, as was revealed by a multi-tracer PET study using [18F]MK-6240 and [11C]UCB-J (Vanhaute et al., 2020).
Discussion
SV2A PET has shown great promise as a biomarker for the non-invasive detection of synaptic density, which can serve as a useful tool for studying synaptopathies in neurodegenerative and psychiatric disorders including AD (Cai et al., 2019; Heurling et al., 2019). Due to the ubiquitous distribution of synaptic vesicles throughout the CNS, a highly selective molecular probe such as SV2A PET tracer can spot the subtle alterations of synaptic density both quantitatively and topographically.
It is anticipated that with the help of multi-target molecular imaging approaches, the correlation between synaptic loss and other pathophysiological factors related to AD can be further elucidated. For instance, the relationship between pathological amyloid and tau protein aggregations, neuronal glucose utility impairment, dysfunctions in various neurotransmitter systems and synaptic loss would be better understood. Since pathological synaptic alterations emerge in the early stage of AD and continue throughout its course, many unanswered questions and controversial issues, e.g., sequence on the emergence of pathologic and physiological biomarkers, energetic, and neurotransmitter system abnormalities, and their relative impacts on disease progression, would be hopefully settled. It is perceivable that SV2A PET has the potential in AD early detection, differential diagnosis and conversion prediction of the prodromal stage subjects. Many studies using SV2A PET imaging in AD and other neurodegenerative diseases are ongoing, and results are emerging rapidly in publications. The introduction of additional 18F-labled SV2A radiotracers with favorable kinetics and high specific binding profiles, such as [18F]SynVesT-1 (also known as [18F]SDM-8 and [18F]MNI-1126) and [18F]SynVesT-2 (also known as [18F]SDM-2), should facilitate these studies since they are suitable for central production and distribution for use in off-site imaging facilities (Figures 8, 9). Further, PET imaging with these SV2A radiotracers will be useful as endpoint measure in AD drug clinical trials (e.g. NCT03493282: Effect of CT1812 treatment on brain synaptic density).
Figure 9.
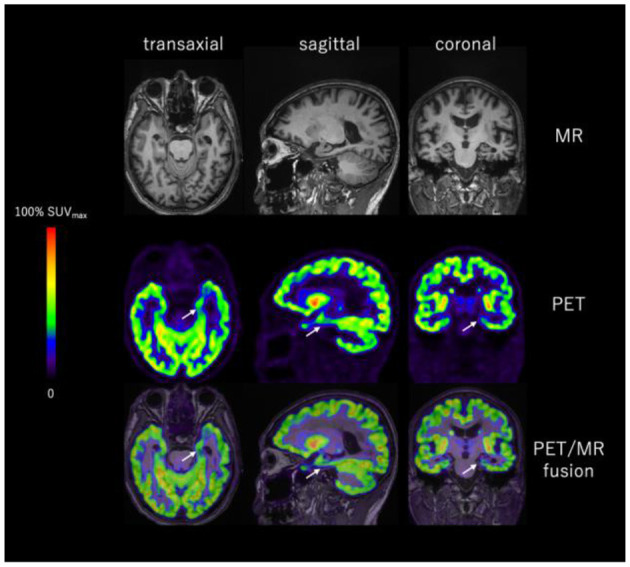
Representative images of T1WI MRI and [18F]SynVesT-1 PET of a [18F]Florbetapir positive AD patient. SV2A binding is significantly reduced in the left hippocampus (white arrow). Images provided by PET Center, Huashan Hospital, Fudan University and Department of Nuclear Medicine, East Hospital, Tongji University.
Summary and Concluding Remarks
Alzheimer's disease, characterized by impairments of cognitive functions and memory loss, is an extremely complex neurodegenerative disorder involving multiple pathophysiological processes, from loss of synapses and neurons, to neuroinflammation, to deposition of Aβ plaques and neurofibrillary tau tangles. Early diagnosis is the key to effectively treat the disease before manifestation of clinical symptoms. The past two decades have witnessed the development of PET imaging agents targeting various pathologic and physiological biomarkers in AD which has greatly contributed to the investigation and diagnosis of this disease.
Radiotracers targeting pathologic hallmarks of AD such as Aβ and tau proteins have allowed the differentiation of AD and non-AD dementia, the tracking of pathological protein burden over the temporal course of the disease, the stratification of patients for clinical trials, and the monitoring of biological effects of therapeutic agents. Nonetheless, there remained a few unanswered questions to be further studied concerning the relationship between these targets and the etiology of AD.
Pathophysiological alterations such as neuroinflammation as well as neurotransmitter and synaptic dysfunction are increasingly thought to commence from the earliest stage of the pathological changes in AD, and PET radiotracers targeting these biomarkers may help to elucidate their roles in AD progression in synergy with the core pathologies.
Measurement of synaptic density through PET imaging of the synaptic biomarker SV2A opens a new avenue for the investigation of neurodegenerative diseases. Initial in vivo imaging results from prodromal AD and AD dementia cohorts were in excellent correlation with cognitive impairment. Preliminary results of healthy aging subjects also demonstrated the early involvement of synaptic density alterations and its relationship to tau aggregation, which is in concordance with autopsy neuropathological findings. SV2A PET imaging, now at its early discovery phase, is expected to provide further important information on synaptic protein alterations, along with other pathological changes, during the entire AD progression continuum, and contribute to the early detection of the disease.
PET imaging of biomarkers has played an important role in AD drug development. This trend will continue, as newer imaging biomarkers are increasingly incorporated into clinical trials of next-generation AD drugs as outcome measures. It is expected that applications of PET imaging biomarkers in drug clinical trials will continue to greatly facilitate the development of AD therapeutics.
The United States National Institute of Aging (NIA) and the Alzheimer's Association (AA) recently put forward a research framework for AD, i.e., the NIA-AA Research Framework, which is based on the current understanding of the AD continuum and grounded on a biomarker-based definition of AD (Jack et al., 2018). Biomarkers are grouped into three categories: Aβ deposition, pathologic tau, and neurodegeneration (ATN) based on the nature of the pathologic process that each measures. Biomarkers of Aβ plaques (labeled “A”) are represented by cortical Aβ plaque burden assessed by amyloid PET imaging or decreased CSF Aβ42. Biomarkers of fibrillar tau (labeled “T”) are elevated CSF phosphorylated tau (p-tau) and/or cortical tau tangles measured by tau PET imaging. Biomarkers of neurodegeneration or neuronal injury (labeled “N”] are based on CSF total tau (t-tau), hypometabolism as revealed by FDG PET imaging, and brain atrophy detected by MRI. It is evident that PET imaging targeting various biomarkers is an essential component of this ATN research framework, as it provides one biomarker measurement for each of the arms: amyloid PET for A, tau PET for T, and FDG PET for N. The recent emergence of synaptic PET imaging targeting SV2A may provide another biomarker for N, neurodegeneration that more closely tracks with the progression of the disease and cognitive impairment, and less sensitive to cofounding factors such as blood glucose level, stimulation and medication which affect FDG PET, a surrogate measure of synaptic/neuronal function (Herholz, 2008; Burns et al., 2013; Ishibashi et al., 2015). These advancements in the development of PET radiotracers for AD biomarkers have been made possible through the efforts of numerous researchers in the PET imaging field in the last two decades. Collectively, AD imaging studies with various radiotracers have greatly increased our knowledge on the etiology and progression of this complex neurodegenerative disorder, and facilitated the research and development of AD therapeutics. It is believed that PET imaging with effective tracers targeting various AD pathophysiological biomarkers will continue to advance our understanding of the disease, and hopefully provides a sensitive and definitive tool for early diagnosis and monitoring of treatment effect.
Author Contributions
WB and YH drafted the manuscript. FX, CZ, and YG participated in revision of manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by grants (Project No. 82071962, 82021002, & 81971641) from the National Natural Science Foundation of China, grant (Project No. IDF151025) from Major Project Pre-research (pilot) Project of Fudan University, and grant 1R01AG065474 from the National Institute on Aging, National Institutes of Health, United States.
References
- Agdeppa E. D., Kepe V., Liu J., Flores-Torres S., Satyamurthy N., Petric A., et al. (2001). Binding characteristics of radiofluorinated 6-dialkylamino-2-naphthylethylidene derivatives as positron emission tomography imaging probes for β-amyloid plaques in Alzheimer's disease. J Neurosci. 21:RC189. 10.1523/JNEUROSCI.21-24-j0004.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghourian M., Legault-Denis C., Soucy J. P., Rosa-Neto P., Gauthier S., Kostikov A., et al. (2017). Quantification of brain cholinergic denervation in Alzheimer's disease using PET imaging with [18F]-FEOBV. Mol. Psychiatry 22, 1531–1538. 10.1038/mp.2017.183 [DOI] [PubMed] [Google Scholar]
- Akiyama H., Barger S., Barnum S., Bradt B., Bauer J., Cole G. M., et al. (2000). Inflammation and Alzheimer's disease. Neurobiol. Aging 21, 383–421. 10.1016/S0197-4580(00)00124-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert M. S., DeKosky S. T., Dickson D., Dubois B., Feldman H. H., Fox N. C., et al. (2011). The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the national institute on aging-alzheimer's association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer Dement. 7, 270–279. 10.1016/j.jalz.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadoru S., Dore V., McLean C. A., Hinton F., Shepherd C. E., Halliday G. M., et al. (2020). Comparison of amyloid PET measured in centiloid units with neuropathological findings in Alzheimer's disease. Alzheimers Res. Ther. 12:22. 10.1186/s13195-020-00587-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbizu J., Festari C., Altomare D., Walker Z., Bouwman F., Rivolta J., et al. (2018). Clinical utility of FDG-PET for the clinical diagnosis in MCI. Eur. J. Nucl. Med. Mol. Imaging 45, 1497–1508. 10.1007/s00259-018-4039-7 [DOI] [PubMed] [Google Scholar]
- Baek M. S., Cho H., Lee H. S., Choi J. Y., Lee J. H., Ryu Y. H., et al. (2020). Temporal trajectories of in vivo tau and amyloid-β accumulation in Alzheimer's disease. Eur. J. Nucl. Med. Mol. Imaging 47, 2879–2886. 10.1007/s00259-020-04773-3 [DOI] [PubMed] [Google Scholar]
- Bahri M. A., Plenevaux A., Aerts J., Bastin C., Becker G., Mercier J., et al. (2017). Measuring brain synaptic vesicle protein 2A with positron emission tomography and [18F]UCB-H. Alzheimers Dement. 3, 481–486. 10.1016/j.trci.2017.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S., Wittenberg R. (2009). Clinical and cost effectiveness of services for early diagnosis and intervention in dementia. Int. J. Geriatr. Psychiatry 24, 748–754. 10.1002/gps.2191 [DOI] [PubMed] [Google Scholar]
- Barger S. W., Harmon A. D. (1997). Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 388, 878–881. 10.1038/42257 [DOI] [PubMed] [Google Scholar]
- Barthel H., Gertz H. J., Dresel S., Peters O., Bartenstein P., Buerger K., et al. (2011). Cerebral amyloid-β PET with florbetaben (18F) in patients with Alzheimer's disease and healthy controls: a multicentre phase 2 diagnostic study. Lancet Neurol. 10, 424–435. 10.1016/S1474-4422(11)70077-1 [DOI] [PubMed] [Google Scholar]
- Bartus R. T., Dean R. L., III., Beer B., Lippa A. S. (1982). The cholinergic hypothesis of geriatric memory dysfunction. Science 217, 408–414. 10.1126/science.7046051 [DOI] [PubMed] [Google Scholar]
- Bastin C., Bahri M. A., Meyer F., Manard M., Delhaye E., Plenevaux A., et al. (2020). In vivo imaging of synaptic loss in Alzheimer's disease with [18F]UCB-H positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 47, 390–402. 10.1007/s00259-019-04461-x [DOI] [PubMed] [Google Scholar]
- Bateman R. J., Aisen P. S., De Strooper B., Fox N. C., Lemere C. A., Ringman J. M., et al. (2011). Autosomal-dominant Alzheimer's disease: a review and proposal for the prevention of Alzheimer's disease. Alzheimers Res. Ther. 3:1. 10.1186/alzrt59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle M. R., Pillay L. C., Lowe V. J., Knopman D., Kemp B., Rowe C. C., et al. (2018). Centiloid scaling for quantification of brain amyloid with [18F]flutemetamol using multiple processing methods. EJNMMI Res. 8:107. 10.1186/s13550-018-0456-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker G., Warnier C., Serrano M. E., Bahri M. A., Mercier J., Lemaire C., et al. (2017). Pharmacokinetic characterization of [18F]UCB-H PET radiopharmaceutical in the rat brain. Mol. Pharm. 14, 2719–2725. 10.1021/acs.molpharmaceut.7b00235 [DOI] [PubMed] [Google Scholar]
- Beeri M. S., Haroutunian V., Schmeidler J., Sano M., Fam P., Kavanaugh A., et al. (2012). Synaptic protein deficits are associated with dementia irrespective of extreme old age. Neurobiol. Aging. 33, 1125.e1–8. 10.1016/j.neurobiolaging.2011.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejanin A., Schonhaut D. R., La Joie R., Kramer J. H., Baker S. L., Sosa N., et al. (2017). Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer's disease. Brain 140, 3286–3300. 10.1093/brain/awx243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett D. A., Wilson R. S., Boyle P. A., Buchman A. S., Schneider J. A. (2012). Relation of neuropathology to cognition in persons without cognitive impairment. Ann. Neurol. 72, 599–609. 10.1002/ana.23654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzinger T. L., Blazey T., Jack C. R., Jr., Koeppe R. A., Su Y., Xiong C., et al. (2013). Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 110, E4502–E4509. 10.1073/pnas.1317918110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betthauser T., Lao P. J., Murali D., Barnhart T. E., Furumoto S., Okamura N., et al. (2017). In vivo comparison of tau radioligands 18F-THK-5351 and 18F-THK-5317. J. Nucl. Med. 58, 996–1002. 10.2967/jnumed.116.182980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierer L. M., Haroutunian V., Gabriel S., Knott P. J., Carlin L. S., Purohit D. P., et al. (1995). Neurochemical correlates of dementia severity in Alzheimer's disease: relative importance of the cholinergic deficits. J. Neurochem. 64, 749–760. 10.1046/j.1471-4159.1995.64020749.x [DOI] [PubMed] [Google Scholar]
- Bischof G. N., Endepols H., van Eimeren T., Drzezga A. (2017). Tau-imaging in neurodegeneration. Methods 130, 114–123. 10.1016/j.ymeth.2017.08.003 [DOI] [PubMed] [Google Scholar]
- Bourgeat P., Dore V., Fripp J., Ames D., Masters C. L., Salvado O., et al. (2018). Implementing the centiloid transformation for 11C-PiB and β-amyloid 18F-PET tracers using CapAIBL. Neuroimage 183, 387–393. 10.1016/j.neuroimage.2018.08.044 [DOI] [PubMed] [Google Scholar]
- Braak H., Braak E. (1997). Frequency of stages of alzheimer-related lesions in different age categories. Neurobiol. Aging 18, 351–357. 10.1016/S0197-4580(97)00056-0 [DOI] [PubMed] [Google Scholar]
- Brugnolo A., Morbelli S., Arnaldi D., De Carli F., Accardo J., Bossert I., et al. (2014). Metabolic correlates of Rey auditory verbal learning test in elderly subjects with memory complaints. J. Alzheimers Dis. 39, 103–113. 10.3233/JAD-121684 [DOI] [PubMed] [Google Scholar]
- Burns C. M., Chen K., Kaszniak A. W., Lee W., Alexander G. E., Bandy D., et al. (2013). Higher serum glucose levels are associated with cerebral hypometabolism in Alzheimer regions. Neurology 80, 1557–1564. 10.1212/WNL.0b013e31828f17de [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagnin A., Brooks D. J., Kennedy A. M., Gunn R. N., Myers R., Turkheimer F. E., et al. (2001). In-vivo measurement of activated microglia in dementia. Lancet 358, 461–467. 10.1016/S0140-6736(01)05625-2 [DOI] [PubMed] [Google Scholar]
- Cai Z., Drake L., Naganawa M., Najafzadeh S., Pracitto R., Lindermann M., et al. (2020b). First-in-human study of [18F]SynVesT-2, a novel SV2A radioligand with fast kinetics and high specific binding signals. J. Nucl. Med. 61 (Suppl. 1):462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z., Li S., Matuskey D., Nabulsi N., Huang Y. (2019). PET imaging of synaptic density: a new tool for investigation of neuropsychiatric diseases. Neurosci. Lett. 691, 44–50. 10.1016/j.neulet.2018.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z., Li S., Zhang W., Pracitto R., Wu X., Baum E., et al. (2020a). Synthesis and preclinical evaluation of an 18F-labeled synaptic vesicle glycoprotein 2A PET imaging probe: [18F]SynVesT-2. ACS Chem. Neurosci. 11, 592–603. 10.1021/acschemneuro.9b00618 [DOI] [PubMed] [Google Scholar]
- Calsolaro V., Edison P. (2016). Neuroinflammation in Alzheimer's disease: current evidence and future directions. Alzheimers Dement. 12, 719–732. 10.1016/j.jalz.2016.02.010 [DOI] [PubMed] [Google Scholar]
- Carter S. F., Scholl M., Almkvist O., Wall A., Engler H., Langstrom B., et al. (2012). Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med. 53, 37–46. 10.2967/jnumed.110.087031 [DOI] [PubMed] [Google Scholar]
- Chen M. K., Mecca A. P., Naganawa M., Finnema S. J., Toyonaga T., Lin S. F., et al. (2018). assessing synaptic density in Alzheimer disease With synaptic vesicle glycoprotein 2A positron emission tomographic imaging. JAMA Neurol. 75, 1215–1224. 10.1001/jamaneurol.2018.1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiotis K., Saint-Aubert L., Savitcheva I., Jelic V., Andersen P., Jonasson M., et al. (2016). Imaging in-vivo tau pathology in Alzheimer's disease with THK5317 PET in a multimodal paradigm. Eur. J. Nucl. Med. Mol. Imaging 43, 1686–1699. 10.1007/s00259-016-3363-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H., Choi J. Y., Lee H. S., Lee J. H., Ryu Y. H., Lee M. S., et al. (2019). Progressive tau accumulation in Alzheimer disease: 2-year follow-up study. J. Nucl. Med. 60, 1611–1621. 10.2967/jnumed.118.221697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark C. M., Pontecorvo M. J., Beach T. G., Bedell B. J., Coleman R. E., Doraiswamy P. M., et al. (2012). Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-b plaques: a prospective cohort study. Lancet Neurol. 11, 669–678. 10.1016/S1474-4422(12)70142-4 [DOI] [PubMed] [Google Scholar]
- Cohen A. D., Price J. C., Weissfeld L. A., James J., Rosario B. L., Bi W., et al. (2009). Basal cerebral metabolism may modulate the cognitive effects of Aβ in mild cognitive impairment: an example of brain reserve. J. Neurosci. 29, 14770–14778. 10.1523/JNEUROSCI.3669-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conejero-Goldberg C., Davies P., Ulloa L. (2008). Alpha7 nicotinic acetylcholine receptor: a link between inflammation and neurodegeneration. Neurosci. Biobehav. Rev. 32, 693–706. 10.1016/j.neubiorev.2007.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin J. M., Du Y., Rosenthal H. B., Slania S., Min Koo S., Park A., et al. (2018). The distribution of the α7 nicotinic acetylcholine receptor in healthy aging: an in vivo positron emission tomography study with [18F]ASEM. Neuroimage 165, 118–124. 10.1016/j.neuroimage.2017.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin J. M., Rubin L. H., Du Y., Rowe S. P., Crawford J. L., Rosenthal H. B., et al. (2019). High availability of the α7 nicotinic acetylcholine receptor in brains of individuals with mild cognitive impairment: a pilot study using 18F-ASEM PET. J. Nucl. Med. 61, 423–426. 10.2967/jnumed.119.230979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft S., Baker L. D., Montine T. J., Minoshima S., Watson G. S., Claxton A., et al. (2012). Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch. Neurol. 69, 29–38. 10.1001/archneurol.2011.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig L. A., Hong N. S., McDonald R. J. (2011). Revisiting the cholinergic hypothesis in the development of Alzheimer's disease. Neurosci. Biobehav. Rev. 35, 1397–1409. 10.1016/j.neubiorev.2011.03.001 [DOI] [PubMed] [Google Scholar]
- Crary J. F., Trojanowski J. Q., Schneider J. A., Abisambra J. F., Abner E. L., Alafuzoff I., et al. (2014). Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 128, 755–766. 10.1007/s00401-014-1349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cselenyi Z., Jonhagen M. E., Forsberg A., Halldin C., Julin P., Schou M., et al. (2012). Clinical validation of 18F-AZD4694, an amyloid-β-specific PET radioligand. J. Nucl. Med. 53, 415–424. 10.2967/jnumed.111.094029 [DOI] [PubMed] [Google Scholar]
- Curtis C., Gamez J. E., Singh U., Sadowsky C. H., Villena T., Sabbagh M. N., et al. (2015). Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol. 72, 287–294. 10.1001/jamaneurol.2014.4144 [DOI] [PubMed] [Google Scholar]
- de Oliveira A. S., Santiago F. E., Balioni L. F., Ferrari Mde F., Almeida M. C., Carrettiero D. C. (2016). BAG2 expression dictates a functional intracellular switch between the p38-dependent effects of nicotine on tau phosphorylation levels via the α7 nicotinic receptor. Exp Neurol. 275 (Pt. 1):69–77. 10.1016/j.expneurol.2015.10.005 [DOI] [PubMed] [Google Scholar]
- de Wilde M. C., Overk C. R., Sijben J. W., Masliah E. (2016). Meta-analysis of synaptic pathology in Alzheimer's disease reveals selective molecular vesicular machinery vulnerability. Alzheimers Dement. 12, 633–644. 10.1016/j.jalz.2015.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Declercq L., Rombouts F., Koole M., Fierens K., Marien J., Langlois X., et al. (2017). Preclinical evaluation of 18F-JNJ64349311, a novel PET tracer for tau imaging. J. Nucl. Med. 58, 975–981. 10.2967/jnumed.116.185199 [DOI] [PubMed] [Google Scholar]
- DeKosky S. T., Scheff S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann. Neurol. 27, 457–464. 10.1002/ana.410270502 [DOI] [PubMed] [Google Scholar]
- DeKosky S. T., Scheff S. W., Styren S. D. (1996). Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration 5, 417–421. 10.1006/neur.1996.0056 [DOI] [PubMed] [Google Scholar]
- Dong M., Yeh F., Tepp W. H., Dean C., Johnson E. A., Janz R., et al. (2006). SV2 is the protein receptor for botulinum neurotoxin A. Science 312, 592–596. 10.1126/science.1123654 [DOI] [PubMed] [Google Scholar]
- Dubois B., Feldman H. H., Jacova C., Dekosky S. T., Barberger-Gateau P., Cummings J., et al. (2007). Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 6, 734–746. 10.1016/S1474-4422(07)70178-3 [DOI] [PubMed] [Google Scholar]
- Dubois B., Feldman H. H., Jacova C., Hampel H., Molinuevo J. L., Blennow K., et al. (2014). Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. 13, 614–629. 10.1016/S1474-4422(14)70090-0 [DOI] [PubMed] [Google Scholar]
- Edison P., Archer H. A., Gerhard A., Hinz R., Pavese N., Turkheimer F. E., et al. (2008). Microglia, amyloid, and cognition in Alzheimer's disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol. Dis. 32, 412–419. 10.1016/j.nbd.2008.08.001 [DOI] [PubMed] [Google Scholar]
- Edison P., Archer H. A., Hinz R., Hammers A., Pavese N., Tai Y. F., et al. (2007). Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology 68, 501–508. 10.1212/01.wnl.0000244749.20056.d4 [DOI] [PubMed] [Google Scholar]
- Edison P., Donat C. K., Sastre M. (2018). In vivo imaging of glial activation in Alzheimer's disease. Front. Neurol. 9:625. 10.3389/fneur.2018.00625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers C., Herholz K., Kalbe E., Heiss W. D. (2006). Cortical acetylcholine esterase activity and ApoE4-genotype in Alzheimer disease. Neurosci. Lett. 408, 46–50. 10.1016/j.neulet.2006.08.061 [DOI] [PubMed] [Google Scholar]
- Elias A., Woodward M., Rowe C. C. (2014). Management impact of FDG-PET in dementia: results from a tertiary center memory clinic. J. Alzheimers Dis. 42, 885–892. 10.3233/JAD-132729 [DOI] [PubMed] [Google Scholar]
- Ellis J. R., Villemagne V. L., Nathan P. J., Mulligan R. S., Gong S. J., Chan J. G., et al. (2008). Relationship between nicotinic receptors and cognitive function in early Alzheimer's disease: a 2-[18F]fluoro-A-85380 PET study. Neurobiol. Learn. Mem. 90, 404–412. 10.1016/j.nlm.2008.05.006 [DOI] [PubMed] [Google Scholar]
- Estrada S., Lubberink M., Thibblin A., Sprycha M., Buchanan T., Mestdagh N., et al. (2016). [11C]UCB-A, a novel PET tracer for synaptic vesicle protein 2A. Nucl. Med. Biol. 43, 325–332. 10.1016/j.nucmedbio.2016.03.004 [DOI] [PubMed] [Google Scholar]
- Finnema S. J., Nabulsi N. B., Eid T., Detyniecki K., Lin S. F., Chen M. K., et al. (2016). Imaging synaptic density in the living human brain. Sci. Transl. Med. 8:348ra96. 10.1126/scitranslmed.aaf6667 [DOI] [PubMed] [Google Scholar]
- Fitzpatrick A. W. P., Falcon B., He S., Murzin A. G., Murshudov G., Garringer H. J., et al. (2017). Cryo-EM structures of tau filaments from Alzheimer's disease. Nature 547, 185–190. 10.1038/nature23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher A. S., Pontecorvo M. J., Devous M. D., Sr., Lu M., Arora A. K., Truocchio S. P., et al. (2020). Positron emission tomography imaging with [18F]flortaucipir and postmortem assessment of alzheimer disease neuropathologic changes. JAMA Neurol. 77, 829–839. 10.1001/jamaneurol.2020.0528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisoni G. B., Boccardi M., Barkhof F., Blennow K., Cappa S., Chiotis K., et al. (2017). Strategic roadmap for an early diagnosis of Alzheimer's disease based on biomarkers. Lancet Neurol. 16, 661–676. 10.1016/S1474-4422(17)30159-X [DOI] [PubMed] [Google Scholar]
- Gamage R., Wagnon I., Rossetti I., Childs R., Niedermayer G., Chesworth R., et al. (2020). Cholinergic modulation of glial function during aging and chronic neuroinflammation. Front. Cell. Neurosci. 14:577912. 10.3389/fncel.2020.577912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garibotto V., Herholz K., Boccardi M., Picco A., Varrone A., Nordberg A., et al. (2017). Clinical validity of brain fluorodeoxyglucose positron emission tomography as a biomarker for Alzheimer's disease in the context of a structured 5-phase development framework. Neurobiol. Aging 52, 183–195. 10.1016/j.neurobiolaging.2016.03.033 [DOI] [PubMed] [Google Scholar]
- Getsios D., Blume S., Ishak K. J., Maclaine G., Hernandez L. (2012). An economic evaluation of early assessment for Alzheimer's disease in the United Kingdom. Alzheimers Dement. 8, 22–30. 10.1016/j.jalz.2010.07.001 [DOI] [PubMed] [Google Scholar]
- Giacobini E., Gold G. (2013). Alzheimer disease therapy–moving from amyloid-β to tau. Nat. Rev. Neurol. 9, 677–686. 10.1038/nrneurol.2013.223 [DOI] [PubMed] [Google Scholar]
- Goedert M., Spillantini M. G. (2006). A century of Alzheimer's disease. Science 314, 777–781. 10.1126/science.1132814 [DOI] [PubMed] [Google Scholar]
- Golla S. S., Boellaard R., Oikonen V., Hoffmann A., van Berckel B. N., Windhorst A. D., et al. (2015). Quantification of [18F]DPA-714 binding in the human brain: initial studies in healthy controls and Alzheimer's disease patients. J. Cereb. Blood Flow Metab. 35, 766–772. 10.1038/jcbfm.2014.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guehl N. J., Wooten D. W., Yokell D. L., Moon S. H., Dhaynaut M., Katz S., et al. (2019). Evaluation of pharmacokinetic modeling strategies for in-vivo quantification of tau with the radiotracer [18F]MK6240 in human subjects. Eur. J. Nucl. Med. Mol. Imaging 46, 2099–2111. 10.1007/s00259-019-04419-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas B., Pavlova E., Kasa P., Gulya K., Bakota L., Varszegi S., et al. (2011). Activated MAO-B in the brain of Alzheimer patients, demonstrated by [11C]-L-deprenyl using whole hemisphere autoradiography. Neurochem. Int. 58, 60–68. 10.1016/j.neuint.2010.10.013 [DOI] [PubMed] [Google Scholar]
- Guo T., Landau S. M., Jagust W. J., Alzheimer's Disease Neuroimaging I. (2020). Detecting earlier stages of amyloid deposition using PET in cognitively normal elderly adults. Neurology 94, e1512–e1524. 10.1212/WNL.0000000000009216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halff A. W., Gomez-Varela D., John D., Berg D. K. (2014). A novel mechanism for nicotinic potentiation of glutamatergic synapses. J. Neurosci. 34, 2051–2064. 10.1523/JNEUROSCI.2795-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall B., Mak E., Cervenka S., Aigbirhio F. I., Rowe J. B., O'Brien J. T. (2017). In vivo tau PET imaging in dementia: pathophysiology, radiotracer quantification, and a systematic review of clinical findings. Ageing Res. Rev. 36, 50–63. 10.1016/j.arr.2017.03.002 [DOI] [PubMed] [Google Scholar]
- Hamelin L., Lagarde J., Dorothee G., Leroy C., Labit M., Comley R. A., et al. (2016). Early and protective microglial activation in Alzheimer's disease: a prospective study using 18F-DPA-714 PET imaging. Brain 139 (Pt. 4), 1252–1264. 10.1093/brain/aww017 [DOI] [PubMed] [Google Scholar]
- Hamos J. E., DeGennaro L. J., Drachman D. A. (1989). Synaptic loss in Alzheimer's disease and other dementias. Neurology 39, 355–361. 10.1212/WNL.39.3.355 [DOI] [PubMed] [Google Scholar]
- Hanseeuw B. J., Betensky R. A., Jacobs H. I. L., Schultz A. P., Sepulcre J., Becker J. A., et al. (2019). Association of amyloid and tau with cognition in preclinical alzheimer disease: a longitudinal study. JAMA Neurol. 76, 915–924. 10.1001/jamaneurol.2019.1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J., Allsop D. (1991). Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol. Sci. 12, 383–388. 10.1016/0165-6147(91)90609-V [DOI] [PubMed] [Google Scholar]
- Hardy J., Duff K., Hardy K. G., Perez-Tur J., Hutton M. (1998). Genetic dissection of Alzheimer's disease and related dementias: amyloid and its relationship to tau. Nat. Neurosci. 1, 355–358. 10.1038/1565 [DOI] [PubMed] [Google Scholar]
- Hardy J., Gwinn-Hardy K. (1998). Genetic classification of primary neurodegenerative disease. Science 282, 1075–1079. 10.1126/science.282.5391.1075 [DOI] [PubMed] [Google Scholar]
- Hardy J. A., Higgins G. A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185. 10.1126/science.1566067 [DOI] [PubMed] [Google Scholar]
- Harrison T. M., La Joie R., Maass A., Baker S. L., Swinnerton K., Fenton L., et al. (2019). Longitudinal tau accumulation and atrophy in aging and alzheimer disease. Ann. Neurol. 85, 229–240. 10.1002/ana.25406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiss W. D., Kessler J., Mielke R., Szelies B., Herholz K. (1994). Long-term effects of phosphatidylserine, pyritinol, and cognitive training in Alzheimer's disease. A neuropsychological, EEG, and PET investigation. Dementia 5, 88–98. 10.1159/000106702 [DOI] [PubMed] [Google Scholar]
- Herholz K. (2008). “Imaging of dementia,” in Diseases of the Brain, Head & Neck, Spine, eds J Hodler, G. K. Von Schulthess, C. L. Zollikofer (Milan: Springer; ), 215–8. 10.1007/978-88-470-0840-3_33 [DOI] [PubMed] [Google Scholar]
- Herholz K. (2012). Use of FDG PET as an imaging biomarker in clinical trials of Alzheimer's disease. Biomark. Med. 6, 431–439. 10.2217/bmm.12.51 [DOI] [PubMed] [Google Scholar]
- Heurling K., Ashton N. J., Leuzy A., Zimmer E. R., Blennow K., Zetterberg H., et al. (2019). Synaptic vesicle protein 2A as a potential biomarker in synaptopathies. Mol. Cell. Neurosci. 97, 34–42. 10.1016/j.mcn.2019.02.001 [DOI] [PubMed] [Google Scholar]
- Hillmer A. T., Li S., Zheng M. Q., Scheunemann M., Lin S. F., Nabulsi N., et al. (2017). PET imaging of α7 nicotinic acetylcholine receptors: a comparative study of [18F]ASEM and [18F]DBT-10 in nonhuman primates, and further evaluation of [18F]ASEM in humans. Eur. J. Nucl. Med. Mol. Imaging 44, 1042–1050. 10.1007/s00259-017-3621-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig L. S., Vellas B., Woodward M., Boada M., Bullock R., Borrie M., et al. (2018). Trial of solanezumab for mild dementia due to Alzheimer's disease. N. Engl. J. Med. 378, 321–330. 10.1056/NEJMoa1705971 [DOI] [PubMed] [Google Scholar]
- Hoover B. R., Reed M. N., Su J., Penrod R. D., Kotilinek L. A., Grant M. K., et al. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. 10.1016/j.neuron.2010.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoozemans J. J., van Haastert E. S., Veerhuis R., Arendt T., Scheper W., Eikelenboom P., et al. (2005). Maximal COX-2 and ppRb expression in neurons occurs during early Braak stages prior to the maximal activation of astrocytes and microglia in Alzheimer's disease. J. Neuroinflammation. 2:27. 10.1186/1742-2094-2-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoozemans J. J., Veerhuis R., Rozemuller J. M., Eikelenboom P. (2011). Soothing the inflamed brain: effect of non-steroidal anti-inflammatory drugs on Alzheimer's disease pathology. CNS Neurol. Disord. Drug Targets 10, 57–67. 10.2174/187152711794488665 [DOI] [PubMed] [Google Scholar]
- Hoyer S. (2002). The brain insulin signal transduction system and sporadic (type II) Alzheimer disease: an update. J. Neural. Transm. 109, 341–360. 10.1007/s007020200028 [DOI] [PubMed] [Google Scholar]
- Hsu J. L., Lin K. J., Hsiao I. T., Huang K. L., Liu C. H., Wu H. C., et al. (2020). The imaging features and clinical associations of a novel Tau PET Tracer-18F-APN1607 in Alzheimer disease. Clin. Nucl. Med. 45, 747–756. 10.1097/RLU.0000000000003164 [DOI] [PubMed] [Google Scholar]
- Iaccarino L., Tammewar G., Ayakta N., Baker S. L., Bejanin A., Boxer A. L., et al. (2018). Local and distant relationships between amyloid, tau and neurodegeneration in Alzheimer's disease. Neuroimage Clin. 17, 452–464. 10.1016/j.nicl.2017.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa M., Lohith T. G., Shrestha S., Telu S., Zoghbi S. S., Castellano S., et al. (2017). 11C-ER176, a radioligand for 18-kDa translocator protein, has adequate sensitivity to robustly image all three affinity genotypes in human brain. J. Nucl. Med. 58, 320–325. 10.2967/jnumed.116.178996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic M. D., Abrahamson E. E., Price J. C., Mathis C. A., Klunk W. E. (2016). [F-18]AV-1451 positron emission tomography retention in choroid plexus: more than “off-target” binding. Ann. Neurol. 80, 307–308. 10.1002/ana.24706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi K., Onishi A., Fujiwara Y., Ishiwata K., Ishii K. (2015). Relationship between Alzheimer disease-like pattern of 18F-FDG and fasting plasma glucose levels in cognitively normal volunteers. J. Nucl. Med. 56, 229–233. 10.2967/jnumed.114.150045 [DOI] [PubMed] [Google Scholar]
- Iyo M., Namba H., Fukushi K., Shinotoh H., Nagatsuka S., Suhara T., et al. (1997). Measurement of acetylcholinesterase by positron emission tomography in the brains of healthy controls and patients with Alzheimer's disease. Lancet 349, 1805–1809. 10.1016/S0140-6736(96)09124-6 [DOI] [PubMed] [Google Scholar]
- Jack C. R., Jr. (2014). PART and SNAP. Acta Neuropathol. 128, 773–776. 10.1007/s00401-014-1362-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R., Jr., Albert M. S., Knopman D. S., McKhann G. M., Sperling R. A., Carrillo M. C., et al. (2011). Introduction to the recommendations from the national institute on aging-alzheimer's association workgroups on diagnostic guidelines for alzheimer's Disease. Alzheimers Dement. 7, 257–262. 10.1016/j.jalz.2011.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R., Jr., Bennett D. A., Blennow K., Carrillo M. C., Dunn B., Haeberlein S. B., et al. (2018). NIA-AA research framework: toward a biological definition of alzheimer's Disease. Alzheimers Dement. 14, 535–562. 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R., Jr., Holtzman D. M. (2013). Biomarker modeling of Alzheimer's disease. Neuron 80, 1347–1358. 10.1016/j.neuron.2013.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R., Jr., Knopman D. S., Chetelat G., Dickson D., Fagan A. M., Frisoni G. B., et al. (2016). Suspected non-Alzheimer disease pathophysiology–concept and controversy. Nat. Rev. Neurol. 12, 117–124. 10.1038/nrneurol.2015.251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R., Jr., Knopman D. S., Jagust W. J., Petersen R. C., Weiner M. W., Aisen P. S., et al. (2013b). Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216. 10.1016/S1474-4422(12)70291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R., Jr., Wiste H. J., Lesnick T. G., Weigand S. D., Knopman D. S., Vemuri P., et al. (2013a). Brain β-amyloid load approaches a plateau. Neurology 80, 890–896. 10.1212/WNL.0b013e3182840bbe [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen W. J., Ossenkoppele R., Knol D. L., Tijms B. M., Scheltens P., Verhey F. R., et al. (2015). Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA 313, 1924–1938. 10.1001/jama.2015.4668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. A., Gregas M., Becker J. A., Kinnecom C., Salat D. H., Moran E. K., et al. (2007). Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann. Neurol. 62, 229–234. 10.1002/ana.21164 [DOI] [PubMed] [Google Scholar]
- Johnson K. A., Schultz A., Betensky R. A., Becker J. A., Sepulcre J., Rentz D., et al. (2016). Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann. Neurol. 79, 110–119. 10.1002/ana.24546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasinen V., Nagren K., Jarvenpaa T., Roivainen A., Yu M., Oikonen V., et al. (2002). Regional effects of donepezil and rivastigmine on cortical acetylcholinesterase activity in Alzheimer's disease. J. Clin. Psychopharmacol. 22, 615–620. 10.1097/00004714-200212000-00012 [DOI] [PubMed] [Google Scholar]
- Kadir A., Darreh-Shori T., Almkvist O., Wall A., Grut M., Strandberg B., et al. (2008). PET imaging of the in vivo brain acetylcholinesterase activity and nicotine binding in galantamine-treated patients with AD. Neurobiol. Aging 29, 1204–1217. 10.1016/j.neurobiolaging.2007.02.020 [DOI] [PubMed] [Google Scholar]
- Kendziorra K., Wolf H., Meyer P. M., Barthel H., Hesse S., Becker G. A., et al. (2011). Decreased cerebral nicotinic acetylcholine receptor availability in patients with mild cognitive impairment and Alzheimer's disease assessed with positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 38, 515–525. 10.1007/s00259-010-1644-5 [DOI] [PubMed] [Google Scholar]
- Kimura Y., Ichise M., Ito H., Shimada H., Ikoma Y., Seki C., et al. (2015). PET Quantification of tau pathology in human brain with 11C-PBB3. J. Nucl. Med. 56, 1359–1365. 10.2967/jnumed.115.160127 [DOI] [PubMed] [Google Scholar]
- Klunk W. E., Engler H., Nordberg A., Wang Y., Blomqvist G., Holt D. P., et al. (2004). Imaging brain amyloid in Alzheimer's disease with pittsburgh compound-B. Ann. Neurol. 55, 306–319. 10.1002/ana.20009 [DOI] [PubMed] [Google Scholar]
- Klunk W. E., Koeppe R. A., Price J. C., Benzinger T. L., Devous M. D., Sr., Jagust W. J., et al. (2015). The centiloid project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 11:1–15.e1–4. 10.1016/j.jalz.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koscik R. L., Betthauser T. J., Jonaitis E. M., Allison S. L., Clark L. R., Hermann B. P., et al. (2020). Amyloid duration is associated with preclinical cognitive decline and tau PET. Alzheimers Dement. 12:e12007. 10.1101/778415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisl W. C., Lyoo C. H., Liow J. S., Snow J., Page E., Jenko K. J., et al. (2016b). Distinct patterns of increased translocator protein in posterior cortical atrophy and amnestic Alzheimer's disease. Neurobiol. Aging 51, 132–140. 10.1016/j.neurobiolaging.2016.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisl W. C., Lyoo C. H., Liow J. S., Wei M., Snow J., Page E., et al. (2016a). 11C-PBR28 binding to translocator protein increases with progression of Alzheimer's disease. Neurobiol. Aging 44, 53–61. 10.1016/j.neurobiolaging.2016.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisl W. C., Lyoo C. H., McGwier M., Snow J., Jenko K. J., Kimura N., et al. (2013). In vivo radioligand binding to translocator protein correlates with severity of Alzheimer's disease. Brain 136 (Pt. 7):2228–2238. 10.1093/brain/awt145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl D. E., Koeppe R. A., Minoshima S., Snyder S. E., Ficaro E. P., Foster N. L., et al. (1999). In vivo mapping of cerebral acetylcholinesterase activity in aging and Alzheimer's disease. Neurology 52, 691–699. 10.1212/WNL.52.4.691 [DOI] [PubMed] [Google Scholar]
- Kuhl D. E., Minoshima S., Frey K. A., Foster N. L., Kilbourn M. R., Koeppe R. A. (2000). Limited donepezil inhibition of acetylcholinesterase measured with positron emission tomography in living Alzheimer cerebral cortex. Ann Neurol. 48, 391–395. [DOI] [PubMed] [Google Scholar]
- Kuwabara H., Comley R. A., Borroni E., Honer M., Kitmiller K., Roberts J., et al. (2018). Evaluation of 18F-RO-948 PET for quantitative assessment of tau accumulation in the human brain. J. Nucl. Med. 59, 1877–1884. 10.2967/jnumed.118.214437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforce R., Jr., Buteau J. P., Paquet N., Verret L., Houde M., Bouchard R. W. (2010). The value of PET in mild cognitive impairment, typical and atypical/unclear dementias: a retrospective memory clinic study. Am. J. Alzheimers Dis. Other Demen. 25:324–332. 10.1177/1533317510363468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau S. M., Harvey D., Madison C. M., Koeppe R. A., Reiman E. M., Foster N. L., et al. (2011). Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol. Aging 32, 1207–1218. 10.1016/j.neurobiolaging.2009.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzy A., Chiotis K., Lemoine L., Gillberg P. G., Almkvist O., Rodriguez-Vieitez E., et al. (2019). Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol. Psychiatry 24, 1112–1134. 10.1038/s41380-018-0342-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey A. I. (1996). Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 93:13541–13546. 10.1073/pnas.93.24.13541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Cai Z., Wu X., Holden D., Pracitto R., Kapinos M., et al. (2019). Synthesis and in vivo evaluation of a novel PET radiotracer for imaging of synaptic vesicle glycoprotein 2A (SV2A) in nonhuman primates. ACS Chem. Neurosci. 10, 1544–1554. 10.1021/acschemneuro.8b00526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister-James J., Pontecorvo M. J., Clark C., Joshi A. D., Mintun M. A., Zhang W., et al. (2011). Florbetapir F-18: a histopathologically validated β-amyloid positron emission tomography imaging agent. Semin. Nucl. Med. 41, 300–304. 10.1053/j.semnuclmed.2011.03.001 [DOI] [PubMed] [Google Scholar]
- Lowe V. J., Curran G., Fang P., Liesinger A. M., Josephs K. A., Parisi J. E., et al. (2016). An autoradiographic evaluation of AV-1451 tau PET in dementia. Acta Neuropathol. Commun. 4:58. 10.1186/s40478-016-0315-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., Bao W., Li M., Li L., Zhang Z., Alberts I., et al. (2020). Associations of [18F]-APN-1607 tau PET binding in the brain of alzheimer's disease patients with cognition and glucose metabolism. Front. Neurosci. 14:604. 10.3389/fnins.2020.00604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas T., Eidenmuller J., Brandt R. (2000). Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. J. Biol. Chem. 275, 15733–15740. 10.1074/jbc.M000389200 [DOI] [PubMed] [Google Scholar]
- Maelicke A., Samochocki M., Jostock R., Fehrenbacher A., Ludwig J., Albuquerque E. X., et al. (2001). Allosteric sensitization of nicotinic receptors by galantamine, a new treatment strategy for Alzheimer's disease. Biol. Psychiatry 49, 279–288. 10.1016/S0006-3223(00)01109-4 [DOI] [PubMed] [Google Scholar]
- Mallik A., Drzezga A., Minoshima S. (2017). Clinical amyloid imaging. Semin. Nucl. Med. 47, 31–43. 10.1053/j.semnuclmed.2016.09.005 [DOI] [PubMed] [Google Scholar]
- Masdeu J., Pascual B., Zanotti-Fregonara P., Yu M., Funk Q., Arbones V., et al. (2020). [11C]MK-6884 PET tracer for M4 muscarinic cholinergic receptors in Alzheimer's disease: comparison with [18F]FDG PET. Neurology 94 (15 Suppl):2640. 10.1016/j.jalz.2019.06.4256 [DOI] [Google Scholar]
- Masliah E., Mallory M., Alford M., DeTeresa R., Hansen L. A., McKeel D. W., Jr., et al. (2001). Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology 56, 127–129. 10.1212/WNL.56.1.127 [DOI] [PubMed] [Google Scholar]
- Masliah E., Mallory M., Hansen L., DeTeresa R., Alford M., Terry R. (1994). Synaptic and neuritic alterations during the progression of Alzheimer's disease. Neurosci. Lett. 174, 67–72. 10.1016/0304-3940(94)90121-X [DOI] [PubMed] [Google Scholar]
- Mattsson N., Smith R., Strandberg O., Palmqvist S., Scholl M., Insel P. S., et al. (2018). Comparing 18F-AV-1451 with CSF t-tau and p-tau for diagnosis of Alzheimer disease. Neurology 90, e388–e95. 10.1212/WNL.0000000000004887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer S. V., Williams C. L. (2017). The Cholinergic system modulates memory and hippocampal plasticity via its interactions with non-neuronal cells. Front. Immunol. 8:1489. 10.3389/fimmu.2017.01489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer E. G., McGeer P. L. (2010). Neuroinflammation in Alzheimer's disease and mild cognitive impairment: a field in its infancy. J. Alzheimers Dis. 19, 355–361. 10.3233/JAD-2010-1219 [DOI] [PubMed] [Google Scholar]
- McGeer P. L., Itagaki S., Boyes B. E., McGeer E. G. (1988). Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 38, 1285–1291. 10.1212/WNL.38.8.1285 [DOI] [PubMed] [Google Scholar]
- McGeer P. L., Kamo H., Harrop R., McGeer E. G., Martin W. R., Pate B. D., et al. (1986). Comparison of PET, MRI, and CT with pathology in a proven case of Alzheimer's disease. Neurology 36, 1569–1574. 10.1212/WNL.36.12.1569 [DOI] [PubMed] [Google Scholar]
- McGeer P. L., Schulzer M., McGeer E. G. (1996). Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology 47, 425–432. 10.1212/WNL.47.2.425 [DOI] [PubMed] [Google Scholar]
- McKhann G. M., Knopman D. S., Chertkow H., Hyman B. T., Jack C. R., Jr., Kawas C. H., et al. (2011). The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on aging-alzheimer's association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7, 263–269. 10.1016/j.jalz.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecca A. P., Chen M. K., O'Dell R. S., Naganawa M., Toyonaga T., Godek T. A., et al. (2020). In vivo measurement of widespread synaptic loss in Alzheimer's disease with SV2A PET. Alzheimers Dement. 16, 974–982. 10.1002/alz.12097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mega M. S., Cummings J. L., O'Connor S. M., Dinov I. D., Reback E., Felix J., et al. (2001). Cognitive and metabolic responses to metrifonate therapy in Alzheimer disease. Neuropsychiatry Neuropsychol. Behav. Neurol. 14, 63–68. [PubMed] [Google Scholar]
- Melancon B. J., Tarr J. C., Panarese J. D., Wood M. R., Lindsley C. W. (2013). Allosteric modulation of the M1 muscarinic acetylcholine receptor: improving cognition and a potential treatment for schizophrenia and Alzheimer's disease. Drug Discov. Today 18, 1185–1199. 10.1016/j.drudis.2013.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier J., Archen L., Bollu V., Carre S., Evrard Y., Jnoff E., et al. (2014). Discovery of heterocyclic nonacetamide synaptic vesicle protein 2A (SV2A) ligands with single-digit nanomolar potency: opening avenues towards the first SV2A positron emission tomography (PET) ligands. ChemMedChem 9, 693–698. 10.1002/cmdc.201300482 [DOI] [PubMed] [Google Scholar]
- Mielke R., Schroder R., Fink G. R., Kessler J., Herholz K., Heiss W. D. (1996). Regional cerebral glucose metabolism and postmortem pathology in Alzheimer's disease. Acta Neuropathol 91, 174–179. 10.1007/s004010050410 [DOI] [PubMed] [Google Scholar]
- Molinuevo J. L., Berthier M. L., Rami L. (2011). Donepezil provides greater benefits in mild compared to moderate Alzheimer's disease: implications for early diagnosis and treatment. Arch. Gerontol. Geriatr. 52, 18–22. 10.1016/j.archger.2009.11.004 [DOI] [PubMed] [Google Scholar]
- Morales I., Guzman-Martinez L., Cerda-Troncoso C., Farias G. A., Maccioni R. B. (2014). Neuroinflammation in the pathogenesis of Alzheimer's disease. A rational framework for the search of novel therapeutic approaches. Front. Cell Neurosci. 8:112. 10.3389/fncel.2014.00112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L., De Santi S., Brys M., Tsui W. H., Pirraglia E., Glodzik-Sobanska L., et al. (2008). Hypometabolism and altered cerebrospinal fluid markers in normal apolipoprotein E E4 carriers with subjective memory complaints. Biol. Psychiatry 63, 609–618. 10.1016/j.biopsych.2007.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser D. M., Edwards J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969. 10.1038/nri2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrak R. E., Griffin W. S. (2005). Glia and their cytokines in progression of neurodegeneration. Neurobiol. Aging 26, 349–354. 10.1016/j.neurobiolaging.2004.05.010 [DOI] [PubMed] [Google Scholar]
- Murray M. E., Lowe V. J., Graff-Radford N. R., Liesinger A. M., Cannon A., Przybelski S. A., et al. (2015). Clinicopathologic and 11C-pittsburgh compound B implications of thal amyloid phase across the Alzheimer's disease spectrum. Brain 138 (Pt. 5):1370–1381. 10.1093/brain/awv050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabulsi N. B., Mercier J., Holden D., Carre S., Najafzadeh S., Vandergeten M. C., et al. (2016). Synthesis and preclinical evaluation of 11C-UCB-J as a PET tracer for imaging the synaptic vesicle glycoprotein 2A in the brain. J. Nucl. Med. 57, 777–784. 10.2967/jnumed.115.168179 [DOI] [PubMed] [Google Scholar]
- Naganawa M., Li S., Nabulsi N. B., Henry S., Zheng M. Q., Pracitto R., et al. (2020b). First-in-human evaluation of 18F-SynVesT-1, a novel radioligand for PET imaging of synaptic vesicle protein 2A. J. Nucl. Med. 10.2967/jnumed.120.249144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganawa M., Nabulsi N. B., Henry S., Matuskey D., Lin S. F., Slieker L., et al. (2020a). First in human assessment of the novel M1 muscarinic acetylcholine Receptor PET radiotracer 11C-LSN3172176. J. Nucl. Med. 10.2967/jnumed.120.246967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navitsky M., Joshi A. D., Kennedy I., Klunk W. E., Rowe C. C., Wong D. F., et al. (2018). Standardization of amyloid quantitation with florbetapir standardized uptake value ratios to the Centiloid scale. Alzheimers Dement. 14, 1565–1571. 10.1016/j.jalz.2018.06.1353 [DOI] [PubMed] [Google Scholar]
- Ng K. P., Pascoal T. A., Mathotaarachchi S., Therriault J., Kang M. S., Shin M., et al. (2017). Monoamine oxidase B inhibitor, selegiline, reduces 18F-THK5351 uptake in the human brain. Alzheimers Res. Ther. 9:25. 10.1186/s13195-017-0253-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg A., Hartvig P., Lilja A., Viitanen M., Amberla K., Lundqvist H., et al. (1990). Decreased uptake and binding of 11C-nicotine in brain of Alzheimer patients as visualized by positron emission tomography. J. Neural Transm. Park. Dis. Dement. Sect. 2, 215–224. 10.1007/BF02257652 [DOI] [PubMed] [Google Scholar]
- Nordberg A., Lilja A., Lundqvist H., Hartvig P., Amberla K., Viitanen M., et al. (1992). Tacrine restores cholinergic nicotinic receptors and glucose metabolism in alzheimer patients as visualized by positron emission tomography. Neurobiol. Aging.13:747–758. 10.1016/0197-4580(92)90099-J [DOI] [PubMed] [Google Scholar]
- Nordberg A., Lundqvist H., Hartvig P., Lilja A., Langstrom B. (1995). Kinetic analysis of regional (S)(-) 11C-nicotine binding in normal and alzheimer brains–in vivo assessment using positron emission tomography. Alzheimer Dis. Assoc. Disord. 9, 21–27. 10.1097/00002093-199505000-00006 [DOI] [PubMed] [Google Scholar]
- Okada H., Ouchi Y., Ogawa M., Futatsubashi M., Saito Y., Yoshikawa E., et al. (2013). Alterations in α4β2 nicotinic receptors in cognitive decline in Alzheimer's aetiopathology. Brain 136 (Pt. 10), 3004–3017. 10.1093/brain/awt195 [DOI] [PubMed] [Google Scholar]
- Okafor M., Nye J. A., Shokouhi M., Shaw L. M., Goldstein F., Hajjar I. (2020). 18F-Flortaucipir PET associations with cerebrospinal fluid, cognition, and neuroimaging in mild cognitive impairment due to Alzheimer's disease. J. Alzheimers Dis. 74, 589–601. 10.3233/JAD-191330 [DOI] [PubMed] [Google Scholar]
- Okamura N., Furumoto S., Harada R., Tago T., Yoshikawa T., Fodero-Tavoletti M., et al. (2013). Novel 18F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J. Nucl. Med. 54, 1420–1427. 10.2967/jnumed.112.117341 [DOI] [PubMed] [Google Scholar]
- Okamura N., Harada R., Ishiki A., Kikuchi A., Nakamura T., Kudo Y. (2018). The development and validation of tau PET tracers: current status and future directions. Clin. Transl. Imaging 6, 305–316. 10.1007/s40336-018-0290-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura N., Suemoto T., Furumoto S., Suzuki M., Shimadzu H., Akatsu H., et al. (2005). Quinoline and benzimidazole derivatives: candidate probes for in vivo imaging of tau pathology in Alzheimer's disease. J. Neurosci. 25, 10857–10862. 10.1523/JNEUROSCI.1738-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okello A., Edison P., Archer H. A., Turkheimer F. E., Kennedy J., Bullock R., et al. (2009b). Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology 72, 56–62. 10.1212/01.wnl.0000338622.27876.0d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okello A., Koivunen J., Edison P., Archer H. A., Turkheimer F. E., Nagren K., et al. (2009a). Conversion of amyloid positive and negative MCI to AD over 3 years: an 11C-PIB PET study. Neurology 73, 754–760. 10.1212/WNL.0b013e3181b23564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M., Sahara N., Kumata K., Ji B., Ni R., Koga S., et al. (2017). Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain 140, 764–780. 10.1093/brain/aww339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R., Jansen W. J., Rabinovici G. D., Knol D. L., van der Flier W. M., van Berckel B. N., et al. (2015). Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA 313, 1939–1949. 10.1001/jama.2015.4669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R., Schonhaut D. R., Scholl M., Lockhart S. N., Ayakta N., Baker S. L., et al. (2016). Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain 139 (Pt. 5), 1551–1567. 10.1093/brain/aww027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R., Yeo A. J., Gunn R. N., Song K., Wadsworth G., Lewis A., et al. (2012). An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab. 32, 1–5. 10.1038/jcbfm.2011.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp K. V., Buckley R., Mormino E., Maruff P., Villemagne V. L., Masters C. L., et al. (2020). Clinical meaningfulness of subtle cognitive decline on longitudinal testing in preclinical AD. Alzheimers Dement. 16, 552–560. 10.1016/j.jalz.2019.09.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passamonti L., Vazquez Rodriguez P., Hong Y. T., Allinson K. S., Williamson D., Borchert R. J., et al. (2017). 18F-AV-1451 positron emission tomography in Alzheimer's disease and progressive supranuclear palsy. Brain 140, 781–791. 10.1093/brain/aww340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry E., Walker M., Grace J., Perry R. (1999). Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends Neurosci. 22, 273–280. 10.1016/S0166-2236(98)01361-7 [DOI] [PubMed] [Google Scholar]
- Petrou M., Dwamena B. A., Foerster B. R., MacEachern M. P., Bohnen N. I., Muller M. L., et al. (2015). Amyloid deposition in Parkinson's disease and cognitive impairment: a systematic review. Mov. Disord. 30, 928–935. 10.1002/mds.26191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham E., Crews L., Ubhi K., Hansen L., Adame A., Cartier A., et al. (2010). Progressive accumulation of amyloid-β oligomers in Alzheimer's disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 277, 3051–3067. 10.1111/j.1742-4658.2010.07719.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps M. E., Huang S. C., Hoffman E. J., Selin C., Sokoloff L., Kuhl D. E. (1979). Tomographic measurement of local cerebral glucose metabolic rate in humans with (F-18)2-fluoro-2-deoxy-D-glucose: validation of method. Ann. Neurol. 6, 371–388. 10.1002/ana.410060502 [DOI] [PubMed] [Google Scholar]
- Pike K. E., Ellis K. A., Villemagne V. L., Good N., Chetelat G., Ames D., et al. (2011). Cognition and β-amyloid in preclinical Alzheimer's disease: data from the AIBL study. Neuropsychologia 49, 2384–2390. 10.1016/j.neuropsychologia.2011.04.012 [DOI] [PubMed] [Google Scholar]
- Pike K. E., Savage G., Villemagne V. L., Ng S., Moss S. A., Maruff P., et al. (2007). β-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer's disease. Brain 130 (Pt. 11), 2837–2844. 10.1093/brain/awm238 [DOI] [PubMed] [Google Scholar]
- Pooler A. M., Noble W., Hanger D. P. (2014). A role for tau at the synapse in Alzheimer's disease pathogenesis. Neuropharmacology 76 (Pt. A):1–8. 10.1016/j.neuropharm.2013.09.018 [DOI] [PubMed] [Google Scholar]
- Potkin S. G., Anand R., Fleming K., Alva G., Keator D., Carreon D., et al. (2001). Brain metabolic and clinical effects of rivastigmine in Alzheimer's disease. Int. J. Neuropsychopharmacol 4, 223–230. 10.1017/S1461145701002528 [DOI] [PubMed] [Google Scholar]
- Querfurth H. W., LaFerla F. M. (2010). Alzheimer's disease. N. Engl. J. Med. 362, 329–344. 10.1056/NEJMra0909142 [DOI] [PubMed] [Google Scholar]
- Rabinovici G. D., Jagust W. J. (2009). Amyloid imaging in aging and dementia: testing the amyloid hypothesis in vivo. Behav. Neurol. 21, 117–128. 10.1155/2009/609839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne J. O., Wong D. F., Wolk D. A., Leinonen V., Arnold S. E., Buckley C., et al. (2012). [18F]Flutemetamol PET imaging and cortical biopsy histopathology for fibrillar amyloid β detection in living subjects with normal pressure hydrocephalus: pooled analysis of four studies. Acta Neuropathol. 124, 833–845. 10.1007/s00401-012-1051-z [DOI] [PubMed] [Google Scholar]
- Robinson J. L., Molina-Porcel L., Corrada M. M., Raible K., Lee E. B., Lee V. M., et al. (2014). Perforant path synaptic loss correlates with cognitive impairment and Alzheimer's disease in the oldest-old. Brain 137 (Pt. 9), 2578–2587. 10.1093/brain/awu190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Vieitez E., Carter S. F., Chiotis K., Saint-Aubert L., Leuzy A., Scholl M., et al. (2016). Comparison of early-phase 11C-Deuterium-l-Deprenyl and 11C-pittsburgh compound B PET for assessing brain perfusion in Alzheimer disease. J. Nucl. Med. 57, 1071–1077. 10.2967/jnumed.115.168732 [DOI] [PubMed] [Google Scholar]
- Rombouts F. J. R., Declercq L., Andres J. I., Bottelbergs A., Chen L., Iturrino L., et al. (2019). Discovery of N-(4-[18F]Fluoro-5-methylpyridin-2-yl)isoquinolin-6-amine (JNJ-64326067), a new promising tau positron emission tomography imaging tracer. J. Med. Chem. 62, 2974–2987. 10.1021/acs.jmedchem.8b01759 [DOI] [PubMed] [Google Scholar]
- Rowe C. C., Ackerman U., Browne W., Mulligan R., Pike K. L., O'Keefe G., et al. (2008). Imaging of amyloid β in Alzheimer's disease with 18F-BAY94-9172, a novel PET tracer: proof of mechanism. Lancet Neurol. 7, 129–135. 10.1016/S1474-4422(08)70001-2 [DOI] [PubMed] [Google Scholar]
- Rowe C. C., Dore V., Jones G., Baxendale D., Mulligan R. S., Bullich S., et al. (2017). 18F-Florbetaben PET β-amyloid binding expressed in Centiloids. Eur. J. Nucl. Med. Mol. Imaging 44, 2053–2059. 10.1007/s00259-017-3749-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe C. C., Ellis K. A., Rimajova M., Bourgeat P., Pike K. E., Jones G., et al. (2010). Amyloid imaging results from the Australian imaging, biomarkers and lifestyle (AIBL) study of aging. Neurobiol. Aging 31, 1275–1283. 10.1016/j.neurobiolaging.2010.04.007 [DOI] [PubMed] [Google Scholar]
- Rowe C. C., Jones G., Dore V., Pejoska S., Margison L., Mulligan R. S., et al. (2016). Standardized expression of 18F-NAV4694 and 11C-PiB β-amyloid PET results with the centiloid scale. J. Nucl. Med. 57, 1233–1237. 10.2967/jnumed.115.171595 [DOI] [PubMed] [Google Scholar]
- Rowe C. C., Villemagne V. L. (2011). Brain amyloid imaging. J. Nucl. Med. 52, 1733–1740. 10.2967/jnumed.110.076315 [DOI] [PubMed] [Google Scholar]
- Sabri O., Becker G. A., Meyer P. M., Hesse S., Wilke S., Graef S., et al. (2015b). First-in-human PET quantification study of cerebral nicotinic acetylcholine receptors using the novel specific radioligand (-)-[18F]Flubatine. Neuroimage 118, 199–208. 10.1016/j.neuroimage.2015.05.065 [DOI] [PubMed] [Google Scholar]
- Sabri O., Kendziorra K., Wolf H., Gertz H. J., Brust P. (2008). Acetylcholine receptors in dementia and mild cognitive impairment. Eur. J. Nucl. Med. Mol. Imaging (35 Suppl. 1), S30–45. 10.1007/s00259-007-0701-1 [DOI] [PubMed] [Google Scholar]
- Sabri O., Meyer P. M., Graf S., Hesse S., Wilke S., Becker G. A., et al. (2018). Cognitive correlates of α4β2 nicotinic acetylcholine receptors in mild Alzheimer's dementia. Brain 141, 1840–1854. 10.1093/brain/awy099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabri O., Sabbagh M. N., Seibyl J., Barthel H., Akatsu H., Ouchi Y., et al. (2015a). Florbetaben PET imaging to detect amyloid β plaques in Alzheimer's disease: phase 3 study. Alzheimers Dement. 11, 964–974. 10.1016/j.jalz.2015.02.004 [DOI] [PubMed] [Google Scholar]
- Salloway S., Sperling R., Fox N. C., Blennow K., Klunk W., Raskind M., et al. (2014). Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N. Engl. J. Med 370, 322–333. 10.1056/NEJMoa1304839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheef L., Spottke A., Daerr M., Joe A., Striepens N., Kolsch H., et al. (2012). Glucose metabolism, gray matter structure, and memory decline in subjective memory impairment. Neurology 79, 1332–1339. 10.1212/WNL.0b013e31826c1a8d [DOI] [PubMed] [Google Scholar]
- Scheff S. W., DeKosky S. T., Price D. A. (1990). Quantitative assessment of cortical synaptic density in Alzheimer's disease. Neurobiol. Aging 11, 29–37. 10.1016/0197-4580(90)90059-9 [DOI] [PubMed] [Google Scholar]
- Scheff S. W., Price D. A. (2006). Alzheimer's disease-related alterations in synaptic density: neocortex and hippocampus. J. Alzheimers Dis. 9 (3 Suppl), 101–115. 10.3233/JAD-2006-9S312 [DOI] [PubMed] [Google Scholar]
- Schilling T., Eder C. (2011). Amyloid-β-induced reactive oxygen species production and priming are differentially regulated by ion channels in microglia. J. Cell. Physiol. 226, 3295–3302. 10.1002/jcp.22675 [DOI] [PubMed] [Google Scholar]
- Schliebs R., Arendt T. (2011). The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 221, 555–563. 10.1016/j.bbr.2010.11.058 [DOI] [PubMed] [Google Scholar]
- Schmidt M. E., Janssens L., Moechars D., Rombouts F. J. R., Timmers M., Barret O., et al. (2020). Clinical evaluation of [18F]JNJ-64326067, a novel candidate PET tracer for the detection of tau pathology in Alzheimer's disease. Eur. J. Nucl. Med. Mol. Imaging 47, 3176–3185. 10.1007/s00259-020-04880-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt R., Ropele S., Pendl B., Ofner P., Enzinger C., Schmidt H., et al. (2008). Longitudinal multimodal imaging in mild to moderate Alzheimer disease: a pilot study with memantine. J. Neurol. Neurosurg. Psychiatry 79, 1312–1317. 10.1136/jnnp.2007.141648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz T. W., Mur M., Aghourian M., Bedard M. A., Spreng R. N., Alzheimer's Disease Neuroimaging I. (2018). Longitudinal alzheimer's degeneration reflects the spatial topography of cholinergic basal forebrain projections. Cell Rep. 24, 38–46. 10.1016/j.celrep.2018.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl M., Almkvist O., Bogdanovic N., Wall A., Langstrom B., Viitanen M., et al. (2011). Time course of glucose metabolism in relation to cognitive performance and postmortem neuropathology in Met146Val PSEN1 mutation carriers. J. Alzheimers Dis. 24, 495–506. 10.3233/JAD-2011-101563 [DOI] [PubMed] [Google Scholar]
- Schuitemaker A., Kropholler M. A., Boellaard R., van der Flier W. M., Kloet R. W., van der Doef T. F., et al. (2013). Microglial activation in Alzheimer's disease: an (R)-[11C]PK11195 positron emission tomography study. Neurobiol. Aging 34, 128–136. 10.1016/j.neurobiolaging.2012.04.021 [DOI] [PubMed] [Google Scholar]
- Sepulcre J., Schultz A. P., Sabuncu M., Gomez-Isla T., Chhatwal J., Becker A., et al. (2016). In vivo tau, amyloid, and gray matter profiles in the aging brain. J. Neurosci. 36, 7364–7374. 10.1523/JNEUROSCI.0639-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada H., Kitamura S., Shinotoh H., Endo H., Niwa F., Hirano S., et al. (2017). Association between Aβ and tau accumulations and their influence on clinical features in aging and Alzheimer's disease spectrum brains: A [11C]PBB3-PET study. Alzheimers Dement. 6, 11–20. 10.1016/j.dadm.2016.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinotoh H., Aotsuka A., Fukushi K., Nagatsuka S., Tanaka N., Ota T., et al. (2001). Effect of donepezil on brain acetylcholinesterase activity in patients with AD measured by PET. Neurology.56:408–410. 10.1212/WNL.56.3.408 [DOI] [PubMed] [Google Scholar]
- Smith G. S., Laxton A. W., Tang-Wai D. F., McAndrews M. P., Diaconescu A. O., Workman C. I., et al. (2012). Increased cerebral metabolism after 1 year of deep brain stimulation in Alzheimer disease. Arch. Neurol. 69, 1141–1148. 10.1001/archneurol.2012.590 [DOI] [PubMed] [Google Scholar]
- Sperling R. A., Aisen P. S., Beckett L. A., Bennett D. A., Craft S., Fagan A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer's disease: recommendations from the national institute on aging-alzheimer's association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7, 280–292. 10.1016/j.jalz.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R. A., Donohue M. C., Raman R., Sun C. K., Yaari R., Holdridge K., et al. (2020). Association of factors with elevated amyloid burden in clinically normal older individuals. JAMA Neurol. 77, 735–745. 10.1001/jamaneurol.2020.0387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini M. G., Goedert M. (2013). Tau pathology and neurodegeneration. Lancet Neurol. 12, 609–622. 10.1016/S1474-4422(13)70090-5 [DOI] [PubMed] [Google Scholar]
- Streit W. J., Mrak R. E., Griffin W. S. (2004). Microglia and neuroinflammation: a pathological perspective. J. Neuroinflammation 1:14. 10.1186/1742-2094-1-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y., Fu J., Yu J., Zhao Q., Guan Y., Zuo C., et al. (2020). Tau PET Imaging with [18F]PM-PBB3 in frontotemporal dementia with MAPT mutation. J. Alzheimers Dis. 76, 149–157. 10.3233/JAD-200287 [DOI] [PubMed] [Google Scholar]
- Suridjan I., Pollock B. G., Verhoeff N. P., Voineskos A. N., Chow T., Rusjan P. M., et al. (2015). In-vivo imaging of grey and white matter neuroinflammation in Alzheimer's disease: a positron emission tomography study with a novel radioligand, [18F]-FEPPA. Mol. Psychiatry 20, 1579–1587. 10.1038/mp.2015.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagai K., Ono M., Kubota M., Kitamura S., Takahata K., Seki C., et al. (2021). High-Contrast in vivo imaging of tau pathologies in alzheimer's and non-alzheimer's disease tauopathies. Neuron 109:42–58.e8. 10.1016/j.neuron.2020.09.042 [DOI] [PubMed] [Google Scholar]
- Tang Y., Nyengaard J. R., De Groot D. M., Gundersen H. J. (2001). Total regional and global number of synapses in the human brain neocortex. Synapse 41, 258–273. 10.1002/syn.1083 [DOI] [PubMed] [Google Scholar]
- Teipel S., Drzezga A., Grothe M. J., Barthel H., Chetelat G., Schuff N., et al. (2015). Multimodal imaging in Alzheimer's disease: validity and usefulness for early detection. Lancet Neurol. 14, 1037–1053. 10.1016/S1474-4422(15)00093-9 [DOI] [PubMed] [Google Scholar]
- Teng E., Ward M., Manser P. T., Sanabria-Bohorquez S., Ray R. D., Wildsmith K. R., et al. (2019). Cross-sectional associations between [18F]GTP1 tau PET and cognition in Alzheimer's disease. Neurobiol. Aging 81, 138–145. 10.1016/j.neurobiolaging.2019.05.026 [DOI] [PubMed] [Google Scholar]
- Terry R. D., Masliah E., Salmon D. P., Butters N., DeTeresa R., Hill R., et al. (1991). Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol 30, 572–580. 10.1002/ana.410300410 [DOI] [PubMed] [Google Scholar]
- Thal D. R., Beach T. G., Zanette M., Heurling K., Chakrabarty A., Ismail A., et al. (2015). [18F]flutemetamol amyloid positron emission tomography in preclinical and symptomatic Alzheimer's disease: specific detection of advanced phases of amyloid-β pathology. Alzheimers Dement. 11, 975–985. 10.1016/j.jalz.2015.05.018 [DOI] [PubMed] [Google Scholar]
- Therriault J., Benedet A., Pascoal T. A., Savard M., Ashton N., Chamoun M., et al. (2021). Determining Amyloid-β positivity using 18F-AZD4694 PET imaging. J. Nucl. Med.62:247–252. 10.2967/jnumed.120.245209 [DOI] [PubMed] [Google Scholar]
- Thompson P. W., Ye L., Morgenstern J. L., Sue L., Beach T. G., Judd D. J., et al. (2009). Interaction of the amyloid imaging tracer FDDNP with hallmark Alzheimer's disease pathologies. J. Neurochem. 109, 623–630. 10.1111/j.1471-4159.2009.05996.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L., Li W., Lo M. M., Gao X., Wai J. M., Rudd M., et al. (2020). Discovery of [11C]MK-6884: a positron emission tomography (PET) Imaging agent for the study of M4 muscarinic receptor positive allosteric modulators (PAMs) in neurodegenerative diseases. J. Med. Chem. 63, 2411–2425. 10.1021/acs.jmedchem.9b01406 [DOI] [PubMed] [Google Scholar]
- Tune L., Tiseo P. J., Ieni J., Perdomo C., Pratt R. D., Votaw J. R., et al. (2003). Donepezil HCl (E2020) maintains functional brain activity in patients with Alzheimer disease: results of a 24-week, double-blind, placebo-controlled study. Am. J. Geriatr. Psychiatry 11, 169–177. 10.1097/00019442-200303000-00007 [DOI] [PubMed] [Google Scholar]
- Tzimopoulou S., Cunningham V. J., Nichols T. E., Searle G., Bird N. P., Mistry P., et al. (2010). A multi-center randomized proof-of-concept clinical trial applying [18F]FDG-PET for evaluation of metabolic therapy with rosiglitazone XR in mild to moderate Alzheimer's disease. J. Alzheimers Dis. 22, 1241–1256. 10.3233/JAD-2010-100939 [DOI] [PubMed] [Google Scholar]
- Van Der Gucht A., Verger A., Yagdigul Y., Poussier S., Joly L., Watfa G., et al. (2015). Complementarity of visual and voxel-based FDG-PET analysis to detect MCI-like hypometabolic pattern in elderly patients with hypertension and isolated memory complaints. Acta Radiol. 56, 980–989. 10.1177/0284185114542366 [DOI] [PubMed] [Google Scholar]
- Vanhaute H., Ceccarini J., Michiels L., Koole M., Sunaert S., Lemmens R., et al. (2020). In vivo synaptic density loss is related to tau deposition in amnestic mild cognitive impairment. Neurology 95, e545–e53. 10.1212/WNL.0000000000009818 [DOI] [PubMed] [Google Scholar]
- Vannini P., Hanseeuw B., Munro C. E., Amariglio R. E., Marshall G. A., Rentz D. M., et al. (2017). Hippocampal hypometabolism in older adults with memory complaints and increased amyloid burden. Neurology 88, 1759–1767. 10.1212/WNL.0000000000003889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varley J., Brooks D. J., Edison P. (2015). Imaging neuroinflammation in Alzheimer's disease and other dementias: recent advances and future directions. Alzheimers Dement. 11, 1110–1120. 10.1016/j.jalz.2014.08.105 [DOI] [PubMed] [Google Scholar]
- Varrone A., Oikonen V., Forsberg A., Joutsa J., Takano A., Solin O., et al. (2015). Positron emission tomography imaging of the 18-kDa translocator protein (TSPO) with [18F]FEMPA in Alzheimer's disease patients and control subjects. Eur. J. Nucl. Med. Mol. Imaging 42, 438–446. 10.1007/s00259-014-2955-8 [DOI] [PubMed] [Google Scholar]
- Vehmas A. K., Kawas C. H., Stewart W. F., Troncoso J. C. (2003). Immune reactive cells in senile plaques and cognitive decline in Alzheimer's disease. Neurobiol. Aging 24, 321–331. 10.1016/S0197-4580(02)00090-8 [DOI] [PubMed] [Google Scholar]
- Vellas B., Andrieu S., Sampaio C., Wilcock G., European Task Force G. (2007). Disease-modifying trials in Alzheimer's disease: a European task force consensus. Lancet Neurol. 6, 56–62. 10.1016/S1474-4422(06)70677-9 [DOI] [PubMed] [Google Scholar]
- Venneti S., Lopresti B. J., Wang G., Hamilton R. L., Mathis C. A., Klunk W. E., et al. (2009). PK11195 labels activated microglia in Alzheimer's disease and in vivo in a mouse model using PET. Neurobiol. Aging 30, 1217–1226. 10.1016/j.neurobiolaging.2007.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne V. L., Dore V., Burnham S. C., Masters C. L., Rowe C. C. (2018). Imaging tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nat. Rev. Neurol. 14, 225–236. 10.1038/nrneurol.2018.9 [DOI] [PubMed] [Google Scholar]
- Villemagne V. L., Pike K. E., Chetelat G., Ellis K. A., Mulligan R. S., Bourgeat P., et al. (2011). Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann. Neurol. 69, 181–192. 10.1002/ana.22248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne V. L., Pike K. E., Darby D., Maruff P., Savage G., Ng S., et al. (2008). Aβ deposits in older non-demented individuals with cognitive decline are indicative of preclinical Alzheimer's disease. Neuropsychologia 46, 1688–1697. 10.1016/j.neuropsychologia.2008.02.008 [DOI] [PubMed] [Google Scholar]
- Wadsworth H., Jones P. A., Chau W. F., Durrant C., Fouladi N., Passmore J., et al. (2012). [18F]GE-180: a novel fluorine-18 labelled PET tracer for imaging translocator protein 18 kDa (TSPO). Bioorg. Med. Chem. Lett. 22, 1308–1313. 10.1016/j.bmcl.2011.12.084 [DOI] [PubMed] [Google Scholar]
- Wang H. Y., Li W., Benedetti N. J., Lee D. H. (2003). a7 nicotinic acetylcholine receptors mediate β-amyloid peptide-induced tau protein phosphorylation. J. Biol. Chem. 278, 31547–31553. 10.1074/jbc.M212532200 [DOI] [PubMed] [Google Scholar]
- Wang Y., Mandelkow E. (2016). Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21. 10.1038/nrn.2015.1 [DOI] [PubMed] [Google Scholar]
- Wei W., Nguyen L. N., Kessels H. W., Hagiwara H., Sisodia S., Malinow R. (2010). Amyloid β from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 13, 190–196. 10.1038/nn.2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels A. M., Tariot P. N., Zimmer J. A., Selzler K. J., Bragg S. M., Andersen S. W., et al. (2020). Efficacy and safety of lanabecestat for treatment of early and mild Alzheimer disease: The AMARANTH and DAYBREAK-ALZ randomized clinical trials. JAMA Neurol. 77, 199–209. 10.1001/jamaneurol.2019.3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell J. L., Graff-Radford J., Tosakulwong N., Weigand S. D., Machulda M. M., Senjem M. L., et al. (2018). Imaging correlations of tau, amyloid, metabolism, and atrophy in typical and atypical Alzheimer's disease. Alzheimers Dement. 14, 1005–1014. 10.1016/j.jalz.2018.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley C. A., Lopresti B. J., Venneti S., Price J., Klunk W. E., DeKosky S. T., et al. (2009). Carbon 11-labeled pittsburgh compound B and carbon 11-labeled (R)-PK11195 positron emission tomographic imaging in Alzheimer disease. Arch. Neurol. 66, 60–67. 10.1001/archneurol.2008.511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson H., Dervenoulas G., Pagano G., Tyacke R. J., Polychronis S., Myers J., et al. (2019). Imidazoline 2 binding sites reflecting astroglia pathology in Parkinson's disease: an in vivo 11C-BU99008 PET study. Brain 142, 3116–3128. 10.1093/brain/awz260 [DOI] [PubMed] [Google Scholar]
- Wolfsgruber S., Kleineidam L., Wagner M., Mosch E., Bickel H., Lupsilonhmann D., et al. (2016). Differential risk of incident Alzheimer's disease dementia in stable versus unstable patterns of subjective cognitive decline. J. Alzheimers Dis. 54, 1135–1146. 10.3233/JAD-160407 [DOI] [PubMed] [Google Scholar]
- Wolters E. E., Ossenkoppele R., Verfaillie S. C. J., Coomans E. M., Timmers T., Visser D., et al. (2020). Regional [18F]flortaucipir PET is more closely associated with disease severity than CSF p-tau in Alzheimer's disease. Eur. J. Nucl. Med. Mol. Imaging 47, 2866–2878. 10.1007/s00259-020-04758-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D. F., Kuwabara H., Horti A. G., Roberts J. M., Nandi A., Cascella N., et al. (2018). Brain PET imaging of α7-nAChR with [18F]ASEM: reproducibility, occupancy, receptor density, and changes in schizophrenia. Int. J. Neuropsychopharmacol. 21, 656–667. 10.1101/245118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D. F., Kuwabara H., Kim J., Brasic J. R., Chamroonrat W., Gao Y., et al. (2013). PET imaging of high-affinity α4β2 nicotinic acetylcholine receptors in humans with 18F-AZAN, a radioligand with optimal brain kinetics. J. Nucl. Med. 54, 1308–1314. 10.2967/jnumed.112.108001 [DOI] [PubMed] [Google Scholar]
- Wooten D. W., Guehl N. J., Verwer E. E., Shoup T. M., Yokell D. L., Zubcevik N., et al. (2017). Pharmacokinetic evaluation of the tau PET radiotracer 18F-T807 (18F-AV-1451) in human subjects. J. Nucl. Med. 58, 484–491. 10.2967/jnumed.115.170910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T., Mucke L. (2002). Inflammation in neurodegenerative disease–a double-edged sword. Neuron 35, 419–432. 10.1016/S0896-6273(02)00794-8 [DOI] [PubMed] [Google Scholar]
- Xia C. F., Arteaga J., Chen G., Gangadharmath U., Gomez L. F., Kasi D., et al. (2013). [18F]T807, a novel tau positron emission tomography imaging agent for Alzheimer's disease. Alzheimers Dement. 9, 666–676. 10.1016/j.jalz.2012.11.008 [DOI] [PubMed] [Google Scholar]
- Yasuno F., Ota M., Kosaka J., Ito H., Higuchi M., Doronbekov T. K., et al. (2008). Increased binding of peripheral benzodiazepine receptor in Alzheimer's disease measured by positron emission tomography with [11C]DAA1106. Biol. Psychiatry 64, 835–841. 10.1016/j.biopsych.2008.04.021 [DOI] [PubMed] [Google Scholar]
- Yokokura M., Mori N., Yagi S., Yoshikawa E., Kikuchi M., Yoshihara Y., et al. (2011). In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer's disease. Eur. J. Nucl. Med. Mol. Imaging 38, 343–351. 10.1007/s00259-010-1612-0 [DOI] [PubMed] [Google Scholar]
- Yuan Y., Gu Z. X., Wei W. S. (2009). Fluorodeoxyglucose-positron-emission tomography, single-photon emission tomography, and structural MR imaging for prediction of rapid conversion to Alzheimer disease in patients with mild cognitive impairment: a meta-analysis. AJNR Am. J. Neuroradiol. 30, 404–410. 10.3174/ajnr.A1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti-Fregonara P., Zhang Y., Jenko K. J., Gladding R. L., Zoghbi S. S., Fujita M., et al. (2014). Synthesis and evaluation of translocator 18 kDa protein (TSPO) positron emission tomography (PET) radioligands with low binding sensitivity to human single nucleotide polymorphism rs6971. ACS Chem. Neurosci. 5, 963–971. 10.1021/cn500138n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Han D., Tan X., Feng J., Guo Y., Ding Y. (2012). Diagnostic accuracy of 18F-FDG and 11C-PIB-PET for prediction of short-term conversion to Alzheimer's disease in subjects with mild cognitive impairment. Int. J. Clin. Pract. 66, 185–198. 10.1111/j.1742-1241.2011.02845.x [DOI] [PubMed] [Google Scholar]
- Zimmer E. R., Leuzy A., Benedet A. L., Breitner J., Gauthier S., Rosa-Neto P. (2014). Tracking neuroinflammation in Alzheimer's disease: the role of positron emission tomography imaging. J. Neuroinflammation 11:120. 10.1186/1742-2094-11-120 [DOI] [PMC free article] [PubMed] [Google Scholar]