

Abstract
Objective: Multiple mechanisms including vascular endothelial cell damage have a critical role in the formation and development of atherosclerosis (AS), but the specific molecular mechanisms are not exactly clarified. This study aims to determine the possible roles of proline-rich tyrosine kinase 2 (Pyk2)/mitochondrial calcium uniporter (MCU) pathway in AS mouse model and H2O2-induced endothelial cell damage model and explore its possible mechanisms.
Approach and Results: The AS mouse model was established using apolipoprotein E-knockout (ApoE–/–) mice that were fed with a high-fat diet. It was very interesting to find that Pyk2/MCU expression was significantly increased in the artery wall of atherosclerotic mice and human umbilical vein endothelial cells (HUVECs) attacked by hydrogen peroxide (H2O2). In addition, down-regulation of Pyk2 by short hairpin RNA (shRNA) protected HUVECs from H2O2 insult. Furthermore, treatment with rosuvastatin on AS mouse model and H2O2-induced HUVEC injury model showed a protective effect against AS by inhibiting the Pyk2/MCU pathway, which maintained calcium balance, prevented the mitochondrial damage and reactive oxygen species production, and eventually inhibited cell apoptosis.
Conclusion: Our results provide important insight into the initiation of the Pyk2/MCU pathway involved in AS-related endothelial cell damage, which may be a new promising target for atherosclerosis intervention.
Keywords: atherosclerosis, mitochondrion, Pyk2/MCU, ApoE–/– mice, HUVECs
Highlights
-
-
Pyk2/MCU regulates mitochondrial Ca2 + uptake, ROS production, mitochondrial membrane potential and apoptotic signaling, which affects the occurrence and development of atherosclerosis at a certain extent.
-
-
Down-regulation of Pyk2 by short hairpin RNA (shRNA) protected HUVECs from H2O2 insult.
-
-
Pyk2/MCU pathway can be inhibited by the treatment of rosuvastatin which showed a protective effect on AS mouse model and H2O2-induced HUVECs injury.
Introduction
Atherosclerosis (AS) is the leading cause of cardiovascular and cerebrovascular disorders such as myocardial infarction and brain stroke (Huang et al., 2019). Dysregulation of lipid metabolism and inflammatory processes that involve the binding of monocytes to dysfunctional endothelium, their recruitment to susceptible areas of the arterial wall, and their differentiation to macrophages and development into foam cells have been associated with atherosclerosis formation (Yu et al., 2018). Several other pathogeneses of atherosclerosis have been suggested, including lipid infiltration theory, thrombosis theory, and smooth muscle cell cloning theory. The theory of endothelial injury response is recently reported as a major factor for this disease, which renders the modulation of endothelial cell (EC) functions a key therapeutic target (Widlansky and Gutterman, 2011; Sandow et al., 2012; Godo and Shimokawa, 2017). The change of intracellular Ca2+ concentration ([Ca2+]i) has an important role in ECs’ functions (Alevriadou et al., 2017). In most cells, fluctuations in Ca2+ concentration are translated into the production of cellular signals, while [Ca2+]i transients are defined and shaped by the mitochondria. Ca2+ homeostasis regulates numerous cell functions, including energy metabolism, reactive oxygen species (ROS) generation, spatiotemporal dynamics of Ca2+ signaling, cell growth, and cell death. The mitochondria are highly specialized in this context because of their sponge-like retentive capacity with calcium balance (Smith and Gallo, 2018).
The cell has a complicated mitochondrial Ca2+ transport mechanism that controls Ca2+ entry to the mitochondrial matrix and exit to the cytosol. Mitochondria have a large negative membrane potential which may reach 180 mV, which facilitates flooding of mitochondria Ca2+ from the cytoplasm (Mallilankaraman et al., 2012). On the other hand, the heterogeneous protein complex with mixed molecular identity assembled into mitochondrial calcium uniporter (MCU) is a new core component that is identified in 2011, which regulates the influx of calcium ions into the mitochondria (Baughman et al., 2011). Previous studies have found that MCU expression regulates the buffering of cytoplasmic Ca2+ during systole in neonatal rat cardiomyocytes (Drago et al., 2012). In pancreatic β cells, MCU regulates cellular glucose sensing capacity (Tarasov et al., 2012). Thus, MCU plays a specific role in the occurrence and development of diseases. However, how MCU activity contributes to EC dysfunction has received considerably less attention. Non-receptor calcium-dependent proline-rich tyrosine kinase 2 (Pyk2) is an important signaling molecule that senses changes of intracellular calcium levels and induces alterations in cell function (Hirschler-Laszkiewicz et al., 2018). Recently, studies have reported that Pyk2 regulates mitochondrial calcium uptake through phosphorylation of the MCU in myocardial cells (O-Uchi et al., 2014). In addition, in our previous study, we did find that the Pyk2/MCU pathway was activated in a rat cerebral ischemia model, which was responsible for mitochondrial dysfunction, calcium balance, and neuronal apoptosis (Zhang et al., 2018). Meanwhile, by reviewing the literature, it was reported that angiotensin II (Ang II) stimulated a calcium-sensitive tyrosine kinase Pyk2 via the type-1 angiotensin II (AT1) receptor in pulmonary vein endothelial cells (PVECs) and vascular smooth muscle cells (VSMCs) (Tang et al., 2000; Satoh et al., 2001; Nakashima et al., 2006). In the endothelial cells, Ang II binds to EC receptors and facilitates the expression of plasminogen activator inhibitor-1 and cell adhesion molecules, which may promote the development of atherosclerosis and myocardial infarction (Luscinskas and Gimbrone, 1996; St Paul et al., 2020), due to promoting ROS generation, increasing the cell-surface expression of cell adhesion molecules, and enhancing leukocyte adhesiveness to ECs, eventually causing endothelium dysfunction. Would the presence of Ang II and EC injury in the initial stage of atherosclerosis activate the Pyk2/MCU pathway (further aggravating the progression of the disease)? We would address the above-mentioned questions in this study.
Therefore, we observed the changes of the Pyk2/MCU pathway in the H2O2-induced endothelial cell injury model and the AS mouse model, and we used rosuvastatin (Rosu) to treat the EC injury model and AS mouse model. Statins have long been used to lower cholesterol levels through inhibition of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme in the mevalonate pathway (Lozano-Cuenca et al., 2020). Recently, their anti-inflammatory and endothelial cells’ protective actions have been proposed independently of their anti-hyperlipidemic effects (Escudero et al., 2015). Meantime, evidence had shown that statins could block Ang II-induced Pyk2 activation in PVECs (Satoh et al., 2001), which provided an idea for us to study the molecular pathways related to Pyk2 and the therapeutic mechanism of rosuvastatin. In the present study, it will be observed if rosuvastatin could inhibit Ang II and the Pyk2/MCU pathway simultaneously in the development process of atherosclerosis, which would further identify the Pyk2/MCU pathway as a new target to prevent EC dysfunction and AS progression as well as Ang II related to Pyk2/MCU at some levels.
Materials and Methods
Animals
Male apolipoprotein E-knockout (ApoE–/–) mice (B6.129P2-Apoetm1Unc/J) were a kind gift of Professor Zheng Bin (Biochemistry Department of Hebei Medical University, Shijiazhuang, China). Male C57BL/6J mice (6 weeks old) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (China). All the animals were housed in an environment with temperature of 22 ± 1°C, relative humidity of 50 ± 1%, and a light/dark cycle of 12/12 h. At 8 weeks of age, randomized ApoE–/– mice and C57BL/6J mice were fed with a high-fat diet (Beijing Keao Xieli Feed Co., Ltd., China) containing 78.85% basic feed, 21% lard, and 0.15% cholesterol, while the control groups (ApoE–/– mice and C57BL/6J mice) were given a general chow diet. Furthermore, all in vivo experiments (including the mice euthanasia procedure) were conducted in accordance with the requirements and principles of the Animal Care and Use Committee of Hebei Medical University as well as the AAALAC and the IACUC guidelines.
Animal Model of Atherosclerosis
An atherosclerotic mouse model was induced in male ApoE–/– mice that were fed with a high-fat diet for 12 weeks. After 12 weeks, three mice were randomly selected to examine the formation of plaque in artery. All the mice were divided into five groups: C57BL/6J+general diet (n = 11), ApoE–/–+general diet (n = 11), C57BL/6J+high-fat diet (n = 13), ApoE–/–+high-fat diet (n = 13), and ApoE–/–+high-fat diet+Rosu (n = 13). After 12 weeks, the drug treatment group (ApoE–/–+high-fat diet+Rosu) received an intragastric administration of 5 mg/kg/day rosuvastatin (MedChemExpress, United States) according to the previous protocol (Calkin et al., 2008) for 4 weeks, while other mice received the same amount of saline solution. After 4 weeks, all the mice were anesthetized with 10% hydrated chlorine aldehyde.
Cell Culture and Treatments
HUVECs were obtained from the BeNa Culture Collection (Beijing, China). The cells were cultured in Dulbecco’s modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, United States), streptomycin (100 μg/ml), and penicillin (100 IU/ml) in a humidified atmosphere containing 5% CO2/95% air at 37°C. The culture medium was replaced every 2 days.
In the experiment, HUVECs were pretreated with rosuvastatin (MedChemExpress, United States; 2.5, 5, and 10 μM) for 2 h. The concentrations were determined prior to the experiment with a drug concentration gradient (Wang et al., 2010). Rosuvastatin was dissolved at a certain concentration in dimethyl sulfoxide (DMSO) (Piconi et al., 2008); the final concentration of DMSO was always lower than 0.01%, which had been shown to have no effect on cell viability (Gao et al., 2008). The cells were then exposed to H2O2 (750 μM) for 24 h (Chen et al., 2014). The final concentrations of H2O2 were determined by the experiment with a H2O2 concentration gradient.
Cell Viability Assay
Cell viability was measured with the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega Corporation, United States). After 24 h exposure to H2O2, 20 μl of MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] was added to each well and the plates were incubated at 37°C for 4 h. After removal of the medium, 100 μl DMEM was added to each well to avoid previous additions affecting absorbance values. Absorbance was measured at 490 nm by a microplate reader (Bioteck, Inc., Winooski, VT, United States).
Serum Lipid and Angiotensin II Detection
After 16 weeks of intervention, all mice were fasted overnight. Blood was collected from retro-bulbar sinus. Blood samples were then centrifuged at 2500 rpm for 5 min, and the serum and the plasma were collected, respectively. Concretely, total cholesterol (TC), triglyceride (TG), high-density lipoprotein cholesterol (HDL-c), low-density lipoprotein cholesterol (LDL-c) of serum, and Ang II of plasma were detected (Yi He Medical Laboratory, Shijiazhuang, China), respectively. The concentrations of Ang II in conditioned HUVECs culture media were measured using human angiopoietin-2 ELISA Kit (MultiSciences, China) according to the manufacturer’s instructions. In brief, cells were collected, washed in phosphate-buffered saline (PBS) three times, and then resuspended. The cells were then shocked and fragmented using Ultrasonic Cell Disruptor. The cell suspension was centrifuged, and the supernatant was taken to determine the concentration of Ang II.
Histopathological Analysis
The mice were perfused through the left ventricle with normal saline and then fixed in situ with 4% paraformaldehyde. For each mouse, the artery from the aorta root to the iliac artery was dissected, dehydrated overnight in 30% sucrose, the length from the aortic root coming out of the heart to before the bifurcation of the aortic arch embedded in optimum cutting temperature compound, and finally frozen. Continuous frozen sections (5-μm-thick) obtained by a Leica CM3050S frozen slicer (Germany Leica Co., Ltd.) were made from the aortic root to the arch of the aorta before it bifurcated. The frozen sections were stained with Oil red O and hematoxylin–eosin (HE) to analyze plaque relative area and tissue damage, respectively, and the results were observed with an optical microscope (Germany Leica Co., Ltd.). The remaining arterial tissues were stained with Oil red O as observed with a Smz-b4 microscope (Chongqing Otter Company, China).
Moreover, we detected the expression of CD31, Pyk2, and MCU in the artery tissues. The frozen sections were placed in a 37°C electrothermostat for 30 min. The sections were then washed with 0.01 M PBS three times, blocked with goat serum, and placed in a moist chamber at room temperature for 1 h. The sections were then incubated with purified rat anti-mouse CD31 antibody (1:10, BD Pharmingen, United States), anti-MCU polyclonal antibody (1:100, Bioworld Technology, United States), and anti-Pyk2 monoclonal antibody (1:60, Abcam, United States) at 4°C overnight, following incubation with the goat anti-rabbit Dylight 594 fluorescence secondary antibody (1:300, Abbkine, United States) and goat anti-rat IgG, FITC conjugated (1:100, CWBIO, China) for 1 h at room temperature. Finally, DAPI (Boster Biological Technology Co., Ltd., United States) was used to dye the nucleus. The immunofluorescent results were photographed with a BX61 universal microscope (Japan Olympus Co., Ltd.). To detect the protein expression in immunofluorescence images, we counted the mean of integrated option density (IOD) in order to reduce artificial errors. The mean of IOD = (IOD)/SUM AREA of the image that was selected in this experiment, that is, the optical density values of each point in the target aorta AREA on the image were first summed up to obtain IOD and then divided by the AREA of the effective target distribution AREA to obtain the mean density, which reflected the concentration per unit AREA of the target substance. An automated image analysis system (Image-Pro Plus 6.0; Media Cybernetics, Silver Spring, MD, United States) was used for all quantitative measurements.
Western Blot Analysis
Harvested cells and artery tissues from the aorta root to the iliac artery were used for western blotting. Cold radioimmunoprecipitation assay lysis buffer and phenylmethylsulfonyl fluoride protease inhibitors (Beijing Solarbio Science & Technology Co., Ltd., China) were used for protein extraction following the manufacturer’s protocol. Protein concentration was determined with the BCA Protein Assay Reagent Kit (Beijing Solarbio Science & Technology Co., Ltd., China). Then, 30 μg total proteins were resolved by electrophoresis and then transferred onto polyvinylidene difluoride membranes (Millipore Corporation, United States). After blocking with 5% skim milk for 1 h at room temperature, the membranes were incubated at 4°C overnight with anti-MCU (1:2,000, Cellsignal), anti-Pyk2 (1:1,000, Abcam), and anti-caspase-3 (1:1,000, Signalway, United States/Proteintech, China) antibodies diluted in 5% skim milk. Polyclonal mouse anti-β-actin antibody (1:3,000, Bioworld Technology) and anti-GAPDH polyclonal antibody (1:3,000, Atagenix, Wuhan, China) were used as an internal control. On the next day, the membranes were incubated with fluorescent-labeled secondary antibodies (Dylight 800, goat anti-rabbit IgG, 1:2,000, Abbkine) for 1 h at room temperature; after washing, they were scanned and analyzed on an Odyssey infrared laser scanner (LICOR Bioscience, Lincoln, NE, United States). Densitometric values were normalized to β-actin or GAPDH immunoreactivity to correct for any loading and transfer differences among samples.
Ultrastructural Observation of Tissues and Cells
Mice were perfused with 0.9% normal saline followed by 4% paraformaldehyde and 4% glutaraldehyde in 0.9% normal saline. The wall of the blood vessel from the aortic root coming out of the heart to before the bifurcation of the aortic arch was cut into 1 × 1-mm pieces and then fixed in 4% glutaraldehyde for 24 h. Next, the samples were post-fixed with 1% osmium tetroxide for 2 h at 4°C. After dehydration with ethyl alcohol, the samples were embedded in EponTM resin. Ultrathin sections were cut and stained with uranyl acetate and lead citrate. To obtain the cell samples, the HUVECs were digested with 0.25% trypsin, washed with PBS three times and collected in 1.5-ml EP tubes. The cell suspension was then centrifuged at 1,000 rpm for 3 min, and the supernatant was discarded. Cell mass was suspended with 4% glutaraldehyde and fixed for 24 h. The further procedures of dehydration, fixation, sectioning, and staining were the same as those used for the collected mice specimens. The specimens were observed and photographed with a JEM-1230 (Japan) electron microscope.
Real-Time Quantitative Polymerase Chain Reaction
The total RNA of HUVECs was extracted with Trizol Reagent (Invitrogen, Carlsbad, CA, United States) according to the manufacturer’s instructions. RNA (2 μg) from each sample was converted to cDNA. The primers were synthesized by Beijing Saibaisheng Gene Technology Co., Ltd. The forward and reverse primers that were used were as follows: 5′-AGGATCGGGGAATTGACAGAG-3′ (for ward) and 5′-GTGTGGTGTATAGTTGCTGGAC-3′ (rev erse) for MCU, 5′-GAAGCCGAGTGGAGGTATG-3′ (forw ard) and 5′-GCGCTGCATGTAGTCGTT-3′ (reverse), 5′-GTGGCCGTCAAGACCTGTAA-3′ (forward) and 5′-AGC TTGACGATGTGAGGGTG-3′ (reverse) for Pyk2, 5′-GAGCTA CGAGCTGCCTGAC-3′ (forward) and 5′-GGTAGTTTCGTGG ATGCCACAG-3′ (reverse) for β-actin, and 5′-TCAAG AAGGTGGTGAAGCAGG-3′ (forward) and 5′-TCAA AGGTGGAGGAGTGGGT-3′ (forward) for GAPDH. All samples were normalized to β-actin or GAPDH. The relative gene copy number for each sample was measured using the comparative threshold cycle number (Ct) method. Each sample was processed in triplicate. Relative gene expression was evaluated using the formula 2–ΔΔCt method as previously described (Plengvidhya et al., 2012).
Mitochondrial Membrane Potential
The HUVECs with different treatments were harvested using 0.25% trypsin without ethylenediaminetetraacetic acid and washed with PBS three times. Finally, we used PBS to resuspend the cells, and Rhodamine 123 (Sigma-Aldrich, Steinheim, Germany) was used to detect the mitochondrial membrane potential (MMP). Rhodamine 123 (2 μM) was added to dissociated cells for 30 min at 37°C, and the cells were then washed three times with PBS. Lastly, fluorescence intensity was measured by flow cytometry (Cytomics FC 500, Beckman Coulter, United States).
Analysis of Reactive Oxygen Species
The production of ROS was determined by using ROS-sensitive fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA) (Beyotime, Shanghai, China), following the manufacturer’s instruction. In brief, the cells were harvested as described above, resuspended in 1 ml PBS with 1 μl DCFH-DA (10 μM), incubated in the dark at 37°C for 20–30 min, and then washed three times with PBS. Fluorescence was then measured with a Cytomics FC 500, Beckman Coulter flow cytometer (United States).
Intracellular Free Ca2+ Concentration
The fluorescent Ca2+ indicator Fluo-4/AM([Ca2+]i) (Invitrogen, United States) was used to detect the intracellular free calcium concentration. The HUVECs were prepared as described above. A total of 2 μM Fluo-4/AM was then added for 30 min at 37°C. The changes in [Ca2+]i were measured with a flow cytometer (CytomicsFC500, Beckman Coulter, United States) and expressed as relative fluorescence intensity.
Cell Apoptosis
The number of apoptotic cells was detected by using Annexin V-FITC Apoptosis Detection Kit (Beyotime, Shanghai, China), following the manufacturer’s instruction. Briefly, HUVECs were collected as previously described. The cells were then resuspended in 200 μl binding buffer, incubated with 5 μl Annexin V-FITC and 10 μl PI, gently mixed, and then placed in the dark at room temperature for 15 min. Finally, the cells were mixed with 300 μl binding buffer and analyzed using flow cytometry (FACSCantoTM II, BD, United States).
Pyk2 Gene Silencing by shRNA
Four short hairpin RNAs (shRNAs) targeting the Pyk2 gene as well as a scramble shRNA (NC, non-specific control) were both designed and synthesized by the company of GeneCopoeia (GeneCopoeia Inc., United States). Several different shRNAs of mRNA at different target sites were selected, and an effective interference sequence was screened out through the preliminary experiment (Figure 7A). As can be seen from Figure 7A, the infection efficiency of the cells that enhanced green fluorescent protein (EGFP) co-expressed with shRNA in the five groups had reached 80–90%. The interference sequence of shRNA and EGFP was initiated by different promoters: the interference sequence of shRNA was initiated by H1, and EGFP was initiated by SV40. Then, real-time quantitative PCR (rt-qPCR) and western blot were used for further identification to determine the virus that made Pyk2 of low expression better.
FIGURE 7.

Pyk2-shRNA lentivirus transfection efficiency observation of human umbilical vein endothelial cells (HUVECs). (A) Fluorescence results of HUVECs infected with different targeting sequences of Pyk2 gene (×200). Scale bar: 100 μm; target sequence (NC: GCTTCGCGCCGTAGTCTTA; Pyk2-shRNA-1: GCTACTTGCCAGAAGACTTCA; Pyk2-shRNA-2: GCAGCATAGAGTCAGACATCT; Pyk2-shRNA-3: GCTGTACTCACTGCAGATATG; Pyk2-shRNA-4: GCGGCAAATCCTGGACAAACA). (B) Protein expression results of HUVECs infected by different Pyk2-shRNA lentivirus. (C) mRNA expression results of HUVECs infected by different Pyk2-shRNA lentivirus. Data are shown as mean ± SD of at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus NC group, one-way ANOVA.
HUVECs were normally cultured, and then the slabs were laid with 2.5 × 105 cells/well (six-well plates) and cultured overnight at 37°C. When the cells were in good condition, HUVECs were infected by lentiviral particles according to a different multiplicity of infection (MOI) on the next day, and repeated infection was conducted 8 h later. Fresh culture medium was changed on the third day. At 3 days after transfection, the expression of green fluorescent protein was observed with a fluorescence microscope, and the transfection efficiency (green fluorescent protein-positive cell rate) was calculated. Meanwhile, western blot and rt-qPCR were performed to examine the low expression effect of the gene. Next, HUVECs were divided into several groups: blank group (control), blank+H2O2 group, NC (non-specific control) +H2O2 group, shRNA+H2O2 group, H2O2+Rosu group, and shRNA+H2O2+Rosu group. MTS was applied to detect the changes of cell viability in different groups, flow cytometry was used to detect changes in intracellular Ca2+ and mitochondrial membrane potential, and western blot was adopted to detect the protein expression of Pyk2, MCU, caspase-3, and GAPDH. The methods had been described above.
Statistical Analysis
All results were shown as mean ± SD. Statistical comparisons were preformed using one-way ANOVA followed by Student–Newman–Keuls test and least significant difference test for multiple comparisons. Student’s t-test and Mann–Whitney nonparametric tests were applied to compare variables between two groups (a = 0.05 as inspection level). Results with P < 0.05 were considered statistically significant. SPSS 23.0 statistical software (IBM SPSS, Chicago, IL, United States) was used for statistical analysis.
Results
Validation of the Atherosclerotic Mouse Model and the Therapeutic Effect of Rosuvastatin on Atherosclerosis
Oil red O and HE staining were used to stain the lipid plaque in aortic lesions (Figure 1A). As expected, we found lipid accumulation in the inner wall of the aortic and the relative plaque area, which was much larger and necrotic in the model group (ApoE–/–+high-fat diet). On the contrary, the plaque area in the group treated with rosuvastatin was significantly smaller and smoother in different degrees compared to the model group (Figures 1B–D).
FIGURE 1.

The lesion degree of atherosclerosis and the effect of rosuvastatin in ApoE– /– mice. (A) HE pathological section, Oil red O pathological section, gross arterial tissue Oil red O staining. Scale bar: 50 μm. (B–D) The relative lesion area of plaques was calculated by Image Pro Plus 6.0. (E) Concentration of lipids and Ang II in different animal groups. Data are shown as mean ± SD of at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle CH; #P < 0.05, ##P < 0.01, ###P < 0.001 versus vehicle model (M), one-way ANOVA. CG, C57BL/6J+general diet; AG, ApoE– /– +general diet; CH, C57BL/6J+high-fat diet; M, ApoE– /– +high-fat diet; M+Rosu, ApoE– /– +high-fat diet+Rosu.
Furthermore, the levels of serum lipids were spontaneously elevated in the model group. Compared with the CH (C57BL/6J+high-fat diet) group, the levels of TC, TG, HDL-c, and LDL-c were all increased in the model group (Figure 1E). In addition, those levels were significantly decreased in the rosuvastatin group compared to the model group except the level of TG. On the other hand, compared with the CH group, the level of Ang II in the model group was slightly increased while it was not significant, and the concentration of Ang II (Figure 1E) revealed a significant decline in the rosuvastatin group compared to the model group.
To observe the ultrastructural changes of vascular wall during atherosclerosis, transmission electron microscopy was used to examine the ultrastructure of the segment from the aortic root coming out of the heart to before the bifurcation of the aortic arch. The endothelial cells of the endodermis from the control group had normal nuclear and cytoplasmic contents, vascular wall lamination was clearly visible, and smooth muscle layer thickness was uniform (Figure 2A). In the AG group (ApoE–/–+general diet), we can see that the endothelial cells of endodermis changed slightly and vascular wall lamination was visible, while the smooth muscle layer thickened slightly (Figure 2B). In the CH group (C57BL/6J+high-fat diet), what we can see are the changes in the morphology of endothelial cells and thickening of the smooth muscle layer (Figure 2C). On the other hand, significant morphological transformation was observed in the model group, which was characterized by thickening of the smooth muscle lay, endothelial cell shape changes (from long fusiform to round and spherical shape) and nuclear pyknosis, cytoplasmic condensation, and cell volume increase (Figure 2D). In addition, the endothelial cells had obvious edema, membrane dissolution, severe intracytoplasmic vacuolization, rare intact organelles, nuclear membrane dissolution, nuclear chromatin edge set, cell body deformation, and nuclear condensation. On the contrary, the morphology of the arterial wall in the rosuvastatin group had less damage compared with the model group; the edema of the endothelial cells was mitigated, the organelles’ structure was recovered almost similarly to the normal, the nucleoli and the nuclear membrane seemed intact (Figure 2E).
FIGURE 2.
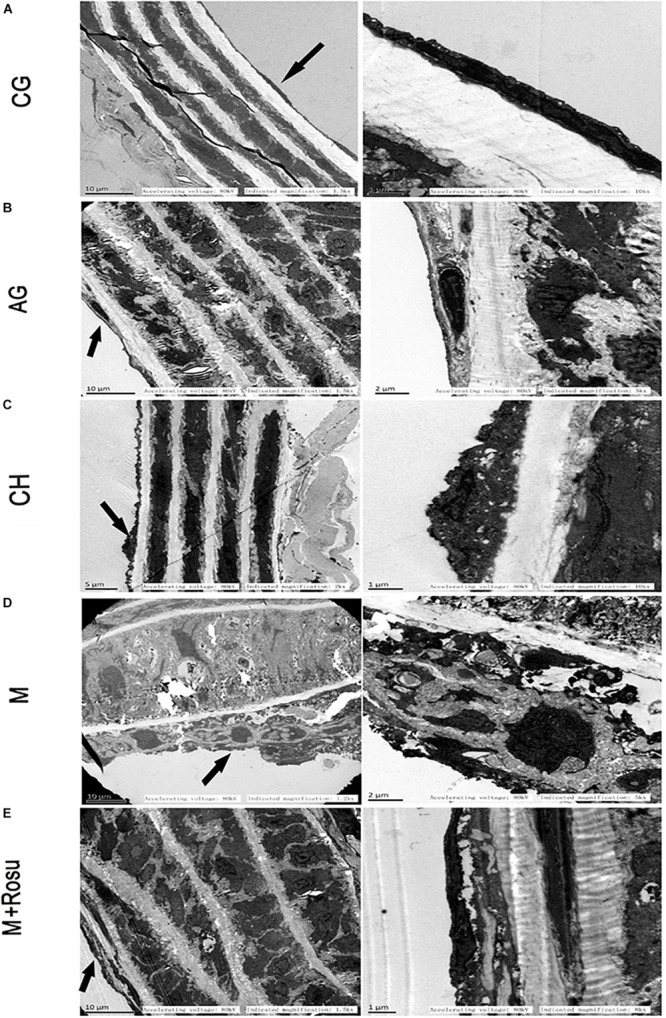
Ultrastructural changes in arterial wall during atherosclerosis. (A) C57BL/6J+general diet. The endothelial cells of endodermis had normal nuclear and cytoplasmic contents, vascular wall lamination was clearly visible, and smooth muscle layer thickness was uniform. (B) ApoE– /– +general diet. The endothelial cells of endodermis changed slightly; vascular wall lamination was visible, while the smooth muscle layer thickened slightly. (C) C57BL/6J+high-fat diet. We can see changes in the morphology of endothelial cells and thickening of the smooth muscle layer. (D) ApoE– /– +high-fat diet. Significant morphological transformation was observed in this group, which was characterized by thickening of the smooth muscle layer, endothelial cell shape changes (from long fusiform to round and spherical shape) and nuclear pyknosis, cytoplasmic condensation, and cell volume increasing. (E) ApoE– /– +high-fat diet+Rosu. The morphology of the arterial wall in the rosuvastatin group had less damage compared with the model group; the edema of the endothelial cells was mitigated, the organelles’ structure was recovered gradually, and the nucleoli and the nuclear membrane seemed intact. The arrow shows the endothelial cell. The area indicated by the arrow is enlarged in the right column. CG, C57BL/6J+general diet; AG, ApoE– /– +general diet; CH, C57BL/6J+high-fat diet; M, ApoE– /– +high-fat diet; M+Rosu, ApoE– /– +high-fat diet+Rosu.
The Expression of Pyk2/MCU in the Artery Wall Underlying Atherosclerosis
Next, we set out to verify the changes of the Pyk2/MCU pathway during AS. To investigate whether the Pyk2/MCU pathway was associated with AS, we examined the related proteins Pyk2, MCU, and caspase-3 by western blot. The results showed that the above-mentioned three proteins were enhanced obviously in the model group compared with the control group, respectively (Figures 3A–D). In addition, in the CH group (C57BL/6J+high-fat diet), the expression of the three proteins revealed upregulation, especially caspase-3 which increased significantly, which together supported that the relevant indicators were activated with the severity of the disease. Moreover, with the treatment of rosuvastatin, these related proteins decreased significantly.
FIGURE 3.

The expression of Pyk2/mitochondrial calcium uniporter (MCU) after atherosclerosis (AS) and rosuvastatin intervention in a mouse model of AS by western blot. (A) The protein expression of Pyk2, MCU, and caspase-3. (B–D) Statistical graph of Pyk2 (B), MCU (C), and caspase-3 (D). Data are shown as mean ± SD of at least three independent experiments. *P < 0.05, **P < 0.01 versus vehicle control (CG); #P < 0.05, ##P < 0.01, ###P < 0.001 versus vehicle model (M), one-way ANOVA and unpaired Student’s t-test (a = 0.05 as inspection level, M versus M+Rosu). CG, C57BL/6J+general diet; AG, ApoE– /– +general diet; CH, C57BL/6J+high-fat diet; M, ApoE– /– +high-fat diet; M+Rosu, ApoE– /– +high-fat diet+Rosu.
We further verified the expression of Pyk2 and MCU in the endothelial layer of the arterial wall that was subjected to AS by immunofluorescence (Figures 4A–E). As expected, both MCU and Pyk2 were obviously expressed in the model group compared to other groups, which was consistent with the western blot data. At the same time, we used CD31 to locate the arterial wall endodermis, which revealed the increased expression of CD31 in the thickened artery intima. CD31 was significantly increased in the model group, while the Pyk2, MCU, and CD31 protein levels showed a downward trend after rosuvastatin intervention. Based on the above-mentioned results, we considered that the Pyk2/MCU pathway activity is upregulated during atherosclerosis and suppressed by rosuvastatin at some levels.
FIGURE 4.

The expression levels of Pyk2/mitochondrial calcium uniporter (MCU) after atherosclerosis in different groups by immunofluorescence. (A,B) Tissue immunofluorescence; arrowhead: the prominent position of the lesion in the intima. Scale bar: 100 μm. (C–E) Mean of integrated option density of Pyk2, MCU, and caspase-3. Data are shown as mean ± SD of at least three independent experiments. *P < 0.05, **P < 0.01 versus vehicle control (CG); #P < 0.05, ##P < 0.01 versus vehicle model (M); one-way ANOVA and Mann–Whitney nonparametric tests were applied to compare variables between two groups (a = 0.05 as inspection level). CG, C57BL/6J+general diet; AG, ApoE– /– +general diet; CH, C57BL/6J+high-fat diet; M, ApoE– /– +high-fat diet; M+Rosu, ApoE– /– +high-fat diet+Rosu.
The Pathological and Ultrastructural Changes of HUVECs Induced by H2O2 Insults
In order to further study the pathological changes related to atherosclerosis, we used HUVECs to establish a H2O2-induced endothelial cell injury model. MTS was used to further examine the HUVECs’ viability after H2O2-induced injury. The results showed that 750 and 1,000 μM H2O2 significantly reduced the cell viability after 24 h (Figure 5A). In addition, the protective effect of rosuvastatin on cell viability increased when the concentration of rosuvastatin was 2.5 and 5 μM. However, it would not show a significantly protective effect compared with the control group when the concentration of rosuvastatin was 10 μM (Figure 5B). We also found that H2O2 stimulation increased intracellular Ca2+ and intracellular ROS and reduced the mitochondrial membrane potential, while rosuvastatin reversed the changes induced by H2O2 (Figures 5C–E). In addition, the data revealed a significant increase in the apoptotic rates in the H2O2 group compared with the control group. H2O2-induced apoptosis of HUVECs was also inhibited by rosuvastatin (Figure 5F).
FIGURE 5.

Rosuvastatin intervention promotes mitochondrial function and reduces apoptotic cells from H2O2-induced human umbilical vein endothelial cells (HUVECs) injury. (A) HUVECs viability was reduced when exposed to H2O2 (750/1,000 μM, 24 h). (B) Rosuvastatin (pretreatment 2 h, 2.5/5 μM) protected cells from H2O2-induced injury in HUVECs. (C–F) H2O2-induced endothelial injury caused an increase in intracellular free Ca2+ (C), and the release of ROS (D), reduction of ΔΨm (E), apoptotic cell augmentation (F), and mitochondrial damage when compared to the control group. Rosuvastatin pretreatment was able to reduce the number of apoptotic cells, maintain ΔΨm, reduce the store of intracellular free Ca2+ as well as ROS production and protect against H2O2-induced endothelial injury simultaneously. (G) Ultrastructural changes of endothelial cells in different groups. A structured endothelial cell with normal morphology in the control group, including an evident nucleolus (thick arrow) and an intact nuclear membrane, cytoplasmic mitochondria (thin arrow), and endoplasmic reticulum (thin arrow). Dilated mitochondria (thin arrow), many folds in the nuclear membrane, and the exposed nucleolus (thick arrow) are shown in the H2O2-induced injury group. Through rosuvastatin pretreating, the cell morphology seemed to be recovering, with the nucleolus less damaged (thick arrow), the nuclear membrane damage being relatively minor, and with reduction in mitochondrial swelling (thin arrow). (H) The concentration of Ang II in different cell groups. Data are shown as mean ± SD of at least three independent experiments. The significance of the four quadrants represented in the picture of apoptosis: Q1 represents the necrotic cells, Q2 represents the late apoptotic cells, Q3 represents the normal cells, and Q4 represents the early apoptotic cells. We counted the total number of Q2+Q4 as the apoptotic cells. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle control; #P < 0.05, ##P < 0.01, ###P < 0.001 versus vehicle H2O2 group, one-way ANOVA.
Furthermore, we observed morphological changes in the ultrastructure of endothelial cells exposed to different treatments. The endothelial cells in the control group had no edema, the nucleus and nucleoli were obvious, the nuclear membrane was complete, and the cytoplasmic mitochondria, endoplasmic reticulum, and other organelles were complete and clearly visible (Figure 5G). In the H2O2 group, the endothelial cells appeared to undergo cytoplasmic condensation, the mitochondria were swollen, and the mitochondrial crest disappeared. Furthermore, vacuolization was observed, and the endoplasmic reticulum and nuclear membrane were dissolved. When the cells were pretreated with rosuvastatin, the damage was significantly reduced compared with the H2O2 group. The nucleoli were clearer than those of the model group, and the nuclear membranes, mitochondria, endoplasmic reticulum as well as other organelles presented less damage, which showed its protective effect on H2O2-induced endothelial cell injury.
We further examined the damage mechanisms of endothelial cells exposed to H2O2 (Figure 5H). The results showed that H2O2 significantly increased Ang II levels, while rosuvastatin inhibited Ang II elevation induced by H2O2. A previous study had reported that the calcium-dependent activation of tyrosine kinase Pyk2 could be induced by Ang II in vascular endothelial cells (Tang et al., 2000; Satoh et al., 2001). Our results demonstrated that endothelial cell injury was accompanied by an increase in Ang II in the in vitro model and took the findings of Tang et al. (2000); Satoh et al. (2001), and Nakashima et al. (2006) into consideration, suggesting that this might provide an interesting evidence between Ang II and Pyk2/MCU signaling. In conclusion, we observed the changes of intracellular free Ca2+ (Figure 5C), ROS (Figure 5D), the change of mitochondrial membrane potential (Figure 5E), and cell apoptosis (Figure 5F) in HUVECs exposed to H2O2 by flow cytometry. These results indicate that rosuvastatin may promote its protective effect through which it reduces Ca2+ overload, ROS generation, and the release of apoptotic proteins and improves mitochondrial membrane potential. Whether or not the specific functions of the above-mentioned descriptions are related to the Pyk2/MCU pathway requires to be further identified.
Inhibition of the Pyk2/MCU Pathway by Rosuvastatin Protects Endothelial Cells From H2O2-Induced Injury
In order to further examine whether the Pyk2/MCU pathway is a target for reversing atherosclerosis, we investigated the changes in Pyk2 and MCU at the transcript level and protein level on cell models (Figure 6). The changes of mRNA expression in Pyk2 and MCU were detected by using real-time quantitative PCR at different treatment groups. As expected, there was a marked increase in MCU mRNA expression after H2O2-induced injury relative to the control group, which was consistent with the protein expression results (Figures 6A,B). In addition, we observed an increase in Pyk2 mRNA expression in H2O2-induced HUVEC injury (Figure 6C), and this difference was statistically significant when compared to the control group. On the contrary, the cells pretreated with rosuvastatin for 2 h showed a significant decrease in Pyk2 and MCU mRNA expression compared to the H2O2 group, which was in agreement with our western blot results.
FIGURE 6.

The change of Pyk2/mitochondrial calcium uniporter (MCU) and caspase-3 protein expression in H2O2-induced human umbilical vein endothelial cells (HUVECs) injury. (A) Protein expression of Pyk2/MCU and caspase-3 in different groups. (B) Statistical graph of panel (A). (C) The level of Pyk2 and MCU mRNA in H2O2-induced HUVEC injury. Data are shown as mean ± SD of at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle control; #P < 0.05, ##P < 0.01 versus vehicle H2O2 group, one-way ANOVA.
On the other hand, the expression of both MCU and Pyk2 proteins increased significantly in H2O2 group than the control and reduced in the high-dose rosuvastatin group compared to the H2O2 group (Figures 6A,B). In addition, we also measured the expression level of caspase-3 (Figures 6A,B), and there is a small increase after H2O2 treatment when compared to the control group (We used unpaired Student’s t-test to confirm that caspase-3 is upregulated with the H2O2 intervention. n = 5, P < 0.05, control vs. H2O2 group). We also found that two rosuvastatin groups reduced caspase-3 protein significantly. Thus, we concluded again that the Pyk2/MCU pathway may be involved in H2O2-induced endothelial cell injury. Rosuvastatin may inhibit the Pyk2/MCU pathway and endothelial cell apoptosis, which protected the endothelial cells from oxidative stress caused by H2O2.
Down-Regulation of Pyk2 by shRNA on H2O2-Induced HUVEC Injury
In order to verify the effect of Pyk2 down-regulation on H2O2-induced HUVEC injury, we conducted the lentivirus transfection (MOI = 50) and used rt-qPCR and western blot technologies to check the interference efficiency of different viruses (Pyk2-shRNA-1, Pyk2-shRNA-2, Pyk2-shRNA-3, and Pyk2-shRNA-4) targeting sequences (Figures 7A–C). Based on the results, the Pyk2-shRNA-3 lentivirus was selected for subsequent formal experiments. To gain insights into the underlying mechanisms regarding whether the Pyk2/MCU pathway was associated with endothelial cell injury, a shRNA technique was used in the next experiments. This section is divided into six groups: blank group, H2O2 group (blank+H2O2), NC group (NC+H2O2), rosuvastatin group (H2O2+Rosu), shRNA group (shRNA+H2O2), and (shRNA+H2O2+Rosu) group. Then, we made a pairwise comparison based on the statistical analysis results, which contain blank+H2O2 vs. shRNA+H2O2, blank+H2O2 vs. H2O2+Rosu, blank+H2O2 vs. NC+H2O2, NC+H2O2 vs. shRNA+H2O2, shRNA+H2O2 vs. shRNA+H2O2+Rosu, and H2O2+Rosu vs. shRNA+H2O2+Rosu. The results revealed that, compared with the H2O2 group (blank+H2O2), cell viability was significantly higher in the shRNA group (shRNA+H2O2), and rosuvastatin group (H2O2+Rosu) can also promote cell viability. Cell viability in the NC group (NC+H2O2) showed no significant changes when compared with the H2O2 group (blank+H2O2). A comparison of cell survival between the NC group (NC+H2O2) and shRNA group (shRNA+H2O2) revealed dramatic differences. In addition, both giving rosuvastatin pretreatment and Pyk2 of low expression (shRNA+H2O2+Rosu) showed a stronger protective effect than using rosuvastatin solely (Figure 8A). The results directly indicated that inhibition of Pyk2 can prevent H2O2-induced endothelial cell injury, while rosuvastatin had a protective role which degenerated Pyk2 expression, consistent with our above-mentioned observations. The changes in mitochondrial membrane potential and intracellular free Ca2+ concentration were detected by flow cytometry. We observed a downward trend in the concentration of intracellular free Ca2+ in the transfected shRNA group (shRNA+H2O2), although no statistical significance was found compared to the H2O2 group (blank+H2O2). The NC group (NC+H2O2) showed no significant changes when compared with the shRNA group (shRNA+H2O2) in intracellular free Ca2+. However, after treatment with rosuvastatin, there was a significant decrease in intracellular Ca2+, which was even more obvious when the gene of Pyk2 was of low expression simultaneously. Both giving rosuvastatin pretreatment and Pyk2-shRNA reduced intracellular free Ca2+ better than rosuvastatin alone or merely Pyk2 of low expression with shRNA technique (Figures 8B,C). In the measurement of MMP, we found that the trend was slightly obvious when intervened with rosuvastatin, although this difference was not statistically significant (Figure 8D and Table 1). Accordingly, it appears that inhibition of Pyk2 expression may reduce Ca2+ influx, and rosuvastatin can also reverse the injury caused by H2O2 stimulation through this pathway.
FIGURE 8.

The effect of Pyk2 down-regulation on H2O2-induced human umbilical vein endothelial cells (HUVECs) injury and regulation of the Pyk2/mitochondrial calcium uniporter (MCU) pathway in endothelial cell. (A) Detection of cell viability by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)- 2H-tetrazolium assay after transfection with shRNA. (B–D) Detection of intracellular free Ca2+ and mitochondrial membrane potential in different groups by flow cytometry. (E) Pyk2, MCU, and caspase-3 protein expression in different groups by western blot. (F) Statistical graph of panel (E). Data are shown as mean ± SD of at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle blank group (control); #P < 0.05, ##P < 0.01, ###P < 0.001 versus vehicle H2O2 group (blank+H2O2); ✩P < 0.05, ✩✩P < 0.01, ✩✩✩P < 0.001, NC+H2O2 vs. shRNA+H2O2, NC+H2O2 vs. shRNA+H2O2+Rosu; H2O2+Rosu vs. shRNA+H2O2+Rosu, shRNA+H2O2 vs. shRNA+H2O2+Rosu, one-way ANOVA. (G) Endothelial angiotensin II involvement-Pyk2-dependent MCU phosphorylation initiates mitochondrial Ca2+ entry, which, in turn, induces reactive oxygen species generation and a decrease in mitochondrial membrane potential (MMP/ΔΨ) and eventually leading to cell death.
TABLE 1.
Mean fluorescence intensity (MFI) of mitochondrial membrane potential in different groups.
| Group | Blank | Blank+H2O2 | NC+H2O2 | shRNA+H2O2 | shRNA+H2O2+Rosu |
| MFI | 1,914.57 ± 1 | 1,801.73 ± 26.49** | 1,809.63 ± 22.49** | 1,805.53 ± 22.06** | 1,835.37 ± 30.96* |
*P < 0.05, **P < 0.01 vs. blank group (mean ± SD; n = 3), one-way ANOVA.
We further evaluated the proteins’ expression of Pyk2, MCU, and caspase-3 by western blot, which showed that the expression of these proteins in the shRNA group (shRNA+H2O2) was notably lower compared with those in the H2O2 group, implying that H2O2 activated the Pyk2/MCU pathway, and apoptotic protein release was prohibited by Pyk2 shRNA (Figures 8E,F). There were marked changes of these proteins’ expression in the shRNA group (shRNA+H2O2) compared with the NC group (NC+H2O2). What we can see in the data is a significant decrease in the expression of the three proteins when rosuvastatin was administered, thus suggesting that the Pyk2/MCU pathway may be the target of rosuvastatin, and the drug protects endothelial cells from H2O2 insult through inhibiting the signaling. Meanwhile, treatment with Pyk2 shRNA and rosuvastatin at the same time showed a stronger inhibition of Pyk2, MCU, and caspase-3 (Figures 8E,F). Collectively, these observations indicate that inhibition of the Pyk2/MCU pathway by shRNA technique and/or rosuvastatin intervention eliminates the excessive apoptotic cells produced, mitochondrial injury, and Ca2+ accumulated in the endothelial cells, thus reducing the occurrence of injury of endothelial cells caused by mitochondrial damage and improving the cells’ viability.
Discussion
Atherosclerosis is associated with a number of life-threatening cardiovascular disorders. The exact mechanisms of action that underlie these associations remain unclear (Lusis, 2000; Lozano et al., 2012). Identification of specific molecules that mediate resistance to AS may contribute to facilitate the development of novel therapies and improve response to currently available therapies. In this study, we identified the Pyk2/MCU pathway as a new regulator for atherosclerosis. Briefly, Pyk2/MCU regulates mitochondrial Ca2+ uptake, ROS production, mitochondrial membrane potential, and apoptotic signaling, which, in turn, affects the occurrence and development of atherosclerosis. On the other hand, we discovered that rosuvastatin, an important lipid-lowering treatment drug, may affect the Pyk2/MCU pathway, thus preventing the development of AS (Figure 8G). This model was proven in vivo, by using mice exposed to high-fat diet, and in vitro, using a H2O2-induced endothelial cell injury model. Notably, these results provide new insights into the molecular basis of the Pyk2/MCU pathway. The inhibition of the Pyk2/MCU pathway which further modulates mitochondrial Ca2+ handling and apoptosis in atherosclerosis may confer a protective effect to the arteries against atherosclerosis.
The Regulation of the Pyk2/MCU Pathway Underlying Endothelial Cell Injury
Many factors are involved in the pathophysiological process of atherosclerosis. Although the molecular details underlying atherosclerotic lesion development have not yet been elucidated, endothelial dysfunction has been recognized as an integral component in the initiation and progression of atherosclerosis (Sun et al., 2011). The role of oxidative stress in the development of endothelial dysfunction and subsequent atherosclerotic disease has been an area of interest over the few decades. Initial work in this field considered ROS as the primarily toxic metabolic by-product with adverse effects on vascular function from direct damage to key cellular proteins (Widlansky and Gutterman, 2011). Mitochondria, as a major cellular source of ROS, have recently garnered increased attention for their contribution to the detrimental effects of cardiovascular and cerebrovascular disease (Walters et al., 2016). The increase of ROS production from mitochondria has been closely associated with excessive mitochondrial Ca2+ uptake (O-Uchi et al., 2014). Furthermore, mitochondrial Ca2+ overload results in mitochondrial membrane potential depolarization that opens a mitochondria permeability transition pore, generates intracellular ROS, and finally releases the proapoptotic proteins into the cytosol, which, in turn, results in cell injury and death (Zhang et al., 2018). Thus, the potential risk of mitochondrial Ca2+ overload leading to apoptosis and the signaling of mitochondrial Ca2+ uptake in endothelial cells need to be precisely regulated. In this experiment, we successfully established an EC injury model with H2O2 and found that H2O2 caused a decrease in cell viability, an increase in ROS production, and a destruction of mitochondrial function. We found similar trends in the ApoE–/– mouse model of atherosclerosis. To sum up, studies are consistent, and we believe that oxidative stress and mitochondrial damage are involved in atherosclerotic endothelial damage and accelerate the process of atherosclerosis.
Recently, it has been discovered that a highly negative membrane potential (ΔΨ) regulates the Ca2+ that enters the mitochondria through a selective inward rectifying MCU channel (Hajnoczky et al., 2006). The channel, which is composed of mitochondrial matrix resident proteins MICUs, MCUb, MCUR1, and EMRE (Kamer and Mootha, 2014; Lee et al., 2015), is transcriptionally regulated by upstream Ca2+ cascade, post-translational modification, and divalent cations. The mode of regulation can either inhibit or enhance MCU channel activity and thus regulate mitochondrial metabolism and cell fate. Under conditions of oxidative stress, C97 at the structure of N-terminal domain of MCU gets S-glutathionylated, promoting MCU oligomerization and increasing mitochondria Ca2+ uptake, leading to Ca2+ overload and swelling of the mitochondria (Nemani et al., 2018). Therefore, mitochondria damage has an important role in atherosclerosis, and calcium imbalance is one of the significant links. However, the precise regulation and molecular mechanism behind this process is still unclear. In this study, we found that an increase in MCU protein is accompanied with the reduction of mitochondrial membrane potential, production of ROS, cell apoptosis induction, and mitochondria swelling in the H2O2-induced endothelial cell injury and AS mouse model, while inhibition of MCU reversed those processes. Therefore, we consider that MCU participates in the pathological process of atherosclerosis.
Pyk2 is a 116-kDa cytoplasmic tyrosine kinase which is a member of the focal adhesion kinase family (Zhang et al., 2018). Previous studies have demonstrated that activation of Pyk2 in PVECs regulates Ca2+ dynamics, which may be responsible for EC death (Satoh et al., 2001). In this study, we want to confirm whether Pyk2 is stimulated by other factors and the relationship between Pyk2 and MCU in the process of atherosclerosis. The existing evidence had clearly shown that Pyk2 modulates mitochondrial calcium uptake through phosphorylation of the MCU in cardiomyocyte (O-Uchi et al., 2014), and the Pyk2/MCU pathway is activated in a rat cerebral ischemia model, which is responsible for mitochondrial dysfunction and neuronal apoptosis (Zhang et al., 2018). Interestingly, a separate study confirmed the calcium dependence of tyrosine kinase Pyk2 can be activated by Ang II in PVECs (Satoh et al., 2001). The role of Pyk2 in the regulation of Ang II-induced signaling pathways was certified in mediating VSMC growth, which was later reported (Rocic et al., 2001). Particularly, Ang II has a crucial role in several vascular pathologies including aortic aneurysm and atherosclerosis (Daugherty et al., 2000, 2010), and Ang II-induced vascular hypertrophy is mediated by intracellularly produced ROS (Nakashima et al., 2006). Meanwhile, by the involvement of Ang II, ROS can mediate the Ca2+-dependent transactivation of epidermal growth factor receptor (Yin et al., 2003). It can be seen that, under some pathological conditions, Ang II and ROS will interact with each other, which are related to the calcium dependence of tyrosine kinase Pyk2. Studies have also shown that Ang II infusion stimulates the progression of atherosclerosis in ApoE–/–mice (Weiss et al., 2001; Rateri et al., 2011). In addition, Ang II cross-talks with several tyrosine kinases via AT1Rs, including receptor tyrosine kinases (EGFR, PDGF, insulin receptor and nonreceptor tyrosine kinases, c-Src family kinases, Ca2+-dependent Pyk2, focal adhesion kinase, and Janus kinases) (Mehta and Griendling, 2007). Given the relationship described above, we then investigated whether the Pyk2/MCU pathway is correlated with the pathological process of AS and Ang II could influence this pathway. In the present study, it is possible that the activation of Pyk2 is connected with the Ang II increase since we observed that the Pyk2/MCU pathway is activated, and Ang II was upregulated simultaneously in the H2O2-induced EC injury model. This is in line with evidence suggesting that Ang II stimulated a calcium-sensitive tyrosine kinase Pyk2 in PVECs (Tang et al., 2000; Satoh et al., 2001). However, we cannot see the obvious change of Ang II in the atherosclerosis mouse model in spite of it being inhibited by the treatment of rosuvastatin, which is a limitation of our study. Therefore, it becomes our next goal which requires to be further demonstrated because we want to completely reflect the in vivo situation and we do not stop in the future. Thereafter, we used shRNA technique to reduce Pyk2 expression and interestingly discovered that MCU expression decreased, the mitochondria were protected, and the apoptosis was reduced, which, in turn, led to an alleviation of H2O2-induced EC injury. Therefore, we consider that the activation of the Pyk2/MCU pathway has a crucial role in the EC injury process and may be involved in the pathological process of atherosclerosis. Next, further verification would address the question whether inhibition of the Pyk2/MCU pathway is likely to be a new therapeutic target in the animal model for preventing the development of atherosclerosis.
Pyk2/MCU Pathway May Be a New Target of Reversing Atherosclerosis
Treatments for atherosclerosis may include healthy lifestyle changes and medicines, such as oral lipid-lowering drugs (Grundy et al., 2019). However, there is no cure for atherosclerosis so far. Statin is important for effective inhibition of cholesterol synthesis (Vale et al., 2014), improving physiological function of vascular endothelium (Duran et al., 2010), anti-inflammatory reaction (Naito et al., 2006), decreasing vascular plaque area (Takayama et al., 2016), anti-apoptosis effect (Geng et al., 2019), anti-oxidant stress, protecting mitochondrial function (Liu et al., 2017), and so on. This study confirmed that rosuvastatin can inhibit the Pyk2/MCU pathway during atherosclerosis process in ApoE–/– mice with high-fat diet and HUVECs exposed to H2O2, further promoting mitochondrial function by maintaining the MMP, blocking intracellular Ca2+ influx, and reducing ROS release. These results suggest that the Pyk2/MCU pathway may serve as a new target for atherosclerosis. The function of rosuvastatin on the AS mouse model was consistent with the effect of the Pyk2-shRNA results in HUVECs, and that drug declined the activation of Pyk2/MCU pathway as well as protected mitochondrial function, which further confirmed that rosuvastatin protected ECs by inhibiting the Pyk2/MCU pathway. However, how do statins inhibit the Pyk2/MCU pathway? Through consulting the literature, we found that atherosclerosis or endothelial injury caused higher Ang II, and statins can inhibit Ang II (Colucci et al., 2013). In our experiment, the results showed that, in the H2O2-induced EC injury model, there was Ang II increase, and it may be correlated to the Pyk2/MCU pathway, while rosuvastatin decreased Ang II in both in vitro and in vivo situations. Although there are few experimental results on the relationship between Ang II and the Pyk2/MCU pathway in our study at present, the present results provide more possibilities for our future research. These data revealed the underlying mechanism of the EC protection conferred by rosuvastatin during atherosclerosis, which may be explained by the inhibition of Ang II involved in the Pyk2/MCU pathway activation, then decreasing intracellular ROS production and Ca2+ influx, protecting the mitochondrial function and ultimately preventing EC apoptosis (Figure 8G).
Conclusion
This study is the first to report the association between the Pyk2/MCU pathway and atherosclerosis. We provide a body of evidence demonstrating that Pyk2/MCU may be a new target for inhibiting or reversing atherosclerosis. However, additional studies are required to elaborate the specific molecular mechanisms for this effect and eventually find new molecules which target on this pathway.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Hebei Medical University institutional animal care.
Author Contributions
XL and PF designed the experiments. YzZ performed and wrote the manuscript. XY, ZL, KB, TL, ZM, BW, LM, HL, KZ, LjL, and YyZ helped with experiments. YpZ, JQ, LL, SL, and JC contributed to the discussion and review of the manuscript. XL obtained the funding, designed the experiments, and modified the manuscript. All authors have read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Professor Zheng Bin at the Biochemistry Department of Hebei Medical University for providing ApoE–/– mice (B6.129P2-Apoetm1Unc/J) for in vivo studies.
Footnotes
Funding. This work was supported primarily by grants to XL from the National Natural Science Foundation of China (81571160) and the Ministry of Human Resources and Social Security of People’s Republic of China (CG2016003003).
References
- Alevriadou B. R., Shanmughapriya S., Patel A., Stathopulos P. B., Madesh M. (2017). Mitochondrial Ca(2+) transport in the endothelium: regulation by ions, redox signalling and mechanical forces. J. R. Soc. Interface 14:20170672. 10.1098/rsif.2017.0672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman J. M., Perocchi F., Girgis H. S., Plovanich M., Belcher-Timme C. A., Sancak Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476 341–345. 10.1038/nature10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkin A. C., Giunti S., Sheehy K. J., Chew C., Boolell V., Rajaram Y. S., et al. (2008). The HMG-CoA reductase inhibitor rosuvastatin and the angiotensin receptor antagonist candesartan attenuate atherosclerosis in an apolipoprotein E-deficient mouse model of diabetes via effects on advanced glycation, oxidative stress and inflammation. Diabetologia 51 1731–1740. 10.1007/s00125-008-1060-6 [DOI] [PubMed] [Google Scholar]
- Chen S., Tang Y., Qian Y., Chen R., Zhang L., Wo L., et al. (2014). Allicin prevents H2O2-induced apoptosis of HUVECs by inhibiting an oxidative stress pathway. BMC Complement. Altern. Med. 14:321. 10.1186/1472-6882-14-321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colucci R., Fornai M., Duranti E., Antonioli L., Rugani I., Aydinoglu F., et al. (2013). Rosuvastatin prevents angiotensin II-induced vascular changes by inhibition of NAD(P)H oxidase and COX-1. Br. J. Pharmacol. 169 554–566. 10.1111/j.1476-5381.2012.02106.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty A., Manning M. W., Cassis L. A. (2000). Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Invest. 105 1605–1612. 10.1172/jci7818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty A., Rateri D. L., Charo I. F., Owens A. P., Howatt D. A., Cassis L. A. (2010). Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE–/– mice. Clin. Sci. (Lond) 118 681–689. 10.1042/cs20090372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago I., De Stefani D., Rizzuto R., Pozzan T. (2012). Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 109 12986–12991. 10.1073/pnas.1210718109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran W. N., Breslin J. W., Sanchez F. A. (2010). The NO cascade, eNOS location, and microvascular permeability. Cardiovasc. Res. 87 254–261. 10.1093/cvr/cvq139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudero P., Martinez de Maranon A., Collado A., Gonzalez-Navarro H., Hermenegildo C., Peiro C., et al. (2015). Combined sub-optimal doses of rosuvastatin and bexarotene impair angiotensin II-induced arterial mononuclear cell adhesion through inhibition of Nox5 signaling pathways and increased RXR/PPARalpha and RXR/PPARgamma interactions. Antioxid. Redox Signal. 22 901–920. 10.1089/ars.2014.5969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M., Zhang W. C., Liu Q. S., Hu J. J., Liu G. T., Du G. H. (2008). Pinocembrin prevents glutamate-induced apoptosis in SH-SY5Y neuronal cells via decrease of bax/bcl-2 ratio. Eur. J. Pharmacol. 591 73–79. 10.1016/j.ejphar.2008.06.071 [DOI] [PubMed] [Google Scholar]
- Geng J., Xu H., Yu X., Xu G., Cao H., Lin G., et al. (2019). Rosuvastatin protects against oxidized lowdensity lipoproteininduced endothelial cell injury of atherosclerosis in vitro. Mol. Med. Rep. 19 432–440. 10.3892/mmr.2018.9666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godo S., Shimokawa H. (2017). Endothelial functions. Arterioscler. Thromb. Vasc. Biol. 37 e108–e114. 10.1161/atvbaha.117.309813 [DOI] [PubMed] [Google Scholar]
- Grundy S. M., Stone N. J., Bailey A. L., Beam C., Birtcher K. K., Blumenthal R. S., et al. (2019). 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. J. Am. Coll. Cardiol. 73 e285–e350. 10.1016/j.jacc.2018.11.003 [DOI] [PubMed] [Google Scholar]
- Hajnoczky G., Csordas G., Das S., Garcia-Perez C., Saotome M., Sinha Roy S., et al. (2006). Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 40 553–560. 10.1016/j.ceca.2006.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschler-Laszkiewicz I., Chen S. J., Bao L., Wang J., Zhang X. Q., Shanmughapriya S., et al. (2018). The human ion channel TRPM2 modulates neuroblastoma cell survival and mitochondrial function through Pyk2, CREB, and MCU activation. Am. J. Physiol. Cell Physiol. 315 C571–C586. 10.1152/ajpcell.00098.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Chambliss K. L., Gao X., Yuhanna I. S., Behling-Kelly E., Bergaya S., et al. (2019). SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature 569 565–569. 10.1038/s41586-019-1140-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer K. J., Mootha V. K. (2014). MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 15 299–307. 10.1002/embr.201337946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y., Min C. K., Kim T. G., Song H. K., Lim Y., Kim D., et al. (2015). Structure and function of the N-terminal domain of the human mitochondrial calcium uniporter. EMBO Rep. 16 1318–1333. 10.15252/embr.201540436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. W., Yang F., Cheng S. Z., Liu Y., Wan L. H., Cong H. L. (2017). Rosuvastatin postconditioning protects isolated hearts against ischemia-reperfusion injury: the role of radical oxygen species, PI3K-Akt-GSK-3beta pathway, and mitochondrial permeability transition pore. Cardiovasc. Ther. 35 3–9. 10.1111/1755-5922.12225 [DOI] [PubMed] [Google Scholar]
- Lozano R., Naghavi M., Foreman K., Lim S., Shibuya K., Aboyans V., et al. (2012). Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet 380 2095–2128. 10.1016/s0140-6736(12)61728-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano-Cuenca J., Valencia-Hernández I., López-Canales O. A., Flores-Herrera H., López-Mayorga R. M., Castillo-Henkel E. F., et al. (2020). Possible mechanisms involved in the effect of the subchronic administration of rosuvastatin on endothelial function in rats with metabolic syndrome. Braz. J. Med. Biol. Res. 53:e9304. 10.1590/1414-431X20199304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscinskas F. W., Gimbrone M. A., Jr. (1996). Endothelial-dependent mechanisms in chronic inflammatory leukocyte recruitment. Annu. Rev. Med. 47 413–421. 10.1146/annurev.med.47.1.413 [DOI] [PubMed] [Google Scholar]
- Lusis A. J. (2000). Atherosclerosis. Nature 407 233–241. 10.1038/35025203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K., Doonan P., Cardenas C., Chandramoorthy H. C., Muller M., Miller R., et al. (2012). MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151 630–644. 10.1016/j.cell.2012.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta P. K., Griendling K. K. (2007). Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 292 C82–C97. 10.1152/ajpcell.00287.2006 [DOI] [PubMed] [Google Scholar]
- Naito Y., Katada K., Takagi T., Tsuboi H., Isozaki Y., Handa O., et al. (2006). Rosuvastatin, a new HMG-CoA reductase inhibitor, reduces the colonic inflammatory response in dextran sulfate sodium-induced colitis in mice. Int. J. Mol. Med. 17 997–1004. [PubMed] [Google Scholar]
- Nakashima H., Suzuki H., Ohtsu H., Chao J. Y., Utsunomiya H., Frank G. D., et al. (2006). Angiotensin II regulates vascular and endothelial dysfunction: recent topics of Angiotensin II type-1 receptor signaling in the vasculature. Curr. Vasc. Pharmacol. 4 67–78. 10.2174/157016106775203126 [DOI] [PubMed] [Google Scholar]
- Nemani N., Shanmughapriya S., Madesh M. (2018). Molecular regulation of MCU: implications in physiology and disease. Cell Calcium 74 86–93. 10.1016/j.ceca.2018.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O-Uchi J., Jhun B. S., Xu S., Hurst S., Raffaello A., Liu X., et al. (2014). Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2-dependent tyrosine phosphorylation of the mitochondrial Ca2+ uniporter. Antioxid. Redox Signal. 21 863–879. 10.1089/ars.2013.5394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piconi L., Corgnali M., Da Ros R., Assaloni R., Piliego T., Ceriello A. (2008). The protective effect of rosuvastatin in human umbilical endothelial cells exposed to constant or intermittent high glucose. J. Diabetes Complications 22 38–45. 10.1016/j.jdiacomp.2007.03.004 [DOI] [PubMed] [Google Scholar]
- Plengvidhya N., Chanprasert K., Tangjittipokin W., Thongnoppakhun W., Yenchitsomanus P. T. (2012). Identification of copy number variation of CAPN10 in Thais with type 2 diabetes by multiplex PCR and denaturing high performance liquid chromatography (DHPLC). Gene 506 383–386. 10.1016/j.gene.2012.06.094 [DOI] [PubMed] [Google Scholar]
- Rateri D. L., Howatt D. A., Moorleghen J. J., Charnigo R., Cassis L. A., Daugherty A. (2011). Prolonged infusion of angiotensin II in ApoE(−/−) mice promotes macrophage recruitment with continued expansion of abdominal aortic aneurysm. Am. J. Pathol. 179 1542–1548. 10.1016/j.ajpath.2011.05.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocic P., Govindarajan G., Sabri A., Lucchesi P. A. (2001). A role for PYK2 in regulation of ERK1/2 MAP kinases and PI 3-kinase by ANG II in vascular smooth muscle. Am. J. Physiol. Cell Physiol. 280 C90–C99. 10.1152/ajpcell.2001.280.1.C90 [DOI] [PubMed] [Google Scholar]
- Sandow S. L., Senadheera S., Grayson T. H., Welsh D. G., Murphy T. V. (2012). Calcium and endothelium-mediated vasodilator signaling. Adv. Exp. Med. Biol. 740 811–831. 10.1007/978-94-007-2888-2_36 [DOI] [PubMed] [Google Scholar]
- Satoh K., Ichihara K., Landon E. J., Inagami T., Tang H. (2001). 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors block calcium-dependent tyrosine kinase Pyk2 activation by angiotensin II in vascular endothelial cells. involvement of geranylgeranylation of small G protein Rap1. J. Biol. Chem. 276 15761–15767. 10.1074/jbc.M009165200 [DOI] [PubMed] [Google Scholar]
- Smith G. M., Gallo G. (2018). The role of mitochondria in axon development and regeneration. Dev. Neurobiol. 78 221–237. 10.1002/dneu.22546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Paul A., Corbett C. B., Okune R., Autieri M. V. (2020). Angiotensin II, hypercholesterolemia, and vascular smooth muscle cells: a perfect trio for vascular pathology. Int. J. Mol. Sci. 21:4525. 10.3390/ijms21124525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C., Wu M. H., Yuan S. Y. (2011). Nonmuscle myosin light-chain kinase deficiency attenuates atherosclerosis in apolipoprotein E-deficient mice via reduced endothelial barrier dysfunction and monocyte migration. Circulation 124 48–57. 10.1161/circulationaha.110.988915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama T., Komatsu S., Ueda Y., Fukushima S., Hiro T., Hirayama A., et al. (2016). Comparison of the effect of rosuvastatin 2.5 mg vs 20 mg on coronary plaque determined by angioscopy and intravascular ultrasound in Japanese with stable angina pectoris (From the aggressive lipid-lowering treatment approach using intensive rosuvastatin for vulnerable coronary artery plaque [ALTAIR] randomized trial). Am. J. Cardiol. 117 1206–1212. 10.1016/j.amjcard.2016.01.013 [DOI] [PubMed] [Google Scholar]
- Tang H., Zhao Z. J., Landon E. J., Inagami T. (2000). Regulation of calcium-sensitive tyrosine kinase Pyk2 by angiotensin II in endothelial cells. Roles of Yes tyrosine kinase and tyrosine phosphatase SHP-2. J. Biol. Chem. 275 8389–8396. 10.1074/jbc.275.12.8389 [DOI] [PubMed] [Google Scholar]
- Tarasov A. I., Semplici F., Ravier M. A., Bellomo E. A., Pullen T. J., Gilon P., et al. (2012). The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic beta-cells. PLoS One 7:e39722. 10.1371/journal.pone.0039722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale N., Nordmann A. J., Schwartz G. G., de Lemos J., Colivicchi F., den Hartog F., et al. (2014). Statins for acute coronary syndrome. Cochrane Database Syst. Rev. 15:CD006870. 10.1002/14651858.CD006870.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters J. W., Amos D., Ray K., Santanam N. (2016). Mitochondrial redox status as a target for cardiovascular disease. Curr. Opin. Pharmacol. 27 50–55. 10.1016/j.coph.2016.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Tao W., Wang Y., Bikow J., Lu B., Keating A., et al. (2010). Rosuvastatin, identified from a zebrafish chemical genetic screen for antiangiogenic compounds, suppresses the growth of prostate cancer. Eur. Urol. 58 418–426. 10.1016/j.eururo.2010.05.024 [DOI] [PubMed] [Google Scholar]
- Weiss D., Kools J. J., Taylor W. R. (2001). Angiotensin II-induced hypertension accelerates the development of atherosclerosis in apoE-deficient mice. Circulation 103 448–454. 10.1161/01.cir.103.3.448 [DOI] [PubMed] [Google Scholar]
- Widlansky M. E., Gutterman D. D. (2011). Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid. Redox Signal. 15 1517–1530. 10.1089/ars.2010.3642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin G., Yan C., Berk B. C. (2003). Angiotensin II signaling pathways mediated by tyrosine kinases. Int. J. Biochem. Cell Biol. 35 780–783. 10.1016/s1357-2725(02)00300-x [DOI] [PubMed] [Google Scholar]
- Yu P., Xiong T., Tenedero C. B., Lebeau P., Ni R., MacDonald M. E., et al. (2018). Rosuvastatin reduces aortic sinus and coronary artery atherosclerosis in SR-B1 (Scavenger receptor class B type 1)/ApoE (Apolipoprotein E) double knockout mice independently of plasma cholesterol lowering. Arterioscler. Thromb. Vasc. Biol. 38 26–39. 10.1161/atvbaha.117.305140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Yan J., Wang L., Tian X., Zhang T., Guo L., et al. (2018). The Pyk2/MCU pathway in the rat middle cerebral artery occlusion model of ischemic stroke. Neurosci. Res. 131 52–62. 10.1016/j.neures.2017.09.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.