

Abstract
Every cell has a highly sophisticated system for regulating heme levels, which is particularly important with regard to turnover. Heme degradation generates carbon monoxide (CO) and while CO has long been viewed as a metabolic waste product, and at higher concentrations cellular lethal, we now know that CO is an indispensable gasotransmitter that participates in fundamental physiologic processes necessary for survival. Irrefutable preclinical data has resulted in concerted efforts to develop CO as a safe and effective therapeutic, but against this notion lies dogma that CO is a poison, especially to the brain. The emergence of this debate is discussed here highlighting the neuroprotective properties of CO through its role on the central circadian clock and ongoing strategies being developed for CO administration for clinical use.
Keywords: Heme, Clock, Circadian Rhythm, Gasotransmitters, Neuroprotection, Sleep/Wake
Heme and Its Role in Health and Disease
Heme and carbon monoxide (CO) molecules are considered dangerous when free in the body and brain. They are also inextricably linked: heme catabolism by the heme oxygenases results in endogenous generation of CO within the cell and the principle cellular targets for CO are hemoproteins and would include guanylate cyclase, nitric oxide synthase, mitochondrial oxidases and transcription factors such as Bach-1 and NPAS2. Emerging data suggest, paradoxically, that both heme and CO are critically involved in regulating a host of cellular processes fundamental to cell survival. For example, CO has emerged as an important neurotransmitter that plays a role in central nervous system functions ranging from the regulation of circadian clock function, to memory processing, as well as stress responses to injury and inflammation. Heme, as a functional moiety of numerous proteins in the cell, likewise contributes to the regulation of circadian rhythms and cellular bioenergetics. While the fundamental importance of CO and heme to cellular function are widely recognized, the mere suggestion that heme and CO, but CO in particular, may represent an underexplored clinically viable therapeutic modality is generally met with great skepticism. Yet, unlike other gaseous signaling molecules, which include nitric oxide, oxygen, and hydrogen sulfide, CO is generally non-reactive. CO also has the potential to modulate cellular responses both locally, i.e., at the site of its endogenous generation, and in a more distal (pseudo-humoral) manner. Here we discuss the cellular biology of CO and heme in the context of it serving as the source of endogenous generation during its catabolism. We include their interrelationship in this discussion since hemoproteins serve as the primary binding target for CO and the breakdown of heme increases endogenous generation of CO. Historically, CO is accepted as a poison to the brain caused by anoxia, but recent preclinical and clinical data suggest that not only is this inaccurate, CO is intimately involved in neurotransmission, fundamental regulation of the circadian clock and is neuroprotective in pathologies as diverse as stroke, traumatic brain injury and neurodegenerative disorders. We therefore focus here on this dichotomy of disparate views of this simple gas when considering the brain and argue that CO should be redefined as an evolutionarily conserved protective gas.
The Cellular Biology of Heme and Endogenous CO
Heme is an integral component of numerous proteins within the cell and is fundamental to proper cellular function by way of its contributions to key oxidative reactions, electron transfer processes, signal transduction, delivery of molecular oxygen to cells and, importantly, gene regulation. In recent years it has become evident that heme, likely along with one or more of its catabolic products, also serves an important role in regulating the central circadian clock, neurotransmission and memory. It has long been known that free heme, or heme that is not incorporated into hemoproteins is cytotoxic [1], and thus all organisms have developed redundant mechanisms to rapidly clear it from the extracellular space. Under non-pathological conditions, however, free heme does not accumulate in tissues, but rather its cellular titer is constrained to a dynamic range dictated by the flux of synthesis and degradation (Box 1).
Box 1. Regulation of Heme in Physiologic and Pathophysiologic Environments.
Synthesis
Heme synthesis is an endogenous process that takes place primarily within the liver and bone marrow, although heme synthesis has also been shown to occur in other tissues, including the nervous system [61, 62]. Heme synthesis begins in the mitochondria during the Krebs cycle with the synthesis of δ-aminolevulinic acid (δALA) from the amino acid glycine and succinyl-CoA. The production of heme is regulated by ALA synthase, which in turn is negatively regulated by the concentration of glucose and heme. Once in the cytoplasm, ALA molecules combine to form porphobilinogen (PBG) and water. Uroporphyrinogen I synthase (PBG deaminase) then combines PBG molecules to form a linear tetrapyrrole called hydroxymethylbilane. Through a series of reactive intermediates the linear tetrapyrrole is ultimately oxidized to protoporphyrin, and the iron is incorporated via ferrochetalase producing the heme molecule [63]. Freshly synthesized heme can then be exported across the mitochondrial membranes by Feline Leukemia Virus Subgroup C Receptor (FLVCR1) and shuttled to the endoplasmic reticulum for incorporation as the active moiety of a multitude of hemoproteins including mitochondrial oxidases, cytochrome p450 enzymes, nitric oxide synthases, transcription factors and signaling molecules such as Bach-1 and guanylate cyclase respectively.
Metabolism
HO-1 can be induced by numerous environmental stimuli, including UV radiation, heavy metals, lipopolysaccharides, thermal shock, hydrogen peroxide, NO, inflammatory cytokines, endotoxins, hyperoxia and hypoxia [64]. In fact, upregulation of HO-1 secondary to oxidative stress occurs so rapidly, that HO-1 was initially designated a heat shock protein. Consistent with this role as a stress-response protein, HO-1 has been shown to modulate cell survival, inflammation and innate immune responses as well as tissue damage control and repair [65, 66]. Absence of HO-1 in humans and mice results in heme accumulation and massive tissue injury. Each of the products resulting from heme catalysis that include the bile pigments biliverdin and bilirubin, iron and CO all exhibit bioactive properties, but CO is the most studied. HO-2 is primarily constitutively expressed in the brain, testicle, spleen, neurons, endothelial cells and glia [67]. Unlike HO-1, HO-2 is not typically inducible and its activity can be influenced by post-translational modifications including that resulting from reactive oxygen and nitrogen species [68, 69]. HO-2 may function as a potential oxygen sensor by regulating calcium-activated potassium channels and the hypoxic response in mammalian cells, which has been shown to occur in part through the generation of CO [70].
Heme is under very tight regulation due to its ability to contribute to oxidative stress through Fenton chemistry. The enzymes responsible for removal of free heme are the Heme Oxygenases (HO) that are the rate-limiting steps in the degradation of heme into biliverdin (BV), free ferrous iron (Fe2+) and CO [2, 3]. These enzymes are widely distributed in the nervous system and exist as one of two isoforms, inducible HO-1 and constitutive HO-2 that regulate diverse processes across multiorgan systems (Figure 1). Although each of these isozymes is expressed in neurons, the distribution of HO-1 in the brain is very limited, with high levels of expression observed only in the hippocampal dentate gyrus and ventromedial hypothalamus [4]. HO-1 expression and therefore CO generation becomes highly induced following deviation from homeostasis due to tissue injury including that which occurs in instances of neurodegeneration, stroke and concussion trauma. One common theme with the above neuropathologies is a sudden increase in heme release from tissue parenchyma and/or erythrocyte rupture [5–10]. In contrast, the constitutive isoform, HO-2, is expressed in neurons throughout the neuraxis, including the hippocampus, midbrain, basal ganglia, thalamus, cerebellum and brain stem. HO-2 is also expressed in the hypothalamic suprachiasmatic (SCN) circadian clock and we and others have proposed that HO-2 may play a fundamental role in the regulation of the clock. Specifically, HO-2 generated CO binds hemoprotein guanylate cyclase and stimulates the cGMP/PKG axis that in turn increases the activity of heme-containing neuronal nitric oxide synthase, which is known, for example, to mediate the effects of light on suprachiasmatic nucleus (SCN) function [7, 11, 12]. In addition, and as described in detail below, many of the so-called ‘core clock genes’ contain heme and hence can be directly modulated by CO. We further propose that increased free heme resulting from a sudden stress or injury to neurons or brain microvasculature, could lead to dysregulation of the SCN clock vis a vis elevated CO. Seemingly consistent with this suggestion is the established fact that individuals who suffer a traumatic brain injury or stroke often exhibit disrupted sleep-wake cycles with clock dysfunction considered a likely contributing etiological factor [13–15]. How free heme is managed therefore is precarious as its accumulation and presence in an inflammatory environment can amplify potent oxidative and inflammatory signaling pathways. Indeed, this may explain in part the development of neurodegenerative diseases such as Alzheimer’s Disease (AD) and Parkinson’s Disease (PD) and even multiple sclerosis, where chronic inflammation plays an important role and has been shown to be intimately linked to the presence of free heme and the heme oxygenases [5, 16, 17]. Treatment with exogenous CO has been shown to abrogate inflammation in the brain and prevent neuronal cell loss. [9,10]
Figure 1. Location and function of HO-1 and HO-2 in the central and peripheral nervous systems in physiologic and pathophysiologic settings.
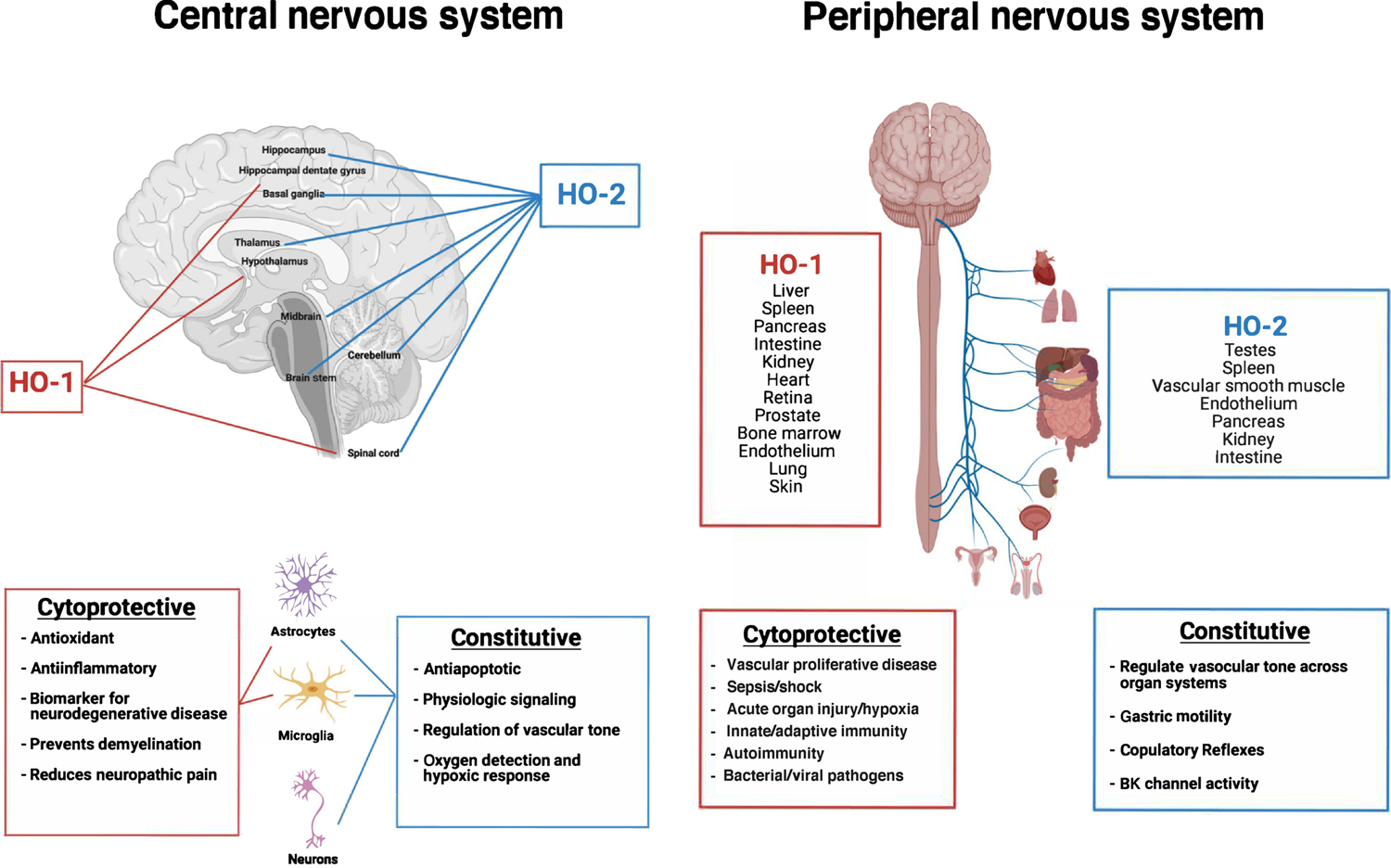
Inducible HO-1 is principally expressed in liver, spleen, pancreas, intestine, kidney, heart, retina, prostate, vascular smooth muscle cells, endothelium, lung, skin, bone marrow, brain, and spinal cord. HO-2, the constitutive isoform, is primarily expressed in spinal cord, testes, vascular smooth muscle cells, endothelium, pancreas, kidney and intestine, but the predominant expression is in the brain. The various known functions associated with the two HO isoforms is also provided.
CO is widely touted as having only harmful attributes with particularly deleterious effects in the brain. Underscoring this point is the fact that CO has been coined ‘the silent killer’ because it is neither visible nor carries a smell. Symptoms associated with suspected overdose include headache, nausea, memory loss, and dyskinesia [18] at carboxyhemoglobin (COHb) levels in the range of 5–10% (baseline is ≤1% in non-smokers). Since CO is a by-product of the incomplete combustion of gas, oil, carbon, and wood, it is tacitly assumed that negative signs of overexposure to combustion products is solely due to CO. In fact, combustion leads to exposure of greater than 500 substances, the majority of which are considered dangerous at high concentrations, especially in the brain. In industrial doses, CO certainly interferes with oxygen delivery through its ability to tightly bind to hemoglobin (Hb) ~ 210 times than that of oxygen. However, at low doses of CO the opposite is true, exogenous administration of CO is potently neuroprotective in models of stroke, TBI, and even AD and PD [19] and likely reflects the fact that at normal physiologic levels CO is a signaling molecule arising from heme degradation. CO, like nitric oxide has been shown to function as a neurotransmitter and because it is highly diffusible can regulate cellular hemoprotein function [20]. There are hundreds of reports in the literature describing the benefits of low-dose CO which has led to numerous ongoing clinical trials (www.clinicaltrials.gov, and Table 1). Healthy human volunteer data shows no adverse events in subjects treated with CO sufficient to generate a COHb of up to 14% with no evidence of headache, nausea or dyskinesia [21]. The mechanism by which CO influences circadian (and sleep) function remains unclear, but it is tempting to speculate a role for CO in the brain and nervous system based on the following: i.) CO is neuroprotective, ii.) CO restores circadian rhythm after hemorrhagic stroke or acute kidney injury [10, 22–25] and iii.) circadian hemoproteins such as NPAS2, nNOS, and Nr1d1 (RevErbα) are modulated by CO [26–28].
Table 1.
Therapeutic Development of Carbon Monoxide
| Clinical Trial | Status | Description/Indication | CO Delivery Modality | Sponsor | NCT/Publication |
|---|---|---|---|---|---|
| Phase 1 | Completed | Single Dose Safety | Inhaled | Ikaria/iNO | Mahan52 |
| Phase 1 | Completed | Repeat Dose (10d) Safety | Inhaled | Ikaria/iNO | Mahan52 |
| Phase 1 | Completed | Single Dose Safety | CORM* | Prolong | Misra78 |
| Phase 1 | Completed | Lung Inflammation | Inhaled | Academic | NCT00094406 |
| Phase 1 | Completed | Mitochondrial Biogenesis | Inhaled | Academic | Rodes79 |
| Phase 1 | Completed | Experimental Shock | Inhaled | Academic | Mayr80 |
| Phase 1 | Completed | Headache/Migraine | Inhaled | Academic | NCT02066558 |
| Phase 1 | Completed | Lung Vascular Function | Inhaled | Academic | NCT03067701 |
| Phase 1 | Completed | Retinal Blood Flow | Inhaled | Academic | Resch81 |
| Phase 1 | Completed | Regulation of Chemoreflex | Inhaled | Academic | Vesely82 |
| Phase 1b | Completed | SCD | Inhaled | Academic | Sirs83 |
| Phase 1b | Completed | SCD | Inhaled | Academic | Beutler84 |
| Phase 1b | Completed | SCD | CORM* | Sangart | Howard |
| Phase 1b | Completed | SCD | CORM* | Prolong | NCT01848925 |
| Phase 1b | Completed | ARDS | Inhaled | Academic | NCT02425579 |
| Phase 1b | Completed | End Stage Renal Disease | CORM* | Prolong | NCT02437422 |
| Phase 1b | Completed | Headache/Migraine | Inhaled | Academic | NCT03385174 |
| Phase 1b | Completed | Migraine Inducing Effects | Inhaled | Academic | NCT03075020 |
| Phase 2 | Completed | COPD | Inhaled | Academic | NCT00122694 |
| Phase 2 | Completed | IPF | Inhaled | Academic | NCT01214187 |
| Phase 2 | Completed | SCD - VoC | CORM* | Prolong | NCT02672540 |
| Phase 2 | Completed | SCD - VoC | CORM* | Prolong | NCT02411708 |
| Phase 2 | Completed | Subarachnoid Hemorrhage | CORM* | Prolong | NCT02323685 |
| Phase 2 | Completed | SCD-ulcer | CORM* | Prolong | NCT02600390 |
| Phase 2/3 | Completed | Kidney Transplant | CORM* | Prolong | NCT02490202 |
| Phase 2 | Ongoing | ARDS | Inhaled | Academic | NCT03799874 |
SCD, sickle cell disease; COPD, chronic obstructive pulmonary disease; IPF, idiopathic pulmonary fibrosis; VoC, vasooclussive crisis;, ARDS, acute respiratory distress syndrome; NCT, national clinical trial (www.clinicaltrials.gov).
Pegylated Hemoglobin
Mechanisms of Action of CO
CO performs its protective functions through various mechanisms of action. The most likely target of CO in the context of ‘protection’ is ferrous iron (Fe II), which is present within the heme coordination complex and represents the key functional moiety of hemoproteins that likely regulate non-heme signaling molecules (see Figure 2). We refer the interested reader to an excellent review that details many of the reported mechanisms and CO targets that have been described [29]. There is also another possibility, which has not been well studied, but involves how the cell responds to changes in oxygen tension within the brain. Circadian rhythms are known to be influenced by oxygen sensing pathways including hypoxia-inducible factors [30–32]. Given the highly conserved heme binding capabilities of CO and O2 and the similarities with how the cell responds to changes in O2 availability, we posit that a plausible mechanism by which CO influences cellular behavior is by ‘duping’ the O2 sensors within the cell. In addition to CO binding directly to ferrous iron in heme to regulate the function of a host of factors, i.e. NPAS2, sGC, NOS, and Nr1d1 (Rev-erbα) CO may influence cellular function indirectly by displacing O2 from heme sources within the cell secondary to different affinity strengths for heme. By changing O2 bioavailability O2 could then be utilized for signaling (reactive species), mitochondrial bioenergetics, and other catalytic processes. We define this phenomenon as pseudohypoxia and this concept may shed light on alternative physiologic mechanisms, in addition to heme binding proteins, that regulate the clock and are directly responsible for neuropreservation after stress or injury.
Figure 2. Principal known mechanisms of action of carbon monoxide (CO) in the nervous system.
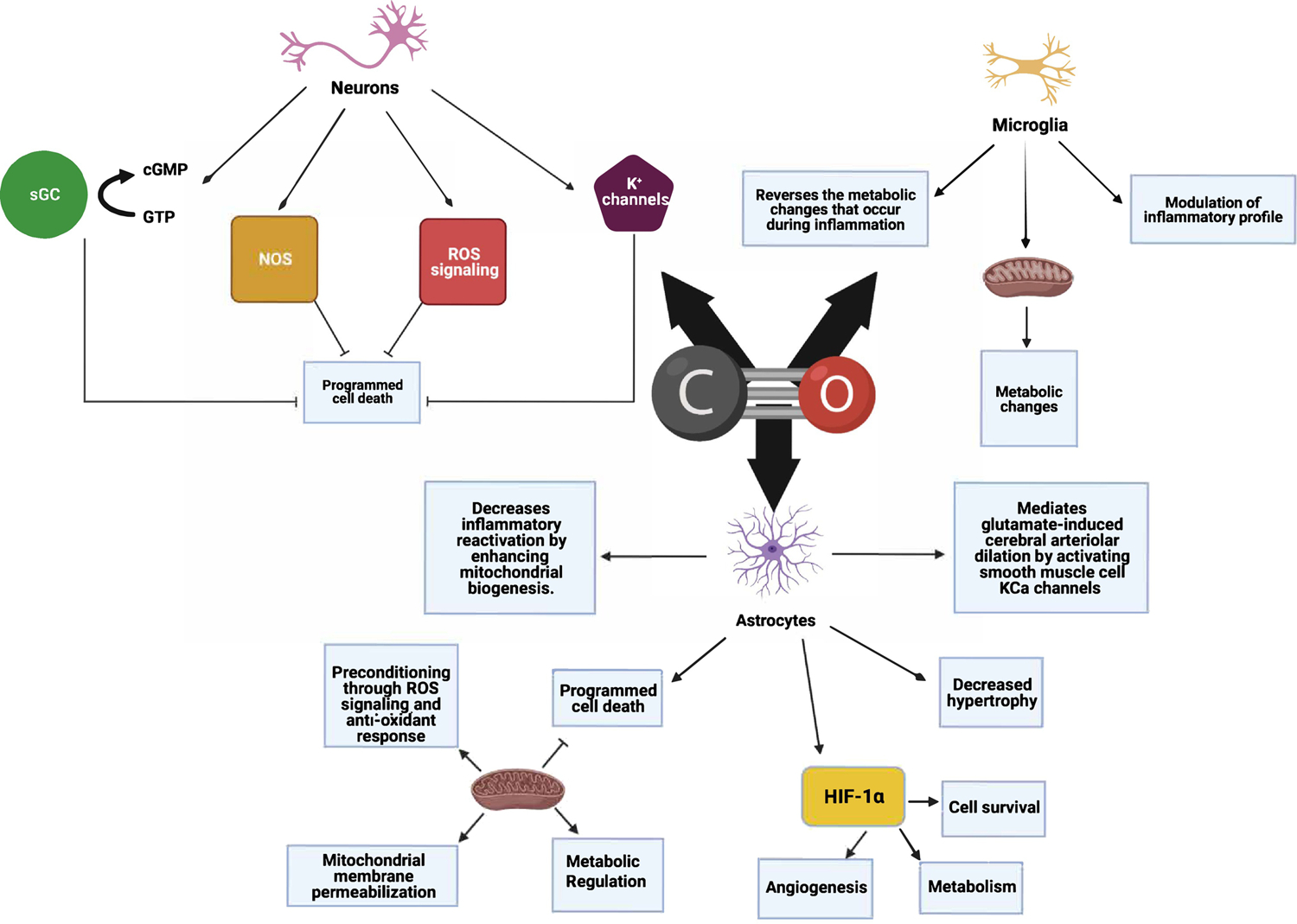
CO is an endogenous gas molecule generated by all cells that functions as a gasotransmitter in the regulation of numerous cellular processes within the nervous system. Among the most well-described include hemoproteins within mitochondria to increase reactive oxygen species (ROS) as well as cytosolic enzymes such including soluble guanylate cyclase (sGC), nitric oxide synthase (NOS) as well as indirect modulation of non-heme proteins including calcium activated K+ channels and hypoxia inducing factor 1- α (HIF-1α). Collectively CO is a critical signaling molecule in the central and peripheral nervous system.
CO is known to increase Hypoxia-Inducible Factor 1-alpha (HIF1α), a non-heme target, and regulate mitochondrial O2 consumption through one of two mechanisms. First CO binds to mitochondrial oxidase heme and increases the generation of reactive oxygen species that stabilize HIF1α [33] and secondly, an indirect effect through CO binding to the iron (Fe II)-containing prolyl hydroxylases (PHD) and preventing HIF1α degradation. Importantly, both mechanisms occur independently of hypoxia. PHD’s do not contain a heme per se, but do contain ferrous iron that binds CO and O2 akin to heme iron. Increased HIF1α-dependent circadian gene expression including Period 1, 2 and CLOCK proteins occurs as a result [33, 34]. Thus CO, through HIF1α modulates an otherwise evolutionarily conserved cellular network that controls metabolic function under stressed conditions, which in turn would increase HO activity and increased availability of endogenous CO. Alternatively, CO may displace O2 from heme, including from the mitochondrial oxidases. Disruption in sleep may result in increased HO-derived CO produced to reestablish a normal cycle. Heme oxygenase deficient animals without the ability to generate carbon monoxide have a clear disruption in normal expression of the clock genes, highlighting the important role the HO/CO axis plays in the regulation of central circadian rhythms, including possibly wake-sleep.
The Nexus of Heme, CO, and Circadian Rhythms
Circadian rhythms are endogenous oscillations that are subserved by an autoregulatory negative feedback transcriptional-translational-posttranslational feedback loop (TTFL) [37, 38] and heme is emerging as being intimately involved in much of the signaling that can be linked to both physiologic and pathophysiologic processes (Figure 3). The basic mechanism of the TTFL involves several transcriptional activators including CLOCK, BMAL-1, Rev-Erb-α, NPAS-2 and two sets of core clock genes: period (per1, per2 and per3 in mice) and cryptochrome (cry1 and cry2 in mice) [39, 40]. [41, 42].
Figure 3. Heme and Regulation of Circadian Rhythm in the Central and Peripheral Nervous System.
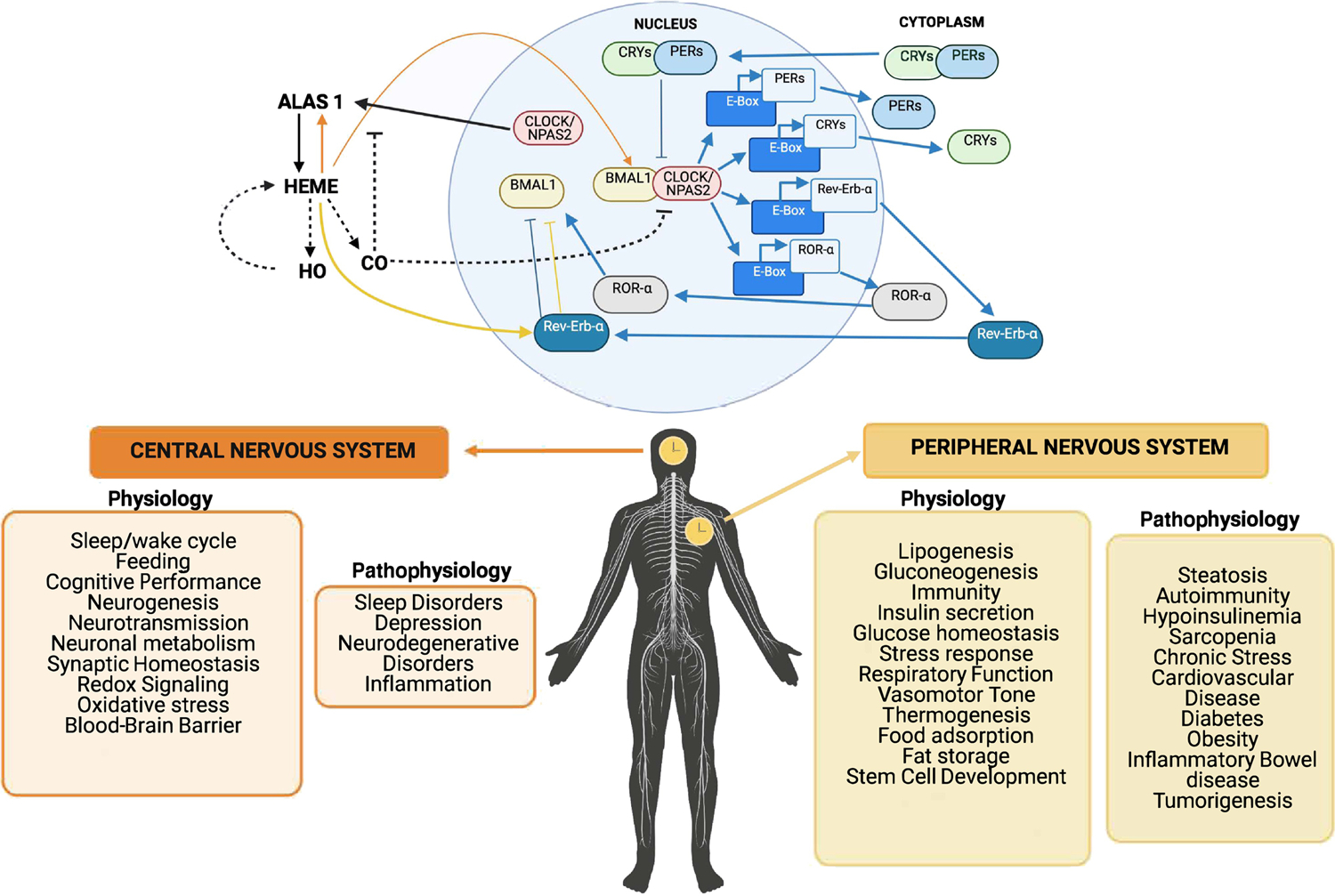
Top Diagram: The circadian clock in mammals is a system composed of transcriptional and translational feedback circuits that oscillate with a cycle of almost 24 hours. The circadian clock controls numerous physiological processes including blood biosynthesis. Some members of the core clock mechanism (NPAS2 and mPER2) contain heme prosthetic groups. Heme synthesis is regulated by the enzyme Alas1 (see Box 1). Heme degradation occurs through the HO enzymes releasing carbon monoxide (CO) as a bioactive product. The principal binding sites for CO are primarily hemoproteins. The heme-containing BMAL1-NPAS2 transcription complex can no longer bind DNA in the presence of CO, thus halting gene transcription. As heme levels decrease and CO is no longer available the quantities of the BMAL1-NPAS2 transcriptional complex can then bind DNA and restart the clock. The loss of circadian cycles in both the CNS and the PNS result in numerous neuropathologies including Alzheimer’s Disease, as well as heart, kidney, digestive and vascular disorders. Lower Diagram: Central versus peripheral circadian synchrony and various associated physiologic and pathophysiologic states that are influenced by the local clocks.
One of the first studies to suggest a link between CO and the circadian clock was from McKnight and colleagues, who reported that low levels of CO could markedly affect the binding (dimerizing) of the circadian transcription factors NPAS2 and Bmal1 [28]. Additionally, the nuclear hormone receptor Nr1d1 is both heme-containing and a target of the core clock genes Bmal1 and Clock and thus may function as a critical CO gas sensor within the SCN [34]. By binding CO, Nr1d1 may in turn modulate the activity of Retinoid related Orphan Receptors (ROR), in particular RORalpha and RORbeta in the SCN that can potently modulate the level of expression of the clock genes [43] [44]. More recently Minegishi et. Al. found that selective removal of endogenous CO in mice, achieved using a highly selective CO scavenger, disrupted rhythmic expression of clock genes [34]. Another recent study found that CO production, secondary to rhythmic heme degradation, strongly attenuates binding of CLOCK-Bmal1 to target promoters [45]. All together, these findings suggest that heme and CO both contribute to normal circadian clock function and therefore likely qualify as important functional “components” of the circadian clock in mammals. Perhaps more intriguing, related and recent work by our group [10, 46] has shown that injury severity in stroke and ischemia reperfusion is correlated with the degree of disruption of clock gene expression, including with the SCN, and that application of exogenous CO largely restores circadian clock gene expression and reduces injury severity. One must ask at this juncture, how can CO be a poison if it is so critically involved in basic physiological processes such as circadian rhythm and when administered in the context of a traumatic injury is highly salutary? It is tempting to speculate, and has in fact been previously proposed, that alterations in heme homeostasis and/or production of CO may underlie sleep and circadian disruption in AD, aging and other neuropathologies [47–49]. Anecdotal reports suggests lower incidence of neurodegenerative disease in smokers. While clearly not something to promote, when presented in the context of a neurologic role for CO, it is tempting to surmise a cause and effect relationship.
Carbon Monoxide Development as a Therapeutic
Delivery of a gaseous molecule is relatively straight forward given the routine use of O2 in every medical center. The challenge was to design a fail-safe device that could accurately dose the correct amount of gas to the patient while not exposing healthcare workers. Currently, there are numerous studies testing inhaled CO in both experimental models as well as FDA-approved phase trials (Table 1). To date, there have been no significant clinical benefits of CO in human trials for fibrotic lung disease or Adult Respiratory Distress Syndrome (ARDS) albeit modulation of one or two biomarkers have been observed when compared to non-CO treated controls [50,51]. In unpublished data an FDA-approved Phase II trial for CO treating kidney transplant recipients showed a trend towards a dose-dependent increase in kidney function in subjects treated intraoperatively with CO through the ventilator. Unfortunately, the trial was never completed for reasons unrelated to the trial data. Inhaled CO is delivered in parts per million (ppm) doses and has been validated in large and small preclinical animal work. Since CO is not metabolized, the pharmacology is extremely well understood with regard to administration and elimination of the gas. CO is transported by hemoglobin (COHb) and principally, if not solely excreted via the lungs and does not accumulate in tissues with a half-life of ~2h in healthy human volunteers. This enables very precise dosing and straightforward monitoring. The challenge is the effective dose necessary in any given disease indication ranging from 4% to 12% COHb and for varying amount of exposure time. FDA Phase 1 data in healthy volunteers determined that 1hr dose escalation showed no adverse events at COHb levels of up to 14% and this has remained as the upper limit [52]. Whether higher levels would also be tolerated has not been tested. These data completely contradict literature reports that COHb of as low as 2–4% had untoward effects including nausea, headache, loss of motor coordination and memory loss. The majority of associated toxicities reported are neurologic in nature [53]. Interestingly, administering CO gas through non-pulmonary routes including gut or abdominal insufflation had completely different effects on toxicity in animals. Studies by Goldbaum and Orellano showed that when comparing COHb greater than 50% achieved via inhalation versus transfusion of saturated erythrocytes versus intraperitoneal injection showed that no toxicity was observed in the latter two routes of delivery [54,55]. This suggests that the therapeutic window for CO may be vastly different depending on how CO is administered and this has led to enormous efforts toward developing small releasing molecules, saturated oral solutions, and hemoglobin carriers for transfusion, which are discussed below and in Hopper et. al.[56].
Pioneered by Motterlini and Foresti as well as Wang, there has been an enormous effort to develop molecules that release CO (termed CORMs or Prodrugs) that are amenable to systemic delivery, and can be titrated, and even tissue targeted. Initially designed as metal carbonyls there has been a remarkable evolution of novel compounds over the last two decades designed to release their CO cargo with exposure to light, i.e. photo-CORMs, changes in pH, alterations in oxygen tension, enzymatic cleavage by esterases and even mitochondrial targeting [57]. The majority of these molecules remain in preclinical development, but hold enormous translational promise as ‘CO in a pill’. Additional modes of delivery that are being developed include oral CO-containing solutions that can be delivered as an oral liquid where CO is saturated in a proprietary formulation (HBI-002) and is absorbed through the stomach and GI epithelium akin to inhaled CO gas crossing lung epithelium. HBI-002 is safe in preclinical GLP toxicology and entering Phase I trials for eventual testing in Sickle Cell Anemia, Organ Transplantation, Inflammatory Bowel Disease and Parkinsons disease. Much like many of the CORMs, HBI-002 mimics efficacy that has been shown with inhaled CO at COHb levels of 4–8% [58]. Finally, there are hemoglobin-based carrier molecules that are furthest along in development where CO-saturated bovine or human hemoglobin requires transfusion and therefore is limited to hospital use only. Sanguinate, is a pegylated bovine COHb that has shown promise in multiple preclinical models is currently in multiple Phase 2/3 trials testing efficacy in hemorrhagic shock and sickle crises as well as subarachnoid hemorrhage and kidney transplantation to prevent delayed graft function (Table 1). The COHb levels achieved are not reported, but a previous human hemoglobin-based carrier was reported to increase the COHb by 2% [59]. There are unique challenges with each of the above modes of delivery that include potency, reliable delivery, safety, untoward side effects and even commercialization concerns related to patent protection. With collectively over one thousand papers supporting exogenous CO use for medical benefit, it seems it is merely a matter of time before conclusive clinical testing is complete across many indications. Where CO will impart its greatest benefit remains unclear as the amount required will certainly depend on pharmacokinetics and pharmacodynamics. The consensus is that targeting a COHb of 6–10% will be the threshold where CO is effective, but if successful tissue targeting strategies are established to deliver pulses of CO locally, systemic COHb may remain at baseline levels. In this same vein, COHb as the only accepted measure of CO presence would become obsolete and the field is desperately seeking alternative biomarkers as indicators of CO exposure and becomes particularly important when considering toxicity.
Concluding Remarks
Historically, CO has been widely viewed as a toxic gas by scientists and non-scientists alike. Over the past two decades however a new view of CO has emerged, which holds that this simple two-atom molecule functions as a critical gasotransmitter in the regulation of numerous biological processes. CO is not only generated constantly in the body but its levels rise during periods of stress [60]. The heme oxygenases regulate CO generation directly through heme metabolism, and are necessary so as to prevent heme from accumulating and contributing to cellular damage. Heme is also essential for the proper function of many proteins including those involved in generating coherent circadian rhythm and sleep wake cycles. In addition, given emerging reports linking the gut microbiome to the regulation of the central clock, the interrelationship between heme, CO and clock function may have profound implications (Box 2). Absence of CO also results in disruption of circadian rhythms and is thought to contribute to development of neurodegenerative disease. The concept that a neurotransmitter, much less a gas, could directly regulate the activity of a transcription factor was a seminal report demonstrating a mechanism for transducing signals both centrally and peripherally [28]. This has now led to many studies of heterodimerization of NPAS2 how it is regulated by a simple gas through a heme-based sensor designed by nature that potently controls both central and peripheral rhythms for reasons yet to be explored (see Outstanding Questions). It is tempting to speculate that the primary mechanism underlying the dramatic salutary effects of CO may, in part involve the restoration of normal function of the SCN circadian pacemaker. In summary, the role of CO and heme in biological processes in the brain, such as circadian rhythms, possibly including the sleep-wake rhythm, is becoming rapidly appreciated. Indeed, an emerging role for CO in the brain is occurring and it is not one of toxicity, but rather challenges this dogma as one that contributes to critical biological and medical processes to ensure life. Given that CO is now safely being tested in a large number of clinical trials as a therapeutic for a wide-range of pathologies based of course on an ever-growing number of preclinical data sets, the view of CO as a poison to the brain needs to be reevaluated. It is unclear why emerging safety data is so vastly different from that reported in the literature, but it might simply reflect the amount and the conditions under which it is evaluated. As Paracelsus originally claimed, ‘all things are toxic, it just depends on the dose’. Reconsideration of the toxicology of CO and particularly with regard to the brain is crucial as CO moves further into clinical therapeutic testing.
Box 2. Gut-Brain Communication and Rhythm.
In recent years it has become clear that visceral information serves a critical role in psychological and emotional regulation that is likely linked to changes in sleep behavior [71–75]. In particular, the mechanisms by which intestinal microbiota exert effects on the brain and therefore on behavior include autonomic signals mediated by the vagus nerve, regulation of stress response and control of immune function [76]. The enteric nervous system (ENS) constitutes an important autonomic division of the nervous system and is critical in the maintenance of homeostasis requiring communication between the gastrointestinal tract and the central nervous system [77].
Given the powerful heme content of gut microbiota it is intriguing to contemplate a signaling mechanism regulating not only local enteric activity, but also remote central nervous system signaling that in turn may feedback to regulate gut function and even microbiome specificity.
Outstanding Questions.
Do the heme oxygenases communicate from the brain to the peripheral clocks through their bioactive products?
Is there a connection between central clock control and CO-induced pseudohypoxia of hemoproteins during a stress response?
Why is a COHb of 5–10% that results from exposure to combustion reported to cause neurological deficits and pathology, yet exposure to pure CO, sufficient to achieve 14% COHb shows no neuropathological consequences?
Is disruption in circadian rhythm influenced by other heme-binding gases including oxygen and nitric oxide?
What disease pathologies can be directly linked to disruption in sleep patterns and heme metabolism?
Is CO used by the clock to communicate timing (phase) information to peripheral tissues?
Could the development of more targeted drugs for modulating heme synthesis prove clinically useful for treating circadian disorders?
Highlights.
Heme is a critical moiety of numerous proteins involved in circadian rhythm. Disruption in one or more of these genes results in disruption of the clock both centrally and peripherally.
When heme is degraded by heme oxygenase it releases bioactive products including carbon monoxide (CO) which have been shown to regulate circadian rhythm in part by acting as a signaling molecule known as a gasotransmitter.
Counter to current dogma, CO is not exclusively a toxic molecule, and in fact is potently neuroprotective in stroke, hemorrhage and neurodegenerative disease.
Administration of CO protects, in part, by regulating circadian rhythm through binding to heme proteins both centrally and peripherally.
Acknowledgements
We acknowledge DoD W81XWH-16-0464, NIH R43GM125430 and the National Football League to L.E.O. NS073613, NS092652 and NS103161 to P.M.F
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclaimer Statement
Leo is a scientific advisor for Hillhurst Biopharmaceuticals.
References
- 1.Maines MD (1988) Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J 2 (10), 2557–68. [PubMed] [Google Scholar]
- 2.Yoshida T and Migita CT (2000) Mechanism of heme degradation by heme oxygenase. J Inorg Biochem 82 (1–4), 33–41. [DOI] [PubMed] [Google Scholar]
- 3.Ryter SW et al. (2006) Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86 (2), 583–650. [DOI] [PubMed] [Google Scholar]
- 4.Ewing JF et al. (1992) Normal and heat-induced patterns of expression of heme oxygenase-1 (HSP32) in rat brain: hyperthermia causes rapid induction of mRNA and protein. J Neurochem 58 (3), 1140–9. [DOI] [PubMed] [Google Scholar]
- 5.Chiziane E et al. (2018) Free Heme and Amyloid-beta: A Fatal Liaison in Alzheimer’s Disease. J Alzheimers Dis 61 (3), 963–984. [DOI] [PubMed] [Google Scholar]
- 6.Portbury SD et al. (2016) A time-course analysis of changes in cerebral metal levels following a controlled cortical impact. Metallomics 8 (2), 193–200. [DOI] [PubMed] [Google Scholar]
- 7.Chang EF et al. (2003) Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury. J Neurosci 23 (9), 3689–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li RC et al. (2009) Heme-hemopexin complex attenuates neuronal cell death and stroke damage. J Cereb Blood Flow Metab 29 (5), 953–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schallner N et al. (2015) Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J Clin Invest 125 (7), 2609–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamat PK, Ahmad AS, Doré S. Carbon monoxide attenuates vasospasm and improves neurobehavioral function after subarachnoid hemorrhage. Arch Biochem Biophys. 2019;676:108117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ewing JF and Maines MD (1997) Histochemical localization of heme oxygenase-2 protein and mRNA expression in rat brain. Brain Res Brain Res Protoc 1 (2), 165–74. [DOI] [PubMed] [Google Scholar]
- 12.Ferreyra GA et al. (1998) Photic control of nitric oxide synthase activity in the hamster suprachiasmatic nuclei. Brain Res 797 (2), 190–6. [DOI] [PubMed] [Google Scholar]
- 13.Gottlieb E et al. (2019) The bidirectional impact of sleep and circadian rhythm dysfunction in human ischaemic stroke: A systematic review. Sleep Med Rev 45, 54–69. [DOI] [PubMed] [Google Scholar]
- 14.Dwyer B and Katz DI (2018) Postconcussion syndrome. Handb Clin Neurol 158, 163–178. [DOI] [PubMed] [Google Scholar]
- 15.Kostyun RO et al. (2015) Sleep disturbance and neurocognitive function during the recovery from a sport-related concussion in adolescents. Am J Sports Med 43 (3), 633–40. [DOI] [PubMed] [Google Scholar]
- 16.Neis VB et al. (2018) Involvement of Heme Oxygenase-1 in Neuropsychiatric and Neurodegenerative Diseases. Curr Pharm Des 24 (20), 2283–2302. [DOI] [PubMed] [Google Scholar]
- 17.Mancuso C et al. (2006) Heme oxygenase and cyclooxygenase in the central nervous system: a functional interplay. J Neurosci Res 84 (7), 1385–91. [DOI] [PubMed] [Google Scholar]
- 18.Gozubuyuk AA et al. (2017) Epidemiology, pathophysiology, clinical evaluation, and treatment of carbon monoxide poisoning in child, infant, and fetus. North Clin Istanb 4 (1), 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi YK (2018) Role of Carbon Monoxide in Neurovascular Repair Processing. Biomol Ther (Seoul) 26 (2), 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mancuso C et al. (2010) Roles of nitric oxide, carbon monoxide, and hydrogen sulfide in the regulation of the hypothalamic-pituitary-adrenal axis. J Neurochem 113 (3), 563–75. [DOI] [PubMed] [Google Scholar]
- 21.Raub JA and Benignus VA (2002) Carbon monoxide and the nervous system. Neurosci Biobehav Rev 26 (8), 925–40. [DOI] [PubMed] [Google Scholar]
- 22.Hanafy KA et al. (2013) Carbon Monoxide and the brain: time to rethink the dogma. Curr Pharm Des 19 (15), 2771–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaiser S et al. (2019) Neuroprotection after Hemorrhagic Stroke Depends on Cerebral Heme Oxygenase-1. Antioxidants (Basel) 8 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wagner KR et al. (2003) Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab 23 (6), 629–52. [DOI] [PubMed] [Google Scholar]
- 25.Bereczki D Jr. et al. (2018) Heme Oxygenase-1: Clinical Relevance in Ischemic Stroke. Curr Pharm Des 24 (20), 2229–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mazzoccoli G et al. (2012) REV-ERBalpha and the clock gene machinery in mouse peripheral tissues: a possible role as a synchronizing hinge. J Biol Regul Homeost Agents 26 (2), 265–76. [PubMed] [Google Scholar]
- 27.Li M et al. (2014) Carbon monoxide induces chromatin remodelling to facilitate endothelial cell migration. Thromb Haemost 111 (5), 951–9. [DOI] [PubMed] [Google Scholar]
- 28.Dioum EM et al. (2002) NPAS2: a gas-responsive transcription factor. Science 298 (5602), 2385–7. [DOI] [PubMed] [Google Scholar]
- 29.Rochette L et al. (2013) Carbon monoxide: mechanisms of action and potential clinical implications. Pharmacol Ther 137 (2), 133–52. [DOI] [PubMed] [Google Scholar]
- 30.Bartman CM and Eckle T (2019) Circadian-Hypoxia Link and its Potential for Treatment of Cardiovascular Disease. Curr Pharm Des 25 (10), 1075–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peek CB et al. (2017) Circadian Clock Interaction with HIF1alpha Mediates Oxygenic Metabolism and Anaerobic Glycolysis in Skeletal Muscle. Cell Metab 25 (1), 86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Depping R and Oster H (2017) Interplay between environmentally modulated feedback loops - hypoxia and circadian rhythms - two sides of the same coin? FEBS J 284 (22), 3801–3803. [DOI] [PubMed] [Google Scholar]
- 33.Chin BY et al. (2007) Hypoxia-inducible factor 1alpha stabilization by carbon monoxide results in cytoprotective preconditioning. Proc Natl Acad Sci U S A 104 (12), 5109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minegishi S et al. (2018) Circadian clock disruption by selective removal of endogenous carbon monoxide. Sci Rep 8 (1), 11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landgraf D et al. (2015) Embryonic development and maternal regulation of murine circadian clock function. Chronobiol Int 32 (3), 416–27. [DOI] [PubMed] [Google Scholar]
- 36.Poss KD and Tonegawa S (1997) Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci U S A 94 (20), 10919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Albrecht U (2012) Timing to perfection: the biology of central and peripheral circadian clocks. Neuron 74 (2), 246–60. [DOI] [PubMed] [Google Scholar]
- 38.Buhr ED and Takahashi JS (2013) Molecular components of the Mammalian circadian clock. Handb Exp Pharmacol (217), 3–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reppert SM and Weaver DR (2002) Coordination of circadian timing in mammals. Nature 418 (6901), 935–41. [DOI] [PubMed] [Google Scholar]
- 40.Harms E et al. (2004) Posttranscriptional and posttranslational regulation of clock genes. J Biol Rhythms 19 (5), 361–73. [DOI] [PubMed] [Google Scholar]
- 41.Preitner N et al. (2002) The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 110 (2), 251–60. [DOI] [PubMed] [Google Scholar]
- 42.Ueda HR et al. (2002) A transcription factor response element for gene expression during circadian night. Nature 418 (6897), 534–9. [DOI] [PubMed] [Google Scholar]
- 43.Tilley SL et al. (2007) Retinoid-related orphan receptor gamma controls immunoglobulin production and Th1/Th2 cytokine balance in the adaptive immune response to allergen. J Immunol 178 (5), 3208–18. [DOI] [PubMed] [Google Scholar]
- 44.Guenthner CJ et al. (2009) Heme reversibly damps PERIOD2 rhythms in mouse suprachiasmatic nucleus explants. Neuroscience 164 (2), 832–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klemz R et al. (2017) Reciprocal regulation of carbon monoxide metabolism and the circadian clock. Nat Struct Mol Biol 24 (1), 15–22. [DOI] [PubMed] [Google Scholar]
- 46.Correa-Costa M et al. (2018) Carbon monoxide protects the kidney through the central circadian clock and CD39. Proc Natl Acad Sci U S A 115 (10), E2302–E2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Atamna H et al. (2002) Heme deficiency may be a factor in the mitochondrial and neuronal decay of aging. Proc Natl Acad Sci U S A 99 (23), 14807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dwyer BE et al. (2009) Heme-a, the heme prosthetic group of cytochrome c oxidase, is increased in Alzheimer’s disease. Neurosci Lett 461 (3), 302–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaasik K and Lee CC (2004) Reciprocal regulation of haem biosynthesis and the circadian clock in mammals. Nature 430 (6998), 467–71. [DOI] [PubMed] [Google Scholar]
- 50.Rosas IO, Goldberg HJ, Collard HR, et al. A Phase II Clinical Trial of Low-Dose Inhaled Carbon Monoxide in Idiopathic Pulmonary Fibrosis. Chest. 2018;153(1):94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fredenburgh LE, Perrella MA, Barragan-Bradford D, et al. A phase I trial of low-dose inhaled carbon monoxide in sepsis-induced ARDS. JCI Insight. 2018;3(23):e124039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mahan VL Neuroprotective, neurotherapeutic, and neurometabolic effects of carbon monoxide. Med Gas Res. 2012;2(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weaver LK. Carbon monoxide poisoning. Undersea Hyperb Med. 2020;47(1):151–169. [PubMed] [Google Scholar]
- 54.Goldbaum LR; Ramirez RG; Absalon KB, What is the mechanism of carbon monoxide toxicity? Aviat Space Environ Med 1975, 46, 1289–91. [PubMed] [Google Scholar]
- 55.Orellano T; Dergal E; Alijani M; Briggs C; Vasquez J; Goldbaum LR; Absolon KB, Studies on the mechanism of carbon monoxide toxicity. J Surg Res 1976, 20, 485–7. [DOI] [PubMed] [Google Scholar]
- 56.Hopper CP, Meinel L, Steiger C, Otterbein LE. Where is the Clinical Breakthrough of Heme Oxygenase-1 / Carbon Monoxide Therapeutics?. Curr Pharm Des. 2018;24(20):2264–2282. [DOI] [PubMed] [Google Scholar]
- 57.Ji X, Damera K, Zheng Y, Yu B, Otterbein LE, Wang B. Toward Carbon Monoxide-Based Therapeutics: Critical Drug Delivery and Developability Issues. J Pharm Sci. 2016;105(2):406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Correa-Costa M, Gallo D, Csizmadia E, et al. Carbon monoxide protects the kidney through the central circadian clock and CD39. Proc Natl Acad Sci U S A. 2018;115(10):E2302–E2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jo H, Swee L, Galacteros F, Adlette Inati A, Reid M, Keipert PE, Small TN, Booth F Safety and Tolerability Of MP4CO: A Dose Escalation Study In Stable Patients With Sickle Cell Disease. Blood. 2013, 122 (21). [Google Scholar]
- 60.Alm S et al. (1999) Urban commuter exposure to particle matter and carbon monoxide inside an automobile. J Expo Anal Environ Epidemiol 9 (3), 237–44. [DOI] [PubMed] [Google Scholar]
- 61.Smith AG et al. (2011) The regulatory role of heme in neurons. Metallomics 3 (10), 955–62. [DOI] [PubMed] [Google Scholar]
- 62.Gozzelino R (2016) The Pathophysiology of Heme in the Brain. Curr Alzheimer Res 13 (2), 174–84. [DOI] [PubMed] [Google Scholar]
- 63.Ponka P (1997) Tissue-specific regulation of iron metabolism and heme synthesis: distinct control mechanisms in erythroid cells. Blood 89 (1), 1–25. [PubMed] [Google Scholar]
- 64.Dennery PA (2000) Regulation and role of heme oxygenase in oxidative injury. Curr Top Cell Regul 36, 181–99. [DOI] [PubMed] [Google Scholar]
- 65.Martin D et al. (2004) Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem 279 (10), 8919–29. [DOI] [PubMed] [Google Scholar]
- 66.Otterbein LE et al. (2016) Heme Oxygenase-1 and Carbon Monoxide in the Heart: The Balancing Act Between Danger Signaling and Pro-Survival. Circ Res 118 (12), 1940–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maines MD (1997) The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol 37, 517–54. [DOI] [PubMed] [Google Scholar]
- 68.He JZ et al. (2010) Enhanced translation of heme oxygenase-2 preserves human endothelial cell viability during hypoxia. J Biol Chem 285 (13), 9452–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Munoz-Sanchez J and Chanez-Cardenas ME (2014) A review on hemeoxygenase-2: focus on cellular protection and oxygen response. Oxid Med Cell Longev 2014, 604981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maines MD and Panahian N (2001) The heme oxygenase system and cellular defense mechanisms. Do HO-1 and HO-2 have different functions? Adv Exp Med Biol 502, 249–72. [DOI] [PubMed] [Google Scholar]
- 71.Allen AP et al. (2017) A psychology of the human brain-gut-microbiome axis. Soc Personal Psychol Compass 11 (4), e12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Christoff K et al. (2011) Specifying the self for cognitive neuroscience. Trends Cogn Sci 15 (3), 104–12. [DOI] [PubMed] [Google Scholar]
- 73.Mayer EA et al. (2014) Gut microbes and the brain: paradigm shift in neuroscience. J Neurosci 34 (46), 15490–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fukui H et al. (2018) Role of Gut Microbiota-Gut Hormone Axis in the Pathophysiology of Functional Gastrointestinal Disorders. J Neurogastroenterol Motil 24 (3), 367–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sarkar A et al. (2018) The Microbiome in Psychology and Cognitive Neuroscience. Trends Cogn Sci 22 (7), 611–636. [DOI] [PubMed] [Google Scholar]
- 76.Palacios-Garcia I and Parada FJ (2019) Measuring the Brain-Gut Axis in Psychological Sciences: A Necessary Challenge. Front Integr Neurosci 13, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Goyette P et al. (2007) Molecular pathogenesis of inflammatory bowel disease: genotypes, phenotypes and personalized medicine. Ann Med 39 (3), 177–99. [DOI] [PubMed] [Google Scholar]
- 78.Misra H, Lickliter J, Kazo F, Abuchowski A. PEGylated carboxyhemoglobin bovine (SANGUINATE): results of a phase I clinical trial. Artif Organs. 2014. 38(8):702–7. [DOI] [PubMed] [Google Scholar]
- 79.Rhodes MA, Carraway MS, Piantadosi CA, Reynolds CM, Cherry AD, Wester TE, Natoli MJ, Massey EW, Moon RE, Suliman HB. Carbon monoxide, skeletal muscle oxidative stress, and mitochondrial biogenesis in humans. Am J Physiol Heart Circ Physiol. 2009. 297: H392–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mayr FB, Spiel A, Leitner J, Marsik C, Germann P, Ullrich R, Wagner O, Jilma B. Effects of carbon monoxide inhalation during experimental endotoxemia in humans. Am J Respir Crit Care Med. 2005. 171: 354–60. [DOI] [PubMed] [Google Scholar]
- 81.Resch H, Zawinka C, Weigert G, Schmetterer L, Garhöfer G. Inhaled carbon monoxide increases retinal and choroidal blood flow in healthy humans. Invest Ophthalmol Vis Sci. 2005. 46: 4275–80. [DOI] [PubMed] [Google Scholar]
- 82.Vesely AE, Somogyi RB, Sasano H, Sasano N, Fisher JA, Duffin J. The effects of carbon monoxide on respiratory chemoreflexes in humans. Environ Res. 2004. 94: 227–33. [DOI] [PubMed] [Google Scholar]
- 83.SIRS JA. The use of carbon monoxide to prevent sickle-cell formation. Lancet. 1963. 1: 971–2. [DOI] [PubMed] [Google Scholar]
- 84.Beutler E The effect of carbon monoxide on red cell life span in sickle cell disease. Blood. 1975. 46: 253–9. [PubMed] [Google Scholar]