

Abstract
All neurodegenerative diseases feature aggregates, which usually contain disease‐specific diagnostic proteins; non‐protein constituents, however, have rarely been explored. Aggregates from SY5Y‐APPSw neuroblastoma, a cell model of familial Alzheimer's disease, were crosslinked and sequences of linked peptides identified. We constructed a normalized “contactome” comprising 11 subnetworks, centered on 24 high‐connectivity hubs. Remarkably, all 24 are nucleic acid‐binding proteins. This led us to isolate and sequence RNA and DNA from Alzheimer's and control aggregates. RNA fragments were mapped to the human genome by RNA‐seq and DNA by ChIP‐seq. Nearly all aggregate RNA sequences mapped to specific genes, whereas DNA fragments were predominantly intergenic. These nucleic acid mappings are all significantly nonrandom, making an artifactual origin extremely unlikely. RNA (mostly cytoplasmic) exceeded DNA (chiefly nuclear) by twofold to fivefold. RNA fragments recovered from AD tissue were ~1.5‐to 2.5‐fold more abundant than those recovered from control tissue, similar to the increase in protein. Aggregate abundances of specific RNA sequences were strikingly differential between cultured SY5Y‐APPSw glioblastoma cells expressing APOE3 vs. APOE4, consistent with APOE4 competition for E‐box/CLEAR motifs. We identified many G‐quadruplex and viral sequences within RNA and DNA of aggregates, suggesting that sequestration of viral genomes may have driven the evolution of disordered nucleic acid‐binding proteins. After RNA‐interference knockdown of the translational‐procession factor EEF2 to suppress translation in SY5Y‐APPSw cells, the RNA content of aggregates declined by >90%, while reducing protein content by only 30% and altering DNA content by ≤10%. This implies that cotranslational misfolding of nascent proteins may ensnare polysomes into aggregates, accounting for most of their RNA content.
Keywords: aggregation, Alzheimer’s disease, apolipoprotein E, beta amyloid, cotranslational misfolding, DNA, endogenous viruses, functional annotation, gene ontology, neurodegeneration, nucleic acid sequence, nucleic acids, protein aggregates, proteomics, retrotransposons, RNA
Nucleic acid‐binding proteins are shown attaching to RNA and DNA, and then undergoing oxidation, thermal denaturation (reversible or irreversible), or other PTMs that destabilize their secondary structure sufficiently to allow aggregation to occur, an essentially irreversible process. Nascent polypeptides as they are synthesized by ribosomes provide another source of transiently misfolded proteins that can aggregate without oxidation or other trigger for denaturation. The sequence shown is not obligatory, in that disordered proteins could also bind nucleic acids after aggregating.
Abbreviations
- AD
Alzheimer's disease
- AMC
age‐matched controls
- APOE
APOE, apolipoprotein E (protein, gene)
- Aβ1–42
Amyloid beta (residues 1–42)
- ChIP‐seq
Chromatin ImmunoPrecipitation ‐ sequencing
- CLEAR
Coordinated Lysosomal Expression and Regulation, a binding motif
- DNA
deoxyribonucleic acid
- EEF2
EEF2, eukaryotic elongation factor 2 (protein, gene)
- G4BP
G‐quadruplex binding protein
- MAP1A
microtubule‐associated protein 1A
- NA
nucleic acids
- NS
not significant (referring to a difference between groups)
- OE
overexpressing
- PTM
post‐translational modification
- RNA
ribonucleic acid
- TF
transcription factor
1. INTRODUCTION
Protein aggregation increases inexorably with aging in all animal species and in all tissues examined (Ayyadevara, Balasubramaniam, Johnson, et al., 2016; Ayyadevara, Balasubramaniam, Suri, et al., 2016; Brignull et al., 2007; Cohen et al., 2009; David et al., 2010; Dillin & Cohen, 2011; Reis‐Rodrigues et al., 2012). Specific aggregate components, diagnostic for each human neurodegenerative disease, are thought to play causal roles because their pathology‐associated mutation and/or overexpression are sufficient to confer heritable neuropathy in human pedigrees and in transgenic‐animal models (Bandyopadhyay et al., 2007; Dillin & Cohen, 2011; Li et al., 2013; Miller et al., 2010; Roodveldt et al., 2009). Proteins that require structural flexibility often incorporate disordered regions, thus rendering them vulnerable to aggregation; other proteins are only susceptible to aggregation after oxidation or specific post‐translational modifications (Ayyadevara et al., 2015, 2017; Ayyadevara, Balasubramaniam, Parcon, et al., 2016).
We recently developed improved click‐chemistry crosslinking reagents and analytical software to identify adjacent proteins in aggregates, based on peptide‐peptide crosslinking, and we applied it to define the protein‐adherence network, or “contactome”, of aggregates. We began with total, sarkosyl‐insoluble aggregates isolated from SY5Y‐APPSw human neuroblastoma cells (Ayyadevara et al., 2017), a model of familial Alzheimer's disease (fAD). This work revealed a complex, non‐random structure of aggregates in which megahubs (very‐high‐connectivity hubs with ≥100 partners) and hub connectors (low‐connectivity proteins linking large hubs) contribute functionally to the assembly of large aggregates (Balasubramaniam et al., 2019). We noted marked enrichment among megahubs for large structural proteins such as titin, ankyrins 1 – 3, nesprins 1 – 3, MAP1A, and other neurofilament proteins, purely as a consequence of their size. We also observed significant enrichment for a variety of nucleic acid‐binding proteins (Balasubramaniam et al., 2019).
2. RESULTS
2.1. The aggregate interactome
To compensate for protein size variation, we reassessed the aggregate interactome with normalization for protein length. The intra‐aggregate contactome, based on length‐normalized connectivity (interaction number per residue), fell into 11 clusters comprising 24 “central hubs” (Figure 1; hubs with >4 edges, indicated by red circles). Four “hub connectors” of low degree (≤4 edges; green circles) bring together large hubs not otherwise connected. Remarkably, all 24 central hubs and 2 of 4 hub connectors are nucleic acid‐binding proteins (Figure 1), revealing a striking enrichment for proteins that bind RNA (N = 16; p < 3E‒150), or bind DNA (N = 6; p < 2E‒20), or both (N = 2). This supports and extends our earlier observation that nucleic acid‐binding proteins are especially susceptible to aggregation (Balasubramaniam et al., 2019) and led us to inquire whether their targets, RNA and DNA, might also be present in the entities we call “protein aggregates”.
FIGURE 1.
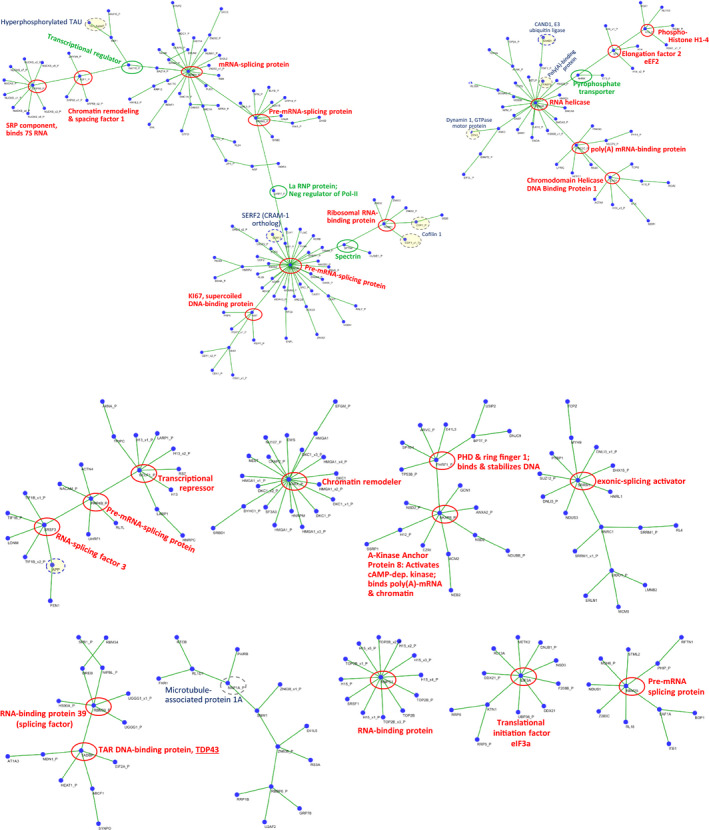
The “aggregate contactome” of proteins isolated from SY5Y‐APPSw human neuroblastoma cells, an in vitro model of familial AD. The contactome was generated from proteomic data for cross‐linked peptide pairs in sarkosyl‐insoluble aggregates, using a modified version of X‐link Identifier (Balasubramaniam et al., 2019; Du et al., 2011), requiring ≥10 spectral hits per protein observed in at least 2 of 3 replicate crosslinking experiments. Hits were normalized to hub length (amino acids in the most abundant isoform). Red circles highlight central hubs with 5 or more large‐hub interactors; green circles show smaller hub‐connectors, which join major hubs not otherwise connected. Other proteins of interest are indicated by dashed gray circles
2.2. Quantitation of aggregate nucleic acids from AD vs. control hippocampus, or human glioma cells
We isolated total sarkosyl‐insoluble aggregates from hippocampal tissue of individuals diagnosed with Alzheimer's disease (AD) and confirmed by histopathological markers, and from age‐matched controls (AMC) without dementia or AD‐diagnostic markers (amyloid deposits or hyperphosphorylated tau). From equal initial weights of hippocampus, quantified recoveries of nucleic acids increased in AD aggregates, over those in controls, by 1.5‐ to 2‐fold for DNA, and ~twofold for RNA (Figure 2A,B). These elevations did not differ significantly from the difference in protein content of total aggregates, which was ~60% higher in AD than in controls, in close agreement with previous results (Ayyadevara, Balasubramaniam, Parcon, et al., 2016). Among normal controls, there was fourfold to sixfold more RNA than DNA in total sarkosyl‐insoluble aggregates (p < 1E–5), regardless of the methods used for separation and quantitation (see Experimental Procedures). For AD samples, nucleic acid recoveries were higher and more variable, with roughly twice as much RNA as DNA (Figure 2B).
FIGURE 2.
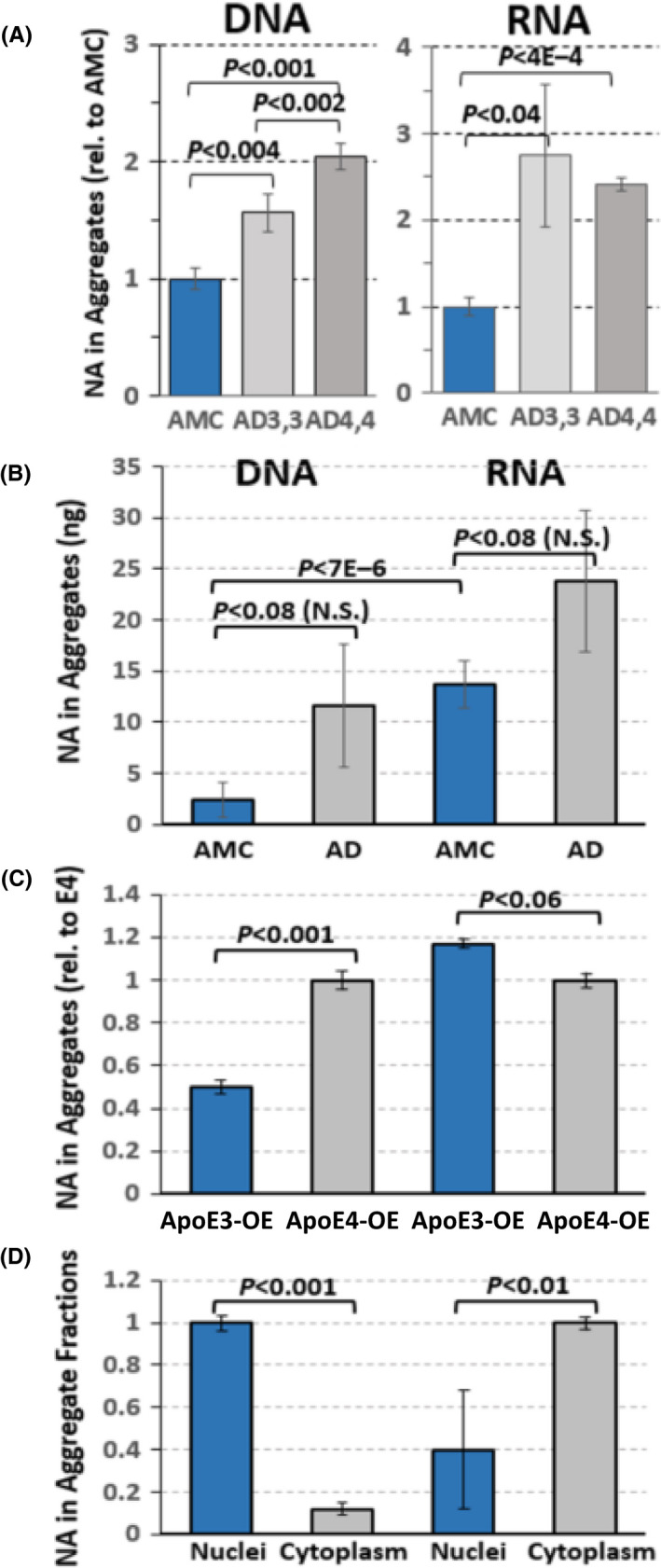
Recovery of DNA and RNA from aggregates. DNA and RNA were extracted and quantified from insoluble aggregates, isolated from hippocampi of AMC (APOE ε3/ε3), or AD (ε3/ε3 or ε4/ε4) individuals (A, each N = 3); or from similar mixes of APOE ε3/ε3 and ε3/ε4 individuals (B, each N = 5–6). (C, D) DNA and RNA were extracted from insoluble aggregates from T98G glioma cells. C, independent cultures of T98G cells overexpressing APOE ε3 from a transgene were compared to cultures expressing ε4. D, T98G cells without any transgene were lysed in 0.5% NP40, and nuclei separated from cytoplasm. DNA was chiefly associated with nuclear aggregates, and RNA with cytoplasmic aggregates, as expected. Means ±SDs are shown. p values reflect 2‐tailed heteroscedastic t tests.
Apolipoprotein E (APOE) gene alleles are the leading genetic risk factors for AD, with at least fourfold increased AD risk for each APOE ε4 allele (abbreviated APOE4, ε4, or E4), and increased severity of aggregate‐associated neuropathology for AD carriers of APOE4 alleles (Neu et al., 2017; Parker et al., 2005). We recently reported that the concerted transcription of autophagy genes is disrupted in the human glioblastoma cell line T98G, when overexpressing an APOE4 transgene rather than APOE3 (Parcon et al., 2018). To assess whether the greater nucleic acid content of aggregates in AD vs. AMC hippocampi may reflect the disruption of autophagy in AD, we separately analyzed aggregates from T98G cells that overexpress APOE ε3 or ε4 from transgenes. The DNA content of T98G/E4 aggregates was about twice that of T98G/E3 aggregates (Figure 2C; p < 0.001), whereas their RNA content declined a little (<15%, N. S.) with the APOE4 allele overexpressed.
Most neuropathic aggregates are cytoplasmic, but may also be nuclear or extracellular. When we separated nuclei from cytoplasm of T98G cells prior to aggregate isolation, similar amounts of aggregate protein were recovered from each fraction. However, nuclear aggregates contained mostly DNA and only 40% as much RNA, while cytoplasmic aggregates contained ~10‐fold more RNA than DNA (Figure 2D).
2.3. Sequencing data for aggregate nucleic acids
To assess whether DNA and RNA fragments in aggregates are a random sampling from the genome and transcriptome, respectively, these nucleic acids were separately extracted from pooled aggregate preparations from either AD or age‐matched control (AMC) individuals (APOE ε3/ε4 heterozygotes; 3 subjects per group), and their sequences determined (UT Southwestern Genomics Core, Dallas TX). DNA fragments were analyzed using a ChIP‐seq protocol, suited to detection of site specificity, and were then mapped to the human genome. RNA fragments underwent a dual screening, comprising a test of peak significance (similar to ChIP‐seq) followed by RNA‐seq analysis of differential abundance, and mapping to the human genome.
Of the 38 loci that showed significant DNA read peaks in at least one pool (each p < 10‒5 that the reads actually came from a uniform distribution), 25 reached that threshold in AD vs. 28 in AMC tissue, and 26 in T98G cells carrying APOE3 vs. 30 in APOE4 cells (Table 1, Supplementary Table S1). Each group contains 17‒20 peak loci with p < 10‒8, and one locus with p < 10‒50. Considering that a random representation of DNA reads would display a flat (uniform) distribution, these data provide decisive evidence that aggregate DNA fragments are not random, but most likely reflect specific binding sites for proteins that ensnared them into aggregates. Only 5 of these 38 DNA peaks (13%) mapped to known genes or ORFs: RP5‐857K21.4, LINC00486, DUX4L26, MAMDC2‐AS1, and ROCK1P1. The other 33 peaks mapped to intergenic regions. Of the 24 chromosomes represented, Y had the most DNA reads (47‒98 reads in each of 6 peaks), followed by chromosomes 4 (3 peaks of 48‒53 reads), 1 (2 peaks of 57‒65 reads), 10 (2 peaks of 35‒45 reads), 16 (2 peaks of 26‒35 reads), 17 (2 peaks of 24‒37 reads), and 21 (4 peaks of 9‒17 reads). Neither the numbers of DNA peaks or reads differed significantly between the groups compared (AD vs. AMC, or E3 vs. E4), with the sole exception of a Y‐chromosome peak mapping at 26,638,004‒ 26,638,595, which yielded only 5 reads in aggregates from AD, but 59 for AMC (Chi2 p < 0.005) and a range of 52‒63 reads for the 3 non‐AD samples.
TABLE 1.
DNA Fragments Recovered from AD, AMC Aggregates (ChIP‐seq analysis)
| Chrom. | Region | Length | 5’ Gene | 5’ distance | 3’ Gene | 3’ Distance | AD Peaks | AMC Peaks | AD Peak p‐value | AMC Peak p‐value | Gene Description (Nearest Gene) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 633735..634300 | 566 | RP5‐857 K21.3 | 36236 | RP5‐857 K21.4 | 0 | 1 | 4 | 4. E−02 | 4. E−06 | Transcribed processed pseudogene; also in all aggregate RNAs |
| 1 | 143202361..143202903 | 543 | RP11‐344P13.1 | 21621836 | CH17‐333 M13.2 | 140276 | 56 | 51 | 3. E−29 | 9. E−31 | |
| 2 | 32916116..32916684 | 569 | LINC00486 | 0 | AL121656.5 | 10442 | 2 | 2 | 4. E−11 | 2. E−13 | Long Intergenic Non‐Protein‐Coding RNA 486; also in all aggregate RNAs |
| 2 | 89830806..89831378 | 573 | 5S_rRNA | 230125 | IGKV2D−40 | 20412 | 11 | 11 | 2. E−05 | 9. E−06 | |
| 3 | 75668953..75669543 | 591 | DUX4L26 | 0 | LINC00960 | 2847 | 1 | 1 | 4. E−11 | 4. E−11 | Double Homeobox 4‐Like 26 |
| 3 | 93470298..93470861 | 564 | HSPE1P19 | 3208589 | RNU6‐488P | 372902 | 2 | 1 | 6. E−57 | 1. E−63 | |
| 4 | 49149228..49149839 | 612 | TPI1P4 | 132073 | AC118282.3 | 47363 | 34 | 27 | 9. E−11 | 1. E−09 | |
| 4 | 49711347..49711912 | 566 | AC119751.4 | 111837 | DCUN1D4 | 2131087 | 13 | 18 | 9. E−16 | 7. E−18 | |
| 4 | 190179314..190179950 | 637 | AC215524.1 | 4089 | ABC7‐42391500H16.3 | 5896 | 3 | 3 | 2. E−13 | 3. E−13 | |
| 5 | 49661354..49661926 | 573 | CTD−2013 M15.1 | 3765383 | EMB | 734265 | 15 | 13 | 2. E−11 | 1. E−10 | |
| 6 | 61322762..61323316 | 555 | RP1‐91 N13.1 | 81450 | MTRNR2L9 | 251011 | 1 | 1 | 3. E−02 | 3. E−06 | |
| 7 | 59995570..59996144 | 575 | RP11‐548 K12.13 | 2159318 | RP11‐715L17.1 | 2279216 | 0 | 9 | no peak | 2. E−03 | |
| 8 | 43237602..43238166 | 565 | VN1R46P | 16792 | RP11‐726G23.2 | 8588 | 4 | 4 | 4. E−08 | 4. E−09 | |
| 9 | 70038064..70038625 | 562 | RN7SL570P | 153409 | MAMDC2‐AS1 | 0 | 1 | 1 | 8. E−07 | 1. E−05 | MAM Domain Containing 2 (antisense strand) |
| 10 | 42070370..42070986 | 617 | RP11‐96F8.1 | 3287261 | KSR1P1 | 78323 | 39 | 35 | 9. E−06 | 2. E−04 | |
| 10 | 133688058..133688661 | 604 | AL845259.2 | 2945 | DUX4L29 | 51944 | 2 | 2 | 2. E−05 | 3. E−04 | |
| 12 | 35614697..35615271 | 575 | AK6P1 | 1364738 | RP11‐125 N22.4 | 1926740 | 1 | 1 | 8. E−03 | 5. E−05 | |
| 13 | 18211736..18212297 | 562 | AL356585.2 | 4266 | RP11‐341D18.4 | 41228 | 1 | 1 | 2. E−11 | 6. E−25 | |
| 16 | 34063731..34064293 | 563 | RP11‐598D12.2 | 12369 | CTD−2522B17.8 | 56176 | 5 | 5 | 7. E−05 | 2. E−04 | |
| 16 | 34587942..34588505 | 564 | BCLAF1P2 | 319292 | CTD−2144E22.9 | 353471 | 15 | 16 | 8. E−16 | 7. E−18 | |
| 16 | 46394505..46395067 | 563 | PPP1R1AP2 | 1E+07 | ANKRD26P1 | 74273 | 16 | 12 | 3. E−23 | 4. E−23 | |
| 17 | 21972876..21973349 | 474 | KCNJ18 | 268263 | AC144838.2 | 83237 | 9 | 7 | 8. E−06 | 2. E−06 | |
| 17 | 26603641..26604283 | 643 | RP11‐846F4.11 | 3897955 | RP11‐260A9.1 | 377410 | 17 | 17 | 3. E−12 | 1. E−09 | |
| 18 | 107991..108643 | 653 | RP11‐683L23.6 | 13660 | ROCK1P1 | 421 | 1 | 2 | 1. E−07 | 9. E−10 | |
| 18 | 110241..110848 | 608 | ROCK1P1 | 0 | MIR8078 | 1407 | 2 | 2 | 9. E−09 | 2. E−08 | Rho‐Assoc'd Coiled‐Coil Prot. Kinase 1 ψgene1 |
| 20 | 31241472..31242039 | 568 | AC104301.2 | 339650 | DEFB115 | 15624 | 17 | 13 | 3. E−18 | 5. E−16 | |
| 21 | 7926020..7926634 | 615 | SNORA11 | 99794 | CH507‐338C24.3 | 174616 | 3 | 3 | 9. E−09 | 4. E−09 | |
| 21 | 8806909..8807490 | 582 | SNX18P10 | 5028 | bP−2189O9.1 | 38075 | 1 | 1 | 2. E−04 | 1. E−05 | |
| 21 | 10269991..10270557 | 567 | CH507‐216 K13.1 | 132986 | AP003900.6 | 57853 | 1 | 4 | 5. E−03 | 8. E−06 | |
| 21 | 10692593..10693162 | 570 | IGHV1OR21‐1 | 42757 | AP001464.4 | 2306513 | 4 | 2 | 4. E−15 | 6. E−15 | |
| 22 | 18896073..18896656 | 584 | AC008103.4 | 11390 | DGCR6 | 9371 | 1 | 1 | 2. E−02 | 4. E−05 | |
| X | 156030320..156030900 | 581 | DDX11L16 | 2442 | 1 | 1 | 1. E−06 | 1. E−06 | |||
| Y | 10746657..10747217 | 561 | RNA5‐8SP6 | 546350 | RP1‐85D24.4 | 295504 | 16 | 8 | 5. E−05 | 1. E−03 | |
| Y | 11305019..11305652 | 634 | AC134878.1 | 120061 | DUX4L16 | 1265 | 6 | 4 | 1. E−09 | 1. E−10 | |
| Y | 11312236..11312823 | 588 | AC134882.1 | 3884 | DUX4L17 | 2097 | 4 | 5 | 1. E−08 | 8. E−09 | |
| Y | 11324443..11324950 | 508 | DUX4L18 | 1619 | DUX4L19 | 7378 | 8 | 8 | 3. E−05 | 4. E−05 | |
| Y | 11721743..11722304 | 562 | RP11‐295P22.2 | 247505 | RCC2P1 | 58747 | 8 | 8 | 1. E−09 | 2. E−08 | |
| Y | 26638004..26638595 | 592 | FAM58CP | 10844 | CTBP2P1 | 3E+07 | 5 | 59 | 8. E−03 | 3. E−13 |
All listed peaks differed significantly from a uniform distribution at p < 0.05 to p < 6E–57; peak‐coincident genes (at zero distance from peaks) are indicated by bold font. Peak counts were not corrected for the 1.6‐fold higher protein and DNA recovery from AD relative to AMC hippocampus, since all reflect normalized data from 1 µg DNA.
RNA differed from DNA in several important respects. Most notably, 81% of RNA peaks mapped to known or putative genes, vs. only 13% of DNA peaks. (Table 2, Supplementary Tables S2, S3; note that intergenic RNA peaks were omitted to conserve space.) The 49 “within‐peak” genes include 39 (80%) that differed significantly in read count between AD and AMC at p < 0.001 (2‐tailed Fisher exact tests), vs. 1 of 38 (2.6%) for DNA. It is noteworthy that 30 significantly differential genes were more abundant in AD, while only 9 (23%) were relatively enriched in AMC. This 3:1 bias is on top of the ~1.8‐fold higher abundance of RNA in AD aggregates (Figure 2A), since all counts were normalized to the source library. Among T98G glioblastoma genes (Supplementary Table S3), 54 of 59 RNA peaks (92%) differ between APOE3 and APOE4 at p < 0.0001, in marked contrast to DNA peaks of which none were significantly differential.
TABLE 2.
RNA Fragments Recovered from AD, AMC Aggregates (RNA‐seq analysis)
| Chrom. | Region | Length | 5’ gene | 5’ dist. | 3’ gene | 3’ dist. | AD reads | AMC reads | AD/AMC reads | Chi2 p< | Transcript or encoded protein |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 633888..634164 | 277 | RP5‐857 K21.3 | 36389 | RP5‐857 K21.4 | 0 | 515 | 1360 | 0.38 | 0.0001 | Transcribed processed pseudogene |
| 1 | 2281959..2282363 | 405 | SKI | 0 | MORN1 | 38889 | 1396 | 1967 | 0.71 | 0.0001 | Ski proto‐oncogene |
| 1 | 109100211..109100485 | 275 | SCARNA2 | 0 | RP5‐1065 J22.4 | 3049 | 131 | 98 | 1.34 | 0.05 | scaRNA, small Cajal‐body specific RNA2, guides mod'n of snRNAs |
| 1 | 244863775..244864066 | 292 | RP11‐11 N7.5 | 8277 | HNRNPU | 0 | 509 | 653 | 0.78 | 0.004 | Heterog. Nuclear Ribonucleoprot. U (transripts 1, 3, 4, 8) |
| 2 | 32916221..32916538 | 318 | LINC00486 | 0 | AL121656.5 | 10588 | 17 | 5 | 3.40 | 0.02 | Long Intergenic Non‐Protein Coding RNA 486 |
| 2 | 47335292..47335571 | 280 | CALM2 | 158690 | AC073283.4 | 0 | 2728 | 2093 | 1.30 | 0.0002 | Uncharacterized transcript |
| 2 | 148961908..148962183 | 276 | KIF5C | 0 | LYPD6B | 75923 | 3055 | 1856 | 1.65 | 0.0001 | Kinesin Family Member 5C (transcript KIF5C_2) |
| 2 | 181678574..181678849 | 276 | ITGA4 | 142386 | CERKL | 0 | 107 | 60 | 1.78 | 0.001 | Ceramide Kinase Like |
| 2 | 231460607..231460888 | 282 | AC017104.2 | 7453 | NCL | 0 | 41 | 27 | 1.52 | N. S. | Ceroid‐Lipofuscinosis Neuronal Protein 5 (neurodegenerative lysosome storage disease) |
| 2 | 233275721..233275994 | 274 | ATG16L1 | 0 | SCARNA5 | 0 | 400/11 | 206/5 | 1.94/2.2 | 0.0001 | Autophagy Related 16‐Like 1 / scaRNA, small Cajal‐body RNA5 |
| 2 | 240713854..240714163 | 310 | RP11‐118 M12.2 | 12168 | KIF1A | 0 | 2806 | 4988 | 0.56 | 0.0001 | Kinesin family member 1A |
| 3 | 160514943..160515216 | 274 | RNU7‐136P | 42526 | KPNA4 | 0 | 536 | 232 | 2.31 | 0.0001 | Karyopherin (Importin) Subunit Alpha 4 |
| 6 | 26204696..26204971 | 276 | HIST1H4E | 0 | HIST1H2BG | 11094 | 164 | 304 | 0.54 | 0.0001 | Histone 1 (AD‐agg‐enriched) |
| 6 | 34071883..34072162 | 280 | MIR1275 | 71831 | GRM4 | 0 | 10689 | 5521 | 1.94 | 0.0001 | Glutamate Metabotropic Receptor 4 |
| 6 | 52995648..52995925 | 278 | RN7SK | 0 | ICK | 5353 | 32569 | 12138 | 2.68 | 0.0001 | 7SK RNA |
| 6 | 99408034..99408309 | 276 | COQ3 | 13829 | PNISR | 0 | 206 | 135 | 1.53 | 0.001 | PNN Interact. Ser & Arg‐Rich Prot. (Pinin, desmosome assoc. prot.) |
| 7 | 5529123..5529444 | 322 | MIR589 | 33205 | ACTB | 0 | 1210 | 1719 | 0.70 | 0.0001 | beta actin (AD‐agg‐enriched) |
| 7 | 26200464..26200741 | 278 | CTB−119C2.1 | 25518 | HNRNPA2B1 | 0 | 168 | 99 | 1.70 | 0.0002 | HnRNP A2/B1 (AD‐agg‐enriched) |
| 8 | 9903986..9904242 | 257 | MIR597 | 162217 | LINC00599 | 0 | 1667 | 1113 | 1.50 | 0.0001 | Long Intergenic Non‐Protein Coding RNA |
| 8 | 27604977..27605252 | 276 | GULOP | 15903 | CLU | 0 | 156 | 105 | 1.49 | 0.005 | Clusterin (a.k.a. ApoJ) (AD‐agg‐enriched) |
| 9 | 35657748..35658022 | 275 | SIT1 | 6797 | RMRP_1 | 0 | 472 | 184 | 2.57 | 0.0001 | RNA Component of Mitoch. RNA Processing Endoribonuclease |
| 10 | 101799051..101799385 | 335 | NPM3 | 15637 | MGEA5 / OGA | 0 | 683 | 432 | 1.58 | 0.0001 | Hyaluronidase =OGA, removes O‐GlcNAc modifications |
| 11 | 35663281..35663611 | 331 | TRIM44 | 0 | KRT18P14 | 196637 | 1147 | 592 | 1.94 | 0.0001 | Tripartite Motif Containing 44 |
| 11 | 62841562..62841839 | 278 | RP11‐727F15.9 | 7518 | WDR74 | 0 | 304 | 149 | 2.04 | 0.0001 | WD Repeat Domain 74 |
| 11 | 65502716..65503064 | 349 | AP000769.7 | 4310 | MALAT1_1 | 0 | 30539 | 16494 | 1.85 | 0.0001 | Metastasis Assoc. Lung Adenocarcinoma Transcr. 1 (Non‐Protein) |
| 11 | 111911504..111911780 | 277 | RPL37AP8 | 22029 | CRYAB | 0 | 228 | 216 | 1.06 | N. S. | Crystalin A beta, a small HSP (AD‐agg‐enriched) |
| 11 | 123061064..123061339 | 276 | RPL31P47 | 9662 | HSPA8 | 0 | 871 | 1512 | 0.58 | 0.0001 | Heat Shock Protein Family A (Hsp70) Member 8 (= HSC70) |
| 12 | 110281619..110281909 | 291 | ATP2A2 | 0 | RN7SL769P | 78542 | 433 | 225 | 1.92 | 0.0001 | SERCA2 ATPase Sarcoplasmic/Endopl. Reticulum Ca2+ Transport2 |
| 13 | 45975357..45975649 | 293 | AL445232.1 | 60203 | ZC3H13 | 0 | 372 | 240 | 1.55 | 0.0001 | Zinc Finger CCCH‐Type Containing 13 |
| 14 | 20343094..20343370 | 277 | SNORD126 | 16567 | RPPH1 | 0 | 2313 | 634 | 3.65 | 0.0001 | Ribonuclease P RNA Component H1 |
| 14 | 23321579..23321860 | 282 | PABPN1 | 0 | SLC22A17 | 24445 | 341 | 261 | 1.31 | 0.01 | Poly(A) Binding Protein Nuclear 1 |
| 14 | 49586595..49586871 | 277 | RNA5SP384 | 33846 | RPS29 | 0 | 22800 | 4958 | 4.60 | 0.0001 | Ribosomal protein, small subunit 29 |
| 14 | 49853648..49853923 | 276 | RNU6‐539P | 13820 | Metazoa_SRP_138 | 0 | 24010 | 7554 | 3.18 | 0.0001 | RNA, cytoplasmic Signal Recognition Particle |
| 14 | 102084750..102085033 | 284 | RN7SL472P | 7277 | HSP90AA1 | 0 | 186 | 94 | 1.98 | 0.0001 | HSP 90 family (AD‐agg‐enriched) |
| 17 | 6452910..6453209 | 300 | FAM64A | 1440 | PITPNM3 | 0 | 671 | 520 | 1.29 | 0.004 | PITPNM 3, Phosphatidylinositol Transfer Protein Memb.‐ Assoc. |
| 17 | 19187969..19188246 | 278 | KYNUP1 | 18300 | SNORD3A | 0 | 342 | 99 | 3.45 | 0.0001 | small nucleolar RNA |
| 17 | 44911190..44911459 | 270 | AC015936.3 | 6643 | GFAP | 0 | 7051 | 8989 | 0.78 | 0.0002 | Glial Fibrillary Acidic Protein, GFAP (AD‐agg‐enriched) |
| 18 | 49814124..49814400 | 277 | ACAA2 | 163 | RP11‐886H22.1 | 0 | 295 | 139 | 2.12 | 0.0001 | Transcribed processed pseudogene |
| 19 | 13298568..13298866 | 299 | CTC−250I14.1 | 139779 | CACNA1A | 0 | 10268 | 5984 | 1.72 | 0.0001 | Calcium Voltage‐Gated Channel Subunit Alpha1 A |
| 19 | 36304598..36304871 | 274 | CTD−3162L10.1 | 0 | LINC00665 | 8195 | 556 | 164 | 3.39 | 0.0001 | Uncharacterized locus |
| 19 | 44907737..44908015 | 279 | APOE | 0 | CTB−129P6.7 | 1359 | 1028 | 1003 | 1.02 | N. S. | ApoE, Apolipoprotein E (AD‐agg‐enriched) |
| 19 | 48645763..48646036 | 274 | DBP | 8324 | CA11 | 0 | 407 | 573 | 0.71 | 0.0002 | Carbonic Anhydrase 11 |
| 19 | 49107777..49108115 | 339 | SNRNP70 | 0 | LIN7B | 6208 | 2096 | 1398 | 1.50 | 0.0001 | Small Nuclear Ribonucleoprotein U1 Subunit 70 |
| 20 | 23637606..23637888 | 283 | CST9 | 31729 | CST3 | 0 | 573 | 742 | 0.77 | 0.003 | Cystatin C, Cystatin 3 |
| 20 | 41533110..41533383 | 274 | RP4‐620E11.5 | 150002 | CHD6 | 0 | 1245 | 864 | 1.44 | 0.0001 | Chromodomain Helicase DNA Binding Protein 6, CHDBP6 |
| 21 | 8258828..8259116 | 289 | RNA5‐8S5 | 1894 | FP671120.6 | 0 | 1200 | 132 | 9.09 | 0.0001 | Uncharacterized transcript |
| 21 | 8435771..8435981 | 211 | FP236383.5 | 0 | FP236383.1 | 96 | 207 | 12 | 17.25 | 0.0001 | Uncharacterized transcript |
| 21 | 26021821..26022101 | 281 | LLPHP2 | 258487 | APP | 0 | 1477 | 702 | 2.10 | 0.0001 | Amyloid precursor protein, APP, A4 (AD‐agg‐enriched) |
| X | 140784311..140784609 | 299 | LINC00632 | 11631 | CDR1 | 0 | 36675 | 62951 | 0.58 | 0.0001 | Cerebellar Degeneration Related Protein 1, CDRP1 |
All peaks shown were significant at p < 0.01; peak‐coincident genes (at zero distance from peaks) are indicated by bold font. Read ratios (AD/AMC) and p values for AD‐AMC differences were not corrected for 1.6‐fold higher RNA recovery from AD relative to AMC hippocampus, since all normalized RNA‐seq libraries used 1 µg RNA. “AD‐agg enriched” indicates proteins that were also found to be significantly enriched in AD aggregates relative to controls (Ayyadevara, Balasubramaniam, Parcon, et al., 2016).
Aggregate RNA reads that were substantially more abundant in AD than controls (Chi‐square or 2‐tailed Fisher exact p < 0.001) include two uncharacterized transcripts on chromosome 21, enriched 17‐fold and 9‐fold beyond other RNAs in AD; ribosomal protein/RPS29, 4.6‐fold; RNAse‐P/RPPH1_2, 3.7‐fold; nucleolar RNA/SNORD3A and long noncoding RNA/LINC00486, enriched 3.5‐ and 3.4‐fold; signal‐recognition‐particle RNAs (SRP_138 and RN7SK), 3.2‐ and 2.7‐fold; mitochondrial RNAse P1/RMRP1, 2.6‐fold; karyopherin/KPNA4, 2.3‐fold; amyloid precursor protein/APP, 2.1‐fold, and SERCA2/ATP2A2, 1.9‐fold (Table 2). It is noteworthy that 2 of the 5 genes identified in aggregate DNA, LINC00486 and RP5‐857K21.4, were also among the AD‐enriched transcripts in aggregates, and 9 of the 39 genes (23%) enriched in AD aggregate RNA, relative to AMC, encode proteins that were also enriched in AD‐specific aggregates (Ayyadevara, Balasubramaniam, Parcon, et al., 2016) (bold font in the rightmost column of Table 2).
Because we had observed roughly twice as much DNA in aggregates isolated from glioblastoma cells overexpressing APOE4, as in identical cells expressing APOE3, we asked whether any particular loci or genes were differentially represented in their aggregates. DNA read counts from E3 and E4 aggregates were in fact quite similar for all DNA loci sequenced (Supplementary Table S1), whereas RNA sequencing data (listed in Supplementary Table S3) show striking increases in aggregate‐entrapped RNA transcripts isolated from APOE3‐overexpressing (OE) cells, relative to isogenic cells overexpressing APOE4. For 53 of the 59 genes that were confidently identified within fragment alignment peaks, the read count in APOE4‐OE cells differed significantly from APOE3‐OE cells at Fisher exact p < 10‒4, with E3/E4 ratios ranging from 1.7‒13.9. Only one gene appeared to be more abundant in the presence of excess APOE4, a long noncoding RNA for which there were too few reads to attain significance. This bias is consistent with evidence that APOE4 protein competes with transcription factor TFEB for the ~400 DNA binding sites containing the CLEAR motif, most of which drive expression of proteins involved in autophagy/lysosome functions (Sardiello, 2016).
Differential RNA‐fragment abundances in glioblastoma aggregates, in which only the APOE allele differs between cell lines, tend to be highly significant (53 of 59 have p < 0.0001) and comprise an interesting set. Examples include α‐enolase (E3/E4 = 1.85), MHC‐II YBX1 (3.8), scaRNA2 (2.3), histones H2B (2.8) and H1 (6.7), HSP60 (1.6), HSP90‐A1 (1.8) and ‐B1 (1.9), HSP‐A8 (2.0) and HSP‐B1 (3.4), IGF‐BP3 (5.4) and ‐BP5 (2.0), NCL (2.0), prothymosin α (3.2), SPARC (2.4), nucleophosmin (2.5), RACK1 (3.0), 7SK small nuclear RNA (6.2), EEF1‐A1 (2.2), β‐actin (3.2), peptidylprolyl isomerase A (2.6), collagen 1A2 (4.1), vimentin (4.2), CD44 (2.7), cofilin 1 (3.6), GAPDH (2.9), α tubulin (2.9), RNAse P component H1 (9.2), ribosomal proteins RPS2 (4.2) and RPS29 (13.9), 7SL RNA 2 (8.7), β2‐microglobulin (4.1), annexin A2 (3.4), pyruvate kinase M (2.4), profilin 1 (3.7), γ‐actin (3.4), APOE (3.6), ferritin light chain 1 (3.0), galectin 1 (5.0), and filamin A (2.1). With lesser significance, we find synapsin 3 (E3/E4 = 3.5; p < 0.0002), sequestosome_1/SQSTM1 (1.3; p < 0.004) and vimentin antisense (15; p < 0.002).
In both the direction and magnitude of the RNA‐abundance shift, the influence of Alzheimer's disease was less consistent and so appeared less pronounced on average, than that of the APOE allele. This is almost certainly due to genetic and environmental variance among AD and AMC subjects (Ayyadevara, Balasubramaniam, Parcon, et al., 2016), in contrast to the single transgene that distinguishes T98G/E3 from T98G/E4 cells. Because all human subjects considered in the present comparison were APOE3/E4 heterozygotes, the AD effect could not have arisen from a difference in APOE genotypes. The prevailing reduction in RNA content of E4 aggregates, for the most differentially expressed genes, may reflect transcriptional suppression of TFEB targets by APOE4 (Parcon et al., 2018), rather than an impact of the APOE allele on aggregation per se.
Mapping the RNA transcripts to the human genome revealed a remarkable cluster of at least 20 intergenic loci in a relatively silent segment (21p11.2‒21p12) of the chromosome 21 short arm (Supplementary Figure S1). While these loci are not differentially represented for the most part, either between AD and AMC or between APOE3 and APOE4, they include 2 loci with the highest AD/AMC ratios we observed, 9.1 and 17.2 (each Chi2 p < 10‒6, Table 2).
2.4. Annotation enrichment meta‐analysis of RNA fragments in aggregates
Although we had expected the RNA fragments embedded in aggregates to comprise a random selection from the transcriptome, gene ontology and pathway term enrichment analysis (functional‐annotation clustering in DAVIDTM, http://david.ncifcrf.gov) revealed highly significant enrichment for specific groups of RNAs. Focusing on genes with RNA reads that map to significant peaks and are differentially abundant in T98G/E3 vs. T98G/E4 glioblastoma cells, DAVID meta‐analysis revealed highly significant enrichment clusters for gene annotations relating to [extracellular exosome + acetylation + phosphoprotein + nucleus, acetylation + poly(A) binding], [Ubl conjugation + cadherin binding + cell‐cell adherens junction ], [glycoprotein binding + protein stabilization], and [myelin sheath + unfolded protein response + protein refolding + stress response + chaperone] (Table 3A).
TABLE 3.
DAVID Meta‐Analysis of Top RNA‐seq Peaks from Aggregates
| A. Genes from E3, E4 reads (55 DAVID IDs, implicating 16 clusters of terms sharing members) | ||||
|---|---|---|---|---|
| Cluster #, GO Term | Cluster Enrich. | Count | Fold Enrich. | Benjamini |
| 1, Extracellular exosome | 10.45 | 37 | 5.5 | 9E−21 |
| Acetylation | 10.45 | 31 | 4.2 | 3E−12 |
| Phosphoprotein | 10.45 | 35 | 2.0 | 8E−6 |
| Nucleus | 10.45 | 27 | 2.1 | 4E−4 |
| 2, Acetylation | 8.65 | 31 | 4.2 | 3E−12 |
| Poly(A) RNA binding | 8.65 | 21 | 7.1 | 1E−10 |
| Ubiquitinlike (Ubl) conjugation | 8.65 | 20 | 5.5 | 3E−8 |
| 3, Ubiquitinlike (Ubl) conjugation | 6.79 | 20 | 5.5 | 3E−8 |
| Cadherin binding, cell‐cell adhesion | 6.79 | 9 | 12 | 6E−5 |
| Cell‐cell adherens junction | 6.79 | 8 | 12 | 2E−5 |
| 4, Glycoprotein binding | 3.58 | 5 | 30 | 6E−4 |
| Protein stabilization | 3.58 | 6 | 17 | 4E−3 |
| 5, Myelin sheath | 2.77 | 8 | 22 | 2E−6 |
| Response to unfolded protein | 2.77 | 5 | 45 | 2E−3 |
| Protein refolding | 2.77 | 4 | 100 | 2E−3 |
| Stress response | 2.77 | 5 | 23 | 1E−3 |
| Chaperone | 2.77 | 6 | 14 | 1E−3 |
| B. Genes from AD, AMC reads (38 DAVID IDs, implicating 9 clusters of terms sharing members) | ||||
|---|---|---|---|---|
| Cluster #, GO Term | Cluster Enrich. | Count | Fold Enrich. | Benjamini |
| 1. Intracellular ribonucleoprotein complex | 3.35 | 6 | 26 | 4E−4 |
| Methylation | 3.35 | 10 | 6.4 | 4E−4 |
| Ubiquitinlike (Ubl) conjugation | 3.35 | 10 | 3.8 | 2E−2 |
| Poly(A) RNA binding | 3.35 | 9 | 4.2 | 5E−2 |
| Acetylation | 3.35 | 12 | 2.3 | 8E−2 |
| 2, Extracellular matrix | 2.92 | 6 | 12 | 4E−3 |
| Chaperone | 2.92 | 4 | 13 | 4E−2 |
| ATPase activity | 2.92 | 4 | 12 | 0.13 |
| 3, Nucleoplasm | 2.82 | 15 | 3.2 | 9E−6 |
| Nucleus | 2.82 | 16 | 2.0 | 4E−5 |
| 4, Myelin sheath | 2.17 | 5 | 19 | 5E−3 |
| Chaperone | 2.17 | 4 | 13 | 4E−2 |
| Unfolded protein binding | 2.17 | 3 | 14 | 0.30 |
| C. Annotations from all RNA reads combined | ||||
|---|---|---|---|---|
| GO Term | p value | Count | Fold Enrich. | Benjamini |
| Extracellular exosome | 2E−17 | 42 | 4.0 | 3E−15 |
| Extracellular matrix | 4E−16 | 18 | 16.3 | 4E−14 |
| Poly(A) RNA binding | 4E−13 | 26 | 5.7 | 8E−11 |
| Phosphoprotein | 4E−11 | 55 | 2.0 | 2E−09 |
| Acetylation | 2E−11 | 36 | 3.2 | 2E−09 |
| Isopeptide bond | 3E−11 | 22 | 5.9 | 2E−09 |
| Ubiquitinlike (Ubl) conjugation | 4E−11 | 26 | 4.6 | 2E−09 |
| Focal Adhesion | 2E−09 | 14 | 9.6 | 1E−07 |
| Myelin sheath | 4E−09 | 10 | 17.6 | 2E−07 |
| Membrane | 2E−08 | 27 | 3.3 | 1E−06 |
| Identical protein binding | 3E−08 | 17 | 5.6 | 3E−06 |
| Intracellular ribonucleoprotein complex | 4E−08 | 9 | 17.7 | 1E−06 |
| Disease mutation | 4E−08 | 27 | 3.2 | 1E−06 |
| Methylation | 1E−07 | 17 | 5.1 | 3E−06 |
| Cytosol | 1E−07 | 32 | 2.6 | 4E−06 |
| Glycoprotein binding | 2E−07 | 7 | 26.7 | 2E−05 |
| Protein binding | 1E−06 | 55 | 1.6 | 6E−05 |
| Oxidation | 2E−06 | 6 | 56 | 2E−05 |
| Chaperone | 4E−06 | 8 | 12 | 9E−05 |
| Unfolded protein binding | 5E−06 | 7 | 15.8 | 2E−04 |
| Neurodegeneration | 5E−06 | 9 | 9.3 | 1E−04 |
| Protein stabilization | 1E−05 | 7 | 13.5 | 9E−03 |
| Response to unfolded protein | 2E−05 | 5 | 31.2 | 7E−03 |
| Cadherin binding, cell‐cell adhesion | 2E−05 | 9 | 7.7 | 7E−04 |
| Protein refolding | 2E−05 | 4 | 70 | 6E−03 |
| Cell‐cell adherens junction | 2E−05 | 9 | 7.5 | 5E−04 |
| MHC class II protein complex binding | 3E−05 | 4 | 62.1 | 1E−03 |
| Amyloidosis | 1E−04 | 4 | 43.2 | 2E−03 |
| Stress response | 3E−04 | 6 | 15 | 5E−03 |
N.B.: Minor terms were omitted from each cluster. Cluster enrichment is the “Enrichment Score” from Functional Annotation Clustering under DAVID; fold enrichment is “Fold Change” per term; Benjamini indicates the false discovery rate, FDR, predicted by the Benjamini‐Hochberg procedure.
Among genes with well‐mapped reads that differ significantly between aggregates from AD vs. AMC, the most enriched clusters include [intracellular ribonucleoprotein complex + methylation + Ubl conjugation + poly(A) RNA binding + acetylation ], [extracellular matrix + chaperone + ATPase activity], [nucleoplasm + nucleus ], and [myelin sheath + unfolded protein binding + chaperone ] (Table 3B). The very existence of these clusters, and their marked overlap between meta‐analyses derived from gene lists of very different origin (aggregates from cultured glioblastoma cells vs. human hippocampi) despite only 11 common members, suggests that the underlying aggregate‐RNA fragments are strikingly nonrandom in nature. The specific annotation terms that were most enriched (Table 3C) are likely to reflect the nature of proteins that coalesce in AD and AD‐model aggregates, which include terms (fold enrichment) such as protein refolding (70), MHC class II protein complex binding (62), oxidation (56), amyloidosis (43), response to unfolded protein (31), glycoprotein binding (27), intracellular ribonucleoprotein complex (18), unfolded protein binding (16), and neurodegeneration (9).
2.5. What mechanisms account for RNA and DNA fragments co‐aggregating with proteins?
What does the inclusion of DNA and RNA fragments imply about aggregates or the mechanism of aggregation? Clearly, there are proteins in aggregates that evolved to bind both nucleic acids and proteins. RNA assumes many transient structures constrained chiefly by its duplex regions, which form A‐helices. DNA, in addition to its repertoire of relatively stable structures (A‐, B‐ and Z‐duplex helices, triplex, and G‐quadruplex forms), in the course of replication and transcription can adopt as wide a range of single‐stranded structures as RNA. Affinity for nucleic acids, as well as the protein constituents of multimeric RNA‐ and DNA‐binding complexes, may require protein structures that are at least partially disordered (Zhang et al., 2013) and/or are highly polar, which in turn may favor aggregation (Babu, 2016; Kovacech et al., 2010). DNA‐binding proteins include histones; high‐mobility‐group (HMG) proteins; constituents of DNA replication, transcription, and repair complexes (e.g. topoisomerases, helicases and polymerases; transcription factors, co‐factors, and repressors); and proteins that stabilize or remodel chromatin (Figure 1) (Li et al., 2006; Mitchell & Tjian, 1989; Stoyanova et al., 2009; Wade, 2001). A key feature shared by many DNA‐binding proteins, in addition to structural instability, is an excess of positively charged residues—allowing formation of electrostatic bonds to the negatively charged phosphates that link DNA‐backbone sugars.
RNA‐binding proteins include splicing factors, translational initiation and elongation factors, ribosomal and associated proteins (e.g., refolding chaperones), signal recognition particles, and proteins involved in the processing and functions of noncoding RNAs (Figure 1) (Castello et al., 2012; Glisovic et al., 2008; Hentze et al., 2018; Turner & Hodson, 2012). There are also diverse proteins that bind both RNA and DNA—including RNA polymerases and other transcription‐complex components, RNA/DNA helicases, and TAR DNA‐binding protein (TDP43/TADBP) (Gao et al., 2019; Hudson & Ortlund, 2014; Kobren & Singh, 2019; Nikpour & Salavati, 2019; Norman et al., 2016; Shi & Berg, 1995; Zacco et al., 2019). Such proteins may account for the presence in aggregates of both RNA and DNA fragments mapping to LINC00486 and RP5‐857K21.4 loci.
G‐quadruplex binding proteins (G4BPs) could be responsible for the presence of certain DNA and RNA segments in aggregates. Of the 39 DNA loci listed in Table 1, 13 (33%) had predicted G‐quadruplex‐forming sequences at >100‐fold higher likelihood than predicted at random, whereas 18 (46%) were <20‐fold above random expectation (Supplementary Table S4 and Figure S2A; note that numbers differ slightly due to binning). This partition into G4‐rich and G4‐poor regions suggests that a subset of DNA fragments may have been “recruited” into aggregates by G4BPs. A similar but less extreme split was observed for RNA fragments listed in this table: 15 of 51 peaks (30%) had ratios >100, vs. 11 (22%) with ratios <20 (Table S2 and Figure S2B).
2.6. Viral RNA and DNA fragments are enriched in AD aggregates relative to AMC
RNA and DNA fragments recovered from aggregates include sequences that do not map to the consensus human genome, but are related to known viral sequences compiled in the VirTect Database (Khan et al., 2019; Xia et al., 2019). After removal of all sequence reads homologous to the human genome, the remainder were mapped to the VirTect virus‐sequence library. Raw viral RNA reads comprise 0.09% of total AMC RNA‐fragment sequences (124,803/132,252,624), and 0.15% of AD RNA‐fragments (236,939/158,235,930). Viral DNA reads comprise 0.33% of total AMC or AD DNA‐fragment sequences (124,805/37,225,119 for AMC, 146,271/44,743,221 for AD). In view of their scarcity, such fragments are unlikely to drive aggregation; moreover, only a small minority of total raw reads met all three of the stringent VirTect thresholds (coverage depth ≥5x, a continuous/contiguous region ≥100 nt, and a read count ≥400) required for positive identification of human viruses. A total of 7 human viruses met all criteria, for a total of >135,000 reads (Table 4), out of >800 viruses or viral fragments detected (271,000 DNA reads, 362,000 RNA reads). As a negative control, the C. elegans genome was screened with identical parameters, yielding zero viral reads.
TABLE 4.
Human viral sequences in RNA and DNA fragments recovered from insoluble hippocampal aggregates
| Viral Genome Sequence (viral classification group) | AMC_RNAf Reads | AD_RNAf Reads | AMC_DNAf Reads | AD_DNAf Reads | AMC RNA/DNA | AD RNA/DNA | RNA (AD/AMC) | DNA (AD/AMC) |
|---|---|---|---|---|---|---|---|---|
| NC_022518.1_HERV_K113 (ssRNA‐RT) | 1,289 | 2,242 | 3,586 | 4,698 | 0.36 | 0.48 | 1.74 | 1.31 |
| NC_001806.1_Human_herpesvirus_1 (dsDNA) |
(162) |
(327) | 192 | 183 | 0.84 | 1.79 | 2.02 | 0.95 |
| NC_001798.1_Human_herpesvirus_2 (dsDNA) | 12,806 | ***28,052 | 1,403 | (1,589) | 9.13 | 17.65 | 2.19 | 1.13 |
| NC_007605.1_Human_herpesvirus_4 (dsDNA) | (137) | 234 | 200 | 178 | 0.69 | 1.31 | 1.71 | 0.89 |
| NC_000898.1_Human_herpesvirus_6B (dsDNA) | (521) | ***1,476 | 2,411 | 2,627 | 0.22 | 0.56 | 2.83 | 1.09 |
| NC_012959.1_Human_adenovirus_54 (dsDNA) | 273 | 440 | (14) | (10) | 19.50 | 44.00 | 1.61 | 0.71 |
| NC_009823.1_Hepatitis_C_virus_type_2 (+ssRNA) | (5,492) | ***(15,501) | 19,181 | (22,028) | 0.29 | 0.70 | 2.82 | 1.15 |
| gi|60955|lcl|HPV6REF.1|_Human_papillomavirus_6_(HPV6) (dsDNA) | (82) | (140) | (151) | 218 | 0.54 | 0.64 | 1.71 | 1.44 |
| gi|9627396|lcl|HPV9REF.1|_Human_papillomavirus_9_(HPV9) (dsDNA) | (121) | ***(405) | 94 | (112) | 1.29 | 3.62 | 3.35 | 1.19 |
| gi|1491683|lcl|HPV72REF.1|_Human_papillomavirus_72_(HPV72) (dsDNA) | 334 | **(692) | (22) | (33) | 15.18 | 20.97 | 2.07 | 1.50 |
| TOTALS | 21,217 | ***49,509 | 27,254 | 31,676 |
Values in parentheses failed to meet one or more thresholds, but are included here for purposes of comparison. AD RNA counts differ from AMC by Chi‐squared test: **p<0.001; ***p<0.0001.
For 5 of the 10 viruses shown in Table 4, viral RNA fragments were significantly enriched in AD over AMC (at p < 0.01 to p < 0.0001), relative to the 1.6‐fold AD enrichment of aggregate proteins (Ayyadevara, Balasubramaniam, Parcon, et al., 2016), for a combined significance of p < 1E–18. Three of these viruses, all linear duplex DNA viruses, were substantially more abundant in RNA than in DNA: Herpesvirus 2 (RNA/DNA = 9.1 in AMC, 17.7 in AD); Human Adenovirus 54 (RNA/DNA = 19.5 in AMC, 44.0 in AD); and Human Papillomavirus 72 (RNA/DNA = 15.2 in AMC, 21.0 in AD). Most of the remaining viruses were more highly represented in DNA than RNA, signifying that they were transcriptionally inactive. The consistently greater RNA/DNA ratios in AD tissue than in AMC is intrinsically “corrected” for relative viral and aggregate abundance, and strongly implies greater transcription and/or aggregation of RNA in AD hippocampus relative to AMC.
2.7. Cotranslational aggregation
As noted above, RNA reads substantially exceeded DNA reads by twofold to fivefold (Figure 2A). The propensity for nucleic acid‐binding proteins to be inherently disordered, suggested above as an explanation for entrapment of nucleic acids in aggregates, is not expected to differ greatly between RNA‐ and DNA‐binding proteins. We propose another mechanism, specific to RNA, that would account for the greater abundance of RNA in aggregates: cotranslational misfolding. Among the RNAs identified in AD‐model aggregates (Table 2), many encode proteins that are themselves enriched in AD aggregates: for example, HnRNP_A2/B1, clusterin/ApoJ, β‐crystallin A (CRYAB), SERCA_2/ATP2A2, GFAP, APOE, and Amyloid Precursor Protein/APP (Ayyadevara, Balasubramaniam, Parcon, et al., 2016). Of the 49 genes with RNA positively identified in aggregates, 9 (18%) encoded proteins that were also identified in aggregates. Twenty‐three (46%) of the same 49 RNAs were significantly more abundant in AD aggregates than in controls, while 6 (12%) were significantly enriched in AD aggregates as both RNA and protein.
During translation, nascent proteins are at high risk for misfolding and aggregation until entire structural domains have emerged from the ribosome. From bacteria to mammals, chaperone complexes that include members of the HSP40, HSP60, and HSP90 families are closely associated with ribosomes, where they counteract misfolding of nascent polypeptides (Deuerling et al., 2019; Zhang & Ignatova, 2011). Nevertheless, the fraction of newly synthesized proteins that is cotranslationally degraded can exceed 50% (Turner & Varshavsky, 2000), indicating that chaperone protection is highly fallible. We wondered whether the remarkable abundance in aggregates of diverse RNA fragments, the great majority of which contain coding sequences, might be a clue that cotranslational aggregation occurs when misfolded, nascent proteins are neither prevented from misfolding nor degraded, prior to their coalescence with other misfolded proteins to form insoluble aggregates.
If this is the case, then interventions that arrest or delay translation should sharply reduce the aggregate content of RNA fragments. We used shRNA knockdown of EEF2 mRNA, reducing its steady‐state level by 33% (Figure 3A,B) to attenuate protein translation in SY5Y‐APPSw human neuroblastoma cells. Suppression of EEF2 has been shown to extend lifespan, reduce stress response, and improve the balance of protein quality control (Anisimova et al., 2018; David et al., 2010; Tavernarakis, 2008; Turner & Varshavsky, 2000). While prior research showed the existence of co‐translational protein misfolding and degradation (G. Zhang & Ignatova, 2011), our results suggest that slowing translation may reduce aggregation of misfolded proteins, both in C. elegans (data not shown) and in cultured human cells as follows. In SY5Y‐APPSw cells, shRNA targeting EEF2 eliminated over 90% of the RNA entrapped in aggregates (p < 0.0001; Figure 3C,D), far exceeding the 33% efficacy of EEF2 knockdown (Figure 3B). At the same time, this RNAi exposure had little or no effect on aggregate DNA content (Figure 3E,F), but reduced aggregate protein by 30% (p < 0.01; Figure 3G,H). In SY5Y‐APPSw cells treated for 4 h with MG132, a cell‐permeant proteasome inhibitor, aggregates increased 20–30%; however, this rise was not accompanied by any increase in aggregate RNA fragments (Figure S3). This suggests that the reduction in aggregate burden per se cannot account for the decline in aggregate RNA after EEF2 knockdown.
FIGURE 3.
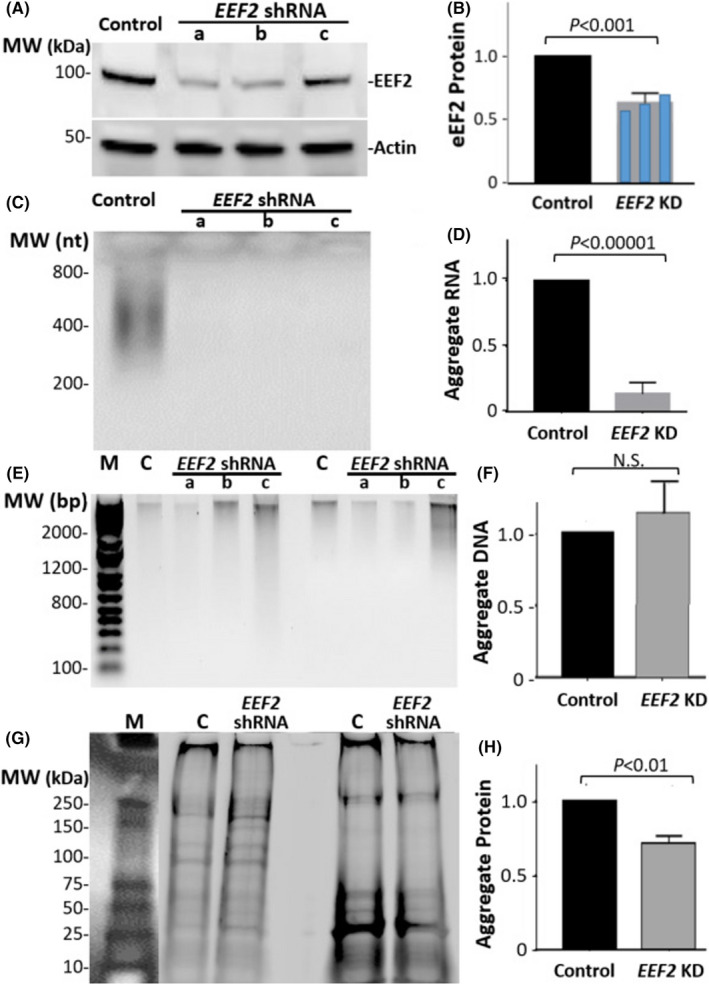
Effects of EEF2 knockdown on the composition of aggregates in SY5Y‐APPSw cells. Results shown in each panel comprise data from 3 independent cell expansions treated with shRNA constructs targeting EEF2, or 2 scrambled RNAs for controls. Replicate experiments produced similar results. (A, B), Western‐blot quantitation of EEF2 knockdown efficacy, evaluated by EEF2 protein; efficacies of individual shRNAs (constructs a, b, c in Methods) are superimposed in B. (C, D), total RNA fragments in aggregates, quantified by gel staining with SYBR Gold. (E, F) Total DNA fragments in aggregates, quantified by ethidium bromide fluorescence. (G, H) Total aggregate protein, quantified by staining with SYPRO Ruby. p values shown here are based on 2‐tailed heteroscedastic t tests, for 3 – 4 experiments, combining data from shRNAs a – c
3. DISCUSSION
Pathognomonic complexes associated with neurodegenerative diseases, including Alzheimer's, Parkinson's, and Huntington's diseases, are widely termed “protein aggregates” because their diagnostic antigenic markers are proteins. Whether these aggregates also contain other components, however, is a question that has not been adequately addressed. We were aware that some amalgamations of cell debris that accumulate with aging, known as lipofuscin granules, contain a complex mixture of oxidized, glycated and carbonylated proteins, lipids, and possibly other carbohydrates; however, nucleic acids were only rarely noted among their constituents (Cindrova‐Davies et al., 2018; Nowotny et al., 2014). Ginsberg et al. (1998, 1999) reported that 80% of neurofibrillary tangles and 55% of senile (amyloid) plaques can be stained with acridine orange, implying the presence of RNA. Numerous studies have implicated nucleic acid binding by mammalian prion‐like protein, PrP (Cordeiro et al., 2014; Gomes et al., 2012; Macedo et al., 2012; Silva et al., 2008), and the evidence that this extends to other neurodegenerative‐disease seed proteins has been reviewed (Cordeiro et al., 2014).
We were led from the results of proteomic “contactome” studies, intended to define the molecular architecture of aggregates (i.e., which proteins adhere to which other proteins), to investigate the nature, extent, and specificity of nucleic acids incorporated into aggregates. In each of these three respects, the results were unexpected. We observed two‐ to fivefold more RNA than DNA in aggregates, whether isolated from AD or control hippocampus (Figure 2A). Many RNA sequences identified in human hippocampal aggregates were differentially abundant in AD‐ vs. control‐derived aggregates; of these, twice as many were enriched significantly in AD aggregates as in non‐AD controls. Proteomic analyses of aggregates from equal‐weight aliquots of AD vs. AMC hippocampus samples indicate an AD/AMC ratio of 1.84 (t test p < 0.01), in reasonable agreement with previous AD/AMC protein ratios of 1.65 and 1.66 for Aβ and tau aggregates, respectively (Ayyadevara, Balasubramaniam, Parcon, et al., 2016), and do not differ significantly from the ratios observed here for RNA and DNA. Interactome complexities of Aβ and tau aggregates (unpublished data) indicate AD/AMC ratios of 1.84 and 1.64, respectively (each t test p < 0.01)—implying more abundant and varied protein interfaces in AD than in AMC hippocampus.
When we compared aggregates isolated from glioblastoma cells overexpressing an APOE3 vs. APOE4 transgene, sequences with the most differential representation were quite consistently more abundant in APOE3‐bearing cells. We believe this very likely reflects the surprising ability of APOE4 to enter nuclei and bind competitively to the CLEAR/E‐box motifs recognized by transcription factor EB (TFEB), thereby inhibiting expression of autophagy and lysosomal genes (Parcon et al., 2018). Not surprisingly, >90% of aggregate DNA originated from nuclear aggregates, while RNA in aggregates was predominantly of cytoplasmic origin.
Only a small fraction of aggregate‐associated nucleic acids (0.09 – 0.15% of RNA reads, 0.33% of a smaller set of DNA reads) appears to be of viral origin, although these totals may be underestimated due to as‐yet‐uncatalogued and mutated viruses or endogenous retroposons (Sanjuan et al., 2010). The striking 2.3‐fold enrichment of viral RNA sequences in AD aggregates relative to controls, vs. only 1.15‐fold for viral DNA fragments (see Table 4), is consistent with possible roles of viral infection and/or transcriptional activation in the etiology of Alzheimer's disease (Balin & Hudson, 2018; Irish et al., 2009; Kreutz, 2002; Kristensson, 1992; Linet al., 1997; Romeo et al., 2019; Steel & Eslick, 2015). It is also possible that the observed enrichments reflect secondary effects of Alzheimer's pathology, including chronic low‐grade inflammation (Majde, 2010), insofar as they may augment viral infection or transcriptional activation in the AD brain. Previous studies have shown that soluble amyloid‐like proteins bind to nucleic acids, which could lead to formation of amyloid fibrils (Di Domizio et al., 2012). Nucleic acid‐containing amyloid fibrils induce interferon and activate innate immune Toll‐like receptors, driving neuroinflammation and synapse loss in AD (Di Domizio et al., 2012; Roy et al., 2020).
Somatically integrated and even endogenous (heritable) viral genomes have highly variable insertion sites. As a result, viral RNAs and DNAs require identification by searching a database of known virus genomes. Quantitation of viral sequences may thus be underestimated due to the many human viruses as yet unidentified, plus the high viral mutation rates impeding sequence alignment. Nevertheless, viral RNA and DNA comprise very small fractions of the nucleic acids recovered from aggregates. From an evolutionary perspective, however, they may ultimately be responsible for the perseverance in our genomes of proteins with high levels of disorder and high probability of aggregation—provided only that disorder contributes to the ability to bind viral nucleic acids and/or to sequester them in aggregates.
The observed data are consistent with a scenario in which endogenous retroviruses—of which HERV K113 is the youngest and only actively transposing exemplar (Boller et al., 2008)—and integrated genomic copies of retroviruses (e.g., Hepatitis C viruses) and DNA viruses (e.g., Herpes viruses) become activated and transcribed into RNA. Darwinian selection might favor protein variants that are predisposed to misfold, provided that they disable replication and transcription of viral genomes within cells by entrapping them in aggregates. Variants that enhanced survival of a pandemic by even a few percent would undergo strong selective pressure to sweep the population, becoming the predominant or sole alleles (Karlsson et al., 2014).
Predicted G‐quadruplex‐forming sequences in both DNA and RNA, the best known and most abundant class of four‐stranded nucleic acid structures, are also markedly enriched in AD aggregates. Sequences with G‐quadruplex‐forming potential can be recognized by their binding proteins based on singular structural features; they thus often serve as recognition sites for critical proteins with key surveillance or regulatory functions, such as telomere‐binding proteins, viral‐replication proteins, and gene promotor regions (Brazda et al., 2014).
The observation of consistent functional‐annotation terms and clusters, both within each aggregate type and between the two sources of aggregates, confirms that the particular RNA species found in aggregates are not a random sampling from the transcriptome—but it does not explain the basis for their enrichment. We propose two routes by which nucleic acids can be incorporated into aggregates that form either as a result of aging per se or due to an age‐dependent pathology such as Alzheimer's disease: (1) “hitchhiker” or “bystander” entrapment of DNA and RNA, when they are bound by proteins that become misfolded and consequently enmeshed in aggregates; and (2) cotranslational misfolding of proteins in the midst of their translation, which might be expected to also ensnare ribosomes and the mRNAs they are translating. The first mechanism is supported by the remarkably high abundance of DNA‐ and RNA‐binding proteins in the aggregate interactome (Figure 1). The second mechanism is most compellingly supported by the decimation (>10‐fold reduction) of aggregate RNA content following shRNA knockdown of the translational procession factor EEF2. We suspect that cotranslational aggregation occurs preferentially in pathways or processes that involve enzymes with multiple partners, and/or several nucleic acid‐binding proteins—thus accounting for the highly significant enrichments observed in aggregated RNA, for genes annotated with specific clusters of descriptive terms. Note that neither of these explanations attributes a primary or causal role to nucleic acids, through which they would “drive” aggregate accrual. Rather, they are collateral casualties due to misfolding of their attached proteins.
Why did EEF2 knockdown have a far greater effect on RNA content than protein content of aggregates? This is actually the expected result if cotranslational aggregation accounts for only a minor fraction of the protein deposition in aggregates, but is responsible for 90% of their RNA content. Nascent proteins may misfold transiently during translation, but even mature proteins can misfold over time, as a consequence of post‐translational disturbances such as oxidation, phosphorylation, or alkylation, and other temperature‐ or time‐dependent processes that favor misfolding of pre‐existing proteins. Such processes would continue with little prospect of reversal, for all previously‐synthesized proteins—unabated by translational arrest. RNA, however, may only appear in aggregates when it is bound by a misfolded (and hence aggregation‐prone) protein, or when the RNA is in the process of translation into a nascent protein that has a high probability of transient misfolding and aggregation. Our observations imply that cotranslational aggregation is the predominant route, accounting for at least 90% of aggregate RNA.
Our data suggest that proteostasis in SY5Y‐APPSw cells, which are subjected to chronic ER stress by continual generation of Aβ1–42, is normally insufficient to prevent cotranslational aggregation. However, even moderate alleviation of that stress appears to shift the balance back to sustainable translational proteostasis. Translational inhibition has been reported to lower chronic inflammation (Mazumder et al., 2010), which may be a consequence of reduced protein aggregation, augmented by a disproportionate decrease in aggregate RNA.
4. CONCLUSIONS
“Protein aggregates” contain nucleic acid constituents that are highly nonrandom in sequence—making it unlikely that they are artifacts, but instead implying that they contain protein‐binding features (including G‐quadruplexes) that might pull them into aggregates. The number and variety of viral sequences found in aggregates suggests that there may be an evolutionary advantage (i.e., antiviral protection) to the synthesis of nucleic acid binding proteins that readily misfold and thus sequester viral genomes in aggregates. Significant enrichment of viral sequences in AD aggregates, relative to controls, is consistent with roles for integrated viruses in AD susceptibility. The preferential enrichment of RNA over DNA in aggregates may implicate a mechanism specific to transcripts: cotranslational aggregation of polysomes during initial misfolding of nascent polypeptides. This process would likely be quite sensitive to the balance between translation rate and chaperone‐mediated refolding capacity. A critical role of cotranslational entrapment is supported by our observation that shRNA knockdown of the translation elongation factor EEF2, although only 25–35% effective, selectively eliminates at least 90% of RNA in aggregates.
5. EXPERIMENTAL PROCEDURES
5.1. Preparation of cells
Cells were grown in 75‐cm2 flasks, with culture medium comprising Dulbecco's Modified Eagles Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) at 37°C, grown in an atmosphere of air supplemented with 5% CO2. Cells were harvested and washed with phosphate buffered saline and then digested with 0.25% (w/v) trypsin (Thermo Fisher) at 37°C for 4 min or until cells detach from the surface.
5.2. EEF‐2 knockdown
For EEF‐2 gene knockdown, RNAi knockdown was performed with 3 distinct EEF‐2 shRNA sequences, targeting human EEF‐2 (SASI_Hs01_00212218 and SASI_Hs_0022218 from Millipore‐Sigma; s4493 from Thermo Fisher), each introduced separately into SH‐SY5Y‐APPSw cells. Cells were harvested and replated at a density sufficient to achieve ~80% confluence 72 h later. RNAiMax (Thermo Fisher) was used as the transfection reagent, following the manufacturer's protocol. MISSION shRNA universal negative controls (SIC001 and SIC002, Millipore‐Sigma) were transfected by the same protocol, as negative controls for the EEF‐2 knockdowns. Cells were harvested and flash frozen 72 hours after transfection.
5.3. Isolation of sarkosyl‐insoluble aggregates
Aggregates were prepared from Alzheimer's Disease (AD) vs. age‐matched control (AMC) hippocampus; T98G/APOE3 or T98G/APOE4 human glial cell pellets; or SY5Y‐APPSw human neuroblastoma cell pellets. Frozen tissues or cells were pulverized with a mortar and pestle (cooled on dry ice) and suspended in lysis buffer containing 20‐mM HEPES pH 7.4, 0.3‐M NaCl, 2‐mM MgCl2, 1% NP40 (w/v), supplemented with phosphatase and protease inhibitors (CalBiochem). Tissue suspensions were lysed in a Teflon homogenizer (2 times 10 s, at 0°C) and sonicated (3 times 10 s, at 0°C). Samples were centrifuged 5 min at 600 × g to remove debris. Supernatant protein was quantified and each sample (0.6–1.0 mg) was centrifuged 15 min at 13,000 × g. Supernatants (soluble protein) were removed, and to each insoluble pellet the same lysis buffer was added plus 1% (v/v) sarkosyl, and mixed well. Samples were centrifuged 20 min at 100,000 × g; supernatants and pellets were recovered as “sarkosyl‐soluble aggregates” and “sarkosyl‐insoluble aggregates”, respectively.
5.4. Immunoprecipitation of amyloid beta and tau aggregates
AD and AMC hippocampal tissue samples were pulverized as described above. After removal of debris (centrifugation for 5 min at 1400 × g), protein was quantified by the Bradford protein assay. Protein was then gently mixed with magnetic beads coated with antibody to either Aβ1–42 (ab11132) or tau (ab80579) for immuno‐pulldown (IP); sarkosyl‐insoluble protein was isolated from the antibody‐bound fractions as described previously (Ayyadevara, Balasubramaniam, Parcon, et al., 2016).
5.5. Aggregate contactome generation
Insoluble aggregates isolated from SY5Y‐APPSw cells as above, were cross‐linked following procedures described previously (Balasubramaniam et al., 2019). In brief, purified aggregates were rinsed, cross‐linked with modified click reagents, digested with trypsin, and the linked peptide pairs were affinity purified using streptavidin‐coated beads to capture the biotin‐coupled crosslinking moiety. Cross‐linked peptide pairs were identified from high‐resolution LC/MS‐MS raw data files, using a modified version of Xlink identifier (Balasubramaniam et al., 2019; Du et al., 2011). Xlink identifier outputs were analyzed with the GePhi software package to calculate the degree (number of interacting partners) of each hub. Because high‐molecular‐weight proteins (e.g., titin) have greater potential to interact with other proteins, spectral hits for each hub were normalized, i.e. divided by the length of that hub protein in amino acids. Identified contactome proteins were categorized by degree, as described previously (Balasubramaniam et al., 2019). Proteins with a high normalized degree (number of interacting partners divided by length in amino acids) or classified as hub‐connectors (connecting 2 or more hub proteins that are not otherwise connected) were pursued by further graph modeling; the Cytoscape package (Shannon et al., 2003) was used with default parameters to construct and visualize graphs.
5.6. Isolation and quantitation of nucleic acids in aggregates
For sequencing of nucleic acid fragments from isolated aggregates, RNA and DNA were extracted from sarkosyl‐insoluble material isolated from cultured cells, or from AD and AMC hippocampus, using the Qiagen AllPrep kit following manufacturer's instructions and a protocol in which this kit was shown to recover even small nucleic acid fragments (Pena‐Llopis & Brugarolas, 2013).
To quantify DNA and RNA trapped in sarkosyl‐insoluble aggregates, nucleic acids were extracted and assayed by multiple protocols, with consistent results. These consisted of (1) separation of RNA and DNA fragments using a Qiagen AllPrep DNA/RNA extraction kit according to the manufacturer's protocol, with recovery assayed by absorbance at 260 nm; (2) separation of RNA and DNA fragments with the Qiagen kit, and quantitation by ethidium bromide and/or SYBR Gold after resolution by acrylamide gel electrophoresis; (3) selective enzymatic digestion with RNAse‐free DNAse (Thermo Fisher, CA) and assay by 260‐nm absorption (RNA directly; DNA by subtraction), and (4) using TRI Reagent (Molecular Research Ctr., TR118) to isolate RNA, DNA, and protein in a single protocol. Figure 2 data were obtained by method (3) above.
5.7. RNA‐seq and ChIP‐seq analyses
All RNA‐seq and ChIP‐seq analyses were performed by the UT Southwestern Genomics Core, analyzed using the CLC Genomics Workbench. We employed ChIP‐seq to evaluate DNA‐fragment specificity; thus, the primary analytic value is the number of significant peaks, with peak validity assessed by an E value relative to a flat distribution (peak absence). RNA‐seq was preceded by peak validation, just as for ChIP‐seq. Subsequently, valid‐peak reads that map uniquely to exons (“unique exon reads”) were summed as our expression metric, and were used to determine differential expression between groups.
The nucleic acid contig assemblies were quite consistent in size, 579 ± 34 (SD) base pairs in length for DNA peaks, and 291 ± 31 (SD) for RNA‐fragment contigs. The efficiency of ChIP‐seq and RNA‐seq fragment cloning protocols, employed prior to sequencing, is quite sensitive to fragment size. Under normal ChIP‐seq protocols, they would be determined by shearing or sonication, size selection by cloning vector, and/or manual size selection. However, in the case of aggregate nucleic acid fragments, other factors may be influential—such as the size, age, and intracellular location of individual aggregates.
5.8. Viral sequence analysis
We employed a modified version of VirTect to scan DNA and RNA fragment sequences from human AD and AMC (age‐matched control) hippocampi. VirTect is a pipeline script that calls a sequence of RNA‐seq pattern‐matching routines (Khan et al., 2019). VirTect retrievals of viral matches to aggregate nucleic acid reads, from 3 AD and 3 AMC brain samples, were filtered using the following parameters (https://github.com/WGLab/VirTect/blob/master/README.md): ≥5x coverage depth, a continuous/contiguous region cutoff of ≥75, and a read count ≥50. Several protocol modifications were made for our pipeline: (1) tophat2 was replaced by hisat2; (2) code was optimized to use all available threads; (3) the internal threshold number of reads was reduced in exploratory runs for the purpose of obtaining AD/AMC read ratios, but recommended thresholds were maintained to eliminate false positives in the assignment of valid hits; (4) the modified script was rewritten in Bash, with unnecessary subroutine calls deleted to reduce run‐time. The database screened by VirTect comprises complete sequences of 757 viruses, as described (Khan et al. 2019). Target sequences were not restricted to human viruses, in recognition of the high frequency of zoonoses and multiple‐host pathogens.
5.9. G‐quadruplex analyses
We employed two programs to screen RNA and DNA sequences for G‐quadruplex‐forming regions: G4CatchAll (Doluca, 2019) and QGRS Mapper (Kikin et al., 2006). Both strands were scanned for each DNA‐fragment sequence, but only strands with G4‐forming potential were pursued in subsequent analyses. The following parameters were used for G4CatchAll: G3L (loop limit) was set to 1.3; G2L (allowing 2‐G loops) was set to 1.3; G4H (enables the G4Hunter algorithm for final evaluation). The following parameters were used for QGRS Mapper: Max. Length 30; Min G‐Group 2; Loop size 0 – 36.
5.10. Statistical analyses
Inter‐group differences were tested for significance by 2‐tailed Behrens–Fisher heteroscedastic t tests, unless otherwise indicated. These conservative tests are appropriate to small‐sample comparisons in which the intra‐group variance is not well estimated. Comparisons of ratios generally employed Yates chi2 (chi‐squared) nondirectional tests, substituting 2‐tailed Fisher Exact tests as required to meet numerical constraints. This conservative replacement is stated in the text but is not made explicit (line by line) in tables to conserve space.
CONFLICTS OF INTEREST
The authors declare no competing or conflicting interests.
AUTHOR CONTRIBUTIONS
RJSR and SA designed the study; MB and SA performed aggregate cross‐linking studies; MB undertook subsequent data analysis and network modeling; MB and AG performed additional proteomics data analyses and contactome construction; JJ undertook G‐quadruplex and human‐virus searches, and checked all RNA and DNA sequencing data analyses (based on data and initial CLC bio/Qiagen analyses provided by the Genomics Core facility, University of Texas Southwest Medical Center); RA and SA performed EEF2‐shRNA knockdown experiments, and RNA and DNA recovery quantitations; RJSR wrote the manuscript with contributions from SA and MB. All authors read and approved the final manuscript.
Supporting information
Fig S1‐S3
Table S1
ACKNOWLEDGMENTS
This work was supported by grants (Merit 2 I01 BX001655 and Senior Research Career Scientist Award IK6 BX004851) to R. J. S. R. from the U. S. Dept. of Veteran Affairs; by R01 grant AG062254‐01 to R. J. S. R. and S. A., and by Program Project grant 2P01AG012411‐17A1 (W. S. T. Griffin, P. I.), both from the National Institute on Aging (NIA/NIH).
Contributor Information
Robert J. Shmookler Reis, Email: rjsr@uams.edu.
Srinivas Ayyadevara, Email: ayyadevarasrinivas@uams.edu.
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this published article and its supplementary information files. Any reasonable request for additional data will be honored.
REFERENCES
- Anisimova, A. S. , Alexandrov, A. I. , Makarova, N. E. , Gladyshev, V. N. , & Dmitriev, S. E. (2018). Protein synthesis and quality control in aging. Aging, 10(12), 4269–4288. 10.18632/aging.101721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyadevara, S. , Balasubramaniam, M. , Gao, Y. , Yu, L. R. , Alla, R. , & Shmookler Reis, R. J. (2015). Proteins in aggregates functionally impact multiple neurodegenerative disease models by forming proteasome‐blocking complexes. Aging Cell, 14(1), 35–48. 10.1111/acel.12296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyadevara, S. , Balasubramaniam, M. , Johnson, J. , Alla, R. , Mackintosh, S. G. , & Shmookler Reis, R. J. (2016). PIP3‐binding proteins promote age‐dependent protein aggregation and limit survival in C. elegans . Oncotarget, 7(31), 48870–48886. 10.18632/oncotarget.10549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyadevara, S. , Balasubramaniam, M. , Kakraba, S. , Alla, R. , Mehta, J. L. , & Shmookler Reis, R. J. (2017). Aspirin‐mediated acetylation protects against multiple neurodegenerative pathologies by impeding protein aggregation. Antioxidants & Redox Signaling, 27(17), 1383–1396. 10.1089/ars.2016.6978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyadevara, S. , Balasubramaniam, M. , Parcon, P. A. , Barger, S. W. , Griffin, W. S. T. , Alla, R. , Tackett, A. J. , Mackintosh, S. G. , Petricoin, E. , Zhou, W. , & Shmookler Reis, R. J. (2016). Proteins that mediate protein aggregation and cytotoxicity distinguish Alzheimer's hippocampus from normal controls. Aging Cell, 15(5), 924–939. 10.1111/acel.12501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyadevara, S. , Balasubramaniam, M. , Suri, P. , Mackintosh, S. G. , Tackett, A. J. , Sullivan, D. H. , Shmookler Reis, R. J. , & Dennis, R. A. (2016). Proteins that accumulate with age in human skeletal‐muscle aggregates contribute to declines in muscle mass and function in Caenorhabditis elegans . Aging, 8(12), 3486–3497. 10.18632/aging.101141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu, M. M. (2016). The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochemical Society Transactions, 44(5), 1185–1200. 10.1042/BST20160172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramaniam, M. , Ayyadevara, S. , Ganne, A. , Kakraba, S. , Penthala, N. R. , Du, X. , & Shmookler Reis, R. J. (2019). Aggregate interactome based on protein cross‐linking interfaces predicts drug targets to limit aggregation in neurodegenerative diseases. iScience, 20, 248–264. 10.1016/j.isci.2019.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balin, B. J. , & Hudson, A. P. (2018). Herpes viruses and Alzheimer's disease: new evidence in the debate. The Lancet Neurology, 17(10), 839–841. 10.1016/S1474-4422(18)30316-8 [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay, B. , Li, G. , Yin, H. , & Kuret, J. (2007). Tau aggregation and toxicity in a cell culture model of tauopathy. Journal of Biological Chemistry, 282(22), 16454–16464. 10.1074/jbc.M700192200 [DOI] [PubMed] [Google Scholar]
- Boller, K. , Schönfeld, K. , Lischer, S. , Fischer, N. , Hoffmann, A. , Kurth, R. , & Tönjes, R. R. (2008). Human endogenous retrovirus HERV‐K113 is capable of producing intact viral particles. Journal of General Virology, 89, 567–572. 10.1099/vir.0.83534-0 [DOI] [PubMed] [Google Scholar]
- Brazda, V. , Haronikova, L. , Liao, J. C. , & Fojta, M. (2014). DNA and RNA quadruplex‐binding proteins. International Journal of Molecular Sciences, 15(10), 17493–17517. 10.3390/ijms151017493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignull, H. R. , Morley, J. F. , & Morimoto, R. I. (2007). The stress of misfolded proteins: C. elegans models for neurodegenerative disease and aging. Advances in Experimental Medicine and Biology, 594, 167–189. 10.1007/978-0-387-39975-1_15 [DOI] [PubMed] [Google Scholar]
- Castello, A. , Fischer, B. , Eichelbaum, K. , Horos, R. , Beckmann, B. M. , Strein, C. , Davey, N. E. , Humphreys, D. T. , Preiss, T. , Steinmetz, L. M. , Krijgsveld, J. , & Hentze, M. W. (2012). Insights into RNA biology from an atlas of mammalian mRNA‐binding proteins. Cell, 149(6), 1393–1406. 10.1016/j.cell.2012.04.031 [DOI] [PubMed] [Google Scholar]
- Cindrova‐Davies, T. , Fogarty, N. M. E. , Jones, C. J. P. , Kingdom, J. , & Burton, G. J. (2018). Evidence of oxidative stress‐induced senescence in mature, post‐mature and pathological human placentas. Placenta, 68, 15–22. 10.1016/j.placenta.2018.06.307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, E. , Paulsson, J. F. , Blinder, P. , Burstyn‐Cohen, T. , Du, D. , Estepa, G. , Adame, A. , Pham, H. M. , Holzenberger, M. , Kelly, J. W. , Masliah, E. , & Dillin, A. (2009). Reduced IGF‐1 signaling delays age‐associated proteotoxicity in mice. Cell, 139(6), 1157–1169. 10.1016/j.cell.2009.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro, Y. , Macedo, B. , Silva, J. L. , & Gomes, M. P. B. (2014). Pathological implications of nucleic acid interactions with proteins associated with neurodegenerative diseases. Biophysical Reviews, 6(1), 97–110. 10.1007/s12551-013-0132-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David, D. C. , Ollikainen, N. , Trinidad, J. C. , Cary, M. P. , Burlingame, A. L. , & Kenyon, C. (2010). Widespread protein aggregation as an inherent part of aging in C. elegans . PLoS Biology, 8(8), e1000450. 10.1371/journal.pbio.1000450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuerling, E. , Gamerdinger, M. , & Kreft, S. G. (2019). Chaperone interactions at the ribosome. Cold Spring Harbor Perspectives in Biology, 11(11), a033977. 10.1101/cshperspect.a033977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domizio, J. , Dorta‐Estremera, S. , Gagea, M. , Ganguly, D. , Meller, S. , Li, P. , Zhao, B. , Tan, F. K. , Bi, L. , Gilliet, M. , & Cao, W. (2012). Nucleic acid‐containing amyloid fibrils potently induce type I interferon and stimulate systemic autoimmunity. Proceedings of the National Academy of Sciences of the United States of America, 109(36), 14550–14555. 10.1073/pnas.1206923109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillin, A. , & Cohen, E. (2011). Ageing and protein aggregation‐mediated disorders: from invertebrates to mammals. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 366(1561), 94–98. 10.1098/rstb.2010.0271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doluca, O. (2019). G4Catchall: A G‐quadruplex prediction approach considering atypical features. Journal of Theoretical Biology, 463, 92–98. 10.1016/j.jtbi.2018.12.007 [DOI] [PubMed] [Google Scholar]
- Du, X. , Chowdhury, S. M. , Manes, N. P. , Wu, S. I. , Mayer, M. U. , Adkins, J. N. , Anderson, G. A. , & Smith, R. D. (2011). Xlink‐identifier: an automated data analysis platform for confident identifications of chemically cross‐linked peptides using tandem mass spectrometry. Journal of Proteome Research, 10(3), 923–931. 10.1021/pr100848a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, X. , Liu, Z. , Zhong, M. , Wu, K. , Zhang, Y. , Wang, H. , & Zeng, T. (2019). Knockdown of DNA/RNA‐binding protein KIN17 promotes apoptosis of triple‐negative breast cancer cells. Oncol Lett, 17(1), 288–293. 10.3892/ol.2018.9597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg, S. D. , Crino, P. B. , Hemby, S. E. , Weingarten, J. A. , Lee, V. M. , Eberwine, J. H. , & Trojanowski, J. Q. (1999). Predominance of neuronal mRNAs in individual Alzheimer's disease senile plaques. Annals of Neurology, 45(2), 174–181. [PubMed] [Google Scholar]
- Ginsberg, S. D. , Galvin, J. E. , Chiu, T. S. , Lee, V. M. , Masliah, E. , & Trojanowski, J. Q. (1998). RNA sequestration to pathological lesions of neurodegenerative diseases. Acta Neuropathologica, 96(5), 487–494. 10.1007/s004010050923 [DOI] [PubMed] [Google Scholar]
- Glisovic, T. , Bachorik, J. L. , Yong, J. , & Dreyfuss, G. (2008). RNA‐binding proteins and post‐transcriptional gene regulation. FEBS Letters, 582(14), 1977–1986. 10.1016/j.febslet.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes, M. P. , Vieira, T. C. , Cordeiro, Y. , & Silva, J. L. (2012). The role of RNA in mammalian prion protein conversion. Wiley Interdisciplinary Reviews: RNA, 3(3), 415–428. 10.1002/wrna.118 [DOI] [PubMed] [Google Scholar]
- Hentze, M. W. , Castello, A. , Schwarzl, T. , & Preiss, T. (2018). A brave new world of RNA‐binding proteins. Nature Reviews Molecular Cell Biology, 19(5), 327–341. 10.1038/nrm.2017.130 [DOI] [PubMed] [Google Scholar]
- Hudson, W. H. , & Ortlund, E. A. (2014). The structure, function and evolution of proteins that bind DNA and RNA. Nature Reviews Molecular Cell Biology, 15(11), 749–760. 10.1038/nrm3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish, B. P. , Khan, Z. K. , Jain, P. , Nonnemacher, M. R. , Pirrone, V. , Rahman, S. , & Wigdahl, B. (2009). Molecular mechanisms of neurodegenerative diseases induced by human retroviruses: A review. American Journal of Infectious Diseases, 5(3), 231–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson, E. K. , Kwiatkowski, D. P. , & Sabeti, P. C. (2014). Natural selection and infectious disease in human populations. Nature Reviews Genetics, 15(6), 379–393. 10.1038/nrg3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, A. , Liu, Q. , Chen, X. , Stucky, A. , Sedghizadeh, P. P. , Adelpour, D. , Zhang, X. I. , Wang, K. , & Zhong, J. F. (2019). Detection of human papillomavirus in cases of head and neck squamous cell carcinoma by RNA‐seq and VirTect. Molecular Oncology, 13(4), 829–839. 10.1002/1878-0261.12435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikin, O. , D'Antonio, L. , & Bagga, P. S. (2006). QGRS Mapper: a web‐based server for predicting G‐quadruplexes in nucleotide sequences. Nucleic Acids Research, 34, W676–W682. 10.1093/nar/gkl253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobren, S. N. , & Singh, M. (2019). Systematic domain‐based aggregation of protein structures highlights DNA‐, RNA‐ and other ligand‐binding positions. Nucleic Acids Research, 47(2), 582–593. 10.1093/nar/gky1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacech, B. , Skrabana, R. , & Novak, M. (2010). Transition of tau protein from disordered to misordered in Alzheimer's disease. Neuro‐Degenerative Diseases, 7(1–3), 24–27. 10.1159/000283478 [DOI] [PubMed] [Google Scholar]
- Kreutz, C. (2002). Molecular, immunological and clinical properties of mutated hepatitis B viruses. Journal of Cellular and Molecular Medicine, 6(1), 113–143. 10.1111/j.1582-4934.2002.tb00317.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensson, K. (1992). Potential role of viruses in neurodegeneration. Molecular and Chemical Neuropathology, 16(1–2), 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. , Shi, J. J. , Mao, C. J. , Liu, S. , Wang, J. D. , Chen, J. , & Liu, C. F. (2013). Alteration of dynein function affects alpha‐synuclein degradation via the autophagosome‐lysosome pathway. International Journal of Molecular Sciences, 14(12), 24242–24254. 10.3390/ijms141224242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Wang, Q. E. , Zhu, Q. , El‐Mahdy, M. A. , Wani, G. , Praetorius‐Ibba, M. , & Wani, A. A. (2006). DNA damage binding protein component DDB1 participates in nucleotide excision repair through DDB2 DNA‐binding and cullin 4A ubiquitin ligase activity. Cancer Research, 66, 8590–8597. 10.1158/0008-5472.CAN-06-1115 [DOI] [PubMed] [Google Scholar]
- Lin, W. R. , Casas, I. , Wilcock, G. K. , & Itzhaki, R. F. (1997). Neurotropic viruses and Alzheimer's disease: a search for varicella zoster virus DNA by the polymerase chain reaction. Journal of Neurology, Neurosurgery and Psychiatry, 62(6), 586–589. 10.1136/jnnp.62.6.586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macedo, B. , Millen, T. A. , Braga, C. A. C. A. , Gomes, M. P. B. , Ferreira, P. S. , Kraineva, J. , Winter, R. , Silva, J. L. , & Cordeiro, Y. (2012). Nonspecific prion protein‐nucleic acid interactions lead to different aggregates and cytotoxic species. Biochemistry, 51(27), 5402–5413. 10.1021/bi300440e [DOI] [PubMed] [Google Scholar]
- Majde, J. A. (2010). Neuroinflammation resulting from covert brain invasion by common viruses ‐ a potential role in local and global neurodegeneration. Medical Hypotheses, 75(2), 204–213. 10.1016/j.mehy.2010.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder, B. , Li, X. , & Barik, S. (2010). Translation control: a multifaceted regulator of inflammatory response. The Journal of Immunology, 184(7), 3311–3319. 10.4049/jimmunol.0903778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, J. , Arrasate, M. , Shaby, B. A. , Mitra, S. , Masliah, E. , & Finkbeiner, S. (2010). Quantitative relationships between huntingtin levels, polyglutamine length, inclusion body formation, and neuronal death provide novel insight into huntington's disease molecular pathogenesis. Journal of Neuroscience, 30(31), 10541–10550. 10.1523/JNEUROSCI.0146-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, P. J. , & Tjian, R. (1989). Transcriptional regulation in mammalian cells by sequence‐specific DNA binding proteins. Science, 245(4916), 371–378. 10.1126/science.2667136 [DOI] [PubMed] [Google Scholar]
- Neu, S. C. , Pa, J. , Kukull, W. , Beekly, D. , Kuzma, A. , Gangadharan, P. , Wang, L.‐S. , Romero, K. , Arneric, S. P. , Redolfi, A. , Orlandi, D. , Frisoni, G. B. , Au, R. , Devine, S. , Auerbach, S. , Espinosa, A. , Boada, M. , Ruiz, A. , Johnson, S. C. , … Toga, A. W. (2017). Apolipoprotein E genotype and sex risk factors for Alzheimer disease: A meta‐analysis. JAMA Neurology, 74(10), 1178–1189. 10.1001/jamaneurol.2017.2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikpour, N. , & Salavati, R. (2019). The RNA binding activity of the first identified trypanosome protein with Z‐DNA‐binding domains. Scientific Reports, 9(1), 5904. 10.1038/s41598-019-42409-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman, M. , Rivers, C. , Lee, Y. B. , Idris, J. , & Uney, J. (2016). The increasing diversity of functions attributed to the SAFB family of RNA‐/DNA‐binding proteins. The Biochemical Journal, 473(23), 4271–4288. 10.1042/BCJ20160649 [DOI] [PubMed] [Google Scholar]
- Nowotny, K. , Jung, T. , Grune, T. , & Hohn, A. (2014). Accumulation of modified proteins and aggregate formation in aging. Experimental Gerontology, 57, 122–131. 10.1016/j.exger.2014.05.016 [DOI] [PubMed] [Google Scholar]
- Parcon, P. A. , Balasubramaniam, M. , Ayyadevara, S. , Jones, R. A. , Liu, L. , Shmookler Reis, R. J. , Barger, S. W. , Mrak, R. E. , & Griffin, W. S. T. (2018). Apolipoprotein E4 inhibits autophagy gene products through direct, specific binding to CLEAR motifs. Alzheimer's & Dementia: the Journal of the Alzheimer's Association, 14(2), 230–242. 10.1016/j.jalz.2017.07.754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, G. R. , Cathcart, H. M. , Huang, R. , Lanham, I. S. , Corder, E. H. , & Poduslo, S. E. (2005). Apolipoprotein gene E4 allele promoter polymorphisms as risk factors for Alzheimer's disease. Psychiatric Genetics, 15(4), 271–275. 10.1097/00041444-200512000-00009 [DOI] [PubMed] [Google Scholar]
- Pena‐Llopis, S. , & Brugarolas, J. (2013). Simultaneous isolation of high‐quality DNA, RNA, miRNA and proteins from tissues for genomic applications. Nature Protocols, 8(11), 2240–2255. 10.1038/nprot.2013.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis‐Rodrigues, P. , Czerwieniec, G. , Peters, T. W. , Evani, U. S. , Alavez, S. , Gaman, E. A. , Vantipalli, M. , Mooney, S. D. , Gibson, B. W. , Lithgow, G. J. , & Hughes, R. E. (2012). Proteomic analysis of age‐dependent changes in protein solubility identifies genes that modulate lifespan. Aging Cell, 11(1), 120–127. 10.1111/j.1474-9726.2011.00765.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo, M. A. , Faggioni, A. , & Cirone, M. (2019). Could autophagy dysregulation link neurotropic viruses to Alzheimer's disease? Neural Regeneration Research, 14(9), 1503–1506. 10.4103/1673-5374.253508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roodveldt, C. , Bertoncini, C. W. , Andersson, A. , van der Goot, A. T. , Hsu, S. T. , Fernandez‐Montesinos, R. , & Dobson, C. M. (2009). Chaperone proteostasis in Parkinson's disease: Stabilization of the Hsp70/alpha‐synuclein complex by Hip. EMBO Journal, 28, 3758–3770. 10.1038/emboj.2009.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, E. R. , Wang, B. , Wan, Y.‐W. , Chiu, G. , Cole, A. , Yin, Z. , Propson, N. E. , Xu, Y. , Jankowsky, J. L. , Liu, Z. , Lee, V.‐Y. , Trojanowski, J. Q. , Ginsberg, S. D. , Butovsky, O. , Zheng, H. , & Cao, W. (2020). Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. Journal of Clinical Investigation, 130(4), 1912–1930. 10.1172/JCI133737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan, R. , Nebot, M. R. , Chirico, N. , Mansky, L. M. , & Belshaw, R. (2010). Viral mutation rates. Journal of Virology, 84(19), 9733–9748. 10.1128/JVI.00694-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello, M. (2016). Transcription factor EB: from master coordinator of lysosomal pathways to candidate therapeutic target in degenerative storage diseases. Annals of the New York Academy of Sciences, 1371(1), 3–14. 10.1111/nyas.13131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N. S. , Wang, J. T. , Ramage, D. , & Ideker, T. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research, 13(11), 2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, Y. , & Berg, J. M. (1995). Specific DNA‐RNA hybrid binding by zinc finger proteins. Science, 268(5208), 282–284. 10.1126/science.7536342 [DOI] [PubMed] [Google Scholar]
- Silva, J. L. , Lima, L. M. , Foguel, D. , & Cordeiro, Y. (2008). Intriguing nucleic‐acid‐binding features of mammalian prion protein. Trends in Biochemical Sciences, 33(3), 132–140. 10.1016/j.tibs.2007.11.003 [DOI] [PubMed] [Google Scholar]
- Steel, A. J. , & Eslick, G. D. (2015). Herpes viruses increase the risk of Alzheimer's disease: A meta‐analysis. Journal of Alzheimer's Disease, 47(2), 351–364. 10.3233/JAD-140822 [DOI] [PubMed] [Google Scholar]
- Stoyanova, T. , Roy, N. , Kopanja, D. , Raychaudhuri, P. , & Bagchi, S. (2009). DDB2 (damaged‐DNA binding protein 2) in nucleotide excision repair and DNA damage response. Cell Cycle, 8(24), 4067–4071. 10.4161/cc.8.24.10109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavernarakis, N. (2008). Ageing and the regulation of protein synthesis: a balancing act? Trends in Cell Biology, 18(5), 228–235. 10.1016/j.tcb.2008.02.004 [DOI] [PubMed] [Google Scholar]
- Turner, G. C. , & Varshavsky, A. (2000). Detecting and measuring cotranslational protein degradation in vivo. Science, 289(5487), 2117–2120. 10.1126/science.289.5487.2117 [DOI] [PubMed] [Google Scholar]
- Turner, M. , & Hodson, D. J. (2012). An emerging role of RNA‐binding proteins as multifunctional regulators of lymphocyte development and function. Advances in Immunology, 115, 161–185. 10.1016/B978-0-12-394299-9.00006-0 [DOI] [PubMed] [Google Scholar]
- Wade, P. A. (2001). Methyl CpG‐binding proteins and transcriptional repression. BioEssays, 23(12), 1131–1137. 10.1002/bies.10008 [DOI] [PubMed] [Google Scholar]
- Xia, Y. , Liu, Y. , Deng, M. , & Xi, R. (2019). Detecting virus integration sites based on multiple related sequencing data by VirTect. BMC Medical Genomics, 12(Suppl 1), 19. 10.1186/s12920-018-0461-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacco, E. , Grana‐Montes, R. , Martin, S. R. , de Groot, N. S. , Alfano, C. , Tartaglia, G. G. , & Pastore, A. (2019). RNA as a key factor in driving or preventing self‐assembly of the TAR DNA‐binding protein 43. Journal of Molecular Biology, 431(8), 1671–1688. 10.1016/j.jmb.2019.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G. , & Ignatova, Z. (2011). Folding at the birth of the nascent chain: coordinating translation with co‐translational folding. Current Opinion in Structural Biology, 21(1), 25–31. 10.1016/j.sbi.2010.10.008 [DOI] [PubMed] [Google Scholar]
- Zhang, T. , Faraggi, E. , Li, Z. , & Zhou, Y. (2013). Intrinsically semi‐disordered state and its role in induced folding and protein aggregation. Cell Biochemistry and Biophysics, 67(3), 1193–1205. 10.1007/s12013-013-9638-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S3
Table S1
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files. Any reasonable request for additional data will be honored.