

SUMMARY
The prion-like spread of tau pathology could underlie a spectrum of clinical syndromes including Alzheimer’s disease (AD). Although evidence indicates that tau is transmissible, it is unclear how pathogenic tau seeds are processed in neurons. Here, we analyze fibrillar wild-type and disease-associated P301L tau seeds by using in vitro and neuronal assays. We show that P301L seeds are uniquely modified by post-translational modifications (PTMs) within the microtubule-binding region (MTBR). Although these modifications do not alter tau seed trafficking or localization, acetylated tau variants show accelerated tau aggregation, enhanced tau PTM priming, and prion-like templating. To explain the enhanced tau seed acetylation, we demonstrate that P301L seeds undergo auto-acetylation. Moreover, tau acts generally to inhibit HDAC6 deacetylase activity by preventing HDAC6 phosphorylation, leading to increased substrate acetylation. Our study highlights complex post-translational regulation of transmissible tau seeds and provides insight into the biological properties of tau strains in AD and other tauopathies.
In brief
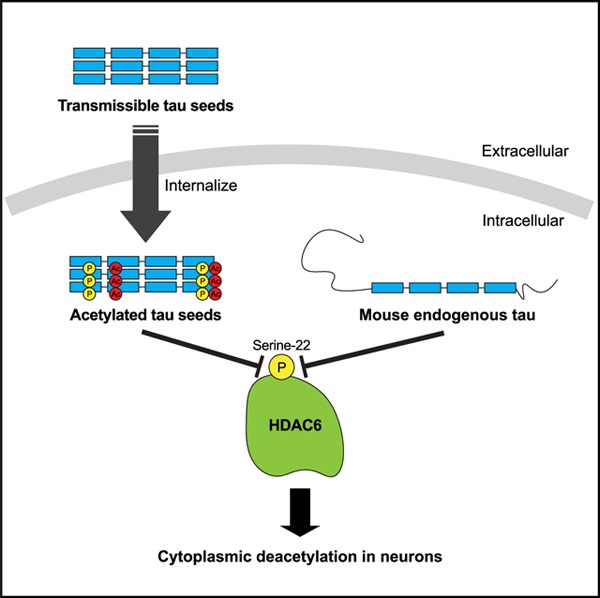
Tseng et al. show tau seeds are abnormally processed when internalized into neurons. Tau seeds undergo a series of modifications that result as a consequence of auto-acetylation events as well as inhibition of the deacetylase HDAC6. Tau acts as an HDAC6 inhibitor by preventing HDAC6 phosphorylation at Ser-22.
Graphical Abstract
INTRODUCTION
Tau pathology is thought to spread through the brain in a defined spatiotemporal manner, potentially originating in the brain stem and spreading outward through synaptic contacts into the entorhinal cortex and higher neocortical regions (Goedert et al., 2017). Aberrant forms of tau are released, taken up by neighboring cells, and in a structurally templated fashion, recruit endogenous tau to propagate tau pathology (Walker et al., 2013). Such transmissible tau seeds may comprise a mixture of different tau strains, or unique tau conformers, that may explain the cell-type specificity and clinical heterogeneity seen among diverse tauopathies including Alzheimer’s disease (AD), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD) (Arakhamia et al., 2020; Gibbons et al., 2018; Kaufman et al., 2016, 2017; Narasimhan et al., 2017). However, the tau-intrinsic factors that dictate tau strain identity and disease progression remain poorly understood.
Recent cryoelectron microscopy (cryo-EM) and mass spectrometry studies strongly support tau post-translational modifications (PTMs) as defining features of tau filaments derived from brains of AD and other tauopathies (Arakhamia et al., 2020; Wesseling et al., 2020). In particular, lysine acetylation has emerged as a tau PTM capable of accelerating tau aggregation and inducing a series of AD-like deficits including synaptic dysfunction, neuronal loss, and cognitive impairments (Min et al., 2015; Tracy et al., 2016). Although a role for tau acetylation in release, uptake, or seeding has not been firmly established, recent studies showed that seed-induced tau pathology correlates with tau acetylation. For example, a bigenic mouse model (PDAPP: PS19) generating both Aβ plaques and tau tangles showed reduced survival and enhanced tau pathology, highlighted by >5-fold more acetylated tau in insoluble brain fractions compared to monogenic PS19 parental mice (Cohen et al., 2011; Hurtado et al., 2010). Similarly, injection of prion-like tau fibrils into the hippocampus of PS19 tau transgenic mice accelerated tau pathology and led to an induction of tau acetylation just 2 weeks after injection, followed by subsequent neuronal loss (Iba et al., 2013, 2015).
Although a detailed understanding of the enzymes regulating tau acetylation is still emerging, in vitro and cell-based experiments suggest that CREB-binding protein (CBP), or the highly homologous p300, acetylates tau with high affinity within the microtubule-binding region (MTBR) (Cohen et al., 2011, 2013; Min et al., 2010). In the absence of CBP/p300, tau also possesses an intrinsic ability to auto-acetylate by cysteine residues located in the MTBR, implying that energetically favorable conditions can promote acetyl group transfer in the absence of a dedicated tau acetyltransferase (Cohen et al., 2013).
Counteracting tau acetylation, HDAC6 and SIRT1 were shown to deacetylate tau in vitro, in cultured cells, and in mice (Cohen et al., 2011; Min et al., 2010). HDAC6 in particular is predominantly cytoplasmic, associated with microtubules (MTs), and physically interacts with tau. We previously found that overexpressed HDAC6 was sufficient to deacetylate tau, and blocking or depleting HDAC6 increased tau acetylation and led to accelerated disease progression in a mouse model of tauopathy (Cohen et al., 2011; Trzeciakiewicz et al., 2020). Although the use of HDAC6 inhibitors as potential neuroprotective agents has been well documented, it is important to note that HDAC6 also has protective roles as a surveillance factor to prevent toxic tau accumulation (Trzeciakiewicz et al., 2020), and therefore, its chronic or long-term inhibition could potentially be linked to proteostasis failure (Lee et al., 2010; Pandey et al., 2007).
Here, we provide evidence that tau seeds undergo aberrant post-translational processing. Although primary neurons internalized both wild-type (WT) and P301L disease-associated tau seeds, P301L seeds were subject to robust acetylation and phosphorylation within the MTBR domain. To explain these observations, we found that P301L tau seeds were subject to auto-acetylation and also preferentially inhibited the deacetylase HDAC6, which further primed a series of pathogenic tau modifications within the MTBR and accelerated tau aggregation. Surprisingly, depleting tau is sufficient to enhance HDAC6 activity toward internalized tau seeds and non-tau substrates. This study supports the formation of distinctly modified tau conformers that may underlie the diversity of tau strains and impact tau spreading in AD and related tauopathies.
RESULTS
Tau seeds harbor unique PTM signatures in neurons
We explored the possibility that internalized tau seeds harbor unique PTM profiles. Recombinant WT or P301L mutant tau seeds comprising the MTBR domain (TauRD) were generated in vitro, and their fibrillar morphology was confirmed by transmission electron microscopy (TEM) (Figure 1A). Tau seeds were added to primary cortical mouse neurons for up to 72 h, followed by analysis of soluble and insoluble neuronal lysate fractions by immunoblotting. The total monomeric (M) and tau seed (S) inputs produced in vitro before addition to neuronal cultures were similar between WT and P301L (Figure 1B). The difference in migration between tau seeds comprising the repeat domain (~15 kD) and endogenous mouse tau (~50 kD) allowed us to distinguish among the tau species present in the treated neurons. Surprisingly, only the internalized P301L seeds became acetylated and phosphorylated within the MTBR (Figure 1C, see 24- to 72-h time points). Although the normal steady-state levels of internalized WT seeds were lower than the more stable P301L seeds, WT seed modifications were nearly undetectable throughout the 72-h time course. Quantification of these differences, which were normalized to the levels of total internalized tau seeds, revealed a significant increase in P301L seed acetylation and phosphorylation, an effect that plateaued ~24 h after addition to neurons (Figure 1D; Figure S1A).
Figure 1. Tau seeds are aberrantly modified in primary neurons.

(A) Transmission electron microscopy (TEM) images of wild-type (WT) and P301L tau fibrils. Tau seeds were generated by sonication of preformed tau fibrils. Scale bar, 200 nm.
(B) Coomassie blue staining of WT and P301L tau monomers (M) and seeds (S) prior to addition to neurons. Band intensity was quantified and normalized to WT monomer.
(C) Primary cortical neurons were treated with WT or P301L tau seeds for 0–72 h, followed by immunoblotting analysis with site-specific tau acetylation (ac-K280 and ac-K369) or phosphorylation (p-S262 and p-S356) and total tau antibodies.
(D) Quantification by densitometry shows that P301L tau seed modifications are significantly increased by 24 h compared with WT tau seed (ac-K280 and p-S262). The extent of tau seed modification was normalized to total tau seed level. Error bars indicate SEM; n = 3 biologically independent experiments. p value was determined by unpaired t test. ***p < 0.001.
(E) WT or P301L (PL) tau monomers and seeds were added to 293A cells with or without lipophilic carrier reagent (Lipofectamine 2000) or primary neurons for 2 days, and lysates were analyzed by immunoblotting to detect aberrant tau modifications as described above.
We performed several additional experiments to confirm these observations. First, no detectable tau seed modifications within the MTBR (ac-K280, ac-K369, p-S262, and p-S356) were observed before neuronal seed uptake, excluding the possibility that recombinant tau proteins become post-translationally modified in bacteria or during the tau protein purification process (at the sites examined) (Figure S1B). To exclude the possibility that these results simply reflected lower levels of internalized WT seeds, we analyzed 3-fold more WT seeds than P301L seeds to achieve comparable levels of total seeds. However, even in this scenario, WT seed modifications were nearly undetectable (Figures S1C and S1D). Second, in addition to the MTBR fragment, a similar increase in acetylated tau was observed with full-length 2N4R-P301L tau seeds compared to 2N4R-WT tau seeds (Figures S1E and S1F), indicating that PTM changes are not specific to the C-terminal tau MTBR fragment. Last, in addition to P301L, the S320F mutation, also located in the 3rd MTBR repeat region, showed similar aberrant PTMs including acetylation, indicating that multiple frontotemporal dementia (FTDP-17) tau mutants within this region are similarly processed in neurons (Figures S1G and S1H).
In contrast to primary neurons, tau seeds were not appreciably modified by either acetylation or phosphorylation when delivered to standard cultured cell lines including 293A cells, either in the absence or presence of a lipophilic carrier reagent (Figure 1E), suggesting that cell-type-specific modifications occur in response to tau seeds. We also examined seed uptake in immune cells, implicated in the release and propagation of tau seeds (Asai et al., 2015; Funk et al., 2015; Hopp et al., 2018). Cultured primary mouse microglia or human-monocyte-derived macrophages (hMDMs) were also able to internalize WT and P301L seeds, but similar to the primary neuron analysis, only P301L seeds were acetylated by double labeling of myc-tagged tau seeds with the ac-K280 antibody (Figures S2A–S2F). These data suggest that primary neurons and immune cells, but not the cultured cell lines analyzed, internalize and process tau seeds into abnormally modified species.
To determine the intracellular localization of internalized WT and P301L seeds, neurons were fractionated into vesicular and cytosolic fractions. The vast majority of the ac-K280- and p-S262-positive P301L seeds were detected in fractions enriched with endosomal and lysosomal markers (Figure 2A). We note that tau seeds were recovered from neurons mostly as monomers (TauRD monomer, ~15 kD) but occasionally formed less abundant dimers (TauRD dimer, ~30 kD) (Figure 2A). Modified seeds were not detected in cytosolic fractions, despite the fact that a pool of total internalized seeds was indeed recovered in the cytoplasm, as detected with a total tau antibody (Figure 2A). Quantification of these results indicates that the vesicular pool of P301L seeds was significantly more ac-K280- and p-S262-positive than WT seeds (Figure 2B).
Figure 2. Disease-associated tau seeds preferentially accumulate in autophagic vesicles.

(A) Primary neurons were treated with WT or P301L tau seeds for 2 days and fractionated into vesicular and cytosolic fractions. The presence of WT and P301L seeds within the biochemical fractions was evaluated by immunoblotting with site-specific tau antibodies and markers of vesicular and cytosolic fractions, including EEA1, LC3, and GAPDH. The tau MTBR fragment is labeled as tauRD, which migrates as monomers and dimers, whereas endogenous full-length mouse tau is labeled as mouse tau.
(B) Quantification of tau seeds (monomer and dimer) present in the vesicular fraction was performed with site-specific or total tau antibodies. The extent of tau modifications was normalized to total tau levels. Error bars indicate SEM; n = 3 biologically independent experiments. p value was determined by unpaired t test. *p < 0.05.
(C) Primary neurons were treated with myc-tagged WT or P301L tau seeds and analyzed by double-labeling with the myc antibody (red) to mark tau seeds in combination with LC3 antibody (green). The arrows in the inset highlight regions of co-localization. Scale bar, 50 mm.
(D) Quantification of co-localization between tau seeds and vesicular markers was determined as a ratio of the number of co-localized tau seeds per total number of tau seeds. Error bars indicate SEM; n = 6 biologically independent experiments. p value was determined by unpaired t test. n.s. p > 0.05, ***p < 0.001.
(E) Primary neurons were treated with WT or P301L tau seeds for 2 days in the presence or absence of 3MA or MG-132 to impair autophagic or proteasome-mediated degradation, followed by immunoblotting with ac-K280 and total tau antibodies.
(F) Quantification of tau seed acetylation normalized to total tau seed levels. Error bars indicate SEM; n = 3 biologically independent experiments. p value was determined by one-way ANOVA with Tukey’s test for multiple comparisons among groups. n.s. p > 0.05, **p < 0.01.
We further analyzed the localization of WT and P301L tau seeds in neurons by using myc-tagged seeds that are suitable for confocal imaging. Tau seeds were imaged and quantified based on their co-localization with endosomes (EEA1 and Rab5), autophagosomes (LC3), or lysosomes (LAMP1) after uptake into primary neurons. WT and P301L seeds did not show differences in targeting to endosomes or lysosomes, whereas P301L seeds were enriched in autophagic vesicles compared to WT seeds (Figures 2C and 2D; Figures S3A–S3C), consistent with their autophagic targeting (Chesser et al., 2013; Wang et al., 2010; Wang and Mandelkow, 2012). To address whether WT seeds are preferentially degraded by autophagy, thus explaining their lower steady-state levels, we pharmacologically inhibited either autophagic or proteasomal degradation pathways by using 3-methyladenine (3MA) or MG-132, respectively. Autophagy inhibition stabilized acetylated WT seeds to a level comparable with P301L seeds (Figures 2E and 2F), indicating that WT seeds are more efficiently targeted by autophagy in neurons.
Acetylation enhances tau seed pathogenesis
To assess the significance of tau seed acetylation in the context of the P301L mutant, we considered three non-mutually exclusive properties that could be affected by excessive acetylation of tau seeds, namely, aggregation, PTM priming, and/or templated seeding of monomeric tau. First, we generated recombinant purified tau proteins containing acetylation-mimic (K→Q) or non-mimic (K→R) substitutions at residue K280 in the context of the P301L mutation, resulting in P301L/K280Q and P301L/K280R double-mutant proteins (Figure 3A). We analyzed their toxicity after addition to neurons by using standard LDH (lactate dehydrogenase) and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-di-phenyltetrazolium bromide) assays, and as expected, 5 days of incubation with WT primary neurons resulted in minimal toxicity (Figure S3D). In vitro heparin-induced sedimentation assays were performed over an 8-h time period. WT tau proteins did not fully transition to the pellet fraction until ~6–8 h of incubation. The P301L/K280Q mutation showed slightly accelerated sedimentation, more so than the single P301L mutant, and was nearly ~100% pelleted by 1 h (Figures 3B and 3C, compare P301L to P301L/K280Q). In contrast, the P301L/K280R mutant showed delayed aggregation and did not fully pellet until ~2 h (Figures 3B and 3C, compare P301L to P301L/K280R). Supporting the sedimentation data, thioflavin-T (ThT) fluorescence showed accelerated P301L/K280Q aggregation at an early time point (0.5 h), whereas P301L/K280R mutant aggregation was delayed throughout the time course (Figure 3D). These data suggest that charge alterations at residue K280 significantly alter the aggregation propensity of the P301L mutant.
Figure 3. Acetylation promotes tau seed pathogenesis.

(A) Schematic representation of P301L tau, acetylation-mimic (P301L/K280Q), and non-mimic (P301L/K280R) tau constructs used in this study.
(B) Fibrillization of WT, P301L, P301L/K280Q, or P301L/K280R was performed for up to 8 h (WT) or 2.5 h (P301L, P301L/K280Q, and P301L/K280R). Samples were sedimented into supernatant (S) and pellet (P) fractions and analyzed by Coomassie blue staining.
(C) Quantification of the sedimentation assay showed increased pelleted fractions with P301L and P301L/K280Q compared to P301L/K280R. Error bars indicate SEM; n = 3 biologically independent experiments. p value was determined by unpaired t test. ***p < 0.001.
(D) In vitro tau aggregation was monitored by thioflavin-T measurements for up to 2 h to analyze the aggregation kinetics among tau seeds. Error bars indicate SEM; n = 3 biologically independent experiments. p value was determined by unpaired t test. ***p < 0.001.
(E) Coomassie blue staining of WT, P301L, or P301L/K280R tau monomers (M) and seeds (S) are depicted as loading controls. Band intensity was quantified and normalized to WT monomer.
(F) Primary neurons were treated with WT, P301L, or P301L/K280R tau seeds and analyzed by immunoblotting with site-specific tau antibodies (ac-K280, ac-K369, p-S262, and p-S356). The asterisks (*) highlight multimeric tau dimers and trimers.
(G) Quantification of the site-specific tau modifications detected on WT, P301L, or P301L/K280R tau seeds after neuron processing. The extent of tau modifications was normalized to total tau levels. Error bars indicate SEM; n = 3 biologically independent experiments. p value was determined by one-way ANOVA with Tukey’s test for multiple comparisons among groups. n.s. p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001.
Next, we asked whether acetylated seeds undergo PTM priming at adjacent sites within the MTBR, including the neighboring KXGS motifs that are targeted by the kinase MARK2. Tau PTM priming is thought to represent a pathogenic mechanism linked to tau-mediated toxicity (Carlomagno et al., 2017; Cho and John-son, 2003; Steinhilb et al., 2007). Insoluble fractions from primary neurons exposed to WT, P301L, or P301L/K280R tau seeds were analyzed by immunoblotting. Although the total seed inputs were comparable (Figure 3E), non-acetylated seeds at residue K280 (P301L/K280R) showed a striking reduction in C-terminal tau acetylation (K369) and phosphorylation within the MTBR (S262 and S356) (Figure 3F, see tau seeds migrating as ~15-kD monomers and ~30-kD dimers), suggesting that K280 acetylation is required to prime nearby modifications within the MTBR domain. We note that P301L/K280R seeds showed similar higher order molecular weight species compared to P301L seeds, as detected by the total tau antibody (e.g., dimers and trimers) (Figure 3F, see bands marked by asterisks) but nonetheless were still not efficiently acetylated or phosphorylated within the MTBR. The relative quantification of all modifications detected among WT, P301L, and P301L/K280R seeds is shown in Figure 3G. Therefore, in addition to enhanced aggregation, tau seed acetylation is a critical determinant of tau’s overall PTM profile.
Last, we evaluated whether acetylation altered seed-induced templating by introducing tau seeds into cells ectopically expressing full-length human P301L tau and the acetyltransferase CBP. CBP acetylated the tau seeds, which resulted in a ~3-fold increase in tau seeding activity, as determined by the conversion of full-length ~65-kD human P301L tau into insoluble hyperphosphorylated tau aggregates (AT8-immunoreactive) (Figure 4A, compare lanes 3–5 to lanes 6–8, and see quantification in Figure 4B). The full-length tau conversion was more robust with P301L seeds rather than WT seeds (Figure 4C). We note that CBP promoted tau acetylation, as expected, but did not significantly alter the expression of full-length tau, excluding any confounding or indirect effects of CBP on general transcription or steady-state tau levels (Figures S4A–S4C).
Figure 4. Acetylation promotes tau-seed-induced templating of full-length tau.

(A–C) 293A cells were transfected with full-length P301L tau in the absence or presence of the acetyltransferase CBP. WT or P301L tau seeds were transduced into transfected cells 10 h post-transfection using Lipofectamine reagent to promote full-length tau templating and seeding. The cells were harvested 48 h after seed transduction and fractionated into soluble (Figure S4A) and insoluble (A) fractions. The extent of tau seeding was evaluated by immunoblotting with phosphorylated tau antibodies (AT8 and p-S396), and ac-K280 and total tau served as readouts for acetylated and total tau expression. Quantification of aberrant full-length tau phosphorylation and templating was analyzed in soluble (Figures S4B and S4C) or insoluble fractions (B, P301L seed; C, WT seed). Error bars indicate SEM; n = 3 biologically independent experiments. p value was determined by unpaired t test. n.s., p > 0.05; *p < 0.05, **p < 0.01, ***p < 0.001.
(D and E) Recombinant P301L tau monomer was left unmodified (mock) or in vitro acetylated, and seeds generated from mock or acetylated P301L tau monomers were transduced into 293A cells that had been transfected prior with full-length P301L tau. Insoluble fractions were analyzed using immunoblotting with phosphorylated, acetylated, and total tau antibodies. The asterisks (*) indicate hyperphosphorylated AT8 immunoreactive bands. The extent of aberrant full-length tau PTMs and templating were quantified in (E). Error bars indicate SEM; n = 3 biologically independent experiments. P value was determined by unpaired t test. n.s., p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001.
Because the introduction of ectopically expressed CBP is expected to acetylate both tau seeds (~15 kD) and the ectopically expressed full-length tau (~65 kD), we next sought to restrict the acetylation by exclusively acetylating tau seeds, followed by subsequent evaluation of full-length tau seeding. We performed recombinant in vitro tau acetylation reactions in the presence of acetyl-coenzyme A (acetyl-CoA) and generated either mock non-acetylated control or acetylated P301L seeds that were subsequently delivered to cells expressing full-length P301L tau. As shown in Figure 4D, restricted acetylation of P301L seeds led to the acetylation of the ~65-kD full-length tau (see asterisks) but did not influence phosphorylated or aggregated tau, as detected by immunoblotting with AT8 and total tau antibodies (Figure 4E). We conclude that an acetylation-enhancing environment (e.g., induced by active tau acetyltransferases) is conducive for the templated conversion of monomeric full-length tau.
Tau seeds are subject to auto-acetylation
Our findings to this point support a model whereby acetylation promotes the pathogenesis of tau seeds. We therefore investigated the relevant tau acetylation/deacetylation machinery responsible for this regulation. Although CBP/p300 has been implicated as one of the major tau acetyltransferases (Min et al., 2010, 2015), its activity is reportedly reduced as a consequence of cognitive decline in AD brain (Bartolotti et al., 2016), suggesting tau seed acetylation can potentially occur through other alternative mechanisms. Indeed, neurons harboring P301L seeds were exposed to C646, a selective CBP/p300 inhibitor, which did not alter tau acetylation, despite the fact that acetylation of histone H3, a known CBP/p300 target, was reduced (Figure 5A). Although we cannot conclusively rule out a role for CBP/p300 in this process, these data prompted us to search for additional mechanisms that could mediate tau seed acetylation.
Figure 5. Tau seeds undergo auto-acetylation and inhibit HDAC6 activity.

(A) Primary neurons were exposed to P301L tau seeds in the presence or absence of a CBP/p300 inhibitor, C646 (20 μM), followed by immunoblotting with acetylated tau (ac-K280), H3K27ac, and total H3 antibodies. Acetylation of tau and H3K27 were quantified on the right. Error bars indicate SEM; n = 3 biologically independent experiments. P value was determined by unpaired t test. n.s. p > 0.05, *p < 0.05.
(B) Primary neurons were exposed to myc-tagged P301L tau seeds and analyzed by double labeling with the mouse tau-specific T49 antibody (red) in combination with myc antibody (green) to illustrate minimal co-localization between endogenous mouse tau and tau seeds. Scale bar, 50 μm.
(C) Primary neurons were exposed to WT, P301L, or P301L/C291A/C322A (P301L-2CA) seeds followed by immunoblotting with acetylated (ac-K280) and total tau antibodies. The asterisks (*) highlight tau dimers. Double cysteine mutants (P301L-2CA) are partly deficient in tau auto-acetylation.
(D) Quantification of tau acetylation among WT, P301L, and P301L-2CA seeds. Error bars indicate SEM; n = 3 biologically independent experiments. p value was determined by one-way ANOVA with Tukey’s test for multiple comparisons among groups. *p < 0.05, **p < 0.01.
(E) Primary neurons were exposed to myc-tagged WT or P301L tau seeds and analyzed by double-labeling with myc antibody (green) in combination with a mouse HDAC6 antibody (red). Co-localization is highlighted by arrows in the inset. Scale bar, 50 μm.
(F) Quantification of co-localization was determined as percent tau seed co-localization among total tau seeds. Error bars indicate SEM; n = 4 biologically independent experiments. p value was determined by unpaired t test. **p < 0.01.
(G) Primary neurons were exposed to WT or P301L tau seeds in the absence or presence of tubastatin A (TubA; 1 μM) followed by immunoblotting with ac-tubulin, total tubulin, and HDAC6 antibodies. GAPDH served as a loading control.
(H) Quantification of ac-tubulin normalized to total tubulin levels in neurons exposed to WT or P301L tau seeds. Error bars indicate SEM; n = 4 (WT) or 5 (P301L) biologically independent experiments. P value was determined by unpaired t test. *p < 0.05.
(I) An HDAC6 in vitro activity assay was used to evaluate the inhibitory capability of WT or P301L monomers and seeds. Trichostatin (TSA) and full-length tau lacking the MTBR (tauFLΔR1–4) served as positive and negative controls, respectively. The different inhibitory capabilities between tau species were analyzed. Error bars indicate SEM; n = 4 (WT seed and P301L seed), 5 (tauFLΔR1–4), or 7 (TSA, WT monomer, and P301L monomer) biologically independent experiments. p value was determined by unpaired t test. *p < 0.05, **p < 0.01.
(J) Coomassie blue staining of WT, P301L, and tauFLΔR1–4 recombinant tau proteins.
We considered that endogenous mouse tau could potentially acetylate tau seeds, as our previous study showed that tau possesses intrinsic acetyltransferase activity (Cohen et al., 2013). To evaluate this possibility, we analyzed the co-localization between endogenous tau and exogenously introduced P301L seeds. Endogenous tau showed minimal co-localization with myc-tagged P301L seeds (Figure 5B). Further excluding tau as its own acetyltransferase, in vitro acetylation assays showed that recombinant full-length tau was unable to directly acetylate tau seeds when incubated with acetyl-CoA (Figure S5A). Finally, we considered the possibility that tau auto-acetylation might occur predominantly in cis, in which individual tau molecules can undergo self-acetylation mediated by tau’s catalytic cysteine residues via the formation of an acetyl-cysteine inter-mediate (Cohen et al., 2013). In support of this latter possibility, mutation of cysteine residues (C291 and C322) in the context of P301L seeds (P301L-2CA) reduced tau seed acetylation (Figures 5C and 5D). Therefore, internalized P301L seeds are more susceptible to undergo auto-acetylation, which is partly responsible for the distinct tau PTM profile.
Tau seeds inhibit the deacetylase HDAC6
Next, we considered the possibility that P301L seeds might inactivate an endogenous deacetylase that would allow for further increases in tau acetylation. We focused on the deacetylase HDAC6, which physically interacts with and deacetylates tau (Cohen et al., 2011; Ding et al., 2008; Perez et al., 2009), and examined whether P301L seeds preferentially associate with and impair HDAC6 activity. WT and P301L seeds were analyzed for their co-localization with HDAC6 in primary neurons. We found preferential co-localization of P301L seeds, but not WT seeds, with mouse HDAC6 within neuronal processes (Figures 5E and 5F). Moreover, upon delivery to neurons, P301L seeds mildly increased acetylated tubulin levels to a greater extent than WT seeds, an indicator of reduced HDAC6 function (Figures 5G and 5H). To determine whether tau seeds directly associate with and inhibit HDAC6, an in vitro deacetylase assay was performed by incubating tau seeds with recombinant HDAC6 and a fluorescent-based reporter. P301L seeds inhibited HDAC6 activity ~2-fold compared to WT seeds, whereas the negative control lacking the entire MTBR domain, a region required for tau-HDAC6 binding (Trzeciakiewicz et al., 2020), did not alter HDAC6 activity (Figures 5I and 5J).
We were surprised to find significant HDAC6 inhibition in response to soluble monomeric tau proteins (Figure 5I), consistent with a previous report (Perez et al., 2009), suggesting that tau-mediated inhibition of HDAC6 is not specific to fibrillar or aggregated tau but can also be achieved with tau monomer. We suspect that the ability of tau to regulate HDAC6 is likely dependent on several factors, including the full extent of tau release/uptake, intracellular tau and HDAC6 concentration, and/or a distinct tau conformation that may favor accessibility or binding to HDAC6. We conclude that P301L tau is not only subject to auto-acetylation but also acts as a more effective inhibitor of HDAC6, suggesting multiple mechanisms likely account for the increased susceptibility of tau to become aberrantly modified.
Tau inhibits HDAC6 to control the lysine acetylation profile
Given that tau monomer showed a surprising inhibition of HDAC6 activity (Figure 5I), we reasoned that a better understanding of the physiological tau-HDAC6 interaction would provide insight into how modified tau seeds evolve. Thus, we sought to more precisely define which tau species are capable of targeting and inhibiting HDAC6. To determine whether normal physiological tau can regulate HDAC6, we used a genetic approach and asked whether endogenous mouse tau inhibits HDAC6 activity in neurons. In this scenario, one would expect mouse tau depletion to enhance HDAC6 deacetylase activity towards internalized tau seeds. To this end, P301L tau seeds were introduced into WT (Tau+/+), heterozygous (Tau+/−), or homozygous (Tau−/−) tau knockout neurons, which was confirmed by the absence of mouse tau (Figure 6A). Strikingly, P301L seeds were significantly less acetylated and phosphorylated in Tau+/− neurons, an effect that was even more pronounced in Tau−/−neurons completely lacking tau (Figures 6A and 6B, see ac-K280 and p-S262). We also note reduced higher order tau dimers and even trimers in Tau−/−neurons (Figure 6A, see asterisks marked by the total tau antibody). The reduced tau seed acetylation observed in Tau−/−neurons was restored by pharmacological inhibition of HDAC6 with tubastatin A (TubA) (Figure 6C), supporting the notion that mouse tau depletion activates the TubA-sensitive endogenous HDAC6, leading to the enhanced deacetylation of internalized P301L seeds.
Figure 6. Tau reduction activates HDAC6.

(A) Primary Tau+/+, Tau+/−, and Tau−/−neurons were exposed to P301L tau seeds, fractionated into soluble and insoluble fractions, and analyzed by immunoblotting with site-specific tau antibodies (ac-K280 and p-S262), as well as a total tau antibody. TauRD dimers and trimers are highlighted with asterisks (*). GAPDH served as loading control.
(B) Quantification of site-specific modifications of P301L tau seeds in Tau+/+, Tau+/−, or Tau−/−neurons. Error bars indicate SEM; n = 3 biologically independent experiments. p value was determined by one-way ANOVA with Tukey’s test for multiple comparisons among groups. n.s. p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001.
(C) Primary Tau−/−neurons were exposed to P301L tau seeds in the absence or presence of TubA (1 μM) and analyzed by immunoblotting with ac-K280 as well as a total tau antibody. The asterisks (*) highlight dimers and trimers. Acetylated tau seeds normalized to total tau levels were quantified on the right. Error bars indicate SEM; n = 3 biologically independent experiments. p value was determined by unpaired t test. *p < 0.05.
(D) Primary Tau+/+, Tau+/−, and Tau−/−neurons were left untreated or exposed to TubA (1 μM) overnight, fractionated into soluble and insoluble fractions, and analyzed by immunoblotting with a pan-acetyl-lysine antibody to reveal acetyl-lysine profile changes. Seven distinct acetylated substrates were detected by acetyl-lysine immunoreactivity and the protein bands were numbered accordingly. GAPDH served as loading control.
(E) The levels of acetylated substrates were quantified and classified as subtypes I–III based on correlations with tau genotype and responsiveness to the HDAC6 inhibitor TubA.
Because mouse tau depletion could conceivably lead to a general enhancement of HDAC6 activity toward many other substrates, beyond the regulation of tau seeds, we extended these observations and evaluated the overall extent of lysine acetylation in Tau+/+, Tau+/−, and Tau−/−neurons by using well-characterized pan-acetyl-lysine antibodies. Surprisingly, overall, lysine acetylation was significantly reduced in both soluble and insoluble fractions of Tau−/−neurons, based on pan-acetyl-lysine immunoreactivity (Figure 6D, see ac-substrates 1–7 highlighted by asterisks), which paralleled the reduced acetylation of P301L tau seeds. The level of acetylated tubulin, a major HDAC6 substrate (Hubbert et al., 2002), was also reduced in Tau−/−neurons, further supporting increased HDAC6 activity in neurons lacking tau. To extend these observations to mice, we analyzed cortical brain homogenates from WT and Tau−/−mice by using acetyl-lysine antibodies and observed a similar reduction in specific acetylated substrates in Tau−/−brain, although we note that acetylated tubulin was not appreciably altered in these mice (Figures S6A and S6B).
To further confirm that mouse tau depletion activates HDAC6, we exposed Tau−/−neurons to HDAC6 antisense oligonucleotides (HDAC6 ASOs), which effectively reduced HDAC6 protein levels. Treatment with either TubA or the HDAC6-specific ASO was sufficient to restore global acetylation levels in Tau−/−neurons back to WT levels (Figures 6D and 6E; Figures S7A–S7D). We found that the acetyl-lysine profile in neurons could be categorized into three distinct subtypes (I–III) based on their sensitivity to either tau depletion or HDAC6 inhibition (Figure 6E; Figure S7B). A subset of acetylated substrates was not tau dependent but responsive to TubA (subtype I). Another subset was both tau dependent and TubA responsive (subtype II), highlighting those particular substrates that are regulated downstream of tau and HDAC6. Finally, a third subset (subtype III) was tau dependent but not responsive to TubA (HDAC6 independent), indicating the possible involvement of other unrelated HDACs and/or sirtuin family members. Therefore, the acetyl-lysine immunoreactivity profile in neurons consists of distinct subsets of acetylated substrates, with subtype II reflecting those that may be coordinately regulated by tau and HDAC6.
Tau inhibits HDAC6 phosphorylation
Our results suggest that tau seeds and even normal physiological tau can target and suppress HDAC6 function. To investigate precisely how this regulation might occur, we examined whether tau controls HDAC6 phosphorylation status because a recent study indicated that phosphorylation enhances HDAC6 deacetylase activity (Du et al., 2015). We began with an unbiased approach to evaluate the full spectrum of HDAC6 phosphorylation sites that are detectable by mass spectrometry. Large-scale transfections were used to immunopurify FLAG-tagged HDAC6 from 293A cells, yielding μg quantities of purified full-length HDAC6, as visualized by Coomassie blue staining (Figure 7A). Gel excision and trypsin digestion followed by nano liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis detected multiple HDAC6 phosphorylation sites with high confidence. The highest scoring peptide 18QNPQSPPQDSSVTSK32 was phosphorylated on residue Ser-22, which was confirmed by the peptide MS/MS spectrum and the number of spectral counts (Figures 7B and 7C; Table S1).
Figure 7. Tau prevents HDAC6 phosphorylation on Ser-22.

(A) 293A cells were transfected with pcDNA5/TO vector control or WT HDAC6 expression plasmids. Cell lysates were harvested, and FLAG-tagged HDAC6 was immunopurified. The Coomassie-blue-stained gel shows purified HDAC6 protein that was analyzed by mass spectrometry (MS).
(B) MS/MS spectrum of the doubly charged ion (m/z 840.3673) corresponding to the HDAC6 peptide QNPQ(S)PPQDSSVTSK. Ser-5 in the peptide is phosphorylated, which corresponds to Ser-22 in human HDAC6. Mass error (ppm) for each fragment is shown.
(C) Protein coverage map of HDAC6. Peptides highlighted in green were identified by LC-MS/MS. Phosphorylation sites identified are indicated, as well as number of spectral counts observed across the samples (n = 2).
(D) Primary neurons were transduced at DIV3 with a lentivirus expressing WT HDAC6. Cell lysates were harvested at DIV10, treated or untreated with λ-phosphatase, and analyzed by immunoblotting with p-HDAC6 (Ser-22) and total HDAC6 antibodies. Quantification shows that the p-HDAC6/total HDAC6 ratio is significantly reduced after λ-phosphatase treatment. Error bars indicate SEM; n = 3 biologically independent experiments. p value was determined by unpaired t test. **p < 0.01.
(E and F) Primary neurons were transduced at DIV3 with a lentivirus expressing WT HDAC6 in combination with either control or full-length tau (tauFL)-expressing lentiviruses. Neurons were harvested at DIV10 and analyzed by immunoblotting with p-HDAC6 (S22), total HDAC6, and total tau antibodies. GAPDH served as loading control. The p-HDAC6/total HDAC6 ratio was quantified in (F), indicating that HDAC6 phosphorylation is reduced in the presence of full-length tau. Error bars indicate SEM; n = 3 biologically independent experiments. p value was determined by unpaired t test. *p < 0.05.
(G and H) Primary Tau+/+ and Tau−/−neurons were transduced with a lentivirus expressing WT HDAC6; followed by treatment with P301L seeds for 2 days; and analyzed by immunoblotting with p-HDAC6 (S22), total HDAC6, and total tau antibodies. GAPDH served as loading control. Two separate experimental replicates are shown (exp.1 and exp.2). p-HDAC6/total HDAC6 was quantified and plotted in (H), highlighting reduced HDAC6 phosphorylation in the presence of endogenous tau (Tau+/+ ) or exogenous tau seeds (P301L seed). Error bars indicate SEM; n = 4 (Tau+/+ and Tau+/+ + P301L seeds) or 5 (Tau−/−and Tau−/−+ P301L seeds) biologically independent experiments. p value was determined by one-way ANOVA with Tukey’s test for multiple comparisons among groups. n.s. p > 0.05, *p < 0.05.
We then evaluated HDAC6 phosphorylation in neurons by using an HDAC6 lentiviral expression system that provides enhanced p-HDAC6 detection sensitivity. Primary cultured cortical neurons at 3 days in vitro (DIV3) were transduced with a lentivirus expressing WT HDAC6 and harvested at DIV10 for subsequent immunoblotting by using a phosphorylation-specific HDAC6 antibody targeting Ser-22 (p-HDAC6). p-HDAC6 was detected in HDAC6 lentiviral-transduced neuron cultures, confirming that active phosphorylated HDAC6 is present in WT neurons (Figure 7D, lanes 1–3). To validate that the p-HDAC6 antibody is indeed phosphorylation specific, lysates were lambda (λ) phosphatase treated, which reduced p-HDAC6 immunoreactivity (Figure 7D, lanes 4–6). Next, we co-transduced HDAC6 with either control or full-length human 2N4R tau expressing lentivirus to evaluate whether ectopic tau is sufficient to suppress HDAC6 phosphorylation. We found that p-HDAC6, but not total HDAC6, was reduced ~80% in the presence of lentiviral tau (Figures 7E and 7F), implying that tau acts to suppress HDAC6 activity, at least in part, by preventing HDAC6 phosphorylation at Ser-22.
To evaluate whether tau depletion conversely activates HDAC6 by Ser-22 phosphorylation, we assayed p-HDAC6 in WT or Tau−/−neurons transduced with WT HDAC6 lentivirus (Figure 7G). Consistent with tau depletion activating HDAC6 function, neurons lacking tau showed a subtle but significant increase in p-HDAC6 (in the absence of tau seeds), supporting a basal tau-mediated suppression of HDAC6 activity (Figure 7H). As expected, in the presence of tau seeds that are capable of inhibiting HDAC6, p-HDAC6 (and therefore HDAC6 activity) was reduced regardless of the genotype in either WT or Tau−/−neurons. These data indicate that normal endogenous tau or internalized tau seeds act as modulators of HDAC6 activity by altering HDAC6 phosphorylation status.
Tau reduction prevents stress-induced acetyl-lysine accumulation
Because tau depletion was shown to be neuroprotective (Roberson et al., 2007), we evaluated whether the loss of tau and the resulting activation of HDAC6 are sufficient to counteract the global increase in lysine acetylation that occurs under vulnerable conditions during neuronal stress. Previous studies showed that oxidative stress (e.g., sodium arsenite) is a potent trigger for lysine acetylation (Cohen et al., 2013, 2015) and that enhanced global lysine acetylation correlates with protein aggregation and general cellular toxicity (Olzscha et al., 2017). The extent of the stress-induced acetylome that occurs in neurons, however, remains poorly characterized. We exposed WT neurons to 0.25 mM arsenite for up to 4 h and observed a gradual increase in acetyl-lysine immunoreactivity, indicating that arsenite is indeed a potent trigger for lysine acetylation in neurons (Figures S8A–S8D).
Having established a stress-induced lysine acetylation paradigm in neurons, we asked whether tau depletion was sufficient to activate HDAC6 and hence protect against aberrant lysine acetylation. Cortical neurons lacking one or both copies of tau (Tau+/− or Tau−/−) were exposed to arsenite, followed by fractionation into soluble and insoluble pools (Figure S8E). In Tau+/− neurons, arsenite converted both tau and HDAC6 into the insoluble fraction (Figures S8F–S8G), which coincided with the robust accumulation of insoluble, aggregated, and acetylated substrates ranging from ~30–250 kD (Figures S8H and S8I). However, in neurons completely lacking tau (Tau−/−), the stress-dependent increase in lysine acetylation was abrogated, as both acetylated tubulin and pan-acetyl-lysine immunoreactivity were blunted in soluble and insoluble fractions of arsenite-treated neurons (Figures S8H and S8I). In particular, insoluble substrates 4 and 5 corresponding to 50 kD and 30 kD bands (Figure 6D), as well as high-molecular-weight aggregated species (Figure S8E, see protein bands within bracket) were reduced in Tau−/−neurons. The protective capacity of Tau−/−neurons to suppress acetyl-lysine accumulation was largely negated by the addition of TubA, again confirming that tau depletion is protective against aberrant lysine acetylation by activation of HDAC6 (Figure S8I). We conclude that tau can act in both physiological and pathological contexts to prevent HDAC6 phosphorylation, impair HDAC6 activity, and thereby control the neuronal stress response.
DISCUSSION
Recent studies have shown that different tauopathies contain unique tau conformations that may represent tau strains. However, the biochemical or structural basis for this observation is poorly understood. Here, we show that tau seeds are differentially processed and marked by unique PTM signatures that may underlie their pathogenicity. Because most tauopathies analyzed to date have detected some degree of acetylated tau pathology (Cohen et al., 2011; Cook et al., 2014; Irwin et al., 2012, 2013; Min et al., 2010), the overall extent of tau acetylation could dictate tau’s conformation, aggregation, and seeding propensity.
Our data reveal that P301L mutant tau seeds have distinct PTM profiles marked by increased lysine acetylation and serine phosphorylation. In prior studies, seed-induced pathology in PS19 tau transgenic mice was strongly ac-K280 immunoreactive (Iba et al., 2013), which was attributed to the aggregation of the overexpressed human tau transgene. Our data suggest that both propagated tau seeds and templated tau are subject to extensive PTM remodeling. Under acetylation-enhancing conditions (e.g., in the presence of active or ectopic tau acetyltransferases), both tau seeds and tau monomer were sensitive to lysine acetylation (Figures 4A–4C; Figures S4A–SC).
Although phosphorylated tau has typically been used to mark seeded tau pathology, recent studies challenge the notion that tau phosphorylation correlates with disease pathogenesis. For example, tau seeding activity in some instances was associated with a lack of phosphorylated tau deposition, suggesting other PTMs could regulate tau spreading (Furman et al., 2017; Kaufman et al., 2017, 2018). Furthermore, studies have even reported neuroprotective roles for tau phosphorylation at epitopes that are commonly used to mark propagated tau pathology (e.g., the AT8, 12E8, and PHF-1 epitopes) (Haj-Yahya et al., 2019; Ittner et al., 2016; Povellato et al., 2014). Thus, it is plausible that tau acetylation (perhaps in combination with tau phosphorylation) facilitates tau spreading, which could occur by promoting the formation of toxic oligomeric tau intermediates (Fá et al., 2016; Hill et al., 2019; Lasagna-Reeves et al., 2011). Indeed, we observed that the more robust acetylated P301L seeds formed monomeric, dimeric, and trimeric species (Figures 2A, 3F, 5C, 6A, and 6C).
Although all tau seeds examined altered HDAC6 activity to some extent, the P301L seeds were more effective HDAC6 inhibitors. We also found, rather surprisingly, that tau monomers were capable of inhibiting HDAC6 activity, warranting future studies into the exact transmissible forms of tau that become internalized and engage HDAC6, as both monomeric and oligomeric tau species have been previously implicated in prion-like spreading (Mirbaha et al., 2015, 2018; Sharma et al., 2018). We suspect that under normal conditions, HDAC6 likely acts in a protective capacity to maintain a deacetylated tau pool. However, once internalized, tau seeds may engage HDAC6 by the MTBR domain, leading to inhibition of HDAC6 activity (Ding et al., 2008; Perez et al., 2009). The enhanced binding and inhibition of HDAC6 would effectively increase aggregate-prone acetylated tau species and also simultaneously impair tau degradation by reduced autophagy, as HDAC6 is required for efficient autophagosome-lysosome fusion (Lee et al., 2010; Wang et al., 2019). We propose that HDAC6 targets and deacetylates tau, which facilitates its autophagic degradation. Indeed, in the absence of functional autophagy, we observed that WT seeds become hyper-acetylated (Figures 2E and 2F). Thus, although HDAC6 inhibition is reportedly neuroprotective by stabilization of axonal MTs or prevention of excitotoxic tau mislocalization (Benoy et al., 2018; Godena et al., 2014; Mo et al., 2018; Tseng et al., 2017), its inhibition in the presence of propagated tau species may lead to aberrantly modified tau species and longer-term proteostasis failure that accelerates tau pathogenesis.
The details regarding how tau seeds initially become acetylated require further investigation. First, tau could act as a scaffold to recruit nearby acetyltransferases and hence drive tau acetylation. However, given the specificity of the primary tubulin acetyltransferase αTAT-1 and the predominant nuclear localization of CBP/p300, this possibility remains uncertain. Second, tau could indirectly increase the local acetyl-CoA pool by influencing metabolic gene expression, which will require additional studies. Last, tau could facilitate its own acetylation by auto-acetylation mediated by tau’s catalytic cysteine residues. Our data support the latter scenario and that tau auto-acetylation may occur in a cell-type-specific manner because we find that tau seeds are not appreciably modified when delivered into several different cultured cell lines in the absence of ectopically overexpressed acetyltransferases (Figure 1E). Tau could undergo auto-acetylation in a manner similar to mitochondria-localized proteins, a process that is reported to occur in a non-enzymatic fashion in the brain (Cheng et al., 2016; Wagner and Payne, 2013). Interestingly, the production of acetyl-CoA could be metabolically coupled to tau auto-acetylation similar to that reported for histone acetylation (Mews et al., 2017). Follow-up studies will be required to test whether metabolic regulation of tau auto-acetylation contributes to prion-like tau spreading.
We were intrigued to find that tau seed processing showed cell-type specificity, which could reflect unique events occurring in the central nervous system (CNS) (neurons and microglia) compared to other cell types (Figures 1E; Figures S2A–S2F). Therefore, seeding assays in immortalized cell lines may not recapitulate the full processing that occurs in the brain. We find that neurons and microglia efficiently internalize and acetylate tau seeds, which could explain their cell-type-specific tau uptake and secretion reported in vivo. If extended to other aggregate-prone paradigms, these results may also explain the selectivity of prion-like spreading in the CNS, as this process could involve the generation of unique PTM signatures that decorate aggregated proteopathic seeds, which could influence CNS susceptibility, disease onset, or progression.
Importantly, our data suggest that tau is an endogenous regulator of HDAC6 activity, an interaction that likely impacts the acetylation of many cytosolic HDAC6 target substrates (i.e., the acetyl-lysine profile) (Figures 6D and 6E; Figures S7A and S7B). The extent of this regulation is best illustrated by the reduction of acetyl-lysine observed in Tau−/−neurons. Because tau binds, co-localizes with, and inhibits HDAC6 activity, our data support a poorly characterized yet critical function for tau as a potential adaptor that controls HDAC6. A recent study suggests that tau may not directly stabilize axonal MTs as previously thought (Qiang et al., 2018), and therefore, tau-mediated inhibition of HDAC6 represents an alternative and perhaps non-traditional role for tau in modulating MT stability indirectly by inhibition of HDAC6, which could impact many processes regulated by HDAC6, including the cytoskeleton, autophagy, and heat shock protein (HSP) function (Yan, 2014).
We found that tau acts to inhibit HDAC6 by impeding HDAC6 phosphorylation at Ser-22 (Figure 7). The reduced HDAC6 phosphorylation may reflect tau-mediated regulation of HDAC6 kinases and/or phosphatase accessibility to HDAC6. A recent study supports this possibility, as tau interacts with PTEN, suppresses PTEN activity, and leads to Akt/mTOR pathway activation in several mouse models of autism (Tai et al., 2020). Future studies are required to identify putative tau-associated HDAC6 kinase(s) and phosphatase(s). However, it is worth noting that several HDAC6 kinases have been identified, including ERK1, ASK1, and PKCζ, that could potentially be regulated in a tau-dependent manner (Du et al., 2015; Ran et al., 2020; Williams et al., 2013; Zhu et al., 2011). Given the prominent Ser-22 phosphorylation site identified by mass spectrometry, subsequent kinase screening could further unravel the molecular pathways leading to HDAC6 activation. Because tau depletion alleviates disease progression in several mouse models of neurodegeneration (Cheng et al., 2014; DeVos et al., 2017; Roberson et al., 2007), it is tempting to speculate that the neuroprotection afforded by tau reduction may result as a consequence of sustained or heightened HDAC6 activity and an overall increase in proteostasis capacity driven by enhanced HDAC6 deacetylase activity. Such a model is consistent with increased global acetylation correlating with protein aggregation stress and toxicity, a phenotype that we suspect could be suppressed by enhanced HDAC6 activity (Olzscha et al., 2017; Wagner and Hirschey, 2014). Therefore, although acute HDAC6 inhibition may confer short-term neuroprotection, it remains possible that genetic or pharmacological approaches to deplete tau and/or activate HDAC6 could be used to modulate lysine acetylation in the brain and, hence, suppress long-term neurotoxicity and cognitive decline.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Todd J.Cohen (toddcohen@neurology.unc.edu).
Materials availability
All unique resources generated in this study are available from the lead contact with a completed materials transfer agreement. HDAC6 ASOs were provided by Ionis Pharmaceuticals, Inc.
Data and code availability
Original data generated by this study are available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
293A cells (Invitrogen) are commercially available and were grown in full DMEM media (supplemented with 10% FBS, 1X L-glutamine, and 1X penicillin/streptomycin). This cell line is a subclone of the standard HEK293 line with a relatively flat morphology, is more slowly growing, and maintains ectopic plasmid expression at more physiological levels.
Primary mouse neurons
Primary neuron cultures were prepared from embryonic day (E) 15-E16 embryos of non-transgenic CD-1 (Charles River), Tau+/+, Tau+/−, or Tau−/−embryos. All procedures were performed in strict compliance with animal protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of North Carolina at Chapel Hill (#19.017). The mice were lethally anesthetized in isoflurane and the uterus was removed and placed in cold HEPES-buffered Hank’s balanced salt solution (HBSS). The fetuses were removed and the brain was harvested from the cranium of fetuses. The cerebral hemispheres were minced and digested in the presence of 20 U/mL papain (Worthington) and 5U/mL DNase (Promega) for 30 min at 37 °C. Tissue was dissociated mechanically using a P1000 pipette. The cell suspension was passed through a cell strainer. Dissociated neurons were counted and plated onto poly-D-lysine (PDL, Sigma)-coated coverslips or plates. Wild-type, P301L, P301L/K280Q, P301L/K280R seeds (5 μg per well for 6-well plates, or 2 μg per well for 12-well plates) were added at days in vitro (DIV) 10 for 2 d when most neurons exhibited a mature neuronal morphology. Cultured neurons were exposed to 3MA (5 μM for 16 h), MG-132 (1 μM for 16 h), tubastatin A (1 μM for 16 h or 5 μM for 5 h) and C646 (20 μM for 16 h), where indicated, and were added 1 h prior to exposure to tau seeds or sodium arsenite. Lentiviruses (control, WT HDAC6 or tau-FL lentivirus) were transduced in primary neurons at DIV3, incubated for 1 week, and cells were harvested at DIV10.
Primary mouse microglia
CD-1 mice (Charles River) at E16 were used for the culture of microglia. All procedures were performed in strict compliance with animal protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of North Carolina at Chapel Hill (#19.017). The pregnant mouse was euthanized in isoflurane, the uterus immediately removed and rinsed briefly in ice cold 70% ethanol followed immediately by sterile HBSS to remove any surface contamination. The uterus was then placed in a 150-mm dish containing ice cold sterile HBSS and transferred to a sterile dissecting hood. The fetuses were removed, placed in fresh HBSS and the brains were collected. Surface membranes and vessels were stripped from the brain and brains were washed twice in HBSS followed by two washes in complete MEM (GIBCO, MEM, 10% FBS, 20 μg/mL gentamicin). The cortex and hippocampus were separated from the subcortical areas. Pieces of brain in complete MEM were then gently pulled into a syringe through a blunt 23 gauge needle with edges sharpened to reduce any trauma or shearing effects. This results in the generation of many long thin cores of tissue that break into small pieces. The tissue fragments formed from the core were then gently transferred to a 15 mL culture tube containing 10 mL of complete MEM. The tube was mixed, the pieces were allowed to settle and the supernatant containing cell debris was aspirated and replaced with 12 mL fresh complete MEM. The pieces were then distributed into the wells of an ultralow adhesion 6-well plate. The pieces form floating “brain balls” from which microglia migrate and attach to the surface of the dish. No other cells stick to the low adhesion surface providing a highly purified (>>98%), confluent population of microglia after about 2–3 weeks. This protocol is much more productive than more widely used protocols for microglia production. Under these conditions the microglia are maintained in the presence of neural tissue to preserve the natural milieu. The pure microglia can then be challenged by simply washing the neural tissue from the plate and adding the test substances. Alternatively, the microglia can be harvested from the low adhesion plates without the addition of proteases and then seeded onto coverslips for imaging studies or other uses.
Human monocyte-derived macrophages (hMDMs)
Human buffy coat leukocytes were purchased from healthy male donors at the New York Blood Center (donor age is unobtainable). Blood was diluted 1:1 with phosphate buffered saline (PBS) and was layered on top of Ficoll-Paque (GE Healthcare). Blood/Ficoll-plaque was centrifuged at 500 xg for 25 min and the peripheral blood mononuclear cells (PBMCs) were collected from the PBS/Ficoll-Paque interface. PBMCs were washed in red blood cell lysis buffer (Sigma) to remove any red blood cell contamination. PBS was added to 40 mL and the PBMCs were centrifuged at 100 xg for 20 min and the pellet was again washed in 40 mL of PBS. PBMCs were centrifuged at 450 xg, the supernatant was aspirated, and the pellet was resuspended in Dulbecco’s modified eagle medium (DMEM, GIBCO) with high glucose, 10% fetal bovine serum (GIBCO) and 20 μg/mL gentamicin (GIBCO). Cells were aliquoted into low adhesion 6-well plates or 10-cm dishes (Corning) at a density of approximately 107 cells/well. PBMCs were cultured for 5–7 days to allow monocyte attachment. Remaining white blood cells were washed, from the plate yielding a pure monocyte/macrophage culture. The adherent cells were differentiated into monocyte-derived macrophages (hMDM) using human GM-CSF (15 ng/mL) in complete DMEM for one week. hMDMs and microglia were counterstained with phalloidin to reveal structural details (e.g., ruffles, podosomes) associated with the phenotype of cells.
METHOD DETAILS
Plasmids and cell culture
Two versions of human tau were used in this study. The seeding experiments used the full-length tau isoform containing both N-terminal inserts and all four repeat domains (2N4R-tau, also designated as tauFL). The purified recombinant tau protein is comprised of only the four repeat domains consisting of the MTBR (designated as tau repeat domain, or tauRD). All tau expression plasmids for transient transfections were cloned into the pcDNA5/TO vector (Life Technologies). The K280Q, K280R, and P301L mutations were created using site-directed mutagenesis (New England BioLabs). WT CBP expression plasmid (generous gift from Dr. Tso-Pang Yao, Duke University) was used in cell-based seeding experiments.
Cell transfection and seed transduction
FuGENE 6 transfection reagent (Promega) and Lipofectamine 2000 (Invitrogen) were used to perform the 293A cell transfections and seed transductions, respectively. For cell-based seeding, cells were first transfected with tauFL-P301L in the presence or absence of CBP. Next, 10 h after the transfection, the cells were transduced with tauRD-WT, tauRD-P301L, or acetylated tauRD-P301L seeds in 6-well plates. Tau seeds were prepared by resuspending pellets from tau fibrillization reactions in OptiMEM (GIBCO) followed by sonication (OptiMEM buffer was used as no seed control). Subsequently, 6 μg of sonicated tauRD-WT, tauRD-P301L, or acetylated tauRD-P301L seeds were then combined with Lipofectamine 2000 (Invitrogen), allowed to incubate at room temperature for 20 min, and the tau seed/Lipofectamine mixture was added to cells overexpressing tauFL-P301L and CBP, where indicated. After an overnight incubation, cell culture media was replaced with fresh DMEM media. Cells were harvested after 48 h using triton lysis buffer (1% Triton X-100, 150 mM NaCl, 50 mM Tris pH 7.6) supplemented with deacetylase, phosphatase and protease inhibitors as described previously, sonicated and centrifuged to generate the soluble fraction. The pellet was resuspended in SDS lysis buffer (1% SDS, 150 mM NaCl, 50 mM Tris pH 7.6) supplemented with deacetylase, phosphatase and protease inhibitors as mentioned above, sonicated and centrifuged to generate insoluble fractions.
Antisense oligonucleotides
HDAC6 antisense oligonucleotides were developed by Ionis Pharmaceuticals and added to neurons (10 μM) at DIV 3 to ensure HDAC6 knockdown. Synthesis and purification of all chemically modified oligonucleotides were performed as previously described (Swayze et al., 2007). The MOE-gaper ASOs are 20 nucleotides in length, wherein the central gap segment comprising ten 2′-deoxy-ribonucleotides that are flanked on the 5′ and 3′ wings by five 2′MOE modified nucleotides. Internucleotide linkages are phosphorothioate interspersed with phosphodiester, and all cytosine residues are 5′-methylcytosines.
Lentivirus cloning and generation
To generate lentiviral expression plasmids for HDAC6, the WT HDAC6 cDNA with a FLAG-tag at the 3′ end was amplified by PCR and inserted into the pUltra vector using AgeI and SalI restriction endonucleases to replace the eGFP cassette. The control pLKO.1-eGFP vector was ordered from the UNC Lentiviral Core Facility. Lentiviral production was performed by co-transfecting 37.5 μg lentivirus plasmid with 25 μg psPAX2, 12.5 μg VSVG, and 6.25 μg REV plasmids per 15 cm culture plates of lenti-X 293T cells (Takara), with three dishes of cells used for each lentiviral production. Two days after transfection, culture media was collected and centrifuged at 450 xg for 10 min. The supernatants were then filtered with a 0.45 μm filter flask. Lentiviral particles were purified using a double-sucrose gradient method. Briefly, the filtered supernatants were loaded onto a 70%–60%−30%–20% sucrose gradient and centrifuged at 70,000 xg for 2 h at 17°C. The 30%–60% fraction containing the viral particles was retrieved, resuspended in PBS, loaded onto a 20% sucrose solution, and centrifuged a second time at 70,000 xg for 2 h at 17°C. The supernatants were carefully discarded and the viral particles present in the pellet were resuspended in PBS, aliquoted and stored at −80° C.
Protein expression and purification
Protein expression, extraction, and purification was performed using chromatography methodology to purify heat stable tau proteins. TauFL and tauRD plasmids were cloned into the pRK172 bacterial expression vector for inducible protein expression. Protein was expressed in BL21-CodonPlus (DE3)-RIL E. coli cells. Bacteria were grown in lysogeny broth, ampicillin was added, and when an OD of 1.0 was reached, protein expression was induced with isopropyl β-D-1-thiogalactopyranoside (IPTG) at a final concentration of 1.0 mM. After continued growth for 2 h, bacterial cultures were then centrifuged at 2,340 xg for 20 min, and pellets were immediately frozen at −80°C. To perform recombinant tau protein purification, bacterial pellets were resuspended in a high salt RAB buffer, pH 7.0 (0.1 M MES, 1 mM EGTA, 0.5 mM MgSO4, 750 mM NaCl, 20 mM NaF, 0.1 mM PMSF, 0.1% protease inhibitor cocktail). This resuspension was homogenized, boiled, and centrifuged at 100,000 g for 45 min. The resulting supernatant, with addition of 0.1 mM PMSF and 0.1% protease inhibitor cocktail, was dialyzed against FPLC buffer, pH 6.5 [20 mM piperazine-N,N’-bis(ethanesulfonic acid), 10 mM NaCl, 1 mM EGTA, 1 mM MgSO4, 0.1 mM PMSF and 2 mM dithiothreitol (DTT)]. After overnight dialysis, the contents were passed through a HiTrap sulfopropyl Sepharose high performance cation exchange column (GE) attached to an ÄKTA Pure chromatography system equilibrated in FLPC buffer. Fractions were eluted over a 0–0.4 M NaCl gradient. Portions of the fractions were separated by SDS-PAGE and stained with Coomassie blue. Fractions containing tau protein were subsequently pooled. The FPLC buffer present in pooled tau protein fractions was exchanged for 100 mM sodium acetate (pH 7.0), and protein was concentrated using Amicon Ultra centrifugal filter devices (Millipore). Resultant protein concentration was determined using bicinchoninic acid assay (Thermo Scientific Pierce) and stored at −80°C.
Tau fibrillization reactions
For sedimentation analysis, thioflavin-T (ThT) analysis, and tau seed preparation, a concentration of 20 μM tauFL or tauRD monomers of interest was incubated at 1,000 rpm with 20 μM heparin (Sigma) and 2 mM DTT in a 100 mM sodium acetate buffer (pH 7.0) at 37°C for the indicated time periods. The reactions can be stored in −80°C for future analysis. To prepare tau seeds, the fibrillization reactions were centrifuged at 21,130 xg for 30 min at 4°C, and the resulting pellet was resuspended in equivalent volume of PBS and sonicated 50 times at 25% intensity using a Qsonica Q125 sonicator.
Sedimentation analysis
Tau fibrillization reactions were centrifuged at 21,130 xg for 30 minutes at 4°C to separate supernatant and pellet fractions. The pellet fraction was resuspended in equivalent volume of PBS. Equal amounts of supernatant and pellet fractions for various reaction times were mixed with 6X gel loading buffer with 100 mM DTT, and separated by SDS-PAGE followed by Coomassie blue staining to analyze the extent of tau protein in different fractions.
Thioflavin-T (ThT) fluorescence assay
The ThT fluorescent intensity of tau fibrillization reactions at the indicated incubation periods was measured at 430 nm (excitation) and 500 nm (emission) in a 10 μM ThT solution using a FLUOstar Omega microplate reader (BMG LABTECH). Tau fibrillization reactions (15 μL) were combined with 10 μM ThT solution (140 μL) in a 96-well plate format followed by 5 s plate vortexing prior to fluorescence analysis.
HDAC6 in vitro activity assay
In vitro recombinant HDAC6 activity was measured using the HDAC6 FLUOR DE LYS fluorometric activity assay kit, per the manufacturer’s detailed protocol and instructions (BML-AK516, Enzo Life Sciences). We assessed the inhibition of HDAC6 activity with the following recombinant tau proteins: tauRD-WT monomer, tauRD-WT seed, tauRD-P301L monomer, tauRD-P301L seed, and tauFLΔR1–4 lacking the MTBR. All tau proteins were added to a final concentration of 7.3 μM, while HDAC6 were preloaded at 74.6 nM per the manufacturer’s protocol. Samples lacking tau but containing HDAC6 and substrate represent negative controls for HDAC6 inhibition (100% HDAC6 activity). Samples lacking HDAC6 was used as a positive control for inhibition of HDAC6 activity (0% HDAC6 activity). Trichostatin A (TSA) was used as another positive control for inhibition of HDAC6 activity and was employed at a final concentration of 10 nM. HDAC6 reactions were carried out for 1 h at 30°C, followed by the addition of developer for 45 min. HDAC6 assay was plotted as normalized HDAC6 activity, which was established as a relative baseline representing maximal inhibition of HDAC6 activity. Fluorescence measurements were acquired using the FLUOstar Omega microplate reader (BMG LABTECH).
In vitro acetylation assay
Reactions containing 3 μg of recombinant tauRD-P301L or tauRD-P301L/C291A/C322A (tauRD-P301L-2CA) seeds were mixed with 3 μg of recombinant tauFL-WT or tauFL-2CA monomers in the presence of 0.4 mM CoA or acetyl-CoA (Sigma) in acetylation reaction buffer (50 mM Tris-HCl pH 8.0, 10% glycerol, 1 mM DTT, 100 μM EDTA) for 1.5 h at 37°C. The reaction was terminated by adding 6X gel loading buffer and analyzed by western blotting. To generate acetylated P301L seeds in vitro, 1 mL reaction consists of 200 μg recombinant tauRD-P301L monomers and 1 mM acetyl-CoA in acetylation reaction buffer, as described above, was incubated for 1.5 h at 37°C. The resulting acetylated tauRD-P301L monomers were fibrillized as described above and sonicated before use.
Biochemical analysis, extraction, and immunoblotting
Biochemical analyses for preparation of lysates were performed as follows. Cells from 6-well plates were sonicated and homogenized in Triton lysis buffer (50 mM Tris pH 7.6, 150 mM NaCl, 1% Triton X-100) containing 1 mM phenylmethylsulfonyl fluoride, a mixture of deacetylase inhibitors (100 μM trichostatin A, and 10 mM nicotinamide), a mixture of protease inhibitors (1 mg/mL pepstatin, leuptin, N-p-tosyl-L-phenylalanine chloromethyl ketone, Nα-Tosyl-L-lysine chloromethyl ketone hydrochloride, trypsin inhibitor; Sigma) and a mixture of phosphatase inhibitors (2 mM imidazole, 1 mM NaF, 1 mM sodium orthovanadate; Sigma). Samples were sonicated and centrifuged at 21,130 xg for 30 min at 4°C. The supernatant was collected as the triton soluble fractions. The resulting pellets were washed and resuspended in SDS lysis buffer (50 mM Tris pH 7.6, 150 mM NaCl, 1% SDS) in the presence of all the protease, deacetylase, and phosphatase inhibitors listed above. Samples were sonicated and centrifuged at 21,130 xg for 30 min at room temperature. The supernatant was collected as the triton insoluble fractions. Both triton soluble and insoluble fractions were analyzed by western blotting using the indicated antibodies. The primary antibodies used are listed in the Key resources table. For analysis of the acetyl-lysine profile and other markers in mouse brain homogenates, the cortex was collected from 12 month-old male Tau+/+ or Tau−/−mice. Tissue was homogenized in 4 vol per g of high-salt buffer (0.1 M MES, 1 mM EGTA, 0.5 mM MgSO4, 0.75 M NaCl, 0.02 M NaF, 1 mM PMSF, and 0.1% protease inhibitor cocktail) and centrifuged at 21,130 xg for 45 min to generate high-salt fractions. Resulting pellets were re-extracted in 4 vol per g of RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40, 5 mM EDTA, 0.5% sodium deoxycholate, 0.1% SDS). Myelin floatation was performed on pellets re-extracted in RIPA buffer supplemented with 20% sucrose. Finally, resultant insoluble pellets were extracted in 1 vol per g of tissue in SDS buffer (1% SDS in 50 mM Tris, 150 mM NaCl, pH 7.6). High-salt (Soluble) and SDS (Insoluble) fractions were analyzed by western blotting using the designated antibodies. Protein band quantification was measured by densitometry using ImageQuant TL (GE Healthcare Life Sciences) or Image Studio Lite (LI-COR Biosciences) on an LAS-4000 imager (GE Healthcare Life Sciences).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-tau p-S202/T205 (AT8) | Invitrogen | Cat#MN1020 RRID: AB_223647 |
| Rabbit polyclonal anti-tau p-S262 | Invitrogen | Cat#44-750G RRID: AB_2533743 |
| Rabbit polyclonal anti-tau p-S356 | Invitrogen | Cat#44-751G RRID: AB_2533744 |
| Rabbit polyclonal anti-tau p-S396 | Invitrogen | Cat#44-752G RRID: AB_2533745 |
| Rabbit polyclonal anti-tau ac-K280 | Dr. Todd Cohen | Cohen et al., 2011 |
| Rabbit polyclonal anti-tau ac-K369 | Dr. Todd Cohen | Cohen et al., 2013 |
| Rabbit polyclonal anti-tau (K9JA) | Dako | Cat#A0024 RRID: AB_10013724 |
| Mouse monoclonal anti-tau (T49) | Millipore | Cat#MABN827 RRID: AB_2848143 |
| Mouse monoclonal anti-c-Myc (9E10) | Santa Cruz | Cat#sc-40 RRID: AB_627268 |
| Rabbit polyclonal anti-c-Myc | Sigma | Cat#C3956 RRID: AB_439680 |
| Rabbit polyclonal anti-mHDAC6 | Dr. Tso-Pang Yao | Gao et al., 2007 |
| Rabbit polyclonal anti-HDAC6 | Millipore | Cat#07-732 RRID: AB_441966 |
| Rabbit polyclonal anti-hHDAC6 (H-300) | Santa Cruz | Cat#sc-11420 RRID: AB_2116634 |
| Rabbit polyclonal anti-HDAC6 p-S22 | Abcam | Cat#ab61058 RRID: AB_942257 |
| Mouse monoclonal anti-acetylated tubulin | Sigma | Cat#T7451 RRID: AB_609894 |
| Mouse monoclonal anti-alpha tubulin (DM1A) | Santa Cruz | Cat#sc-32293 RRID: AB_628412 |
| Rabbit polyclonal anti-acetylated-lysine | Cell Signaling | Cat#9441 RRID: AB_331805 |
| Mouse monoclonal anti-acetylated-lysine | Cell Signaling | Cat#9681 RRID: AB_331799 |
| Rabbit polyclonal anti-MAP2 | Millipore | Cat#AB5622 RRID: AB_91939 |
| Mouse monoclonal anti-TUBB3 (TUJ1) | BioLegend | Cat#801202 RRID: AB_10063408 |
| Rabbit polyclonal anti-acetyl Histone H3 (acetyl K27) | Abcam | Cat#ab177178 RRID: AB_2828007 |
| Rabbit polyclonal anti-Histone H3 | Cell Signaling | Cat#2650 RRID: AB_2115124 |
| Mouse monoclonal anti-LAMP1 | Enzo | Cat#ADI-VAM-EN001 RRID: AB_10630197 |
| Rabbit polyclonal anti-Rab5A | Santa Cruz | Cat#sc-309 RRID: AB_632295 |
| Chicken polyclonal anti-EEA1 | Invitrogen | Cat#405700 RRID: AB_596712 |
| Rabbit polyclonal anti-LC3B | Cell Signaling | Cat#2775S RRID: AB_915950 |
| Rabbit polyclonal anti-GAPDH | Millipore | Cat#ABS16 RRID: AB_10806772 |
| Alexa Fluor 594 Phalloidin | Invitrogen | Cat#A12381 |
| Bacterial and virus strains | ||
| BL21-CodonPlus (DE3)-RIL E. coli | Agilent | Cat#230245 |
| NEB® Stable Competent E. coli | New England Biolabs | Cat#C3040I |
| NEB® 5-alpha F’Iq Competent E. coli | New England Biolabs | Cat#C2992I |
| NEB® 5-alpha Competent E. coli | New England Biolabs | Cat#C2988J |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant hTauRD-P301L-K280Q | This paper | N/A |
| Recombinant hTauRD-P301L-K280R | This paper | N/A |
| Recombinant hTauFL-ΔR1-4 | Dr. Todd Cohen | Trzeciakiewicz et al., 2020 |
| Recombinant hTauRD | Dr. Todd Cohen | Trzeciakiewicz et al., 2017 |
| Recombinant hTauRD-P301L | Dr. Todd Cohen | Trzeciakiewicz et al., 2017 |
| Heparin | Sigma | Cat#H3393 |
| Thioflavine T | MP Biomedicals | Cat#156877 |
| Sodium arsenite | Sigma | Cat#S7400 |
| 3-Methyladenine | Sigma | Cat#M9281 |
| MG-132 | Sigma | Cat#M7449 |
| Tubastatin A hydrochloride | Sigma | Cat#SML0044 |
| C646 | Sigma | Cat#SML0002 |
| Critical commercial assays | ||
| FLUOR DE LYS® HDAC6 fluorometric drug discovery kit | Enzo | Cat#BML-AK516 |
| Lambda Protein Phosphatase (Lambda PP) | New England Biolabs | Cat#P0753 |
| CytoTox 96® Non-Radioactive Cytotoxicity Assay | Promega | Cat#G1780 |
| CellTiter 96® Non-Radioactive Cell | Promega | Cat#G4000 |
| Proliferation Assay | ||
| Experimental models: Cell lines | ||
| 293A Cell Line | Invitrogen | Cat#R70507 |
| Lenti-X 293T Cell Line | TaKaRa | Cat#632180 |
| Experimental models: Organisms/strains | ||
| B6.129X1-Mapttm1Hnd/J | Jackson Lab | Cat#007251 |
| Oligonucleotides | ||
| pUltra-hHDAC6 FW primer: ttataccggtgccaccatgacctcaaccggccag |
This paper | N/A |
| pUltra-hHDAC6 RV primer: ggccgtcgacgggccctctagtttatttatcatcatcatc |
This paper | N/A |
| pUltra-hTau FW primer: ccgaccggtgccaccatggctgagccccgccag; |
This paper | N/A |
| pUltra-hTau RV primer: cgcgtcgactcacaaaccctgcttggccagg |
This paper | N/A |
| Recombinant DNA | ||
| pRK172-hTauRD-P301L-K280Q | This paper | N/A |
| pRK172-hTauRD-P301L-K280R | This paper | N/A |
| pRK172-hTauRD-P301L | Dr. Virginia Lee | N/A |
| pRK172-hTauRD | Dr. Virginia Lee | N/A |
| pcDNA5/TO-hTauFL | Dr. Virginia Lee | N/A |
| pcDNA5/TO-hTauFL-P301L | Dr. Virginia Lee | N/A |
| Software and algorithms | ||
| Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| LI-COR Image Studio | LI-COR | https://www.licor.com/bio/image-studio-lite/ |
| ImageQuant TL | Cytiva | https://www.cytivalifesciences.com/en/us/shop/protein-analysis/molecular-imaging-for-proteins/imaging-software/imagequant-tl-8-2-image-analysis-software-p-09518 |
| Photoshop | Adobe | https://www.adobe.com/products/photoshop.html |
Subcellular fractionation
Primary neurons treated with wild-type or P301L tau seeds were harvested using the subcellular fractionation buffer (250 mM sucrose, 20 mM HEPES pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA) containing a mixture of deacetylase inhibitors, protease inhibitors, phosphatase inhibitors, as described previously. The cell lysates were passed through a 25 Ga needle 10 times followed by a centrifugation at 720 xg for 5 min. The resulting pellet was then washed, dispersed by the subcellular fractionation buffer, passed through a 25 Ga needle 10 times, and centrifuged again at 720 xg for 10 min. The pellet was suspended in the vesicular buffer (subcellular fractionation buffer supplemented with 10% glycerol and 0.1% SDS) to obtain the vesicular fraction. The supernatant was centrifuged again at 10,000 xg for 10 min. The resulting supernatant represents the cytosolic fraction.
Double-labeling immunofluorescence
Primary cortical neurons were grown on poly-D-lysine (PDL)-coated coverslips, cultured as described above, and untreated or treated for the indicative tau seeds for 48 h. Cells were fixed in 4% paraformaldehyde for 15 min, rinsed three times in PBS, and permeabilized with 0.2% Triton X-100 in PBS for 10 min. Cells were then blocked in 2% milk for 1 h and subsequently incubated with primary antibodies of interest overnight at 4°C. Cells were washed in PBS and incubated with Alexa 488- or Alexa 594-conjugated secondary antibody. Cells were analyzed using an Olympus IX83 inverted microscope or a Zeiss LSM7−80° Confocal microscope. The primary antibodies used for immunofluorescence are listed in the Key resource table.
Neuronal viability assays
Cell viability was quantitatively evaluated by measuring lactate dehydrogenase (LDH) in medium and cellular metabolic function using MTT assays. Primary cortical neurons in 24-well dishes were treated with various tau seeds at DIV 10 for 5 days. For LDH assays, after 5 days of treatment, cell-free culture medium was collected, and the amount of LDH released into the media was measured by using the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega) according to the manufacturer’s protocol. Results were analyzed on a FLUOstar Omega Microplate Reader (BMG Labtech) at 490 nm. Percent cell injury was determined as experimental LDH release/total LDH release after lysis buffer-induced death after correcting for baseline absorbance. All assays were repeated in triplicate from three independent experiments. For MTT assays, the CellTiter 96 Non-Radioactive Cell Proliferation Assay Kit (Promega) was employed to assess metabolic viability of the cells. Briefly, at the end of the treatment, dye solution containing the MTT tetrazolium component was added to the cells. After 4 h of incubation, the formazan product was converted by living cells. The stop solution was then added to solubilize the formazan product, and the absorbance at 570 nm was recorded with a background correction at 630 nm.
In vitro HDAC6 dephosphorylation assay
In vitro de-phosphorylation assay was performed using the lambda (λ) protein phosphatase kit (New England BioLabs). 15 μL of primary neuronal lysates transduced with a lentivirus expressing WT HDAC6 were treated with or without 1 μL λ-phosphatase in the presence of 2 μL 10X NEBuffer and 2 μL 10X MnCl2 for 30 min at 30°C. The reaction was terminated by adding 6X gel loading buffer and the results were analyzed by western blotting with the p-HDAC6 (S22) antibody.
Mass spectrometry analysis
293A cells were transfected with either WT HDAC6 or pcDNA5/TO. The cell lysates were subjected to SDS-PAGE and stained with Coomassie blue. The band corresponding to HDAC6 was excised and the proteins were reduced, alkylated, and in-gel digested with trypsin overnight at 37°C. Peptides were extracted, desalted with C18 spin columns (Pierce) and dried via vacuum centrifugation. Peptide samples were stored at −80°C until further analysis.
The peptide samples were analyzed in duplicate by LC/MS/MS using an Easy nLC 1200 coupled to a QExactive HF mass spectrometer (Thermo Scientific). Samples were injected onto an Easy Spray PepMap C18 column (75 μm id 3 25 cm, 2 μm particle size) (Thermo Scientific) and separated over a 60 min method. The gradient for separation consisted of 5%–40% mobile phase B at a 250 nl/min flow rate, where mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in 80% ACN. The QExactive HF was operated in data-dependent mode where the 15 most intense precursors were selected for subsequent fragmentation. Resolution for the precursor scan (m/z 350–1600) was set to 120,000 with a target value of 3 × 106 ions. MS/MS scans resolution was set to 15,000 with a target value of 1 × 105 ions. The normalized collision energy was set to 27% for HCD. Dynamic exclusion was set to 30 s, peptide match was set to preferred, and precursors with unknown charge or a charge state of 1 and ≥ 8 were excluded.
Raw data files were processed using Proteome Discoverer version 2.1 (Thermo Scientific). Peak lists were searched against a reviewed Uniprot human database, appended with a common contaminants database, using Sequest. The following parameters were used to identify tryptic peptides for protein identification: 10 ppm precursor ion mass tolerance; 0.02 Da product ion mass tolerance; up to two missed trypsin cleavage sites; (C) carbamidomethylation was set as a fixed modification; (M) oxidation, (S, T, Y) phosphorylation, and (K, N terminus) acetylation were set as variable modifications. The ptmRS node was used to localize the sites of phosphorylation and acetylation. Peptide false discovery rates (FDR) were calculated by the Percolator node using a decoy database search and data were filtered using a 1% FDR cutoff. MS/MS spectrum was annotated using IPSA (Brademan et al., 2019).
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism 9 software was used for all statistical analyses. Results were pooled from a minimum of three independent experiments and presented as average ± standard error of the mean (SEM). Comparisons between two groups were analyzed using unpaired Student’s t test. Multiple comparisons between > 2 groups were analyzed using one-way ANOVA with Tukey’s post hoc test. Significance is presented as n.s. p > 0.05, * p < 0.05, ** p < 0.01, or *** p < 0.001. To analyze the degree of tau seed uptake (Figures S2B and S2E) or tau seed acetylation (Figures S2C and S2F) in microglia or macrophage, at least three isolated areas of primary microglia or macrophage on coverslips were chosen at random within regions containing seeds. The percent tau seed-positive microglia or macrophage was determined as a percentage of the number of microglia or macrophage containing tau seeds per the total number of microglia or macrophage present in Figures S2B or S2E. The percent tau seed acetylation was determined as a ratio of ac-K280-positive tau seeds to total tau seeds in Figures S2C or S2F. To determine the extent of co-localization between tau seeds and vesicular markers (Figure 2D) or HDAC6 (Figure 5F), at least three isolated regions of primary neurons on coverslips were chosen at random within areas harboring tau seeds. Percent co-localization was determined as a ratio of the number of co-localized tau seeds per total number of tau seed-positive foci present in Figures 2D and 5F.
Supplementary Material
Highlights.
Tau seeds are internalized and aberrantly modified in neurons
Acetylated tau seeds display pathological properties
HDAC6 is associated with and inhibited by tau seeds
Tau prevents HDAC6 phosphorylation on Ser-22
ACKNOWLEDGMENTS
Support for this work was provided by the National Institutes of Health (NIH) grants R01AG061188 and R21AG058080, Alzheimer’s Association grant NIRG-14-321219, Association for Frontotemporal Degeneration (AFTD) pilot grant, and American Federation for Aging Research (AFAR) grant RAG15247 (all to T.J.C.). This research is based in part upon work conducted using the UNC Proteomics Core Facility, which is supported in part by P30CA016086 Cancer Center Core support grant to the UNC Lineberger Comprehensive Cancer Center. We thank Dr. Tso-Pang Yao (Duke University) for providing anti-mouse HDAC6 antibodies. We thank Dr. Brian Strahl (University of North Carolina) for providing H3K27ac and total H3 antibodies. We thank the UNC Neuroscience Microscopy core for providing excellent technical support.
Footnotes
DECLARATION OF INTERESTS
K.L. and F.R. are paid employees of Ionis Pharmaceuticals, Inc.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109037.
REFERENCES
- Arakhamia T, Lee CE, Carlomagno Y, Duong DM, Kundinger SR, Wang K, Williams D, DeTure M, Dickson DW, Cook CN, et al. (2020). Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell 180, 633–644.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kügler S, and Ikezu T. (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci 18, 1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolotti N, Bennett DA, and Lazarov O. (2016). Reduced pCREB in Alzheimer’s disease prefrontal cortex is reflected in peripheral blood mononuclear cells. Mol. Psychiatry 21, 1158–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoy V, Van Helleputte L, Prior R, d’Ydewalle C, Haeck W, Geens N, Scheveneels W, Schevenels B, Cader MZ, Talbot K, et al. (2018). HDAC6 is a therapeutic target in mutant GARS-induced Charcot-Marie-Tooth disease. Brain 141, 673–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brademan DR, Riley NM, Kwiecien NW, and Coon JJ (2019). Interactive Peptide Spectral Annotator: A Versatile Web-based Tool for Proteomic Applications. Mol. Cell. Proteomics 18, S193–S201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlomagno Y, Chung DC, Yue M, Castanedes-Casey M, Madden BJ, Dunmore J, Tong J, DeTure M, Dickson DW, Petrucelli L, and Cook C. (2017). An acetylation-phosphorylation switch that regulates tau aggregation propensity and function. J. Biol. Chem 292, 15277–15286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JS, Craft R, Yu GQ, Ho K, Wang X, Mohan G, Mangnitsky S, Ponnusamy R, and Mucke L. (2014). Tau reduction diminishes spatial learning and memory deficits after mild repetitive traumatic brain injury in mice. PLoS One 9, e115765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Yang Y, Zhou Y, Maharana C, Lu D, Peng W, Liu Y, Wan R, Marosi K, Misiak M, et al. (2016). Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise and Metabolic and Excitatory Challenges. Cell Metab. 23, 128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesser AS, Pritchard SM, and Johnson GV (2013). Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front. Neurol 4, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, and Johnson GV (2003). Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. J. Biol. Chem 278, 187–193. [DOI] [PubMed] [Google Scholar]
- Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, and Lee VM (2011). The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun 2, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen TJ, Friedmann D, Hwang AW, Marmorstein R, and Lee VM (2013). The microtubule-associated tau protein has intrinsic acetyltransferase activity. Nat. Struct. Mol. Biol. 20, 756–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen TJ, Hwang AW, Restrepo CR, Yuan CX, Trojanowski JQ, and Lee VM (2015). An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun 6, 5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Carlomagno Y, Gendron TF, Dunmore J, Scheffel K, Stetler C, Davis M, Dickson D, Jarpe M, DeTure M, and Petrucelli L. (2014). Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum. Mol. Genet 23, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, Chen G, Shen T, Tran H, Nichols B, et al. (2017). Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med 9, eaag0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Dolan PJ, and Johnson GV (2008). Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem 106, 2119–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Seibenhener ML, Yan J, Jiang J, and Wooten MC (2015). aPKC phosphorylation of HDAC6 results in increased deacetylation activity. PLoS One 10, e0123191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fá M, Puzzo D, Piacentini R, Staniszewski A, Zhang H, Baltrons MA, Li Puma DD, Chatterjee I, Li J, Saeed F, et al. (2016). Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci. Rep 6, 19393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk KE, Mirbaha H, Jiang H, Holtzman DM, and Diamond MI (2015). Distinct Therapeutic Mechanisms of Tau Antibodies: Promoting Microglial Clearance Versus Blocking Neuronal Uptake. J. Biol. Chem 290, 21652–21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman JL, Vaquer-Alicea J, White CL 3rd, Cairns NJ, Nelson PT, and Diamond MI (2017). Widespread tau seeding activity at early Braak stages. Acta Neuropathol. 133, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YS, Hubbert CC, Lu J, Lee YS, Lee JY, and Yao TP (2007). Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol. Cell. Biol 27, 8637–8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons GS, Banks RA, Kim B, Changolkar L, Riddle DM, Leight SN, Irwin DJ, Trojanowski JQ, and Lee VMY (2018). Detection of Alzheimer Disease (AD)-Specific Tau Pathology in AD and NonAD Tauopathies by Immunohistochemistry With Novel Conformation-Selective Tau Antibodies. J. Neuropathol. Exp. Neurol 77, 216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godena VK, Brookes-Hocking N, Moller A, Shaw G, Oswald M, Sancho RM, Miller CC, Whitworth AJ, and De Vos KJ (2014). Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat. Commun 5, 5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Eisenberg DS, and Crowther RA (2017). Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci 40, 189–210. [DOI] [PubMed] [Google Scholar]
- Haj-Yahya M, Gopinath P, Rajasekhar K, Mirbaha H, Diamond MI, and Lashuel HA (2019). Site-specific hyperphosphorylation inhibits, rather than promotes, tau fibrillization, seeding capacity and its microtubule binding. Angew. Chem. Int. Ed. Engl 59, 4059–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill E, Karikari TK, Moffat KG, Richardson MJE, and Wall MJ (2019). Introduction of tau oligomers into cortical neurons alters action potential dynamics and disrupts synaptic transmission and plasticity. eNeuro 6, ENEURO.0166–19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopp SC, Lin Y, Oakley D, Roe AD, DeVos SL, Hanlon D, and Hyman BT (2018). The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflammation 15, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, and Yao TP (2002). HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458. [DOI] [PubMed] [Google Scholar]
- Hurtado DE, Molina-Porcel L, Iba M, Aboagye AK, Paul SM, Trojanowski JQ, and Lee VM (2010). Abeta accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am. J. Pathol 177, 1977–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, and Lee VM (2013). Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J. Neurosci 33, 1024–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba M, McBride JD, Guo JL, Zhang B, Trojanowski JQ, and Lee VM (2015). Tau pathology spread in PS19 tau transgenic mice following locus co-eruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol. 130, 349–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, Cohen TJ, Grossman M, Arnold SE, Xie SX, Lee VM, and Trojanowski JQ (2012). Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 135, 807–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, Cohen TJ, Grossman M, Arnold SE, McCarty-Wood E, Van Deerlin VM, Lee VM, and Trojanowski JQ (2013). Acetylated tau neuropathology in sporadic and hereditary tauopathies. Am. J. Pathol 183, 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner A, Chua SW, Bertz J, Volkerling A, van der Hoven J, Gladbach A, Przybyla M, Bi M, van Hummel A, Stevens CH, et al. (2016). Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer’s mice. Science 354, 904–908. [DOI] [PubMed] [Google Scholar]
- Kaufman SK, Sanders DW, Thomas TL, Ruchinskas AJ, Vaquer-Alicea J, Sharma AM, Miller TM, and Diamond MI (2016). Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron 92, 796–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman SK, Thomas TL, Del Tredici K, Braak H, and Diamond MI (2017). Characterization of tau prion seeding activity and strains from formaldehyde-fixed tissue. Acta Neuropathol. Commun 5, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman SK, Del Tredici K, Thomas TL, Braak H, and Diamond MI (2018). Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phosphor-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol. 136, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, and Kayed R. (2011). Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, et al. (2010). HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29, 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mews P, Donahue G, Drake AM, Luczak V, Abel T, and Berger SL (2017). Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, Shirakawa K, Minami SS, Defensor E, Mok SA, et al. (2015). Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med 21, 1154–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirbaha H, Holmes BB, Sanders DW, Bieschke J, and Diamond MI (2015). Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation. J. Biol. Chem 290, 14893–14903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirbaha H, Chen D, Morazova OA, Ruff KM, Sharma AM, Liu X, Goodarzi M, Pappu RV, Colby DW, Mirzaei H, et al. (2018). Inert and seed-competent tau monomers suggest structural origins of aggregation. eLife 7, e36584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Z, Zhao X, Liu H, Hu Q, Chen XQ, Pham J, Wei N, Liu Z, Zhou J, Burgess RW, et al. (2018). Aberrant GlyRS-HDAC6 interaction linked to axonal transport deficits in Charcot-Marie-Tooth neuropathy. Nat. Commun 9, 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV, He Z, Zhang B, Gathagan RJ, Trojanowski JQ, and Lee VMY (2017). Pathological Tau Strains from Human Brains Recapitulate the Diversity of Tauopathies in Nontransgenic Mouse Brain. J. Neurosci 37, 11406–11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzscha H, Fedorov O, Kessler BM, Knapp S, and La Thangue NB (2017). CBP/p300 Bromodomains Regulate Amyloid-like Protein Aggregation upon Aberrant Lysine Acetylation. Cell Chem. Biol 24, 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, et al. (2007). HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863. [DOI] [PubMed] [Google Scholar]
- Perez M, Santa-Maria I, Gomez de Barreda E, Zhu X, Cuadros R, Cabrero JR, Sanchez-Madrid F, Dawson HN, Vitek MP, Perry G, et al. (2009). Tau–an inhibitor of deacetylase HDAC6 function. J. Neurochem 109, 1756–1766. [DOI] [PubMed] [Google Scholar]
- Povellato G, Tuxworth RI, Hanger DP, and Tear G. (2014). Modification of the Drosophila model of in vivo Tau toxicity reveals protective phosphorylation by GSK3β. Biol. Open 3, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang L, Sun X, Austin TO, Muralidharan H, Jean DC, Liu M, Yu W, and Baas PW (2018). Tau Does Not Stabilize Axonal Microtubules but Rather Enables Them to Have Long Labile Domains. Curr. Biol 28, 2181–2189.e4. [DOI] [PubMed] [Google Scholar]
- Ran J, Liu M, Feng J, Li H, Ma H, Song T, Cao Y, Zhou P, Wu Y, Yang Y, et al. (2020). ASK1-Mediated Phosphorylation Blocks HDAC6 Ubiquitination and Degradation to Drive the Disassembly of Photoreceptor Connecting Cilia. Dev. Cell 53, 287–299.e5. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, and Mucke L. (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754. [DOI] [PubMed] [Google Scholar]
- Sharma AM, Thomas TL, Woodard DR, Kashmer OM, and Diamond MI (2018). Tau monomer encodes strains. eLife 7, e37813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhilb ML, Dias-Santagata D, Fulga TA, Felch DL, and Feany MB (2007). Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol. Biol. Cell 18, 5060–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swayze EE, Siwkowski AM, Wancewicz EV, Migawa MT, Wyrzykiewicz TK, Hung G, Monia BP, and Bennett CF (2007). Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 35, 687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai C, Chang CW, Yu GQ, Lopez I, Yu X, Wang X, Guo W, and Mucke L. (2020). Tau Reduction Prevents Key Features of Autism in Mouse Models. Neuron 106, 421–437.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy TE, Sohn PD, Minami SS, Wang C, Min SW, Li Y, Zhou Y, Le D, Lo I, Ponnusamy R, et al. (2016). Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss. Neuron 90, 245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzeciakiewicz H, Ajit D, Tseng JH, Chen Y, Ajit A, Tabassum Z, Lobrovich R, Peterson C, Riddick NV, Itano MS, et al. (2020). An HDAC6-dependent surveillance mechanism suppresses tau-mediated neurodegeneration and cognitive decline. Nat. Commun 11, 5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzeciakiewicz H, Tseng JH, Wander CM, Madden V, Tripathy A, Yuan CX, and Cohen TJ (2017). A Dual Pathogenic Mechanism Links Tau Acetylation to Sporadic Tauopathy. Sci. Rep 7, 44102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng JH, Xie L, Song S, Xie Y, Allen L, Ajit D, Hong JS, Chen X, Meeker RB, and Cohen TJ (2017). The Deacetylase HDAC6 Mediates Endogenous Neuritic Tau Pathology. Cell Rep. 20, 2169–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, and Hirschey MD (2014). Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol. Cell 54, 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, and Payne RM (2013). Widespread and enzyme-independent Nε-acetylation and Nε-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem 288, 29036–29045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC, Diamond MI, Duff KE, and Hyman BT (2013). Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol. 70, 304–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, and Mandelkow E. (2012). Degradation of tau protein by autophagy and proteasomal pathways. Biochem. Soc. Trans 40, 644–652. [DOI] [PubMed] [Google Scholar]
- Wang Y, Krüger U, Mandelkow E, and Mandelkow EM (2010). Generation of tau aggregates and clearance by autophagy in an inducible cell model of tauopathy. Neurodegener. Dis 7, 103–107. [DOI] [PubMed] [Google Scholar]
- Wang R, Tan J, Chen T, Han H, Tian R, Tan Y, Wu Y, Cui J, Chen F, Li J, et al. (2019). ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome-lysosome fusion. J. Cell Biol 218, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesseling H, Mair W, Kumar M, Schlaffner CN, Tang S, Beerepoot P, Fatou B, Guise AJ, Cheng L, Takeda S, et al. (2020). Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease. Cell 183, 1699–1713.e1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KA, Zhang M, Xiang S, Hu C, Wu JY, Zhang S, Ryan M, Cox AD, Der CJ, Fang B, et al. (2013). Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J. Biol. Chem 288, 33156–33170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J. (2014). Interplay between HDAC6 and its interacting partners: essential roles in the aggresome-autophagy pathway and neurodegenerative diseases. DNA Cell Biol. 33, 567–580. [DOI] [PubMed] [Google Scholar]
- Zhu J, Coyne CB, and Sarkar SN (2011). PKC alpha regulates Sendai virus-mediated interferon induction through HDAC6 and β-catenin. EMBO J. 30, 4838–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data generated by this study are available upon request.