

Abstract
Paclitaxel-induced peripheral neuropathy (PIPN) is a common and dose-limiting adverse event. The role of P-glycoprotein (P-gp) in the neuronal efflux of paclitaxel was assessed using a translational approach. SH-SY5Y cells were differentiated to neurons and paclitaxel toxicity in the absence and presence of a P-gp inhibitor was determined. Paclitaxel caused marked dose-dependent toxicity in SH-SY5Y-derived neurons. Paclitaxel neurotoxicity was exacerbated with concomitant P-gp inhibition by valspodar and verapamil, consistent with increased intracellular accumulation of paclitaxel. Cancer patients treated with paclitaxel and P-gp inhibitors had a 2.4-fold (95% confidence interval (CI): 1.3–4.3) increased risk of peripheral neuropathy-induced dose modification, a 4.7-fold (95% CI: 1.9–11.9) increased risk for patients treated with strong P-gp inhibitors and a 7.0-fold (95% CI: 2.3–21.5) increased risk in patients treated with atorvastatin. Atorvastatin also increased neurotoxicity by paclitaxel in SH-SY5Y-derived neurons. Clinicians should be aware that co-medication with P-gp inhibitors may lead to increased risk of PIPN.
Keywords: Chemotherapy-induced peripheral neuropathy, paclitaxel, P-glycoprotein, drug-drug interaction
Introduction
Paclitaxel is a cytotoxic anticancer drug used in the treatment of solid organ tumors such as breast cancer and ovarian cancer [1]. While effective, paclitaxel provokes multiple side effects in patients. A common and potentially serious adverse reaction to treatment with paclitaxel is peripheral neuropathy, which occurs in up to 50% of all patients [2]. Clinical signs of paclitaxel-induced peripheral neuropathy (PIPN) include sensory loss, paresthesia, dysesthesia, numbness and tingling and these symptoms often lead to neuropathic pain [3,4]. PIPN can be very serious, but even mild to moderate symptoms may persist for several years after treatment cessation, significantly impairing quality of life [5]. With more than 80% of breast cancer patients expected to be long-term survivors, this severe and long-lasting adverse reaction is particularly concerning and of public health interest. Further, since treatment success with paclitaxel is correlated to dose [2] and PIPN is the major reason for dose reduction [6], the development of PIPN may influence survival rates among cancer patients. Thus, in order to increase quality of life and improve treatment outcomes among patients treated with paclitaxel, more knowledge regarding risk factors for developing PIPN and methods for treating and preventing this adverse reaction are needed.
Paclitaxel is mainly metabolized by CYP2C8 with a minor contribution of CYP3A4. Distribution of paclitaxel is mediated by a number of ATP-binding cassette transporters (ABC) and solute carriers (SLC). Paclitaxel is a well-known substrate for P-glycoprotein (P-gp, MDR1, ABCB1) [7–10] which limits the oral absorption of the drug. Thus, concomitant administration of a P-gp inhibitor with oral paclitaxel increased the bioavailability of the drug up to 7–8-fold in rats [11]. Paclitaxel is also a substrate for the efflux transporters multidrug resistance associated protein 1 (MRP1, ABCC1) [10], MRP2 (ABCC2) [7,12] and MRP7 (ABCC10) [13]. Paclitaxel uptake is mediated by hepatic OATP1B3 (SLCO1B3) and OATP1B1 (SLCO1B1) [14,15]. Recently, murine OATP1B2 was shown to play a key role in regulating paclitaxel distribution to sensory neurons and neurotoxicity [16] but the role of efflux transporters on intraneuronal accumulation of paclitaxel is unknown. Interestingly, pharmacogenetic studies have identified several associations between genetic variants in ABCB1 and risk of peripheral neuropathy, although these findings are not validated [17–21]. The reported genetic associations were initially attributed to altered plasma pharmacokinetics of paclitaxel, but the studies assessing this are inconsistent and only minor changes in paclitaxel plasma concentrations have been detected, suggesting negligible impact of genetic variants in ABCB1 on paclitaxel pharmacokinetics [22–24].
We hypothesized that inhibiting P-gp function would increase accumulation of paclitaxel in sensory neurons, leading to increased risk of PIPN. To examine this hypothesis, a translational approach using a cellular neuronal model together with clinical data from paclitaxel-treated patients was applied. SH-SY5Y-derived neurons were used to determine the effect of P-gp inhibition on paclitaxel neuronal toxicity. Additionally, a retrospective cohort of paclitaxel-treated breast and ovarian cancer patients was queried to determine the impact of concomitant ingestion of P-gp inhibitors on the risk of dose modification due to sensory neuropathy.
Methods
Cell culture
SH-SY5Y cells (94030304, Sigma-Aldrich) were kept in DMEM/F:12 (11320074, ThermoFisher) with 15% heat-inactivated fetal bovine serum (hiFBS, F9665 Sigma-Aldrich), 1% penicillin/streptomycin (p/s) and 2 mM glutamine. Cells were split 1:10–1:20 using 0.05% trypsin-EDTA (10779413, Fisher Scientific) at 70–80% confluence. At high passage number the epithelial cell type takes over the culture and causes drastically reduced efficiency of the differentiation. Thus, cells were only used below passage 15.
Reagents and antibodies
Paclitaxel (T7402, Sigma-Aldrich) was diluted serially in DMSO (D2650, Sigma-Aldrich) and these stock solutions diluted 1:500 to ensure 0.2% DMSO in all samples. These concentrations were selected based on the clinical pharmacokinetic profile of paclitaxel [25]. Valspodar (SML0572, Sigma-Aldrich) was used at a final concentration of 4 μM and verapamil (V4629, Sigma-Aldrich) was used at 5 μM and 50 μM. Simvastatin (S6196, Sigma-Aldrich) and atorvastatin (PHR1422, Sigma-Aldrich) were used at clinically relevant concentrations based on their clinical pharmacokinetic profile. The Cmax,,ss of atorvastatin can reach 0.7 μM after an 80 mg dose after reaching steady-state [26]. To account for extensive plasma protein binding and interindividual variability of atorvastatin pharmacokinetics concentrations of 10 nM and 100 nM were chosen to reflect unbound concentrations of atorvastatin. Accordingly, a similar approach was used to select simvastatin concentrations of 5 nM and 50 nM. Antibody against TUBB3 (MA1–118, ThermoFisher Scientific, RRID: AB_2536829) was used at a 1:1000 dilution. Human dorsal root ganglion RNA for qPCR analysis was purchased from Clontech Laboratories (636150, Clontech laboratories, Inc, Mountain View, California, USA). Human dorsal root ganglion for protein quantification of ABCB1 was obtained from National Disease Research Interchange (Philadelphia, PA, USA).
Differentiation of SH-SY5Y cells
SH-SY5Y cells (RRID: CVCL_0019) were differentiated as previously described in detail by Shipley et al. [27]. Briefly, SH-SY5Y cells below passage 15 were seeded at 5–6 ×103 cells/cm2 in uncoated 6-well plates (day 0 (D0)) and left overnight in growth media. The next day (D1) differentiation was initiated using DMEM/F:12 media with 2.5% hiFBS, 1% p/s, 2 mM glutamine and 10 μM retinoic acid (RA, R2625 Sigma-Aldrich) and media was replaced at D3 and D5. On D7 cells were split 1:1 using 0.05% trypsin-EDTA to uncoated dishes and on D8 hiFBS content was reduced to 1%. On D10 the cells were split 1:1 to extracellular matrix-coated dishes (E0282, Sigma-Aldrich). On D11 media was replaced with the final differentiation media consisting of neurobasal media (21103049, ThermoFisher) with 1X B27 supplement (17504044, ThermoFisher), 20 mM KCl (10697623, Fisher Scientific), 1% p/s, 1X Glutamax, 50 ng/ml brain-derived neurotrophic factor (BDNF, CST-3897S, Peprotech), 2 mM dibutyryl cyclic AMP (db-cAMP, D0627 Sigma-Aldrich) and 10 μM RA. Media was replaced on D14 and D17 and cells were tested and confirmed as fully differentiated on D18 and used for downstream applications as outlined below.
Real time quantitative polymerase chain reaction (RT-qPCR)
Total RNA from SH-SY5Y cell lines was isolated using a RNeasy Mini kit (Qiagen, Valencia, CA). Total RNA (1 μg) was reverse transcribed into cDNA using a SuperScript VILO cDNA Synthesis kit (Life Technologies, CA). Quantitative real-time PCR was carried out in 384-well reaction plates using 2X Taqman Fast Universal Master Mix (Applied Biosystems, Foster City, CA), 20X Taqman specific gene expression probes for each transporter, and 10 ng of the cDNA template. The reactions were carried out on an Applied Biosystems 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA). The relative expression level of each mRNA transcript was calculated by the comparative method (ΔCt method), normalized to the housekeeping gene, hypoxanthine phosphoribosyl transferase (HPRT).
Immunolabelling
Both differentiated and undifferentiated SH-SY5Y cells were fixed using 4% paraformaldehyde. Cells were permeabilized using 0.25% Triton X-100 and unspecific binding was blocked using bovine serum albumin (BSA). Primary antibodies were incubated overnight at 4°C and secondary antibodies were incubated for one hour at room temperature. Nuclei were stained using 1.5 μM DAPI for 5 minutes. Imaging was performed using a Leica DMI4000B microscope. Neurite morphology was assessed using the ImageJ modules CellCounter and Simple Neurite Tracer. To determine number of neurites per cell, CellCounter was used to count the number of cells with 0, 1, 2… n neurites. Neurite length was determined using Simple Neurite Tracer. For both endpoints, at least five images of each condition from three separate differentiations were used. ImageJ analysis was blinded to the person assessing length and number of neurites to ensure no bias. To assess the impact of verapamil, atorvastatin and simvastatin on paclitaxel-induced neurotoxicity fully differentiated SH-SY5Y-derived neurons were stained with Alexa 488-conjugated phalloidin (A12379, ThermoFisher Scientific) and DAPI. Images were acquired using an ImageXpress Pico (Molecular Devices, San Jose, CA, USA) and a total of nine images/condition were acquired at 10X from three separate differentiations. Total number of neurites/image was manually counted in ImageJ and adjusted for cell count assessed using Cell Reporter Xpress software (Molecular Devices, San Jose, CA, USA).
LC-MS/MS method to determine intracellular paclitaxel concentrations
Fully differentiated SH-SY5Y cells were exposed to 1–10 μM paclitaxel for 1 hour with and without 1 hour pre-incubation with efflux transporter inhibitors as optimized in preliminary experiments. After this, media was aspirated, and cells were lysed using radioimmunoprecipitation assay (RIPA) buffer with 4% protease inhibitors for 10 minutes on ice. Cells were collected using a cell scraper, vortexed and sonicated (1 sec X2), centrifuged and the supernatant was stored at −80°C until liquid chromatography and tandem mass spectrometry (LC-MS/MS) analysis. The concentration of paclitaxel in the cell lysate was determined at the Department of Clinical Pharmacology and Pharmacy, Institute of Public Health, University of Southern Denmark by use of LC-MS/MS. The LC-MS/MS system consisted of an Ultimate 3000 UHPLC system connected to a TSQ Quantiva Triple Quadropole Mass Spectrometer with heated electrospray ionization (H-ESI) (Thermo Scientific, San Jose, CA). The ionization was performed in positive mode with a spray voltage of 3000 V, sheath gas 40 (AU), aux gas 9 (AU), sweep gas 1 (AU) and a vaporizer temperature of 400°C. The ion transfer tube temperature was 350°C. Data acquisition was performed in single reaction monitoring (SRM) mode. Paclitaxel was quantified at the transition from (m/z) 876.18 – 308.00, and with (m/z) 876.18 – 591.11 and (m/z) 876.19 – 531.11 as qualifier traces. The analytical separation was performed by use of a Hypersil GOLD (C18) 50 × 2.1 mm (1.9 μm) column (Thermo Scientific, San Jose, CA) with a mobile phase of acetonitrile: 0.1 M formic acid (40:60 v/v) at a flow rate of 0.4 mL/min. The sample preparation of the cell lysate consisted of a single dilution step. A volume of 100 μL cell lysate and 100 μL acetonitrile was pipetted into a 1.5 mL polypropylene micro tube (Sarsted, Nürnbrecht, Germany). The sample was vortex mixed for 5 min, and thereafter centrifuged at 15,000 g for 15 min. A volume of 10 μL of the supernatant was injected onto the LC-MS/MS system. Calibration curves ranging from 25 nM to 3000 nM as well as quality control samples were prepared and included in each batch of analysis. The within-batch imprecision was < 8%. The limit of detection (LOD) for the method was 1 nM and lower limit of quantification (LLOQ) was 5 nM. Finally, paclitaxel concentrations were adjusted for protein concentration in each well as assessed using a Pierce BCA protein assay kit.
LC-MS/MS to determine transporter expression
Transporter protein expression levels were analyzed by TXP targeted proteomic analysis, which has been described before (Table S2) [28,29]. In short, cell pellets were incubated for one hour with lysis buffer containing 1% NP-40 (Thermo Fisher), 0.01% SDS (ThermoFisher), 0.15M NaCl (Merck), 0.01M di-sodium hydrogen phosphate dihydrate (Merck), 2mM EDTA, and 2.5 units/mL Benzonase (Novagen). The protein concentration in the lysate was determined by BCA assay (Thermo Fisher Scientific) according to the manufacturer’s manual. Subsequently, 70 μg protein was proteolyzed with trypsin overnight. Stable isotope labelled peptides and TXP- antibodies (custom produced by Pineda) were added to 20 or 40 μg of the digest and incubated for one hour. Peptide-antibody complexes were precipitated, washed and denatured by using protein G-coated magnetic beads (ThermoFisher) and a magnetic particle processor (KingFisher 96, ThermoFisher). The precipitated peptides were subsequently quantified using the previously described 10 min LC-MS method (UltiMate 3000 RSLCnano and tSIM - QExactive Plus™ Thermo Scientific, Waltham, MA, USA) [29]. LC gradients were optimized for each multiplex. Raw data were processed with Skyline v4.1. Peak areas of isotopically-labeled peptides representing known peptide amounts and endogenous signals were set in relation to one another on parent ion level.
P-gp inhibitors and paclitaxel-induced peripheral neuropathy in patients
To assess if P-gp inhibition leads to increased risk of peripheral neuropathy we used a previously described database of 503 paclitaxel treated patients with breast or ovarian cancer [30]. Briefly, dose modifications due to peripheral neuropathy were collected from medical records and multivariate logistic regression was performed to assess if users of P-gp inhibitors had a higher risk of dose modifications due to neuropathy. P-gp inhibitors were selected based on two literature reviews [31,32]. From these reviews, we identified drugs listed as P-gp inhibitors (drugs classified as inhibitor in either review), and, within these, strong P-gp inhibitors (drugs classified as inhibitor in both reviews). A full list of P-gp inhibitors is shown in Table S3.
Statistics
Analysis of variance (ANOVA) was used to assess dose-dependent effects of paclitaxel on number and length of neurites and intracellular accumulation of paclitaxel. Data were log-transformed to ensure normality of data. When log-transformation was not sufficient to normalize data, the non-parametric Kruskall-Wallis test was used in lieu of ANOVA. STATA software version 15 was used to perform the statistical analysis. The logistic regression to assess risk of dose modification of paclitaxel due to P-gp inhibitors was adjusted for age, body surface, tumor type, cancer stage, treatment schedule and previous chemotherapy. SPSS software version 19 was used to perform the logistic regression analysis.
Results
Paclitaxel affects neuronal morphology
SH-SY5Y cells were effectively differentiated to neurons using the protocol of Shipley et al. [27] (Figure S1). After 18 days of differentiation the cells form a complex neuronal network with high TUBB3 expression (Figure 1) and the majority of cells (>97.5%) were negative for the proliferation marker Ki67, indicating that the cells are post-mitotic (Figure S2). Neuronal morphology was assessed by measuring neurite length and the number of neurites/cell in fully differentiated SH-SY5Y cells following 24 hours paclitaxel treatment. Paclitaxel induced a dose-dependent neurotoxicity to differentiated SH-SY5Y cells (Figure 1) and significantly reduced both the number of neurites (p<0.001) and their length (p<0.001) (Figure 2).
Figure 1.
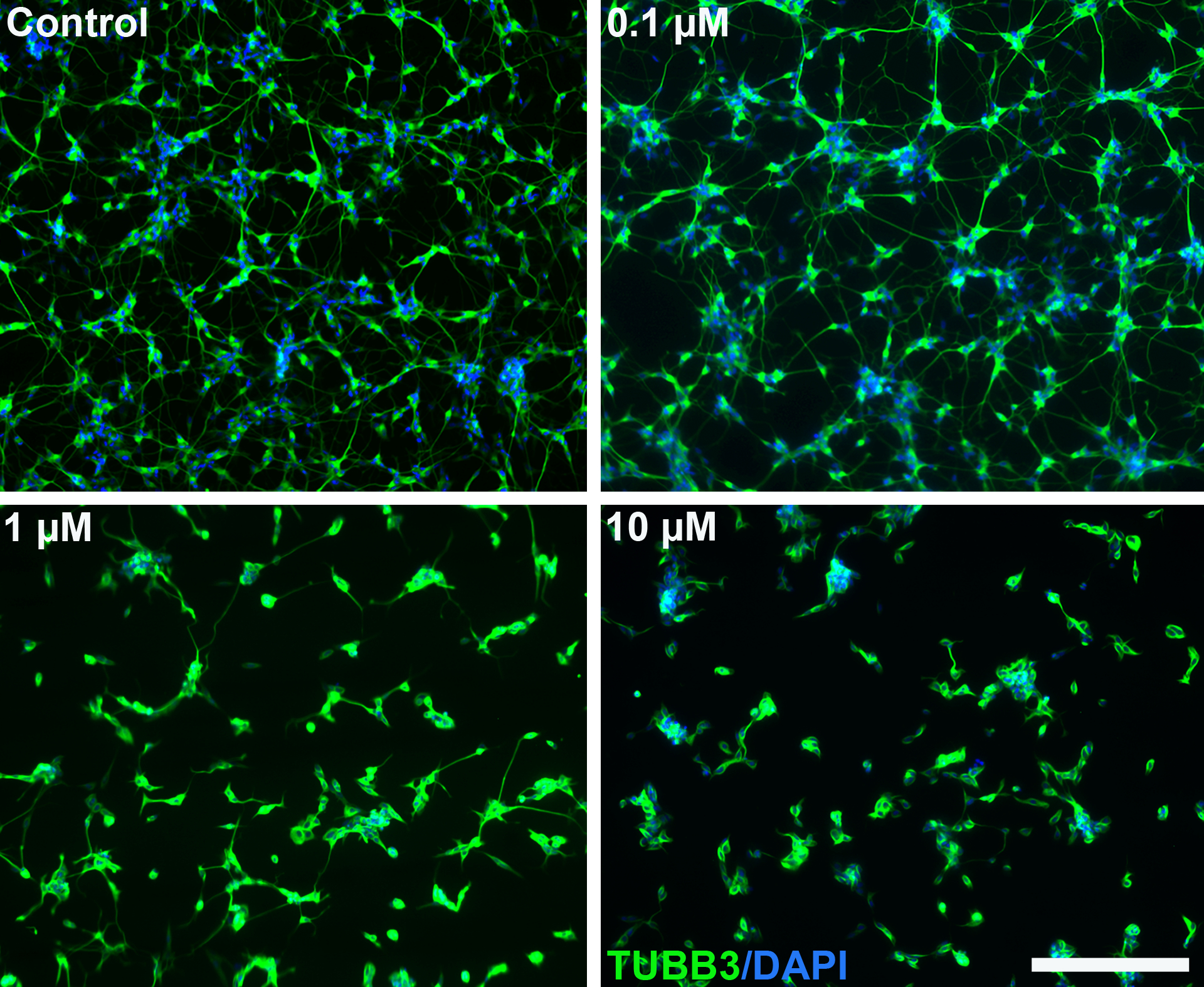
Paclitaxel causes dose-dependent reduced neuronal networks in SH-SY5Y-derived neurons. Fully differentiated SH-SY5Y cells were treated with the indicated concentrations of paclitaxel for 24 hours. The cells were stained for β-tubulin (TUBB3) and nuclei (DAPI). Scale bar represents 200 μm.
Figure 2.
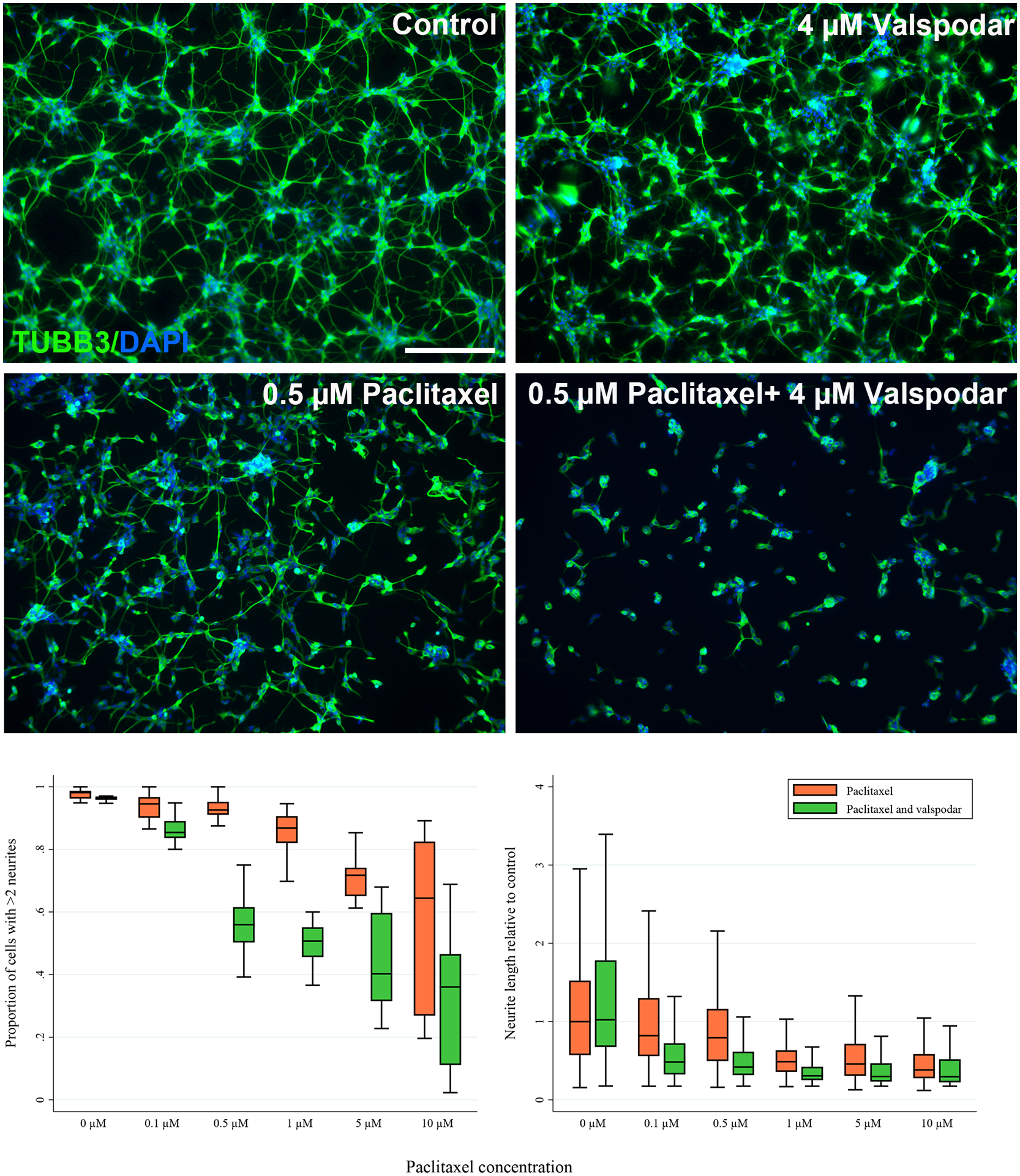
Paclitaxel reduces the length and number of neurites in SH-SY5Y-derived neurons and this effect is exacerbated by inhibition of P-glycoprotein by valspodar. Fully differentiated SH-SY5Y cells were treated with the indicated concentrations of paclitaxel for 24 hours in the absence and presence of 4 μM valspodar. A) The cells were stained for β-tubulin (TUBB3) and nuclei (DAPI) and representative images are shown. Exposure to low concentrations of paclitaxel causes minimal effects on neuronal networks compared to the 0.2% DMSO control, but concomitant treatment with 4 μM of the P-gp inhibitor valspodar causes increased neuronal toxicity of paclitaxel. Scale bar represents 200 μm. B) The proportion of cells with more than two neurites (left panel) and the neurite length (right panel) were quantified using ImageJ. At least five images from three separate differentiations were assessed. Valspodar significantly exacerbated the paclitaxel effect on both phenotypes (p<0.001 for both figures, ANOVA).
Inhibition of efflux transporters exacerbates paclitaxel neuronal toxicity
mRNA and protein expression of relevant paclitaxel transporters were assessed by qPCR and LC-MS/MS and indicated expression of the efflux transporters P-gp and MRP1 in both SH-SY5Y-derived neurons and human dorsal root ganglia (Table 1). To assess the role of P-gp on the neuronal accumulation and neurotoxicity of paclitaxel, P-gp was inhibited with 4 μM valspodar. Inhibiting P-gp with valspodar substantially exacerbated the neuronal toxicity of paclitaxel as indicated by a decrease in the number and length of neurites (p<0.001, ANOVA, Figure 2). Valspodar did not affect the neuronal morphology without paclitaxel present (Figure 2). The P-gp inhibitor verapamil (5 μM and 50 μM) also increased neuronal toxicity of paclitaxel at 0.1 μM and 0.5 μM (p<0.001, Kruskall-Wallis, Figure 3) further supporting the role of P-gp in paclitaxel in vitro neurotoxicity.
Table 1.
Expression of drug transporters relevant for paclitaxel disposition in SH-SY5Y-derived neurons and human dorsal root ganglion.
| Transporter gene | qPCR Expression relative to SH-SY5Y ABCB1 | LC-MS/MS fmol/μg protein | ||
|---|---|---|---|---|
| SH-SY5Y-derived neurons | Human dorsal root ganglion | SH-SY5Y-derived neurons | Human dorsal root ganglion | |
| ABCB1 | 1.0 | 0.04 | 3.1 | 0.06 |
| ABCC1 | 0.2 | 0.06 | Below LoQ | 0.05 |
| ABCC2 | 0.002 | Not detected | Below LoQ | Below LoQ |
| ABCG2 | Not detected | 0.04 | Not measured | Not measured |
| SLCO1B1 | Not detected | Not detected | Not measured | Not measured |
| SLCO1B3 | Not detected | 0.006 | Not measured | Not measured |
Figure 3.
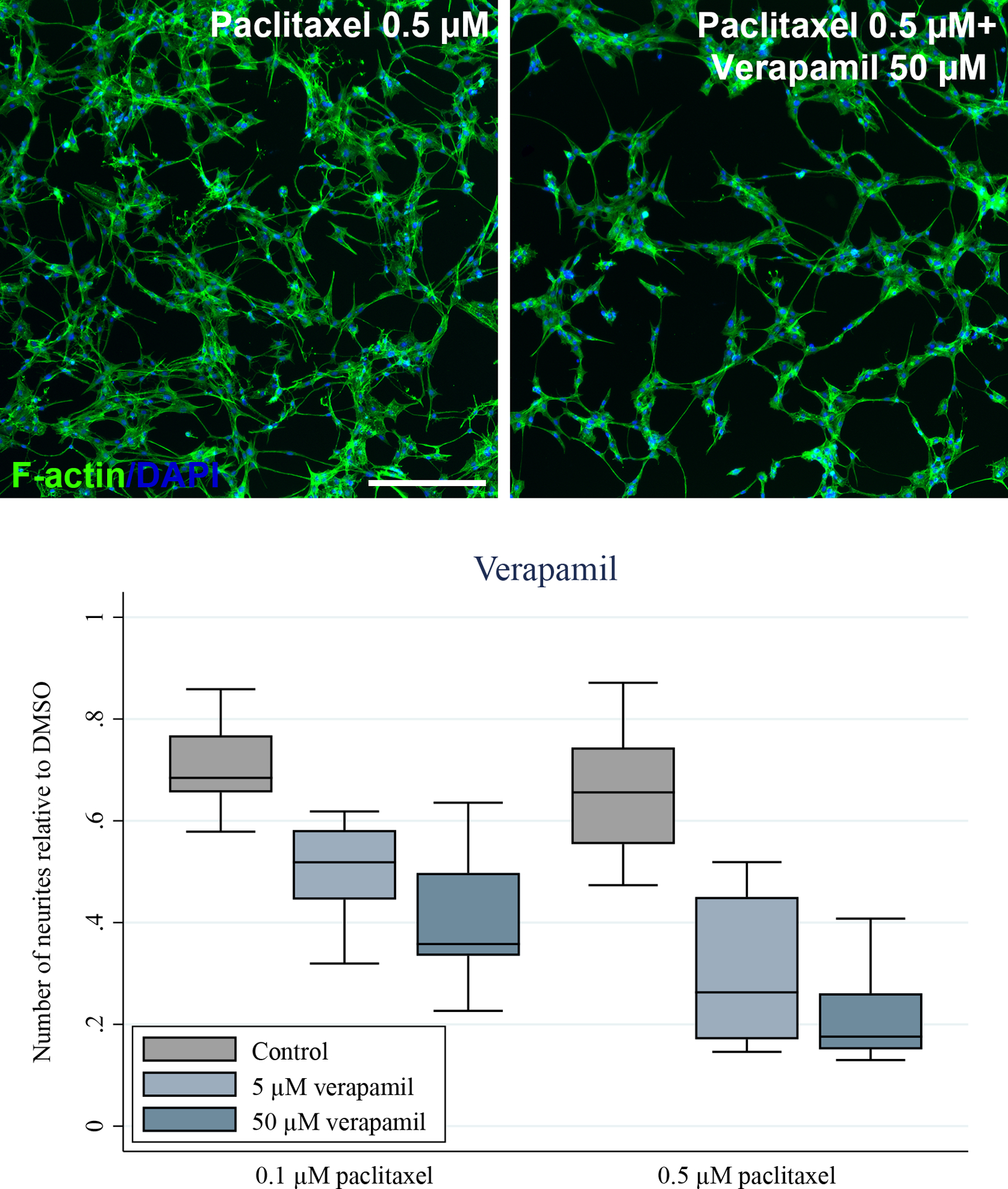
Paclitaxel reduces the number of neurites in SH-SY5Y-derived neurons and this effect is exacerbated by inhibition of P-glycoprotein by verapamil. Fully differentiated SH-SY5Y cells were treated with the indicated concentrations of paclitaxel for 24 hours in the absence and presence of 5 μM and 50 μM verapamil. A) The cells were stained for F-actin and nuclei (DAPI) and representative images are shown. Exposure to 0.5 μM of paclitaxel causes minimal effects on neuronal networks compared to the 0.2% DMSO control, but concomitant treatment with 50 μM of the P-gp inhibitor verapamil causes increased neuronal toxicity of paclitaxel. Scale bar represents 200 μm. B) The number of neurites were quantified using ImageJ. At least five images from three separate differentiations were assessed. Verapamil (5 μM and 50 μM) significantly exacerbated the paclitaxel (0.1 μM and 0.5 μM) effect on number of neurites (p<0.001, ANOVA).
Intracellular concentrations of paclitaxel
To confirm that the increased effect of paclitaxel on neuronal morphology under conditions of P-gp inhibition was related to increased exposure, the intracellular concentrations of paclitaxel were quantified. Cells were treated with paclitaxel with and without concomitant P-gp inhibition by valspodar and intracellular levels were measured 1 hour after treatment. Inhibition of P-gp caused a >3-fold increase in intracellular accumulation of paclitaxel at 10 μM (p<0.001, Figure 4).
Figure 4.
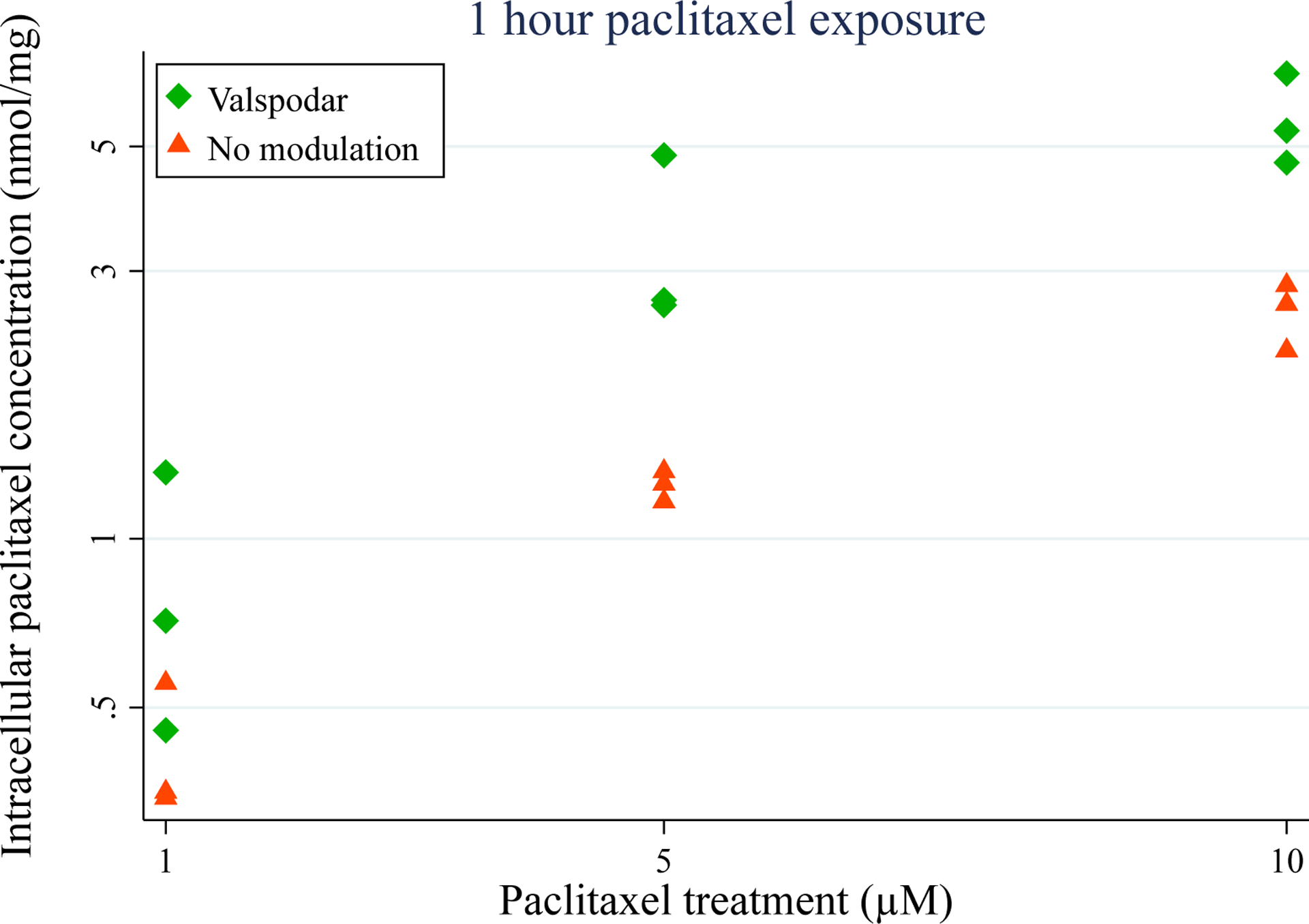
P-gp inhibition causes increased accumulation of paclitaxel in SH-SY5Y-derived neurons. Fully differentiated SH-SY5Y cells were treated with the indicated concentrations of paclitaxel for 1 hour in the absence and presence of 4 μM valspodar. The values shown are triplicates from one differentiation and are increased in the presence of valspodar (p<0.001, ANOVA).
P-gp inhibitors and paclitaxel-induced peripheral neuropathy in patients
To assess if concomitant ingestion of P-gp inhibitors causes increased risk of peripheral neuropathy among patients treated with paclitaxel, a database of 503 paclitaxel-treated patients with breast and ovarian cancer was queried for paclitaxel-induced dose modifications due to peripheral neuropathy [30]. A total of 428 patients had concomitant drug data recorded and thus included in this association analysis. Paclitaxel was administered once weekly for breast cancer patients (n=350) and every 21 days for most ovarian cancer patients (n=78), with the exception of 6 patients who were dosed weekly. Breast cancer patients received paclitaxel in monotherapy and ovarian cancer patients received paclitaxel in combination with carboplatin. Ninety-eight percent of patients received paclitaxel as first-line therapy. The association from univariate and multivariate analysis between paclitaxel dose modification due to peripheral neuropathy and concomitant P-gp inhibitor use are shown in Tables 2 and S1. Patients treated with any P-gp inhibitor had a 2.4-fold (95% confidence interval (CI): 1.3–4.3) increased risk of dose modification due to peripheral neuropathy. The risk of dose modification increased to 4.7-fold (95% CI: 1.9–11.9) in patients treated with strong P-gp inhibitors and to 7.0-fold (95% CI: 2.3–21.5) in patients treated with atorvastatin; users of simvastatin were not at increased risk of paclitaxel dose modifications. When accumulated paclitaxel dose was also added as a covariate, there was no substantial change in the association (e.g. odds-ratio (OR) 2.3 (95% CI: 1.3–4.3) for any P-gp inhibitor and OR 3.5 (95% CI: 1.3–9.1) for strong inhibitors).
Table 2.
Paclitaxel-treated patients using P-gp inhibitors have a higher risk of dose modification due to peripheral neuropathy.
| P-gp inhibitorsa | n | Univariate analysis | Multivariate analysisb |
|---|---|---|---|
| Odds-ratio (95% confidence interval) | Odds-ratio (95% confidence interval) | ||
| Any inhibitor | 117 | 2.55 (1.51–4.31) | 2.36 (1.31–4.25) |
| Strong inhibitor | 24 | 5.73 (2.46–13.36) | 4.73 (1.88–11.90) |
| Atorvastatin | 16 | 9.41 (3.30–26.82) | 7.01 (2.28–21.53) |
| Simvastatin | 26 | 0.63 (0.18–2.16) | 0.33 (0.09–1.19) |
Atorvastatin and simvastatin exacerbate neuronal toxicity of paclitaxel
Finally, we aimed to assess if the clinical impact of atorvastatin on paclitaxel neurotoxicity could be reverse translated to our cell model. SH-SY5Y-derived neurons were treated with atorvastatin and simvastatin concomitantly with paclitaxel over 24 hours and the number of neurites were counted to determine neuronal network complexity. Atorvastatin exacerbated 0.1 μM and 0.5 μM paclitaxel neurotoxicity at concentrations of 0.01 μM and 0.1 μM (p<0.001 for both, Figure 5). Simvastatin did not affect paclitaxel neurotoxicity at 0.1 μM paclitaxel but caused a substantially increased toxicity at 0.5 μM paclitaxel (p=0.002, Figure S3). Importantly, neither statin had a neurotoxic effect when used without paclitaxel (Figures S4–S5, p>0.05).
Figure 5.
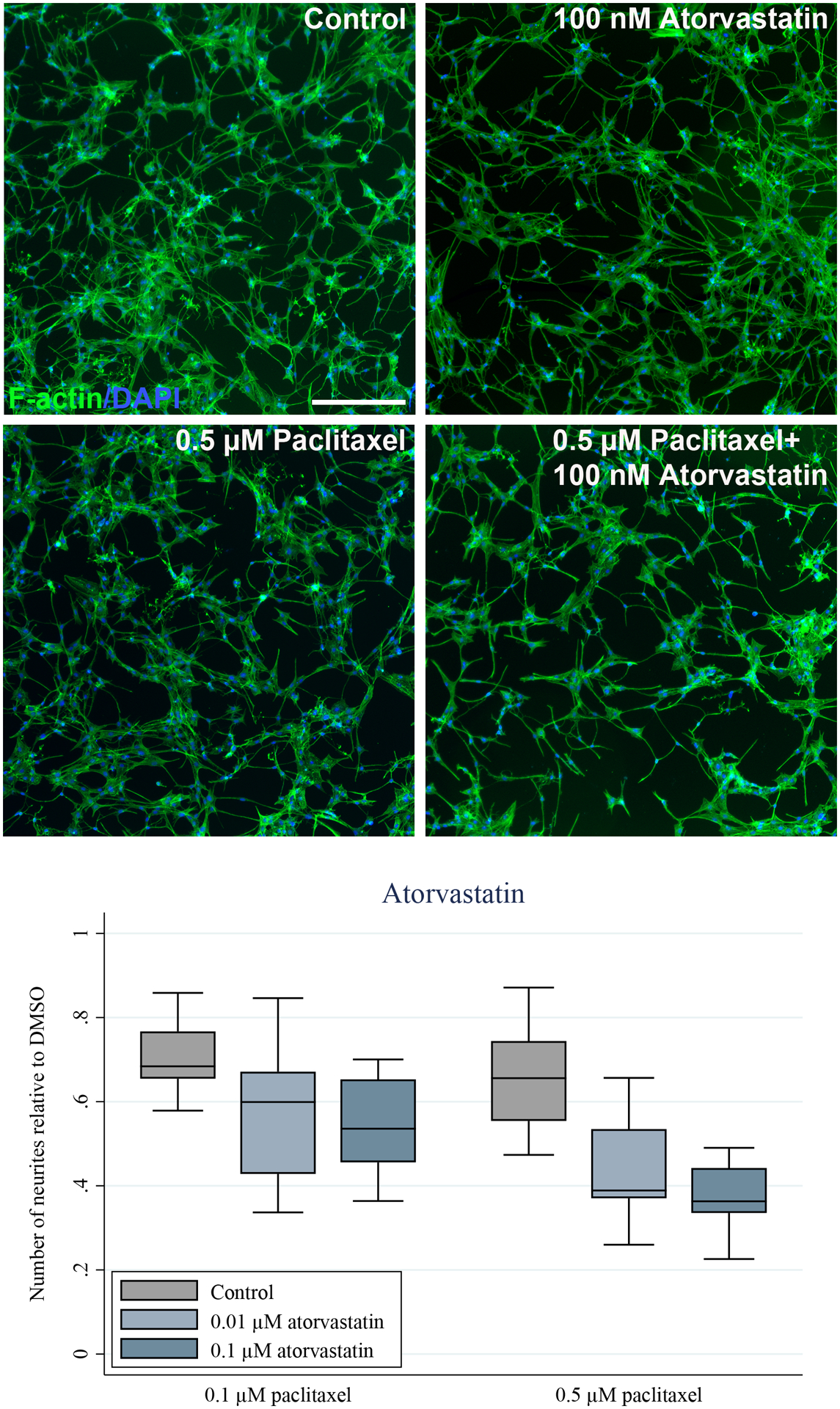
Paclitaxel reduces the number of neurites in SH-SY5Y-derived neurons and this effect is exacerbated by inhibition of P-glycoprotein by atorvastatin. Fully differentiated SH-SY5Y cells were treated with the indicated concentrations of paclitaxel for 24 hours in the absence and presence of 10 nM and 100 nM atorvastatin. A) The cells were stained for F-actin and nuclei (DAPI) and representative images are shown. Exposure to 0.5 μM of paclitaxel causes minimal effects on neuronal networks compared to the 0.2% DMSO control, but concomitant treatment with 100 nM of atorvastatin causes increased neuronal toxicity of paclitaxel. Scale bar represents 200 μm. B) The number of neurites were quantified using ImageJ. At least five images from three separate differentiations were assessed. Atorvastatin (10 nM and 100 nM) significantly exacerbated the paclitaxel (0.1 μM and 0.5 μM) effect on number of neurites (p<0.001, ANOVA).
Discussion
The role of P-gp in paclitaxel-induced peripheral neuropathy was studied using a forward and reverse translational approach. Expression of P-gp in the peripheral nervous system was confirmed in humans and P-gp inhibition in SH-SY5Y-derived neurons led to increased intraneuronal accumulation and exacerbated neurotoxicity of paclitaxel. Cancer patients treated with P-gp inhibitors concomitantly with paclitaxel had a higher risk of dose modification of paclitaxel due to peripheral neuropathy.
This study provides a plausible biological mechanism for the recurring links between polymorphisms in ABCB1 and risk of peripheral neuropathy from treatment with paclitaxel [17–21]. Reduced function of P-gp (encoded by ABCB1) might lead to increased intraneuronal accumulation of paclitaxel in sensory neurons and in turn confer increased risk of peripheral neuropathy. Though P-gp inhibition may lead to altered plasma pharmacokinetics of paclitaxel, studies have shown that treatment with the selective and potent P-gp inhibitor valspodar causes minor changes in paclitaxel pharmacokinetics [33–36]. Interpretation of the pharmacokinetic impact of valspodar is complicated as paclitaxel dose is reduced among patients receiving valspodar in all clinical studies assessing this drug-drug interaction. Paclitaxel pharmacokinetics are nonlinear [25] and thus comparison of pharmacokinetics across different dose levels requires proper adjustment. Paclitaxel displays linear pharmacokinetics with respect to unbound plasma concentrations and valspodar did not affect the unbound clearance of paclitaxel [37], highlighting that P-gp inhibition plays a minor role in plasma pharmacokinetics of paclitaxel. Further supporting this finding, P-gp knockout mice only have 27% higher paclitaxel AUC than wild type mice [7]. Evidence from rodent models are conflicting regarding the role of P-gp in distribution of drugs to the peripheral nervous system. Thus, Mdr1a(−/−) mice had increased accumulation of vinblastine in sciatic nerve [38], while recent studies did not support a role of P-gp in drug distribution to the peripheral nervous system in Mdr1a(−/−) rats [39,40]. Whether these discordant data are caused by species differences or other factors is unknown.
The proposed mechanism in this study may extend to other P-gp dependent chemotherapeutics that cause peripheral neuropathy. ABCB1 polymorphisms have been linked to both vincristine [41] and docetaxel neurotoxicity [42] and future studies should examine whether this might be caused by accumulation of the drugs in the peripheral nervous system. These results also highlight the importance of considering drug transporters in the peripheral nervous system. Multidrug resistance conveyed by efflux transporters in tumors is a recurring problem in treating cancers [43]. Inhibition of efflux transporters to increase chemotherapy accumulation and efficacy in tumor cells remains a potential strategy for improved treatment response, particularly in a personalized medicine setting where tumors are selected for overexpression of efflux transporters [44]. The current findings suggest that for paclitaxel this may lead to off-target effects in the peripheral nervous system and increased risk of peripheral neuropathy. Inhibition of efflux transporters to increase drug efficacy should be performed with caution and a robust assessment of peripheral nervous system adverse effects.
Interestingly, users of atorvastatin were at substantial increased risk of dose-modification due to sensory neuropathy compared to users of simvastatin. Both drugs have been shown to inhibit P-gp to varying degrees though the extent and clinical relevance are unknown [45–48]. Atorvastatin caused a pronounced exacerbation of paclitaxel neurotoxicity while simvastatin had a more modest effect in our cellular model, consistent with the clinical observations of paclitaxel dose reductions during treatment with these statins. Additionally, distinct differences in the clinical pharmacokinetics of the drugs may impact the clinical relevance of their P-gp inhibition properties. Simvastatin is ingested before bedtime due to its short elimination half-life (t½ = 2–5 hours [49]) and increased cholesterol production during fasting periods [50] while atorvastatin has a longer elimination half-life (t½ = 14–30 hours [49]) and thus is not required to be administered at bedtime. Considering the different elimination half-lives of these statins, during paclitaxel infusion there will be no appreciable simvastatin concentrations in plasma while atorvastatin will still be circulating. Therefore, statin-induced P-gp inhibition may be more clinically relevant for atorvastatin than for simvastatin. This is based on the assumption that statin-mediated P-gp inhibition is competitive instead of non-competitive, which would lead to longer inhibition. Whether users of atorvastatin are at increased risk for paclitaxel-induced peripheral neuropathy needs further clinical validation.
The primary strength of this work is the translational approach. Initial studies in SH-SY5Y-derived neurons led to the hypothesis that P-gp might play a key role in paclitaxel-induced peripheral neuropathy by regulating paclitaxel exposure to the peripheral nervous system. P-gp inhibition was also shown to increase risk of dose modification due to peripheral neuropathy in a sample of cancer patients treated with paclitaxel. This bench-to-bedside approach provides a mechanistic basis for paclitaxel distribution to the peripheral nervous system that might have a direct impact on patient response to treatment with this chemotherapeutic. Based on the clinical data, switching atorvastatin to simvastatin among paclitaxel-treated patients is predicted to reduce the risk of peripheral neuropathy. Importantly, this requires comprehensive replication in additional clinical cohorts.
The primary limitation of the cell studies is the use of SH-SY5Y cells as a model representative for the peripheral nervous system. While it is widely established and shown that SH-SY5Y-derived neurons express key neuronal markers such as TUBB3, BRN3A and MAP2 [27] they are not sensory neurons and thus the direct translation of these results to clinical relevance is limited. However, expression of uptake and efflux drug transporters in these neuronal cells is similar to human dorsal root ganglion, supporting the use of these cells as a neuronal model for transporter experiments. Additionally, the retrospective evaluation of dose modification of paclitaxel due to sensory neuropathy is by nature exploratory. The association of treatment with P-gp inhibitors or the strong association of atorvastatin with risk of neuropathy requires validation and replication in additional cohorts of paclitaxel-treated patients, in which a detailed longitudinal evaluation of the neuropathy would be critical for an in-depth association analysis. It is unethical to assess potentially harmful drug-drug interactions in clinical trials, supporting the use of retrospective data as previously done for the drug-drug interaction between clopidogrel and paclitaxel [51].
In conclusion, paclitaxel was shown to cause neuronal toxicity in SH-SY5Y-derived neurons. Additionally, these neurons express the efflux transporter P-gp and inhibition of this transporter exacerbated neuronal toxicity of paclitaxel. These findings might provide a mechanistic basis for previous links between genetic variants in ABCB1 and paclitaxel-induced peripheral neuropathy and highlight a novel mechanism for drug-drug interactions in the peripheral nervous system. The finding that users of P-gp inhibitors and atorvastatin are potentially at a higher risk of paclitaxel-induced peripheral neuropathy highlight the need for further clinical studies of the mechanisms underlying this common and dose-limiting neurotoxicity.
Supplementary Material
Study Highlights.
What is the current knowledge on the topic?
The efflux transporter P-glycoprotein (P-gp) is well characterized for its ability to efflux chemotherapeutics from cancer cells and contribute to multidrug resistance. Genetic association studies suggest that P-gp might also play a role in paclitaxel-induced peripheral neuropathy.
What question did this study address?
This study characterized the role of P-gp in paclitaxel-induced neurotoxicity using neuronal cells in culture and examination of paclitaxel dose modifications in cancer patients concomitantly treated with P-gp inhibitors.
What does this study add to our knowledge?
The results of these studies support a critical role for P-gp in limiting paclitaxel-induced peripheral neuropathy. Inhibition of P-gp increasd paclitaxel-induced toxicity in SH-SY57-derived neurons, and patients treated with a P-gp inhibitor were at an increased risk of neuropathy-related modifications of paclitaxel dose.
How might this change clinical pharmacology or translational science?
Concomitant use of P-gp inhibitors and paclitaxel may increase the risk of paclitaxel-induced peripheral neuropathy in patients and highlights the importance of considering co-medications when assessing the risk of this dose-limiting toxicity.
Acknowledgements
We would like to acknowledge laboratory technician Birgitte Damby Sørensen for her work on determining intracellular paclitaxel concentrations in SH-SY5Y-derived neurons and Alex Sparreboom and Shuying Hu for supplying the human dorsal root ganglia for protein analysis and providing valuable input to the paper.
Funding:
This work was supported by grants from the Independent Research Fund Denmark (5053–00042B), A.P. Møller Foundation, National Cancer Institute (R01CA192156), Danish Cancer Society grant (R231-A13918) and Lundbeck Foundation (R307-2018-2980).
Footnotes
Conflicts of interest: Helen S. Hammer and Oliver Poetz are employees of Signatope. A provisional patent application related to chemotherapy-induced peripheral neuropathy has been filed by University of Southern Denmark with Tore Bjerregaard Stage listed as inventor. All other authors declared no competing interest for this work.
References
- 1.Bishop JF, Dewar J, Toner GC, Smith J, Tattersall MH, Olver IN, et al. Initial paclitaxel improves outcome compared with CMFP combination chemotherapy as front-line therapy in untreated metastatic breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 1999. August;17(8):2355–64. [DOI] [PubMed] [Google Scholar]
- 2.Lee JJ, Swain SM. Peripheral neuropathy induced by microtubule-stabilizing agents. J Clin Oncol Off J Am Soc Clin Oncol. 2006. April 1;24(10):1633–42. [DOI] [PubMed] [Google Scholar]
- 3.Park SB, Krishnan AV, Lin CS-Y, Goldstein D, Friedlander M, Kiernan MC. Mechanisms underlying chemotherapy-induced neurotoxicity and the potential for neuroprotective strategies. Curr Med Chem. 2008;15(29):3081–94. [DOI] [PubMed] [Google Scholar]
- 4.Jaggi AS, Singh N. Mechanisms in cancer-chemotherapeutic drugs-induced peripheral neuropathy. Toxicology. 2012. January 27;291(1–3):1–9. [DOI] [PubMed] [Google Scholar]
- 5.Hershman DL, Weimer LH, Wang A, Kranwinkel G, Brafman L, Fuentes D, et al. Association between patient reported outcomes and quantitative sensory tests for measuring long-term neurotoxicity in breast cancer survivors treated with adjuvant paclitaxel chemotherapy. Breast Cancer Res Treat. 2011. February;125(3):767–74. [DOI] [PubMed] [Google Scholar]
- 6.Mielke S, Sparreboom A, Mross K. Peripheral neuropathy: a persisting challenge in paclitaxel-based regimes. Eur J Cancer Oxf Engl 1990. 2006. January;42(1):24–30. [DOI] [PubMed] [Google Scholar]
- 7.Lagas JS, Vlaming ML, van Tellingen O, Wagenaar E, Jansen RS, Rosing H, et al. Multidrug resistance protein 2 is an important determinant of paclitaxel pharmacokinetics. Clin Cancer Res Off J Am Assoc Cancer Res. 2006. October 15;12(20 Pt 1):6125–32. [DOI] [PubMed] [Google Scholar]
- 8.Fellner S, Bauer B, Miller DS, Schaffrik M, Fankhänel M, Spruss T, et al. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J Clin Invest. 2002. November;110(9):1309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brooks TA, Minderman H, O’Loughlin KL, Pera P, Ojima I, Baer MR, et al. Taxane-based reversal agents modulate drug resistance mediated by P-glycoprotein, multidrug resistance protein, and breast cancer resistance protein. Mol Cancer Ther. 2003. November;2(11):1195–205. [PubMed] [Google Scholar]
- 10.Allen JD, Brinkhuis RF, van Deemter L, Wijnholds J, Schinkel AH. Extensive contribution of the multidrug transporters P-glycoprotein and Mrp1 to basal drug resistance. Cancer Res. 2000. October 15;60(20):5761–6. [PubMed] [Google Scholar]
- 11.Woo JS, Lee CH, Shim CK, Hwang S-J. Enhanced oral bioavailability of paclitaxel by coadministration of the P-glycoprotein inhibitor KR30031. Pharm Res. 2003. January;20(1):24–30. [DOI] [PubMed] [Google Scholar]
- 12.Huisman MT, Chhatta AA, van Tellingen O, Beijnen JH, Schinkel AH. MRP2 (ABCC2) transports taxanes and confers paclitaxel resistance and both processes are stimulated by probenecid. Int J Cancer. 2005. September 20;116(5):824–9. [DOI] [PubMed] [Google Scholar]
- 13.Hopper-Borge EA, Churchill T, Paulose C, Nicolas E, Jacobs JD, Ngo O, et al. Contribution of Abcc10 (Mrp7) to in vivo paclitaxel resistance as assessed in Abcc10−/− mice. Cancer Res [Internet]. 2011. May 15 [cited 2019 Jan 11];71(10):3649–57. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3096848/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith NF, Acharya MR, Desai N, Figg WD, Sparreboom A. Identification of OATP1B3 as a high-affinity hepatocellular transporter of paclitaxel. Cancer Biol Ther. 2005. August;4(8):815–8. [DOI] [PubMed] [Google Scholar]
- 15.Nieuweboer AJM, Hu S, Gui C, Hagenbuch B, Ghobadi Moghaddam-Helmantel IM, Gibson AA, et al. Influence of drug formulation on OATP1B-mediated transport of paclitaxel. Cancer Res. 2014. June 1;74(11):3137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leblanc AF, Sprowl JA, Alberti P, Chiorazzi A, Arnold WD, Gibson AA, et al. OATP1B2 deficiency protects against paclitaxel-induced neurotoxicity. J Clin Invest. 2018. February 1;128(2):816–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sissung TM, Mross K, Steinberg SM, Behringer D, Figg WD, Sparreboom A, et al. Association of ABCB1 genotypes with paclitaxel-mediated peripheral neuropathy and neutropenia. Eur J Cancer Oxf Engl 1990. 2006. November;42(17):2893–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abraham JE, Guo Q, Dorling L, Tyrer J, Ingle S, Hardy R, et al. Replication of genetic polymorphisms reported to be associated with taxane-related sensory neuropathy in patients with early breast cancer treated with Paclitaxel. Clin Cancer Res Off J Am Assoc Cancer Res. 2014. May 1;20(9):2466–75. [DOI] [PubMed] [Google Scholar]
- 19.Kus T, Aktas G, Kalender ME, Demiryurek AT, Ulasli M, Oztuzcu S, et al. Polymorphism of CYP3A4 and ABCB1 genes increase the risk of neuropathy in breast cancer patients treated with paclitaxel and docetaxel. OncoTargets Ther. 2016;9:5073–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanabe Y, Shimizu C, Hamada A, Hashimoto K, Ikeda K, Nishizawa D, et al. Paclitaxel-induced sensory peripheral neuropathy is associated with an ABCB1 single nucleotide polymorphism and older age in Japanese. Cancer Chemother Pharmacol. 2017. June;79(6):1179–86. [DOI] [PubMed] [Google Scholar]
- 21.Boora GK, Kanwar R, Kulkarni AA, Abyzov A, Sloan J, Ruddy KJ, et al. Testing of candidate single nucleotide variants associated with paclitaxel neuropathy in the trial NCCTG N08C1 (Alliance). Cancer Med. 2016. January 14; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakajima M, Fujiki Y, Kyo S, Kanaya T, Nakamura M, Maida Y, et al. Pharmacokinetics of paclitaxel in ovarian cancer patients and genetic polymorphisms of CYP2C8, CYP3A4, and MDR1. J Clin Pharmacol. 2005. June;45(6):674–82. [DOI] [PubMed] [Google Scholar]
- 23.Yamaguchi H, Hishinuma T, Endo N, Tsukamoto H, Kishikawa Y, Sato M, et al. Genetic variation in ABCB1 influences paclitaxel pharmacokinetics in Japanese patients with ovarian cancer. Int J Gynecol Cancer Off J Int Gynecol Cancer Soc. 2006. June;16(3):979–85. [DOI] [PubMed] [Google Scholar]
- 24.Gréen H, Söderkvist P, Rosenberg P, Mirghani RA, Rymark P, Lundqvist EA, et al. Pharmacogenetic studies of Paclitaxel in the treatment of ovarian cancer. Basic Clin Pharmacol Toxicol. 2009. February;104(2):130–7. [DOI] [PubMed] [Google Scholar]
- 25.Stage TB, Bergmann TK, Kroetz DL. Clinical Pharmacokinetics of Paclitaxel Monotherapy: An Updated Literature Review. Clin Pharmacokinet [Internet]. 2017. June 13 [cited 2017 Jun 14];1–13. Available from: https://link.springer.com/article/10.1007/s40262-017-0563-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cilla DD, Whitfield LR, Gibson DM, Sedman AJ, Posvar EL. Multiple-dose pharmacokinetics, pharmacodynamics, and safety of atorvastatin, an inhibitor of HMG-CoA reductase, in healthy subjects. Clin Pharmacol Ther. 1996. December;60(6):687–95. [DOI] [PubMed] [Google Scholar]
- 27.Shipley MM, Mangold CA, Szpara ML. Differentiation of the SH-SY5Y Human Neuroblastoma Cell Line. JoVE J Vis Exp [Internet]. 2016. February 17 [cited 2017 Jan 4];(108):e53193–e53193. Available from: http://www.jove.com/video/53193/differentiation-of-the-sh-sy5y-human-neuroblastoma-cell-line [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wegler C, Gaugaz FZ, Andersson TB, Wiśniewski JR, Busch D, Gröer C, et al. Variability in Mass Spectrometry-based Quantification of Clinically Relevant Drug Transporters and Drug Metabolizing Enzymes. Mol Pharm. 2017. 05;14(9):3142–51. [DOI] [PubMed] [Google Scholar]
- 29.Weiss F, Hammer HS, Klein K, Planatscher HP, Zanger UM, Noren A, et al. Direct Quantification of Cytochromes P450 and Drug Transporters - A Rapid, Targeted Mass Spectrometry-Based Immunoassay Panel for Tissues and Cell Culture Lysates. Drug Metab Dispos [Internet]. 2018. January 1 [cited 2018 May 24];dmd.117.078626. Available from: http://dmd.aspetjournals.org/content/early/2018/01/17/dmd.117.078626.1 [DOI] [PubMed] [Google Scholar]
- 30.Sánchez-Barroso L, Apellaniz-Ruiz M, Gutiérrez-Gutiérrez G, Santos M, Roldán-Romero JM, Curras M, et al. Concomitant Medications and Risk of Chemotherapy-Induced Peripheral Neuropathy. The Oncologist. 2018. November 23; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lund M, Petersen TS, Dalhoff KP. Clinical Implications of P-Glycoprotein Modulation in Drug-Drug Interactions. Drugs. 2017. May;77(8):859–83. [DOI] [PubMed] [Google Scholar]
- 32.Wessler JD, Grip LT, Mendell J, Giugliano RP. The P-glycoprotein transport system and cardiovascular drugs. J Am Coll Cardiol. 2013. June 25;61(25):2495–502. [DOI] [PubMed] [Google Scholar]
- 33.Patnaik A, Warner E, Michael M, Egorin MJ, Moore MJ, Siu LL, et al. Phase I dose-finding and pharmacokinetic study of paclitaxel and carboplatin with oral valspodar in patients with advanced solid tumors. J Clin Oncol Off J Am Soc Clin Oncol. 2000. November 1;18(21):3677–89. [DOI] [PubMed] [Google Scholar]
- 34.Advani R, Fisher GA, Lum BL, Hausdorff J, Halsey J, Litchman M, et al. A phase I trial of doxorubicin, paclitaxel, and valspodar (PSC 833), a modulator of multidrug resistance. Clin Cancer Res Off J Am Assoc Cancer Res. 2001. May;7(5):1221–9. [PubMed] [Google Scholar]
- 35.Chico I, Kang MH, Bergan R, Abraham J, Bakke S, Meadows B, et al. Phase I study of infusional paclitaxel in combination with the P-glycoprotein antagonist PSC 833. J Clin Oncol Off J Am Soc Clin Oncol. 2001. February 1;19(3):832–42. [DOI] [PubMed] [Google Scholar]
- 36.Fracasso PM, Westervelt P, Fears CL, Rosen DM, Zuhowski EG, Cazenave LA, et al. Phase I study of paclitaxel in combination with a multidrug resistance modulator, PSC 833 (Valspodar), in refractory malignancies. J Clin Oncol Off J Am Soc Clin Oncol. 2000. March;18(5):1124–34. [DOI] [PubMed] [Google Scholar]
- 37.ten Tije AJ, Synold TW, Spicer D, Verweij J, Doroshow JH, Sparreboom A. Effect of valspodar on the pharmacokinetics of unbound paclitaxel. Invest New Drugs. 2003. August;21(3):291–8. [DOI] [PubMed] [Google Scholar]
- 38.Saito T, Zhang ZJ, Ohtsubo T, Noda I, Shibamori Y, Yamamoto T, et al. Homozygous disruption of the mdrla P-glycoprotein gene affects blood-nerve barrier function in mice administered with neurotoxic drugs. Acta Otolaryngol (Stockh). 2001. September;121(6):735–42. [DOI] [PubMed] [Google Scholar]
- 39.Huang L, Li X, Roberts J, Janosky B, Lin M-HJ. Differential role of P-glycoprotein and breast cancer resistance protein in drug distribution into brain, CSF and peripheral nerve tissues in rats. Xenobiotica Fate Foreign Compd Biol Syst. 2015;45(6):547–55. [DOI] [PubMed] [Google Scholar]
- 40.Liu H, Chen Y, Huang L, Sun X, Fu T, Wu S, et al. Drug Distribution into Peripheral Nerve. J Pharmacol Exp Ther. 2018;365(2):336–45. [DOI] [PubMed] [Google Scholar]
- 41.Ceppi F, Langlois-Pelletier C, Gagné V, Rousseau J, Ciolino C, De Lorenzo S, et al. Polymorphisms of the vincristine pathway and response to treatment in children with childhood acute lymphoblastic leukemia. Pharmacogenomics. 2014. June;15(8):1105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sissung TM, Baum CE, Deeken J, Price DK, Aragon-Ching J, Steinberg SM, et al. ABCB1 genetic variation influences the toxicity and clinical outcome of patients with androgen-independent prostate cancer treated with docetaxel. Clin Cancer Res Off J Am Assoc Cancer Res. 2008. July 15;14(14):4543–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wijdeven RH, Pang B, Assaraf YG, Neefjes J. Old drugs, novel ways out: Drug resistance toward cytotoxic chemotherapeutics. Drug Resist Updat Rev Comment Antimicrob Anticancer Chemother. 2016;28:65–81. [DOI] [PubMed] [Google Scholar]
- 44.Kathawala RJ, Gupta P, Ashby CR, Chen Z-S. The modulation of ABC transporter-mediated multidrug resistance in cancer: A review of the past decade. Drug Resist Updat [Internet]. 2015. January 1 [cited 2019 Mar 1];18:1–17. Available from: http://www.sciencedirect.com/science/article/pii/S1368764614000788 [DOI] [PubMed] [Google Scholar]
- 45.Wang E, Casciano CN, Clement RP, Johnson WW. HMG-CoA reductase inhibitors (statins) characterized as direct inhibitors of P-glycoprotein. Pharm Res. 2001. June;18(6):800–6. [DOI] [PubMed] [Google Scholar]
- 46.Hochman JH, Pudvah N, Qiu J, Yamazaki M, Tang C, Lin JH, et al. Interactions of Human P-glycoprotein with Simvastatin, Simvastatin Acid, and Atorvastatin. Pharm Res [Internet] 2004. September 1 [cited 2019 Apr 3];21(9):1686–91. Available from: 10.1023/B:PHAM.0000041466.84653.8c [DOI] [PubMed] [Google Scholar]
- 47.Chen C, Mireles RJ, Campbell SD, Lin J, Mills JB, Xu JJ, et al. Differential interaction of 3-hydroxy-3-methylglutaryl-coa reductase inhibitors with ABCB1, ABCC2, and OATP1B1. Drug Metab Dispos Biol Fate Chem. 2005. April;33(4):537–46. [DOI] [PubMed] [Google Scholar]
- 48.Sakaeda T, Fujino H, Komoto C, Kakumoto M, Jin J-S, Iwaki K, et al. Effects of acid and lactone forms of eight HMG-CoA reductase inhibitors on CYP-mediated metabolism and MDR1-mediated transport. Pharm Res. 2006. March;23(3):506–12. [DOI] [PubMed] [Google Scholar]
- 49.Goard CA, Mather RG, Vinepal B, Clendening JW, Martirosyan A, Boutros PC, et al. Differential interactions between statins and P-glycoprotein: implications for exploiting statins as anticancer agents. Int J Cancer. 2010. December 15;127(12):2936–48. [DOI] [PubMed] [Google Scholar]
- 50.Wallace A, Chinn D, Rubin G. Taking simvastatin in the morning compared with in the evening: randomised controlled trial. BMJ [Internet]. 2003. October 4 [cited 2019 Apr 2];327(7418):788. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC214096/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Agergaard K, Mau-Sørensen M, Stage TB, Jørgensen TL, Hassel RE, Steffensen KD, et al. Clopidogrel paclitaxel drug-drug interaction: A pharmacoepidemiologic study. Clin Pharmacol Ther. 2017. February 22; [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.