

Abstract
Background
Left ventricular (LV) fibrofatty infiltration in arrhythmogenic right ventricular (RV) dysplasia/cardiomyopathy (ARVD/C) has been reported, however, detailed cardiovascular magnetic resonance (CMR) characteristics and association with outcomes are uncertain. We aim to describe LV findings on CMR in ARVD/C patients and their relationship with arrhythmic outcomes.
Methods
CMR of 73 subjects with ARVD/C according to the 2010 Task Force Criteria (TFC) were analyzed for LV involvement, defined as ≥ 1 of the following features: LV wall motion abnormality, LV late gadolinium enhancement (LGE), LV fat infiltration, or LV ejection fraction (LVEF) < 50%. Ventricular volumes and function, regional wall motion abnormalities, and the presence of ventricular fat or fibrosis were recorded. Findings on CMR were correlated with arrhythmic outcomes.
Results
Of the 73 subjects, 50.7% had CMR evidence for LV involvement. Proband status and advanced RV dysfunction were independently associated with LV abnormalities. The most common pattern of LV involvement was focal fatty infiltration in the sub-epicardium of the apicolateral LV with a “bite-like” pattern. LGE in the LV was found in the same distribution and most often had a linear appearance. LV involvement was more common with non-PKP2 genetic mutation variants, regardless of proband status. Only RV structural disease on CMR (HR 3.47, 95% CI 1.13–10.70) and prior arrhythmia (HR 2.85, 95% CI 1.33–6.10) were independently associated with arrhythmic events.
Conclusion
Among patients with 2010 TFC for ARVD/C, CMR evidence for LV abnormalities are seen in half of patients and typically manifest as fibrofatty infiltration in the subepicardium of the apicolateral wall and are not associated with arrhythmic outcomes.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12968-020-00702-3.
Introduction
Arrhythmogenic right ventricular (RV) dysplasia/cardiomyopathy (ARVD/C) is a rare, inherited non-ischemic cardiomyopathy characterized predominantly by progressive fibrofatty RV replacement, ventricular arrhythmias, and an increased risk for sudden cardiac death. The pathogenesis of ARVD/C has been linked to defects in genes that encode structural proteins necessary for normal myocyte cell–cell adhesion, the most common of which in North-America is the gene for plakophilin-2 (PKP2) [1]. Varying degrees of left-ventricular (LV) involvement can be seen in ARVD/C, which can take the form of decreased LV systolic function, chamber dilation, wall motion abnormalities (WMA, defined as regional dyskinesia, akinesia, or dyssynchronous contraction), fat infiltration, and fibrosis [2–4].
Prior studies have shown a link between reduced LV function on echocardiography and either cardiac mortality or arrhythmias in ARVC [3, 5–7]. However, the significance of fibrofatty changes in the LV wall is uncertain. Both fat replacement and late-gadolinium enhancement (LGE), indicating fibrosis, have been reported in the LV on cardiovascular magnetic resonance imaging (CMR). However, detailed description of the typical patterns and distributions of these findings and their association with outcomes are lacking. The purpose of this study was to investigate the prevalence, patterns, and clinical significance, with respect to arrhythmic outcomes, of LV fibrofatty replacement on CMR in a large North American cohort of ARVD/C patients.
Methods
Study population
This is a retrospective study of consecutive subjects enrolled in our institutional ARVD/C registry from 2002–2012. Detailed information on phenotyping, definitions of clinical variables, family history collection, and follow-up has been previously described [8]. Briefly, this is an observational, prospective, cohort study initiated in 1999 with annual follow-up. All individuals provided written informed consent. The study protocol was Health Insurance Portability and Accountability Act compliant and approved by the Johns Hopkins Institutional Review Board. All registry subjects with a diagnosis of ARVD/C based on 2010 Task Force Criteria (TFC) [9] and available CMR imaging (n = 102) were reviewed; 29 patients were excluded because they were missing essential CMR sequences for evaluation of both fat and LGE (Additional file 1: Table S1). Genotype and variant adjudication are summarized in the Online Supplement. CMR studies were scored as major, minor, or no TFC based on qualitative and quantitative results as defined by the 2010 TFC. The total number of TFC points were tallied, with 2 points for each major criteria and 1 point for each minor criteria. Details on CMR imaging protocol are included in Additional file 1.
CMR analysis
LV and RV volumes and function were analyzed on dedicated software QMASS (Medis, Leiden, The Netherlands). Measurements were performed by a single reader with two years of CMR experience blinded to clinical information. Endocardial margins of LV and RV were manually contoured on end- systolic and end- diastolic images. Papillary muscles were included in the blood pool volume. Volumes were indexed to body surface area (BSA). End systolic volume index (ESVI), end diastolic volume index (EDVI), and ejection fraction (EF) for both ventricles were obtained. The ventricles were also assessed for WMA, fatty infiltration and LGE in consensus by two blinded CMR readers with ten and five years of experience, respectively. The location of cardiac abnormalities was recorded based on a 17 segment American Heart Association LV model and a 5 segment RV model previously described [10]. LV fat infiltration was identified by high signal within the LV myocardium on fast spin echo proton density/T1 weighted images or high signal with India-ink etching artifact on balanced steady-state free precession images as seen in Fig. 1 [11, 12]. In regions with suspected sub-epicardial fat infiltration, a contour deformity on the epicardial side of the LV wall with regional LV wall thinning were required to distinguish epicardial fat outside the LV from fatty infiltration involving the myocardium. LGE was identified by regions of increased signal relative to nulled myocardium. The presence of increased fast spin echo and LGE signal in the same segment were considered areas of overlap of fat and LGE. For fat infiltration and LGE, both the intra-mural location within the myocardium (transmural, subendocardial, mid-myocardial, sub-epicardial) and the pattern of involvement (patchy, linear, scalloped/bite-like), were recorded (Fig. 1). Patients with more than one intramural location or pattern of involvement were scored for both. LV involvement was defined as the presence of ≥ 1 of the following features: LV wall motion abnormality, LV-LGE, LV fat infiltration or LV-EF < 50%.
Fig. 1.
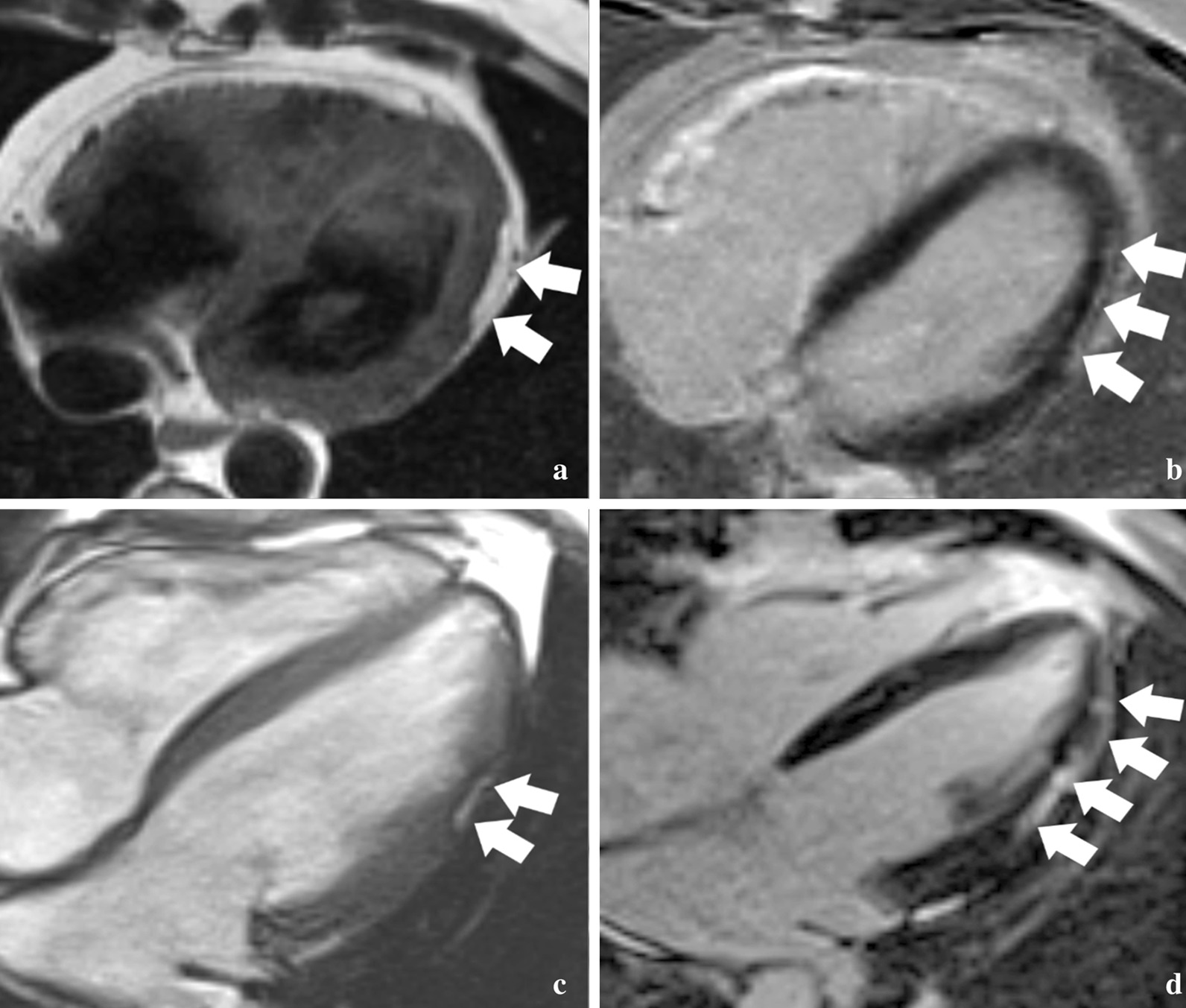
Patterns of left ventricular (LV) fat and fibrosis in arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C). a Axial dark blood fast spin echo image obtained in a 46 year-old male proband with a PKP2 variant shows a typical bite-like pattern of epicardial fat infiltration in the apicolateral LV (arrows). b Axial late gadolinium enhancement (LGE) image in the same patient shows linear subepicardial enhancement suggesting fibrosis in the same location. c Axial balanced steady-state free precession cine image from a 28 year-old male proband with a PKP2 variant shows a typical “bite-like” pattern of LV fat infiltration on bright blood images, with the associated dark etching artifact between the fat and the myocardium. d Axial LGE image in the same patient shows more extensive enhancement indicating fibrosis in a predominantly linear subepicardial pattern along the lateral wall
Outcomes
The primary (arrhythmic) outcome was the time-to-occurrence (from baseline CMR) of sustained ventricular arrhythmia, defined as a composite of spontaneous sustained ventricular tachycardia (lasting ≥ 30 s at ≥ 100 bpm or with hemodynamic compromise requiring cardioversion), aborted sudden cardiac death, sudden cardiac death, or appropriate implantable cardioverter-defibrillator (ICD) intervention (shock or anti-tachycardia pacing) for a ventricular arrhythmia. Follow-up duration was calculated from the date of CMR to the date of reaching the endpoint as above or censoring, which was defined as death from any other cause, heart transplantation, or the most recent follow-up at which the endpoint could be ascertained through review of medical records. Outcomes were adjudicated at our Precision Medicine Center of Excellence in Arrhythmogenic Right Ventricular Cardiomyopathy and Complex Arrhythmias. In patients without an ICD, ventricular tachycardia (VT)/ventricular fibrillation (VF) outcome was adjudicated based on reviewing electrocardiograms (ECGs) and medical records; in patients with an ICD, the device-stored ECGs were reviewed for appropriateness of ICD therapy using standard device interrogation according to following definitions: VF or ventricular flutter was defined as an irregular or regular tachycardia with a mean cycle length ≤ 240 ms. VT was defined as a regular tachycardia arising from the ventricle with a cycle length > 240 and < 600 ms. Decisions regarding ICD programming were made by the managing cardiovascular specialist. When complete ICD interrogation information or ECG tracings were not available, interpretation by the referring electrophysiologist was used to classify arrhythmic events. In patients with multiple endpoints, time-to-outcome was measured at the first event.
Statistical analysis
All continuous variables were reported as mean ± standard deviation and categorical variables as numbers (percentages). Continuous variables were compared using the independent Student t-test for normally distributed variables and Mann–Whitney U test for non-normally distributed variables. Categorical variables were compared using the Ficher’s exact test given sample size. Cumulative freedom from the composite arrhythmic outcome after the CMR exam was determined using multivariable Cox proportional-hazards models. All statistical analyses were performed using STATA (version 13, Stata Corporation, College Station, Texas, USA). A two-sided p-value of < 0.05 was considered statistically significant.
Results
Patient characteristics
The study population contained 73 patients with ARVD/C. Table 1 shows their clinical characteristics. Mean age of the study population was 34.2 ± 13.5 years, and 37 (50.7%) were male. A slight majority (n = 41, 56.2%) were probands, and the median number of TFC points was 6 (IQR 5–7). Of the study cohort, 35 (48%) carried an ARVD/C-associated pathogenic mutation, with PKP2 the most common (n = 30) mutation. Additional file 1: Table S2 summarizes genetic characteristics of study participants with an ARVC/D-associated mutation.
Table 1.
Demographic, clinical, and CMR data for the entire cohort, comparing subjects with and without left ventricular (LV) involvement at CMR
| All (n = 73) | No LV Involvement (n = 36) | LV Involvement (n = 37) | p-value | |
|---|---|---|---|---|
| Demographics and clinical data | ||||
| Age (years) | 34.2 ± 13.5 | 34.8 ± 15.7 | 33.6 ± 11.2 | 0.81 |
| Male sex (%) | 37 (50.7) | 16 (44.4) | 21 (56.8) | 0.35 |
| Mutation positive (%) | 35 (48.0) | 17 (47.2) | 18 (48.7) | 1.00 |
| PKP2 (%) | 30 (41.1) | 17 (47.2) | 13 (35.1) | 0.39 |
| DSP (%) | 1 (1.4) | 0 (0) | 1 (2.7) | |
| DSC2 (%) | 0 (0) | 0 (0) | 0 (0) | |
| DSG2 (%) | 1 (1.4) | 0 (0) | 1 (2.7) | |
| PLN (%) | 1 (1.4) | 0 (0) | 1 (2.7) | |
| Compounda (%) | 2 (2.7) | 0 (0) | 2 (5.4) | |
| Proband (%) | 41 (56.2) | 14 (39.0) | 27 (73.0) | 0.005 |
| TFC points | 6 (IQR 5–7) | 5 (IQR 4–6.5) | 7 (IQR 5–8) | 0.001 |
| CMR findings | ||||
| RVEDVI (mL/m2) | 106.7 ± 31.0 | 101.5 ± 26.1 | 111.9 ± 34.6 | 0.19 |
| RVEF (%) | 41.7 ± 10.0 | 46.3 ± 8.7 | 37.2 ± 9.2 | < 0.001 |
| LVEDVI (mL/m2) | 89.3 ± 19.8 | 87.9 ± 17.8 | 90.5 ± 21.6 | 0.45 |
| LVEF (%) | 54.1 ± 6.6 | 57.9 ± 4.3 | 50.3 ± 6.5 | < 0.001 |
| RV-LGE (% with any RV LGE) | 13 (17.8) | 6 (16.7) | 7 (18.9) | 1.00 |
| RV Fat (% with any RV fat) | 22 (30.1) | 9 (25.0) | 13 (35.1) | 0.45 |
| RV-WMA (% with any RV WMA) | 54 (74.0) | 20 (55.6) | 34 (91.9) | < 0.001 |
LV left ventricle, TFC task force criteria, CMR cardiovascular magnetic resonance imaging, RVEDVI right ventricular end-diastolic volume indexed to body surface area, RVEF right ventricular ejection fraction, LVEDVI LV end-diastolic volume indexed to body surface area, LVEF LV ejection fraction, RV-LGE right ventricular late gadolinium enhancement, RV fat right ventricular fat, RV-WMA right ventricular wall motion abnormality
aOne patient had both a PKP2 and a DSP mutation. A second patient had two different DSG2 mutations in trans (compound heterozygous)
Prevalence of LV involvement on CMR
Among the overall population, 37 (50.7%) patients had LV involvement. As seen in Table 1, patients with and without LV involvement were similar in age (p = 0.81) and sex (p = 0.35). Those with LV involvement were more likely to be probands and had significantly higher number of TFC points (5 [IQR 4–6.5] vs. 7 [IQR 5–8], p = 0.001), lower RV-EF (36.9 ± 9.4% vs. 45.2 ± 9.0%, p < 0.001) and more prevalent RV-WMAs (91.9% vs. 55.6%, p < 0.001). There was no difference in the likelihood of carrying an ARVD/C-associated genotype variant between patients with and without LV involvement, even after controlling for proband status.
Patterns of LV fat and LGE
Fat and LGE were most often localized in the apico-lateral, infero-basal and lateral walls of the LV (Fig. 2). LGE was typically sub-epicardial and linear in location and pattern, whereas fat was sub-epicardial but often had a scalloped/bite-like pattern (Fig. 1 and Table 2). The mid-myocardium was involved only half as frequently as the sub-epicardium with both fat and LGE, while the sub-endocardium was never involved. LV segments with isolated LGE (70/527, 13.2%) were more frequent than isolated fat (23/527, 4.3%), and LGE-fat overlap occurred in 53/527 (10.1%) of segments in patients with LV involvement.
Fig. 2.

Bullseye plot of the distribution of left ventricular (LV) fat (A) and late gadolinium enhancement (LGE) (B) by American Heart Association segments among the 31 ARVD/C subjects with LV abnormalities at CMR. As seen in the figures, Fat and LGE were most commonly localized in the apico-lateral, infero-basal and lateral walls of the LV
Table 2.
Typical patterns of left ventricular abnormalities among 37 subjects with LV involvement
| LGE | Fat | WMA | Low EF | |
|---|---|---|---|---|
| Subjects (%) | 27 (73.0) | 25 (67.6) | 9 (24.0) | 18 (48.7) |
| Most frequent American Heart Association segment involved | Apico-lateral (n = 18) | Apico-lateral (n = 19) | Mid-inferolateral (n = 6) | – |
| Myocardium involved | ||||
| Sub-endocarium (%) | 0 (0) | 0 (0) | – | – |
| Mid-myocardium (%) | 14 (45.2) | 8 (25.8) | – | – |
| Sub-epicardium (%) | 24 (77.4) | 17 (54.8) | – | – |
| Trans-mural (%) | 2 (6.5) | 0 (0) | – | – |
| Pattern | ||||
| Linear (%) | 21 (67.7) | 9 (29.0) | – | – |
| Patchy (%) | 6 (19.4) | 2 (6.5) | – | – |
| Bite/scalloped (%) | 5 (16.1) | 19 (61.3) | – | – |
EF ejection fraction, LGE late gadolinium enhancement, WMA wall motion abnormality
Genotype-specific LV changes
As seen in Table 3, there were no differences in RV size or function on CMR between all 3 groups (no mutation vs. PKP2 vs. non-PKP2 variants), but patients in the latter group had lower LVEF, more prevalent LV-LGE as well as LV-WMAs. With regards to fat patterns, 7/14 patients with “bite-like” fatty infiltration carried a mutation, all were PKP2 mutations. Only two patients had isolated LV involvement (without RV abnormalities), one with an extra-desmosomal mutation in the phospholamban (PLN) gene, and the other with a desmoplakin (DSP) mutation. Interestingly, both of these patients had extensive LV-LGE but no LV fat. Characteristics of the two patients are summarized in Additional file 1: Table S3.
Table 3.
Demographic, clinical, and CMR data for the entire cohort, comparing subjects with and without PKP2 variant
| All (n = 73) | No mutation (n = 38) | PKP2 mutation (n = 30) | Other mutationsa (n = 5) | p-value** | |
|---|---|---|---|---|---|
| Demographics and clinical data | |||||
| Age (years) | 34.2 ± 13.5 | 34.8 ± 13.6 | 33.7 ± 14.0 | 33.1 ± 12.8 | 0.934 |
| Male sex (%) | 37 (50.7) | 19 (50.0) | 18 (60.0) | 0 (0) | 0.045 |
| Proband (%) | 41 (56.2) | 25 (65.8) | 13 (43.3) | 3 (60.0) | 0.177 |
| VT prior to CMR | 29 (39.7) | 16 (42.1) | 12 (40.0) | 1 (20.0) | 0.637 |
| VT during follow-up | 37 (50.7) | 20 (52.6) | 16 (53.3) | 1 (20.0) | 0.363 |
| CMR findings | |||||
| RVEDVI (mL/m2) | 106.7 ± 31.0 | 111.8 ± 30.9 | 104.0 ± 30.7 | 84.8 ± 25.7 | 0.152 |
| RVEF (%) | 41.7 ± 10.0 | 41.6 ± 9.8 | 42.0 ± 10.7 | 40.0 ± 8.9 | 0.918 |
| LVEDVI (mL/m2) | 89.3 ± 19.8 | 93.2 ± 23.1 | 85.0 ± 14.5 | 87.2 ± 20.0 | 0.245 |
| LVEF (%) | 54.1 ± 6.6 | 54.7 ± 6.7 | 54.5 ± 5.9 | 47.3 ± 7.8 | 0.057 |
| LV-LGE (%) | 27 (37.0) | 15 (39.5) | 7 (23.3) | 5 (100.0) | 0.004 |
| LV Fat (%) | 25 (34.3) | 14 (36.8) | 9 (30.0) | 2 (40.0) | 0.808 |
| LV-WMA (%) | 9 (12.3) | 3 (7.9) | 2 (6.7) | 4 (80.0) | < 0.001 |
| Low LV-EF (< 50%) (%) | 18 (24.7) | 9 (23.7) | 6 (20.0) | 3 (60.0) | 0.155 |
LV left ventricle, TFC Task Force Criteria, RV-EDVI right ventricular end-diastolic volume indexed to body surface area, RVEF right ventricular ejection fraction, LV-EDVI LV end-diastolic volume indexed to body surface area, LVEF LV ejection fraction, RV-LGE right ventricular late gadolinium enhancement, RV fat right ventricular fat, RV-WMA right ventricular wall motion abnormality, VT ventricular tachycardia
**p-value derived from ANOVA test. No significant difference was found between the No Mutation and PKP2 Mutation groups on Rank-Sum or Ficher’s Exact tests for all the above demographic and CMR variables
aMutations include DSP, DSG2, DSC2, PLN, and compound non-PKP2 mutations. One patient had both a PKP2 and a DSP mutation and was included in PKP2 + ve group
Arrhythmic outcomes
Patients were followed for a mean 4.6 ± 2.9 years after baseline CMR. The majority (57/73, 78.1%) of patients had an ICD implanted during the follow-up period. The arrhythmic outcome was reached by 37 patients, all of whom underwent ICD implantation during the follow-up period. The most common outcome was ICD therapy which occurred in 25/73 (34.2%), followed by sustained VT (11/73, 15.1%), and a single life-vest shock (2.7%). Mean cycle length was available in 25 patients with arrhythmic outcomes and was 277.8 ± 56.5 ms. No deaths occurred during follow up.
Patients with arrhythmic outcomes were more often males, and had a history of VT (Table 4). No genotype differences were seen between patients with vs. without arrhythmias on follow-up. On CMR analysis, RV- and LVEDVI were greater and RV-EF lower in patients with arrhythmias, while LVEF was similar between the two groups. LV fat, RV-WMAs and RV-LGE were significantly more common in patients with arrhythmias, whereas RV fat and LV-WMAs were not. LV-LGE was more common in patients with arrhythmias although this did not achieve statistical significance. As such, patients experiencing arrhythmias were significantly more likely to fulfill major CMR-TFC compared to those without arrhythmias (92.2% vs. 35.9%, p < 0.001). On univariate Cox proportional-hazards analysis, male sex, history of prior sustained ventricular arrhythmia, CMR-TFC status (a marker of RV disease severity), and LV-LGE were associated with ventricular arrhythmias on follow-up. However, on multivariate analysis shown in Table 5, only a history of prior sustained ventricular arrhythmias and CMR-TFC were significantly associated with the outcome. Kaplan–Meier curves for arrhythmic outcomes stratified by CMR-TFC status, VT history at baseline, and LV involvement on CMR are shown in Fig. 3.
Table 4.
Comparison of demographic and CMR data for subjects with and without ventricular arrhythmias during follow-up
| Ventricular arrhythmia (n = 37) | No ventricular arrhythmia (n = 36) | p-value | |
|---|---|---|---|
| Demographics and clinical | |||
| Age (years) | 34.3 ± 13.6 | 34.2 ± 13.6 | 0.85 |
| Male sex (%) | 25 (67.6) | 12 (33.3) | 0.005 |
| Gene positive (%) | 17 (46.0) | 18 (50.0) | 0.82 |
| VT prior to CMR (%) | 25 (67.6) | 4 (11.1) | < 0.001 |
| RV CMR findings | |||
| RV WMA (%) | 34 (91.9) | 20 (55.6) | < 0.001 |
| RV Fat (%) | 14 (37.8) | 8 (22.2) | 0.20 |
| RV LGE (%) | 11 (29.7) | 2 (5.6) | 0.012 |
| RVEDVI (mL/m2) | 119.7 ± 30.2 | 93.4 ± 25.9 | < 0.001 |
| RVEF (%) | 37.5 ± 7.8 | 46.0 ± 10.3 | < 0.001 |
| LV CMR findings | |||
| LV WMA (%) | 5 (13.5) | 4 (11.1) | 1.000 |
| LV fat (%) | 17 (45.9) | 8 (22.2) | 0.048 |
| LV fat in a bite pattern (%) | 12 (32.4) | 2 (5.6) | 0.006 |
| LV LGE (%) | 18 (48.7) | 9 (25.0) | 0.052 |
| LV fat and/or LGE (%) | 20 (54.1) | 11 (30.6) | 0.059 |
| LVEDVI (mL/m2) | 93.8 ± 21.7 | 84.7 ± 16.6 | 0.011 |
| LVEF (%) | 53.8 ± 6.8 | 54.4 ± 6.5 | 0.45 |
| Low LVEF (< 50%) (%) | 10 (27.0) | 8 (22.2) | 0.79 |
| Any LV Involvement | 22 (59.5) | 15 (41.7) | 0.16 |
| CMR task force criteria | |||
| Any TFC for CMR (%) | 34 (92.2) | 13 (35.9) | < 0.001 |
| TFC for CMR major (%) | 30 (81.1) | 10 (27.8) | < 0.001 |
| TFC for CMR minor (%) | 4 (11.1) | 3 (8.1) | 1.000 |
| Total TFC Score | 7 (IQR 6–8) | 5 (IQR 5–6) | < 0.001 |
RV right ventricle, VT ventricular tachycardia, LV left ventricle, WMA wall motion abnormality, LGE late gadolinium enhancement, RVEDVI right ventricular end-diastolic volume indexed to body surface area, RVEF right ventricular ejection fraction, LVEDVI LV end-diastolic volume indexed to body surface area, LVEF LV ejection fraction, TFC task force criteria
Table 5.
Univariate and multivariate Cox regression analysis of the relationship between ventricular tachycardia events in follow-up and demographic and CMR findings
| Univariate | Multivariate | |||
|---|---|---|---|---|
| HR (95% CI) | p-value | HR (95% CI) | p-value | |
| Age | 0.99 (0.97–1.02) | 0.491 | – | – |
| Male sex | 2.20 (1.09–4.41) | 0.027 | – | – |
| Prior VT prior to CMR | 4.54 (2.24–9.22) | < 0.001 | 2.85 (1.33–6.10) | 0.007 |
| TFC for CMR (major or minor) | 5.85 (2.06–16.59) | 0.001 | 3.47 (1.13–10.70) | 0.030 |
| LV Fat | 1.75 (0.90–3.38) | 0.098 | – | – |
| LV Fat in a bite pattern | 1.99 (0.97–4.08) | 0.059 | – | – |
| LV LGE | 1.99 (1.03–3.84) | 0.040 | – | – |
| Low LVEF (< 50%) | 0.99 (0.47–2.11) | 0.979 | – | – |
TFC task force criteria, LV left ventricle, WMA wall motion abnormality, LGE late gadolinium enhancement
Fig. 3.

Kaplan–Meier survival curves for sustained ventricular arrhythmias in the study population stratified according to a history of sustained ventricular arrhythmias; b CMR Task Force Criteria (TFC) status (as a marker of right ventricular (RV) disease severity), and c LV Involvement on CMR. Survival curves illustrate the findings of our multivariate analysis that a history of prior arrhythmia as well as advanced RV—rather than LV—structural disease at baseline CMR independently portray increased risk of arrhythmic events during follow-up
We subdivided patients with preserved LVEF according to the presence or absence of tissue abnormalities on CMR. As seen in Supplemental Table 4, patients with detectable LV fibrofatty infiltrates on CMR had higher TFC score, and were more likely to be probands. The Kaplan–Meier Survival curve (Additional file 1: Figure S1) shows that they had higher rates of VT throughout the follow-up period without achieving statistical significance (log-rank p = 0.08).
Discussion
In this retrospective analysis of LV abnormalities in a large cohort of North American patients with ARVD/C we describe several important results. First, regional abnormalities in the LV, including fatty infiltration and fibrosis, were found in 51% of ARVD/C patients, were most often localized to the apico-lateral portion of the LV in a sub-epicardial distribution, were associated with more advanced disease, and were most prevalent in patients with non-PKP2 ARVD/C-associated variants. Second, these LV abnormalities had no significant independent association with arrhythmic events after adjusting for clinical variables and extent of RV disease. Third, the strongest risk factors for future arrhythmic events in this cohort were the presence of CMR evidence for RV structural abnormalities to meet CMR TFC and a history of ventricular arrhythmia.
LV fibrofatty infiltration—prevalence and patterns
The presence of fibrofatty infiltration in the LV of ARVD/C patients is a well-known phenomenon, initially described in the pathology literature from autopsy specimens. In the largest, autopsy-based multicenter study of 202 ARVD/C hearts after sudden cardiac death, there was histopathological LV involvement in 87% of cases, of which 17% had isolated LV changes [13]. Similarly, in another study of 42 ARVD/C hearts, LV fibrofatty changes were identified in 76% of cases, with a predilection for involvement of the sub-epicardial region of the LV wall [2]. This high frequency of LV abnormalities has led to the suggestion that the more generic and encompassing term, “arrhythmogenic cardiomyopathy” be used to describe this disease [1]. However, current TFC diagnostic criteria for CMR rely solely right-sided abnormalities, and as such we have continued to use the term ARVD/C for these patients. In the current study, we found LV-LGE in 37% of our cohort. In these cases, LV-LGE most often had a linear, sub-epicardial distribution, or, less frequently, a patchy, midmyocardial pattern. Previous LGE-CMR studies have non-invasively confirmed the sub-epicardial-predominant distribution of fibrosis in ARVD/C, with a variable prevalence in patients ranging from 14–84% [14–16]. Discrepancy in prevalence between CMR-based and autopsy-based studies may be due to lower sensitivity and resolution of CMR to detect microscopic fibrofatty infiltration of the cardiac muscle, as well as a selection bias in autopsy studies towards patients with more advanced disease who died of sudden cardiac death.
In most cases in our present study, fat and LGE were seen in the same segment, suggesting the process of fibrofatty replacement as reported by autopsy studies [2, 17, 18]. However, in some cases without regional overlap, fat identify and fibrosis were identified as separate entities. In general, LV fat on CMR has received less attention than LGE, being reported in a relatively small number of studies [16, 19]. Our study shows that similar to LGE, LV fat is most often found in the apico-lateral wall with a sub-epicardial distribution, in accord with prior reports [19, 20]. Interestingly, we found fat infiltration in the LV most often showed a unique scalloped or bite-like pattern. CMR patterns of LV-LGE and LV fat may have important relevance to the diagnosis of ARVD/C. Sarcoidosis is an important mimic of ARVD/C and can have significant overlap in clinical presentation, electrical findings, and extent of structural heart disease [21]. Indeed, the pattern of LV-LGE we have observed in this cohort (sub-epicardial location, linear pattern) has substantial overlap with the imaging pattern expected for sarcoidosis. To further add to potential diagnostic confusion, it is well known that in addition to the LV, the RV is frequently involved in sarcoidosis, manifested by both systolic dysfunction and LGE, and indeed is an important negative prognosticator for these patients [22]. In contradistinction to LGE, the bite-like pattern of fat infiltration we describe has never been reported for any disease besides ARVD/C, and as such, we believe it may have utility in distinguishing ARVD/C from sarcoidosis and other mimics. It is important to contrast this concept of LV fat at CMR in ARVD/C, which we hypothesize could prove a specific disease marker, with RV fat at CMR, which has been more thoroughly studied and is known to lack diagnostic sensitivity and specificity [23, 24], due to both overlap with non-ARVD/C entities [25] and challenges with imaging of the thin RV wall [26].
LV fibrofatty infiltration—genotype–phenotype determination
Previous studies have shown that certain ARVD/C variants are strongly associated with LV fibrofatty changes, regardless of proband status [27–29]. In mutation positive ARVD/C, DSP carriers are associated with four-fold higher likelihood of sudden cardiac death, LV dysfunction, and heart failure compared to PKP2 carriers [27]. Similarly, PLN, DSP, DSG2 and DSC2 carriers were found to have higher rates of LV dysfunction compared to PKP2 [27, 29, 30]. Similarly, in a study by Groeneweg et al., non-desmosomal PLN variants were more likely to have LV involvement compared with desmosomal mutation carriers [31]. In a study of 289 ARVC/D patients, Gilotra et al. did not find an association between odds of heart failure and presence of a genetic mutation; interestingly, all patients with more than one mutation in this study had CHF, and the DSP variant was more common in patients with LV dysfunction [5]. Despite the small number of non-PKP2 variants in our cohort, our findings align with prior reports such that non-PKP2 variants carriers had a higher likelihood of exhibiting LV disease such as WMAs, low LVEF, LV-LGE and LV-fat (Table 3). Interestingly, in 3 patients, a predominantly fibrotic pattern was seen with isolated LV-LGE and no LV fat. All were positive for gene variants that have been associated with the “left-sided” ARVD/C phenotype (PLN, DSP, DSG2) [28, 29, 32]. These findings further reinforce the concept of genotype–phenotype correlations for LV involvement in ARVD/C, with subepicardial fibrofatty LV changes seen among PKP2 mutation carriers and diffuse LV fibrosis and dysfunction among DSP mutation carriers [33].
Arrhythmic outcomes
LV abnormalities have been inconsistently associated with adverse outcomes in ARVD/C. Several European groups have shown a strong, independent association of LV dysfunction with cardiac death or life-threatening arrhythmias [2, 3, 7]. However, other studies of North American populations have not replicated these findings [8, 31, 34]. In 365 patients with definite ARVC/D from The Johns Hopkins ARVD/C Registry, LV dysfunction was not found to be a significant predictor of sustained VT/VF during follow-up [31]. In the largest multicenter and multinational cohort study to date (n = 976 patients), Cadrin-Tourigny et al. retrospectively demonstrated that LV EF on echocardiography is not a significant predictor of sustained ventricular arrhythmias after adjusting for RV dysfunction [35]. Similar to these findings, we found no difference in LV systolic function between patients with or without arrhythmic events at follow-up. Genotypic differences between European and North American ARVD/C populations may be driving these disparate findings with increased prevalence of patients with DSP variants in European cohorts [36]. These results reinforce a growing body of evidence suggesting that RV structural disease on CMR is of critical importance when assessing arrhythmic risk. In a recent study of ARVD/C mutation carriers, arrhythmic events only occurred in patients with structural disease in the RV, whereas those with isolated electrical abnormalities did not experience arrhythmic events at follow-up [37]. In another study from Deac et al. RV abnormalities identified on CMR had significant prognostic value; with a negative predictive value for future of events of 98.8% for a normal CMR study [38].
Pathophysiology of LV abnormalities in ARVC/D
The significance of these regional abnormalities of the LV wall, independent of global LV dysfunction, has not been previously described in imaging studies. A prior study has also reported inducible LV-based VTs during electrophysiological studies performed in ARVD/C patients. The VTs localized to the LV posterolateral wall, corresponding to the most common location of involvement of LV fat and fibrosis on CMR [10]. These findings raise the question of whether the identification of LV involvement by CMR has prognostic significance for determining future arrhythmic risk. Interestingly, in this current study we show that, although fibrosis and fat in the LV wall were seen more often in patients with arrhythmic events, there was no association with arrhythmic outcomes at follow-up after controlling for RV disease severity and history of arrhythmias prior to CMR. Pathophysiologically, it is important to distinguish between extensive LV scar involving a large portion of the LV and leading to heart failure, as opposed to localized and focal fibrofatty replacement leading to abnormal electrical conduction and sustained ventricular arrhythmias. On subgroup analysis, participants with preserved LV-EF and tissue abnormalities had higher rates of VT throughout follow-up compared to those with preserved LV-EF without tissue abnormalities, albeit not reaching statistical significance. As such, further and larger studies are needed to compare risks of life-threatening arrhythmias independently incurred by focal LV fibrofatty infiltration in patients with ARVD/C independent of global systolic dysfunction.
Limitations
Our study has several limitations. First, this is a retrospective study of a cohort of ARVD/C patients from a tertiary care center, which may result in referral bias, however, this is unavoidable given the rarity of this disease. Second, given the known regional/ethnic variability in ARVD/C-associated pathogenic mutations leading to unbalanced predominance of PKP2-positive patients in our North-American cohort, we are limited in our ability to draw statistically significant conclusions with regards to any genetic association in terms of LV involvement. Third, we do not have pathologic proof the LGE identified at CMR represents myocardial fibrosis, although this concept has been supported by a large body of literature in ischemic and non-ischemic cardiomyopathies [3]. Fourth, fat infiltration may result in increased signal on LGE-based CMR imaging, therefore some cases with presumed overlap of fat and fibrosis could represent predominantly fat infiltration. Fifth, we did not report the prevalence of focal "crinkling" of the RV wall (also known as “accordion sign”). This finding was described in mutation positive, asymptomatic first degree relatives of ARVC patients, highlighting the utility of this finding as a potential early sign of the disease [40]. The utility of this minor finding in probands with advanced global/regional structural disease is very limited, and the reproducibility of identifying the accordion sign in the presence of moderate to severe myocardial dilation, aneurysms and wall motion abnormalities is unknown. Finally, given that we have not performed endomyocardial biopsies we cannot exclude the possibility of prior subclinical myocarditis to explain prevalent LV tissue abnormalities.
Conclusions
In a North American cohort of ARVD/C patients, LV involvement was found in half of the patients. LV disease is most often characterized by fibrofatty infiltration in the sub-epicardium of the apico-lateral wall. Fat infiltration was often seen in a bite-like pattern, which may be a unique marker of ARVD/C. Although LV abnormalities were more common in patients experiencing arrhythmic events, there was no significant independent association with outcomes on multivariable analysis. History of prior arrhythmic event and extent of RV disease are the only significant predictors of arrhythmic events after CMR.
Supplementary information
Additional file 1. Cardiac MRI protocol and supplemental tables and figures.
Acknowledgements
The authors are grateful to the patients and families who made this work possible.
Abbreviations
- ARVD/C
Arrhythmogenic right ventricular dysplasia/cardiomyopathy
- BSA
Body surface area
- CMR
Cardiovascular magnetic resonance imaging
- DSG2
Desmoglein-2
- DSP
Desmoplakin
- EDVI
End diastolic volume index
- EF
Ejection fraction
- ECG
Electrocardiogram
- ESVI
End systolic volume index
- ICD
Implantable cardioverter defibrillator
- LGE
Late gadolinium enhancement
- LV
Left ventricle/left ventricular
- LVEF
Left ventricular ejection fraction
- PKP2
Plakophilin-2
- PLN
Phospholamban
- RV
Right ventricle/right ventricular
- RVEF
Right ventricular ejection fraction
- TFC
Task Force Criteria
- VF
Ventricular fibrillation
- VT
Ventricular tachycardia
- WMA
Wall motion abnormality(s)
Authors' contributions
ATR, CJ, NR, HT, HC, IK and SZ designed the study. TZ, ATR, CJ, NR, BM, CT, IK and SZ acquired the data. TZ, ATR, CJ, and SZ analyzed and interpreted the data. TZ, ATR, CJ and SZ were major contributors to writing the manuscript. MH, DB, HT, HC, IK and SZ provided substantial revisions to the manuscript. All authors read and approved the final manuscript.
Funding
The Johns Hopkins ARVD/C Program is supported the Dr. Francis P. Chiaramonte Private Foundation and Foundation Leducq (16 CVD 02) (all to HC). The Johns Hopkins ARVD/C Program is supported by the Leyla Erkan Family Fund for ARVD Research, the Dr. Francis P. Chiramonte Private Foundation, the Dr. Satish, Rupal, and Robin Shah ARVD Fund at Johns Hopkins, the Bogle Foundation, the Healing Hearts Foundation, the Campanella family, the Patrick J. Harrison Family, the Peter French Memorial Foundation, and the Wilmerding Endowments. The authors also wish to acknowledge funding from Foundation Leducq (16 CVD 02 to HC), the Netherlands Heart Foundation (2015T058 to AtR) and the Cardiovascular Research Netherlands CVON-PREDICT (Young Talent Program to AtR). Stefan L Zimmerman receives salary support as an advisor to Siemens Healthcare. The Johns Hopkins arrhythmogenic right ventricular cardiomyopathy/dysplasia program receives research support from Boston Scientific. The rest of the authors have no financial disclosures related to this present work. The sponsors or funders had no role in the design and conduct of the study, in the collection, analysis, and interpretation of the data, and in the preparation, review, or approval of the manuscript.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Saffitz JE. The pathobiology of arrhythmogenic cardiomyopathy. Annu Rev Pathol Mech Dis. 2011;6:299–321. doi: 10.1146/annurev-pathol-011110-130151. [DOI] [PubMed] [Google Scholar]
- 2.Silvestri F, Corrado D, McKenna WJ, Davies MJ, Wlodarska EK, Blomstrom-Lundqvist C, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 2002;30:1512–20. doi: 10.1016/s0735-1097(97)00332-x. [DOI] [PubMed] [Google Scholar]
- 3.Hulot JS, Jouven X, Empana JP, Frank R, Fontaine G. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2004;110:1879–84. doi: 10.1161/01.CIR.0000143375.93288.82. [DOI] [PubMed] [Google Scholar]
- 4.Rastegar N, Zimmerman SL, Te Riele ASJM, James C, Burt JR, Bhonsale A, et al. Spectrum of biventricular involvement on CMR among carriers of ARVD/C-associated mutations. JACC Cardiovasc Imag. 2015;8:863–4. doi: 10.1016/j.jcmg.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Gilotra NA, Bhonsale A, James CA, Te Riele ASJ, Murray B, Tichnell C, et al. Heart failure is common and under-recognized in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ Hear Fail. 2017;10:e003819. doi: 10.1161/CIRCHEARTFAILURE.116.003819. [DOI] [PubMed] [Google Scholar]
- 6.Hauer RNW, Te Riele ASJM, Mast TP, Teske AJ, Velthuis BK, Groeneweg JA, et al. Left ventricular involvement in arrhythmogenic right ventricular dysplasia/cardiomyopathy assessed by echocardiography predicts adverse clinical outcome. J Am Soc Echocardiogr. 2015;28(1103–1113):e9. doi: 10.1016/j.echo.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 7.Pinamonti B, Dragos AM, Pyxaras SA, Merlo M, Pivetta A, Barbati G, et al. Prognostic predictors in arrhythmogenic right ventricular cardiomyopathy: Results from a 10-year registry. Eur Heart J. 2011;32:1105–13. doi: 10.1093/eurheartj/ehr040. [DOI] [PubMed] [Google Scholar]
- 8.Bhonsale A, James CA, Tichnell C, Murray B, Madhavan S, Philips B, et al. Risk stratification in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. Circ Arrhythmia Electrophysiol. 2013;6:569–78. doi: 10.1161/CIRCEP.113.000233. [DOI] [PubMed] [Google Scholar]
- 9.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur Heart J. 2010;31:806–14. doi: 10.1093/eurheartj/ehq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Te Riele ASJM, James CA, Philips B, Rastegar N, Bhonsale A, Groeneweg JA, et al. Mutation-positive arrhythmogenic right ventricular dysplasia/ cardiomyopathy: The triangle of dysplasia displaced. J Cardiovasc Electrophysiol. 2013;24:1311–20. doi: 10.1111/jce.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aquaro GD, Nucifora G, Pederzoli L, Strata E, De Marchi D, Todiere G, et al. Fat in left ventricular myocardium assessed by steady-state free precession pulse sequences. Int J Cardiovasc Imaging. 2012;28:813–21. doi: 10.1007/s10554-011-9886-2. [DOI] [PubMed] [Google Scholar]
- 12.Strata E, Di Bella G, Lombardi M, Todiere G, Barison A, Aquaro GD. Usefulness of india ink artifact in steady-state free precession pulse sequences for detection and quantification of intramyocardial fat. J Magn Reson Imaging. 2013;40:126–32. doi: 10.1002/jmri.24335. [DOI] [PubMed] [Google Scholar]
- 13.Miles C, Finocchiaro G, Papadakis M, Gray B, Westaby J, Ensam B, et al. Sudden death and left ventricular involvement in arrhythmogenic cardiomyopathy. Circulation. 2019;118:037230. doi: 10.1161/CIRCULATIONAHA.118.037230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bisbal F, Alarcón F, Ferrero-de-Loma-Osorio A, González-Ferrer JJ, Alonso C, Pachón M, et al. Left atrial geometry and outcome of atrial fibrillation ablation: results from the multicentre LAGO-AF study. Eur Hear J. 2018;12:5. doi: 10.1093/ehjci/jey060. [DOI] [PubMed] [Google Scholar]
- 15.Moon J, Lee H-J, Yu J, Pak H-N, Ha J-W, Lee M-H, et al. Prognostic implication of left atrial sphericity in atrial fibrillation patients undergoing radiofrequency catheter ablation. Pacing Clin Electrophysiol. 2017;40:713–20. doi: 10.1111/pace.13088. [DOI] [PubMed] [Google Scholar]
- 16.Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115:1710–20. doi: 10.1161/CIRCULATIONAHA.106.660241. [DOI] [PubMed] [Google Scholar]
- 17.te Rijdt WP, de Weger RA, van der Heijden JF, Chamuleau SAJ, Harakalova M, van Es R, et al. High resolution systematic digital histological quantification of cardiac fibrosis and adipose tissue in phospholamban p.Arg14del mutation associated cardiomyopathy. PLoS One. 2014;9:e94820. doi: 10.1371/journal.pone.0094820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burke A, Oliveira JB, Cresswell N, Li L, Franco M, Fowler D, et al. Distribution of biventricular disease in arrhythmogenic cardiomyopathy: an autopsy study. Hum Pathol. 2011;43:592–6. doi: 10.1016/j.humpath.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 19.Judge DP, Amat N, Daly A, Tichnell C, Russell SD, Bluemke DA, et al. Morphologic variants of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2009;53:1289–99. doi: 10.1016/j.jacc.2008.12.045. [DOI] [PubMed] [Google Scholar]
- 20.Pillois X, Amraoui S, Frontera A, Wielandts J-Y, Haïssaguerre M, Sellal J-M, et al. Characterization of the left-sided substrate in arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythmia Electrophysiol. 2015;8:1403–12. doi: 10.1161/CIRCEP.115.003213. [DOI] [PubMed] [Google Scholar]
- 21.Madhavan S, Calkins H, Murray B, Tandri H, Nazarian S, Philips B, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy and cardiac sarcoidosis. Circ Arrhythmia Electrophysiol. 2014;7:230–6. doi: 10.1161/CIRCEP.113.000932. [DOI] [PubMed] [Google Scholar]
- 22.Greulich S, Mahrholdt H. Prognosis of myocardial damage in sarcoidosis patients with preserved left ventricular ejection fraction. Circ Cardiovasc Imag. 2016;9:e003738. doi: 10.1161/CIRCIMAGING.115.004417. [DOI] [PubMed] [Google Scholar]
- 23.Tandri H, Castillo E, Ferrari VA, Nasir K, Dalal D, Bomma C, et al. Magnetic resonance imaging of arrhythmogenic right ventricular dysplasia. sensitivity, specificity, and observer variability of fat detection versus functional analysis of the right ventricle. J Am Coll Cardiol. 2006;48:2277–84. doi: 10.1016/j.jacc.2006.07.051. [DOI] [PubMed] [Google Scholar]
- 24.Tandri H, Macedo R, Calkins H, Marcus F, Cannom D, Scheinman M, et al. Role of magnetic resonance imaging in arrhythmogenic right ventricular dysplasia: Insights from the North American arrhythmogenic right ventricular dysplasia (ARVD/C) study. Am Heart J. 2008;155:147–53. doi: 10.1016/j.ahj.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 25.Macedo R, Prakasa K, Tichnell C, Marcus F, Calkins H, Lima JAC, et al. Marked lipomatous infiltration of the right ventricle: MRI findings in relation to arrhythmogenic right ventricular dysplasia. AJR Am J Roentgenol. 2007;188:W423–7. doi: 10.2214/AJR.06.0161. [DOI] [PubMed] [Google Scholar]
- 26.Nasir K, Castillo E, Rutberg J, Lima JAC, Tandri H, Bluemke DA, et al. Arrhythmogenic right ventricular dysplasia: ex vivo and in vivo fat detection with black-blood MR imaging. Radiology. 2007;232:38–48. doi: 10.1148/radiol.2321030688. [DOI] [PubMed] [Google Scholar]
- 27.Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JDH, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J. 2015;36:847–55. doi: 10.1093/eurheartj/ehu509. [DOI] [PubMed] [Google Scholar]
- 28.Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, et al. Left-dominant arrhythmogenic cardiomyopathy. an under-recognized clinical entity. J Am Coll Cardiol. 2008;52:2175–87. doi: 10.1016/j.jacc.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 29.Syrris P, Ward D, Asimaki A, Evans A, Sen-Chowdhry S, Hughes SE, et al. Desmoglein-2 mutations in arrhythmogenic right ventricular cardiomyopathy: a genotype-phenotype characterization of familial disease. Eur Heart J. 2007;28:581–8. doi: 10.1093/eurheartj/ehl380. [DOI] [PubMed] [Google Scholar]
- 30.Fressart V, Duthoit G, Donal E, Probst V, Deharo JC, Chevalier P, et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace. 2010;12:861–8. doi: 10.1093/europace/euq104. [DOI] [PubMed] [Google Scholar]
- 31.Groeneweg JA, Van Der Zwaag PA, Olde Nordkamp LRA, Bikker H, Jongbloed JDH, Jongbloed R, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy according to revised 2010 task force criteria with inclusion of non-desmosomal phospholamban mutation carriers. Am J Cardiol. 2013;112:1197–206. doi: 10.1016/j.amjcard.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 32.Groeneweg JA, Van Der Zwaag PA, Jongbloed JDH, Cox MGPJ, Vreeker A, De Boer RA, et al. Left-dominant arrhythmogenic cardiomyopathy in a large family: associated desmosomal or nondesmosomal genotype? Hear Rhythm. 2013;10:548–59. doi: 10.1016/j.hrthm.2012.12.020. [DOI] [PubMed] [Google Scholar]
- 33.Te Riele ASJM, Bhonsale A, Burt JR, Zimmerman SL, Tandri H. Genotype-specific pattern of LV involvement in ARVD/C. JACC Cardiovasc Imag. 2012;5:849–51. doi: 10.1016/j.jcmg.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 34.McNitt S, Estes NAM, Gear K, Marcus F, Link MS, Zareba W, et al. Ventricular Arrhythmias in the North American Multidisciplinary Study of ARVC. J Am Coll Cardiol. 2014;64:119–25. doi: 10.1016/j.jacc.2014.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cadrin-Tourigny J, Bosman LP, Nozza A, Wang W, Tadros R, Bhonsale A, et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. Narnia. 2019;40:1850–8. doi: 10.1093/eurheartj/ehz103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basso C, Bauce B, Corrado D, Thiene G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2012;9:223–33. doi: 10.1038/nrcardio.2011.173. [DOI] [PubMed] [Google Scholar]
- 37.Madhavan S, Calkins H, Kamel IR, Tichnell C, Zimmerman SL, Bhonsale A, et al. Incremental value of cardiac magnetic resonance imaging in arrhythmic risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy–associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1761–9. doi: 10.1016/j.jacc.2012.11.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deac M, Alpendurada F, Fanaie F, Vimal R, Carpenter JP, Dawson A, et al. Prognostic value of cardiovascular magnetic resonance in patients with suspected arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol. 2013;168:3514–21. doi: 10.1016/j.ijcard.2013.04.208. [DOI] [PubMed] [Google Scholar]
- 39.Ordovas KG, Higgins CB. Delayed contrast enhancement on MR images of myocardium: past, present future. Radiology. 2011;261:358–74. doi: 10.1148/radiol.11091882. [DOI] [PubMed] [Google Scholar]
- 40.Dalal D, Tandri H, Judge DP, Amat N, Macedo R, Jain R, et al. Morphologic variants of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. A genetics-magnetic resonance imaging correlation study. J Am Coll Cardiol. 2009;53:1289–99. doi: 10.1016/j.jacc.2008.12.045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Cardiac MRI protocol and supplemental tables and figures.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.