

SUMMARY
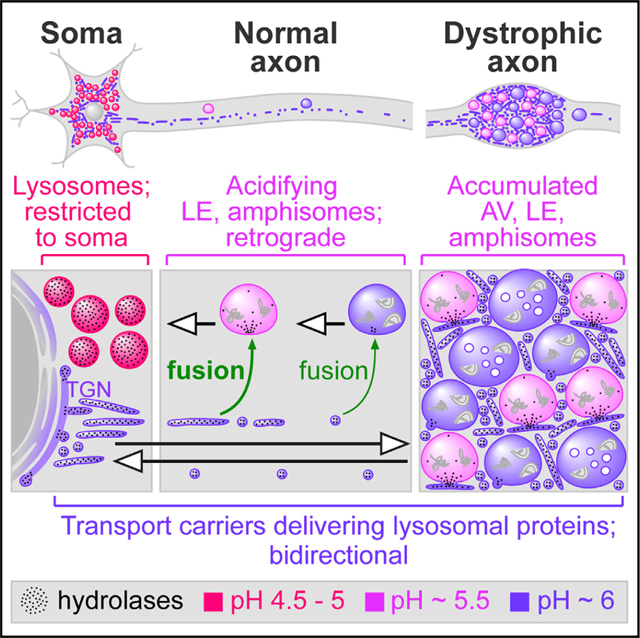
Lysosomal trafficking and maturation in neurons remain poorly understood and are unstudied in vivo despite high disease relevance. We generated neuron-specific transgenic mice to track vesicular CTSD acquisition, acidification, and traffic within the autophagic-lysosomal pathway in vivo, revealing that mature lysosomes are restricted from axons. Moreover, TGN-derived transport carriers (TCs), not lysosomes, supply lysosomal components to axonal organelles. Ultrastructurally distinctive TCs containing TGN and lysosomal markers enter axons, engaging autophagic vacuoles and late endosomes. This process is markedly upregulated in dystrophic axons of Alzheimer models. In cultured neurons, most axonal LAMP1 vesicles are weakly acidic TCs that shuttle lysosomal components bidirectionally, conferring limited degradative capability to retrograde organelles before they mature fully to lysosomes within perikarya. The minor LAMP1 subpopulation attaining robust acidification are retrograde Rab7+ endosomes/amphisomes, not lysosomes. Restricted lysosome entry into axons explains the unique lysosome distribution in neurons and their vulnerability toward neuritic dystrophy in disease.
In brief
Lie et al. generate mouse models to demonstrate that, in vivo, fully mature lysosomes are restricted from axons. Instead, TGN-derived transport carriers supply lysosomal components to retrogradely transported pre-degradative vesicular compartments in axons for their maturation toward lysosomal identity, revealing insights into fundamental neurobiology of lysosomes in health and disease.
Graphical Abstract
INTRODUCTION
Survival of neurons requires the efficient clearance of substrates in lysosomal degradative processes, including autophagy and endocytic pathways (Damme et al., 2015; Hara et al., 2006; Komatsu et al., 2006; Menzies et al., 2015; Nixon, 2013; Yamamoto and Yue, 2014). The lysosomal system has emerged as a prominent target in the etiology of neurodegenerative diseases (Abeliovich and Gitler, 2016; Colacurcio and Nixon, 2016; Lie and Nixon, 2019). Classically defined lysosomes are fully mature with complete degradative capability and have unique vulnerability to functional disruptions (Nixon, 2016). Importantly, their disease relevance is distinct from that of partially degradative organelles with limited hydrolase activity and destined to mature toward lysosomal identity. Despite active research on the neurobiology of lysosomes, much of the current knowledge remains conjectural and molecular mechanisms of lysosomal dysfunction underlying neurodegenerative diseases are poorly understood.
Functionally mature lysosomes are classically defined based on four cardinal features, namely, a high degree of acidification (pH 4.5—5), high levels of lysosomal membrane proteins and lysosomal hydrolases, and a lack of mannose 6-phosphate receptors (MPRs) (Saftig and Klumperman, 2009). Substrates targeted for lysosomal degradation traverse the autophagy- or endo-lysosomal pathways via progressively maturing autophagic vacuoles (AVs) and late endosomes (LEs), respectively, which receive lysosomal components from the biosynthetic pathway via post-Golgi trafficking routes that may be direct (Golgi-to-endosome) or indirect (via plasma membrane and re-internalization) (Saftig and Klumperman, 2009).
In neurons, axonal endosomes and autophagosomes acquire Rab7 and LAMP1 while maturing into LEs and amphisomes, which then undergo progressive acidification and further maturation toward lysosomal identity during their retrograde transport (Cheng et al., 2015; Deinhardt et al., 2006; Kuijpers et al., 2020; Lee et al., 2011; Maday et al., 2012; Overly and Hollenbeck, 1996). This is consistent with the observed gradation of increasing acidification of endocytic organelles from distal to proximal axon (Overly and Hollenbeck, 1996) and enriched localization of fully mature lysosomes in the perinuclear area of neuronal cell bodies (Overly et al., 1995; Parton et al., 1992).
While lysosomal biogenesis and maturation in neurons share considerable similarities with the corresponding processes in non-neuronal cells, surprisingly little is known about key neuron-specific features. Potential adaptations in biosynthetic and lysosomal trafficking routes and steady-state organelle distribution that accommodate the unique architecture of neurons remain unclear. These fundamental issues are obscured by unvalidated concepts based on inadequate experimental designs or inconsistent adherence to stringent criteria for identifying lysosomes.
There is no unanimous agreement on lysosomal distribution and motility in axons. Studies in non-neuronal cells have uncovered an impressive amount of mechanistic details on the regulation of lysosomal positioning by BORC, SKIP, Arl8, and kinesin-mediated anterograde trafficking of lysosomes (Pu et al., 2015, 2017). These findings have recently been extended to neurons. Lysosomes are proposed to undergo bidirectional axonal transport with a large proportion of anterograde vesicles (Farías et al., 2017; Snouwaert et al., 2018), conflicting with the observation that acidic compartments undergo predominant retrograde transport (Gowrishankar et al., 2017). It is unclear what fraction (if any) of the anterograde “lysosomes” are authentic functionally mature degradative lysosomes, because most of those findings are based on the use of markers with poor specificity/selectivity. Especially problematic is the common use of LAMP1 as the sole lysosomal marker because LAMP1 is present on many non-lysosomal compartments (Vukoja et al., 2018; Yu et al., 2010), and LAMP1 acquisition by autophagosomes is temporally separate from acidification (Evans and Holzbaur, 2020). Adding to the confusion, many studies have over-simplified the nomenclature by generalizing a category of endosomal-lysosomal compartments to be equivalent to “lysosomes,” and their inaccurate identity as “lysosomes” is often inadvertently propagated by other investigators.
In addition to the lysosomal misidentification issue, another important but often overlooked issue is the reliance on in vitro analyses of cultured neurons and the assumption that they accurately represent intact, fully developed adult brains. Lysosomal maturation and trafficking have not been systematically studied in intact mammalian brains. The potential differences between vesicular dynamics defined in immature cultured neurons with pathfinding axons and those occurring in the mature intact brain have been rarely addressed. Our study addresses these two issues by using a unique combination of in vivo and in vitro approaches to comprehensively characterize the distribution, axonal trafficking, and functional features of both biosynthetic and degradative organelles in the process of lysosomal maturation in neurons. Our findings reveal insights into the fundamental neurobiology of lysosomes, which have important implications for understanding neurodegenerative disease processes such as the hallmark neuritic dystrophy of Alzheimer disease (AD).
RESULTS
Mature autolysosomes and lysosomes are restricted from axons in vivo
We assessed the distribution pattern of organelles in the autophagy-lysosomal pathway of neurons in vivo by visualizing autophagosome/autolysosome acidification and lysosomal hydrolase acquisition in intact adult mouse brains using two mouse models: (1) human cathepsin D transgenic mice (hCTSD Tg) expressing hCTSD specifically in neurons under the Thy1 promoter (Figures 1 and S1) and (2) hCTSD Tg crossed with our recently developed TRGL mice (Lee et al., 2019) expressing tandem fluorescent mRFP-eGFP-LC3 under Thy1 (Figure 1). The rationale for examining exogenous hCTSD expressed by neurons instead of endogenous CTSD is to circumvent the difficulty in distinguishing endogenous CTSD puncta within axons from those in glia, especially oligodendrocytes that are closely associated with axons. The expression of hCTSD in Tg mouse brains was confirmed by the CD5 antibody that specifically reacts with hCTSD (Figure S1A) but not mouse CTSD (mCTSD) (Figures S1A, S1B, S1D, and S1E). Normal function of the expressed hCTSD was demonstrated by CTSD enzyme activity assay (Figure S1C). Comparable targeting of hCTSD to that of endogenous mCTSD was verified by a high colocalization between hCTSD and mCTSD (Figure S1E). Besides minor enhancement in lysosomal clearance of LC3-II (Figure S1F), hCTSD expression did not affect levels of other cathepsins, autophagy induction, or survival of neurons and other cell types (Figures S1B and S1F–S1H), validating hCTSD Tg as a useful model for accurate tracking of CTSD-containing compartments in neurons in vivo. In hCTSD Tg mouse brains, we visualized hCTSD and endogenous mCTSD by DAB immunostaining. Both hCTSD and mCTSD puncta were highly enriched in somatodendritic regions in layer 5 cortical neurons and hippocampal CA1 neurons (Figures 1A and 1B), demonstrating that CTSD is efficiently detected by these antibodies and labeling conditions. Surprisingly, hCTSD puncta were not readily detectable in axonal fibers within corpus callosum (Figures 1A and 1B), striatum, and cerebellum (Figure 1C). In contrast, numerous endogenous mCTSD puncta were found near or overlapping with axon fibers in corpus callosum (Figures 1A and 1B), indicating that analyses of endogenous CTSD in axons would yield many false-positive puncta from the abundant glia-derived CTSD (Xue et al., 2014). It is noted that we deliberately used stringent labeling conditions to ensure signal fidelity for differentiating neuronal hCTSD from endogenous mCTSD. The intention is to highlight the pitfall of falsely identifying glia-derived endogenous mCTSD signals as axonal signals. Under these conditions, CTSD-containing transport carriers (TCs) in axons were not readily detectable although their presence will be demonstrated by more sensitive methods below: namely, immunofluorescence labeling (Figure 1F) and immuno-electron microscopy (Figures 2B–2D). Importantly, lysosomes, which are readily and comparably detectable in glial cells under the different labeling conditions, are not detectable in axons.
Figure 1. Fully mature autolysosomes and lysosomes are restricted from axons in vivo.
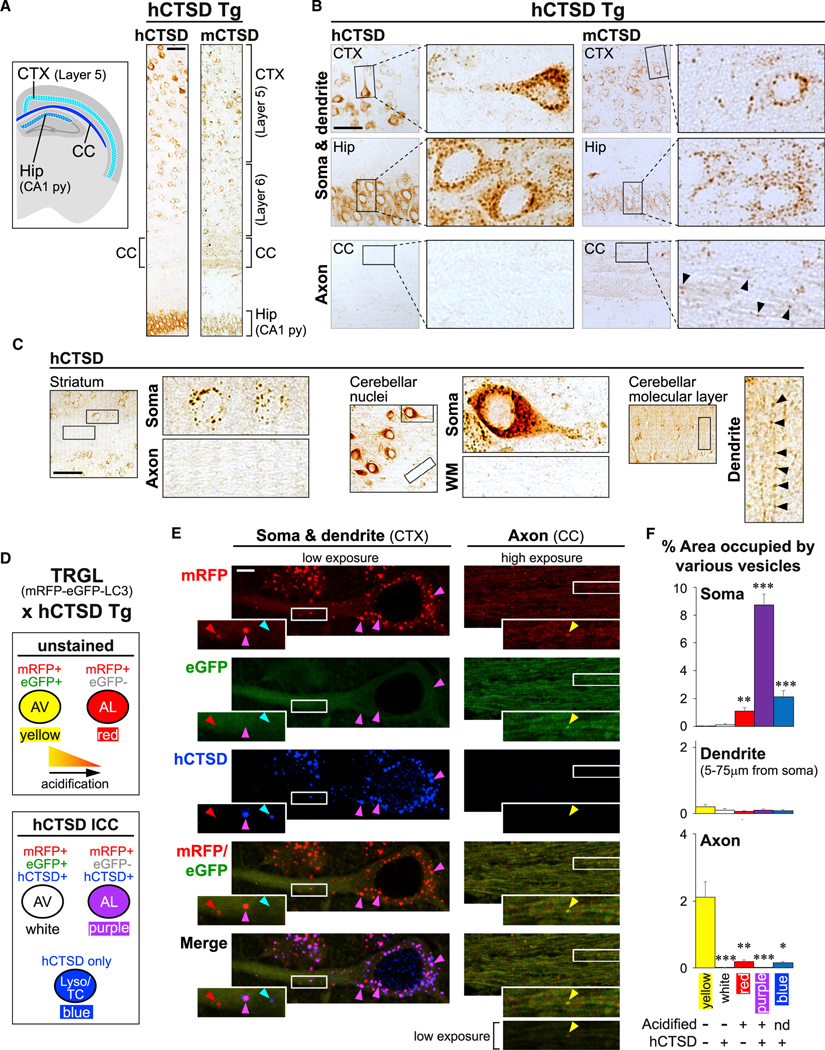
(A–C) Human CTSD transgenic (hCTSD Tg) mouse model expressing hCTSD specifically in neurons. (A) Schematic shows brain regions of interest. Tiled images on the right show DAB immunostaining against hCTSD or endogenous mouse CTSD (mCTSD) in hCTSD Tg brain sections. Scale bar, 100 μm. CTX, cortex; CC, corpus callosum; Hip, hippocampus; py, pyramidal cell layer. (B and C) Images showing hCTSD or mCTSD signals in specified brain regions. Magnified views of boxed areas are shown next to each low-magnification image. Arrowheads mark punctate signals within neurites. Scale bars, 100 μm. WM, white matter.
(D–F) Double transgenic TRGL × hCTSD Tg mouse model expressing mRFP-eGFP-LC3 and hCTSD specifically in neurons. (D) Key for (E) and (F) explaining colors of different vesicle types after merging mRFP (red), eGFP (green, quenched by acidification), and immunostained hCTSD (blue). AV, autophagic vacuole; AL, autolysosome; Lyso, lysosome; TC, transport carrier. (E) Images of mRFP-eGFP-LC3 and immunostained hCTSD in CTX and CC of double transgenic brain sections. Magnified views of boxed areas are shown as insets. All axon images are shown at increased brightness (consistent across all channels) except for bottom image (original brightness shown as reference). Scale bar, 5 μm. (F) Graphs showing percentage of area in soma, dendrite, and axon occupied by different vesicle types. Bars, mean ± SEM (n = 28 soma, 25 dendrites, 24 high-magnification axonal images: dimensions = 53 × 18 μm, each contains >20 axonal segments). *p < 0.05, **p < 0.01, ***p < 0.0001, Kruskal-Wallis test followed by Dunn’s multiple comparisons against “yellow.” See also Figure S1.
Figure 2. Lysosomes with classic ultrastructure are restricted from axons in vivo.

(A–D) Electron micrographs of immunogold labeling of TGN38 (10 nm gold, cyan arrowheads) alone or co-labeled with CTSD or hCTSD (6 nm gold, magenta arrowheads) in wild-type and hCTSD Tg mouse brains, respectively. (A) Top left, overview of a soma within the cortex (CTX). Magnified view of white box (inset) and additional image on right show Golgi cisternae and associated vesicles. Black boxes (a, b) are magnified below (a’, b’). Scale bars, white = 500 nm; black = 100 nm. N, nucleus; Lyso, lysosome; LE, late endosome, MVB, multivesicular body. (B, D) Low-magnification top images show regions within myelinated axons in corpus callosum (CC). Magnified views of white boxes and additional high-magnification images show TGN-derived transport carriers (TCs) with TGN38 and CTSD labeling. Scale bars, 100 nm. (C) Quantification of CTSD and TGN38 immunogold densities on various organelles. Bars, mean ± SEM (n = no. of organelles indicated above bars; pooled data from single and double labeling). **p < 0.01, Kruskal-Wallis test followed by Dunn’s multiple comparisons against mitochondria (Mito) as negative Ctrl. AV, autophagic vacuole.
(E–H) Electron micrographs of histochemical ACPase labeling (green arrowheads/asterisks) in wild-type mouse brains. (E) Overview of a somal region within the CTX with magnified view of white boxes in insets and an additional image on right showing an ACPase signal in lysosomes and Golgi. Scale bars, 500 nm. (F) Tiled images on top show overview of a somal region with the attached neurite. White boxes (a–d) are magnified below (a’–d’). Scale bars, 500 nm. (G and H) Low-magnification images on top show regions within myelinated axons in CC. Magnified views of white boxes and additional high-magnification images show TCs and AVs with an ACPase signal. Scale bars,100 nm. (H) White arrows, double membrane of AVs; green arrowheads, ACPase engaged to exterior of AVs; green asterisks, ACPase incorporated into AVs. See also Figure S2.
In TRGL × hCTSD Tg mice, upon acidification and maturation of mRFP-eGFP-LC3-containing immature AVs (autophagosomes and early amphisomes) into autolysosomes, mRFP fluorescence remains stable, but eGFP fluorescence is quenched by the acidic lumenal pH, with only ~10% signal retention at pH 5 and complete quenching at or below pH 4 (Bomati et al., 2014) (Figure 1D). Integrity of the mRFP-eGFP-LC3 protein in brains has been demonstrated previously (Lee et al., 2019). We visualized mRFP-eGFP-LC3 and hCTSD signals in somatodendritic regions of layer 5 cortical neurons and their long axons within the corpus callosum (Figure 1E). Most somal LC3 vesicles were acidified and hCTSD+ autolysosomes (purple puncta) (Figures 1D–1F). In contrast, corpus callosal axons did not contain autolysosomes but were occupied predominantly by poorly acidified autophagosomes/amphisomes without an hCTSD signal (yellow puncta) (Figures 1D–1F). In addition, somas were enriched in hCTSD vesicles lacking an LC3 signal (blue puncta, lysosomes, and CTSD-containing TCs as we later show). In axons, puncta with only an hCTSD signal were much rarer, although a small number could be found (Figure 1F), and these will later be demonstrated to be TCs delivering CTSD, and not lysosomes. Proximal dendrites contained relatively few autophagic compartments at various stages of maturation (Figures 1D–1F), reflecting a continuous transition between soma and dendrites. Quantification in Figure 1F reflects the percentage volume within each subcellular region (i.e., soma, dendrite, and axon) being occupied by these different types of puncta. This is represented by percentage of area occupied in confocal images with the same optical section thickness. These results demonstrate that mature autolysosomes and lysosomes are restricted from axons of intact adult brains.
In vivo, axons lack lysosomes identified by their classic ultrastructural features but contain TGN-derived TCs and maturing AVs and LEs
By electron microscopy, we examined somal and axonal organelles containing lysosomal hydrolases CTSD (immunogold, Figures 2A–2D) and lysosomal acid phosphatase (ACPase) (histochemistry, Figures 2E–2H) in mouse brains. In soma, CTSD and ACPase were not only enriched in lysosomes with classic ultrastructural features (round shaped with amorphous electron-dense lumenal content and diameter of ~0.2–0.5 μm) but also in Golgi cisternae and vesicles budding from the TGN (Figures 2A, 2C, and 2E). Thus, we co-immunolabeled CTSD with TGN38 (murine ortholog of human TGN46), a TGN marker and putative cargo adaptor cycling between the TGN and the plasma membrane or endosomes (Banting and Ponnambalam, 1997) (Figures 2A–2D). The two types of gold particles (6 and 10 nm) used for co-labeling each showed a range of apparent diameters with a minor overlap between the two populations (Figure S2). Particles with an ambiguous diameter were excluded from our analyses in co-labeling experiments. Specificity of CTSD and TGN38 labeling was verified by the density of immunogold particles on various organelles (Figure 2C). TGN38 colocalized with CTSD on somal TGN-associated structures but not lysosomes (Figures 2A and 2C). In corpus callosum, axons lacked lysosomes but CTSD (endogenous mCTSD in wild-type [WT] or hCTSD in Tg brains) colocalized with TGN38 in tubulovesicular structures with morphology resembling TGN-derived TCs (Figures 2B–2D) (Luini et al., 2008). These structures were 30– 60 nm wide with variable lengths up to a few microns. Similarly, ACPase-containing lysosomes were occasionally detected in the most proximal axonal segment immediately adjacent to the cell body (Figure 2F) but not at more distal levels within the corpus callosum (Figures 2G and 2H). Instead, axonal ACPase was localized to tubulovesicular compartments (Figures 2F and 2G) with similar dimensions as TCs containing CTSD and TGN38 (Figures 2B and 2D). We were unable to perform double labeling of ACPase and TGN38 due to the lack of suitable ACPase antibody. LEs and AVs at different stages of maturation were also decorated by CTSD, TGN38, and ACPase in both soma and axon (Figures 2A, 2C, and 2H). Unlike the complete infiltration of ACPase in lysosomal lumens (Figure 2E), labeled AVs showed patchy ACPase staining that suggests the incomplete introduction of newly acquired lysosomal hydrolase within these maturing organelles (Figure 2H). These AVs were globular in shape with a diameter of at least 100 nm, making them easily distinguishable from the tubulovesicular TCs. In some images (Figure 2H, green arrowheads), TC puncta appeared to engage the outer membrane surface of AVs. These ultrastructural results further demonstrate that classic fully mature lysosomes are restricted from axons under physiological conditions in intact brains. Instead, TCs are mobilized to supply axonal pre-/partially degradative organelles with lysosomal hydrolases.
We further examined the modulation of ACPase distribution under the pathological condition of neuritic dystrophy in PS/ APP mouse brains (Figure 3). PS/APP mice, which express the AD-linked PSEN1 M146L and APP Swedish mutations, manifest characteristic AD pathology of focal swellings in dystrophic neurites with massive accumulation of AVs containing undegraded substrates (Yang et al., 2008). PS/APP neurons showed an upregulated expression of Golgi-associated ACPase, which could be detected in multiple cisternae in each Golgi stack, as opposed to WT neurons that usually showed ACPase reactivity in only one cisterna (Figure 3A) (Cataldo et al., 2004). Accordingly, the percentage of area in Golgi stacks occupied by an ACPase signal was significantly increased (Figure 3A). ACPase also accumulated in focal swellings of dystrophic neurites associated with plaques within cortical neuropil and in myelinated dystrophic axons within the corpus callosum (Figures 3B and 3C). Higher magnification of dystrophic neurites revealed that many ACPase+ tubulovesicular TCs were tightly packed between accumulated AVs and occasionally LEs, engaging with and often partially fused with these degradative organelles containing undigested substrates; however, no mature lysosomes were detected (Figure 3C). By immuno-electron microscopy, we demonstrated fusion between accumulated AVs and tubulovesicular TCs containing TGN38 and CTSD, further supporting the TGN origin of TCs delivering lysosomal components (Figure 3D). These results demonstrate that neurons respond to massive AV accumulation in dystrophic neurites by increasing the transcription (Bordi et al., 2016; Cataldo et al., 1995) and production (Figure 3A) (Cataldo et al., 2004) of lysosomal hydrolases and lysosomal components and increasing their delivery via TCs to stalled AVs and LEs (Figures 3B–3D).
Figure 3. TGN-derived transport carriers (TCs), not lysosomes, accumulate in dystrophic axons in PS/APP mouse brains.
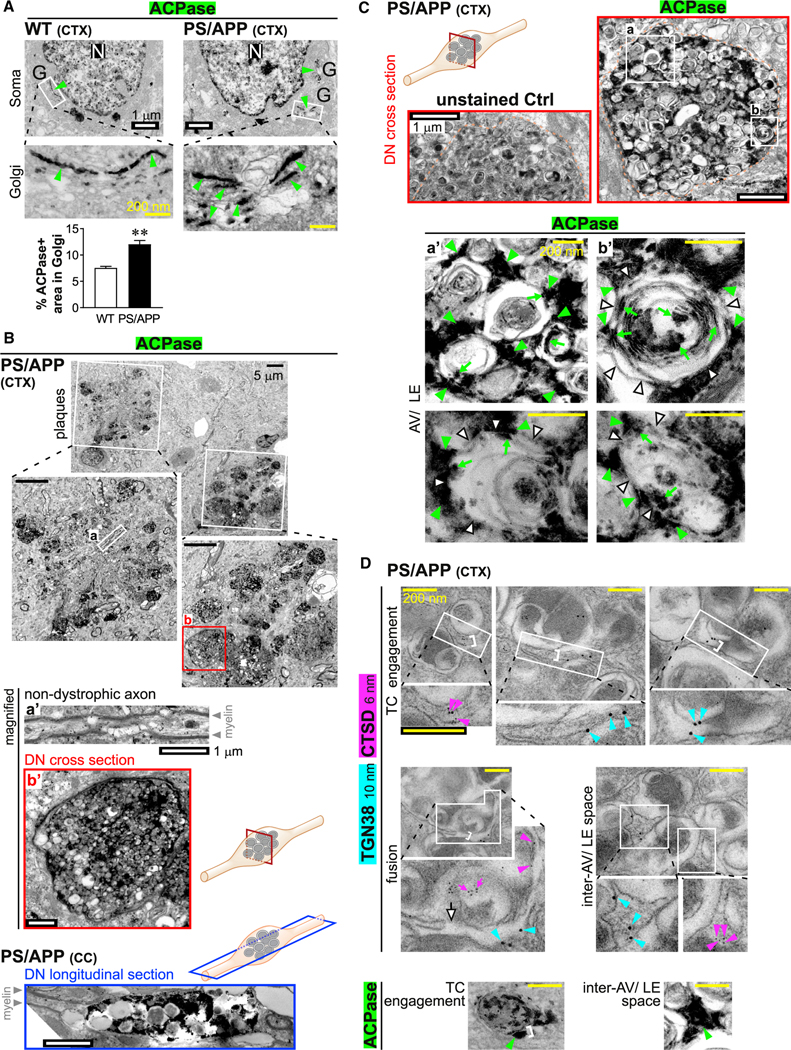
Electron micrographs of (A–C) histochemical ACPase labeling and (D) immunogold labeling of TGN38 and CTSD in PS/APP mouse brains.
(A) Top images show overviews of soma in wild-type (WT) and PS/APP cortex (CTX). White boxes (magnified below) mark Golgi stacks with an ACPase signal (green arrowheads). Scale bars, white = 1 μm; yellow = 200 nm. N, nucleus; G, Golgi. Graph shows percentage of area in Golgi occupied by ACPase. Bar, mean ± SEM (n = 46 WT somas; 37 PS/APP somas). **p < 0.01, Student’s t test.
(B) Top image shows overview of a neuropil region in PS/APP CTX with a plaque-associated ACPase signal. Plaques marked by white boxes are magnified below. Boxed regions (a) and (b) are further magnified below: (a’) shows a non-dystrophic axon; (b’) shows a dystrophic neurite (DN) (cross-section; corresponding to red box in schematic). Bottom image shows a DN (longitudinal section; corresponding to blue box in schematic) in PS/APP corpus callosum (CC). Scale bars, black, 5 μm; white, 1 μm.
(C) Top low-magnification electron micrographs show unstained Ctrl and ACPase-labeled DN (cross-sections; corresponding to red box in schematic) in PS/APPCTX. Orange dotted lines mark boundaries of DN. Boxes (a, b) are magnified in (a’, b’). High-magnification images show an ACPase signal associated with accumulated autophagic vacuoles (AVs) and late endosomes (LEs). White arrowheads, AVs/LEs limiting membrane; green arrowheads, ACPase engaged to exterior of AVs/LEs; green arrows, ACPase incorporated within AVs/LEs. Scale bars, white, 1 μm; yellow, 200 nm.
(D) Immunogold labeling of TGN38 (10 nm gold; cyan arrowheads) and CTSD (6 nm gold; magenta arrowheads) in TCs engaged to or fusing with accumulated AVs/LEs within DN. White brackets show the consistent width (~50 nm) of tubulovesicular TCs. Each white box is magnified in the image below (all magnified views are at the same magnification). Scale bars, 200 nm. White arrow, TC-AV membrane fusion; magenta arrows, incorporated CTSD. Bottom images show similar profiles with histochemical ACPase labeling (green arrowheads; previously shown in Figure 2H and Figure 3C a’).
Lysosomal components co-transport with a TGN marker bidirectionally in axons of cultured neurons
We proceeded to investigate distribution and trafficking of organelles relevant to lysosomal maturation in live mouse neuronal cultures. The general distribution pattern of organelles of interest was preserved in vitro. Consistent with adult mouse brains, acidified LAMP1 vesicles with a pHrodo-dextran signal that marked acidic endocytic organelles were mostly concentrated in soma and were uncommon in axons (Figure S3A). Similarly, acidified mRFP-eGFP-LC3 vesicles with only red fluorescence were also highly concentrated in perikarya (Figure S3B). The proportion of acidified vesicles in axons increased with proximity to the soma, comparable to previous observations (Maday et al., 2012).
By time-lapse imaging of axonal segments, we examined the transport dynamics of fluorescently tagged TGN marker and several lysosomal markers including lysosomal membrane proteins LAMP1 and TRPML1, and the lysosomal hydrolase acid phosphatase 2 (ACP2) (Figure 4). Motile vesicles were defined as having average velocities of at least 0.1 μm/s in either anterograde or retrograde direction. Vesicles below this threshold were collectively termed “non-motile.” LAMP1 vesicles were highly motile in both anterograde and retrograde directions (Figures 4B and 4C). Importantly, over 80% of LAMP1 vesicles were labeled with the TGN marker TGOLN2 (TGN38 homolog) (Figures 4B and 4C), demonstrating their non-lysosomal identity because TGN38 was not found on classic electron-dense lysosomes in soma (Figures 2A and 2C). Based on the literature and our electron microscopy results, LAMP1+ TGOLN2+ vesicles could potentially be TCs containing newly synthesized LAMP1 that are emerging from Golgi (Chen et al., 2017) (Figures 2B–2D) or LEs/amphisomes (fused LEs and AVs) that have gained TGN proteins via fusion with TCs (Figure 3D). Other examined lysosomal proteins were co-transported extensively with LAMP1 in both directions (Figures 4D and 4E). Besides ACP2 shown here, puncta containing CTSD also undergo bidirectional transport (Farías et al., 2017). We further showed the non-lysosomal identity of axonal LAMP1 vesicles by co-immunostaining of LAMP1, cation-independent (CI)-MPR, and cation-dependent (CD)-MPR (Figure S4). Lysosomes with only an LAMP1 signal but that lack CI-MPR and CD-MPR were enriched in soma (Figures S4A and S4C) but were rarely found in axons (Figures S4B and S4C). Instead, axons contained a heterogeneous population of LAMP1 vesicles containing either or both CI-MPR and CD-MPR, which could potentially be TCs or endosomes/amphisomes but not lysosomes (Figures S4B and S4C), consistent with in vivo observations (Figures 1, 2, and 3). Notably, however, as might be predicted in immature axons of cultured developing neurons, the restriction of highly acidified puncta is not as stringent (Figure S3B) as in brains (Figures 1E and 1F).
Figure 4. LAMP1 co-transports with other lysosomal proteins bidirectionally in axons.
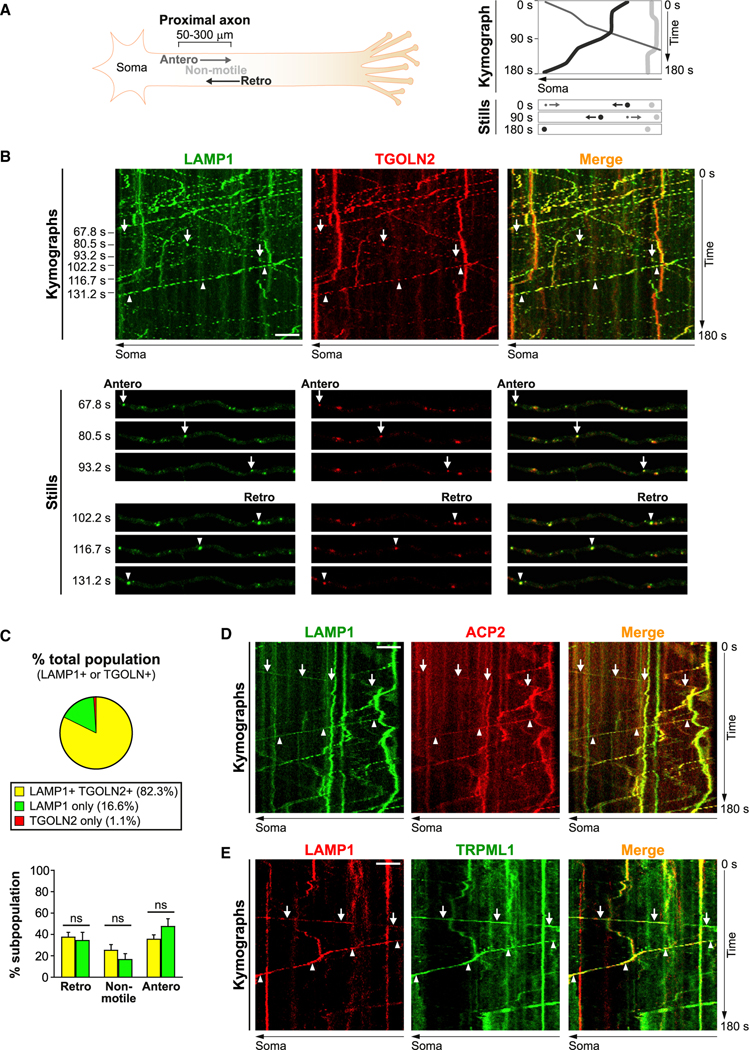
(A) Schematic of axonal time-lapse imaging experiments followed by analyses of kymographs. Antero, anterograde; retro, retrograde.
(B–E) Time-lapse imaging in axonal segments of neurons co-expressing LAMP1-YFP and TGOLN2-mCherry (B-C), LAMP1-YFP and ACP2-mCherry (D), or LAMP1-mCherry and TRPML1-eGFP (E). (B, D, E) Representative kymographs and still images. Complete arrows and arrowheads respectively denote anterograde and retrograde motility. Scale bar, 5 μm. (C) Top panel, pie chart showing percentage of vesicles with LAMP1 and/or TGOLN2 positivity (mean of 10 cells). Bottom panel, percentage of retrograde, non-motile (<0.1 μm/s) and anterograde vesicles in each LAMP1 vesicle subpopulation. Bars, mean ± SEM (n = 10 cells; ns, not significant, Mann-Whitney U test). See also Figures S3, S4, and S6.
Axonal LEs/amphisomes with at least partial acidification undergo exclusive retrograde transport
To more accurately determine the identities of axonal organelles and their transport dynamics, we performed live imaging using multiple combinations of lysosomal markers (LAMP1 and ACP2), LE marker Rab7, TGN marker TGOLN2 (TGN38 homolog), and indicators of acidification and hydrolase activity (Figures 5, 6, and S5). In neurons expressing LAMP1-YFP and co-labeled with LysoTracker-Red (LT) (Figures 5A–5C), acidic LAMP1+ LT+ vesicles constituted only a minor subpopulation (21.3%) of axonal LAMP1 vesicles, and they were either non-motile or exclusively transported retrogradely toward the soma (Figure 5C) at relatively low velocity (Figure 5B). Similarly, LAMP1 or ACP2 vesicles containing active cathepsins were restricted to slow retrograde transport with rare exceptions (Figures S5A–S5D). In contrast, the predominant poorly acidified LAMP1 subpopulation lacking an LT signal was highly motile in both directions and traveled at a wide range of velocities (Figures 5B and 5C). It was not surprising that the majority of axonal LAMP1 vesicles did not show detectable acidification because LAMPs and other lysosomal proteins are present on a wide range of non-endosomal non-lysosomal organelles (Cheng et al., 2018; Pols et al., 2013; Vukoja et al., 2018; Yu et al., 2010), including TCs exporting LAMP1 from Golgi (Chen et al., 2017). These studies, together with the known mild acidification of TGN (Demaurex et al., 1998) and the high colocalization between LAMP1 and the TGN marker (Figures 4B and 4C) present on tubulovesicular TCs (Figures 2B–2D), suggest that poorly acidified LAMP1 vesicles are TCs.
Figure 5. Acidic LAMP1 vesicles undergo retrograde axonal transport.

Time-lapse imaging in axonal segments of neurons expressing LAMP1-YFP co-labeled with LysoTracker (LT)-Red (A–C) or expressing LAMP1-mCherry co-labeled with dextran-Oregon Green 488 (OG488) and dextran-Alexa Fluor 647 (AF647) (D–F).
(A) Representative kymographs and still images. Scale bar, 5 μm. White outlines in stills mark the axonal profile. Complete arrows and arrowheads respectively denote anterograde (Antero) and retrograde (Retro) motility.
(B) Histogram showing the distribution of average velocities in LAMP1 vesicle subpopulations (n = 1,048 vesicles in 19 cells).
(C) Percentage of retrograde, non-motile (<0.1 μm/s) and anterograde vesicles in each LAMP1 vesicle subpopulation. Bars, mean ± SEM (n = 19 cells; **p < 0.01, Mann-Whitney U test).
(D) Schematic and kymographs showing trajectories of several representative vesicles. Identical adjustments have been applied to all dextran images.
(E) Box-and-whisker plots showing effect on OG488/AF647 ratio in endocytically derived organelles in soma of neurons equilibrated to designated pH (n ≥ 8 cells; *p < 0.05, **p < 0.01 against “pH 4,” #p < 0.01 against “pH 7,” Kruskal-Wallis test followed by Dunn’s multiple comparisons).
(F) Box-and-whisker plots with individual data points showing average velocity versus OG488/AF647 ratio (n = 195 vesicles from 11 cells; **p < 0.01, Kolmogorov-Smirnov test). See also Figures S3–S6.
Figure 6. Axonal late endosomes are acidic and undergo retrograde axonal transport.

Time-lapse imaging in axonal segments of neurons expressing LAMP1-YFP and Rab7-DsRed (A–C), or expressing Rab7-eGFP co-labeled with LysoTracker (LT)-Red (D–F), or co-expressing LAMP1-YFP, TGOLN2-mCherry, and co-labeled with LT-Deep Red (G and H).
(A, D, and G) Representative kymographs and still images. Scale bar, 5 μm. Complete arrows, anterograde; closed arrowheads, retrograde: open arrowheads, non-motile.
(B and E) Histograms showing the distribution of average velocities in LAMP1 vesicle subpopulations (n = 335 vesicles in 7 cells) and Rab7 vesicle subpopulations (n = 208 vesicles in 10 cells). Retro, retrograde; Antero, anterograde.
(C, F, and H) Percentage of retrograde, non-motile (<0.1 μm/s) and anterograde vesicles in specified LAMP1 vesicle subpopulations (C) (n = 7 cells), Rab7+ LT+ vesicle subpopulation (F) (n = 10 cells) and LAMP1+ TGOLN+ LT+ vesicle subpopulation (H) (n = 14 cells). Bars, mean ± SEM (*p < 0.05, **p < 0.01, Mann-Whitney U test). See also Figures S3–S6.
We confirmed this drastic shift from bidirectional to retrograde axonal transport of acidic LAMP1 vesicles with another approach involving pH measurement (Figures 5D–5F). Lumenal acidification status of endocytically derived vesicles was ratiometrically measured in neurons loaded with equal amounts of dextrans conjugated with pH-insensitive Alexa Fluor 647 (AF647) and pH-sensitive Oregon green 488 (OG488), which is quenched at low pH (Johnson et al., 2016). OG488/AF647 intensity ratio was positively correlated with pH value of standard buffers, as expected, with a low ratio reflecting a low pH (Figure 5E). In neurons transfected with LAMP1-mCherry and loaded with dextrans, we analyzed motility and dextran signal intensities of axonal LAMP1 vesicles containing dextran-AF647 (Figures 5D and 5F). Robustly acidic LAMP1 vesicles (ratio ≤1.24) were either non-motile or restricted to retrograde transport with very few anterograde outliers. In contrast, poorly acidic LAMP1 vesicles (ratio >1.24) traveled bidirectionally at a wide range of velocities in both retrograde and anterograde directions. These poorly acidic dextran-containing vesicles were presumably not exclusively newly synthesized TGN TCs but, instead, likely included non-acidic structures derived from lysosome reformation (Yu et al., 2010).
We hypothesized the identity of the retrograde LAMP1 vesicle subpopulation to be LEs/amphisomes because axonal LC3 vesicles rapidly acquire LAMP1 and Rab7 and are predominantly retrograde (Lee et al., 2011). This was verified by the co-transport of LAMP1-YFP and Rab7-DsRed (Figure 6). Similar to acidic LAMP1 vesicles (Figures 5A and 5B), LAMP1+ Rab7+ vesicles comprised a minor subset of the total LAMP1 population (Figures 6A and 6B) and were restricted to slow retrograde transport as opposed to the non-Rab7-labeled vesicles that were highly motile in either direction (Figures 6B and 6C). These properties, and the known role of Rab7 in assisting assembly of the vATPase mediating endo-lysosomal acidification (De Luca et al., 2015), suggest that LT and Rab7 mark essentially the same subpopulation of LAMP1 vesicles comprising acidifying LEs/amphisomes. Consistently, almost all Rab7 vesicles were LT+ and they (Figures 6D and 6E) showed exclusive retrograde motility and similar velocity distribution (Figures 6D–6F) to that of LAMP1+ Rab7+ (Figure 6B) and LAMP1+ LT+ vesicles (Figure 5B). Furthermore, acidic LAMP1+ TGOLN2+ LT+ vesicles were also exclusively retrograde (Figures 6G and 6H), supporting their identity as LEs/amphisomes that have gained significant acidification and TGN proteins after fusion with TCs (Figure 3D).
TCs shuttling lysosomal components bidirectionally include a distinct subset of anterogradely biased elongated profiles
We determined that a poorly acidic, bidirectional subpopulation of lysosomal protein-containing vesicles consists of TCs (Figures 5, 6, and S5). In this TC subpopulation, elongated vesicles were occasionally observed (Figure 5A). This is consistent with the reported tubulovesicular morphology of TCs (Luini et al., 2008) and the presence of axonal elongated ultrastructures containing both the TGN marker, TGN38, and lysosomal proteins (Figure 2). We then examined the dimensions, motility, and other features of elongated LAMP1 vesicles (Figures 7 and S5; Table S1). Due to limited resolution of light microscopy, elongated vesicles were only screened from larger vesicles of ≥1 μm diameter (in parallel to the axon). The aspect ratio (long axis: short axis) of these larger vesicles were measured, and elongated vesicles were defined as having aspect ratios of ≥2, while the remaining ones were classified as small/globular (Figure 7A). Remarkably, elongated LAMP1 vesicles were significantly biased to anterograde transport, with 71% of vesicles in this subpopulation traveling anterogradely, while only 17% were retrograde (Figure 7B).
Figure 7. Elongated LAMP1 vesicles are anterograde biased and enzymatically inactive.

(A) Schematic showing criteria for classification of LAMP1 vesicles into small/globular and elongated categories.
(B–D) Time-lapse imaging in axonal segments of neurons expressing LAMP1-YFP or LAMP1-mCherry, either co-expressing other fluorescently tagged lysosomal proteins or co-labeled with vital dyes as specified. Representative kymographs and still images are shown. Arrowheads show examples of elongated and anterograde vesicles. Scale bars, 5 μm. LT, LysoTracker-Red labeled; active CTSB, Magic Red-cathepsin B labeled; active CTSD, Bodipy-Pepstatin A labeled. (B) Graph on right shows percentage of retrograde (retro), non-motile (<0.1 μm/s), and anterograde (antero) vesicles in each LAMP1 vesicle subpopulation. Bars, mean ± SEM (n = 8 cultures with a total of 114 elongated and 2,295 small/globular LAMP1 vesicles; **p < 0.01, Mann-Whitney U test). See also Figures S5 and S6 and Table S1.
(E) Hypothetical model showing maturation, trafficking, and interactions of organelles of autophagy/endo-lysosomal and biosynthetic pathways in axons. Immature early endosomes (EE) and autophagosomes (AP) are concentrated in distal axon and mature into late endosomes (LEs) and amphisomes (Amp), which undergo continuous maturation and retrograde transport while gaining lysosomal components from bidirectional TGN-derived transport carriers (TCs). LEs/amphisomes attain partial acidification and degradative capability before reaching full maturation into lysosomes (Lyso) in perikarya. Organelle colors depict lumenal acidification (purple, mildly acidic; pink, moderately acidic; magenta, fully acidic).
The TC identity of elongated LAMP1 vesicles were confirmed by their weak acidification and undetectable cathepsin activity (negative LT, Magic Red-cathepsin B, or Bodipy-pepstatin A) (Figure 7C), and the presence of TGN marker TGOLN2 (Figure 7D) and other lysosomal proteins ACP2 and TRPML1 (Figures S5E and S5F). Out of all 95 elongated LAMP1 vesicles, only one was acidic and classified as an LE/amphisome (Table S1). This illustrates that an elongated shape is a reliable feature of TCs, consistent with our ultrastructural analyses (Figure 2) although the exact dimensions were likely over-estimated in a non-proportional manner due to limited resolution of light microscopy. Axonal LAMP1 vesicle subpopulations (i.e., LEs/amphisomes, elongated TCs, and globular TCs) display several key differences summarized in Table S1. First, globular TCs were significantly smaller than LEs/amphisomes, but, again, note that measurements for all puncta were likely over-estimated. Second, while globular TCs were bidirectional with almost equal proportions of anterograde and retrograde vesicles, elongated TCs were anterograde-biased evidenced by 4 times more anterograde vesicles than retrograde vesicles. Third, both types of TCs moved at a greater velocity than LEs/amphisomes. In summary, we have demonstrated that multiple lysosomal components are transported together within poorly acidic TGN-derived TCs that are either small vesicles or elongated tubules, both of which are bidirectionally transported at high velocity with the elongated subset being especially anterograde biased.
DISCUSSION
Despite recent active research on lysosomal maturation and trafficking in neurons and cells in general, our current knowledge is much less established than commonly believed owing to abundant misinformation based on studies in which lysosomes have not been definitively identified. Many studies also rely solely on in vitro culture models, which is problematic because upregulated vesicular trafficking and possibly less stringent regulation are not surprising considering that cultured maturing neurons undergo constant growth and retraction and also face stresses from the unnatural cell culture environment. Cautious interpretation of in vitro results is thus warranted and best guided by in vivo results whenever possible. Although the heterogeneity of axonal LAMP1 vesicles has been recognized (Cheng et al., 2018), little is known about the behavior of the full spectrum of lysosomal protein-containing organelles (fully mature lysosomes, other pre-/partially degradative structures and non-degradative biosynthetic precursors), especially in vivo. Aiming to integrate concepts and resolve inconsistencies in the field, we have clarified the mechanism of maturation of axonal pre- and partially degradative organelles into lysosomes and defined the distribution and dynamics of relevant organelles in vivo as well as in vitro, as depicted diagrammatically in Figure 7E.
Fully mature classic lysosomes are restricted from axons in mature intact brain in vivo
Our robust functional and ultrastructural evidence demonstrates that fully mature lysosomes are restricted from axons in intact brains. In myelinated axons, we instead detected AVs, LEs, and most notably tubulovesicular structures containing both lysosomal hydrolases and the TGN marker TGN38. The dimensions, morphology, and the TGN38 positivity of these tubulovesicular compartments indicate their identity as TGN-derived TCs (Banting and Ponnambalam, 1997; Luini et al., 2008; Nakata et al., 1998), directly demonstrating the trafficking of lysosomal components in TGN38+ TCs. Restriction of lysosomes from axons under normal conditions was maintained in the AD mouse model, PS/APP. In AD mouse models and human AD brains, neurons attempt to clear accumulated AVs by upregulating the expression of lysosomal components (Bordi et al., 2016; Cataldo et al., 1995, 2004). Here, we showed significant upregulation and mobilization of Golgi-derived ACPase to dystrophic swellings via TCs but not lysosomes. Similarly, immature LAMP1- and MPR-containing structures accumulate in 5xFAD mouse brains (Gowrishankar et al., 2015). In human AD brains, however, dystrophic swellings persist over a much longer period than in mice. Their contents can thus be more heterogeneous with occasional lysosomes (Nixon et al., 2005; Suzuki and Terry, 1967) fully matured from accumulated AVs remaining static in axonal swellings for long periods and subjected to continuous TC fusions. We also do not preclude the possibility that organelles within dystrophic swellings proximal to soma may have limited access to somal lysosomes.
These in vivo observations were established as the standard that guided our subsequent in vitro studies. The lack of mature lysosomes in axons was further strengthened by our in vitro observation that most axonal LAMP1 vesicles were labeled by TGN38 homolog while carrying other lysosomal proteins TRPML1 and ACP2, indicating their identity as either TCs or LEs/amphisomes but not lysosomes. This phenomenon is likely applicable to dendrites where degradative compartments are confined to proximal dendrites (Yap et al., 2018).
Acidifying LEs/amphisomes undergo exclusive retrograde axonal transport
We showed that robustly acidic LEs/amphisomes commit to exclusively retrograde long-range axonal transport even though they often display instantaneous, small-scale bidirectional movements (Hendricks et al., 2010; Lalli and Schiavo, 2002), consistent with LT vesicle dynamics observed by another group (Gowrishankar et al., 2017). Their retrograde motility is in line with the LE-dependent acquisition of dynein-snapin complex (Cheng et al., 2015). Simultaneous retrograde transport and progressive maturation of acidifying LEs/amphisomes can readily explain the gradient of decreasing pH in endocytically derived organelles from distal to proximal axon (Overly and Hollenbeck, 1996) and the high concentration of mature lysosomes in the perinuclear area. LE/amphisome maturation is driven in part by the acidification-promoting activities of Rab7 effectors. For instance, RILP facilitates assembly of the proton pump vATPase (De Luca et al., 2015), whose activity is enhanced by phosphatidylinositol-3,5-bisphosphate (Li et al., 2014) enriched on LE membrane via a Rab7-dependent process (Liu et al., 2017).
TCs deliver lysosomal constituents bidirectionally
The major subset of axonal LAMP1 vesicles consisted of poorly acidic, fast-moving bidirectional TGN-derived TCs. Our findings are consistent with the early ultrastructural observation of retrogradely and anterogradely transported organelle populations. Retrograde organelles were mainly LE like, while the anterograde pool consisted of tubulovesicular structures of 50- to 80-nm diameter similar to TCs (Tsukita and Ishikawa, 1980). A substantial body of evidence has documented the emergence of LAMP1+ tubulovesicular structures from Golgi (Chen et al., 2017), and the presence of LAMP1 on TCs (Carlsson and Fukuda, 1992; Pols et al., 2013) that also carry other cargoes, including transferrin receptor (Chen et al., 2017), the ion channel TRPM8 (Ghosh et al., 2016), and synaptic constituents (Vukoja et al., 2018). LAMP1-containing TCs travel via both direct (TGN-to-endosome) and indirect post-Golgi trafficking routes (Carlsson and Fukuda, 1992; Janvier and Bonifacino, 2005; Pols et al., 2013). This is enabled by the recognition of sorting signals on LAMP1 and other cargo proteins by adaptor protein complexes such as AP-1, which mediates bidirectional transport between TGN and endosomes. AP-1 recognizes various lysosomal membrane proteins (e.g., LAMP1, TRPML1, and V0a1) and MPR that binds and delivers nascent lysosomal hydrolases (Bagh et al., 2017; Janvier and Bonifacino, 2005; Meyer et al., 2000; Miedel et al., 2006), thus enabling the biosynthetic supply of multiple lysosomal components to endosomes for their maturation toward lysosomal identity. The TGN origin of TCs and their outbound trafficking into axons implicate a higher availability of lysosomal components delivered by TCs to degradative compartments near the perikarya than to distally located ones. This is consistent with a study in non-neuronal cells showing that peripheral endosomes only have limited access to the lysosomal membrane protein LIMP2 (Johnson et al., 2016). Thus, LEs/ amphisomes would have increasing access to lysosomal components to support their progressive maturation as they travel retrogradely toward the soma. This trafficking paradigm, together with the acidification promoting activities of Rab7 effectors discussed above, can explain the increasing acidification of degradative organelles from distal to proximal axon. Indirect TGN-to-plasma membrane trafficking followed by re-internalization, on the other hand, is unlikely to play a major role given the long distance between the soma and the axon terminal. These biosynthetic trafficking routes have been neglected in many studies that misidentify a substantial subpopulation of LAMP1+ TCs as LEs/lysosomes. TC-mediated axonal transport may offer potential advantages over direct delivery of lysosomes from soma to the distal axon by enabling rapid delivery of lysosomal components in bulk over a long distance, without spending extra energy on maintaining membrane potential and lumenal homeostasis as in lysosomes (Xu and Ren, 2015).
We observed a class of elongated TCs and speculate that these are nascent carriers of lysosomal constituents newly delivered from the TGN in the outbound anterograde direction. As they engage with endosomes/amphisomes in the axon, they conceivably undergo either complete or kiss-and-run fusion and consequently lose a portion of their membrane, gradually becoming less elongated and more globular while continuing to shuttle lysosome constituents bidirectionally. Both elongated and small globular TCs traveled at higher velocities than LEs/amphisomes, likely due to a high membrane to volume ratio in TCs that increases the accessibility of microtubule-based motors, in contrast to the globular but larger LEs/amphisomes that have a relatively higher internal volume.
In addition to TGN-derived TCs, it is possible that part of the poorly acidic bidirectional LAMP1 vesicles are tubulovesicular “protolysosomes” derived from autolysosome reformation (Yu et al., 2010), i.e., the budding and recycling of lysosomal membrane components to support new rounds of lysosomal maturation. Interestingly, an early ultrastructural study in mouse brains demonstrated that salt stress induced tubular protrusions from somal lysosomes concomitant to the appearance of ACPase+ tubules in axons (Broadwell and Cataldo, 1984). It is therefore possible that, besides TCs, reformed protolysosomes could serve as an additional source of lysosomal components being shuttled in axons, but this remains to be investigated.
Insufficient specificity or selectivity of lysosomal markers may yield conflicting results
Seemingly contradictory to our results, another study suggested that LAMP1 vesicles in proximal axons are mostly acidified lysosomes that move bidirectionally, and that anterograde trafficking of lysosomes into axons is mediated by BORC/kinesin-1 (Farías et al., 2017). A plausible explanation is that the superecliptic pHlorin probe used in their study was overly acid sensitive, being almost completely quenched at pH ~6 (Sankaranarayanan et al., 2000; Tanida et al., 2014) and therefore would not be able to distinguish lysosomes (pH 4.5–5) or even LEs/amphisomes (pH ~5.5) from mildly acidic organelles like TGN vesicles (pH ~6) (Demaurex et al., 1998). Here, using a ratiometric pH measurement, we identified a critical threshold below which axonal LAMP1 vesicles drastically shifted from bidirectional motility to almost exclusive retrograde transport. These findings highlight the importance of lysosomal marker selectivity, including the extent of vesicle acidification. Notably, in BORCS7 mutant mouse brains, electron microscopy revealed dystrophic axons filled with tubulovesicular structures (Snouwaert et al., 2018). It will be of interest to examine whether these BORC-regulated structures (Farías et al., 2017; Snouwaert et al., 2018) are in fact tubulovesicular TCs delivering lysosomal components rather than lysosomes.
In addition, a recent study has shown frequent anterograde delivery of activated, soma-derived lysosomal hydrolases to distal axons (Farfel-Becker et al., 2019). The authors claimed these axonal compartments to be lysosomes based on vital labeling agents that detect activated lysosomal enzymes. Despite the almost complete reliance on these probes, key aspects of their properties have not been fully considered for proper data interpretation. Given that 3 out of 4 probes in their study targeted lysosomal enzymes can be activated at a relatively high pH (i.e., cathepsins B and L and GCase), it is unclear what proportion of marked organelles had comparable degradative capability to fully mature lysosomes. Furthermore, detailed characterization of the acidification status, extent of enzyme activation and ultrastructural morphology of labeled compartments were lacking, making it impossible to unequivocally determine their maturity and organelle identity. Future investigations will be needed for reconciling this study (Farfel-Becker et al., 2019) with conflicting findings from us and other ones in the field (Li et al., 2019). For example, CTSD was found in pre-synaptic endosomes primarily as the 48 kDa intermediate precursor form but not the 34 kDa mature form (Li et al., 2019), consistent with our hypothesis that lysosomal hydrolases are delivered to distal axons as immature precursors via TCs instead of lysosomes. The 48 kDa precursor, while not fully activated, has detectable specific activity (Benes et al., 2008) that can cause mislabeling of endosomes as lysosomes. Although some limitations of vital labeling agents also apply to the current study, our extensive functional and ultrastructural evidence have established that TCs, but not lysosomes, are transported anterogradely barring rare exceptions observed in vitro.
Conclusions and perspectives
As an important implication of our findings, we recommend that future investigations employ rigorous criteria to distinguish between fully mature lysosomes, maturing compartments and TCs carrying lysosomal components. Although all of these organelles contain lysosomal components including hydrolases, they have fundamental differences in degradative capability, motility, distribution, functional role, and disease relevance. The critical requirement for full lysosomal maturation is exemplified by the PSEN1 KO model, in which lysosomal pH is elevated from 4.7 to 5.4 (Lee et al., 2010). The latter pH level is comparable to that of LEs, which have limited degradative capability in processes such as protease activation, peptide hormone inactivation, and antigen processing (Authier et al., 1996). Accordingly, the degree of abnormal pH elevation in lysosomes of PSEN1 KO mice results in proteolysis deficits and autophagy impairment, indicating substantial functional differences between fully mature lysosomes and maturing LEs/amphisomes. Disease models that specifically disrupt LE maturation (CHMP2B or CHMP5 loss of function) further illustrate the insufficient degradative capability of maturing compartments for maintaining cellular homeostasis (Shim et al., 2006; Urwin et al., 2010).
Axons have limited lysosomal degradative capability and are prone to developing focal swellings with massive AV accumulation in neurodegenerative diseases (Nixon et al., 2005). In PS/APP brains, there is apparently no shortage in lysosomal hydrolase delivery via TCs to dystrophic swellings but the accumulated AVs still cannot successfully mature into lysosomes and degrade their contents. These accumulated vesicles are AVs, as originally characterized in AD brain (Nixon et al., 2005), rather than “cathepsin deficient lysosomes,” as recently proposed (Gowrishankar et al., 2015), despite that occasional lysosomes could indeed develop in dystrophic neurites in human AD brain after years of AV accumulation and continual access to delivered TCs (Nixon et al., 2005; Suzuki and Terry, 1967). To fully elucidate the disease mechanism, we have yet to define the stimulus for TC upregulation such as general autophagy/lysosomal induction as we see in AD (Bordi et al., 2016; Cataldo et al., 1995) and possibly multiple reasons behind lysosomal maturation deficits. These may include the lack of delivery of certain lysosomal components, acidification failure (Colacurcio and Nixon, 2016) and impaired retrograde transport (Lee et al., 2011; Tammineni et al., 2017). Although extensive evidence demonstrates that retrograde transport and access to somal lysosomes are necessary for efficient degradation of axonal organelles targeted to autophagy/endo-lysosomal pathways, including mitophagy (Han et al., 2020; Zheng et al., 2019), whether these are absolute requirements is still under debate (Ashrafi et al., 2014). It is evident that the distal axon is capable of a certain degree of local degradation, as suggested by the enrichment of CTSD intermediate precursors in synaptosomes (Li et al., 2019), the presence of acidified “hubs” at axon terminals capable of partially degrading a dual-fluorescent reporter (Jin et al., 2018), and the detection of cathepsin and GCase activities at axon terminals (Farfel-Becker et al., 2019). The extent and functions of these local degradative compartments, compared to fully mature lysosomes in soma, remain open to future investigations. These fundamental issues, which are central to the pathogenesis of AD as well as other neurological conditions implicating autophagy/lysosomal impairments, can only be addressed by further studies that accurately define the identities of involved organelles and their dysfunction and/or mistrafficking.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Ralph Nixon (nixon@nki.rfmh.org).
Materials availability
Unique reagents and mouse lines generated in this study are available from the lead contact and may require completion of a materials transfer agreement.
Data and code availability
No large datasets or custom code were generated in this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal procedures were performed following the National Institutes of Health Guidelines for the Humane Treatment of Animals, with approval from the Institutional Animal Care and Use Committee at the Nathan Kline Institute for Psychiatric Research. Three transgenic mouse models were used: (1) hCTSD Tg, a single transgenic mouse model expressing human CTSD (hCTSD) under the neuron-specific Thy1 promoter (genetic background: mixed FVB/N x C57/BL6); (2) TRGL x hCTSD Tg, a double transgenic mouse model expressing both hCTSD and tandem-fluorescent mRFP-eGFP-LC3 under individual Thy1 promoters (genetic background: mixed FVB/N x C57/BL6); (3) PS/APP: a double transgenic mouse model expressing mutant APPK670N/M671L and PSEN1M146L (Holcomb et al., 1998). All mice were genotyped by PCR. Mice were group-housed under 12 h light/dark cycle at constant room temperature and provided with food and water ad libitum. Adult mice (4–12 months) of either sex were used for experiments. Age-and sex-matched WT mice of comparable background were used as controls. Littermate controls were used whenever possible.
Primary neuron cultures
E17–18 WT embryos of either sex were obtained from timed-pregnant C57/BL6 female mice. Neurons were isolated from embryonic brains and maintained in culture for 5–10 DIV at 37°C in a humidified environment with 95% air and 5% CO2.
METHOD DETAILS
Generation of transgenic mouse lines
For the generation of hCTSD Tg mice, the full length coding sequence of hCTSD cDNA (accession# Genbank: BC016320) from a hCTSD/pcDNA3 plasmid was subcloned into the XhoI site of pTSC21, a mouse Thy1.2 expression cassette (Andrä et al., 1996). The Thy1.2-hCTSD transgene fragment was isolated by digestion with NdeI and AflIII followed by gel purification. DNA microinjection into FVB/N mouse embryos and embryo transfer were performed under standard procedures at Skirball Transgenics/ES Cell Chimera Facility, New York University. hCTSD-expressing founders, identified by PCR and Southern blotting, were bred with C57BL/6 mice to produce the hCTSD Tg mouse line. For the generation of double transgenic TRGL x hCTSD Tg mice, hCTSD Tg mice were bred with our previously described TRGL6 (Thy1 mRFP-eGFP-LC3, Line 6) transgenic mice expressing tandem fluorescent mRFP-eGFP-LC3 (Lee et al., 2019). This mRFP-eGFP-LC3 fusion protein contains a terminally located mRFP and an internal eGFP to avoid premature degradation of eGFP prior to mRFP in mRFP+ vesicles (Lee et al., 2019).
Generation of monoclonal hCTSD antibody
Purified hCTSD was used as the antigen for monoclonal antibody generation. hCTSD was purified from postmortem human brain by acid precipitation at pH 3.5, followed by ion-exchange chromatography using DEAE-Biogel at pH 7.4, and affinity chromatography using pepstatin-aminohexyl Sepharose 6B at pH 4.5 and elution at pH 8.6 (Nixon and Marotta, 1984). Purified hCTSD was identified by mass spectrometry, amino acid sequencing, and immunoblotting using a sheep anti-hCTSD polyclonal antibody (Cataldo et al., 1990). Monoclonal mouse anti-hCTSD antibody was generated using BALB/c mice under standard protocols at McLean Hospital (Belmont, MA). Ascites was custom-produced by Rockland Immunochemicals (Limerick, PA).
Adult mouse brain tissue preparation
Adult mouse brains were harvested from mice following anesthetization with a mixture of ketamine (100 mg/kg BW) and xylazine (10 mg/kg BW). For light microscopic analyses, mice were transcardially perfused with fixative (4% PFA in 0.1 M sodium cacodylate buffer, pH 7.4), and the brains were immersion-fixed in the same fixative overnight at 4°C. For electron microscopic analyses, perfusion and fixation conditions are specified in the section “Electron Microscopy.” For biochemical analyses including immunoblotting and enzymatic activity assay, mice were transcardially perfused with physiological saline, and the brains or isolated hippocampi were flash frozen on dry ice and stored at –70°C. Alternatively, mice were briefly perfused with saline if their brains were subjected to both morphological and biochemical analyses. One hemisphere or the isolated hippocampus was flash frozen and the other hemisphere was immersion-fixed in 4% PFA for 3 days at 4°C.
Immunostaining of brain sections for light microscopy
Immunostaining was performed as described (Yang et al., 2009). Fixed brains were processed into either 40-μm vibratome sections or 6-μm paraffin-embedded sections, which were deparaffinized and rehydrated prior to staining. For DAB immunocytochemistry, endogenous peroxidase activity was first blocked by treating brain sections with 1%–3% H2O2 in Tris-buffered saline (TBS, pH 7.4) containing 10% methanol. Sections were then permeabilized in Dilution Buffer (0.4% Triton X-100 in TBS containing 1% BSA and 1% normal goat serum), blocked with 20% normal goat serum, and incubated with primary antibodies diluted in Diluting Buffer at 4°C overnight. Subsequent staining steps were performed according to manufacturer’s instructions (Vector Laboratories), which include incubation with biotinylated secondary antibodies, complex formation with avidin and biotinylated HRP using Vectorstain ABC Kit, and reaction with Vector DAB Peroxidase Substrate. If specified, sections were counterstained with cresyl violet. Sections were mounted on gelatin-coated slides (for vibratome sections), dehydrated in a graded series of ethanol and xylene, and coverslipped with Permount. Images were acquired with an Axiovert 200M microscope (Zeiss). For tiled images, overlapping images were taken and images were stitched manually in photoshop. For quantitative analysis, four sagittal brain sections were used from each mouse, evenly spaced (400 μm) from the region between Lateral 0.48 mm and 2.16 mm containing the hippocampus. Three 40X digital images from the CA1 area or three 20X images from the cortex were taken from each section and quantified with AutoMeasure software (Zeiss).
For immunofluorescent staining, 40-μm vibratome sections were permeabilized in Dilution Buffer, blocked with Blocking Solution from M.O.M. Immunodetection Kit, and sequentially incubated with hCTSD primary antibody diluted in the Working Solution from M.O.M. Immunodetection Kit at 4°C overnight and Alexa Fluor 647-conjugated secondary antibodies diluted in the Working Solution. Sections were mounted on gelatin-coated slides and coverslipped with Prolong Diamond Antifade Mountant. Images were acquired with laser scanning confocal microscopy with an LSM880 confocal microscope (Zeiss). Images were processed with a 1-pixel median filter in ImageJ and quantified by “line scan,” “particle analysis” and “measure” functions or “Just Another Colocalization Plugin” in ImageJ. All measurements were performed on unadjusted images. Brightness/contrast of displayed images was adjusted in ImageJ and/or Photoshop.
Immunoblotting
Samples for immunoblotting were prepared by homogenizing brains in a tissue-homogenizing buffer (20 mM Tris, pH 7.4, containing 250 nM sucrose, 1 mM EDTA, 1 mM EGTA) supplemented with protease and phosphatase inhibitors. Following electrophoresis, proteins were transferred onto 0.2 μm-pore nitrocellulose membranes (Whatman, Florham Park, NJ) at 100 mA for 8–12 hours depending on the target protein. The blots were blocked for 1 hour in 5% non-fat milk in TBS, rinsed in TBST (TBS + 0.1% Tween-20), then incubated with a primary antibody in 1% BSA/TBST overnight at 4°C. The membrane was washed and incubated in a HRP conjugated secondary antibodies (Jackson ImmunoResearch) diluted 1:5000 in 5% milk for 1 h at room temperature. The membrane was again washed and then incubated in a Novex ECL (Thermo Fisher) for 1 min and exposed to film.
Enzymatic activity assay for CTSD
CTSD activity was assayed at 37°C at pH 4.0 by measuring the release of fluorescent aminomethyl coumarin (AMC)-containing peptide from the quenching Dnp group following Phe-Phe bond cleavage in 7-methoxycoumarin-4-acetyl-Gly-Lys-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Lys(Dnp)-D-Arg-NH2 as previously described (Yasuda et al., 1999). Assays were performed in white microplates in a total volume of 100 μl with or without 3 μg of pepstatin for one hour. Released fluorescence was measured in a Wallac Victor-2 Spectrofluorimetric plate reader with a filter optimized for detection of AMC standard solution with excitation at 365 nm and emission at 440 nm. However, instead of using AMC standard, a quenched standard 7-methoxycoumarin-4-acetyl-Pro-Leu-OH was used for expressing enzyme activity to account for the release of AMC-containing peptide instead of free AMC. Enzyme activity was expressed as the relative amount of quenched standard released per hour per mg protein.
Electron microscopy
Ultrastructural analyses were performed in adult mouse brain sections following immunogold labeling or acid phosphatase histochemistry. Post-embedding immunogold labeling was performed as described (Yang et al., 2009). Mice were transcardially perfused with fixative (4% PFA, 2% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4). Mouse brains were harvested and immersion-fixed in the same fixative overnight at 4°C. Mouse brains were processed into 60-μm vibratome sections, which were dehydrated in a graded series of ethanol, embedded in Spurr Resin (Electron Microscopy Sciences), and cut into 60-nm ultrathin sections. Ultrathin sections were mounted on grids and etched for 5 min with 1% sodium metaperiodate in PBS. Ultrathin sections were then incubated sequentially in blocking solution (5% horse serum), primary antibodies (overnight at 4°C) and secondary antibodies conjugated with 6 nm or 10 nm gold. Acid phosphatase histochemistry was performed as described (Cataldo et al., 1991; Yang et al., 2009) using cytidine 5’-monophosphate (CMP) as the substrate. Mice were transcardially perfused with fixative (4% PFA, 1% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, containing 0.025% calcium chloride, 5% sucrose and 0.075% CMP). Mouse brains were harvested and immersion-fixed in 4% PFA for 4 h at 4°C. Mouse brains were processed into 50-μm vibratome sections, rinsed and incubated in reaction medium (0.02% M Tris-maleate buffer containing 0.1% CMP, 2% sucrose, 0.005 M manganese chloride, 0.12% lead nitrate, pH 5.0, filtered with #50 paper) for 1 h at 37°C and treated with 1% sodium sulfide. Sections were post-fixed in 1% osmium tetroxide, dehydrated in a graded series of ethanol, embedded in Epon (Electron Microscopy Sciences), and cut into 60-nm ultrathin sections. Both immunogold-labeled and histochemically labeled ultrathin sections were briefly stained with uranyl acetate and lead citrate. Images were acquired with a Thermo Fisher Talos L120C transmission electron microscope equipped with a Ceta camera. Electron micrographs of immunogold-labeling were processed with a 2-pixel median filter in ImageJ. Brightness/contrast of displayed images was adjusted in ImageJ and/or Photoshop.
Primary neuronal culture
Primary cortical neurons were isolated as described (Boland et al., 2008) from E17–18 mouse embryos. Cerebral cortices harvested from mouse embryos were digested in Hibernate E medium containing 10 units/ml papain and 0.05 mg/ml DNase I for 15 min. The reaction was stopped by adding equal volumes of 10% heat-inactivated fetal bovine serum (FBS) in Hibernate E. Cells were then pelleted at 1000 rpm for 5 min, and were resuspended in DMEM/F12 containing HEPES, 5% heat-inactivated FBS and 5% heat-inactivated horse serum, followed by the removal of tissue debris with 40 μm nylon filter. The resulting single cell suspensions were seeded on poly-D-lysine-coated culture dishes or glass coverslips. Unless otherwise specified, for live imaging experiments, neurons were plated at a density of 0.24 × 106 cells/cm2 on glass bottom dishes with 14 mm microwells (Cellvis). These neurons were allowed to attach for 1.5 h, followed by feeding with fresh culture medium: Neurobasal supplemented with B27 and Glutamax, without antibiotics. Thereafter, half of the culture media were replaced every 3 days. All treatments and transfections were performed in culture media containing at least 50% conditioned media until neurons were imaged on 5–7 DIV.
Alternatively, neurons were seeded in microfluidics devices with 450 μm microgrooves. Microfluidics devices were pre-treated with pentane, xylenes, ethanol and sterile water as described (Millet et al., 2007). Dried devices were sterilized under UV for 10 min and were attached to poly-D-lysine-coated glass bottom dishes with 30 mm microwells (Cellvis). Interior surfaces of the devices were pre-wetted with the plating medium (Neurobasal culture medium containing 10% heat-inactivated FBS) for 3–24 h before cell plating. Aliquots of cells suspended in DMEM/F12 described above were centrifuged at 1000 rpm for 7 min. These pellets were resuspended with the plating medium to a concentration of 5 × 106 cells/ml, from which 15 μl was seeded in the somal compartment of microfluidics devices. This resulted in a relatively low cell density to help prevent the growth of multiple axons through a single microgroove. After 40 min, extra plating medium was added. Somal wells were maintained at a higher fluid level than axonal wells. Plating media were completely replaced with conditioned culture media from feeder neurons after 24 h. Thereafter, neurons were fed with a mixture of 50% conditioned and 50% fresh culture media every 3 days. All treatments were performed in culture media containing at least 50% conditioned media until neurons were imaged or harvested on 7–10 DIV.
Construction of plasmids for transfection in neurons
For the construction of LAMP1/pmCherry-N1, LAMP1 cDNA was obtained by EcoRI and BamHI digestion of LAMP1/pEYFP-N1 and cloned in-frame into the EcoRI and BamHI sites of pmCherry-N1 vector. For the construction of ACP2/pmCherry-N1, ACP2 cDNA was amplified from ACP2/pCMV6-Entry by PCR using primers 5’-ATCTCGAGATTCGTCGACTGGATCCG-3’ (forward) and 5’-ATACCGGTGTACGCGTAGCATGGTCT-3’ (reverse), digested with XhoI and AgeI, and cloned in-frame into the XhoI and AgeI sites of pmCherry-N1 vector. All expression plasmids used in this study were transformed into either XL10-Gold Ultracompetent Cells (Agilent) or DH5α Competent Cells (Thermo Fisher) and purified with EndoFree Plasmid Maxi Kit (QIAGEN) according to manufacturer’s instructions.
Transfection and treatments of cultured neurons
For the expression of fluorescent tagged proteins in neurons cultured in 14 mm glass bottom microwells, transfection was performed on 3–5 DIV using Lipofectamine 2000 (Thermo Fisher) according to manufacturer’s instructions with modifications. For the transfection of a single plasmid, neurons were incubated with a transfection mix containing Opti-MEM, 0.368 μg DNA and 1.05 μl Lipofectamine for 50 min. For the co-transfection of two plasmids, neurons were incubated with a transfection mix containing Opti-MEM, 0.375 μg DNA and 0.625 μl Lipofectamine for 24 h. Thereafter, cells were washed and fed with a mixture of 50% conditioned and 50% fresh culture media. Live imaging was performed 48 h after transfection starts. In select experiments, neurons were incubated with one or more of the following reagents diluted in culture medium prior to live imaging or fixation: LysoTracker-Red, 0.25 μM for 22 min; LysoTracker-Deep Red, 0.025 μM for 10 min; dextran-Oregon Green 488 and dextran-Alexa Fluor 647, each at 0.1 mg/ml for 16 h; Bodipy FL-Pepstatin A, 0.96 μM for 30 min; Magic Red cathepsin B, 1:2000 for 30 min; pHrodo Red dextran, 50 μg/ml for 16 hr. For the equilibration of organelles to standard pH, neurons were treated with dextran-Oregon Green 488 and dextran Alexa Fluor 647 as described above, and were subsequently rinsed and incubated for 1.5 hr with 10 μM monensin and 10 μM nigericin in MES buffer (5 nM NaCl, 115 mM KCL, 1.3 mM MgSO4, 25 mM MES) adjusted to desired pH.
Live cell imaging
For imaging of organelles equilibrated to standard pH, neurons were imaged in MES buffer as described above. For all other live imaging, neurons were washed twice in the imaging medium (Hibernate E-low fluorescence supplemented with B27 and Glutamax) and maintained in imaging medium at 37°C. Laser scanning confocal microscopy was performed with a Zeiss LSM 880 or Zeiss LSM 510 confocal microscope. Multi-track line scanning was used when scanning more than 1 channel. Spectrums of detection were adjusted to minimize crosstalk between channels. For axonal transport studies, unless otherwise specified, time-lapse imaging was performed in 3 random segments (~55 μm each) of the proximal-mid axon (50–300 μm from cell body) in each neuron. Axons were identified based on their morphology (long and untapered processes). Each time-lapse series comprises at least 200 images from each channel acquired in a 3-min period. All live imaging data were pooled from at least 3 independent experiments. Subsequent analyses of vesicle movements, signal intensities and/or vesicle dimensions were performed in ImageJ. Time-lapse images were processed with a 1-pixel median filter, followed by the construction of kymographs with the Multiple Kymograph plugin along manually traced axonal profiles. Displayed kymographs were shown at X scale = 1 and Y scale = 4. The entire track of each vesicle was analyzed to calculate average velocity (μm/s) defined as total net displacement/total duration. Vesicles with average velocities of < 0.1 μm/s were classified as “non-motile.” Motile vesicles displaced toward the axon terminal were classified as “anterograde,” and those displaced toward the cell body were classified as “retrograde.” Signal intensities were measured in unadjusted images. Brightness/contrast of displayed images was adjusted in ImageJ and/or Photoshop.
Immunostaining of cultured neurons
Neurons were fixed in methanol at −20°C for 10 min. Thereafter, neurons were permeabilized in 0.02% Triton X-100/ TBS, pH 7.4, and blocked with 10% normal horse serum. Neurons were then incubated with primary antibodies diluted in 1% BSA/ TBS overnight at 40°C and Alexa Fluor-conjugated secondary antibodies. Neurons were mounted with Fluoro-Gel. Soma and random segments of axons were imaged by laser scanning confocal microscopy (multi-track line scanning) using Zeiss LSM 880 confocal microscope. Imaging of dendrites was avoided by ensuring that the imaged neurites had extended beyond microgrooves into the axonal chamber. Images were processed with a 1-pixel median filter and quantified by “particle analysis” function in ImageJ. All measurements were performed on unadjusted images. Brightness/contrast of displayed images was adjusted in ImageJ and/or Photoshop.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data were collected from 3 or more independent experiments with sample sizes specified in figure legends. Analyses were not blinded. All data were included. Normality of data was tested with Shapiro-Wilk test in IBM SPSS Statistics 24. For comparison between 2 groups of normally distributed data, unpaired two-tailed Student’s t test (equal variance not assumed) was performed in Microsoft Excel. For non-parametric comparison between 2 groups, unpaired two-tailed Mann-Whitney U test or Kolmogorov-Smirnov test was performed in IBM SPSS Statistics 24. For non-parametric comparisons of multiple groups, Kruskal-Wallis test followed by Dunn’s multiple comparisons test was performed in GraphPad Prism 8.0.1. Power analysis of sensitivity was performed in G*Power 3.1.9.7 (Faul et al., 2007). Observed effect size d was computed using the standardized test statistic Z from Mann-Whitney U test where , and N = total sample size (Kornbrot et al., 2018; Rosenthal, 1994).
STAR+METHODS
Supplementary Material
KEY RESOURCES TABLE
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-human CTSD | This study | N/A |
| Sheep anti-human CTSD | Cataldo et al., 1990 | N/A |
| Rabbit anti-mouse CTSD | Cataldo et al., 2004 | N/A |
| Sheep anti-TGN38 | Bio-rad | Cat# AHP499G; RRID: AB_2203272 |
| Rabbit anti-CTSD | Scripps Laboratories | Cat# RC245 |
| Goat anti-CTSB | Neuromics | Cat# GT15047; RRID: AB_2737184 |
| Rabbit anti-CTSL | Athens Research & Technology | Cat# 01–12-030112; RRID: AB_11190245 |
| Mouse anti-alpha-Tubulin | Sigma | Cat# T9026; RRID: AB_477593 |
| Rabbit anti-mTOR | Cell Signaling | Cat# 2983s; RRID: AB_2105622 |
| Rabbit anti-p-mTOR | Cell Signaling | Cat# 2974s; RRID: AB_2262884 |
| Rabbit anti-p-ATG14 | Cell Signaling | Cat# 13155; RRID: AB_2798133 |
| Rabbit anti-TFEB | Bethyl Laboratories | Cat# A303–673A; RRID: AB_11204751 |
| Rabbit anti-TFE3 | Sigma | Cat# HPA023881; RRID: AB_1857931 |
| Rabbit anti-ubiquitin | Dako | Cat# Z0458; RRID: AB_2315524 |
| Rabbit anti-LC3 | Novus Biologicals | Cat# NB100–2220; RRID: AB_10003146 |
| Murine anti-NeuN | Millipore | Cat# MAB377MI; RRID: AB_2298772 |
| Rabbit anti-GFAP | Sigma | Cat# G9269; RRID: AB_477035 |
| Rabbit anti-Iba-1 | Waco Chemicals | Cat# 019–19741; RRID: AB_839504 |
| Rat anti-LAMP1 | DSHB | Cat# 1D4B; RRID: AB_2134500 |
| Rabbit anti-IGF2R (CI-MPR) | Sigma | Cat# HPA011332; RRID: AB_1846276 |
| Sheep anti-mouse M6PR (CD-MPR) | LSBio | Cat# LS-C94323; RRID: AB_1935265 |
| Goat anti-mouse IgG, Alexa Fluor 647-conjugated | Thermo Fisher | Cat# A-21236; RRID: AB_2535805 |
| Goat anti-mouse IgG, Alexa Fluor 488-conjugated | Thermo Fisher | Cat# A-11029; RRID: AB_138404 |
| Goat anti-rabbit IgG, Alexa Fluor 568-conjugated | Thermo Fisher | Cat# A-11036; RRID: AB_10563566 |
| Donkey anti-rat IgG, Alexa Fluor 647-conjugated | Jackson ImmunoReseach | Cat# 712–605-153; RRID: AB_2340694 |
| Donkey anti-rabbit IgG, Alexa Fluor 546-conjugated | Thermo Fisher | Cat# A-10040; RRID: AB_2534016 |
| Donkey anti-sheep IgG, Alexa Fluor 488-conjugated | Thermo Fisher | Cat# A-11015; RRID: AB_141362 |
| Goat anti-rabbit IgG, biotinylated | Vector Laboratories | Cat# BA-1000; RRID: AB_2313606 |
| Goat anti-mouse IgG, biotinylated | Vector Laboratories | Cat# BA-2000; RRID: AB_2313581 |
| Donkey anti-rabbit IgG, 6 nm gold-conjugated | Electron Microscopy Sciences | Cat# 25703 |
| Donkey anti-mouse IgG, 6 nm gold-conjugated | Electron Microscopy Sciences | Cat# 25813 |
| Donkey anti-sheep IgG, 10 nm gold-conjugated | Electron Microscopy Sciences | Cat# 25825 |
| Chemicals, peptides, and recombinant proteins | ||
| Sodium cacodylate buffer | Electron Microscopy Sciences | Cat# 11652 |
| Permount | Electron Microscopy Sciences | Cat# 17986 |
| Fluoro-Gel | Electron Microscopy Sciences | Cat# 17985–10 |
| Vectorstain ABC Kit | Vector Laboratories | Cat# PK-4000 |
| Vector DAB Peroxidase Substrate | Vector Laboratories | Cat# SK-4100 |
| M.O.M. Immunodetection Kit | Vector Laboratories | Cat# BMK-2202 |
| Prolong Diamond Antifade Mountant | Thermo Fisher | Cat# P36970 |
| 7-methoxycoumarin-4-acetyl-Gly- Lys-Pro- Ile-Leu-Phe-Phe-Arg-Leu-Lys(Dnp)-D-Arg-NH2 |
Enzo Life Sciences | Cat# BML-P145–0001 |
| Hibernate E | Brainbits | Cat# HE |
| Hibernate E low fluorescence | Brainbits | Cat# HELF |
| Papain | Worthington Biochemical | Cat# LK003178 |
| DNase | Sigma | Cat# DN25 |
| Poly-D-lysine | Sigma | Cat# P0899 |
| DMEM/F12 | Thermo Fisher | Cat# 10565–018 |
| HEPES | Thermo Fisher | Cat# 05630 |
| FBS | Thermo Fisher | Cat# 26140 |
| Horse serum | Thermo Fisher | Cat# 16050 |
| Neurobasal medium | Thermo Fisher | Cat# 12348–017 |
| B27 | Thermo Fisher | Cat# 17504–044 |
| Glutamax | Thermo Fisher | Cat# 35050–061 |
| Lipofectamine 2000 transfection reagent | Thermo Fisher | Cat# 11668–030 |
| Opti-MEM I | Thermo Fisher | Cat# 51985–034 |
| LysoTracker-Red | Thermo Fisher | Cat# L7528 |
| LysoTracker-Deep Red | Thermo Fisher | Cat# L12492 |
| Dextran-Alexa Fluor 647, 10,000 MW | Thermo Fisher | Cat# D22914 |
| Dextran-Oregon Green 488, 10,000 MW | Thermo Fisher | Cat# D7170 |
| Bodipy FL-pepstatinA | Thermo Fisher | Cat# P12271 |
| pHrodo Red dextran, 10,000 MW | Thermo Fisher | Cat# P10361 |
| Magic Red cathepsin B | ImmunoChemistry Technologies | Cat# 937 |
| Endofree Plasmid Maxi Kit | QIAGEN | Cat# 12362 |
| Experimental models: organisms/strains | ||
| Mouse: hCTSD Tg | This study | N/A |
| Mouse: TRGL6 | Lee et al., 2019 | N/A |
| Mouse: TRGL x hCTSD Tg | This study | N/A |
| Mouse: PS/APP | Holcomb et al., 1998 | N/A |
| Mouse: C57/BL6, timed-pregnant | Charles River | N/A |
| Oligonucleotides | ||
| Forward primer for genotyping hCTSD: 5’-TTC CCC ACC ACA GAA TCC-3’ |
This study | N/A |
| Reverse primer for genotyping hCTSD: 5’-CTT TCA GAC GGG GCC TTT GGC-3’ |
This study | N/A |
| Forward primer for genotyping TRGL: 5’-CTT TCC CCA CAG AAT CCA AGT CGG AAC-3’ |
Lee et al., 2019 | N/A |
| Reverse primer for genotyping TRGL: 5’-GCA CGA ATT CGG GCG CCG GTG GAG TGG CGG-3’ |
Lee et al., 2019 | N/A |
| Fabpi-200 forward primer (control for genotyping): 5’-TGG ACA GGA CTG GAC CTC TGC TTT CCT AGA-3’ |
Lee et al., 2019 | N/A |
| Fabpi-200 reverse primer (control for genotyping): 5’-TAG AGC TTT GCC ACA TCA CAG GTC ATT CAG-3’ |
Lee et al., 2019 | N/A |
| Forward primer for amplifying ACP2: 5’-ATCTCGAGATTCGTCGACTGGATCCG-3’ |
This study | N/A |
| Reverse primer for amplifying ACP2: 5’-ATACCGGTGTACGCGTAGCATGGTCT-3’ |
This study | N/A |
| Recombinant DNA | ||
| hCTSD/pcDNA3 | A gift from Dr. Paul Mathews, Nathan Kline Institute, Orangeburg, NY, USA |
N/A |
| pTSC21 (Thy1.2 expression cassette) | A gift from Dr. Matthias Staufenbiel (Andrä et al., 1996) |
N/A |
| LAMP1/pEYFP-N1 | A gift from Dr. J Swanson, University of Michigan, Ann Arbor, MI, USA |
N/A |
| TGOLN2/pmCherry-N1 | A gift from Dr. Michael Davidson | Addgene Cat# 55145; RRID: Addgene_55145 |
| ACP2/pCMV6-Entry | Origene | Cat# MR206733 |
| ACP2/pmCherry-N1 | This study | N/A |
| pmCherry-N1 | Clontech | Cat# 632523 |
| LAMP1/pmCherry-N1 | This study | N/A |
| MCOLN1/pEGFP-C3 (TRPML1) | A gift from Dr. Paul Luzio | Addgene Cat# 62960; RRID: Addgene_62960 |
| Rab7/pDsRed-C1 | A gift from Dr. Richard Pagano | Addgene Cat# 12661; RRID: Addgene_12661 |
| Rab7/pEGFP | A gift from Dr. Anne Cataldo (Lee et al., 2011) |
N/A |
| mRFP-eGFP-LC3 | A gift from Dr. Tamotsu Yoshimori (Kimura et al., 2007) |
N/A |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Adobe Photoshop CS3 | Adobe | N/A |
| IBM SPSS Statistics 24 | IBM | N/A |
| GraphPad Prism 8.0.1 | GraphPad Software | N/A |
| GPower 3.1.9.7 | Faul et al., 2007 | N/A |
| Other | ||
| Microfluidic devices 450 μm | Xona | Cat# RD450 |
Highlights.
Fully mature classic lysosomes are restricted from axons in intact brains
Transport carriers (TCs) deliver lysosomal components from TGN to axonal organelles
TC delivery of lysosomal components is upregulated in dystrophic axons of AD models
Non-degradative bidirectional TCs in axons are commonly misidentified as lysosomes
ACKNOWLEDGMENTS
We thank Dr. Monika Pawlik, Dr. Efrat Levy, Dr. Mala V. Rao, and Chunfeng Huo for maintenance of mouse colonies. This work was supported by NIH P01AG017617 and R01AG062376 to R.A.N.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109034.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abeliovich A, and Gitler AD (2016). Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539, 207–216. [DOI] [PubMed] [Google Scholar]
- Andrä K, Abramowski D, Duke M, Probst A, Wiederhold KH, Bürki K, Goedert M, Sommer B, and Staufenbiel M. (1996). Expression of APP in transgenic mice: a comparison of neuron-specific promoters. Neurobiol. Aging 17, 183–190. [DOI] [PubMed] [Google Scholar]
- Ashrafi G, Schlehe JS, LaVoie MJ, and Schwarz TL (2014). Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol 206, 655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Authier F, Posner BI, and Bergeron JJ (1996). Endosomal proteolysis of internalized proteins. FEBS Lett. 389, 55–60. [DOI] [PubMed] [Google Scholar]
- Bagh MB, Peng S, Chandra G, Zhang Z, Singh SP, Pattabiraman N, Liu A, and Mukherjee AB (2017). Misrouting of v-ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model. Nat. Commun 8, 14612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banting G, and Ponnambalam S. (1997). TGN38 and its orthologues: roles in post-TGN vesicle formation and maintenance of TGN morphology. Biochim. Biophys. Acta 1355, 209–217. [DOI] [PubMed] [Google Scholar]
- Benes P, Vetvicka V, and Fusek M. (2008). Cathepsin D–many functions of one aspartic protease. Crit. Rev. Oncol. Hematol 68, 12–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, and Nixon RA (2008). Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci 28, 6926–6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomati EK, Haley JE, Noel JP, and Deheyn DD (2014). Spectral and structural comparison between bright and dim green fluorescent proteins in Amphioxus. Sci. Rep 4, 5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S, Ginsberg SD, and Nixon RA (2016). Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 12, 2467–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadwell RD, and Cataldo AM (1984). The neuronal endoplasmic reticulum: its cytochemistry and contribution to the endomembrane system. II. Axons and terminals. J. Comp. Neurol 230, 231–248. [DOI] [PubMed] [Google Scholar]
- Carlsson SR, and Fukuda M. (1992). The lysosomal membrane glycoprotein lamp-1 is transported to lysosomes by two alternative pathways. Arch. Biochem. Biophys 296, 630–639. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Thayer CY, Bird ED, Wheelock TR, and Nixon RA (1990). Lysosomal proteinase antigens are prominently localized within senile plaques of Alzheimer’s disease: evidence for a neuronal origin. Brain Res. 513, 181–192. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Paskevich PA, Kominami E, and Nixon RA (1991). Lysosomal hydrolases of different classes are abnormally distributed in brains of patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 88, 10998–11002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztajn S, Lippa C, and Nixon RA (1995). Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron 14, 671–680. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Schmidt SD, Terio NB, Duff K, Beard M, Mathews PM, and Nixon RA (2004). Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J. Neuropathol. Exp. Neurol 63, 821–830. [DOI] [PubMed] [Google Scholar]
- Chen Y, Gershlick DC, Park SY, and Bonifacino JS (2017). Segregation in the Golgi complex precedes export of endolysosomal proteins in distinct transport carriers. J. Cell Biol 216, 4141–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng XT, Zhou B, Lin MY, Cai Q, and Sheng ZH (2015). Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J. Cell Biol 209, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng XT, Xie YX, Zhou B, Huang N, Farfel-Becker T, and Sheng ZH (2018). Characterization of LAMP1-labeled nondegradative lysosomal and endocytic compartments in neurons. J. Cell Biol 217, 3127–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colacurcio DJ, and Nixon RA (2016). Disorders of lysosomal acidificationThe emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev 32, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damme M, Suntio T, Saftig P, and Eskelinen EL (2015). Autophagy in neuronal cells: general principles and physiological and pathological functions. Acta Neuropathol. 129, 337–362. [DOI] [PubMed] [Google Scholar]
- De Luca M, Cogli L, Progida C, Nisi V, Pascolutti R, Sigismund S, Di Fiore PP, and Bucci C. (2015). RILP regulates vacuolar ATPase through interaction with the V1G1 subunit. J. Cell Sci 128, 2565. [DOI] [PubMed] [Google Scholar]
- Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, Bucci C, and Schiavo G. (2006). Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 52, 293–305. [DOI] [PubMed] [Google Scholar]
- Demaurex N, Furuya W, D’Souza S, Bonifacino JS, and Grinstein S. (1998). Mechanism of acidification of the trans-Golgi network (TGN). In situ measurements of pH using retrieval of TGN38 and furin from the cell surface. J. Biol. Chem 273, 2044–2051. [DOI] [PubMed] [Google Scholar]
- Evans CS, and Holzbaur EL (2020). Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. eLife 9, e50260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfel-Becker T, Roney JC, Cheng XT, Li S, Cuddy SR, and Sheng ZH (2019). Neuronal Soma-Derived Degradative Lysosomes Are Continuously Delivered to Distal Axons to Maintain Local Degradation Capacity. Cell Rep. 28, 51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farías GG, Guardia CM, De Pace R, Britt DJ, and Bonifacino JS (2017). BORC/kinesin-1 ensemble drives polarized transport of lysosomes into the axon. Proc. Natl. Acad. Sci. USA 114, E2955–E2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Lang AG, and Buchner A. (2007). G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 39, 175–191. [DOI] [PubMed] [Google Scholar]
- Ghosh D, Pinto S, Danglot L, Vandewauw I, Segal A, Van Ranst N, Benoit M, Janssens A, Vennekens R, Vanden Berghe P, et al. (2016). VAMP7 regulates constitutive membrane incorporation of the cold-activated channel TRPM8. Nat. Commun 7, 10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar S, Yuan P, Wu Y, Schrag M, Paradise S, Grutzendler J, De Camilli P, and Ferguson SM (2015). Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc. Natl. Acad. Sci. USA 112, E3699–E3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar S, Wu Y, and Ferguson SM (2017). Impaired JIP3-dependent axonal lysosome transport promotes amyloid plaque pathology. J. Cell Biol 216, 3291–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Jeong YY, Sheshadri P, Su X, and Cai Q. (2020). Mitophagy regulates integrity of mitochondria at synapses and is critical for synaptic maintenance. EMBO Rep. 21, e49801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, and Mizushima N. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. [DOI] [PubMed] [Google Scholar]
- Hendricks AG, Perlson E, Ross JL, Schroeder HW 3rd, Tokito M, and Holzbaur EL (2010). Motor coordination via a tug-of-war mechanism drives bidirectional vesicle transport. Curr. Biol 20, 697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, et al. (1998). Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med 4, 97–100. [DOI] [PubMed] [Google Scholar]
- Janvier K, and Bonifacino JS (2005). Role of the endocytic machinery in the sorting of lysosome-associated membrane proteins. Mol. Biol. Cell 16, 4231–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin EJ, Kiral FR, Ozel MN, Burchardt LS, Osterland M, Epstein D, Wolfenberg H, Prohaska S, and Hiesinger PR (2018). Live Observation of Two Parallel Membrane Degradation Pathways at Axon Terminals. Curr. Biol 28, 1027–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DE, Ostrowski P, Jaumouillé V, and Grinstein S. (2016). The position of lysosomes within the cell determines their luminal pH. J. Cell Biol 212, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Noda T, and Yoshimori T. (2007). Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescenttagged LC3. Autophagy 3, 452–460. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, and Tanaka K. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. [DOI] [PubMed] [Google Scholar]
- Kornbrot DE, Wiseman R, and Georgiou GJ (2018). Quality science from quality measurement: The role of measurement type with respect to replication and effect size magnitude in psychological research. PLoS ONE 13, e0192808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers M, Azarnia Tehran D, Haucke V, and Soykan T. (2020). The axonal endolysosomal and autophagic systems. J. Neurochem. Published online December 28, 2020. 10.1111/jnc.15287. [DOI] [PubMed] [Google Scholar]
- Lalli G, and Schiavo G. (2002). Analysis of retrograde transport in motor neurons reveals common endocytic carriers for tetanus toxin and neurotrophin receptor p75NTR. J. Cell Biol 156, 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sato Y, and Nixon RA (2011). Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J. Neurosci 31, 7817–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Rao MV, Yang DS, Stavrides P, Im E, Pensalfini A, Huo C, Sarkar P, Yoshimori T, and Nixon RA (2019). Transgenic expression of a ratiometric autophagy probe specifically in neurons enables the interrogation of brain autophagy in vivo. Autophagy 15, 543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SC, Diakov TT, Xu T, Tarsio M, Zhu W, Couoh-Cardel S, Weisman LS, and Kane PM (2014). The signaling lipid PI(3,5)P2 stabilizes V1-V(o) sector interactions and activates the V-ATPase. Mol. Biol. Cell 25, 1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Qin L, Li Y, Yu H, Zhang Z, Tao C, Liu Y, Xue Y, Zhang X, Xu Z, et al. (2019). Presynaptic Endosomal Cathepsin D Regulates the Biogenesis of GABAergic Synaptic Vesicles. Cell Rep. 28, 1015–1028. [DOI] [PubMed] [Google Scholar]
- Lie PPY, and Nixon RA (2019). Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis 122, 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Xing R, Jian Y, Gao Z, Ma X, Sun X, Li Y, Xu M, Wang X, Jing Y, et al. (2017). WDR91 is a Rab7 effector required for neuronal development. J. Cell Biol 216, 3307–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luini A, Mironov AA, Polishchuk EV, and Polishchuk RS (2008). Morphogenesis of post-Golgi transport carriers. Histochem. Cell Biol 129, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Wallace KE, and Holzbaur EL (2012). Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol 196, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies FM, Fleming A, and Rubinsztein DC (2015). Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci 16, 345–357. [DOI] [PubMed] [Google Scholar]
- Meyer C, Zizioli D, Lausmann S, Eskelinen EL, Hamann J, Saftig P, von Figura K, and Schu P. (2000). mu1A-adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. EMBO J. 19, 2193–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miedel MT, Weixel KM, Bruns JR, Traub LM, and Weisz OA (2006). Posttranslational cleavage and adaptor protein complex-dependent trafficking of mucolipin-1. J. Biol. Chem 281, 12751–12759. [DOI] [PubMed] [Google Scholar]
- Millet LJ, Stewart ME, Sweedler JV, Nuzzo RG, and Gillette MU (2007). Microfluidic devices for culturing primary mammalian neurons at low densities. Lab Chip 7, 987–994. [DOI] [PubMed] [Google Scholar]
- Nakata T, Terada S, and Hirokawa N. (1998). Visualization of the dynamics of synaptic vesicle and plasma membrane proteins in living axons. J. Cell Biol 140, 659–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA (2013). The role of autophagy in neurodegenerative disease. Nat. Med 19, 983–997. [DOI] [PubMed] [Google Scholar]
- Nixon RA (2016). The lysosome in aging-related neurodegenerative diseases. In Lysosomes: Biology, Diseases, and Therapeutics, Maxfield FR, Willard JM, and Lu S, eds. (John Wiley & Sons, Inc.). [Google Scholar]
- Nixon RA, and Marotta CA (1984). Degradation of neurofilament proteins by purified human brain cathepsin D. J. Neurochem 43, 507–516. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, and Cuervo AM (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol 64, 113–122. [DOI] [PubMed] [Google Scholar]
- Overly CC, and Hollenbeck PJ (1996). Dynamic organization of endocytic pathways in axons of cultured sympathetic neurons. J. Neurosci 16, 6056–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overly CC, Lee KD, Berthiaume E, and Hollenbeck PJ (1995). Quantitative measurement of intraorganelle pH in the endosomal-lysosomal pathway in neurons by using ratiometric imaging with pyranine. Proc. Natl. Acad. Sci. USA 92, 3156–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG, Simons K, and Dotti CG (1992). Axonal and dendritic endocytic pathways in cultured neurons. J. Cell Biol 119, 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pols MS, van Meel E, Oorschot V, ten Brink C, Fukuda M, Swetha MG, Mayor S, and Klumperman J. (2013). hVps41 and VAMP7 function in direct TGN to late endosome transport of lysosomal membrane proteins. Nat. Commun 4, 1361. [DOI] [PubMed] [Google Scholar]
- Pu J, Schindler C, Jia R, Jarnik M, Backlund P, and Bonifacino JS (2015). BORC, a multisubunit complex that regulates lysosome positioning. Dev. Cell 33, 176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu J, Keren-Kaplan T, and Bonifacino JS (2017). A Ragulator-BORC interaction controls lysosome positioning in response to amino acid availability. J. Cell Biol 216, 4183–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R. (1994). Parametric measures of effect size. In Handbook of Research Synthesis, Cooper H. and Hedges LV, eds. (Russell Sage Foundation; ), pp. 231–244. [Google Scholar]
- Saftig P, and Klumperman J. (2009). Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol 10, 623–635. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, De Angelis D, Rothman JE, and Ryan TA (2000). The use of pHluorins for optical measurements of presynaptic activity. Biophys. J 79, 2199–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim JH, Xiao C, Hayden MS, Lee KY, Trombetta ES, Pypaert M, Nara A, Yoshimori T, Wilm B, Erdjument-Bromage H, et al. (2006). CHMP5 is essential for late endosome function and down-regulation of receptor signaling during mouse embryogenesis. J. Cell Biol 172, 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snouwaert JN, Church RJ, Jania L, Nguyen M, Wheeler ML, Saintsing A, Mieczkowski P, Manuel de Villena FP, Armao D, Moy SS, et al. (2018). A Mutation in the Borcs7 Subunit of the Lysosome Regulatory BORC Complex Results in Motor Deficits and Dystrophic Axonopathy in Mice. Cell Rep. 24, 1254–1265. [DOI] [PubMed] [Google Scholar]
- Suzuki K, and Terry RD (1967). Fine structural localization of acid phosphatase in senile plaques in Alzheimer’s presenile dementia. Acta Neuropathol. 8, 276–284. [DOI] [PubMed] [Google Scholar]
- Tammineni P, Ye X, Feng T, Aikal D, and Cai Q. (2017). Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. eLife 6, e21776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Ueno T, and Uchiyama Y. (2014). A super-ecliptic, pHluorinmKate2, tandem fluorescent protein-tagged human LC3 for the monitoring of mammalian autophagy. PLoS ONE 9, e110600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, and Ishikawa H. (1980). The movement of membranous organelles in axons. Electron microscopic identification of anterogradely and retrogradely transported organelles. J. Cell Biol 84, 513–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urwin H, Authier A, Nielsen JE, Metcalf D, Powell C, Froud K, Malcolm DS, Holm I, Johannsen P, Brown J, et al. ; FReJA Consortium (2010). Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum. Mol. Genet 19, 2228–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukoja A, Rey U, Petzoldt AG, Ott C, Vollweiter D, Quentin C, Puchkov D, Reynolds E, Lehmann M, Hohensee S, et al. (2018). Presynaptic Biogenesis Requires Axonal Transport of Lysosome-Related Vesicles. Neuron 99, 1216–1232. [DOI] [PubMed] [Google Scholar]
- Xu H, and Ren D. (2015). Lysosomal physiology. Annu. Rev. Physiol 77, 57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Wang LR, Sato Y, Jiang Y, Berg M, Yang DS, Nixon RA, and Liang XJ (2014). Single-walled carbon nanotubes alleviate autophagic/lysosomal defects in primary glia from a mouse model of Alzheimer’s disease. Nano Lett. 14, 5110–5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, and Yue Z. (2014). Autophagy and its normal and pathogenic states in the brain. Annu. Rev. Neurosci 37, 55–78. [DOI] [PubMed] [Google Scholar]
- Yang DS, Kumar A, Stavrides P, Peterson J, Peterhoff CM, Pawlik M, Levy E, Cataldo AM, and Nixon RA (2008). Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer’s disease. Am. J. Pathol 173, 665–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DS, Lee JH, and Nixon RA (2009). Monitoring autophagy in Alzheimer’s disease and related neurodegenerative diseases. Methods Enzymol. 453, 111–144. [DOI] [PubMed] [Google Scholar]
- Yap CC, Digilio L, McMahon LP, Garcia ADR, and Winckler B. (2018). Degradation of dendritic cargos requires Rab7-dependent transport to somatic lysosomes. J. Cell Biol 217, 3141–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda Y, Kageyama T, Akamine A, Shibata M, Kominami E, Uchiyama Y, and Yamamoto K. (1999). Characterization of new fluorogenic substrates for the rapid and sensitive assay of cathepsin E and cathepsin D. J. Biochem 125, 1137–1143. [DOI] [PubMed] [Google Scholar]
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. (2010). Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Zhang X, Wu X, Jiang L, Ahsan A, Ma S, Xiao Z, Han F, Qin ZH, Hu W, and Chen Z. (2019). Somatic autophagy of axonal mitochondria in ischemic neurons. J. Cell Biol 218, 1891–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No large datasets or custom code were generated in this study.