

Abstract
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic is undeniably the most severe global health emergency since the 1918 Influenza outbreak. Depending on its evolutionary trajectory, the virus is expected to establish itself as an endemic infectious respiratory disease exhibiting seasonal flare-ups. Therefore, despite the unprecedented rally to reach a vaccine that can offer widespread immunization, it is equally important to reach effective prevention and treatment regimens for coronavirus disease 2019 (COVID-19). Contributing to this effort, we have curated and analyzed multi-source and multi-omics publicly available data from patients, cell lines and databases in order to fuel a multiplex computational drug repurposing approach. We devised a network-based integration of multi-omic data to prioritize the most important genes related to COVID-19 and subsequently re-rank the identified candidate drugs. Our approach resulted in a highly informed integrated drug shortlist by combining structural diversity filtering along with experts’ curation and drug–target mapping on the depicted molecular pathways. In addition to the recently proposed drugs that are already generating promising results such as dexamethasone and remdesivir, our list includes inhibitors of Src tyrosine kinase (bosutinib, dasatinib, cytarabine and saracatinib), which appear to be involved in multiple COVID-19 pathophysiological mechanisms. In addition, we highlight specific immunomodulators and anti-inflammatory drugs like dactolisib and methotrexate and inhibitors of histone deacetylase like hydroquinone and vorinostat with potential beneficial effects in their mechanisms of action. Overall, this multiplex drug repurposing approach, developed and utilized herein specifically for SARS-CoV-2, can offer a rapid mapping and drug prioritization against any pathogen-related disease.
Keywords: multiplex drug repurposing, multi-omics integrative analysis, COVID-19
Introduction
The coronavirus disease 2019 (COVID-19) has claimed, at the time of writing, 1.83 million lives with >83.3 million cases worldwide [World Health Organization—Weekly Situation Report 5 January 2021—https://www.who.int/]. Despite the unprecedented number of ongoing independent efforts, this global health emergency remains unanswered in terms of effective treatment(s) against COVID-19 and the responsible virus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
The short-term challenge that needs to be addressed by the scientific community is the realization of therapeutics that can withhold the onslaught of SARS-CoV-2, until vaccines enable mass immunization. The time scale required for identifying, testing and approving new therapeutic compounds is prohibiting for the urgency of the unfolding situation, albeit more relevant as a medium- to long-term response. Thus, the focus is on drug repurposing (DR) of existing therapeutics. Yet, even for the case of existing compounds, the experimental preclinical screening followed by human safety and efficacy trials is a laborious and time-consuming process. Therefore, prioritization of the candidate drugs entering the testing pipeline is necessary. To this effect, computational approaches can expedite the candidate drug prioritization process through a multitude of methodologies [1] that can exhaustively analyze and integrate the available information on a given drug.
Therapeutics can be categorized roughly into (a) those that target the virus itself by blocking its replication and cell entry, leading to the reduction of the viral load of the infected patient or reducing transmission, (b) those aiming to reverse the effects of the disease such as the acute immune response of the host, leading to the well-documented cytokine storm and organ damage [2–4] and (c) both of the above. Therefore, equally important is the analysis of the disease itself towards deciphering the mechanisms through which COVID-19 develops and progresses.
As the amount of available data accumulates, novel avenues of analysis and informed decisions on drug prioritization emerge. The arsenal of high-throughput sequencing (HTS) methodologies has already been utilized to produce a diverse space of omics data, originating from clinical and preclinical setups from samples derived from human, animal hosts and cell lines. Several computational DR strategies were developed for COVID-19. Most of these were focused on network-based approaches by combining the viral and the human interactome and through network-based strategies prioritized the existing drugs [5, 6]. On the other hand, several studies adopted transcriptomic-based in silico DR approaches, utilizing publicly available gene expression data from SARS-CoV-2 and other infectious viruses, to propose candidate repurposed drugs against COVID-19 [7–9]. As over 500 structures of SARS-CoV-2 proteins or protein complexes are available [10], more and more structure-based DR approaches are being developed using molecular docking and dynamic simulations to identify antiviral compounds against COVID-19 [11–17].
The remaining challenge is on how to integrate the heterogeneous data and extract the critical information needed for selecting potentially repurposable drugs against COVID-19.
In this work, we present a multiplex DR scheme against COVID-19 via three discrete approaches, stepping on the analysis of multi-source and multi-omics publicly available data from patients, cell lines and databases. Following the DR approaches, we used a network-based multi-omic data integration approach to prioritize the most important genes related to COVID-19 and subsequently re-rank the identified candidate drugs. By combining drug structural diversity filtering along with experts’ curation and drug–target mapping on the depicted molecular pathways, we compiled an informed integrated shortlist of candidate drugs against COVID-19. The proposed candidates include compounds that were recently found to generate promising results in clinical trials, but also compounds that are presented for the first time in the literature as potential COVID-19 therapeutics.
Methods
Pipeline overview
The workflow of the proposed pipeline in this paper can be described in the following main steps (as illustrated in detail in Figure 1):
Figure 1 .
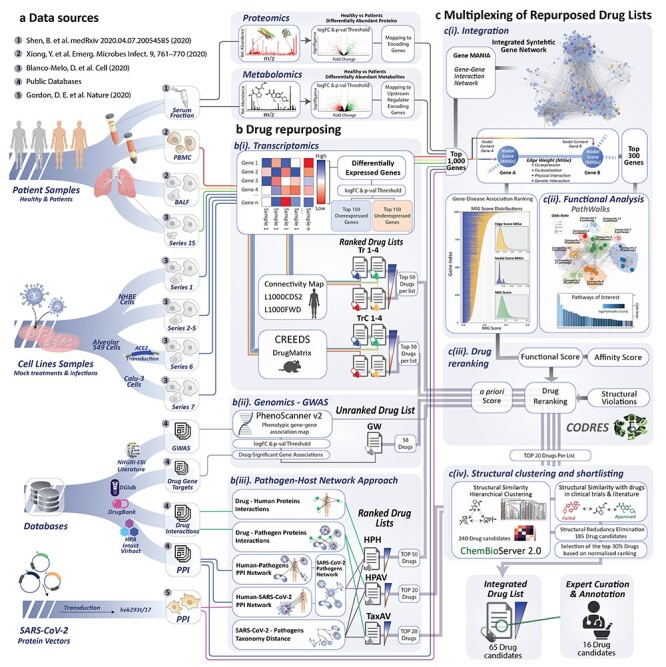
Overview of the workflow. (A) Data sources: multi-omics datasets were selected from various sources in order to identify differentially expressed genes, differentially abundant metabolites and proteins along with protein–protein interactions (PPIs) between SARS-CoV-2 and other pathogens with the human host. This was followed by (B) drug repurposing approaches based on (i) transcriptomics, (ii) genomics—GWAS–phenotype association analysis and (iii) pathogen–host interaction network analysis. (C) Multiplexing of repurposed drug lists: (i) integration of the multi-omic data from patients, (ii) functional analysis, (iii) drug re-ranking based on the calculated target–disease association of the integration map and (iv) structural clustering and shortlisting of drug candidates. Finally, the integrated drug list comprises 65 drugs out of which 16 were manually curated by experts for further annotation.
(A) Multi-omics and protein–protein interaction (PPI) data selection and preprocessing: multi-omics datasets were selected from various available sources in order to identify differentially expressed genes (DEGs), differentially abundant metabolites and proteins along with PPIs between SARS-CoV-2 and other pathogens with the human host.
(B) DR pipelines: multiplex DR approaches were implemented based on (i) transcriptomics analysis, (ii) GWAS–phenotype association analysis and (iii) pathogen–host interaction network analysis in order to generate an initial list of repurposed drugs for COVID-19.
(C) Multi-omic data integration: we developed a ‘network-based multi-source data integration’ methodology to integrate multi-omic data from COVID-19 patients.
(D) Drug re-ranking: the re-ranking of drug candidates was driven by a disease association score of the calculated from the synthetic integration network.
(E) Drug filtering: the structural similarity of candidate drugs was calculated to cluster the top scoring compounds. Finally, we proposed an integrated drug list that comprises 65 drugs out of which 16 were manually curated by experts for further annotation.
Multi-omics and PPI data selection and preprocessing
We selected datasets from multiple sources available at the time of conducting this research. Their description including the number of DEGs, proteins and metabolites with their corresponding selection criteria are shown in Table 1 and their analysis is presented in following sections.
Table 1.
Summary of the data along with the source study
| Source/study method | Dataset name | Sample description | Controls versus infected | Selected differentially expressed genes proteins and metabolites | ||||
|---|---|---|---|---|---|---|---|---|
| Con. | Inf. | Adjusted P-value threshold | logFC | Over | Under | |||
| RNA-Seq/Transcriptomics GSE147507 [18] | Series 1 | Independent biological triplicates of primary human lung epithelium (NHBE) were mock treated or infected with SARS-CoV-2 | 3 | 3 | <0.05 | ≥1 | 94 | 15 |
| Series 2 and 5 | Both Series 2 and 5 have independent biological triplicates of transformed lung alveolar (A549) cells that were mock treated or infected with SARS-CoV-2 | 6 | 6 | <0.05 | ≥1 | 147 | 9 | |
| Series 6 | Independent biological triplicates of transformed lung alveolar (A549) transduced with a vector expressing human ACE2 were mock treated or infected with SARS-CoV-2 | 3 | 3 | <0.05 | ≥1 | 1083 | 352 | |
| Series 7 | Independent biological triplicates of transformed lung-derived Calu-3 cells were mock treated or infected with SARS-CoV-2 | 3 | 3 | <0.05 | ≥1 | 1264 | 660 | |
| Series 15 | Uninfected human lung biopsies from two patients were used as biological replicates. In addition, lung samples derived from a single male COVID-19 deceased patient processed in technical replicates | 2 | 2 | <0.05 | ≥1 | 515 | 3838 | |
| RNA-Seq/Transcriptomics [20] | BALF | Bronchoalveolar lavage fluid (BALF) samples from Zhongnan Hospital of Wuhan University | 3 | 2 | <1(e-10) | ≥2 | 679 | 325 |
| PBMC | Peripheral blood mononuclear cells (PBMC) specimens of COVID-19 patients | 3 | 3 | <0.01 | ≥1 | 707 | 316 | |
| LC–MS/Proteomics [22] | SerumP | COVID-19 patients’ sera and healthy individuals | 28 | 28 | <0.05 | >0.25 | 62 | 58 |
| GC–MS/Metabolomics [22] | SerumM | COVID-19 patients’ sera and healthy individuals | 28 | 28 | <0.05 | >0.25 | 97 | 317 |
Transcriptomics
Transcriptomic (expression profiling by HTS) data were obtained from Gene Expression Omnibus (GEO) accessed on 9 April 2020 with ID GSE147507 [18]. This dataset consists of transcriptional profiling of different cell lines with SARS-CoV-2 infection and of transcriptional profiling of COVID-19 lung biopsies. In our study, we used independent biological triplicates of primary human lung epithelium Normal Human Bronchial Epithelial (NHBE) cells (Series 1), transformed lung alveolar A549 cells (Series 2–5) and transformed lung-derived Calu-3 cells (Series-7), which were mock treated or infected with SARS-CoV-2. We also used independent biological copies of the transformed alveolar lung A549 cells (Series 6) that were transported with a carrier expressing human angiotensin I converting enzyme 2 (ACE2), which were also mock treated or were infected with SARS-CoV-2. Finally, we included uninfected human lung biopsies from two patients that were used as biological replicates and lung samples derived from a single COVID-19 deceased patient that was treated in technical replicates (Series 15). Differential expression analysis of the RNA sequencing (RNA-Seq) data was performed in R programming language using the EdgeR package [19]. We removed low-expressed genes by retaining only those genes that are represented at least in two samples of each group. The messenger RNA counts were normalized using the trimmed mean of M-values method. Finally, we used EdgeR’s negative binomial model to perform the differential expression analysis of infected compared with control samples. From the comparisons, we selected the DEGs by applying selection thresholds of adjusted P-value < 0.05 and log fold-change (logFC) ≥ 1.
In addition, we included RNA-Seq data of RNAs isolated from bronchoalveolar lavage fluid (BALF) and peripheral blood mononuclear cells (PBMC) specimens of COVID-19 patients [20]. Specifically, this dataset included two BALF samples from Zhongnan Hospital of Wuhan University and three BALF samples from healthy controls, downloaded from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database with accession numbers SRR10571724, SRR10571730 and SRR10571732. PBMC samples from three COVID-19 patients and three healthy donors were obtained from Zhongnan Hospital of Wuhan University (Table 1). From these sets, DEGs were selected based on the original publication analysis as described by Xiong et al. [20]. Specifically, DEGs had been identified using the DESeq2 package (v1.26.0) [21]. Selection thresholds on BALF-derived DEGs were set at FC ≥ 4, adjusted P-value < 1e-10 and at least 10 read counts on average across all samples. For the PBMC data, due to lower sequencing depth, the DEGs were selected using FC ≥ 2, adjusted P-value < 0.01 and over 100 read counts on average across all samples.
Due to the heterogeneity of experimental methodologies and samples, the selection thresholds used were different between BALF, PBMC and Series 1, 2–5, 6, 7, 15. For BALF and PBMC, the thresholds were chosen according to the selection criteria applied in the corresponding published studies. For Series 1, 2–5, 6, 7, 15, we analyzed the available raw data and we applied uniform selection criteria.
Proteomics and metabolomics
To further enrich our study, we included proteomic and metabolomic data from Shen et al. [22]. In this study, the authors performed proteomic (SerumP) and metabolomic (SerumM) profiling from sera of COVID-19 and healthy individuals. These samples were procured from 65 patients who visited Taizhou Hospital from January to March 2020. They were diagnosed as COVID-19 according to the Chinese Government Diagnosis and Treatment Guideline. From the analyzed proteomic and metabolomic profiles, 28 severe COVID-19 patients and 28 healthy controls were used in our analysis, and more specifically, the differentially abundant proteins and metabolites, based on adjusted P-value < 0.05 and absolute logFC > 0.25, reported by the authors using a two-sided unpaired Welch’s t-test.
Mapping between metabolites and genes was performed using the Ingenuity Pathway Analysis (IPA) software by Qiagen [23] (http://www.ingenuity.com). Human Metabolome Database (HMDB) Identifiers (IDs), logFC and P-value were used as input into the IPA software, which identified the upstream regulators (Supplementary File S1) that may upregulate or downregulate the input metabolites. The right-tailed Fisher’s exact test was used to obtain the P-values of the identified regulatory genes.
PPI data selection
A set of 336 SARS-CoV-2 human proteins, available in the Human Protein Atlas (HPA) repository, have been further employed in the study. Specifically, the set involves 332 high-confidence SARS-CoV-2–human PPIs, identified by Gordon et al. [24], who had cloned, tagged and expressed 26 SARS-CoV-2 proteins in human cells by means of affinity purification mass spectrometry. Four additional proteins were included: the ACE2 receptor used by SARS-CoVs for host cell entry [25, 26], the serine protease TMPRSS2 involved in the S protein priming [25] and the endosomal cysteine proteases cathepsins CTSB and CTSL needed for viral cell entry, which have been widely expressed in human tissues [25, 26].
PPIs between host and pathogens other than SARS-CoV-2 were downloaded from IntAct [27], PHISTO [28] and VirHostNet [29], amounting to a total of 42 684 unique PPIs from a total of 625 organisms.
DR pipelines
Transcriptomic-based DR
The transcriptomic-based DR was performed based on human cell lines and ex vivo (rat liver) DR tools. The 150 over- and underexpressed genes (based on their FC value) from the BALF, PBMC, Series 15, 1, 2, 5, 6 and 7 were used as transcriptomic signatures. The expression data from Series 2 and 5 were pooled and analyzed prior to the DR rans in a single set since they were generated from the same cell lines and treatments. Next, each set was used as an input in three different transcriptomic-based computational DR tools: Connectivity Map [30], L1000CDS2 [31] and L1000FWD [32]. These tools use transcriptional expression data from multiple human cell lines to probe relationships between diseases and therapeutic agents. Drugs are sorted according to their ‘enrichment’ score, which characterizes if a drug can enhance or reverse the expression levels of a disease based on a given set of genes. Drugs with high negative scores are those that can reverse the gene expression profile towards the normal state.
For each input set, we obtained a candidate list of drugs predicted by each of the three tools, ranked based on their respective reverse enrichment score (inhibition score). Since the output of L1000CDS2 is limited to 50 drugs, we applied the same cutoff for all the other repurposed drug lists. Hence, the top 50 drugs from each of the three tools were combined as their union of unique drugs and ranked by calculating the weighted sum of normalized average rankings and the normalized number of appearances according to Equation (1):
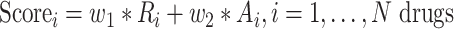 |
(1) |
where 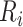 is the average ranking score from each of the three tools,
is the average ranking score from each of the three tools, 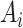 the number of appearances of each drug in the three DR tools, and
the number of appearances of each drug in the three DR tools, and 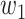 and
and 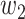 are 0.7 and 0.3, respectively.
are 0.7 and 0.3, respectively.
The drug lists obtained from Series 1, 2 and 5, 6 and 7 sets were combined and re-ranked again using Equation (1) in order to conclude to a single drug list from all cell line-derived RNA-Seq data.
The same gene expression sets were used as input to the CRowd Extracted Expression of Differential Signatures (CREEDS) gene and drug perturbation database [33]. The CREEDS database consists of a list of 4295 single drug perturbations and 8620 single gene perturbations obtained using gene expression data from different tissue types of rat, collected from GEO. The CREEDS database was used herein to extract drugs from the DrugMatrix [33] that are predicted to be able to reverse the disease expression profile of the gene sets of interest. The drugs are ranked by the tool based on Fisher’s exact test-derived P-value. Like above, the top 50 drugs based on the enrichment score returned by CREEDS. Again, the candidate drugs obtained from Series 1, 2 and 5, 6 and 7 were combined and ranked according to Equation (1).
Table 2 shows a summary of the eight drug lists (T1–4, TC1–4) obtained from transcriptomics data.
Table 2.
Summary of the 12 drug lists generated from all repurposing pipelines
| Drug list name | Source DATA | Drug DB | Main pipeline methods and tools | Description |
|---|---|---|---|---|
| Tr1 | RNA-Seq—Series 1, 2 and 5, 6, 7 | LINCS L1000 | Connectivity Map, L1000CDS2, L1000FWD | Drugs aiming at reversing the gene expression profile induced by COVID-19 |
| TrC1 | RNA-Seq—Series 1, 2 and 5, 6, 7 | Drug Matrix (GSE5992) | CREEDS | |
| Tr2 | RNA-Seq—Series 15 | LINCS L1000 | Connectivity Map, L1000CDS2, L1000FWD | |
| TrC2 | RNA-Seq—Series 15 | Drug Matrix (GSE5992) | CREEDS | |
| Tr3 | RNA-Seq—BALF | LINCS L1000 | Connectivity Map, L1000CDS2, L1000FWD | |
| TrC3 | RNA-Seq—BALF | Drug Matrix (GSE5992) | CREEDS | |
| Tr4 | RNA-Seq—PBMC | LINCS L1000 | Connectivity Map, L1000CDS2, L1000FWD | |
| TrC4 | RNA-Seq—PBMC | Drug Matrix (GSE5992) | CREEDS | |
| GW | GWAS | DGIdb | Phenoscanner | Drugs targeting host proteins from genes with significant SNP–phenotypic association with COVID-19 |
| HPH | PPI, NCBI | DrugBank | In-house methods | Drugs targeting host proteins found to interact with SARS-CoV-2 proteins and/or other pathogens |
| HPAV | PPI, NCBI | DrugBank | In-house methods | Drugs targeting pathogen proteins—direct antiviral activity |
| TaxAV | NCBI | DrugBank | In-house methods |
GWAS/phenotype-related COVID-19 DR
The 40 strong SARS-CoV-2 interactors according reported by Gordon et al. [24] were used as an input to PhenoScanner [34, 35]. PhenoScanner returns genotype–phenotype associations across traits and proxies that are collected from various other online databases such as GWAS catalog [36] and CHARGE [37].
Phenotype-associated genes were extracted automatically from PhenoScanner and were used to search for drug interactors. More specifically, the genes were used as input to the Drug–Gene Interaction Database (DGIdb) (www.dgidb.org) [38], which consolidates, organizes and presents drug–gene interactions and gene druggability information from papers, databases and web resources.
Pathogen network-based DR
Pathogen identification and taxonomy-based distances
From the polypeptide targets data file of DrugBank, we extracted the NCBI taxonomy [39] IDs of all organisms other than Homo sapiens contained in DrugBank [40, 41] and have at least one protein that is targeted by a drug. This set was filtered to identify and remove nonpathogens like Metazoa and plants using a custom written script in R in combination with the taxize package to retrieve the taxonomy classification of the organisms. The final list of pathogens included viruses, bacteria and known human parasites from eukaryotes. From the resulting pathogen tax ID list, we constructed their taxonomy tree using R’s taxize package [42]. The distance between each node was calculated using a variable step size between taxonomy ranks proportional to the loss of information at each level as previously proposed [42–44].
DR using the taxonomy distance matrix
Our working assumption was that drugs with a direct inhibitory effect against a given pathogen are more likely to have a similar effect to closely related pathogens in terms of taxonomic distance. Based on this assumption, drugs with protein targets across a broad and diverse range of taxonomy distances are expected to have a less taxon-specific effect, raising the prospect that they are repurposable against SARS-CoV-2, as opposed to drugs known to target only a specific group of distant pathogens. This broad spectrum antipathogenic activity can be captured by the maximum distance of organisms affected by the same drug, whereas the diversity in the inter-taxon distances can be captured using the Shannon Index H, i.e. entropy.
Using the above, we have scored drugs based on function (2):
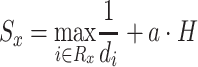 |
(2) |
where Rx is the set of n pathogens with known protein targets for drug x, di is the taxonomy distance of the ith pathogen targeted by drug x and α is the maximum difference of distances d across the pathogens in set Rx, which can be written as:
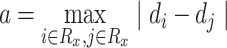 |
(3) |
Finally, the Shannon diversity [45, 46] of distances H can be expressed as:
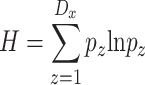 |
(4) |
where Dx is the set of discrete distance values observed in Rx, whereas pz is the ratio of the distance dz frequency over the total number of measured distances in Rx (equal to n–1).
Sx is maximized as di approaches 0, but for larger distances, a drug is favored over other equidistant drugs if it is found to have protein targets over a broader and diverse range of pathogens.
Pathogen–host PPI network-based DR
The approach in this section draws from the assumption that similarities between SARS-CoV-2 and other pathogens at host–protein level may provide the appropriate framework to identify and rank candidate drugs to be used against COVID-19. Specifically, a database repository was initially developed by collecting and combining all the protein and host–protein interactions from all organisms found in IntAct [27], PHISTO [28] and VirHostNet [29] data repositories. The pathogens were identified from the combined unique list of NCBI taxonomy [39] IDs from the aforementioned databases and filtered for nonpathogenic organisms like above. For the case of SARS-CoV-2 pathogen, the 366 host–proteins found in HPA [47] were used, where a pairwise pathogen-to-pathogen network was developed based on common host–proteins in-between pathogens. Herein, the node size represents the number of host–proteins related to a specific pathogen, and the edge weight represents the number of common host–proteins in-between two pathogens. The underlying network revealed 189 pathogens that share common host–proteins with SARS-CoV-2, forming a network of first-neighbors. Based on the assumption that the relation between two pathogens cannot be approximated only by their observed commonality at host–protein level but also by the previously mentioned taxonomy distance D in-between them, the latter network was further enriched with an additional edge score (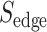 ), according to the following equation:
), according to the following equation:
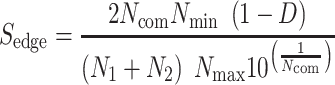 |
(5) |
where 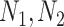 are the number of host–proteins interacting with the two pathogens forming a single edge,
are the number of host–proteins interacting with the two pathogens forming a single edge, 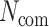 is the number of common host–proteins that two pathogens share,
is the number of common host–proteins that two pathogens share, 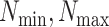 are the maximum and minimum estimations of
are the maximum and minimum estimations of 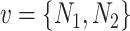 vector, and
vector, and 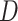 is the taxonomy distance between two pathogens.
is the taxonomy distance between two pathogens.
Herein, the final ranked drug list comes from a specific methodology that scans the edges of the underlying SARS-CoV-2 subnetwork, one by one. Specifically, for each pair of pathogens, drugs were obtained from DrugBank, which target the common host–proteins that form the specific edge. Each drug in the list was ranked according to the following generalized 4-fold equation:
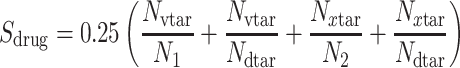 |
(6) |
where 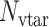 is the number of drug targets included in SARS-CoV-2 host-proteins,
is the number of drug targets included in SARS-CoV-2 host-proteins, 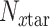 is the number of drug targets included in the x-pathogen that forms an edge with SARS-CoV-2,
is the number of drug targets included in the x-pathogen that forms an edge with SARS-CoV-2, 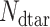 is the total number of drug targets that derive from DrugBank repository [40, 41], and
is the total number of drug targets that derive from DrugBank repository [40, 41], and 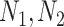 are the total number of host–proteins included in SARS-CoV-2 and the x-pathogen accordingly. Combining Equations (5) and (6) yields the overall drug score (Sd) as follows:
are the total number of host–proteins included in SARS-CoV-2 and the x-pathogen accordingly. Combining Equations (5) and (6) yields the overall drug score (Sd) as follows:
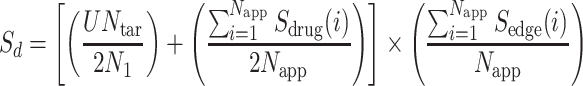 |
(7) |
where 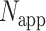 is the number of times a specific drug appeared through the scanning process,
is the number of times a specific drug appeared through the scanning process, 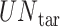 is the number of unique targets appeared in the scan process,
is the number of unique targets appeared in the scan process,  is the number of SARS-CoV-2 host–proteins used and
is the number of SARS-CoV-2 host–proteins used and 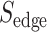 ,
, 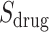 are the estimated scores per edge obtained from Equations (5) and (6) accordingly.
are the estimated scores per edge obtained from Equations (5) and (6) accordingly.
Multi-omic data integration
Towards the integration of multi-source data from patient samples, we developed a ‘network-based multi-source data integration’ methodology based on the integration scheme presented by Zachariou et al. [48].
Data preprocessing
We integrated data from multiple sources in the form of gene lists with two columns, corresponding to gene identity and gene score from the following sources and shown in Supplementary File S1:
Ranked genes lists in terms of absolute logFC from three serum transcriptomic (
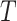 ) datasets (Series 15, BALF, PBMC) [18];
) datasets (Series 15, BALF, PBMC) [18];Ranked genes lists in terms of absolute log fold from one proteomics (
 ) dataset [22];
) dataset [22];Ranked genes lists in terms of P-value from one metabolomics (
 ) dataset [22];
) dataset [22];Unranked list of host proteins (PPI), which interact with SARS-CoV-2 from Gordon et al. [24];
Unranked unique gene list from HPA, excluding the genes identified by Gordon et al.
The gene symbols in all the gene lists were converted to entrez ID for consistent merging to a single gene ID using the R package org.Hs.eg.db [49] for genome-wide annotation for human. For the three transcriptomics and the proteomics dataset, the gene score 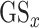 per list was calculated based on
per list was calculated based on
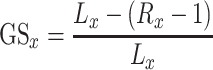 |
(8) |
where 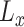 is the total number of genes per list and
is the total number of genes per list and 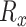 is the rank of each gene based on their absolute logFC for
is the rank of each gene based on their absolute logFC for 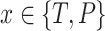 and for
and for 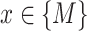 based on their P-value.
based on their P-value. 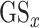 is in canonical form as it takes values
is in canonical form as it takes values 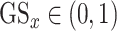 for all gene lists. For the two lists from HPA and Gordon et al. (PPI), their gene score was assigned to 1.0 for all genes.
for all gene lists. For the two lists from HPA and Gordon et al. (PPI), their gene score was assigned to 1.0 for all genes.
The three transcriptomic gene lists were merged and the final score per gene was assigned to be the average 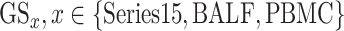 across the three datasets.
across the three datasets.
Gene–gene synthetic network and gene prioritization
We calculated a characteristic score per gene, known as the Multi-source Information Gain (MIG) comprised by two parts:
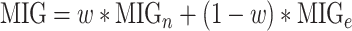 |
(9) |
where 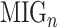 represents the normalized integrated nth gene-specific information (i.e. node characteristics) and
represents the normalized integrated nth gene-specific information (i.e. node characteristics) and 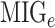 represents the normalised integrated gene–gene information (based on the topology of the multi-integrated super network) and corresponds to the weighted degree of the multi-integrated super network. We considered equal contribution to the score of the gene-specific information and of the topology of the integrated gene–gene super network (
represents the normalised integrated gene–gene information (based on the topology of the multi-integrated super network) and corresponds to the weighted degree of the multi-integrated super network. We considered equal contribution to the score of the gene-specific information and of the topology of the integrated gene–gene super network (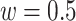 ).
).
The gene-specific information is given by
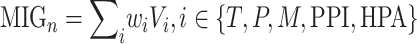 |
(10) |
where 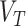 is a vector corresponding to the average gene score of the three transcriptomic datasets (Series 15, BALF, PBMC),
is a vector corresponding to the average gene score of the three transcriptomic datasets (Series 15, BALF, PBMC), 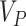 is a vector corresponding to the scored gene list from the proteomics,
is a vector corresponding to the scored gene list from the proteomics, 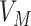 is a vector corresponding to the ranked genes from the dataset,
is a vector corresponding to the ranked genes from the dataset, 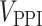 is a list of unranked genes corresponding to the set of proteins as identified from Gordon et al. [24] and
is a list of unranked genes corresponding to the set of proteins as identified from Gordon et al. [24] and 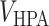 is a list of unranked genes corresponding to the unique proteins annotated to be key in the HPA, excluding the
is a list of unranked genes corresponding to the unique proteins annotated to be key in the HPA, excluding the 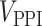 ones.
ones.
The weights 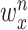 for the respective sources were set to ensure that at least 20% of each gene list is included in the top 500 genes. In addition, the weights were set so that their sum satisfies the condition:
for the respective sources were set to ensure that at least 20% of each gene list is included in the top 500 genes. In addition, the weights were set so that their sum satisfies the condition:
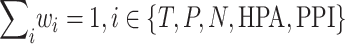 |
(11) |
The integrated gene list contained in total 1351 genes. We filtered to retain only the genes which were recognizable by the GeneMANIA tool, resulting in a list of 1118 genes. From that, we selected the top 1000 top-scored genes based on their integration score to build networks using the GeneMANIA tool [50] in Cytoscape [51]. Note that 1001 genes were selected as three genes shared the same score at the bottom of the list. We selected four networks in GeneMANIA based on co-expression, co-localization, genetic interaction and physical interaction.
The Multi-source Information (MI) super network was constructed based on the weighted sum of the pairwise weighted edge vectors (for each pair of genes) for these four types of networks (the edge weight automatically calculated by GeneMANIA). The total number of connected nodes out of the 1001 genes was 995, and the total number of edges was 45 486.
We calculated the final gene score MIG based on the combination of nodal score and topological information for each gene using equation (10), and ranked the genes with respect to their importance and involvement in COVID-19 (Supplementary File S4).
The igraph package [52] in R [53] was used to generate and analyze the MI super network and to compile the MIG score.
Functional analysis and pathway community identification
PathWalks [54], a map-driven random walk-based methodology on a pathway-to-pathway network, was applied to reveal communities of connected pathways. PathWalks exploits a map that we construct in the form of a synthetic gene network, containing integrated information regarding a disease of interest. For our calculations, we used the multi-thread version of PathWalks with 15 walkers, 10 000 steps per walker and restart every 50 steps run in the infrastructure provided by the National Initiatives for Open Science in Europe—NI4OS Europe (https://ni4os.eu/ni4os-europe-vs-covid19/). Using the resulting pathway frequencies, i.e. number of visits per pathway, we performed an odds ratio (OR) analysis with respect to a random walk of the pathway network using only the topology of the network without any gene guidance. Specifically, we defined the OR between guided to nonguided walks as:
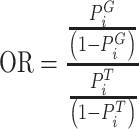 |
(12) |
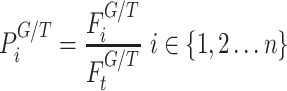 |
(13) |
where index G and T denote the variables corresponding to the guided and topology-only runs, respectively. 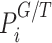 is the visiting probability of the ith pathway calculated as the frequency Fi ratio over the total visits FtT on pathways. According to this analysis pathways, with OR values >1 are the ones with higher relative visiting frequency compared with the topology-only runs and are thus more likely to be involved in the disease of interest. On the other hand, OR values <1 correspond to pathways that are less relevant.
is the visiting probability of the ith pathway calculated as the frequency Fi ratio over the total visits FtT on pathways. According to this analysis pathways, with OR values >1 are the ones with higher relative visiting frequency compared with the topology-only runs and are thus more likely to be involved in the disease of interest. On the other hand, OR values <1 correspond to pathways that are less relevant.
Pathways with OR values >1 were visualized as a network using R’s igraph package [52], highlighting specific pathways of interest.
CoDRes re-ranking and drug lists unification and scoring
The top 50 host-targeting drugs from the 10 lists (Supplementary File S3) were used as input to the CoDReS (Computational Drug Repositioning Score) tool [55]. CoDReS combines an initial drug ranking that may be the repurposing score or an a priori score (aS), with a functional score (FS) of each drug results from the analysis of the disease of interest, as well as with a structural score (StS) derived from drugability violations. Specifically, the initial ranking score of each host-targeted drug from the 10 lists divided with the absolute maximum ranking score was used as the normalized a priori repurposing score for CoDReS. Furthermore, for the case of FS and StS, the 1001 genes from the integration analysis were used with their disease association scores and the structures (SMILES format) of each drug, respectively. A composite score (CoDRes score) was calculated, for each drug, as the normalized-weighted sum of the initial aS with an FS and an StS. The weights that determine the desired influence of each part to the final score were defined as waS = 0.45, wFS = 0.45 and wStS = 0.1. These parameters were chosen such that the contribution of the initial ranking from the individual approaches (aS) and the integrated disease–gene network (FS) are equally weighted, but the influence of the StS score, which pertains to chemical structural violations, is significantly reduced. Although the latter score is relevant for libraries of novel chemical compounds, the drugs of interest in this work are approved drugs with characterized chemical behavior.
Finally, the top 20 drugs from each re-ranked list were selected for chemical structure diversity analysis. The top 20 cutoff was chosen arbitrarily in order to limit and focus the downstream analyses to approximately one-third of the 600 drugs selected from all individual DR approaches.
Chemical structure diversity analysis and clustering
We searched and downloaded the structures of the 240 drugs from PubChem [56], CLUE—The Drug Repurposing Hub [57] (https://clue.io/repurposing#download-data) and from the literature. We removed (i) duplicates entries (drugs found in more than one list), (ii) drugs for which we did not find a structure and (iii) elemental entries (i.e. copper). We then used the OpenBabel software [58] to convert the structures of the remaining 210 drugs to a single Structure data file (SDF) library file, which was then used as input in the ChemBioServer 2.0 tool [59] for calculating the distance matrix of their chemical and structural similarity. Drugs were clustered using a minimum Tanimoto similarity of 80%, which corresponds to a 0.2 distance cutoff.
Rank normalization and shortlisting
The rank of each drug was normalized according to Equation (14):
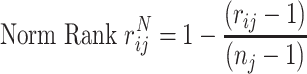 |
(14) |
where 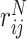 is the normalized rank of the ith drug of the jth drug list, whereas nj is the total number of drugs selected. nj was set to 20 since we selected the top 20 drugs from all lists following the CoDReS re-ranking step.
is the normalized rank of the ith drug of the jth drug list, whereas nj is the total number of drugs selected. nj was set to 20 since we selected the top 20 drugs from all lists following the CoDReS re-ranking step.
The highest normalized rank value of a given drug across all lists was assigned as its maximum normalized value (Max Rank). The Max Rank of drugs introduced exclusively from one list was set equal to the corresponding normalized rank value.
Finally, from the list of 185 unique drugs obtained after removing duplicates and structurally redundant drugs, we shortlisted the top ~30% (65 drugs) with respect to their Max Rank.
Comparison with running clinical trials
All listed clinical studies related to the COVID-19 were collected from the ClinicalTrials.gov. Small-molecule drugs curated through the reported clinical studies and the 2D structures of the drugs (SDF files) were obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/), where available.
The SDF file containing the chemical structures of the integrated list of 65 drugs and all the available structures of the drugs reported in currently running clinical trials was used as an input to Chembioserver 2.0 in order to obtain the corresponding Tanimoto distance matrix. The latter was analyzed in R to identify which proposed drugs have the same or similar compounds in clinical trials using a distance threshold of ≤0.2.
Results
The workflow adopted in this study outlines five mains steps described in detail below. The overall process entails the analysis of COVID-19-related omics datasets to identify significant genes and PPIs of interest with the subsequent identification and shortlisting of candidate repurposed drugs (Figure 1).
Source datasets
During the first part of the analysis, we collected publicly available multi-omics datasets released from February to May 2020. These included proteomics and metabolomics datasets from blood serum [22] and seven transcriptomic datasets—four from cultured cell lines (Series 1—NHBE, Series 2–5—A549, Series 6—A549 with a transduced ACE2 expressing vector and Series 7—Calu-3) [18] and three patient-derived tissue samples (Series 15—lung Biopsy, BALF and PBMC) [20]. A summary of the data along with the source study is available in Table 1 (see Methods).
Using the logFC value and P-value thresholds (see Methods) from the proteomic dataset, we identified an approximately equal number of over- and underexpressed proteins, 62 and 58, respectively. The selected metabolites from the metabolomics dataset included 97 overabundant compounds, whereas the underabundant were ~3-fold higher—317. The analysis of the RNA-Seq datasets yielded four sets of DEGs. The PBMC and BALF sets included approximately equal numbers of ~700 over- and ~300 underexpressed DEGs. On the contrary, from the Series 15 set, we obtained a relatively large number of 3838 DEGs that were under- against 515 overexpressed DEGs. Finally, the DEG sets from cell lines (Series 1, 2 and 5, 6 and 7) included 2588 over- and 1036 underexpressed DEGs in total (Table 1).
In addition, we collected the available information on PPIs between the SARS-CoV-2 and host proteins as reported by Gordon et al. [24] and HPA database [47]. This comprised a set of 336 human proteins found to interact with SARS-CoV-2 proteins. For the host–pathogen-based approaches (see Methods—PPI Data Selection), we collected 42.7 K PPIs across a total of 625 pathogens and human proteins. From the latter, 189 pathogens were found to interact with at least one common host protein with SARS-CoV-2.
DR approaches
We applied three discrete DR approaches based on (i) transcriptomics, generating eight lists of candidate drugs, (ii) GWAS–phenotype association (one list) and (iii) pathogen–host PPIs network analysis, generating three lists. A summary of all 12 generated lists of candidate drugs is shown in Table 2, whereas all drugs per list are reported in Supplementary File S3.
DR based on transcriptomics
Using the RNA-Seq-derived DEG sets, we performed a series of in silico DR analyses with existing computational tools. By selecting the top 150 over- and underexpressed genes with respect to their logFC value (300 in total, shown in Supplementary File S1), we obtained two lists of candidate drugs using each DEG set as an input to:
(1) An ensemble of DR tools: connectivity map [30], L1000CDS2 [31] and L1000FWD [32], followed by a scoring process yielding four lists with 50 drugs each, namely, Tr1 (from Series 1, 2 and 5, 6, 7), Tr2 (from Series 15), Tr3 (from BALF) and Tr4 (from PBMC).
(2) CREEDS [33] using the DrugMatrix-based repurposing feature of the tool, which maps the expression profile of the DEGs of interest to the effect of various drugs as measured in rats. By applying the same cutoff of 50 drugs per run, we obtained four additional lists—TrC1 (from Series 1, 2 and 5, 6, 7), TrC2 (from Series 15), TrC3 (from BALF) and TrC4 (from PBMC).
DR based on GWAS–phenotype association analysis
We used 44 genes as input to the PhenoScanner database [34, 35]. These genes correspond to 40 strong SARS-CoV-2 interactors according to the evolutionary analysis as reported by Gordon et al. [24] and four highlighted proteins, ACE2, TMPRSS2, CTSB and CTSL, from the HPA database [47].
PhenoScanner identified 480 genetic associations between the input COVID-19-related genes and genes—or proxy genes—previously associated with various phenotypes with a default P-value ≤ 1e-05. Out of these, 186 associations were of genome-wide significance (P-value ≤ 5e-08), with cardiovascular diseases (CVD) being the predominant associated phenotype (Supplementary File S2). Overall, the identified Single-nucleotide polymorphism (SNP) associations corresponded to a set of 83 genes, which we used to search for potential drugs in the DGIdb [38]. Following this approach, we compiled a list of 58 drugs (list GW) able to target the 83 genes of interest.
DR based on pathogen–host interaction network analysis
We worked on the assumption that genetic, functional and morphological similarities between SARS-CoV-2 and other pathogens can be approximated by their taxonomic classification. Drugs with a direct inhibitory effect against a given pathogen are more likely to have a similar effect to closely related pathogens in terms of taxonomic distance. To repurpose drugs based on the above assumption, we derived a taxonomy distance matrix (see Methods), which was used to identify antiviral compounds able to target directly a pathogen proteins according to DrugBank’s polypeptide target file [40, 41]. This process yielded a list of DrugBank compounds scored as a function of (a) the taxonomic proximity of known target–pathogens to SARS-CoV-2 and (b) a metric of the broad spectrum activity of a given drug. This approach resulted in a ranked list (TaxAV) of 841 antiviral drugs (Supplementary File S5) that were found in DrugBank [40, 41] to target a total of 345 unique pathogen NCBI taxonomy IDs. The taxonomy tree for all the pathogens considered in this work and their associated drugs is illustrated in Figure 2.
Figure 2 .
Taxonomy tree of all the pathogens used in the host–pathogen interaction drug repurposing approaches. The main taxonomic groups are highlighted with light blue slices showing the ancestral taxonomy groups for SARS-CoV-2 (green leaf). The yellow to red colored bars represent the number of drugs obtained from various taxa. The circular blue to yellow heat map represents the number of pathogen–host protein interactions retrieved from various databases.
Building on the above approach, we included the identified PPIs between pathogen and host proteins in an effort to obtain a more informative scoring scheme regarding the functional interactions between pathogens and human. To this effect, we constructed a pathogen-to-pathogen network where edges describe the commonality of each pathogen to SARS-CoV-2 with respect to (a) their common interactions with host proteins and (b) the taxonomy distance (see Methods). Herein, the antiviral drugs were scored based on the proximity of target pathogens to SARS-CoV-2, resulting in 1178 (Supplementary File S5) scored drugs (list HPAV).
For both TaxAV and HPAV list, we selected the top 20 drugs for forwarding to the structural similarity analysis step (Supplementary File S3).
We used the underlying network to screen further for drugs targeting host proteins. This was based on the assumption that drugs targeting a host protein set with wide interaction overlap between SARS-CoV-2 and other pathogens are deemed more likely to exhibit similar antiviral effects. Herein, drugs were scored by means of a generalized 4-fold equation that accounts for both the host and pathogen protein targets, resulting in a list of 301 drugs. The top 50 drugs (list HPH) shown in Supplementary File S3 were selected and forwarded to the drug re-ranking step, similarly with the other host protein targeting drug lists.
Multiplexing of repurposed drug lists
Integration of multi-omic data from patients
We integrated the available multi-omic data from patient samples based on a previously proposed scheme [48]. Specifically, we integrated the following data: (1) ranked DEGs lists in terms of absolute logFC resulting from the transcriptomics data analysis, (2) a ranked gene list in terms of absolute logFC from the proteomics data analysis, (3) a ranked gene list in terms of P-value derived from the metabolomics data analysis, (4) an unranked list of host-proteins relevant to viral cell entry highlighted in HPA [47] and (5) an unranked list of host-proteins, which interact with SARS-CoV-2 from Gordon et al. [24].
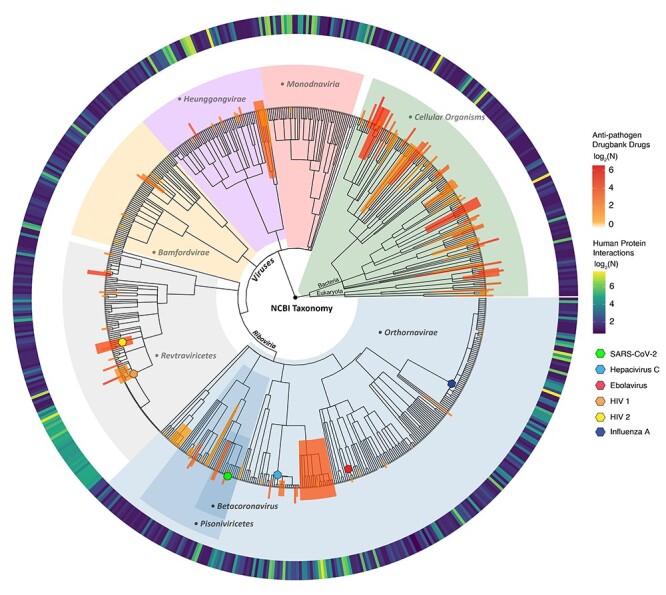
We calculated the MIG score, a characteristic score per gene comprising the integrated gene-specific information, and the local-weighted degree from a synthetic gene-to-gene network based on co-expression, genetic interactions, physical interactions and co-localization extracted from GeneMANIA [50], as described in Methods. We then derived the integrated MI network illustrated in Figure 3 along with the relevant score distributions. The network comprises the top 1000 genes as nodes (specifically 1001 genes due to score ties) originating from all sources, with a total of 45 486 edges prorating the gene-to-gene relationships.
Figure 3 .
The integrated multi-source information (MI) network along with the distributions of the weighted degree, weighted gene-specific information and the combined MIG scores. Node size represents the MIG score bin for each gene, whereas colors indicate the originating omics dataset. Nodes with mixed colors show genes commonly identified from different datasets. The bar plot shows the MIG score for each gene as the weighted sum of Edge (yellow) and Nodal (blue) scores. The subplots show the corresponding distributions.
Finally, for all downstream functional analysis and drug re-ranking, we used the genes ranked based on their MIG score, which represents the integrated gene–disease association.
Highlighting connected pathway communities related to COVID-19 using the generated integration map
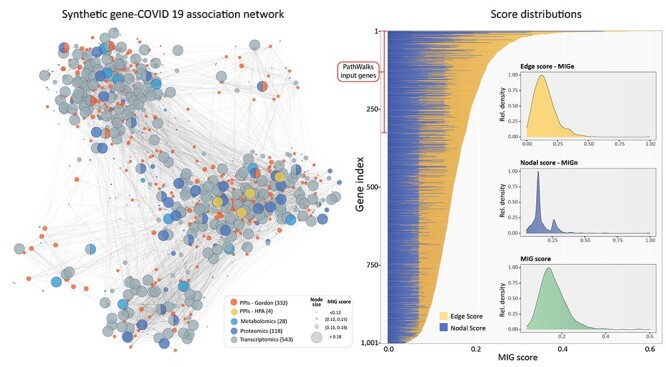
We used the top 300 MIG-ranked genes (as described above) to create a map of significant gene–disease associations. Under the guidance of this integrated map, we used PathWalks [54] to allow walkers to cross a pathway-to-pathway network derived from Kyoto Encyclopedia of Genes and Genomes (KEGG). The most frequent trajectories highlighted communities of pathways predicted to be widely involved in COVID-19. Using the resulting pathway frequencies, i.e. number of visits per pathway, we performed an OR analysis with respect to a random walk using only the topology of the pathway network without any gene map guidance. OR values >1 correspond to higher relative visiting frequency compared with the nonguided runs and are thus more likely to be involved in COVID-19. The network of highlighted pathway communities is shown in Figure 4.
Figure 4 .
Connected pathway communities related to COVID-19 on a pathway-to-pathway network as highlighted by PathWalks. The bar plot shows the odds ratio value for the top 30 KEGG pathways, found to be involved in COVID-19. The bar plot color scale represents the visit counts from less (dark blue) to more (light) frequently visited pathways.
Re-ranking of drug candidates based on the calculated target–disease association
The CoDReS tool [55] was used to re-rank the candidate drugs based on (a) an FS combining the drug targets’ relevance to the disease (as captured by the MIG score above), and the binding affinity to its target genes, (b) an aS defined as the normalized initial drug ranking from each list and (c) an StS representing drugability violations.
We performed the CoDReS re-ranking on the top 50 drugs with respect to their weighted normalized score, from each of the 10 lists of drugs that target host proteins: eight from RNA-Seq data, one from GWAS and one from host–pathogen interactions. The re-ranked lists are given in Supplementary File S3. Next, we selected the top 20 CoDReS drugs from each list (200 in total) for further analysis.
Filtering drugs with diverse chemical structures and shortlisting
The 200 re-ranked drugs together with the top 20 candidate antiviral drugs originating from the two pathogen–host interaction-based approaches, 240 in total, were screened for chemical structure similarity. Using the ChemBioServer 2.0, we calculated the structural distance matrix based on the Tanimoto index [59] for all pairwise combinations of candidate drugs.
A hierarchical clustering analysis revealed that the input drugs spanned a broad range of chemical structure diversity. Specifically, by applying a distance threshold of <0.2 for highly similar compounds, we obtained 185 clusters, from which 25 comprise more than one drug. We eliminated the structural redundancy within our drug list by selecting the top scoring drug from each cluster, yielding a list of 185 drugs. Alcohol was manually excluded as it was deemed inappropriate for pharmacological use against COVID-19.
Furthermore, in order to assess the contribution of each list to the final nonredundant set of 185 drugs, we performed a redundancy analysis given in Supplementary File S7. The analysis suggests that there is a limited overlap between lists in terms of their member drugs, but all lists are contributing a unique set of drugs. The highest overlap was observed between TaxAV and HPAV, while TaxAV had the lowest contribution of unique drugs (seven drugs) and Tr4 and TrC4 the highest (18 drugs each).
Finally, we shortlisted the top one-third candidate drugs based on their maximum normalized rank (Max Rank), amounting to 65 candidate drugs shown in Table 3.
Table 3.
The integrated list of 65 drugs sorted by their maximum normalized rank (Max Rank)
| Drug | Max Rank | Classifications | Related clinical trials | Expert selection |
|---|---|---|---|---|
| Dexamethasone | 1.00 | MeSH: anti-inflammatory; corticosteroid TBC: steroid hormone receptor agonist Chem: steroid |
Clinical trials: 26 Structurally similar in clinical trials: methylprednisolone |
|
| Atorvastatin | 1.00 | MeSH: anticholesteremic TBC: cholesterol synthesis inhibitor Chem: pyrrole |
Clinical trials: 4 | |
| Beta-estradiol | 1.00 | MeSH: estrogen TBC: estrogen receptor agonist Chem: steroid |
Clinical trial: 1 | |
| Vorinostata | 1.00 | MeSH: antineoplastic TBC: histone deacetylase inhibitor Chem: anilide |
✓ | |
| Olanzapine | 1.00 | MeSH: atypical antipsychotic TBC: 5-HT ligand; D2 dopamine receptor ligand; histamine receptor ligand Chem: heterocyclic; benzodiazepine |
||
| Cyclosporin-aa | 1.00 | MeSH: immunosuppressive TBC: calcineurin inhibitor Chem: cyclic peptide |
Clinical trials: 6 | |
| Rosiglitazone | 1.00 | MeSH: hypoglycemic agent TBC: peroxisome proliferator-activated receptor Chem: thiazole; thiazolidinedione |
||
| Bosutinib | 1.00 | MeSH: antineoplastic agent TBC: Src tyrosine kinase inhibitor Chem: aromatic amine; aniline aminoquinoline |
✓ | |
| Zinc acetate | 1.00 | MeSH: antibacterial agent TBC: – Chem: acetate |
||
| Benzyl (2-oxopropyl) carbamate | 1.00 | MeSH: – TBC: SARS-CoV replicase polyprotein 1ab inhibitor; 3 clpro protease inhibitor Chem: benzyloxycarbonyl; ketone |
✓ | |
| Remdesivir | 1.00 | MeSH: antiviral TBC: adenine nucleotide analog Chem: aromatic amine |
Clinical trials: 48 | ✓ |
| Dasatinib | 0.95 | MeSH: antineoplastic TBC: Src tyrosine kinase inhibitor Chem: aromatic amine; thiazoles, aminopyrimidine |
✓ | |
| Mercaptopurine | 0.95 | MeSH: antineoplastic; immunomodulatory agent; antimetabolite TBC: purine analog; enzyme inhibitor Chem: sulfhydryl compound; aryl thiol; purine analog |
||
| D3F | 0.95 | MeSH: enzyme inhibitor TBC: SARS replicase polyprotein 1a inhibitor Chem: nitrobenzene; toluene; trifluoromethylbenzene |
✓ | |
| Raloxifene | 0.95 | MeSH: antineoplastic; bone density conservation agent TBC: hormone; selective estrogen receptor modulator; estrogen antagonist Chem: aromatic cyclic amino compound; benzylidene |
||
| Cytarabine | 0.95 | MeSH: antineoplastic; immunosuppressive; antimetabolite; antiviral TBC: pyrimidine analog; tyrosine protein kinase Src inhibitor; protease inhibitor Chem: heterocycle; pyrimidine; arabinonucleoside |
✓ | |
| Imatinib | 0.95 | MeSH: antineoplastic TBC: tyrosine kinase inhibitor Chem: benzamides; aromatic amine |
Clinical trials: 5 | ✓ |
| Selumetinib | 0.89 | MeSH: antineurofibromatosis type 1 TBC: MAP kinase inhibitor; MEK inhibitor Chem: aecondary amino; benzimidazole |
||
| Azd-8055 | 0.89 | MeSH: experimental antiproliferative TBC: phosphatidyl inositol 3′ kinase-related kinase inhibitor; voltage-gated potassium channel ligand; rapamycin kinase inhibitor Chem: oxazine; morpholine |
||
| Avrainvillamide-analog-3 | 0.89 | MeSH: antiproliferative TBC: nuclear chaperone nucleophosmin ligand Chem: alkaloid |
||
| Linsitinib | 0.89 | MeSH: antineoplastic TBC: insulin receptor inhibitor; insulin-like growth factor-I receptor inhibitor; Type II receptor tyrosine kinase inhibitor Chem: pyrazines; quinoline |
||
| Fluocinoloneacetonide | 0.89 | MeSH: anti-inflammatory TBC: steroid hormone receptor agonist Chem: steroid; alcohol; ketone |
Structurally similar in clinical trials: budesonide |
|
| Dobutamine | 0.89 | MeSH: sympathomimetic; cardiac stimulant TBC: adrenergic beta-1 receptor agonist Chem: catecholamine; secondary amine |
||
| Bromfenac | 0.89 | MeSH: NSAID TBC: COX inhibitor Chem: ketone; benzophenones; aromatic amine; anilines |
||
| Testosterone | 0.89 | MeSH: hormone, androgen, anabolic TBC: sex hormone receptor agonist Chem: steroid; androgen |
Structurally similar in clinical trials: hydrocortisone |
|
| Hydroxychloroquine | 0.89 | MeSH: antiparasitic agent, antimalarial, antirheumatic agent TBC: enzyme inhibitor, heme polymerase inhibitor, endosomes modulator, toll-like receptors inhibitor Chem: aromatic amine; aminoquinoline |
Clinical trials: 250 Structurally similar in clinical trials: hydroxychloroquine sulfate, chloroquine, chloroquine phosphate |
|
| Hydroxyurea | 0.89 | MeSH: antineoplastic agent, antimetabolite, antisickling agent TBC: nucleic acid synthesis inhibitor; ribonucleoside-diphosphate reductase inhibitor Chem: urea |
||
| F3F | 0.89 | MeSH: – TBC: SARS-CoV replicase polyprotein 1ab inhibitor Chem: furan; triazole |
✓ | |
| Daunorubicin | 0.84 | MeSH: antineoplastic; antibacterial TBC: topoisomerase II inhibitor Chem: aminoglycosides; anthracyclines; cyclic ketone, acenoquinone |
||
| PD-0325901 | 0.84 | MeSH: experimental antineoplastic TBC: MAPK inhibitor Chem: benzamide |
||
| Zidovudine | 0.84 | MeSH: antiviral agent, antimetabolite TBC: nucleoside and nucleotide reverse transcriptase inhibitor Chem: glycoside; pyrimidine; dideoxynucleosides |
||
| GRL0617 | 0.84 | MeSH: – TBC: SARS-CoV replicase polyprotein 1ab inhibitor Chem: carboxylic acid nitrile; cyano compound; peptide |
✓ | |
| AG-14361 | 0.84 | MeSH: – TBC: poly ADP-ribose polymerase 1 inhibitor Chem: benzodiazepine; benzimidazole |
||
| Dactolisib (nvp-bez235) | 0.84 | MeSH: antineoplastic TBC: phosphatidyl inositol 3′ kinase-related kinase inhibitor; lipid modifying kinase inhibitor Chem: imidazole; imidazoquinoline |
Clinical trial: 1 | ✓ |
| Carboplatin | 0.84 | MeSH: antineoplastic TBC: aquation agent Chem: coordination complex; platinum compound |
||
| Zileuton | 0.84 | MeSH: NSAID TBC: leukotriene antagonist; lipoxygenase inhibitor Chem: urea; benzothiophene |
||
| Staurosporine | 0.84 | MeSH: multi-enzyme inhibitor TBC: protein kinase C inhibitor; CAMK2 inhibitor; death-associated kinase inhibitor; phosphorylase kinase inhibitor; tyrosine-(Y)-phosphorylation-regulated kinase inhibitor; acetylcholine receptor ligand Chem: indole; indolocarbazole alkaloid |
||
| Fasudil | 0.79 | MeSH: peripheral vasodilators TBC: ROCK inhibitor; DMPK kinase inhibitor; calcium channel blocker Chem: sulfonamides |
||
| GSK-1059615 | 0.79 | MeSH: – TBC: enzyme inhibitor, PI3K inhibitor Chem: pyridine; quinoline; thiazolidinone |
||
| PI 103 hydrochloride | 0.79 | MeSH: – TBC: AGC protein kinase inhibitor; PKC inhibitor; CMGC protein kinase inhibitor; tyrosine protein kinase inhibitor Chem: pyrimidine |
||
| Hexachlorophene | 0.79 | MeSH: anti-infective agent, antiseptic TBC: – Chem: chlorobenzene; chlorophenol |
||
| Ofloxacin | 0.79 | MeSH: antibacterial; antineoplastic TBC: topoisomerase IV inhibitor Chem: aromatic amine; N-arylpiperazine; quinolone |
Clinical trial: 1 | |
| Leflunomide | 0.79 | MeSH: antirheumatic agent TBC: nucleoside synthesis inhibitor; oxidoreductase inhibitor Chem: isoxazole |
Clinical trials: 2 | |
| Prasterone | 0.79 | MeSH: anabolic steroid, hormone TBC: estrogen receptor agonist; androgen receptor agonist; GABAa antagonist, NMDA agonist Chem: steroid |
||
| N-(2-aminoethyl)-1-aziridineethanamine | 0.79 | MeSH: experimental antiviral agent TBC: ACE2 inhibitor Chem: aziridine |
✓ | |
| Triptolide | 0.79 | MeSH: antineoplastic alkylating agent; antispermatogenic agent; immunosuppressive agent, TBC: multi-protein inhibitor Chem: terpene; phenanthrenes; diterpenes; diterpenoid |
||
| WR1 | 0.79 | MeSH: – TBC: inhibitor for the major protease of the SARS coronavirus Chem: benzyloxycarbonyl; benzyloxycarbonyl |
✓ | |
| Y-27632 | 0.74 | MeSH: neuromuscular; muscle relaxant; antihypertensive agent; enzyme inhibitor TBC: rho kinase inhibitor, protein kinase N inhibitor Chem: pyridine; aromatic amide |
||
| DG8735000 | 0.74 | MeSH: – TBC: inhibitor of ultraviolet-mediated damage Chem: aminobenzoate |
||
| Saracatinib | 0.74 | MeSH: antineoplastic agent; enzyme inhibitor TBC: Src inhibitor; tyrosine kinase inhibitor Chem: benzodioxoles; quinazoline |
✓ | |
| LY-255283 | 0.74 | MeSH: antiasthmatic agent TBC: leukotriene antagonists Chem: tetrazoles; aromatic ketone |
||
| Rofecoxib | 0.74 | MeSH: NSAID; analgesic nonnarcotic agent TBC: COX2 inhibitor Chem: lactone; furan; butanolide |
||
| Simvastatin | 0.74 | MeSH: antimetabolite; anticholesteremic; hypolipidemic TBC: hydroxymethylglutaryl-CoA reductase inhibitor Chem: polycyclic aromatic; naphthalene; fatty acid ester |
Clinical trials: 4 | |
| Ebastine | 0.74 | MeSH: antihistamine TBC: H1 antagonist Chem: piperidine; ketone; butyrophenone |
||
| Cisplatina | 0.74 | MeSH: antineoplastic; radiation-sensitizing agent TBC: DNA replication inhibition Chem: platinum compound |
||
| Hydroquinone | 0.74 | MeSH: antioxidant; radiation protective agent TBC: melanin synthesis inhibitor; HDAC inhibitor Chem: phenol; benzenediol |
✓ | |
| Calcium citrate | 0.74 | MeSH: anticoagulant TBC: calcium chelating agent Chem: tricarboxylic acid derivative |
||
| JAK3-IN-1 | 0.68 | MeSH: – TBC: AGC protein kinase inhibitor; tyrosine kinase inhibitor Chem: heterocyclic compound |
||
| Pracinostat | 0.68 | MeSH: experimental antineoplastic TBC: histone deacetylase inhibitor Chem: benzimidazole |
||
| Blebbistatin | 0.68 | MeSH: – TBC: myosin inhibitor Chem: tertiary alcohol; ketone; pyrroloquinoline |
||
| Pioglitazone | 0.68 | MeSH: hypoglycemic agent (antidiabetic) TBC: peroxisome proliferator-activated receptor-gamma activator Chem: azole; thiazole; thiazolidinedione |
Clinical trials: 2 | |
| Tegafur | 0.68 | MeSH: antimetabolite; antineoplastic; immunosuppressive TBC: thymidylate synthase inhibitor Chem: pyrimidine; fluorouracil |
||
| Choline salicylate | 0.68 | MeSH: NSAID, analgesic nonnarcotic TBC: COX inhibitor Chem: acid; benzoate; salicylate, phenol; hydroxybenzoic acid |
||
| Methotrexate | 0.68 | MeSH: antimetabolite; antineoplastic; abortifacient; immunosuppressive; antirheumatic TBC: folic acid metabolism inhibitor; nucleic acid synthesis inhibitor Chem: acid; pterin; aminopterin |
Clinical trials: 3 Structurally similar in clinical trials: folic acid |
✓ |
| Clofibrate | 0.68 | MeSH: hypolipidemic; anticholesteremic TBC: peroxisome proliferator-activated receptor-gamma inhibitor Chem: clofibric acid; ethyl ester |
Notes: The classification of the listed drugs was based on three classification systems: MeSH (Medical Subject Headings), TBC (Target-Based Classification) and Chem (Chemical classification). Currently, active clinical trials that invodatalve drugs in this list are noted (clinicaltrials.gov, last accessed on 24 August 2020). A group of drugs that fulfilled specific criteria was selected by expert curation. D3F: 2-[(2,4-Dichloro-5-Methylphenyl)Sulfonyl]-1,3-Dinitro-5-(Trifluoromethyl)Benzene; F3F: S-[5-(Trifluoromethyl)-4H-1,2,4-Triazol-3-YL] 5-(Phenylethynyl)Furan-2-Carbothioate; GRL0617: 5-amino-2-methyl-N-[(1R)-1-naphthalen-1-ylethyl]benzamide; WR1: Nalpha-[(Benzyloxy)Carbonyl]-N-[(1R)-4-Hydroxy-1-Methyl-2-Oxobutyl]-L-Phenylalaninamide; DG8735000: 4-(Dimethylamino)benzoic acid.
aDrugs found also by using null models.
Evaluation of the integrated drug list with respect to ongoing clinical trials
We evaluated further the list of 65 drugs by comparing it against the drugs that are currently in clinical trials against COVID-19 as obtained from clinicaltrials.gov. Specifically, 5 out of the 11 top-scoring drugs (dexamethasone, beta-estradiol, atorvastatin, cyclosporin A and remdesivir) are already in clinical trials. Also, eight drugs with a lower normalized ranking score (imatinib, hydroxychloroquine, dactolisib, ofloxacin, leflunomide, simvastatin, pioglitazone and methotrexate) were also found in ongoing clinical trials.
From the remaining drugs, we identified, through structural similarity analysis, two more drugs, which have similar compounds (Tanimoto distance <0.2) in clinical trials. In particular, fluocinoloneacetonide and testosterone were found structurally similar to budesonide and hydrocortisone, which are currently in clinical trials, respectively. The integrated list is shown in Table 3. The detailed table of the integrated list of 65 drugs is presented in Supplementary File S6.
To assess potential biases towards particular drugs in our lists, we repeated the transcriptomic-based DR pipeline using as an input 10 random gene lists. We found 3 out of the 65 drugs could potentially be randomly selected when keeping the same number of drugs from each list as in the followed shortlisting procedure. The three drugs are vorinostat, cyclosporin A and cisplatin, and are marked with (*) in Table 3. Further details on the null models used and the corresponding results can be found in the Supplementary File S7.
The 65 drugs included in the produced integrated list (Table 3) present a diverse group of compounds from a chemical, pharmacological and clinical perspective. We used three major classification systems to categorize these compounds: the Medical Subject Headings classification (MeSH), the Target-Based Classification (TBC) and the general Chemical classification system (Chem). Although the integrated list contains a versatile group of drugs from a wide range of therapeutic areas (such as anticoagulants, antihistamines and hypolipidemics), the majority of these are antineoplastic agents, followed by immunosuppressive drugs, antivirals and antibacterials. Nevertheless, 16 drugs from the integrated list are experimental drugs, which either are in clinical trials for a number of conditions or are still under preclinical investigation. The majority of the drugs in the integrated list are enzyme or protein inhibitors, in terms of their established mechanism of action. Regarding their chemistry, the list is inclusive of important chemical classes of drug molecules such as triazoles, pyrimidines, platinum-containing compounds and benzimidazoles. However, the most prominent general chemical groups in the integrated list were aromatic amines (nine drugs), drugs with at least one piperazine ring (seven drugs), steroids (five drugs), thiazoles (four drugs) and quinolines (four drugs).
A characteristic representation of the drug candidates’ classification can be found within the top 5% of their maximum ranking score (between 0.95 and 1), from which 11 (65%) are protein inhibitors, seven (41%) are antineoplastic agents, five (30%) are aromatic amines and four (24%) include a piperazine ring.
Expert curation and annotation of the integrated drug shortlist
From the integrated list of 65 drugs, a thorough expert curation has highlighted a set of 16 drugs (Table 3 and Figure 5). The curation was based on following three main criteria: (a) drugs that have exhibited evidence of efficacy against COVID-19 in Phase 3 clinical trials, (b) drugs that bear pharmacological evidence of direct targeting of SARS-CoVs molecular components and (c) clinically approved drugs that have activity in molecular pathways that have been shown in the literature to be implicated in SARS-CoV-2 biology.
Figure 5 .
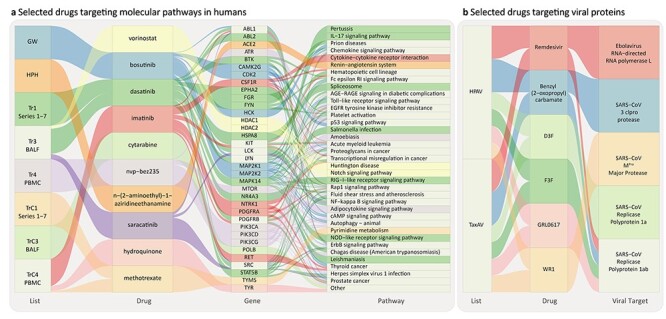
Sankey plots of the drugs highlighted after expert curation of integrated list in Table 3. Drugs are ordered based on their normalized ranking and pathways based on their OR value obtained from PathWalks. (A) Drugs targeting human pathways, their originating list, their target genes and corresponding pathways. (B) Antiviral drugs targeting viral proteins, their originating list and known target pathogens.
The first criterion was fulfilled by two drugs from the integrated list, dexamethasone and remdesivir, which are the only drugs that have shown to be effective against COVID-19. Nevertheless, from these two, only remdesivir has shown a direct effect against SARS-CoV-2, whereas dexamethasone showed an effect in the reduction of the associated inflammation of the disease rather than the biology of the virus.
The second criterion of the curation was fulfilled by six experimental drugs from the integrated list, which have shown a direct effect on SARS-CoVs in various assays (mostly in vitro). Benzyl (2-oxopropyl) carbamate, D3F, F3F, GRL0617 and WR1 have shown direct inhibition of the coronavirus replicase polyprotein 1ab [60–64] (https://pubchem.ncbi.nlm.nih.gov/bioassay/[Bioassay Number], Bioassay Numbers: 977608, 1811, 977608, 977610), which is a major viral protein for the viral replication machinery [24]. N-(2-aminoethyl)-1-aziridineethanamine has shown direct inhibition of the human ACE2 receptor [65, 66].
The third criterion was fulfilled by nine clinical drugs from the integrated list. These drugs were shown to be effective in targeting SARS-CoV-2 replication cycle and more specifically processes implicated in the generation of virally encoded nonstructural proteins (NSPs), which are essential for the assembly of the viral replicase complex. Vorinostat and hydroquinone are histone deacetylase (HDAC) inhibitors; it has been proposed that the main viral protease (Nsp5) may inhibit HDAC2 transport into the nucleus, and therefore HDAC2 inhibitors may be able to disrupt this interaction and suppress the HDAC2-mediated inflammation and interferon activation [24].
Some HDAC inhibitors have been shown to have antiviral activity (such as HDAC6 inhibitors against Influenza A Virus) [67], and there is considerable literature that links HDAC inhibitors with T-cell biology and the immune response [68, 69]. Bosutinib, dasatinib, imatinib and saracatinib are tyrosine kinase inhibitors; tyrosine kinase-linked pathways have been implicated in SARS-CoV-2 biology and activation [70], with various inhibitors having a potential anti-SARS-CoV-2 efficacy. Interestingly, dasatinib and imatinib have previously shown inhibitory effects against the Middle East respiratory syndrome (MERS) and SARS-CoV viruses in vitro at micromolar concentrations [71], as well as against SARS-CoV-2 [72, 73]. Their wide antiviral efficacy has also been showcased against human immunodeficiency virus (HIV) [74]. Methotrexate is a drug with a strong effect in nucleic acid synthesis and a multi-facet role as antineoplastic, antimetabolite and antirheumatic drug. Interestingly, it was recently shown to present a submicromolar activity against SARS-CoV-2, in vitro [73]. Finally, dactolisib is a drug with a dual activity as a PI3K/mTOR inhibitor, a pathway that we know is important in SARS-CoV biology [75, 76]. Although there is evidence of the antiviral activity of the above drugs against HIV [77], their potential efficacy against SARS-CoVs is yet to be determined.
Discussion
Mounting evidence indicate that the clinical manifestations of COVID-19 are systemic, affecting mainly the respiratory and digestive tract, the cardiovascular but also the central nervous system (CNS) [78–82]. Thus, we pursued an approach that integrates multi-omics data from different types of tissue (BALF, PBMC, alveolar, NHBE, Calu-2 cell lines and blood serum) aimed to build a comprehensive molecular profile of the disease to obtain a more informative basis for DR.
Across the most significant DEG sets selected for the integration step, we observed minimal overlap (24 out of 1 K selected—2.4%), indicating a complex perturbation scheme involving a broad range of biological pathways across different tissues and further supporting the need of a multi-source approach.
Coming from patients with severe COVID-19, the identified DEG sets were as expected enriched with genes that are involved in acute immunoresponse and inflammation. Specifically, the top MIG scoring inflammation factors SAA1/2 and CRP were previously proposed as severity biomarkers for the disease [83] and as indicators of inflammatory tissue damage. Cytokine release-related factors, such as CCL3/4, CXCL10, CLC, IL2R and IL10, were also highly ranked along with p53 apoptosis pathway-related genes NTRK1, CTSL, CTSB and IGFB [20]. Genes involved in complement activation and coagulation cascades such as C4a and C5 were also highly perturbed likewise with past Betacoronaviruses-related outbreaks (SARS and MERS) [84].The FDCSP, KCNJ2 and ST20 reported to be highly dysregulated from lung biopsies [85] along with FLNA and EGFR point to significant effects of COVID-19 in lung and gastrointestinal epithelia cell proliferation signaling. Specifically the latter (EGFR) has sparked a wider discussion on the involvement of growth factor receptors in viral infections and potential DR avenues of related antineoplastic compounds [86, 87].
The composition of the identified DEG sets and the functional analysis performed in PathWalks highlighted several pathways related to immune and inflammatory response pathways. Namely, in the top 30 scoring KEGG pathways (Figure 4) with respect to their OR values, we highlighted the ‘Complement and coagulation cascades, IL-17 signaling pathway, Chemokine signaling pathway, Cytokine-cytokine receptor interaction’ and ‘Fc epsilon RI signaling pathway’. The ‘Renin-angiotensin system’ and ‘Renin secretion’ pathways were also highlighted as a result of the central role of ACE2 in viral entry along with the genes involved in the coagulation cascades [25]. In addition, we found a number of mechanisms involved in more generic KEGG disease terms that are related to infections and respiratory conditions like in ‘Pertussis, Asthma, Staphylococcus aureus infection, Prion diseases, Malaria, Systemic lupus erythematosus and African trypanosomiasis’.
In line with the above COVID-19 molecular profile, nine candidate drugs in our proposed integrated list are classified (MeSH) as anti-inflammatory and immunosuppressors such as dexamethasone, cyclosporin A, mercaptopurine, cytarabine, fluocinoloneacetonide, triptolide, LY-255283, tegafur and methotrexate. These were complemented by the nonsteroid anti-inflamatory drugs, bromfenac, zileuton, rofecoxib and choline salicylate. The anticoagulants, renin–angiotensin system targeting drugs in the list were Y-27632, calcium citrate and the ACE2 targeting N-(2-aminoethyl)-1-aziridineethanamine and hydroxychloroquine. Several drugs from the integrated shortlist are classified as antineoplastic/antiproliferative agents (20 in total) including vorinostat, bosutinib, dasatinib and others. Although the antineoplastic drugs are expected to arise from such repurposing approaches, which inevitably identify key genes in proliferation signaling (e.g. EGFR) and apoptosis (p53 signaling pathway), there is a growing interest in the use of this class against viral infections [86, 87].
Focusing on the experts’ curated shortlist from a pathophysiological perspective, a group of four drugs, bosutinib, dasatinib, cytarabine and saracatinibare, are of particular interest as their primary mechanism of action is mediated by the inhibition of Src tyrosine kinase. Inhibiting the latter could help COVID-19 patients who enter a severe clinical trajectory through a number of pathophysiological mechanisms recognized to be involved from preclinical and clinical studies either directly involving COVID-19 or based on previous observations and experiments:
(a) Bruton’s tyrosine kinase signaling has been associated with the production of pro-inflammatory cytokines that can contribute to COVID-19 immunopathology, T-cell differentiation, function and survival; hence, it might be beneficial in treating COVID-19-related immunopathology and lymphopenia [88]. This has been already tested in clinical trials [89], and for dasatinib, it has been confirmed from seminal studies in chronic myelogenous leukemia [90].
(b) Tyrosine kinase inhibitors have been used to inhibit platelet function (antithrombotic activity) through several mechanisms including novel mechanisms like GAS6/TAM signal inhibition, targeting the MERTK tyrosine kinase active site with a highly potent and bioavailable MERTK inhibitor, UNC2025 [91].
(c) Mechanical ventilation (MV) has been extensively used to support patients with COVID-19 pneumonia who are developing respiratory failure; however, mortality is unexpectedly high, higher than current Acute respiratory distress syndrome (ARDS) mortality from other etiologies. It has been confirmed from in vitro, ex vivo and in vivo studies that MV itself can induce lung injury through inflammatory Src tyrosine kinase signaling, ICAM-1 expression, leukocyte infiltration and vascular hyperpermeability [92]. Pulmonary vascular endothelial barrier function is indeed partially regulated by Src kinase-dependent phosphorylation of caveolin-1 and intercellular adhesion molecule-1. Blocking these mechanisms with Src tyrosine inhibition (i.e. nintedanib [93]) has proved effective in preventing acute lung injury (ALI) in animal studies including the potentiation of bleomycin-induced ALI [94].
(d) Pulmonary ischemia–reperfusion, which is associated with a wide range of clinical events [95], including lung transplantation, cardiopulmonary bypass, trauma, resuscitation for circulatory arrest, atherosclerosis and pulmonary embolism, the latter being widely recognized as a detrimental complication of severe COVID-19 [96].
(e) A final mechanism by which tyrosine kinase inhibitors could possibly exert anti-COVID-19 action is by eliminating excess soluble fms-like tyrosine kinase (in blood), which has been correlated with endothelial dysfunction and organ failure in critically ill COVID-19 patients [97, 98].
Complementary to the Src tyrosine kinase inhibitors, the interest on hydroquinone and vorinostat stems from the fact that melanin synthesis inhibition (e.g. by hydroquinone) involves similar pathways with tyrosine kinase. Both drugs are inhibitors of HDAC and have been used to treat pediatric brain cancers [99] and might exert a beneficial effect through inhibition of the pro-inflammatory cytokine storm progression. Although vorinostat has been used against HPV [100], we have not found any specific references in the current literature linking this drug category to COVID-19.
Inhibitors of class IA phosphatidyl inositol-3 kinases, such as dactolisib, are targeting immune responses, particularly through co-stimulation by CD28 and ICOS and have been reported to suppress clinical symptoms in ongoing experimental autoimmune encephalomyelitis and inhibited MOG-specific responses in vitro [101]. Hence, the latter class of drugs can be beneficial in treating COVID-19 effects to the CNS, albeit the underlying mechanisms and the pathology are not well understood. On the other hand, activation of PI3K/Akt pathway has been reported to cause attenuation of Endoplasmic reticulum (ER) stress-induced myocardial apoptosis, facilitating the NGF-induced heart protection [102]. Given that myocardial involvement in COVID-19 was observed in a subset of patients and has detrimental consequences, there might be a reason to be cautious how we interpret this finding.
Despite being less studied, the identified compounds that target directly the viral entry and replication mechanisms and related proteins are of interest. Through our pathogen network-based approaches, the integrated list of antivirals comprises remdesivir, the first drug to obtain Food and Drug Administration (FDA) approval for emergency use in patients with severe COVID-19 [103], along with a number of experimental compounds developed during the SARS and MERS outbreaks with promising in vitro results such as benzyl (2-oxopropyl) carbamate [104, 105], D3F, F3F, GRL0617 and WR1. This list is also complemented by N-(2-aminoethyl)-1-aziridineethanamine that can be effective via its inhibition of ACE2, which is being used by SARS-CoVs for cell entry in order to initiate membrane fusion and cell infection [106]. As expected, the top scoring repurposed antivirals based on commonality and available information from other pathogens were the ones known to be effective against the closest taxa, e.g. members of the SARS-CoV species. However, our full lists of antiviral drug rankings (HPAV and TaxAV available in Supplementary File S5) not only identified and re-ranked drugs nearly from all main phyla of viruses, but also from bacteria that may share interactions with the same host proteins or share genetic similarity. In fact, this approach that was developed and utilized herein specifically for SARS-CoV-2 introduces a rapid mapping and prioritization method of all the known antiviral compounds against any pathogen of interest.
Naturally a multi-source approach like the one proposed in this work comes with certain limitations such as (a) the lack of harmonization across selection criteria applied for the DEGs across several datasets. Ideally data selection should be based on identical criteria/thresholds. This was possible for the cases where raw data were available and the data were produced from the same or similar experimental methodology (e.g. in Series 1, 2–5, 6, 7, 15) as opposed to BALF, PBMC, proteomics and metabolomics datasets; (b) another limitation was the fact that selection biases among drugs might exist as observed in our null model analysis. Although we detected this possible bias for a limited number of drugs, we deemed more appropriate to keep them and highlight them as potential biased results, rather than excluding them from the final list; (c) finally, the redundancy analysis revealed some overlap between specific drug lists, mainly between HPAV and TaxAV. The small size of the initial pool of this category of drugs (compared with the other approaches) and common components (i.e. the taxonomy distance) in the scoring functions of these approaches expectantly yielded a higher degree of overlap. However, since both lists introduced unique drugs, we deemed appropriate to keep both methods in our workflow. It is also worth noting that as the number of known viral–host protein interactions in the literature increases, in a future application of this workflow (for SARS-CoV-2 or other pathogens), the lists are likely to diverge even more since TaxAV is insensitive to these interactions while the opposite applies for HPAV.
Concluding, we have identified a shortlist of drugs generated from multi-source, multi-omics data utilizing a multiplex DR approach. Note that, we do not anticipate that a single drug can lead to a sufficiently effective treatment of severe COVID-19 and especially in patients with comorbidities. Based on the multi-faceted nature of COVID-19, an effective treatment will require combination(s) of drugs, allowing to target multiple pathways to provide (a) anti-inflammatory effects to prevent collateral tissue damage, (b) inhibition of the viral replication mechanisms and (c) where required, protective effects, which account for other underlying conditions like CVD. We anticipate that the proposed integrated list of drugs will enhance the underlying rational and provide insights from a molecular point of view for the compounds that have already entered clinical trials and more importantly will point towards novel therapeutic avenues involving the newly proposed ones.
Key Points
We proposed a protocol for multiplex DR approach against COVID-19 based on the molecular profile of patients, extracted from a multi-source multi-omics integrative analysis.
We have focused on utilizing the publicly available multi-source and multi-omics datasets towards devising a multiplex DR approach yielding a highly informed shortlist of drug candidates against COVID-19 and its causative virus.
We initially extracted all the available drugs relevant to the molecular profile observed in COVID-19 through an ensemble of existing computational tools as well as newly tailored methods. Through a network-based integration approach, we have generated a gene–disease association map, which we exploited further for (a) re-ranking the initial identified therapeutics and (b) identifying the biological mechanisms and pathway communities that are involved in the onset and progression of the disease.
The proposed drug list not only comprises both drugs aiming to reverse COVID-19-induced perturbations (such as immunomodulatory and anti-inflammatory drugs), but also compounds with a direct antiviral activity. Several of these drugs are already in clinical trials.
Through the functional analysis of the identified molecular profiles, we found the Src tyrosine kinase to be involved in multiple COVID-19-related pathophysiological mechanisms. Hence, Src tyrosine kinase inhibitors such as bosutinib, dasatinib, cytarabine and saracatinib could hold a promising potential against COVID-19.
Supplementary Material
Marios Tomazou is a Postdoctoral Researcher at The Cyprus Institute of Neurology and Genetics (CING) and an Associate Faculty at the Cyprus School of Molecular Medicine (CSMM).
Marilena M. Bourdakou is a Visiting Scientist in the Bioinformatics Department at the Cyprus Institute of Neurology and Genetics (CING) and Postdoctoral Researcher in the Laboratory of Pharmacology, Faculty of Medicine, Democritus University of Thrace, Greece.
George Minadakis is an Associate Scientist in the Bioinformatics Department at the Cyprus Institute of Neurology and Genetics (CING) and an Associate Faculty at the Cyprus School of Molecular Medicine (CSMM).
Margarita Zachariou is an Associate Scientist in the Bioinformatics Department at the Cyprus Institute of Neurology and Genetics (CING) and an Associate Faculty at the Cyprus School of Molecular Medicine (CSMM).
Anastasis Oulas is an Associate Scientist in the Bioinformatics Department at the Cyprus Institute of Neurology and Genetics (CING) and an Associate Faculty at the Cyprus School of Molecular Medicine (CSMM).
Evangelos Karatzas is a Research Associate in the Bioinformatics Department at the Cyprus Institute of Neurology and Genetics (CING) and a Postdoctoral Researcher at the Institute for Fundamental Biomedical Research, BSRC “Alexander Fleming”, Vari, Greece.
Eleni M. Loizidou is a Research Associate in the Bioinformatics Department at the Cyprus Institute of Neurology and Genetics (CING) and a Postdoctoral Researcher at the Institute for Fundamental Biomedical Research, BSRC “Alexander Fleming”, Greece and the University of Ioannina, Greece.
Andrea C. Kakouri is a PhD Student at the Cyprus School of Molecular Medicine (CSMM) at The Cyprus Institute of Neurology and Genetics (CING).
Christiana C. Christodoulou is a PhD Student at the Cyprus School of Molecular Medicine (CSMM) at The Cyprus Institute of Neurology and Genetics (CING).
Kyriaki Savva is a PhD Student at the Cyprus School of Molecular Medicine (CSMM) at The Cyprus Institute of Neurology and Genetics (CING).
Maria Zanti is a PhD Student at the Cyprus School of Molecular Medicine (CSMM) at The Cyprus Institute of Neurology and Genetics (CING).
Anna Onisiforou is a PhD Student at the Cyprus School of Molecular Medicine (CSMM) at The Cyprus Institute of Neurology and Genetics (CING).
Sotiroula Afxenti is a PhD Student at the Cyprus School of Molecular Medicine (CSMM) at The Cyprus Institute of Neurology and Genetics (CING).
Jan Richter is a Scientist in the Department of Molecular Virology at the Cyprus Institute of Neurology and Genetics (CING).
Christina G. Christodoulou is the Head of the Department of Molecular Virology at the Cyprus Institute of Neurology and Genetics (CING) and Professor at CSMM.
Theodoros Kyprianou is Associate Professor of Medicine at the University of Nicosia Medical School and Consultant in Respiratory and Intensive Care Medicine at the University Hospitals Bristol & Weston NHS Foundation Trust, UK.
George Kolios is Professor of Pharmacology, Laboratory of Pharmacology, Faculty of Medicine, Democritus University of Thrace, Greece.
Nikolas Dietis is Assistant Professor of Pharmacology and the Head of Experimental Pharmacology Laboratory, Medical School, University of Cyprus.
George M. Spyrou is the Bioinformatics ERA Chair, Head of the Bioinformatics Department at the Cyprus Institute of Neurology and Genetics (CING) and Professor at CSMM.
Contributor Information
Marios Tomazou, Bioinformatics Department, The Cyprus Institute of Neurology and Genetics, Cyprus; The Cyprus School of Molecular Medicine, Cyprus; Neurogenetics Department, The Cyprus Institute of Neurology and Genetics, Cyprus.
Marilena M Bourdakou, Bioinformatics Department, The Cyprus Institute of Neurology and Genetics, Cyprus; Laboratory of Pharmacology, Faculty of Medicine, Democritus University of Thrace, Greece.
George Minadakis, Bioinformatics Department, The Cyprus Institute of Neurology and Genetics, Cyprus; The Cyprus School of Molecular Medicine, Cyprus.
Margarita Zachariou, Bioinformatics Department, The Cyprus Institute of Neurology and Genetics, Cyprus; The Cyprus School of Molecular Medicine, Cyprus.
Anastasis Oulas, Bioinformatics Department, The Cyprus Institute of Neurology and Genetics, Cyprus; The Cyprus School of Molecular Medicine, Cyprus.
Evangelos Karatzas, Bioinformatics Department, The Cyprus Institute of Neurology and Genetics, Cyprus; Institute for Fundamental Biomedical Research, BSRC “Alexander Fleming”, Vari, Greece.
Eleni M Loizidou, Bioinformatics Department, The Cyprus Institute of Neurology and Genetics, Cyprus; Institute for Fundamental Biomedical Research, BSRC “Alexander Fleming”, Vari, Greece.
Andrea C Kakouri, Bioinformatics Department, The Cyprus Institute of Neurology and Genetics, Cyprus; The Cyprus School of Molecular Medicine, Cyprus; Neurogenetics Department, The Cyprus Institute of Neurology and Genetics, Cyprus.
Christiana C Christodoulou, Bioinformatics Department, The Cyprus Institute of Neurology and Genetics, Cyprus; The Cyprus School of Molecular Medicine, Cyprus; Neuroepidemiology Department, The Cyprus Institute of Neurology and Genetics, Cyprus.
Kyriaki Savva, Bioinformatics Department, The Cyprus Institute of Neurology and Genetics, Cyprus; The Cyprus School of Molecular Medicine, Cyprus.
Maria Zanti, Bioinformatics Department, The Cyprus Institute of Neurology and Genetics, Cyprus; The Cyprus School of Molecular Medicine, Cyprus; Cancer Genetics, Therapeutics … Ultrastructural Pathology, The Cyprus Institute of Neurology and Genetics, Cyprus.
Anna Onisiforou, Bioinformatics Department, The Cyprus Institute of Neurology and Genetics, Cyprus; The Cyprus School of Molecular Medicine, Cyprus.
Sotiroula Afxenti, Bioinformatics Department, The Cyprus Institute of Neurology and Genetics, Cyprus; The Cyprus School of Molecular Medicine, Cyprus; Neuroimmunology Department, The Cyprus Institute of Neurology and Genetics, Cyprus.
Jan Richter, Department of Molecular Virology, The Cyprus Institute of Neurology and Genetics, Cyprus; The Cyprus School of Molecular Medicine, Cyprus.
Christina G Christodoulou, Department of Molecular Virology, The Cyprus Institute of Neurology and Genetics, Cyprus; The Cyprus School of Molecular Medicine, Cyprus.
Theodoros Kyprianou, Medical School, University of Nicosia, Cyprus; University Hospitals Bristol and Weston NHS Foundation Trust, United Kingdom.
George Kolios, Laboratory of Pharmacology, Faculty of Medicine, Democritus University of Thrace, Greece.
Nikolas Dietis, Experimental Pharmacology Laboratory, Medical School, University of Cyprus, Cyprus.
George M Spyrou, Bioinformatics Department, The Cyprus Institute of Neurology and Genetics, Cyprus; The Cyprus School of Molecular Medicine, Cyprus.
Data Availability
All the R scripts developed and used in this work along with their input files and details on the use of other computational tools are available for academic use at https://github.com/CING-BIG/DAVID_vs_COVID-Project.
Authors’ Contributions
G.M.S. conceived and supervised the study. M.M.B., A.C.K., C.C.C. and G.M.S. collected, curated and analyzed the RNA-Seq, proteomics and metabolomics datasets. E.M.L. performed and analyzed the GWAS–phenotypic analysis. M.Zach and G.M.S. developed and performed the multi-omic integration approach. G.M., M.T., M.M.B. and G.M.S. developed, performed and analyzed the pathogen–host interaction and taxonomy-based drug repurposing approach. K.S., M.M.B., M.T. and G.M.S. collected the chemical structures for all compounds of interest, performed the structural diversity analysis and comparison of the identified drugs with the ongoing clinical trials. E.K., M.T. and G.M.S. performed and analyzed the PathWalks results for the functional analysis of the genes of interest. N.D. annotated the integrated drug list with the relevant chemical data along with the MeSH, TBC and Chem classifications. Expert’s curation, annotation and discussion on highlighted drugs was performed by N.D. and T.K. with the support of G.K. Figures, tables and supplementary information were prepared by M.T., M.M.B., M.Zach, G.M., N.D. and G.M.S.; M.T., M.M.B., M.Zach, G.M., E.M.L., A.K., C.C.C., K.S., N.D., T.K. and G.M.S. wrote the manuscript, whereas all authors contributed in editing and reviewing. A.Oul, S.A., M. Zanti, A. Onis, J.R., C.G.C., N.D. and G.K. provided feedback on various aspects of the work. All authors have read and approved the submitted version of the manuscript.
Funding
M.T., G.M., M.Zach., A.Oul., A.C.K., M.Zant., C.C.C., and G.M.S. were funded by the European Commission Research Executive Agency Grant BIORISE (number 669026), under the Spreading Excellence, Widening Participation, Science with and for Society Framework.
E.K. was funded by the State Scholarships Foundation (IKY) scholarship, under the Action ‘Strengthening Human Resources, Education and Lifelong Learning’, 2014–20; co-funded by the European Social Fund (ESF) and the Greek State (MIS-5000432). M.M.B. is funded by the State Scholarships Foundation (IKY) scholarship; co-financed by Greece and the European Union (ESF) through the Operational Program ‘Human Resources Development, Education and Lifelong Learning’ in the context of the project ‘Reinforcement of Post-doctoral Researchers - 2nd Cycle’ (MIS-5033021), implemented by the State Scholarships Foundation (ΙΚY).
A part of the computational work of this project was supported by the European Union’s Horizon 2020 European research infrastructures, ‘National Initiatives for Open Science in Europe – NI4OS Europe’ project (grant agreement no. 857645). Specifically, the PathWalks parallel version was run on the PARADOX-IV supercomputing facility at the Scientific Computing Laboratory, National Center of Excellence for the Study of Complex Systems, Institute of Physics Belgrade, supported in part by the Ministry of Education, Science, and Technological Development of the Republic of Serbia under project No. ON171017.
References
- 1. Pushpakom S, Iorio F, Eyers PA, et al. Drug repurposing: Progress, challenges and recommendations. Nat Rev Drug Discov 2018;18:41–58. [DOI] [PubMed] [Google Scholar]
- 2. Jose RJ, Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir Med 2020;8:e46–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou S, Wang Y, Zhu T, et al. CT Features of Coronavirus Disease 2019 (COVID-19) Pneumonia in 62 Patients in Wuhan, China. Am J Roentgenol 2020;214:1287–94. [DOI] [PubMed] [Google Scholar]
- 4. Guo T, Fan Y, Chen M, et al. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol 2020;5:811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gysi DM, Do Valle Í, Zitnik M, et al. Network Medicine Framework for Identifying Drug Repurposing Opportunities for COVID-19 ArXiv. 2020. [DOI] [PMC free article] [PubMed]
- 6. Zhou Y, Hou Y, Shen J, et al. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARSCoV-2. Cell Discov 2020;6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Das S, Camphausen K, Shankavaram U. In silico Drug Repurposing to combat COVID-19 based onPharmacogenomics of Patient Transcriptomic Data. Res Sq 2020. [Google Scholar]
- 8. Loganathan T, Ramachandran S, Shankaran P, et al. Host transcriptome-guided drug repurposing for COVID-19 treatment: a meta-analysis based approach. PeerJ 2020;8:e9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Duarte RR, CopertinoDC, Jr, Iñiguez LP, et al. Repurposing fda-approved drugs for covid-19 using a datadriven approach ChemRxiv. 2020.
- 10. Dotolo S, Marabotti A, Facchiano A, et al. A review on drug repurposing applicable to COVID-19. Brief Bioinform 2021;22:726–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Elmezayen AD, Al-Obaidi A, Şahin AT, et al. Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J Biomol Struct Dyn 2020;1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peele KA, Potla Durthi C, Srihansa T, et al. Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: A computational study. Informatics Med Unlocked 2020;19:100345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Islam R, Parves MR, Paul AS, et al. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J Biomol Struct Dyn 2020;1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kandeel M, Abdelrahman AHM, Oh-Hashi K, et al. Repurposing of FDA-approved antivirals, antibiotics, anthelmintics, antioxidants, and cell protectives against SARS-CoV-2 papain-like protease. J Biomol Struct Dyn 2020;1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lobo-Galo N, Terrazas-López M, Martínez-Martínez A, et al. FDA-approved thiol-reacting drugs that potentially bind into the SARS-CoV-2 main protease, essential for viral replication. J Biomol Struct Dyn 2020;1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shah B, Modi P, Sagar SR. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci 2020;252:117652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu C, Liu Y, Yang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020;10, 766–8839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blanco-Melo D, Nilsson-Payant BE, Liu W-C, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020;181:1036–1045.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xiong Y, Liu Y, Cao L, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect 2020;9:761–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shen B, Yi X, Sun Y, et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020;182:59–72.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krämer A, Green J, Pollard J, et al. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014;30:523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gordon DE, Jang GM, Bouhaddou M, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020;583:459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020;181:271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gkogkou E, Barnasas G, Vougas K, et al. Expression profiling meta-analysis of ACE2 and TMPRSS2, the putative anti-inflammatory receptor and priming protease of SARS-CoV-2 in human cells, and identification of putative modulators. Redox Biol 2020;36:101615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kerrien S, Aranda B, Breuza L, et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res 2012;40:D841–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Durmuş Tekir S, Çakır T, Ardıç E, et al. PHISTO: pathogen–host interaction search tool. Bioinformatics 2013;29:1357–8. [DOI] [PubMed] [Google Scholar]
- 29. Guirimand T, Delmotte S, Navratil V. VirHostNet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res 2015;43:D583–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Subramanian A, Narayan R, Corsello SM, et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017;171:1437–1452.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Duan Q, Reid SP, Clark NR, et al. L1000CDS2: LINCS L1000 characteristic direction signatures search engine. npj Syst Biol Appl 2016;2:16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Z, Lachmann A, Keenan AB, et al. L1000FWD: fireworks visualization of drug-induced transcriptomic signatures. Bioinformatics 2018;34:2150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Z, Monteiro CD, Jagodnik KM, et al. Extraction and analysis of signatures from the Gene Expression Omnibus by the crowd. Nat Commun 2016;7:12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Staley JR, Blackshaw J, Kamat MA, et al. PhenoScanner: a database of human genotype–phenotype associations. Bioinformatics 2016;32:3207–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kamat MA, Blackshaw JA, Young R, et al. PhenoScanner V2: an expanded tool for searching human genotype–phenotype associations. Bioinformatics 2019;35:4851–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Buniello A, MacArthur JAL, Cerezo M, et al. The NHGRI-EBI GWAS Catalog of published genome-wide 40 association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 2019;47:D1005–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Psaty BM, O’Donnell CJ, Gudnason V, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium. Circ Cardiovasc Genet 2009;2:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cotto KC, Wagner AH, Feng Y-Y, et al. DGIdb 3.0: a redesign and expansion of the drug–gene interaction database. Nucleic Acids Res 2018;46:D1068–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Federhen S. The NCBI Taxonomy database. Nucleic Acids Res 2012;40:D136–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wishart DS. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 2006;34:D668–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018;46:D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chamberlain SA. Szöcs E. taxize: taxonomic search and retrieval in R. F1000Research 2013;2:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. CLARKE KR, WARWICK RM. A taxonomic distinctness index and its statistical properties. J Appl Ecol 1998;35:523–31. [Google Scholar]
- 44. Clarke K, Warwick R. A further biodiversity index applicable to species lists: variation in taxonomic distinctness. Mar Ecol Prog Ser 2001;216:265–78. [Google Scholar]
- 45. Spellerberg IF, Fedor PJ. A tribute to Claude Shannon (1916-2001) and a plea for more rigorous use of species richness, species diversity and the ‘Shannon-Wiener’ Index. Glob Ecol Biogeogr 2003;12:177–9. [Google Scholar]
- 46. Hockett CF, Shannon CL, Weaver W. The Mathematical Theory of Communication. Language (Baltim) 1953;29:69. [Google Scholar]
- 47. Thul PJ, Åkesson L, Wiking M, et al. A subcellular map of the human proteome. Science (80-) 2017;356:eaal3321. [DOI] [PubMed] [Google Scholar]
- 48. Zachariou M, Minadakis G, Oulas A, et al. Integrating multi-source information on a single network to detect disease-related clusters of molecular mechanisms. J Proteomics 2018;188:15–29. [DOI] [PubMed] [Google Scholar]
- 49. Carlson M. org. Hs. eg. db: Genome Wide Annotation for Human. R package version 2019;2(3):3. [Google Scholar]
- 50. Zuberi K, Franz M, Rodriguez H, et al. GeneMANIA Prediction Server 2013 Update. Nucleic Acids Res 2013;41:W115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hunter JE, Cohen SH. Package: igraph. Educ Psychol Meas 2007;29:697–700. [Google Scholar]
- 53. R Development Core Team. R . A language and environment for statistical computing. Vienna, Austria, 2018. [Google Scholar]
- 54. Karatzas E, Zachariou M, Bourdakou MM, et al. PathWalks: identifying pathway communities using a disease-related map of integrated information. Bioinformatics 2020;36:4070–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Karatzas E, Minadakis G, Kolios G, et al. A Web Tool for Ranking Candidate Drugs Against a Selected Disease Based on a Combination of Functional and Structural Criteria. Comput Struct Biotechnol J 2019;17:939–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bolton EE, Wang Y, Thiessen PA, et al. PubChem: Integrated Platform of Small Molecules and Biological 41Activities. Annu Rep Comput Chem 2008;217–41. [Google Scholar]
- 57. Corsello SM, Bittker JA, Liu Z, et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med 2017;23:405–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. O’Boyle NM, Banck M, James CA, et al. Open Babel: An open chemical toolbox. J Chem 2011;3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Karatzas E, Zamora JE, Athanasiadis E, et al. ChemBioServer 2.0: an advanced web server for filtering, clustering and networking of chemical compounds facilitating both drug discovery and repurposing. Bioinformatics 2020;36:2602–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schomburg D, Schomburg I. SARS coronavirus main proteinase 3.4.22.69. Cl. 3.4–6 Hydrolases. Lyases, Isomerases, Ligases 2013;65–97. [Google Scholar]
- 61. Lu I-L, Mahindroo N, Liang P-H, et al. Structure-Based Drug Design and Structural Biology Study of Novel Nonpeptide Inhibitors of Severe Acute Respiratory Syndrome Coronavirus Main Protease. J Med Chem 2006;49:5154–61. [DOI] [PubMed] [Google Scholar]
- 62. Ghosh AK, Takayama J, Aubin Y, et al. Structure-Based Design, Synthesis, and Biological Evaluation of a Series of Novel and Reversible Inhibitors for the Severe Acute Respiratory Syndrome−Coronavirus Papain-Like Protease. J Med Chem 2009;52:5228–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chaudhuri R, Tang S, Zhao G, et al. Comparison of SARS and NL63 Papain-Like Protease Binding Sites and Binding Site Dynamics: Inhibitor Design Implications. J Mol Biol 2011;414:272–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jacobs J, Grum-Tokars V, Zhou Y, et al. Discovery, Synthesis, And Structure-Based Optimization of a Series of N -( tert -Butyl)-2-( N -arylamido)-2-(pyridin-3-yl) Acetamides (ML188) as Potent Noncovalent Small Molecule Inhibitors of the Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) 3. J Med Chem 2013;56:534–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Deaton DN, Graham KP, Gross JW, et al. Thiol-based angiotensin-converting enzyme 2 inhibitors: P1′ modifications for the exploration of the S1′ subsite. Bioorg Med Chem Lett 2008;18:1681–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Huentelman MJ, Zubcevic J, Hernández Prada JA, et al. Structure-Based Discovery of a Novel Angiotensin-Converting Enzyme 2 Inhibitor. Hypertension 2004;44:903–6. [DOI] [PubMed] [Google Scholar]
- 67. Husain M, Cheung C-Y. Histone Deacetylase 6 Inhibits Influenza A Virus Release by Downregulating the Trafficking of Viral Components to the Plasma Membrane via Its Substrate, Acetylated Microtubules. J Virol 2014;88:11229–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Triplett TA, Holay N, Kottapalli S, et al. Elucidating the Role of HDACs in T Cell Biology and Comparing Distinct HDAC Inhibitors in Augmenting Responses to Cancer Immunotherapy. J Immunol 2020;204: 165.23 LP-165.23. [Google Scholar]
- 69. Akimova T, Beier UH, Liu Y, et al. Histone/protein deacetylases and T-cell immune responses. Blood 2012;119:2443–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Weisberg E, Parent A, Yang PL, et al. Repurposing of Kinase Inhibitors for Treatment of COVID-19. Pharm Res 2020;37:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dyall J, Coleman CM, Hart BJ, et al. Repurposing of Clinically Developed Drugs for Treatment of Middle East Respiratory Syndrome Coronavirus Infection. Antimicrob Agents Chemother 2014;58:4885–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mulgaonkar N, Wang H, Mallawarachchi S, et al. Bcr-Abl tyrosine kinase inhibitor imatinib as a potential drug for COVID-19 bioRxiv. 2020; 2020.06.18.158196 42.
- 73. Xing J, Shankar R, Drelich A, et al. Reversal of Infected Host Gene Expression Identifies Repurposed Drug Candidates for COVID-19. bioRxiv Prepr Serv Biol 2020. [Google Scholar]
- 74. Bermejo M, López-Huertas MR, García-Pérez J, et al. Dasatinib inhibits HIV-1 replication through the interference of SAMHD1 phosphorylation in CD4+ T cells. Biochem Pharmacol 2016;106:30–45. [DOI] [PubMed] [Google Scholar]
- 75. Ramaiah MJ. mTOR inhibition and p53 activation, microRNAs: The possible therapy against pandemic COVID-19. Gene Reports 2020;20:100765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Omarjee L, Janin A, Perrot F, et al. Targeting T-cell senescence and cytokine storm with rapamycin to prevent severe progression in COVID-19. Clin Immunol 2020;216:108464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Campbell GR, Bruckman RS, Herns SD, et al. Induction of autophagy by PI3K/MTOR and PI3K/MTOR/BRD4 inhibitors suppresses HIV-1 replication. J Biol Chem 2018;293:5808–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mao L, Jin H, Wang M, et al. Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol 2020;77:683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kopel J, Perisetti A, Gajendran M, et al. Clinical Insights into the Gastrointestinal Manifestations of COVID-19. Dig Dis Sci 2020;65:1932–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ma L, Song K, Huang Y. Coronavirus Disease-2019 (COVID-19) and Cardiovascular Complications. J Cardiothorac Vasc Anesth 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zheng Y-Y, Ma Y-T, Zhang J-Y, et al. COVID-19 and the cardiovascular system. Nat Rev Cardiol 2020;17:259–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ye Z, Zhang Y, Wang Y, et al. Chest CT manifestations of new coronavirus disease 2019 (COVID-19): a pictorial review. Eur Radiol 2020;30:4381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Li H, Xiang X, Ren H, et al. Serum Amyloid A is a biomarker of severe Coronavirus Disease and poor prognosis. J Infect 2020;80:646–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lo MW, Kemper C, Woodruff TM. COVID-19: Complement, Coagulation, and Collateral Damage. J Immunol 2020;205:1488–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Saxena A, Chaudhary U, Bharadwaj A, et al. A Lung Transcriptomic Analysis for Exploring Host Response in COVID-19. J Pure Appl Microbiol 2020;14:1077–81. [Google Scholar]
- 86. Hondermarck H, Bartlett NW, Nurcombe V. The role of growth factor receptors in viral infections: An opportunity for drug repurposing against emerging viral diseases such as COVID-19? FASEB BioAdvances 2020;2:296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tutuncuoglu B, Cakir M, Batra J, et al. The Landscape of Human Cancer Proteins Targeted by SARS-CoV-2. Cancer Discov 2020;10:916–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. McGee MC, August A, Huang W. BTK/ITK dual inhibitors: Modulating immunopathology and lymphopenia for COVID-19 therapy. J Leukoc Biol 2021;109:49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Roschewski M, Lionakis MS, Sharman JP, et al. Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci Immunol 2020;5:eabd0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rivera-Torres J, San José E. Src Tyrosine Kinase Inhibitors: New Perspectives on Their Immune, Antiviral, and Senotherapeutic Potential. Front Pharmacol 2019;10:1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Branchford BR, Stalker TJ, Law L, et al. The small-molecule MERTK inhibitor UNC 2025 decreases platelet 43 activation and prevents thrombosis. J Thromb Haemost 2018;16:352–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Piegeler T, Dull RO, Hu G, et al. Ropivacaine attenuates endotoxin plus hyperinflation-mediated acute lung injury via inhibition of early-onset Src-dependent signaling. BMC Anesthesiol 2014;14:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Li L-F, Kao K-C, Liu Y-Y, et al. Nintedanib reduces ventilation-augmented bleomycin-induced epithelialmesenchymal transition and lung fibrosis through suppression of the Src pathway. J Cell Mol Med 2017;21:2937–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Li L-F, Liu Y-Y, Kao K-C, et al. Mechanical ventilation augments bleomycin-induced epithelial–mesenchymal transition through the Src pathway. Lab Invest 2014;94:1017–29. [DOI] [PubMed] [Google Scholar]
- 95. Oyaizu T, Fung S-Y, Shiozaki A, et al. Src tyrosine kinase inhibition prevents pulmonary ischemia–reperfusion-induced acute lung injury. Intensive Care Med 2012;38:894–905. [DOI] [PubMed] [Google Scholar]
- 96. Grillet F, Behr J, Calame P, et al. Acute Pulmonary Embolism Associated with COVID-19 Pneumonia Detected with Pulmonary CT Angiography. Radiology 2020;296:E186–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Dupont V, Kanagaratnam L, Goury A, et al. Excess soluble fms-like tyrosine kinase 1 correlates with endothelial dysfunction and organ failure in critically ill COVID-19 patients. Clin Infect Dis 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Marchetti M. COVID-19-driven endothelial damage: complement, HIF-1, and ABL2 are potential pathways of damage and targets for cure. Ann Hematol 2020;99:1701–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Perla A, Fratini L, Cardoso PS, et al. Histone Deacetylase Inhibitors in Pediatric Brain Cancers: Biological Activities and Therapeutic Potential. Front Cell Dev Biol 2020;8:546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Banerjee NS, Moore DW, Broker TR, et al. Vorinostat, a pan-HDAC inhibitor, abrogates productive HPV-18 DNA amplification. Proc Natl Acad Sci 2018;115:E11138–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Acosta YY, Montes-Casado M, Aragoneses L. Erratum: Suppression of CD4+ T lymphocyte activation ‘in vitro’ and experimental encephalomyelitis ‘in vivo’ by the phosphatidyl inositol 3-kinase inhibitor PIK-75 (International Journal of Immunopathology and Pharmacology (2014) 27:1 (53-67)). Int J Immunopathol Pharmacol 2015. [DOI] [PubMed] [Google Scholar]
- 102. Wei K, Liu L, Xie F, et al. Nerve Growth Factor Protects the Ischemic Heart via Attenuation of the Endoplasmic Reticulum Stress Induced Apoptosis by Activation of Phosphatidylinositol 3-Kinase. Int J Med Sci 2015;12:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Eastman RT, Roth JS, Brimacombe KR, et al. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent Sci 2020;6:672–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Verschueren KHG, Pumpor K, Anemüller S, et al. A Structural View of the Inactivation of the SARS Coronavirus Main Proteinase by Benzotriazole Esters. Chem Biol 2008;15:597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bacha U, Barrila J, Gabelli SB, et al. Development of Broad-Spectrum Halomethyl Ketone Inhibitors Against Coronavirus Main Protease 3CL pro. Chem Biol Drug Des 2008;72:34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhang H, Penninger JM, Li Y, et al. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med 2020;46:586–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the R scripts developed and used in this work along with their input files and details on the use of other computational tools are available for academic use at https://github.com/CING-BIG/DAVID_vs_COVID-Project.