

Abstract
Background
Convalescent plasma and hyperimmune immunoglobulin may reduce mortality in patients with viral respiratory diseases, and are being investigated as potential therapies for coronavirus disease 2019 (COVID‐19). A thorough understanding of the current body of evidence regarding benefits and risks of these interventions is required.
Objectives
Using a living systematic review approach, to assess whether convalescent plasma or hyperimmune immunoglobulin transfusion is effective and safe in the treatment of people with COVID‐19; and to maintain the currency of the evidence.
Search methods
To identify completed and ongoing studies, we searched the World Health Organization (WHO) COVID‐19 Global literature on coronavirus disease Research Database, MEDLINE, Embase, the Cochrane COVID‐19 Study Register, the Epistemonikos COVID‐19 L*OVE Platform, and trial registries. Searches were done on 17 March 2021.
Selection criteria
We included randomised controlled trials (RCTs) evaluating convalescent plasma or hyperimmune immunoglobulin for COVID‐19, irrespective of disease severity, age, gender or ethnicity. For safety assessments, we also included non‐controlled non‐randomised studies of interventions (NRSIs) if 500 or more participants were included.
We excluded studies that included populations with other coronavirus diseases (severe acute respiratory syndrome (SARS) or Middle East respiratory syndrome (MERS)), as well as studies evaluating standard immunoglobulin.
Data collection and analysis
We followed standard Cochrane methodology.
To assess bias in included studies, we used the Cochrane 'Risk of Bias 2' tool for RCTs, and for NRSIs, the assessment criteria for observational studies, provided by Cochrane Childhood Cancer. We rated the certainty of evidence, using the GRADE approach, for the following outcomes: all‐cause mortality, improvement and worsening of clinical status (for individuals with moderate to severe disease), development of severe clinical COVID‐19 symptoms (for individuals with asymptomatic or mild disease), quality of life (including fatigue and functional independence), grade 3 or 4 adverse events, and serious adverse events.
Main results
We included 13 studies (12 RCTs, 1 NRSI) with 48,509 participants, of whom 41,880 received convalescent plasma. We did not identify any completed studies evaluating hyperimmune immunoglobulin. We identified a further 100 ongoing studies evaluating convalescent plasma or hyperimmune immunoglobulin, and 33 studies reporting as being completed or terminated.
Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease
Eleven RCTs and one NRSI investigated the use of convalescent plasma for 48,349 participants with moderate to severe disease. Nine RCTs compared convalescent plasma to placebo treatment or standard care alone, and two compared convalescent plasma to standard plasma (results not included in abstract).
Effectiveness of convalescent plasma
We included data on nine RCTs (12,875 participants) to assess the effectiveness of convalescent plasma compared to placebo or standard care alone.
Convalescent plasma does not reduce all‐cause mortality at up to day 28 (risk ratio (RR) 0.98, 95% confidence interval (CI) 0.92 to 1.05; 7 RCTs, 12,646 participants; high‐certainty evidence). It has little to no impact on clinical improvement for all participants when assessed by liberation from respiratory support (RR not estimable; 8 RCTs, 12,682 participants; high‐certainty evidence). It has little to no impact on the chance of being weaned or liberated from invasive mechanical ventilation for the subgroup of participants requiring invasive mechanical ventilation at baseline (RR 1.04, 95% CI 0.57 to 1.93; 2 RCTs, 630 participants; low‐certainty evidence). It does not reduce the need for invasive mechanical ventilation (RR 0.98, 95% CI 0.89 to 1.08; 4 RCTs, 11,765 participants; high‐certainty evidence). We did not identify any subgroup differences.
We did not identify any studies reporting quality of life, and therefore, do not know whether convalescent plasma has any impact on quality of life. One RCT assessed resolution of fatigue on day 7, but we are very uncertain about the effect (RR 1.21, 95% CI 1.02 to 1.42; 309 participants; very low‐certainty evidence).
Safety of convalescent plasma
We included results from eight RCTs, and one NRSI, to assess the safety of convalescent plasma. Some of the RCTs reported on safety data only for the convalescent plasma group.
We are uncertain whether convalescent plasma increases or reduces the risk of grade 3 and 4 adverse events (RR 0.90, 95% CI 0.58 to 1.41; 4 RCTs, 905 participants; low‐certainty evidence), and serious adverse events (RR 1.24, 95% CI 0.81 to 1.90; 2 RCTs, 414 participants; low‐certainty evidence).
A summary of reported events of the NRSI (reporting safety data for 20,000 of 35,322 transfused participants), and four RCTs reporting safety data only for transfused participants (6125 participants) are included in the full text.
Individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease
We identified one RCT reporting on 160 participants, comparing convalescent plasma to placebo treatment (saline).
Effectiveness of convalescent plasma
We are very uncertain about the effect of convalescent plasma on all‐cause mortality (RR 0.50, 95% CI 0.09 to 2.65; very low‐certainty evidence). We are uncertain about the effect of convalescent plasma on developing severe clinical COVID‐19 symptoms (RR not estimable; low‐certainty evidence).
We identified no study reporting quality of life.
Safety of convalescent plasma
We do not know whether convalescent plasma is associated with a higher risk of grade 3 or 4 adverse events (very low‐certainty evidence), or serious adverse events (very low‐certainty evidence).
This is a living systematic review. We search weekly for new evidence and update the review when we identify relevant new evidence. Please refer to the Cochrane Database of Systematic Reviews for the current status of this review.
Authors' conclusions
We have high certainty in the evidence that convalescent plasma for the treatment of individuals with moderate to severe disease does not reduce mortality and has little to no impact on measures of clinical improvement. We are uncertain about the adverse effects of convalescent plasma. While major efforts to conduct research on COVID‐19 are being made, heterogeneous reporting of outcomes is still problematic. There are 100 ongoing studies and 33 studies reporting in a study registry as being completed or terminated. Publication of ongoing studies might resolve some of the uncertainties around hyperimmune immunoglobulin therapy for people with any disease severity, and convalescent plasma therapy for people with asymptomatic or mild disease.
Plain language summary
Is plasma from people who have recovered from COVID‐19 an effective treatment for people with COVID‐19?
Key messages
• We are very confident that convalescent plasma has no benefits for the treatment of people with moderate to severe COVID‐19.
• We are uncertain about the effects of convalescent plasma for treating people with mild COVID‐19 or who have no symptoms.
• We found about 130 ongoing, unpublished and recently published studies. We will update our review with evidence from these studies as soon as possible. New evidence may answer our remaining questions.
What is convalescent plasma?
The body produces antibodies as one of its defences against infection. Antibodies are found in part of the blood called plasma. Plasma from people who have recovered from the COVID‐19 virus contains COVID‐19 antibodies, and can be used to make two preparations. Firstly, it can be used to make convalescent plasma, which is plasma that contains these antibodies. Secondly, it can be used to make hyperimmune immunoglobulin, which is more concentrated, and therefore contains more antibodies.
Convalescent plasma and hyperimmune immunoglobulin have been used successfully to treat some viruses. These treatments (given by a drip or injection) are generally well‐tolerated, but can cause unwanted effects.
What did we want to find out?
We wanted to find out whether convalescent plasma or hyperimmune immunoglobulin are effective treatments for people with confirmed COVID‐19. We looked at:
• deaths from any cause after treatment with convalescent plasma or hyperimmune immunoglobulin;
• improvement or worsening of patients’ condition, measured by the number of people who needed help from a ventilator (a machine that helps people breathe if they cannot breathe on their own);
• quality of life; and
• unwanted effects.
What did we do?
We searched for studies that investigated convalescent plasma or hyperimmune immunoglobulin to treat people with COVID‐19. Studies could take place anywhere in the world and include participants of any age, gender or ethnicity, with mild, moderate or severe COVID‐19.
Where possible we pooled the studies’ results to analyse them. We rated our confidence in the evidence, based on factors such as study methods and sizes.
What did we find?
We found 13 studies with 48,509 participants that investigated convalescent plasma. All but one of the studies included participants with moderate to severe COVID‐19. We did not find any studies that investigated hyperimmune immunoglobulin. Studies mainly took place in hospitals, in countries all over the world.
Moderate to severe COVID‐19 Convalescent plasma compared to placebo or standard care:
• convalescent plasma makes no difference to deaths from any cause at up to 28 days after treatment. About 237 in 1000 people given placebo or standard care died, compared to 233 in 1000 people who had been given convalescent plasma (7 studies, 12,646 people);
• convalescent plasma makes little to no difference to the improvement of patients’ condition in terms of needing less breathing support for the overall population needing any breathing support before the start of treatment (8 studies, 12,682 people), and also not for the people that were ventilated at the beginning of the study (2 studies, 630 people);
• convalescent plasma makes no difference to the worsening of patients’ condition. About 126 in 1000 people given placebo or standard care needed invasive mechanical ventilation, compared to 123 in 1000 people who had been given convalescent plasma (4 studies, 11,765 people);
• convalescent plasma may make no difference to unwanted effects. The 8 studies that reported unwanted effects measured and reported their results very differently, so we are unable to draw any conclusions.
None of the studies reported quality of life.
Mild COVID‐19 We do not know if convalescent plasma compared to placebo or standard care makes a difference to number of deaths, improvement or worsening of patients’ condition, quality of life or unwanted effects. We found only one study with 160 participants that assessed people with mild COVID‐19.
What are the limitations of the evidence?
• We are very confident in the evidence for deaths from any cause and improvement or worsening of patients’ condition in people with moderate to severe COVID‐19.
• Our confidence in the other evidence for people with moderate and severe, and mild COVID‐19 is very limited because the studies were very different and did not measure and record their results using consistent methods.
• We found little useful evidence on unwanted effects and none on quality of life.
How up to date is this evidence?
This is the fourth version of our review. The evidence is up to date to 17 March 2021.
Summary of findings
Background
‐
Description of the condition
The clinical syndrome coronavirus disease 2019 (COVID‐19) is a new, rapidly emerging zoonotic infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2; WHO 2020a). On 22 March 2020, the World Health Organization (WHO) declared the current COVID‐19 outbreak to be a pandemic, with the outbreak resulting in more than 119 million confirmed cases and over 2.5 million deaths worldwide (WHO 2020b; WHO 2021a). Although there are similarities with historic coronavirus epidemics, with severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) responsible for 813 and 858 deaths respectively, the scale and impact of the COVID‐19 pandemic presents unprecedented challenges to health facilities and healthcare workers all over the world (WHO 2007; WHO 2019).
Approximately 5% of patients with COVID‐19 and 20% of those hospitalised experience severe disease requiring intensive care (Wiersinga 2020). Early reports suggested case fatality rates between 0.7% and 4% (WHO 2020a; WHO 2020c). More recent reports estimate wide‐ranging case fatality rates, as low as 0.0% in Singapore and up to 9.0% in Mexico (Johns Hopkins 2021). However, these numbers should be interpreted with great care due to testing availability, underreporting of cases and delays from confirmation of a case to time of death (Kim 2020), ethnicity, underlying health conditions, access to health care and socioeconomic status (Williamson 2020).
The median incubation period of SARS‐CoV‐2 was reported to be five days, with 97.5% of cases developing symptoms within 11.5 days of infection (Lauer 2020). Common signs and symptoms can include fever, dry cough, fatigue and sputum production (WHO 2020a). Postviral olfactory dysfunction is reported in 5% to 85% of cases, with loss of both smell and taste reported (Izquierdo‐Dominguez 2020). Other less commonly reported signs and symptoms are shortness of breath, sore throat, headache, myalgia or arthralgia, chills, nausea or vomiting, nasal congestion, diarrhoea, haemoptysis and conjunctival congestion (WHO 2020a). Of the reported cases, 80% are estimated to have a mild or asymptomatic course of infection, and an estimated 5% of cases are admitted to the ICU with acute respiratory distress syndrome (ARDS), septic shock, multiple organ failure, or all three conditions (Team 2020; WHO 2020a). A risk factor for developing infection and progressing to severe disease is old age, with people aged over 80 years at highest risk of mortality. Other risk factors are cardiovascular disease, obesity, hypertension, diabetes, chronic respiratory disease, cancer and compromised immune status (Chen 2020a; Huang 2020; Liang 2020; WHO 2020a; Wu 2020a). Early reports have suggested that people who are immune‐compromised may not have an increased risk of being hospitalised with severe COVID‐19 symptoms (D'Antiga 2020). However, evidence has been conflicting, with patients with malignancy and recipients of solid organ and allogeneic transplants reported to have an increased risk of severe COVID‐19 disease (Fung 2020; Sharma 2021).
SARS‐CoV‐2 is a positive‐sense, single‐stranded ribonucleic acid (RNA) virus with a large genome. There are indications that the virus is capable of inducing an excessive immune reaction in the host, with highly activated but decreased numbers of CD4+ and CD8+ T cells detected in the peripheral blood of people with COVID‐19 (Xu 2020a). Early reports also showed that people critically ill with COVID‐19 frequently exhibit a hypercoagulable state and endothelial inflammation, which is hypothesised to lead to the high burden of thromboembolic events seen in this population (Driggin 2020). SARS‐CoV‐2 binds to the angiotensin‐converting enzyme 2 (ACE2). ACE2 is a protein that functions as the receptor, facilitating entry of SARS‐CoV‐2 into the host cell, and is most abundant on type II alveolar cells in the lungs (Tolouian 2020; Van de Veerdonk 2020).
Description of the intervention
Convalescent plasma, convalescent serum and hyperimmune immunoglobulin prepared from convalescent plasma are interventions that have been used in the past to treat conditions when no vaccine or pharmacological interventions were available. Diphtheria, pneumococcal pneumonia, hepatitis A and B, mumps, polio, measles and rabies are conditions where convalescent plasma has been shown to be effective (Eibl 2008).
A systematic review has shown that convalescent plasma may have clinical benefit for people with influenza and SARS (Mair‐Jenkins 2015). This systematic review included observational studies and randomised controlled trials (RCTs) investigating the use of convalescent plasma, serum or hyperimmune immunoglobulin for treating severe acute respiratory infections of laboratory‐confirmed or suspected viral aetiology, and included investigations with patients of any age and sex. Control interventions consisted of sham or placebo therapy and no therapy. Although the included studies were generally small and of low quality, with a moderate to high risk of bias, the review authors concluded that the use of convalescent plasma may reduce mortality, and appears safe (Mair‐Jenkins 2015). The authors also suggested that the effectiveness of convalescent plasma in reducing hospital length of stay is dependent on early administration of the therapy, and use as prophylaxis is more likely to be beneficial than treating severe disease. However, the optimal timing and dosage of convalescent plasma therapy is unknown.
There is conflicting evidence about the effect of convalescent plasma or hyperimmune immunoglobulin for treating severe acute respiratory infections. Studies investigating the effectiveness of hyperimmune immunoglobulin for influenza have been contradictory, with some RCTs showing effectiveness (Hung 2013), whereas others show no benefit (Beigel 2017; Beigel 2019; Davey 2019).
Although convalescent plasma is generally thought to be a safe and well‐tolerated therapy, adverse events can occur. Limited information is available about specific adverse events related to convalescent plasma therapy, but symptoms that have been reported are similar to those for other types of plasma blood components, including fever or chills, allergic reactions, and transfusion‐related acute lung injury (TRALI) (Beigel 2019; Chun 2016; Luke 2006). Furthermore, the transfer of coagulation factors present in plasma products is potentially harmful for people with COVID‐19, who are already at an increased risk of thromboembolic events (Driggin 2020). Plasma transfusions are also known to cause transfusion‐associated circulatory overload (TACO). TACO and TRALI are especially important to consider, because COVID‐19 patients with comorbidities, who might be eligible for experimental treatment with convalescent plasma therapy, are at an increased risk of these adverse events. There are risk‐mitigation strategies that can be implemented to prevent TRALI. These include limiting donations from female donors, especially those with a history of pregnancy, and screening of donors for antibodies that are implicated in TRALI (Otrock 2017). In addition to the aforementioned adverse events, transfusion‐transmitted infections, red blood cell allo‐immunisation and haemolytic transfusion reactions have also been described following plasma transfusion, although they are less common (Pandey 2012). Pathogen inactivation can be implemented to decrease the risk of transmitting infections by transfusion (Rock 2011).
When compared to convalescent plasma, hyperimmune immunoglobulin has the advantage of preventing transfer of potentially harmful coagulation factors that are present in plasma products. The amount and antibody concentration can be more accurately dosed compared to convalescent plasma, and hyperimmune immunoglobulin can be prepared in a consistent manner (Hung 2013). Not many studies have reported on adverse events of hyperimmune immunoglobulin, but the safety profile of standard intravenous immunoglobulin is known and the adverse events reported here are also likely to occur in hyperimmune immunoglobulin therapy. Common adverse events of intravenous immunoglobulin that occur immediately after administration are: infusion site pain; swelling and erythema; and immediate systemic reactions, such as head and body aches, chills and fever (Stiehm 2013). Other, less common early adverse reactions to immunoglobulin therapy are pulmonary complications, such as pulmonary embolism, pulmonary oedema and pleural effusion, with TRALI also reported (Baudel 2020; Stiehm 2013). Anaphylactic and anaphylactoid reactions to immunoglobulin therapy are rare (Brennan 2003; Stiehm 2013). Delayed adverse events of immunoglobulin therapy, which occur within hours to days of initiation of immunoglobulin therapy, are persistent headaches (common), aseptic meningitis, renal failure, thromboembolic events, and haemolytic reactions (Sekul 1994; Stiehm 2013). Transmission of infectious agents has been described after administration of intravenous immunoglobulin, but this risk is considered to be low (Stiehm 2013). Other severe adverse events that occur late after administration are lung disease, enteritis and dermatological disorders (Stiehm 2013).
A theoretical risk related to virus‐specific antibodies, which are transferred with convalescent plasma and hyperimmune immunoglobulin administration, is antibody‐dependent enhancement of infection (Morens 1994). Here, virus‐binding antibodies facilitate the entry and replication of virus particles into monocytes, macrophages and granulocytic cells and thereby increase the risk of more severe disease in the infected host. Although antibody‐dependent enhancement has not been demonstrated in COVID‐19, it has been seen with previous coronavirus infections when the antibodies given targeted a different serotype of the virus (Wan 2020; Wang 2014). A mechanism for antibody‐dependent enhancement in COVID‐19 has recently been proposed, with non‐neutralising antibodies to variable S domains potentially enabling an alternative infection pathway via Fc receptor‐mediated uptake (Ricke 2020). Antibody‐dependent enhancement is therefore a potentially harmful consequence of convalescent plasma and hyperimmune immunoglobulin therapy for COVID‐19. Safety of convalescent plasma for treatment of COVID‐19 has been investigated in a large cohort from the USA Food and Drug Administration (FDA) Expanded Access Program (Joyner 2020). Here, convalescent plasma did not clearly cause an excessive risk of adverse events within seven days of treatment, nor did it show an exceptionally high mortality rate at seven days (8.6%) (Joyner 2020).
In summary, the benefits of the intervention, both for convalescent plasma or hyperimmune immunoglobulin, should be carefully considered in view of the risks of adverse events.
How the intervention might work
Convalescent plasma contains pathogen‐specific neutralising antibodies, which can neutralise viral particles, and treatment with convalescent plasma or hyperimmune immunoglobulins confers passive immunity to recipients. The duration of conferred protection can differ depending on the timing of administration, ranging from weeks to months after treatment (Casadevall 2020).
By neutralising SARS‐CoV‐2 particles, early treatment with convalescent plasma is postulated to increase the patient’s own capacity to clear the initial inoculum (Casadevall 2020; Robbins 1995). This could lead to a reduction in mortality and fewer hospitalised patients progressing to the ICU. Furthermore, convalescent plasma may reduce the length of ICU stay in critically ill patients (Mair‐Jenkins 2015), thus helping to lift pressure from global healthcare systems and increasing ICU capacity.
Preliminary evidence in humans and rhesus macaques has shown that reinfection with SARS‐CoV‐2 is not likely, with most (but not all) patients who recovered from COVID‐19 producing sufficient amounts of neutralising antibodies to protect against reinfection (Bao 2020a; Wu 2020b). This implies that convalescent plasma from people who have recovered from SARS‐CoV‐2 infection may be capable of conferring passive immunity. Retrospective studies also observed a potential correlation between the level of antibody titres in convalescent plasma and recovery after treatment (Joyner 2021; Shen 2020). It is important to note, however, that research in other coronavirus species has shown that immunity may not be long‐lasting, with two to three years of protection estimated from work with SARS and MERS (Mo 2006; Payne 2016). Furthermore, there are indications that the severity of infection has an impact on antibody titres, with less‐severe disease leading to lower neutralising antibody response in people with SARS and COVID‐19 (Ho 2005; Zhao 2020a). And, it is unclear exactly how often reinfection occurs, with the burden of reinfection likely to be underestimated, while at the same time a number of case reports of severe reinfection have been published (Iwasaki 2021).
Why it is important to do this review
There is a clear, urgent need for more information to guide clinical decision‐making for COVID‐19 patients. Pharmacological treatment options are being investigated in many ongoing trials, with currently only treatment of dexamethasone and tocilizumab proven to be effective in reducing mortality (Horby 2020; Horby 2021), and remdesivir shown to reduce time to recovery (Beigel 2020). Current treatment further consists of supportive care with extracorporeal membrane oxygenation in severe cases and oxygen supply in less severe cases (CDC 2020b; WHO 2020d). Despite these treatments, people hospitalised with COVID‐19 are still at a high risk of mortality.
Ongoing vaccination programmes will aid in inducing immunity in the population and may prevent transmission to those who are at risk for severe disease. Several vaccines have been approved, and many more are in development (WHO 2020g). Mass vaccination programmes have been underway since late 2020 (WHO 2021b). Until these vaccines are globally distributed, convalescent plasma may be a potential therapy for COVID‐19 patients. Even with effective vaccines, not everyone can be effectively vaccinated; for example, people who are temporarily or permanently immune‐compromised, and very young children. Convalescent plasma, and hyperimmune immunoglobulin to a certain extent, can be prepared and made rapidly available by blood banks and hospitals when enough potential donors have recovered from the infection, using readily available materials and methods (Bloch 2020). However, its safety and efficacy are not well‐characterised, and there are costs associated with pursuing the use of convalescent plasma for treatment of COVID‐19.
A multitude of clinical trials investigating the safety and effectiveness of convalescent plasma or hyperimmune immunoglobulins have been announced, and their results will need to be interpreted with care. Thus, there needs to be a thorough understanding of the current body of evidence regarding the use of convalescent plasma for people with COVID‐19, and an extensive review of the available literature is required.
Objectives
Using a living systematic review approach, to assess whether convalescent plasma or hyperimmune immunoglobulin transfusion is effective and safe in the treatment of people with COVID‐19; and to maintain the currency of the evidence.
Methods
Criteria for considering studies for this review
Types of studies
The main description of methods is based on a template from the Cochrane Haematology review group and in line with a series of Cochrane Reviews investigating treatments and therapies for COVID‐19. The protocol for this review was registered with the Center for Open Science on 17 April 2020 (Piechotta 2020a). Amendments that have been made since are summarised in Differences between protocol and review and Table 4.
1. Summary of PICO development from protocol stage to current review version.
| Publication date | Participants | Interventions | Comparators | Outcomes | Study designs | |
|
Protocol Piechotta 2020a |
17 April 2020 | Inclusion
Exclusion
|
Inclusion
|
Inclusion
|
All criteria based on Core Outcome Measures in Effectiveness Trials (COMET) Initiative for COVID‐19 patients (COMET 2020) Primary outcomes
Secondary outcomes
|
Planned inclusion priority, determined by availability of sufficient evidence:
|
|
Version 1 Valk 2020 |
14 May 2020 | see above | Inclusion
Exclusion
|
see above | All criteria based onCOMET Initiative for COVID‐19 patients (COMET 2020) Primary outcomes
Secondary outcomes
|
Inclusion
No evidence available for
|
| Changesb | None | Added exclusion criteria
|
None | Revised secondary outcome "Improvement of clinical symptoms, assessed through need for respiratory support":
|
none | |
|
Version 2 Piechotta 2020b |
10 July 2020 | see above | see above |
Inclusion
|
All criteria based on COMET Initiative for COVID‐19 patients (COMET 2020) Primary outcomes
Secondary outcomes
|
Inclusion
Further inclusion
According to originally planned inclusion priorities |
| Changesb | None | None | Added eligible control treatment:
|
Added a secondary outcome:
|
Added inclusion criteria for safety data:
|
|
|
Version 3 Chai 2020 |
12 October 2020 | see above | see above | see above | All criteria based on COMET Initiative for COVID‐19 patients (COMET 2020) Primary outcomes
Secondary outcomes
|
Inclusion
Further inclusion
According to originally planned inclusion priorities Exclusion
|
| Changesb | None | None | None | Revised and renamed secondary outcome “Improvement of clinical symptoms”
Added secondary outcome:
|
Added exclusion criteria:
|
|
| Version 4 | (Current version) | Inclusion
Exclusion
|
see above |
Inclusion
|
All criteria based on COMET Initiative for COVID‐19 patients (COMET 2020), and outcomes prioritised by consumer representatives, referees of previous versions of this review, and the German guideline panel for inpatient therapy of people with COVID‐19. Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe diseaseEffectiveness of convalescent plasma Prioritised outcomes
Additional outcomes
Safety of convalescent plasma
Individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease Effectiveness of convalescent plasma Prioritised outcomes
Additional outcomes
Safety of convalescent plasma
|
Inclusion
|
| Changesb | Introduced separate populationsc
|
None | Added eligible control treatment
Added specifications on placebo treatment
|
Changed primary and secondary outcomes to prioritised (included in 'Summary of findings' table) and additional outcomes (not included in 'Summary of findings' table). Revised and specified outcomes per population. Individuals with moderate to severe disease
Added outcomes for individuals with asymptomatic or mild disease |
Added inclusion criteria:
Added exclusion criteria
|
aIncluding changes in study designs and methodology. bChanges in PICO compared to the previously published version. cAccording to the latest WHO clinical progression score (WHO 2020e).
Abbreviations: CBA: controlled before‐and‐after; COMET: Core Outcome Measures in Effectiveness Trials; ECMO: extracorporeal membrane oxygenation; ICU: intensive care unit; MV: mechanical ventilation; NIV: non‐invasive ventilation; ITS: interrupted time series; NRSI: non‐randomised studies of interventions; RCT: randomised controlled trial; WHO: World Health Organization; WHOQOL‐100: WHO Quality of Life scale.
To assess the benefits and safety of convalescent plasma therapy and hyperimmune immunoglobulins for the treatment of COVID‐19, we included RCTs, as such studies, if performed appropriately, give the best evidence for experimental therapies in highly controlled therapeutic settings. For RCT data, we used the methods recommended by the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2019a), as specified in the description of the methods. If we had identified non‐standard RCT designs, such as cluster‐randomised trials and cross‐over trials, we had planned to include those and to apply the methods recommended in Chapter 23 of the Cochrane Handbook (Higgins 2019b). We had planned to consider only the results from the first cycle of cross‐over RCTs.
In case of insufficient evidence available from RCTs we had planned to include prospective and retrospective controlled non‐randomised studies of interventions (NRSIs), and prospective and retrospective registered non‐controlled NRSIs in a top‐down approach as outlined in Appendix 1.
Because large‐scale or expanded access studies could still provide valuable information on the safety of convalescent plasma or hyperimmune immunoglobulins, we decided to include prospectively registered single‐arm studies, even if upcoming RCTs report safety data for both groups. However, single‐arm studies are mostly of lower quality and too heterogeneous to be pooled. We therefore considered prospectively registered single‐arm studies for safety assessment only, if 500 or more participants with COVID‐19 were included. We followed the methodology as specified in the protocol (Piechotta 2020a).
We followed the suggestions specified in the Cochrane Handbook (Higgins 2019a), as far as possible, and applied the methodology outlined in the following sections. We considered RCTs as specified above, and for safety outcomes considered registered single‐arm studies including 500 or more participants with COVID‐19.
We included full‐text publications, pre‐print articles, abstract publications, and results published in trials registries, if sufficient information was available on study design, characteristics of participants, interventions and outcomes. We did not apply any limitation with respect to the length of follow‐up.
Types of participants
We included individuals with a confirmed diagnosis of COVID‐19, with no age, gender or ethnicity restrictions.
We included trials that included participants with any disease severity. We performed separate analyses for populations with ambulatory mild disease and for hospitalised participants with moderate to severe disease, according to the latest WHO clinical progression score (see Table 5; WHO 2020e).
2. WHO clinical progression scale .
| Patient State | Descriptor | Score |
| Uninfected | Uninfected; no viral RNA detected | 0 |
| Ambulatory mild disease | Asymptomatic; viral RNA detected | 1 |
| Symptomatic; independent | 2 | |
| Symptomatic; assistance needed | 3 | |
| Hospitalised: moderate disease | Hospitalised; no oxygen therapya | 4 |
| Hospitalised; oxygen by mask or nasal prongs | 5 | |
| Hospitalised: severe disease | Hospitalised; oxygen by non‐invasive mechanical ventilation or high flow | 6 |
| Intubation and mechanical ventilation; pO2/FiO2 ≥ 150 or SpO2/FiO2 ≥ 200 | 7 | |
| Invasive mechanical ventilation; pO2/FiO2 < 150 (SpO2/FiO2 < 200) or vasopressors | 8 | |
| Invasive mechanical ventilation; pO2/FiO2 < 150 and vasopressors, dialysis or ECMO | 9 | |
| Dead | Dead | 10 |
World Health Organization (WHO) clinical progression scale from: WHO 2020e
aIf hospitalised for isolation only, record status as for ambulatory patient.
Abbreviations: ECMO: extracorporeal membrane oxygenation; FiO2: fraction of inspired oxygen; pO2: partial pressure of oxygen; SpO2: oxygen saturation
We excluded studies including populations with other coronavirus diseases (SARS or MERS). We also excluded studies that included populations with mixed viral diseases (e.g. influenza), unless the trial authors provided subgroup data for people with COVID‐19.
Types of interventions
We included the following interventions.
Convalescent plasma from people who had recovered from SARS‐CoV‐2 infection
Hyperimmune immunoglobulin therapy
We did not include studies on standard immunoglobulin.
We included the following comparisons for studies with a control arm.
Convalescent plasma therapy versus control treatment, for example, drug treatments (including but not limited to hydroxychloroquine, remdesivir), standard immunoglobulin. Co‐interventions were allowed, but must have been comparable between intervention groups.
Convalescent plasma versus standard care or placebo (i.e. saline solution)
Convalescent plasma versus standard plasma (i.e. fresh frozen plasma)
We had planned to additionally include the following comparisons for studies with a control arm, but did not identify any completed studies.
Convalescent plasma therapy versus hyperimmune immunoglobulin
Hyperimmune immunoglobulin versus standard care or placebo
Hyperimmune immunoglobulin versus control treatment, for example, drug treatments (including but not limited to hydroxychloroquine, remdesivir). Co‐interventions were allowed, but must have been comparable between intervention groups.
Types of outcome measures
We evaluated core outcomes as predefined by the Core Outcome Measures in Effectiveness Trials (COMET) Initiative for COVID‐19 patients (COMET 2020), and additional outcomes that have been prioritised by consumer representatives, referees of previous versions of this review, and the German guideline panel for inpatient therapy of people with COVID‐19.
We defined outcome sets for two populations: individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease, and individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease, according to the WHO clinical progression scale (WHO 2020e).
We assessed disease severity with need for respiratory support according to the WHO clinical progression scale (WHO 2020e).
Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease
Effectiveness of convalescent plasma
Prioritised outcomes (included in the 'Summary of findings' table)
All‐cause mortality at day 28, day 60, time‐to‐event, and at hospital discharge
-
Clinical status, assessed by need for respiratory support with standardised scales (e.g. WHO Clinical Progression Scale (WHO 2020e), WHO Ordinal Scale for Clinical Improvement (WHO 2020f)) at up to day 28, day 60, and up to longest follow‐up) including the following.
-
Improvement of clinical status:
liberation from supplemental oxygen in surviving patients i.e. WHO ≤ 4 on the Clinical Progression Scale (WHO 2020e) (for the subgroup of participants requiring any supplemental oxygen or ventilator support at baseline, i.e WHO ≥ 5);
weaning or liberation from invasive mechanical ventilation in surviving patients i.e. WHO ≤ 6 (for the subgroup of participants requiring invasive mechanical ventilation at baseline, i.e WHO ≥ 7).
-
Worsening of clinical status:
need for invasive mechanical ventilation i.e. WHO 7‐9 (for the subgroup of participants not requiring invasive mechanical ventilation at baseline, i.e WHO ≤ 6);
need for non‐invasive mechanical ventilation or high flow i.e. WHO = 6 (for the subgroup of participants not requiring non‐invasive or non‐invasive mechanical ventilation, or high flow oxygen at baseline, i.e WHO ≤5);
need for oxygen by mask or nasal prongs i.e. WHO = 5 (for the subgroup of participants not requiring any supplemental oxygen or ventilator support at baseline, i.e WHO ≤ 4).
-
Quality of life, including fatigue and functional independence; assessed with standardised scales (e.g. WHOQOL‐100) at up to 7 days, up to 30 days, and longest follow‐up available
Additional outcomes (not included in the 'Summary of findings' table)
Duration of hospitalisation, or time‐to‐discharge from hospital
Admission to the intensive care unit (ICU)
Length of stay on the ICU, or time to discharge from ICU
Viral clearance, assessed with reverse transcription polymerase chain reaction (RT‐PCR) test for SARS‐CoV‐2 at baseline, up to 3, 7, and 15 days
Need for dialysis (at up to 28 days)
Safety of convalescent plasma
Adverse events (any grade, grade 1‐2, grade 3‐4), defined as the number of participants with any event and including potential relationship between intervention and adverse reaction (e.g. TRALI, transfusion‐transmitted infection, TACO, transfusion‐associated dyspnoea (TAD), acute transfusion reactions)
Serious adverse events, defined as the number of participants with any event
Individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease
Effectiveness of convalescent plasma
Prioritised outcomes (included in the 'Summary of findings' table)
All‐cause mortality at day 28, day 60, time‐to‐event, and at longest follow‐up.
-
Development of moderate to severe clinical COVID‐19 symptoms, defined as WHO Clinical Progression Scale ≥ 4 (WHO 2020e), up to longest follow‐up
-
Need for invasive mechanical ventilation, non‐invasive mechanical ventilation or high flow i.e. WHO ≥ 6, severe disease:
need for invasive mechanical ventilation i.e. WHO 7‐9;
need for non‐invasive mechanical ventilation or high flow i.e. WHO = 6.
-
Need for hospitalisation with or without supplemental oxygen i.e. WHO = 4‐5, moderate disease:
need for oxygen by mask or nasal prongs i.e. WHO = 5;
need for hospitalisation without oxygen therapy i.e. WHO = 4.
-
Quality of life, including fatigue and functional independence; assessed with standardised scales (e.g. WHOQOL‐100) at up to 7 days, up to 30 days, and longest follow‐up available
Additional outcomes (not included in the 'Summary of findings' table)
Admission to hospital
Time to symptom onset
Length of hospital stay, for subgroup of participants hospitalised during course of disease
Admission to the ICU
Viral clearance, assessed with RT‐PCR test for SARS‐CoV‐2 at baseline, up to 3, 7, and 15 days
Safety of convalescent plasma
Adverse events (any grade, grade 1‐2, grade 3‐4), defined as the number of participants with any event and including potential relationship between intervention and adverse reaction (e.g. TRALI, transfusion‐transmitted infection, TACO, TAD, acute transfusion reactions)
Serious adverse events, defined as the number of participants with any event
Timing of outcome measurement
For time‐to‐event outcomes, such as mortality, discharge from hospital, and improvement of clinical symptoms, we included outcome measures representing the longest follow‐up time available.
We included all other outcome categories for the observational periods that the study publications reported. We included those adverse events occurring during active treatment and had planned to include long‐term adverse events as well. If sufficient data had been available, we planned to group the measurement time points of eligible outcomes, for example, adverse events and serious adverse events, into those measured directly after treatment (up to seven days after treatment), medium‐term outcomes (15 days after treatment) and longer‐term outcomes (over 30 days after treatment).
Search methods for identification of studies
We carry out weekly searches for completed and ongoing studies. Studies reported in all languages are eligible, in order to limit language bias. We check weekly for newly emerging hyperimmune immunoglobulins and review search methods and strategies approximately monthly, to ensure they reflect any terminology changes in the topic area, or in the databases. We adapt the strategy where necessary.
Electronic searches
We designed and tested search strategies for electronic databases according to methods suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Lefebvre 2019). One review author (CD) developed the original strategies and Cochrane Haematology's Information Specialist (IM) peer‐reviewed and revised them at various times, to reflect the current state of knowledge. In this emerging field, we expected that at least study abstracts would be in English. If studies were published in other languages than those our review team could accommodate (English, Dutch, German, French, Italian, Malay and Spanish), we involved Cochrane TaskExchange to identify people within Cochrane to translate these studies.
As publication bias might influence all subsequent analyses and conclusions, we searched all potential relevant trials registries in detail to detect ongoing as well as completed studies, but not‐yet‐published studies. Because nowadays, it is mandatory to provide results at least in the trials registry, we had planned to extract and analyse these data, in case results were not published elsewhere. However, no outcome data have yet been added to the trials registries.
We searched the following databases and sources from 1 January 2019 to 17 March 2021.
-
Databases of medical literature
MEDLINE (Ovid, 1 January 2019 to 17 March 2021; Appendix 2)
Embase (Ovid, 1 January 2019 to 17 March 2021; Appendix 3)
Cochrane COVID‐19 Study Register (covid-19.cochrane.org; inception to 17 March 2021; Appendix 4)*
PubMed (for epublications ahead of print only; 1 January 2019 to 17 March 2021; Appendix 5)
World Health Organization COVID‐19 Global literature on coronavirus disease (bvsalud.org/global-literature-on-novel-coronavirus-2019-ncov; inception to 17 March 2021) without references of MEDLINE and PubMed; Appendix 6)
Epistemonikos, L*OVE List Coronavirus disease (COVID‐19) (app.iloveevidence.com/loves; inception to 17 March 2021; Appendix 7)
*The Cochrane COVID‐19 Study Register is a specialised register built within the Cochrane Register of Studies (CRS) and is maintained by Cochrane Information Specialists. Complete data sources and search methods for the register are available at: community.cochrane.org/about-covid-19-study-register. The register contains study reports from several sources, including:
daily searches of PubMed;
daily searches of ClinicalTrials.gov;
weekly searches of Embase.com;
weekly searches of the WHO International Clinical Trials Registry Platform (ICTRP);
monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL).
Searching other resources
We handsearched the reference lists of all identified studies, relevant review articles and current treatment guidelines for further literature. We also contacted experts in the field, drug manufacturers and regulatory agencies in order to retrieve information on unpublished studies.
Data collection and analysis
Selection of studies
Using Covidence software, two review authors (from among SJV, KLC, VP, CK, CI and NS) independently screened the results of the search strategies for eligibility, by reading the abstracts. We coded the abstracts as either 'retrieve' or 'do not retrieve'. In the case of disagreement, or if it was unclear whether we should retrieve the abstract or not, we obtained the full‐text publication for further discussion. Two review authors assessed the full‐text articles of selected studies. If the two review authors were unable to reach a consensus, they consulted a third review author to reach a final decision.
We documented the study selection process in a flow chart, as recommended in the PRISMA statement (Moher 2009), and show the total numbers of retrieved references and the numbers of included and excluded studies. We list all studies that we excluded after full‐text assessment and the reasons for their exclusion in the Characteristics of excluded studies table.
Data extraction and management
Two review authors (from among SJV, KLC, VP, CK, CI and ED) independently assessed eligible studies obtained in the process of study selection (as described above) for methodological quality and risk of bias. If the review authors were unable to reach a consensus, we consulted a third review author.
Two review authors (from among SJV, KLC, CK, CI, ED and VP) extracted data using a customised data extraction form, developed in Microsoft Excel (Microsoft Corporation 2018). Another review author (CI, VP, or NS) verified the accuracy and (where applicable) the plausibility of extractions and assessment. We conducted data extraction according to the guidelines proposed by Cochrane (Li 2019). If the review authors were unable to reach a consensus, we consulted a third review author.
We collated multiple reports of one study so that the study, and not the report, is the unit of analysis.
We extracted the following information.
General information: author, title, source, publication date, country, language, duplicate publications
Quality assessment: study design, confounding, definition of risk estimates, bias arising from the randomisation process, due to deviations from the intended interventions, due to missing outcome data, in measurement of the outcome, and in selection of the reported results
Study characteristics: trial design, setting and dates, source of participants, inclusion/exclusion criteria, comparability of groups, treatment cross‐overs, compliance with assigned treatment, length of follow‐up
Participant characteristics: age, gender, ethnicity, number of participants recruited/allocated/evaluated, disease, severity of disease, additional diagnoses, previous treatments (e.g. experimental drug therapies, oxygen therapy, ventilation), whether the donors were tested by nasal swabs or whether the plasma was tested
-
Interventions: convalescent plasma therapy or hyperimmune immunoglobulin therapy, concomitant therapy, duration of follow‐up, donors' disease severity, how donations were tested for neutralising antibody
For studies including a control group: comparator (type)
Outcomes: as specified in Types of outcome measures
Assessment of risk of bias in included studies
Randomised controlled trials
We used the 'Risk of Bias 2' (RoB 2) tool to analyse the risk of bias in the underlying study results (Sterne 2019). Of interest for this review was the effect of the assignment to the intervention (the intention‐to‐treat (ITT) effect) and we performed all assessments with RoB 2 on this effect. The outcomes that we addressed are those specified for inclusion in 'Summary of findings' table 1. Accordingly, the outcomes had been prioritised according to the COMET Initiative for COVID‐19 patients (COMET 2020).
Two review authors (from among SJV, KLC, VP, CK, CI and NS) independently assessed the risk of bias for each study result. In case of discrepancies among their judgements or inability to reach consensus, we consulted a third review author to reach a final decision. We assessed the following types of bias as outlined in Chapter 8 of the Cochrane Handbook (Higgins 2019c).
Bias arising from the randomisation process
Bias due to deviations from the intended interventions
Bias due to missing outcome data
Bias in measurement of the outcome
Bias in selection of the reported result
For cluster‐RCTs, we had planned to add an additional domain to assess bias arising from the timing of identification and recruitment of participants in relation to timing of randomisation, as recommended in the archived RoB 2 guidance for cluster‐randomised trials (Eldridge 2016), and in Chapter 23 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2019b).
To address these types of bias we used the signalling questions recommended in RoB 2 and made a judgement using the following options:
'yes': if there is firm evidence that the question is fulfilled in the study (i.e. the study is at low or high risk of bias given the direction of the question);
'probably yes': a judgement has been made that the question is fulfilled in the study (i.e. the study is at low or high risk of bias given the direction of the question);
'no': if there is firm evidence that the question is unfilled in the study (i.e. the study is at low or high risk of bias for the given the direction of the question);
'probably no': a judgement has been made that the question is unfilled in the study (i.e. the study is at low or high risk of bias given the direction of the question);
'no information': if the study report does not provide sufficient information to allow any judgement.
We used the algorithms proposed by RoB 2 to assign each domain one of the following levels of bias:
low risk of bias;
some concerns;
high risk of bias.
Subsequently we derived a 'Risk of bias' rating for each pre‐specified outcome in each study in accordance with the following suggestions.
'Low risk of bias': we judged the trial to be at low risk of bias for all domains for this result.
'Some concerns': we judged the trial to raise some concerns in at least one domain for this result, but not to be at high risk of bias for any domain.
'High risk of bias': we judge the trial to be at high risk of bias in at least one domain for the result or we judge the trial to have some concerns for multiple domains in a way that substantially lowers confidence in the results.
We used the RoB 2 Excel tool to implement RoB 2 (available on the riskofbiasinfo.org website), added our judgements to the analysis for each assessed study and outcome, and stored our detailed RoB 2 assessments as supplementary online material (Piechotta 2021). We used the overall 'risk of bias' judgement, derived from the RoB 2 Excel tool, to inform our GRADE decision on downgrading for risk of bias.
Controlled non‐randomised studies of interventions
As reported above, we had planned to include NRSI trials if there was insufficient evidence from RCTs. Please refer to Appendix 1 for detailed information on how we would have assessed risk of bias for controlled NRSIs.
Non‐controlled non‐randomised studies of interventions
As specified in the 'Types of studies' section, we also included safety data from prospective non‐controlled NRSIs, if 500 or more participants with COVID‐19 were included.
Because we only included safety data from non‐controlled NRSIs, we only assessed methodological quality and risk of bias for studies reporting any safety data.
Two review authors (VP, NS) assessed eligible studies for methodological quality and risk of bias (using the 'Risk of bias' assessment criteria for observational studies tool provided by Cochrane Childhood Cancer (see Table 6; Mulder 2019). We performed and presented any 'Risk of bias' judgements per outcome per study.
3. 'Risk of bias' assessment criteria for observational studies.
| Heading | Internal validity | External validity |
| Study group |
Selection bias (representative: yes/no)
or
|
Reporting bias (well defined: yes/no)
and
|
| Follow‐up |
Attrition bias (adequate: yes/no)
or
|
Reporting bias (well defined: yes/no)
|
| Outcome |
Detection bias (blind: yes/no)
|
Reporting bias (well defined: yes/no)
|
| Risk estimation |
Confounding (adjustment for other factors: yes/no)
|
Analyses (well defined: yes/no)
|
The quality assessment strongly depends upon information on the design, conduct and analysis of the study. The two review authors (VP, NS) resolved any disagreements regarding the quality assessments by discussion; in case of disagreement they would have consulted a third review author (SJV, KLC or CK).
We assessed the following domains of bias.
-
Internal validity
Unrepresentative study group (selection bias)
Incomplete outcome assessment/follow‐up (attrition bias)
Outcome assessors unblinded to investigated determinant (detection bias)
Important prognostic factors or follow‐up not taken adequately into account (confounding)
-
External validity
Poorly defined study group (reporting bias)
Poorly defined follow‐up (reporting bias)
Poorly defined outcome (reporting bias)
Poorly defined risk estimates (analyses)
For every criterion, risk of bias judgements are 'high', 'unclear' or 'low'.
We used the 'Risk‐of‐bias VISualization' tool ('robvis') to generate risk of bias summary figures for non‐controlled NRSIs (McGuinness 2020).
Measures of treatment effect
Randomised controlled trials
For continuous outcomes, we recorded the mean, standard deviation and total number of participants in both the treatment and control groups. For dichotomous outcomes, we recorded the number of events and total number of participants in both the treatment and control groups.
For continuous outcomes using the same scale we performed analyses using the mean difference (MD) with 95% confidence intervals (CIs). For continuous outcomes measured with different scales we performed analyses using the standardised mean difference (SMD). For interpreting SMDs, we re‐expressed SMDs in the original units of a particular scale with the most clinical relevance and impact.
If available, we extracted and reported hazard ratios (HRs) for time‐to‐event outcomes (e.g. discharge from hospital). If HRs were not available, we made every effort to estimate the HR as accurately as possible using the available data and a purpose‐built method based on the Parmar and Tierney approach (Parmar 1998; Tierney 2007). If sufficient studies provided HRs, we used HRs rather than risk ratios (RRs) or MDs in a meta‐analysis.
For dichotomous outcomes, we had planned to report the pooled RR with a 95% CI (Deeks 2019). If the number of observed events had been small (less than 5% of sample per group), and if studies had balanced treatment groups, we planned to report the Peto odds ratio (OR) with 95% CI (Deeks 2019).
Controlled non‐randomised studies of interventions
Please refer to Appendix 1 for detailed information on how we had planned to extract and report different treatment measures of outcome data from controlled NRSIs.
Non‐controlled non‐randomised studies of interventions
For non‐controlled NRSIs, we did not carry out an analysis using quantitative data from indirect controls, as we are aware of the difficulties of indirect comparisons of participant groups with varying baseline characteristics, especially in the absence of individual patient data. Because authors of non‐controlled NRSIs often discuss their findings using information from other intervention and observational studies as implicit controls, we discussed our findings extensively in the context of what is known about the outcome of 'comparable' patients receiving other experimental treatments, but not convalescent plasma therapy or hyperimmune immunoglobulin therapy. We did not meta‐analyse the data but provided information from individual studies within tables.
Unit of analysis issues
We did not combine any data from different study designs. Meta‐analysis was not appropriate for the non‐controlled NRSIs, as described above. Instead, we reported and presented results narratively.
As recommended in Chapter 6 of the Cochrane Handbook (Higgins 2019d), for studies with multiple treatment groups, we had planned to combine arms if they could be regarded as subtypes of the same intervention.
When arms could not be pooled this way, we had planned to compare each arm with the common comparator separately. For pair‐wise meta‐analysis, we had planned to split the ‘shared’ group into two or more groups with smaller sample sizes, and include two or more (reasonably independent) comparisons. For this purpose, for dichotomous outcomes, both the number of events and the total number of participants would be divided up, and for continuous outcomes, the total number of participants would be divided up with unchanged means and standard deviations (SDs).
Dealing with missing data
Chapter 6 of the Cochrane Handbook for Systematic Reviews of Interventions suggests a number of potential sources for missing data, which we needed to take into account: at study level, at outcome level and at summary data level (Higgins 2019d). In the first instance, it is of the utmost importance to differentiate between data 'missing at random' and 'not missing at random'.
We requested missing data from the study authors. We contacted 11 principal investigators from included studies (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Bajpai 2020; Gharbharan 2020; Hamdy Salman 2020; Horby 2021; Li 2020; Libster 2020; Ray 2020; Simonovich 2020). We received six responses: one each from Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Gharbharan 2020; Horby 2021 and Li 2020, providing all requested information; one from Balcells 2020 providing breakdowns of adverse events that occurred in the study; and one from Rasheed 2020, stating that all or most requested information will be included in the journal publication. Partly based on the additional requested data, we decided to exclude Balcells 2020 and Rasheed 2020.
As we received all outcome data from included studies that we had requested, we did not have to make any assumptions. For updates of this review, if data are still missing, we will have to make explicit assumptions of any methods used in the included studies. For example, we will assume that the data were missing at random, or we will assume that missing values had a particular value, such as a poor outcome.
We further contacted all principal investigators from ongoing studies, asking for their prospective completion dates, as well as completed studies without published results, and invited them to share their data with us for this update. We received 11 responses: one each from Beltran 2021 and NCT04438694, informing us that their trials were completed and that they are willing to share their data for this update (no data were received from the investigators until submission of our review, however, Beltran 2021 published a preprint of their anticipated journal publication after we submitted the review); one each from NCT04433910, ISRCTN85216856 and NCT04397757 informing us that randomisation was completed at the end of December 2020, and that they are willing to share their data with us once analysed (no data were received from the investigators until submission of our review); one from NCT04429854, informing us that their trial was completed by January 2021, and one each from NCT04388410 and NCT04428021, informing us that their trials were completed by February 2021, and that they are willing to share their data with us once analysed (no data were received from the investigators until submission of our review); and one each from Bennett‐Guerrero 2021, NCT04348656 and NCT04377568, informing us that there is not yet any information to share, however, Bennett‐Guerrero 2021 published a preprint of their anticipated journal publication after we submitted the review. We will contact the principal investigators of ongoing and completed studies without published results again for our next update.
Assessment of heterogeneity
We did not combine any data from different study designs. Meta‐analysis was not appropriate for the non‐controlled NRSIs, as described above. Instead, we reported and presented results in tables.
We assessed heterogeneity of treatment effects between trials using a Chi2 test with a significance level at P < 0.1, and visual examination. We used the I2 statistic (Higgins 2003), to quantify possible heterogeneity (I2 > 30% to signify moderate heterogeneity, I2 > 75% to signify considerable heterogeneity; Deeks 2019). If heterogeneity had been above 80%, we would have explored potential causes through sensitivity and subgroup analyses. If we had not found a reason for heterogeneity, we would not have performed a meta‐analysis, but would have only commented on results from all studies and presented these in tables.
Assessment of reporting biases
As mentioned above, we searched trials registries to identify completed studies that have not been published elsewhere, to minimise or determine publication bias. We included studies irrespective of their publication status, as recommended in Cochrane Handbook for Systematic Reviews of Interventions (McKenzie 2019).
In an update of this review, for meta‐analyses involving at least 10 studies, we intend to explore potential publication bias by generating a funnel plot and statistically testing this by conducting a linear regression test (Sterne 2019). We will consider P < 0.1 as significant for this test.
Data synthesis
If the clinical and methodological characteristics of individual studies were sufficiently homogeneous, we pooled the data in meta‐analysis. We performed separate analyses for populations with ambulatory mild disease and for hospitalised participants with moderate to severe disease, according to the latest WHO clinical progression score (WHO 2020e). We performed analyses according to the recommendations of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2019). We did not conduct meta‐analyses that included different study designs. We conducted separate meta‐analyses for each comparison.
We used the Review Manager Web software for analyses (Review Manager Web). One review author entered the data into the software, and a second review author checked the data for accuracy.
We used the random‐effects model for all analyses, as we anticipated that for included studies, true effects would be related, but would not be the same. For binary outcomes, we based the estimation of the between‐study variance using the Mantel‐Haenszel method. We used the inverse variance method for continuous outcomes, outcomes that include data from cluster‐RCTs, or outcomes where HRs are available. We planned to explore heterogeneity above 80% with subgroup analyses. If we could not find a cause for the heterogeneity or if study outcomes were too clinically heterogenous to be combined, we did not perform a meta‐analysis, but commented on the results in a narrative analysis, with the results from all studies presented in tables.
Please see Appendix 1 for detailed information on how we had planned to synthesise data from controlled NRSIs.
We did not meta‐analyse data from non‐controlled NRSIs, as there might be no additional benefit in meta‐analysing data without a control group. We reported outcome data of each included trial within tables.
Subgroup analysis and investigation of heterogeneity
We performed subgroup analyses of the following characteristics for our prioritised outcomes, as specified in the 'Summary of Findings' section.
Severity of condition (divided into moderate and severe disease, assessed with need for respiratory support according to WHO clinical progression scale (WHO 2020e))
Duration since symptom onset (divided into up to 7 days and more than 7 days)
Antibodies in recipients detected at baseline (divided into detected in a maximum of 20% of recipients versus detected in at least 80% of recipients)
For the outcome domain of clinical status, we used additional subdivisions to analyse the changes of the need for respiratory support more precisely, and targeted to the baseline need (see Types of outcome measures).
We used the tests for interaction to test for differences between subgroup results.
We had further planned to perform additional subgroup analyses of the following characteristics.
Age of participants (divided into applicable age groups, e.g. children; 18 to 65 years, 65 years and older)
Pre‐existing conditions (diabetes, respiratory disease, hypertension, immunosuppression)
Level of antibody titre in donors (divided into high and low titres, using the studies definition)
SARS‐CoV‐2 variants (e.g. B1.1.7, B.1.351, P.1, and other variants that may occur in the future)
Sensitivity analysis
We performed sensitivity analyses for the following.
'Risk of bias' assessment components (studies with a low risk of bias or some concerns versus studies with a high risk of bias)
Influence of completed, but not published studies
Influence of premature termination of studies
Summary of findings and assessment of the certainty of the evidence
We used the GRADE approach to assess the certainty of the evidence for the following outcomes, and prepared one 'Summary of findings' table per population.
Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease
All‐cause mortality; all‐cause mortality at hospital discharge most favourable. If not reported, all‐cause mortality day 60, followed by day 28, or time‐to‐event estimate, will be included in the 'Summary of findings' table.
-
Improvement of clinical status; assessed with liberation from respiratory support, i.e. supplemental oxygen support or invasive mechanical ventilation, in accordance with WHO Clinical Progression Scale (WHO 2020e) at longest follow‐up available
For all participants requiring any supplemental oxygen or ventilator support at baseline (WHO ≥5 at baseline on the WHO Clinical Progression Scale (WHO 2020e)): Liberation from supplemental oxygen in surviving patients
For all participants requiring invasive mechanical ventilation at baseline (WHO ≥7 at baseline on the WHO Clinical Progression Scale (WHO 2020e)): Liberation from invasive mechanical ventilation in surviving patients
Worsening of clinical status; assessed with the need for invasive mechanical ventilation i.e. WHO 7‐9 (only for participants not requiring invasive mechanical ventilation at baseline, i.e WHO≤6) on the WHO Clinical Progression Scale (WHO 2020e) at longest follow‐up available
Quality of life, including fatigue and functional independence; assessed with standardised scales (e.g. WHOQOL‐100) at longest follow‐up available
Grade 3 or 4 adverse events
Serious adverse events
Individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease
All‐cause mortality; all‐cause mortality at longest follow‐up and greater than 60 days most favourable. If not reported, all‐cause mortality day 60, followed by day 28, or time‐to‐event estimate, will be included in the 'Summary of findings' table.
Development of severe clinical COVID‐19 symptoms, defined as WHO Clinical Progression Scale ≥ 6 (WHO 2020e) at longest follow‐up available
Quality of life, including fatigue and functional independence; assessed with standardised scales (e.g. WHOQOL‐100) at longest follow‐up available
Grade 3 or 4 adverse events
Serious adverse events
We followed the current GRADE guidance for these assessments in its entirety, as recommended in Chapter 14 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2020). We used GRADEpro GDT software to create a 'Summary of findings' table (Schünemann 2020). For RCTs, we used the overall 'risk of bias' judgement, derived from the RoB 2 Excel tool, to inform our decision on downgrading for risk of bias. We assessed the certainty of the evidence for non‐controlled NRSIs as reported in the GRADE guidance 3, starting from low‐certainty evidence (Balshem 2011). For time‐to‐event outcomes we calculated absolute effects at specific time points, as recommended in the GRADE guidance 27 (Skoetz 2020). We phrased the findings and certainty of the evidence as suggested in the informative statement guidance (Santesso 2020).
Results
Description of studies
Results of the search
For this update, we identified 14,714 new records, in addition to the 7856 potentially relevant records from the previous versions (altogether 22,570 references). After removing duplicates, we screened 10,312 new records for this update (altogether 15,812 records) based on their titles and abstracts, and we excluded 15,492 records that did not meet the prespecified inclusion criteria. We evaluated the remaining 316 records and screened the full texts, or, if these were not available, abstract publications or trials registry entries. See Figure 1 for the study flow diagram (Moher 2009).
1.
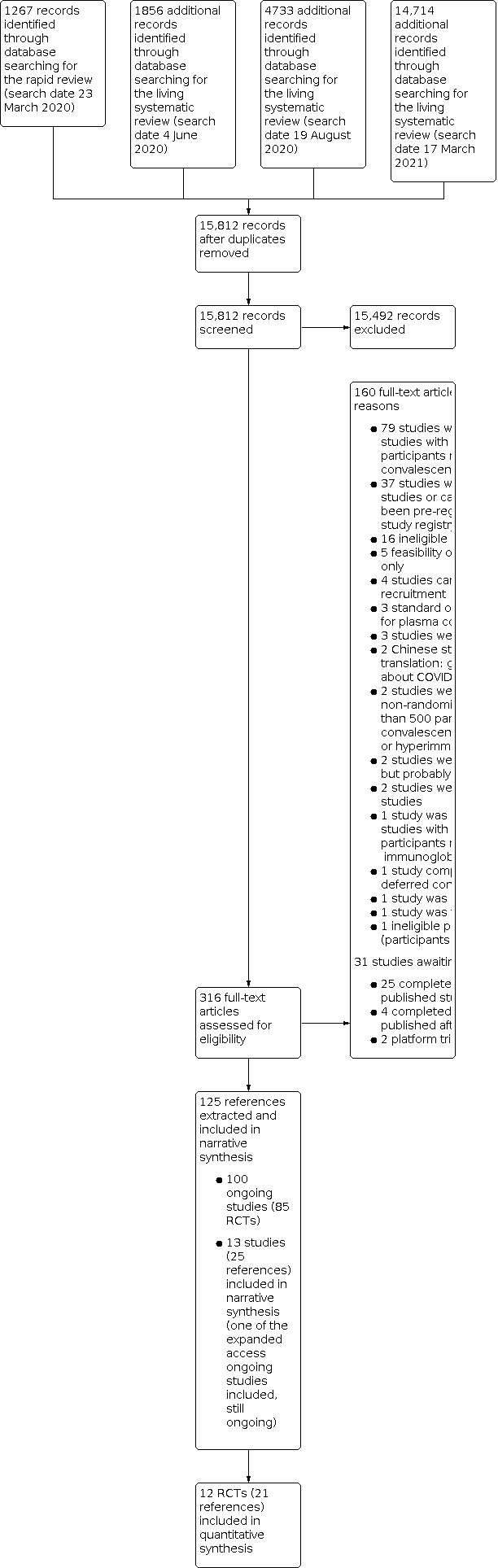
Study flow diagram
We identified 113 potentially eligible studies within 125 citations: 13 included studies (25 records) (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Bajpai 2020; Gharbharan 2020; Hamdy Salman 2020; Horby 2021; Joyner 2020; Li 2020; Libster 2020; O’Donnell 2021; Ray 2020; Simonovich 2020) and 100 ongoing studies (see 'Ongoing studies' below).
Included studies
We included 13 studies reporting on 48,509 participants, of whom 41,880 received convalescent plasma (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Bajpai 2020; Gharbharan 2020; Hamdy Salman 2020; Horby 2021; Joyner 2020; Li 2020; Libster 2020; O’Donnell 2021; Ray 2020; Simonovich 2020).
Design and sample size
We included 12 RCTs (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Bajpai 2020; Gharbharan 2020; Hamdy Salman 2020; Horby 2021; Li 2020; Libster 2020; O’Donnell 2021; Ray 2020; Simonovich 2020) and one non‐controlled NRSI (Joyner 2020).
Setting
The included studies differed considerably in their settings.
Among the RCTs, three were conducted in India (Agarwal 2020; Bajpai 2020; Ray 2020), one was in Bahrain (AlQahtani 2020), one was conducted in Spain (Avendano‐Sola 2020), one was in the Netherlands (Gharbharan 2020), one was conducted in Egypt (Hamdy Salman 2020), one was from the UK (Horby 2021), one was done in China (Li 2020), two were conducted in Argentina (Libster 2020; Simonovich 2020), and one was conducted partly in the USA and partly in Brazil (O’Donnell 2021). The non‐controlled NRSI that we included for safety outcomes was conducted in the USA (Joyner 2020).
Three studies are single‐centre studies (Bajpai 2020; Hamdy Salman 2020; Ray 2020) and nine are multi‐centre studies (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Gharbharan 2020; Joyner 2020; Li 2020; Libster 2020; O’Donnell 2021; Simonovich 2020), with a minimum of two centres for AlQahtani 2020 and a maximum of 2807 centres for Joyner 2020.
Among the RCTs, 11 were performed in an inpatient setting (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Bajpai 2020; Gharbharan 2020; Hamdy Salman 2020; Horby 2021; Li 2020; O’Donnell 2021; Ray 2020; Simonovich 2020). One study was performed in an outpatient setting, for the recruitment of participants; but after randomisation, all participants were admitted to hospital for administration of convalescent plasma (Libster 2020).
Participants
The RCTs by Agarwal 2020, AlQahtani 2020, Avendano‐Sola 2020 and Simonovich 2020 included participants with moderate disease and the RCTs by Gharbharan 2020 and Li 2020 included individuals with severe disease, according to the latest WHO clinical progression score (WHO 2020e). The RCTs by Bajpai 2020, Horby 2021, O’Donnell 2021, Ray 2020 and Hamdy Salman 2020 included individuals with both moderate and severe disease, according to the latest WHO clinical progression score (WHO 2020e). The RCT by Libster 2020 included populations with mild disease.
The non‐controlled NRSI by Joyner 2020 transfused convalescent plasma in individuals with severe or life‐threatening disease.
Interventions
All included RCTs evaluated convalescent plasma in comparison to a control arm, but not all studies had the same comparisons. We did not identify any completed studies evaluating hyperimmune immunoglobulin (IgG). All of the included studies that we evaluated for efficacy and safety outcomes transfused different doses and volumes of convalescent plasma.
Randomised controlled trials
Ten RCTs compared convalescent plasma with standard care, with or without placebo (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Gharbharan 2020; Hamdy Salman 2020; Horby 2021; Li 2020; Libster 2020; Ray 2020; Simonovich 2020), and two RCTs compared convalescent plasma with standard plasma (Bajpai 2020; O’Donnell 2021).
In these 11 RCTs that we evaluated for efficacy and safety, the dose and volume of plasma also varied. The total volume of convalescent plasma transfused varied between 200 mL and 600 mL of plasma, with participants receiving between one dose of plasma (Avendano‐Sola 2020; Hamdy Salman 2020; Libster 2020; O’Donnell 2021; Simonovich 2020), and two or more doses of plasma (Agarwal 2020; AlQahtani 2020; Bajpai 2020; Gharbharan 2020; Horby 2021; Li 2020; Ray 2020).
Non‐controlled, non‐randomised studies of interventions
In the one non‐controlled NRSI by Joyner 2020, that we evaluated for safety outcomes, a volume of 200 mL convalescent plasma was transfused, in one or more doses. The antibody titre test in donors was not performed.
Plasma donors
All included RCTs determined antibody titres in donors, of which five RCTs reported antibody titres in donors' plasma (AlQahtani 2020; Horby 2021; Libster 2020; Ray 2020; Simonovich 2020), and eight RCTs reported neutralising antibody titres in donors' plasma (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Bajpai 2020; Gharbharan 2020; Hamdy Salman 2020; Li 2020; O’Donnell 2021).
Of the included studies, 11 RCTs reported the donors' eligibility criteria (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Bajpai 2020; Gharbharan 2020; Hamdy Salman 2020; Li 2020; Libster 2020; O’Donnell 2021; Ray 2020; Simonovich 2020 ). They also reported some descriptive information about donors, such as their age, gender, disease severity, or their timing from disease recovery and/or the RT‐PCR virus detection. Among those RCTs reporting the sex of donors, in Agarwal 2020, Avendano‐Sola 2020, Gharbharan 2020 and O’Donnell 2021, most of the donors were male (94%, 88%, 91% and 66%, respectively). In Bajpai 2020, all donors were male.
Please refer to the Characteristics of included studies for more detailed information.
Outcomes
We evaluated efficacy and safety outcomes from 12 RCTs (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Bajpai 2020; Gharbharan 2020; Hamdy Salman 2020; Horby 2021; Li 2020; Libster 2020; O’Donnell 2021; Ray 2020; Simonovich 2020) with 12,878 participants, of whom 6555 received convalescent plasma. For the safety outcomes, we also evaluated one non‐controlled NRSI (Joyner 2020) with 35,322 participants, all having received convalescent plasma.
Efficacy outcomes
Different efficacy outcomes were prioritised, based on the setting and the disease severity in participants of the included RCTs (see Types of outcome measures).
Among the RCTs that included individuals with moderate to severe disease, nine studies reported 28‐day mortality (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Bajpai 2020; Horby 2021; Li 2020; O’Donnell 2021; Ray 2020; Simonovich 2020), and three studies also reported all‐cause mortality at hospital discharge (Agarwal 2020; AlQahtani 2020; Gharbharan 2020). Improvement of clinical status, assessed by liberation from supplemental oxygen, was reported in one RCT (Gharbharan 2020). Clinical improvement, assessed by liberation from invasive mechanical ventilation, was reported in two RCTs (Gharbharan 2020; Horby 2021). Worsening of clinical status, assessed by the need for invasive mechanical ventilation, was reported in five RCTs (Agarwal 2020; AlQahtani 2020; Bajpai 2020; Horby 2021; Simonovich 2020). None of the included RCTs reported quality of life. The safety outcome for any grade adverse event was reported in two RCTs (Simonovich 2020; O’Donnell 2021); the outcome of grade 3 or 4 adverse events was reported in four RCTs (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Simonovich 2020); and serious adverse events were reported in three RCTs (Avendano‐Sola 2020; O’Donnell 2021; Simonovich 2020).
The sole RCT that included individuals with asymptomatic or mild disease reported only all‐cause mortality (Libster 2020), at an undefined time point.
Safety outcomes
For safety outcomes, we also evaluated the non‐controlled NRSI (Joyner 2020). Five RCTs (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; O’Donnell 2021; Simonovich 2020) and the one non‐controlled NRSI (Joyner 2020) reported adverse events or serious adverse events, or both, for all the participants. From these six studies, we extracted safety data from 21,154 participants, with safety data for 20,687 participants who received convalescent plasma and 464 participants who did not receive convalescent plasma. All the other included RCTs reported transfusion‐related adverse events, for the participants receiving convalescent plasma (Bajpai 2020; Gharbharan 2020; Hamdy Salman 2020; Horby 2021; Li 2020; Libster 2020; Ray 2020) and for the participants receiving standard plasma (Bajpai 2020). From these eight studies, we extracted safety data from 6266 participants who received convalescent plasma only and 15 participants who received the standard plasma.
Two RCTs were terminated early, Gharbharan 2020 because most of the participants were found to have SARS‐CoV‐2 antibodies present at baseline and Li 2020 because there were no more eligible participants, due to containment of the epidemic in Wuhan, China. Neither of these studies cited safety concerns as the reasons for early termination.
Please refer to the Characteristics of included studies for more detailed information.
Ongoing studies
Of the 100 ongoing studies, seven are expanded access studies from the USA (NCT04338360; NCT04360486; NCT04363034; NCT04374370; NCT04472572; NCT04420988; NCT04445207). As the NCT04338360 study has reported on the first 35,322 participants (with safety data for 20,000 participants; Joyner 2020), and has enrolled a further 86,805 participants (of whom 57,630 received convalescent plasma) into the study as of 7 August 2020 (US Covid Plasma 2020), we decided to treat this record as an ongoing study.
Eighty‐five studies are RCTs (see Table 7 and Table 8), and 10 of these are investigating the effect of hyperimmune immunoglobulin (see Table 7). Of the 85 RCTs, 18 were scheduled to be completed in 2020 and planned to evaluate between 15 and 414 participants, but according to the trial registry, three are not yet recruiting, and 15 are still recruiting. Forty‐nine RCTs are expected to be completed in 2021, and plan to evaluate between 16 and 1200 participants. Of these, 11 were scheduled to be completed by the time of writing, but according to the trial registry, five are not yet recruiting, three are still recruiting and three are active, but not recruiting. Four further, large RCTs are planned to be completed in 2021: NCT04418518, randomising 1200 participants; NCT04345289, evaluating 1100 participants; NCT02735707, evaluating 7100 participants; and NCT04483960, randomising 2400 participants. Another eight RCTs are expected to be completed in 2022 and plan to evaluate between 115 and 1344 participants. One study is expected to be completed in 2023 and plans to evaluate 1000 participants (NCT04364737). Please refer to Table 7 and Table 8 for further details on the planned completed dates and planned number of participants per study.
4. Summary of ongoing hyperimmune immunoglobulin studies: design and planned completion date.
| Study ID | Title | Link | Design | Planned number of participants | Planned completion date | Results available | Other study ID |
| IRCT20200508047346N1 | Evaluation of the efficacy and safety of rabbit polyclonal antibody (CoviGlobulin) in patients with coronavirus COVID‐19 virus moderate to severe | en.irct.ir/trial/47953 | RCT | 124 | NR | No | |
| NCT04366245 | Clinical trial to evaluate the efficacy of treatment with hyperimmune plasma obtained from convalescent antibodies of COVID‐19 infection | clinicaltrials.gov/show/NCT04366245 | RCT | 72 | 1 December 2021 | No | |
| NCT04395170 | Convalescent plasma compared to anti‐COVID‐19 human immunoglobulin and standard treatment (TE) in hospitalized patients | clinicaltrials.gov/show/NCT04395170 | RCT | 75 | 1 June 2021 | No | |
| NCT04453384 | A randomized, double‐blind, placebo‐controlled phase 2a and 2b study to evaluate the safety and efficacy of XAV‐19 in patients with COVID‐19 induced moderate pneumonia |
www.clinicaltrialsregister.eu/ctr‐search/trial/2020‐002574‐27/FR
clinicaltrials.gov/ct2/show/NCT04453384 |
RCT | 414 | 31 December 2021 | No | EUCTR2020‐002574‐27 |
| NCT04468958 | Safety, tolerability, and pharmacokinetics of SAB‐185 in healthy participants | clinicaltrials.gov/ct2/show/NCT04468958 | RCT | 28 | 23 February 2021 | No | SAB‐185‐101 |
| NCT04469179 | Safety, tolerability, and pharmacokinetics of SAB‐185 in ambulatory participants with COVID‐19 | clinicaltrials.gov/ct2/show/NCT04469179 | RCT | 21 | 31 December 2021 | No | SAB‐185‐102 |
| NCT04514302 | Safety and efficacy of anti‐SARS‐CoV‐2 equine antibody fragments (INOSARS) for hospitalized patients with COVID‐19 | clinicaltrials.gov/ct2/show/NCT04514302 | RCT | 51 | 20 June 2021 | No | |
| NCT04546581 | Inpatient treatment with anti‐coronavirus immunoglobulin (ITAC) | clinicaltrials.gov/ct2/show/NCT04546581 | RCT | 500 | 30 July 2021 | No | EUCTR2020‐002542‐16 |
| NCT04555148 | COVIDIG (COVID‐19 Hyper‐ImmunoGlobulin) | clinicaltrials.gov/show/NCT04555148 | RCT | 60 | 30 August 2021 | No | KCT0005649 |
| NCT04573855 | Treatment with anti‐SARS‐CoV‐2 immunoglobulin in patients with COVID‐19 | clinicaltrials.gov/show/NCT04573855 | RCT | 41 | 31 March 2021 | No |
NR: not reported; RCT: randomised controlled trial
5. Summary of ongoing convalescent plasma studies: design and planned completion date.
| Study ID | Title | Link | Design | Planned number of participants | Planned completion date | Results available | Other study ID |
| ChiCTR2000030010 | A randomized, double‐blind, parallel‐controlled, trial to evaluate the efficacy and safety of anti‐SARS‐CoV‐2 virus inactivated plasma in the treatment of severe novel coronavirus pneumonia patients (COVID‐19) | www.chictr.org.cn/showproj.aspx?proj=49777 | RCT | 100 | 31 May 2020 | no | |
| ChiCTR2000030179 | Experimental study of novel coronavirus pneumonia rehabilitation plasma therapy severe novel coronavirus pneumonia (COVID‐19) | www.chictr.org.cn/showproj.aspx?proj=50059 | RCT | 100 | 24 April 2020 | no | |
| ChiCTR2000030627 | Study on the application of convalescent plasma therapy in severe COVID‐19 | www.chictr.org.cn/showproj.aspx?proj=50727 | RCT | 30 | 30 May 2020 | no | |
| ChiCTR2000030702 | Convalescent plasma for the treatment of common COVID‐19: a prospective randomized controlled trial | www.chictr.org.cn/showproj.aspx?proj=50537 | RCT | 30 | 15 August 2020 | no | |
| ChiCTR2000030929 | A randomized, double‐blind, parallel‐controlled trial to evaluate the efficacy and safety of anti‐SARS‐CoV‐2 virus inactivated plasma in the treatment of severe novel | www.chictr.org.cn/showproj.aspx?proj=50696 | RCT | 30 | 16 June 2020 | no | |
| CTRI/2020/04/024915 | A phase II, open label, randomized controlled trial to assess the safety and efficacy of convalescent plasma to limit COVID‐19 associated complications | www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=43332 | RCT | 100 | 9 May 2021 | no | |
| CTRI/2020/05/025346 | A phase II, open label, randomized controlled trial to assess the safety and efficacy of convalescent plasma in severe COVID‐19 patients | www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=43005 | RCT | 90 | 1 June 2022 | no | |
| CTRI/2020/06/025803 | Effect of convalescent plasma in COVID‐19 patients | www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=44478 | RCT | 400 | 18 June 2021 | no | |
| CTRI/2020/06/026123 | Plasma therapy in corona patients (severe COVID‐19) | www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=44667 | RCT | 472 | 25 December 2020 | no | |
| EUCTR2020-001632-10 | A randomized open label phase‐II clinical trial with or without infusion of plasma from subjects after convalescence of SARS‐CoV‐2 infection in high‐risk patients with confirmed severe SARS‐CoV‐2 disease | www.clinicaltrialsregister.eu/ctr‐search/trial/2020‐001632‐10/DE | RCT | 174 | NR | no | NCT04433910 |
| EUCTR2020-001936-86 | A prospective, randomized, open label phase 2 clinical trial to evaluate superiority of anti‐SARS‐CoV‐2 convalescent plasma versus standard‐of‐care in hospitalized patients with mild COVID‐19 | www.clinicaltrialsregister.eu/ctr‐search/trial/2020‐001936‐86/DE | RCT | 340 | NR | no | |
| EUCTR2020-002122-82 | Prospective open‐label randomized controlled phase 2b clinical study in parallel groups for the assessment of efficacy and safety of immune therapy with COVID‐19 convalescent plasma plus standard treatment vs. standard treatment alone of subjects with severe COVID‐19 | www.clinicaltrialsregister.eu/ctr‐search/trial/2020‐002122‐82/DE | RCT | 58 | NR | no | |
| EUCTR2020-005410-18 | Multicentre, randomized, double‐blind, placebo‐controlled, non‐commercial clinical trial to evaluate the efficacy and safety of specific anti‐SARS‐CoV‐2 immunoglobulin in the treatment of COVID‐19 | www.clinicaltrialsregister.eu/ctr‐search/trial/2020‐005410‐18/PL | RCT | 480 | NR | no | |
| IRCT20200501047258N1 | Investigation of the effects of COVID‐19 convalescent plasma in acute respiratory distress syndrome due to COVID‐19 | en.irct.ir/trial/47629 | RCT | 120 | NR | no | |
| NCT02735707 | Randomized, embedded, multifactorial adaptive platform trial for community‐ acquired pneumonia (REMAP‐CAP) | clinicaltrials.gov/ct2/show/NCT02735707 | RCT | 7100 | December 2023 | no | |
| NCT04333251 | Evaluating convalescent plasma to decrease coronavirus associated complications. A phase I study comparing the efficacy and safety of high‐titer anti‐SARS‐CoV‐2 plasma vs best supportive care in hospitalized patients with interstitial pneumonia due to COVID‐19. | clinicaltrials.gov/show/NCT04333251 | RCT | 115 | 31 December 2022 | no | |
| NCT04338360 | Expanded access to convalescent plasma for the treatment of patients with COVID‐19 | clinicaltrials.gov/show/NCT04338360 | expanded access | NR | NR | no | |
| NCT04345289 | Efficacy and safety of novel treatment options for adults with COVID‐19 pneumonia (CCAP) | clinicaltrials.gov/show/NCT04345289 | RCT | 1500 | 15 June 2021 | no | EUCTR2020‐001367‐88 |
| NCT04345991 | Efficacy of convalescent plasma to treat COVID‐19 patients, a nested trial in the CORIMUNO‐19 cohort | clinicaltrials.gov/show/NCT04345991 | RCT | 120 | 1 June 2020 | no | |
| NCT04348656 | Convalescent plasma for hospitalized adults with COVID‐19 respiratory illness (CONCOR‐1) | clinicaltrials.gov/show/NCT04348656 | RCT | 1200 | 31 December 2020 | no | |
| NCT04352751 | Experimental use of convalescent plasma for passive immunization in current COVID‐19 pandemic in Pakistan in 2020 | clinicaltrials.gov/show/NCT04352751 | non‐RCT, single | 2000 | April 2021 | no | |
| NCT04358783 | Convalescent plasma compared to the best available therapy for the treatment of SARS‐CoV‐2 pneumonia | clinicaltrials.gov/show/NCT04358783 | RCT | 30 | 30 May 2021 | no | |
| NCT04360486 | Treatment of COVID‐19 with anti‐SARS‐CoV‐2 convalescent plasma (ASCoV2CP) | clinicaltrials.gov/show/NCT04360486 | expanded access | NR | NR | no | |
| NCT04361253 | Evaluation of SARS‐CoV‐2 (COVID‐19) antibody‐containing plasma therapy | clinicaltrials.gov/show/NCT04361253 | RCT | 220 | December 2021 | no | |
| NCT04362176 | Passive Immunity Trial for Our Nation to Treat COVID‐19 in Hospitalized Adults (PassItOn) | clinicaltrials.gov/show/NCT04362176 | RCT | 1000 | April 2021 | ||
| NCT04363034 | Arkansas expanded access COVID‐19 convalescent plasma treatment program | clinicaltrials.gov/ct2/show/NCT04363034 | expanded access | NR | NR | no | |
| NCT04364737 | Convalescent plasma to limit COVID‐19 complications in hospitalized patients | clinicaltrials.gov/show/NCT04364737 | RCT | 1000 | 30 April 2023 | no | |
| NCT04372979 | Efficacy of convalescent plasma therapy in the early care of COVID‐19 patients | clinicaltrials.gov/show/NCT04372979 | RCT | 80 | May 2021 | no | |
| NCT04373460 | Convalescent plasma to limit SARS‐CoV‐2 associated complications | clinicaltrials.gov/show/NCT04373460 | RCT | 1344 | 31 January 2023 | no | |
| NCT04374370 | SARS‐CoV‐2 (COVID‐19) convalescent plasma (CP) expanded access protocol (EAP) | clinicaltrials.gov/show/NCT04374370 | expanded access | NR | NR | no | |
| NCT04374487 | A phase II, open label, randomized controlled trial to assess the safety and efficacy of convalescent plasma to limit COVID‐19 associated complications | clinicaltrials.gov/show/NCT04374487 | RCT | 100 | 9 May 2021 | no | |
| NCT04374526 | Early transfusIon of convalescent plasma in elderly COVID‐19 patients to prevent disease progression | clinicaltrials.gov/show/NCT04374526 | RCT | 182 | 30 June 2021 | no | |
| NCT04376788 | Exchange transfusion versus plasma from convalescent patients with methylene blue in patients with COVID‐19 | clinicaltrials.gov/show/NCT04376788 | RCT | 15 | 1 June 2020 | no | |
| NCT04377568 | Efficacy of human coronavirus‐immune convalescent plasma for the treatment of COVID‐19 disease in hospitalized children | clinicaltrials.gov/show/NCT04377568 | RCT | 100 | 1 May 2022 | no | |
| NCT04380935 | Effectiveness and safety of convalescent plasma therapy on COVID‐19 patients with acute respiratory distress syndrome | clinicaltrials.gov/show/NCT04380935 | RCT | 60 | 31 August 2020 | no | |
| NCT04385043 | Hyperimmune plasma in patients with COVID‐19 severe infection | clinicaltrials.gov/show/NCT04385043 | RCT | 400 | 15 May 2021 | no | |
| NCT04385186 | Inactivated convalescent plasma as a therapeutic alternative in patients CoViD‐19 | clinicaltrials.gov/show/NCT04385186 | RCT | 60 | 30 November 2020 | no | |
| NCT04385199 | Convalescent plasma for patients with COVID‐19 | clinicaltrials.gov/show/NCT04385199 | RCT | 30 | 1 August 2020 | no | |
| NCT04388410 | Safety and efficacy of convalescent plasma transfusion for patients with SARS‐CoV‐2 infection | clinicaltrials.gov/show/NCT04388410 | RCT | 410 | 31 December 2020 | no | |
| NCT04390503 | Convalescent plasma for COVID‐19 close contacts | clinicaltrials.gov/ct2/show/NCT04390503 | RCT | 150 | 1 April 2021 | no | |
| NCT04391101 | Convalescent plasma for the treatment of severe SARS‐CoV‐2 (COVID‐19) | clinicaltrials.gov/show/NCT04391101 | RCT | 231 | 31 December 2021 | no | |
| NCT04397757 | COVID‐19 convalescent plasma for the treatment of hospitalized patients with pneumonia caused by SARS‐CoV‐2 | clinicaltrials.gov/show/NCT04397757 | RCT | 80 | 13 November 2020 | no | |
| NCT04403477 | Convalescent plasma therapy in severe COVID‐19 infection | clinicaltrials.gov/show/NCT04403477 | RCT | 20 | 30 October 2020 | no | |
| NCT04408040 | Use of convalescent plasma for COVID‐19 | clinicaltrials.gov/show/NCT04408040 | non‐RCT, single | 700 | 1 June 2022 | no | |
| NCT04415086 | Treatment of patients with COVID‐19 with convalescent plasma | clinicaltrials.gov/show/NCT04415086 | RCT | 120 | 22 May 2022 | no | |
| NCT04418518 | A trial of convalescent plasma for hospitalized adults with acute COVID‐19 respiratory illness | clinicaltrials.gov/show/NCT04418518 | RCT | 1200 | 31 December 2021 | no | |
| NCT04420988 | Investigational COVID‐19 convalescent plasma infusion for severely or life‐threateningly ill COVID‐19 patients | clinicaltrials.gov/show/NCT04420988 | expanded access | NR | NR | no | |
| NCT04421404 | Effects of COVID‐19 convalescent plasma (CCP) on coronavirus‐associated complications in hospitalized patients | clinicaltrials.gov/show/NCT04421404 | RCT | 50 | 30 April 2021 | no | |
| NCT04425837 | Effectiveness and safety of convalescent plasma in patients with high‐risk COVID‐19 | clinicaltrials.gov/show/NCT04425837 | RCT | 236 | 28 February 2021 | no | |
| NCT04425915 | Efficacy of convalescent plasma therapy in patients with COVID‐19 | clinicaltrials.gov/show/NCT04425915 | RCT | 400 | 31 May 2021 | no | |
| NCT04428021 | Standard or convalescent plasma in patients with recent onset of COVID‐19 respiratory failure | clinicaltrials.gov/show/NCT04428021 | RCT | 180 | 15 December 2021 | no | |
| NCT04429854 | Donated antibodies working against nCoV | clinicalTrials.gov/show/NCT04429854 | RCT | 483 | 2 November 2021 | no | |
| NCT04432272 | Antibody level‐based analysis of COVID‐19 convalescent serum (ABACCuS) | clinicaltrials.gov/ct2/show/NCT04432272 | non‐RCT | 500 | June 2021 | no | |
| NCT04438057 | Evaluating the efficacy of convalescent plasma in symptomatic outpatients infected with COVID‐19 | clinicaltrials.gov/ct2/show/NCT04438057 | RCT | 150 | 6 July 2021 | no | |
| NCT04442191 | Convalescent plasma as a possible treatment for COVID‐19 | clinicaltrials.gov/show/NCT04442191 | RCT | 50 | 31 May 2021 | no | |
| NCT04445207 | Experimental expanded access treatment with convalescent plasma for the treatment of patients with COVID‐19 | clinicaltrials.gov/ct2/show/NCT04445207 | expanded access | NR | NR | no | |
| NCT04452812 | Statistical and epidemiological study based on the use of convalescent plasma for the management of patients with COVID‐19 | clinicaltrials.gov/ct2/show/NCT04452812 | RCT | 15 | 1 April 2021 | no | |
| NCT04456413 | Convalescent plasma as treatment for subjects with early COVID‐19 infection | clinicaltrials.gov/show/NCT04456413 | RCT | 306 | 31 July 2021 | no | |
| NCT04463823 | "NORPLASMA" COVID‐19 convalescent plasma treatment monitoring study | clinicaltrials.gov/ct2/show/NCT04463823 | non‐RCT, single | 500 | 31 May 2022 | no | |
| NCT04468009 | Treatment of critically ill patients with COVID‐19 with convalescent plasma | clinicaltrials.gov/ct2/show/NCT04468009 | RCT | 36 | 30 June 2021 | no | |
| NCT04472572 | Expanded access to convalescent plasma for treatment of COVID‐19 | clinicaltrials.gov/show/NCT04472572 | expanded access | NR | NR | no | |
| NCT04483960 | Australasian COVID‐19 trial (ASCOT) | clinicaltrials.gov/ct2/show/NCT04483960 | RCT | 2400 | 12 July 2022 | no | ACTRN12620000445976 |
| NCT04497324 | Peruconplasma: evaluating the use of convalescent plasma as management of COVID‐19 | clinicaltrials.gov/ct2/show/NCT04497324 | RCT | 100 | 30 April 2021 | no | PER‐016‐20 |
| NCT04497779 | Analysis of coronavirus disease 19 (COVID‐19) convalescent plasma | clinicaltrials.gov/show/NCT04497779 | non‐RCT, single | 800 | 21 August 2021 | no | |
| NCT04516811 | Therapeutic use of convalescent plasma in the treatment of patients with moderate to severe COVID‐19 | clinicaltrials.gov/ct2/show/NCT04516811 | RCT | 600 | 31 July 2022 | no | |
| NCT04521036 | Convalescent plasma for COVID‐19 patients (CPCP) | clinicaltrials.gov/show/NCT04521036 | RCT | 44 | 30 October 2021 | no | |
| NCT04524507 | COVID‐19 antibody plasma research study in hospitalized patients | clinicaltrials.gov/ct2/show/NCT04524507 | RCT | 56 | 30 May 2021 | no | |
| NCT04528368 | Convalescent plasma for treating patients with COVID‐19 pneumonia without iIndication of ventilatory support | clinicaltrials.gov/ct2/show/NCT04528368 | RCT | 60 | 31 December 2020 | no | |
| NCT04539275 | COVID‐19 (VA CURES‐1) | clinicaltrials.gov/show/NCT04539275 | RCT | 702 | 30 June 2022 | no | |
| NCT04542967 | Study on the safety and efficacy of convalescent plasma in patients with severe COVID‐19 disease | clinicaltrials.gov/ct2/show/NCT04542967 | RCT | 150 | 30 September 2020 | no | |
| NCT04545047 | Observational study of convalescent plasma for treatment of veterans with COVID‐19 | clinicaltrials.gov/ct2/show/NCT04545047 | non‐RCT, controlled | 4000 | 30 June 2022 | no | |
| NCT04558476 | Efficacy of convalescent plasma in patients with COVID‐19 treated with mechanical ventilation | clinicaltrials.gov/ct2/show/NCT04558476 | RCT | 500 | 30 September 2022 | no | |
| NCT04567173 | Convalescent plasma as adjunctive therapy for hospitalized patients with COVID‐19 | clinicaltrials.gov/show/NCT04567173 | RCT | 136 | 30 June 2021 | no | |
| NCT04589949 | Early convalescent plasma therapy for high‐risk patients with COVID‐19 in primary care (the CoV‐early study) | clinicaltrials.gov/show/NCT04589949 | RCT | 690 | 1 November 2023 | no | |
| NCT04600440 | Convalescent plasma in the treatment of COVID‐19 | clinicaltrials.gov/show/NCT04600440 | RCT | 100 | 28 February 2022 | no | |
| NCT04621123 | Plasma for early treatment in non‐hospitalised mild or moderate COVID‐19 patients | clinicaltrials.gov/show/NCT04621123 | RCT | 474 | 1 October 2021 | no | |
| NCT04634422 | Plasma exchange (PLEX) and convalescent plasma (CCP) in COVID‐19 patients with multiorgan failure (COVID‐PLEX) | clinicaltrials.gov/ct2/show/NCT04634422 | RCT | 220 | 30 June 2021 | no | |
| NCT04642014 | Application of convalescent plasma in the treatment of SARS CoV‐2 disease (COVID‐19) with evaluation of therapy effectiveness (EPIC‐19) | clinicaltrials.gov/ct2/show/NCT04642014 | single‐arm study | 500 | 21 February 2022 | no | |
| NCT04649879 | Convalescent plasma for treatment of COVID‐19: an open randomised controlled trial | clinicaltrials.gov/ct2/show/NCT04649879 | RCT | 920 | 1 February 2022 | no | |
| NCT04669990 | Remdesivir and convalescent plasma therapy for treatment of COVID‐19 infection in Nepal: a registry study | clinicaltrials.gov/ct2/show/NCT04669990 | non‐RCT, single (registry) | 2000 | 31 October 2021 | no | 749‐2020 |
| NCT04681430 | Reconvalescent plasma/camostat mesylate early in SARS‐CoV‐2 Q‐PCR (COVID‐19) positive high‐risk individuals (RES‐Q‐HR) | clinicaltrials.gov/ct2/show/NCT04681430 | RCT | 1094 | 1 November 2021 | no | EUCTR2020‐004695‐18 |
| NCT04712344 | Assessment of efficacy and safety of therapy with COVID‐19 convalescent plasma in subjects with severe COVID‐19 (IPCO) (IPCO) | clinicaltrials.gov/ct2/show/NCT04712344 | RCT | 58 | 30 September 21 | no | |
| NCT04716556 | Transfusion of convalescent plasma for the early treatment of pneumonIa in COVID‐19 patients | clinicaltrials.gov/ct2/show/NCT04716556 | RCT | 474 | 1 May 2021 | no | TSUNAMI |
| NCT04730401 | Convalescent plasma in the treatment of COVID‐19 (CP_COVID‐19) | clinicaltrials.gov/ct2/show/NCT04730401 | RCT | 390 | 31 December 2021 | no | |
| NL8633 | A randomized, double blinded clinical trial of convalescent plasma compared to standard plasma for treatment of hospitalized non‐ICU patients with COVID‐19 infections | www.trialregister.nl/trial/8633 | RCT | 430 | 1 May 2021 | no | |
| PACTR202006760881890 | Lagos COVID‐19 convalescent plasma trial (LACCPT) | pactr.samrc.ac.za/TrialDisplay.aspx?TrialID=12168 | RCT | 100 | 30 November 2020 | no | |
| PACTR202007653923168 | A clinical trial comparing use of convalescent plasma therapy plus standard treatment to standard treatment alone in patients with severe COVID‐19 infection | pactr.samrc.ac.za/TrialDisplay.aspx?TrialID=11047 | RCT | 206 | 31 December 2021 | no | |
| PER-013-20 | Convalescent plasma as treatment for COVID‐19 | www.ins.gob.pe/ensayosclinicos/rpec/recuperarECPBNuevoEN.asp?numec=013‐20 | RCT | 192 | 30 June 2021 | no | |
| PER-060-20 | Randomized phase 2 clinical trial to evaluate safety and efficacy of the use of plasma from convalescent plasma with the Coronavirus disease (COVID‐19) for the experimental treatment of patients hospitalized in the Centro Médico Naval "Cirujano Mayor Santiago Távara" | www.ins.gob.pe/ensayosclinicos/rpec/recuperarECPBNuevoEN.asp?numec=060‐20 | RCT | 100 | 7 March 2021 | no | |
| RBR-7jqpnw | Effect of COVID‐19 convalescent plasma produced by HEMOPE: a randomized study, with a comparative group in several centers | www.ensaiosclinicos.gov.br/rg/RBR‐7jqpnw/ | RCT | 110 | 30 July 2021 | no |
Two single‐arm studies, including between 500 and 2000 participants, are expected to be completed in 2021 (NCT04352751) and in 2022 (NCT04642014). Two non‐RCTs, including between 500 and 700 participants, are expected to be completed in 2021 (NCT04432272) and in 2022 (NCT04408040). Four studies are pre‐registered observational studies, including between 500 and 10,000 participants, of which three are expected to be completed in 2021 (NCT04497779; NCT04545047; NCT04669990). One is expected to be completed in 2022 (NCT04463823).
Please refer to Characteristics of ongoing studies and Table 7 and Table 8 for more detailed information.
Studies awaiting assessment
In the process of finalising the review, one of our ongoing studies was terminated early for futility and the trial stopped recruiting participants (NCT04355767). As this study was not yet completed, we decided to keep it under 'Awaiting classification'.
According to the trial registry, 24 studies have been completed, or had their recruitment completed, but no results have yet been published (IRCT20200404046948N1; IRCT20200409047007N1; IRCT20200413047056N1; ISRCTN85216856; NCT04332835; NCT04392414; NCT04405310; NCT04442958; CTRI/2020/05/025299; CTRI/2020/05/025328; CTRI/2020/09/027903; IRCT20120215009014N353; IRCT20150808023559N21; IRCT20200503047281N1; IRCT20200525047562N1; IRCT20201004048922N1; jRCT2031200174; NCT04521309; NCT04433910; NCT04492501; NCT04542941; NCT04547127; NCT04547660; NCT04610502). For this reason, we have placed these studies in the category of 'Awaiting classification'. Of these, one study was a non‐randomised controlled study with 600 participants (NCT04492501). All the others were RCTs.
According to the trial registry, expanded access is no longer available for two studies, but no results have been published yet and we are waiting until the sample size is available, which is why we have kept them under 'Awaiting classification' (NCT04358211; NCT04372368).
Two studies are platform trials, which are not yet completed and do not include our intended interventions. A platform trial is an adaptive, multistage study design in which numerous interventions can be evaluated through interim analyses. Additionally, in a platform trial new study arms can be added within the study period to examine further interventions (Park 2020). However, we want to track these studies, in case they add arms on convalescent plasma or hyperimmune immunoglobulin (NCT04501978; NCT04315948).
We identified full texts of four ongoing studies in our weekly searches, after the submission of the current review version (Beltran 2021; Bennett‐Guerrero 2021; Lopardo 2021; Pouladzadeh 2021). We have categorised them as 'Awaiting classification' and will include them in the next version of this living systematic review. Of these four studies, one is investigating the effect of hyperimmune immunoglobulin (Lopardo 2021).
Excluded studies
We excluded 157 references that did not match our inclusion criteria as follows.
Seventy‐seven studies were single‐arm studies with fewer than 500 participants receiving convalescent plasma (Abdullah 2020; Abolghasemi 2020; Bradfute 2020; ChiCTR2000029850; ChiCTR2000030039; ChiCTR2000031501; ChiCTR2000033798; CTRI/2020/04/024804; CTRI/2020/08/027285; Donato 2020; Duan 2020; Dulipsingh 2020; Ibrahim 2020; IRCT20151228025732N53; IRCT20200406046968N2; IRCT20200414047072N1; IRCT20200416047099N1; Jin 2020; Liu 2020; Madariaga 2020; NCT04264858; NCT04292340; NCT04321421; NCT04327349; NCT04332380; NCT04333355; NCT04345679; NCT04346589; NCT04348877; NCT04353206; NCT04354831; NCT04355897; NCT04356482; NCT04365439; NCT04374565; NCT04376034; NCT04377672; NCT04384497; NCT04388527; NCT04389710; NCT04389944; NCT04390178; NCT04392232; NCT04397523; NCT04407208; NCT04408209; NCT04411602; NCT04412486; NCT04418531; NCT04432103; NCT04458363; NCT04462848; NCT04471051; NCT04474340; NCT04476888; NCT04502472; NCT04513158; NCT04516954; NCT04535063; NCT04554992; NCT04565197; NCT04569188; NCT04570982; NCT04614012; NCT04616976; NCT04622826; NCT04644198; Olivares‐Gazca 2020; PER‐031‐20; Perotti 2020; RBR‐4vm3yy; RPCEC00000323; Salazar 2020a; Xia 2020; Zeng 2020; NCT04383548; NCT04438694).
Thirty‐seven studies were single‐arm studies or case series that had not been pre‐registered in a clinical study registry (Ahn 2020; Anderson 2020; Bao 2020b; Bobek 2020; Cantore 2020; Clark 2020; Enzmann 2020; Erkurt 2020; Fan 2020; Figlerowicz 2020; Grisolia 2020; Im 2020; Jamous 2020; Jiang 2020a; Karatas 2020; Kong 2020; Liu 2020a; Martinez‐Resendez 2020; McCuddy 2020; Mira 2020; Niu 2020; Pei 2020; Peng 2020; Salazar 2020b; Shen 2020; Soleimani 2020; Taher 2020; Tan 2020; Wang 2020; Wright 2020; Xu 2020b; Yang 2020; Ye 2020; Zhang 2020a; Zhang 2020b; Zhang 2020c; Çınar 2020).
Sixteen studies were performed with an intervention other than convalescent plasma or hyperimmune immunoglobulin (Cao 2020a; Chen 2020b; Chen 2020c; Díez 2020; Hu 2020; ISRCTN86534580; Jiang 2020b; Lin 2020; NCT04261426; NCT04344379; NCT04350580; NCT04368013; Robbiani 2020; Shi 2020; Xie 2020; de Assis 2020; CTRI/2020/10/028547).
Five studies pertained to feasibility of collection of convalescent plasma only (Budhai 2020; Hashim 2020; NCT04344015; NCT04344977; NCT04360278).
Four studies were cancelled by the investigator before recruiting participants into the study (ChiCTR2000030312; ChiCTR2000030381; ChiCTR2000030442; NCT04325672; NCT04467151).
Three studies reported standard operating procedure related to plasma donation (Brasil Ministerio 2020; Franchini 2020; Ministerio de Salud 2020).
Two references were in Chinese (Qiu 2020; Tu 2020). Both were translated and assessed by Rujan Shrestha and Ya‐Ying Wang via Cochrane TaskExchange. The papers reported on a generalised collection of information about the COVID‐19 infection of two participants relating to aetiology, pathology, symptoms, clinical presentation and some generalised pharmacological treatment methods.
Two studies were controlled, non‐randomised studies with fewer than 500 participants receiving convalescent plasma or hyperimmune immunoglobulin (NCT04347681; NCT04384588).
Two studies were controlled studies, but probably not truly randomised (Baklaushev 2020; Rasheed 2020).
Two studies were pharmacokinetics studies (NCT04638634; NCT04661839).
One study included an irrelevant participant population (participants exposed to COVID‐19) (NCT04323800).
One study was a single‐arm study with fewer than 500 participants receiving hyperimmune immunoglobulin (NCT04721236).
One study compared early to deferred convalescent plasma (Balcells 2020).
One study was on plasma donors (NCT04555109).
One study was terminated early and stopped because the sponsor was changed and a new study on convalescent plasma sponsored by the Italian Medicines Agency (AIFA) was started in Italy (NCT04393727).
Risk of bias in included studies
We assessed methodological quality and risk of bias for 12 RCTs (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Bajpai 2020; Gharbharan 2020; Hamdy Salman 2020; Horby 2021; Li 2020; Libster 2020; O’Donnell 2021; Ray 2020; Simonovich 2020), using the 'Risk of Bias 2' (RoB 2) tool recommended in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2019c). For one non‐controlled NRSI (Joyner 2020), we used the 'Risk of bias' assessment criteria tool for observational studies provided by Cochrane Childhood Cancer (see Table 6; Mulder 2019). For the non‐controlled NRSI, we only assessed risk of bias for safety outcomes. The completed RoB 2 tool with responses to all assessed signalling questions is available online at zenodo.org/record/4715089#.YIKvBOj7RaQ (Piechotta 2021).
Overall judgements for studies including individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease
All‐cause mortality
Among those studies reporting a mortality outcome, we rated the overall risk of bias to be of some concern in Agarwal 2020, AlQahtani 2020, Gharbharan 2020 and Ray 2020. We assessed this outcome on a study level at day 28, day 60, time‐to‐event, and at hospital discharge. For Agarwal 2020, there were some inconsistencies in the adherence to the allocated interventions, which could be due to awareness of the intervention in this open‐label trial (see Table 151; Table 153). Gharbharan 2020 provided insufficient information on whether co‐interventions were balanced across arms (see Table 152; Table 153). In one study (AlQahtani 2020), the produced result analysed was not in accordance with the pre‐specified analysis plan, as the time point of the mortality outcome was not specified in the study protocol (see Table 151; Table 153). In Ray 2020, the preprint available gave no clear information on allocation concealment; only a preliminary statement of concealment via case record numbers could be retrieved form the trial registry, which led to a high risk of bias from the randomisation process (see Table 151; Table 152).
Risk of bias for analysis 1.1 All‐cause mortality at up to day 28.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.1.1 Individuals with moderate disease | ||||||||||||
| Agarwal 2020 | Low risk of bias | The allocation sequence was random and concealed and there were no differences between intervention groups suggesting a problem with the randomization process. | Some concerns | Five participants in convalescent plasma arm and four in control arm did not receive the allocated intervention and this is probably a deviation from the intended intervention that arose because of the trial context. The analysis was appropriate. | Low risk of bias | Data for this outcome was available for 453 out of 464 participants. Data of six participants in the convalescent plasma arm and of five participants in the control arm were missing. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The data were collected in structured paper case record forms. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Some concerns | For the outcome "mortality" in this study, there is a low risk of bias from the randomization process, missing outcome data, measurement of the outcome and in selection of the reported result. However, there are some concerns for bias due to deviations from intended interventions. |
| AlQahtani 2020 | Low risk of bias | Participants were block randomized by computer‐generated random numbering in a 1:1 ratio to receive either convalescent plasma in addition to the standard therapy or standard of care alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 40 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Some concerns | The data that produced this result was not analysed in accordance with the predefined outcomes stated in the protocol, as the time point of the outcome measurement was not pre‐specified in the study protocol. | Some concerns | For the outcome "mortality", there are some concerns for bias in selection of reported results. However, for all the other domains, there is a low risk of bias. |
| Avendano‐Sola 2020 | Low risk of bias | Participants were randomized through a web‐based eCRF system (ORACLE clinical) in a 1:1 ratio to receive either convalescent plasma in addition to the standard therapy or standard of care alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 81 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Low risk of bias | For the outcome "mortality at up to day 28", there is a low risk of bias for all the domains. |
| Simonovich 2020 | Low risk of bias | Participants were randomized through a randomization program (REDCap) in a 2:1 ratio to receive either convalescent plasma or a placebo. The allocation sequence was random and concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and carers and people delivering the intervention were unaware of the assigned intervention received and the analysis was appropriate. | Low risk of bias | From the 334 participants randomized data was available for 228 participants in the convalescent plasma group and 105 participants in the placebo group. The data of one participant in control group was missing, due to withdrawal of consent after randomisation and therefore the participant was excluded from the analysis. | Low risk of bias | Mortality is an objective outcome measure, appropriately measured and it is unlikely that the measurement differed between intervention groups. The outcome assessors were probably not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "mortality", all the domains have a low risk of bias. |
| Subgroup 1.1.2 Individuals with severe disease | ||||||||||||
| Li 2020 | Low risk of bias | Participants were block randomized via computer‐generated random numbering in a 1:1 ratio to receive standard treatment coupled with convalescent plasma transfusion or standard treatment alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for nearly all participants randomized, a total of 101 out of 103 participants were included in the analysis. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "mortality at up to day 28", all the domains have a low risk of bias. |
| Subgroup 1.1.3 Individuals with moderate or severe disease | ||||||||||||
| Horby 2021 | Low risk of bias | Participants were randomized through web‐based simple randomization with allocation concealment in a 1:1:1 ratio to a platform trial in a factorial design, receiving either convalescent plasma in addition to the standard therapy or standard of care alone. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 11,558 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were probably not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "28‐day mortality", there was a low risk of bias for all the domains. |
| Ray 2020 | Some concerns | Participants were probably allocated randomly to either the standard of care group alone or standard of care with convalescent plasma group and there were no further information given on the randomization process. There is no information on the allocation concealment, the trial registry only indicates concealment through "Case Record Numbers". There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 80 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Some concerns | For the outcome "mortality at up to day 28" in this study, there is a low risk of bias due to deviations from intended interventions, due to missing outcome data, in measurement of the outcome and in selection of the reported result. There are some concerns for bias from the randomization process. |
Risk of bias for analysis 1.3 All‐cause mortality at hospital discharge.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.3.1 Individuals with moderate disease | ||||||||||||
| Agarwal 2020 | Low risk of bias | The allocation sequence was random and concealed and there were no differences between intervention groups suggesting a problem with the randomization process. | Some concerns | Five participants in convalescent plasma arm and four in control arm did not receive the allocated intervention. This could be due to awareness of the intervention, because this trial was open‐label. | Low risk of bias | Data for this outcome was available for 451 out of 464 participants. Data of eight participants in the convalescent plasma arm and of five participants in the control arm were missing. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The data were collected in structured paper case record forms. | Low risk of bias | There was no information on whether the data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration, as the data was provided on request by the study investigators. | Some concerns | For this outcome "mortality", there is a low risk of bias from the randomization process, missing outcome data, measurement of the outcome and in selection of the reported result. However, there are some concerns for bias due to deviations from intended interventions. |
| AlQahtani 2020 | Low risk of bias | Participants were block randomized by computer‐generated random numbering in a 1:1 ratio to receive either convalescent plasma in addition to the standard therapy or standard of care alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 40 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Low risk of bias | For this outcome, there was no information on whether the data that produced this result was analysed in accordance with the predefined protocol, as the data was provided on request by the study investigators. | Low risk of bias | For the outcome "All‐cause mortality at hospital discharge" all the other domains were at low risk of bias. |
| Subgroup 1.3.2 Individuals with severe disease | ||||||||||||
| Gharbharan 2020 | Low risk of bias | Participants were randomized via a web‐based system in a 1:1 ratio to the current standard of care at each hospital with or without the addition of convalescent plasma. There are no baseline differences that would suggest a problem with randomisation. | Some concerns | Both participants and those delivering the intervention were aware of intervention received, but there was no information on deviations from intended intervention, as insufficient information was provided on whether treatments were balanced across arms. The analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 86 participants randomized. | Low risk of bias | The outcome assessors aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement or that the measurement differed between intervention groups. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Some concerns | For the outcome "all‐cause mortality at hospital discharge" there is a low risk of bias from the randomization process, due to missing outcome data, in measurement of the outcome and in selection of the reported result. However, there are some concerns for bias due to deviations from intended interventions. |
Risk of bias for analysis 1.2 Mortality (time to event).
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.2.1 Individuals with moderate disease | ||||||||||||
| Simonovich 2020 | Low risk of bias | Participants were randomized through a randomization program (REDCap) in a 2:1 ratio to receive either convalescent plasma or a placebo. The allocation sequence was random and concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and carers and people delivering the intervention were unaware of the assigned intervention received and the analysis was appropriate. | Low risk of bias | From the 334 participants randomized data was available for 228 participants in the convalescent plasma group and 105 participants in the placebo group. The data of one participant in control group was missing, due to withdrawal of consent after randomisation and therefore the participant was excluded from the analysis. | Low risk of bias | Mortality is an objective outcome measure, appropriately measured and it is unlikely that the measurement differed between intervention groups. The outcome assessors were probably not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "mortality", all the domains have a low risk of bias. |
| Subgroup 1.2.2 Individuals with severe disease | ||||||||||||
| Gharbharan 2020 | Low risk of bias | Participants were randomized via a web‐based system in a 1:1 ratio to the current standard of care at each hospital with or without the addition of convalescent plasma. There are no baseline differences that would suggest a problem with randomisation. | Some concerns | Both participants and those delivering the intervention were aware of intervention received, but there was no information on deviations from intended intervention, as insufficient information was provided on whether treatments were balanced across arms. The analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 86 participants randomized. | Low risk of bias | The outcome assessors aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement or that the measurement differed between intervention groups. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Some concerns | For the outcome "Mortality (time to event)", there is a low risk of bias from the randomization process, due to missing outcome data, in measurement of the outcome and in selection of the reported result. However, there are some concerns for bias due to deviations from intended interventions. |
| Li 2020 | Low risk of bias | Participants were block randomized via computer‐generated random numbering in a 1:1 ratio to receive standard treatment coupled with convalescent plasma transfusion or standard treatment alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 103 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "mortality (time to event)", all the domains have a low risk of bias. |
| Subgroup 1.2.3 Individuals with moderate or severe disease | ||||||||||||
| Horby 2021 | Low risk of bias | Participants were randomized through web‐based simple randomization with allocation concealment in a 1:1:1 ratio to a platform trial in a factorial design, receiving either convalescent plasma in addition to the standard therapy or standard of care alone. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 11,558 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were probably not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "mortality (time to event)", there was a low risk of bias for all the domains. |
| Ray 2020 | Some concerns | Participants were probably allocated randomly to either the standard of care group alone or standard of care with convalescent plasma group and there were no further information given on the randomization process. There is no information on the allocation concealment, the trial registry only indicates concealment through "Case Record Numbers". There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 80 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Some concerns | The data that produced this result was not analysed in accordance with the predefined outcomes stated in the trial registration. The "time‐to‐event" measurement for mortality was not mentioned in the trial registry. | Some concerns | For the outcome "mortality (time to event)", there is a low risk of bias due to deviations from intended interventions, due to missing outcome data and in measurement of the outcome. There are some concerns for bias in selection of the reported result and from the randomization process. |
Clinical status
Among those studies reporting at least one of the outcomes addressing clinical status and progression of disease, we rated the overall risk of bias to be of some concern for Agarwal 2020 and Gharbharan 2020. We assessed clinical status on a study level, in accordance with the WHO Clinical Progression Scale (WHO 2020e), and including both improvement of clinical status and worsening of clinical status. For Gharbharan 2020, we judged the risk of bias for improvement of clinical status to be of some concern, as the study provided insufficient information on whether co‐interventions were balanced across arms (see Table 154; Table 155). For Agarwal 2020, we judged the risk of bias for worsening of clinical status to be of some concern, because of some inconsistencies in the adherence to the allocated interventions, which could be due to awareness of the intervention in this open‐label trial,as well as because the outcome analysed was not prespecified in the trial registry and a study protocol was not available (see Table 156).
Risk of bias for analysis 1.4 Clinical improvement: liberation from supplemental oxygen in surviving patients, for subgroup of participants requiring any supplemental oxygen or ventilator support at baseline.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Gharbharan 2020 | Low risk of bias | Participants were randomized via a web‐based system in a 1:1 ratio to the current standard of care at each hospital with or without the addition of convalescent plasma. There are no baseline differences that would suggest a problem with randomisation. | Some concerns | Both participants and those delivering the intervention were aware of intervention received, but there was no information on deviations from intended intervention, as insufficient information was provided on whether treatments were balanced across arms. The analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants requiring any supplemental oxygen or ventilator support at baseline, being 77 out of 86 participants randomized. | Low risk of bias | The outcome assessors aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement or that the measurement differed between intervention groups. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Some concerns | For the outcome "liberation from supplemental oxygen", there is a low risk of bias from the randomization process, due to missing outcome data, in measurement of the outcome and in selection of the reported result. However, there are some concerns for bias due to deviations from intended interventions. |
Risk of bias for analysis 1.5 Clinical improvement: weaning or liberation from invasive mechanical ventilation in surviving patients, for subgroups of participants requiring invasive mechanical ventilation at baseline.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Gharbharan 2020 | Low risk of bias | Participants were randomized via a web‐based system in a 1:1 ratio to the current standard of care at each hospital with or without the addition of convalescent plasma. There are no baseline differences that would suggest a problem with randomisation. | Some concerns | Both participants and those delivering the intervention were aware of intervention received, but there was no information on deviations from intended intervention, as insufficient information was provided on whether treatments were balanced across arms. The analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants requiring invasive mechanical ventilation at baseline, which was 13 out of 86 participants randomized. | Low risk of bias | The outcome assessors aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement or that the measurement differed between intervention groups. | Low risk of bias | There was no information on whether the data that produced this result was analysed in accordance with a pre‐specified analysis plan, as the data was provided on request by the study investigators. | Some concerns | For the outcome "liberation from invasive mechanical ventilation", there is a low risk of bias from the randomization process, due to missing outcome data, in measurement of the outcome and in selection of the reported result. However, there are some concerns for bias due to deviations from intended interventions. |
| Horby 2021 | Low risk of bias | Participants were randomized through web‐based simple randomization with allocation concealment in a 1:1:1 ratio to a platform trial in a factorial design, receiving either convalescent plasma in addition to the standard therapy or standard of care alone. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants requiring invasive mechanical ventilation at baseline, a total of 617 out of 11,558 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were probably not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "Weaning or liberation from invasive mechanical ventilation", there was a low risk of bias for all the domains. |
Risk of bias for analysis 1.6 Clinical worsening: need for invasive mechanical ventilation, for subgroup of participants not requiring invasive mechanical ventilation at baseline.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Agarwal 2020 | Low risk of bias | The allocation sequence was random and concealed and there were no differences between intervention groups suggesting a problem with the randomization process. | Some concerns | Five participants in convalescent plasma arm and four in control arm did not receive the allocated intervention and this is probably a deviation from the intended intervention that arose because of the trial context. According to the study, a per protocol analysis was performed for this secondary outcome, but there was no potential for a substantial impact (on the result) of the failure to analyse randomized participants. | Low risk of bias | Data for this outcome was available for 451 out of 451 participants included in the per protocol analysis. Data of eight participants in the convalescent plasma arm and of five participants in the control arm were missing. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Some concerns | There was a trial register entry, which provided details of the pre‐specified outcomes, but "need for invasive ventilation" was not pre‐specified at trial registration. | Some concerns | For this outcome "Clinical worsening: Need for invasive mechanical ventilation", there is a low risk of bias from the randomization process, missing outcome data and measurement of the outcome. However, there are some concerns for bias due to deviations from intended interventions and for bias in selection of the reported result. |
| AlQahtani 2020 | Low risk of bias | Participants were block randomized by computer‐generated random numbering in a 1:1 ratio to receive either convalescent plasma in addition to the standard therapy or standard of care alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 40 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "Need for invasive mechanical ventilation", there was a low risk of bias for all the domains. |
| Horby 2021 | Low risk of bias | Participants were randomized through web‐based simple randomization with allocation concealment in a 1:1:1 ratio to a platform trial in a factorial design, receiving either convalescent plasma in addition to the standard therapy or standard of care alone. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants not on invasive mechanical ventilation at baseline, a total of 10,901 out of 11,558 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were probably not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "Need for invasive mechanical ventilation", there was a low risk of bias for all the domains. |
| Simonovich 2020 | Low risk of bias | Participants were randomized through a randomization program (REDCap) in a 2:1 ratio to receive either convalescent plasma or a placebo. The allocation sequence was random and concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and carers and people delivering the intervention were unaware of the assigned intervention received and the analysis was appropriate. | Low risk of bias | From the 334 participants randomized, data was available for 228 participants in the convalescent plasma group and 105 participants in the placebo group. The data of one participant in control group was missing, due to withdrawal of consent after randomisation and therefore the participant was excluded from the analysis. | Low risk of bias | The outcome measurement is an appropriate measure and it is unlikely that the measurement differed between intervention groups. The outcome assessors were not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "Clinical worsening: Need for invasive mechanical ventilation", all the domains have a low risk of bias. |
Quality of life
We could not assess the risk of bias for quality of life, as none of the studies reported this outcome.
Safety
Risk of bias in randomised controlled trials
Among those studies reporting at least one of the safety outcomes, we rated the overall risk of bias to be of some concern for Agarwal 2020 and AlQahtani 2020 (see Table 162). We assessed safety outcomes on a study level and included any adverse events, grade 3 to 4 adverse events and serious adverse events. For Agarwal 2020, we judged the risk of bias for grade 3 to 4 adverse events to be of some concern, because of some inconsistencies in the adherence to the allocated interventions, which could be due to awareness of the intervention in this open‐label trial. For both studies (Agarwal 2020; AlQahtani 2020), the safety data was provided on our request by the study investigator, and it is not clear whether data for this outcome was collected from all, or nearly all the participants randomised. There is also no information available on how safety was measured.
Risk of bias for analysis 1.14 Grade 3 and 4 adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.14.1 Individuals with moderate disease | ||||||||||||
| Agarwal 2020 | Low risk of bias | The allocation sequence was random and concealed and there were no differences between intervention groups suggesting a problem with the randomization process. | Some concerns | Five participants in convalescent plasma arm and four in control arm did not receive the allocated intervention and this is probably a deviation from the intended intervention that arose because of the trial context. There is no information on whether the analysis was appropriate. | Low risk of bias | Data for this outcome was available for 451 participants out of 464 participants randomized and the data of eight participants in the convalescent plasma arm and of five participants in the control arm were missing. | Some concerns | There was no information on the method of measuring for this outcome, as the data was provided on request by the study investigator. The outcome assessors were aware of the intervention received and the knowledge of intervention received could have affected outcome measurement, but it is probably not likely. | Low risk of bias | There was no information on whether the data that produced this result was analysed in accordance with a pre‐specified trial registration, as the data was provided on request by the study investigators. | Some concerns | For the outcome "Grade 3 and 4 adverse events", there is a low risk of bias arising from the randomization process and due to missing outcome data and in the in selection of the reported result. But, there are some concerns for bias due to deviations from intended interventions and in measurement of the outcome. |
| AlQahtani 2020 | Low risk of bias | Participants were block randomized by computer‐generated random numbering in a 1:1 ratio to receive either convalescent plasma in addition to the standard therapy or standard of care alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate.. | Low risk of bias | Data for this outcome was available for all 40 participants randomized. | Some concerns | There is no information available on whether the method of measuring the outcome was appropriate or whether the measurement could have differed between groups , as the data was provided on request by the study investigator and for safety different measurements are possible. The outcome assessors were aware of the intervention received, which is why the knowledge of intervention received could have affected safety outcome assessments, but this is unlikely. | Low risk of bias | For this outcome, there was no information on whether the data that produced this result was analysed in accordance with the predefined protocol, as the data was provided on request by the study investigators. | Some concerns | For the outcome "Grade 3 and 4 adverse events", there is a low risk of bias arising from the randomization process, due to deviations from intended interventions, due to missing outcome data and in the in selection of the reported result. But, there are some concerns for bias in measurement of the outcome. |
| Avendano‐Sola 2020 | Low risk of bias | Participants were randomized through a web‐based eCRF system (ORACLE clinical) in a 1:1 ratio to receive either convalescent plasma in addition to the standard therapy or standard of care alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 81 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Low risk of bias | For this outcome "Grade 3 and 4 adverse events", there is a low risk of bias for all the domains. |
| Simonovich 2020 | Low risk of bias | Participants were randomized through a randomization program (REDCap) in a 2:1 ratio to receive either convalescent plasma or a placebo. The allocation sequence was random and concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and carers and people delivering the intervention were unaware of the assigned intervention received and the analysis was appropriate. | Low risk of bias | From the 334 participants randomized, data was available for 228 participants in the convalescent plasma group and 105 participants in the placebo group. The data of one participant in control group was missing, due to withdrawal of consent after randomisation and therefore the participant was excluded from the analysis. | Low risk of bias | The outcome measurement is an appropriate measure and it is unlikely that the measurement differed between intervention groups. The outcome assessors were not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For this outcome "Grade 3 and 4 adverse events", there is a low risk of bias for all the domains. |
Risk of bias in non‐controlled, non‐randomised studies of interventions
In addition to the high risk of bias due to the non‐randomised and non‐controlled study design, we rated the overall risk of bias within the study (Joyner 2020) to be low. Please refer to our detailed assessment in Appendix 8.
Overall judgements for studies including individuals with a confirmed diagnosis of COVID‐19 and asymptomatic or mild disease
We assessed risk of bias for one outcome, and did not identify any concerns suggesting a risk of bias for Libster 2020, the only study including individuals with asymptomatic or mild disease.
Effects of interventions
See: Table 1; Table 2; Table 3
Summary of findings 1. Summary of Findings Table ‐ Convalescent plasma compared to placebo or standard care alone for individuals with moderate to severe disease.
| Convalescent plasma compared to placebo or standard care alone for individuals with moderate to severe disease | ||||||
| Patient or population: individuals with moderate to severe disease Setting: Intervention: convalescent plasma Comparison: placebo or standard care alone | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo or standard care alone | Risk with convalescent plasma | |||||
| All‐cause mortality at up to day 28 ‐ total | 237 per 1,000 | 233 per 1,000 (218 to 249) | RR 0.98 (0.92 to 1.05) | 12646 (7 RCTs) | ⊕⊕⊕⊕ HIGH | Convalescent plasma does not reduce all‐cause mortality at up to day 28. |
| Clinical improvement, assessed by liberation from respiratory support | Reporting of the clinical status or course of the disease was very heterogeneous across studies and it was not possible to pool data in a meaningful way. The reported evidence in all studies did not suggest any differences in the odds for clinical improvement or time to clinical improvement. a | 12682 (8 RCTs) | ⊕⊕⊕⊕ HIGH | Convalescent plasma has little to no impact on clinical improvement. | ||
| Clinical improvement: weaning or liberation from invasive mechanical ventilation in surviving patients, for subgroups of participants requiring invasive mechanical ventilationat baseline | 362 per 1,000 | 377 per 1,000 (206 to 699) | RR 1.04 (0.57 to 1.93) | 630 (2 RCTs) | ⊕⊕⊝⊝ LOW b,c | Convalescent plasma may have little to no impact on clinical improvement, if assessed with weaning or liberation from invasive mechanical ventilation in surviving patients. |
| Clinical worsening: need for invasive mechanical ventilation,for subgroup of participants not requiring invasive mechanical ventilationat baseline | 126 per 1,000 | 123 per 1,000 (112 to 136) | RR 0.98 (0.89 to 1.08) | 11765 (4 RCTs) | ⊕⊕⊕⊕ HIGH | Convalescent plasma does not reduce clinical worsening if assessed with the need for invasive mechanical ventilation. |
| Quality of life, including fatigue and neurological functioning; assessed with standardised scales up to longest follow‐up | We did not identify any study reporting quality of life on a standardised scale. One study (Agarwal 2020, reporting on 309 participants) assessed resolution of fatigue on day 7 (RR 1.21, 95% CI 1.02 to 1.42, estimated absolute effect with convalescent plasma: 727 of 1000) | 309 (1 RCT) | ⊕⊝⊝⊝ VERY LOW d,e | We do not know whether convalescent plasma has any impact on quality of life, and are very uncertain about the effect on resolution of fatigue. | ||
| Grade 3 and 4 adverse events ‐ total | 64 per 1,000 | 57 per 1,000 (37 to 90) | RR 0.90 (0.58 to 1.41) | 905 (4 RCTs) | ⊕⊕⊝⊝ LOW c,f | We are uncertain whether convalescent plasma reduces or increases the risk of grade 3 and 4 adverse events. |
| Serious adverse events ‐ total | 176 per 1,000 | 218 per 1,000 (142 to 334) | RR 1.24 (0.81 to 1.90) | 414 (2 RCTs) | ⊕⊕⊝⊝ LOW c,f | We are uncertain whether convalescent plasma reduces or increases the risk of serious adverse events. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
| See interactive version of this table: https://gdt.gradepro.org/presentations/#/isof/isof_question_revman_web_423533352409190077. | ||||||
a. One study, reporting on 77 participants, provided sufficient data to calculate a relative effect estimate for our predefined outcome "liberation from supplemental oxygen"; for the subgroup of participants that received any supplemental oxygen or ventilator support. We did not identify any evidence for a difference (RR 1.10, 95% CI 0.81 to 1.48, estimated absolute effect with convalescent plasma: 724 per 1000). b. Downgraded one level for serious inconsistency because direction of effect was not consistent in both studies c. Downgraded one level for serious imprecision, because of few participants and wide confidence intervals d. Downgraded two levels for very serious indirectness, because only one symptom impacting quality of life was assessed, not measured on a standardised scale, and after a short observation period. e. Downgraded one level for serious imprecision, because of few participants f. Downgraded one level for publication bias, because safety outcomes were assessed and reported in most studies for convalescent plasma group only
Summary of findings 2. Summary of Findings Table ‐ Convalescent plasma compared to standard plasma for individuals with moderate to severe disease.
| Convalescent plasma compared to standard plasma for individuals with moderate to severe disease | ||||||
| Patient or population: individuals with moderate to severe disease Setting: Intervention: convalescent plasma Comparison: standard plasma | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with standard plasma | Risk with convalescent plasma | |||||
| All‐cause mortality at up to day 28 | 216 per 1,000 | 205 per 1,000 (37 to 1,000) | RR 0.95 (0.17 to 5.29) | 252 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW a,b | We do not know whether convalescent plasma has any effect on all‐cause mortality at up to day 28. |
| Clinical improvement, assessed by the need for respiratory support | Reporting of the clinical status or course of the disease was heterogenous across studies. One study reported the odds of a one‐point improvement on a six‐point scale in clinical status at day 28 (OR 1.5, 95% CI 0.83 to 2.68). The other study reported the median improvement of O2 saturation: 10% (8.2 to 11) in CP group versus 7.5% (4.75 to 9.25) in SP group; and the median improvement in PaO2/FiO2: 231.15 (183.37 to 245.2) in CP group versus 77.01 (56.93 to 96.20) in SP group on up to day 7, respectively. c | 252 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW d,e | We do not know whether convalescent plasma has any effect on clinical improvement. | ||
| Clinical worsening: need for invasive mechanical ventilation | 67 per 1,000 | 214 per 1,000 (25 to 1,000) | RR 3.21 (0.38 to 27.40) | 29 (1 RCT) | ⊕⊕⊝⊝ LOW b | Convalescent plasma may increase the need for invasive mechanical ventilation. |
| Quality of life, assessed with standardised scales at longest follow‐up | not pooled | not pooled | ‐ | (0 studies) | ‐ | We did not identify any study reporting this outcome. |
| Grade 3 and 4 adverse events | The identified studies did not report the number of participants experiencing any grade 3 or 4 adverse events. One study (reporting on 219 participants) reported the number of participants experiencing any event of grade 3 (27/147 in CP group versus 17/72 in SP group), or grade 4 (26/147 in CP group versus 15/72 in SP group). The study also reported the number of participants experiencing at least one event of any grade (96/147 in CP group versus 40/72 in SP group, RR 1.18, 95% CI 0.93 to 1.49). Both identified studies reported on observed transfusion‐related events no severe side effects observed in one study (reporting on 29 participants); 4/147 in CP group versus 3/72 in SP group in the other study). c | 248 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW d,e | We are very uncertain whether or whether not convalescent plasma increases the risk for grade 3 and 4 adverse events. | ||
| Serious adverse events | 361 per 1,000 | 264 per 1,000 (177 to 401) | RR 0.73 (0.49 to 1.11) | 219 (1 RCT) | ⊕⊕⊝⊝ LOW e | Convalescent plasma may decrease the risk of serious adverse events. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; SMD: Standardised mean difference | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
| See interactive version of this table: https://gdt.gradepro.org/presentations/#/isof/isof_question_revman_web_423575562054934976. | ||||||
a. Downgraded one level for serious inconsistency, because direction of effect is not consistent in both studies b. Downgraded two levels for very serious imprecision, because of few participants, few events and very wide confidence intervals c. CP: convalescent plasma; SP: standard plasma d. Downgraded one level for serious indirectness, because definition of outcomes was different to the definition used in our review e. Downgraded two levels for very serious imprecision, because of few participants, and few events
Summary of findings 3. Summary of Findings Table ‐ Convalescent plasma compared to placebo or standard care alone for individuals with mild disease.
| Convalescent plasma compared to placebo or standard care alone for individuals with mild disease | ||||||
| Patient or population: individuals with mild disease Setting: Intervention: convalescent plasma Comparison: placebo or standard care alone | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo or standard care alone | Risk with convalescent plasma | |||||
| All‐cause mortality | 50 per 1,000 | 25 per 1,000 (5 to 133) | RR 0.50 (0.09 to 2.65) | 160 (1 RCT) | ⊕⊝⊝⊝ VERY LOW a,b | We are very uncertain about the effect of convalescent plasma on all‐cause mortality. |
| Development of severe clinical COVID‐19 symptoms assessed with: WHO Clinical Progression Scale ≥ 6 | We did not identify any study reporting development of severe clinical COVID‐19 symptoms, when assessed according to WHO Clinical Progression Scale. One study (reporting on 160 participants) reported the need for invasive mechanical ventilation (RR 0.50, 95% CI 0.09 to 2.65, estimated absolute effect with convalescent plasma: 25 of 1000), the risk for developing severe respiratory disease (RR 0.52, 95% CI 0.29 to 0.94, estimated absolute effect with convalescent plasma: 313 of 1000), or critical illness with life‐threatening disease (RR 0.83, 95% CI 0.27 to 2.62, estimated absolute effect with convalescent plasma: 63 of 1000) c,d,e | 160 (1 RCT) | ⊕⊕⊝⊝ LOW b | We are uncertain about the effect of convalescent plasma on developing severe clinical COVID‐19 symptoms. | ||
| Quality of life, assessed with standardised scales at longest follow‐up | not pooled | not pooled | ‐ | (0 studies) | ‐ | We did not identify any study reporting this outcome. |
| Grade 3 and 4 adverse events | We did not identify any study reporting grade 3 or 4 adverse events. One study (reporting on 160 participants) reported that no "solicited" adverse events were observed in any group. | 160 (1 RCT) | ⊕⊝⊝⊝ VERY LOW f,g | We do not know whether convalescent plasma is asscociated with a higher risk of grade 3 or 4 adverse events. | ||
| Serious adverse events | We did not identify any study reporting serious adverse events. One study (reporting on 160 participants) reported that no serious "solicited" adverse events were observed in any group. | 160 (1 RCT) | ⊕⊝⊝⊝ VERY LOW f,g | We do not know whether convalescent plasma is associated with a higher risk of serious adverse events. | ||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; SMD: Standardised mean difference | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
| See interactive version of this table: https://gdt.gradepro.org/presentations/#/isof/isof_question_revman_web_423575913930302502. | ||||||
a. Downgraded one level for serious indirectness, because outcome definition did not exactly match our definition (defined as death associated with COVID‐19); and because all patients were admitted to hospital after recruitment b. Downgraded two levels for very serious imprecision, because CI includes zero effect line, few events, and few participants c. Severe respiratory disease defined as a respiratory rate of 30 breaths per minute or more, an oxygen saturation of less than 93% while the person was breathing ambient air, or both. d. Life‐threatening respiratory disease defined as oxygen supplementation at a fraction of inspired oxygen of 100%, noninvasive or invasive ventilation, admission to an intensive care unit, or any combination of these e. Critical systemic illness defined as respiratory failure with a ratio of the partial pressure of oxygen to Fio2 ≤200 mmHg, shock, multiple organ dysfunction syndrome, or any combination of these f. Downgraded one level for serious indirectness, because outcome definition did not match our definition g. Downgraded two levels for very serious imprecision, because of few participants and only one study was identified
Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease
We present the summary of findings and the certainty of the evidence for our prioritised outcomes for individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease in Table 1 and Table 2. Table 1 includes the comparison of convalescent plasma versus placebo or standard care alone. Table 2 includes the comparison of convalescent plasma versus standard plasma. We assessed disease severity with the need for respiratory support according to the WHO clinical progression scale (WHO 2020e).
We have not yet identified any completed studies on hyperimmune immunoglobulins, but continue to closely monitor ongoing studies (see Table 7).
Effectiveness of convalescent plasma
Prioritised outcomes (included in the 'Summary of findings' table)
All‐cause mortality
We assessed all‐cause mortality at day 28, day 60, time‐to‐event, and at hospital discharge.
Convalescent plasma versus placebo or standard care alone
Eight studies reported all‐cause mortality for 12,550 participants. Considering the reported event rates across studies, we estimated that 237 of 1000 participants die at up to 28 days when treated with placebo or standard care alone. Treatment with convalescent plasma results in little to no difference in all‐cause mortality at up to day 28 (RR 0.98, 95% CI 0.92 to 1.05; 233 per 1000; 7 studies, 12,646 participants; I² = 0%; high‐certainty evidence; Analysis 1.1); when measured over time (HR 0.99, 95% CI 0.92 to 1.07; 5 studies, 11,827 participants; I² = 0%; Analysis 1.2); or at hospital discharge (RR 0.90, 95% CI 0.53 to 1.53; 3 studies, 577 participants; I² = 18%; Analysis 1.3). All‐cause mortality at day 60 was not reported in any study.
1.1. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 1: All‐cause mortality at up to day 28
1.2. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 2: Mortality (time to event)
1.3. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 3: All‐cause mortality at hospital discharge
Subgroup analyses
Severity of disease
We identified no evidence for a difference in the effectiveness of convalescent plasma with regard to all‐cause mortality for people with moderate disease (WHO score 4‐5) or severe disease (WHO score ≥ 6), according to the WHO Clinical Progression Scale (WHO 2020e); see Analysis 1.1; Analysis 1.2; Analysis 1.3.
Duration since symptom onset
We identified no evidence for a difference in the effectiveness of convalescent plasma with regard to the duration since symptom onset; see Analysis 4.1.
4.1. Analysis.

Comparison 4: Subgroup analysis: duration since symptom onset for the comparison of convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 1: All‐cause mortality at up to day 28
Antibodies in recipients detected at baseline
We identified no evidence for a difference in the effectiveness of convalescent plasma with regard to the detection of antibodies in the recipients at baseline; see Analysis 5.1.
5.1. Analysis.

Comparison 5: Subgroup analysis: antibodies in recipients detected at baseline for the comparison of convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 1: All‐cause mortality at up to day 28
Other subgroups
We could not perform any other of our planned subgroup analyses for the comparison of convalescent plasma versus standard plasma and the outcome all‐cause mortality. We had planned to investigate potential differences in the age of participants (children; age 18 to 65 years; and age 65 years and older), but none of the studies performed subgroup analyses, using our cut‐offs. Horby 2021 reported subgroup analyses for participants up to 70 years of age, 70 to 79 years of age, and 80 years or older, and did not identify any evidence for a difference. Ray 2020 reported subgroup analyses for participants up to 67 years of age, and 67 years or older, and noted a potential survival benefit for the younger age group when receiving convalescent plasma. However, the study size was small (80 participants) and it was unclear how many participants informed subgroup analyses.
In addition to our pre‐planned subgroup analyses, Horby 2021 conducted further analyses for the following characteristics: sex, ethnicity, respiratory support received, and use of corticosteroids. The principal investigators did not identify evidence for a difference for any of the performed subgroup analyses.
Sensitivity analyses
We summarised the effects of sensitivity analyses in Table 9. Reported effects of our main analysis were robust when removing studies at high risk of bias, preprint articles, or studies that were stopped early.
6. Sensitivity analyses for the comparison of convalescent plasma versus placebo or standard care alone for the population of individuals with moderate to severe disease.
| Outcome | Main analysis | RoB (excluding studiesa at high RoB) | Publication status (excluding preprintsb) | Study termination (excluding premature termination studiesc) |
| All‐cause mortality at up to day 28 | RR 0.98 (95% CI 0.92 to 1.05); including 12,646 participants from 7 studies | RR 0.98 (95% CI 0.92 to 1.05); including 12,646 participants from 7 studies | RR 0.95 (95% CI 0.68 to 1.34); including 887 participants from 3 studiesb | RR 0.99 (95% CI 0.93 to 1.05); including 12,464 participants from 5 studiesc |
| Clinical improvement: liberation from supplemental oxygen (for the subgroup of participants requiring any supplemental oxygen or ventilator support at baseline, i.e. WHO ≥ 5) | RR 1.10 (95% CI 0.81 to 1.48); including 77 participants from 1 study | RR 1.10 (95% CI 0.81 to 1.48); including 77 participants from 1 study | /b | /c |
| Clinical improvement: liberation from invasive mechanical ventilation (for the subgroup of participants requiring invasive mechanical ventilation at baseline, i.e. WHO ≥ 7) | RR 1.04 (95% CI 0.57 to 1.93); including 630 participants from 2 studies | RR 1.04 (95% CI 0.57 to 1.93); including 630 participants from 2 studies | /b | RR 0.81 (95% CI 0.64 to 1.02); including 617 participants from 1 study |
| Clinical worsening: need for invasive mechanical ventilation (for the subgroup of participants not requiring invasive mechanical ventilation at baseline, i.e. WHO ≤ 6) | RR 0.98 (95% CI 0.89 to 1.08); including 11,765 participants from 4 studies | RR 0.98 (95% CI 0.89 to 1.08); including 11,765 participants from 4 studies | RR 1.11 (95% CI 0.79 to 1.56); including 784 participants from 2 studiesb | RR 0.98 (95% CI 0.89 to 1.08); including 11,765 participants from 4 studies |
| Quality of life | NR | NR | NR | NR |
| Grade 3 or 4 adverse events | RR 0.90 (95% CI 0.58 to 1.41); including 905 participants from 4 studies | RR 0.90 (95% CI 0.58 to 1.41); including 905 participants from 4 studies | RR 0.88 (95% CI 0.55 to 1.41); including 784 participants from 2 studiesb | RR 0.88 (95% CI 0.55 to 1.41); including 824 participants from 3 studiesc |
| Serious adverse events | RR 1.24 (95% CI 0.81 to 1.90); including 414 participants from 2 studies | RR 1.24 (95% CI 0.81 to 1.90); including 414 participants from 2 studies | RR 1.31 (95% CI 0.82 to 2.09); including 333 participants from 1 studyb | RR 1.31 (95% CI 0.82 to 2.90); including 333 participants from 1 studyc |
aExcluded studies with high risk of bias (RoB): no study. bExcluded preprints: AlQahtani 2020; Avendano‐Sola 2020; Gharbharan 2020; Horby 2021; Ray 2020
cExcluded studies with premature termination: Gharbharan 2020; Li 2020; Avendano‐Sola 2020
Abbreviations: CI: confidence interval; NR: not reported; RoB: risk of bias; RR: risk ratio; WHO: World Health Organization.
Convalescent plasma versus standard plasma
Two studies reported all‐cause mortality for 252 participants. Considering the reported event rates within the study, we estimated that 216 of 1000 participants die at up to 28 days when treated without convalescent plasma. We are uncertain about the effect of convalescent plasma on all‐cause mortality at up to day 28 when compared to standard plasma (RR 0.95, 95% CI 0.17 to 5.29; 205 per 1000; I² = 62%, very low‐certainty evidence, see Analysis 2.1). Our main reasons for downgrading were serious inconsistency because direction of effect was not consistent in both studies, and very serious imprecision due to few participants, few events, and wide confidence intervals. All‐cause mortality at day 60, over time, or at hospital discharge were not reported in the study.
2.1. Analysis.

Comparison 2: Convalescent plasma versus standard plasma for individuals with moderate to severe disease, Outcome 1: All‐cause mortality at up to day 28
Subgroup analyses
We could not perform any of our planned subgroup analyses for the comparison of convalescent plasma versus standard plasma and the outcome all‐cause mortality.
Sensitivity analyses
We could not perform any of our planned sensitivity analyses for the comparison of convalescent plasma versus standard plasma and the outcome all‐cause mortality.
Clinical status
We assessed the clinical status of participants by the need for respiratory support in accordance with standardised scales (e.g. WHO Clinical Progression Scale (WHO 2020e), WHO Ordinal Scale for Clinical Improvement (WHO 2020f)) at up to day 28, day 60, and up to longest follow‐up). We targeted outcomes that were included in this domain to subgroups of our population to assess clinical improvement and clinical worsening (see Types of outcome measures).
Convalescent plasma versus placebo or standard care alone
Reporting of the clinical status or progression of disease was very heterogeneous across studies. We summarised all reported or additionally received data per study in Table 10.
7. Overview of clinical status and progression of disease.
| Study ID (sample size analysed) | Disease and ventilation status at baseline | Study outcomes (time point, definition) | Reported outcome data |
| Convalescent plasma (CP) versus placebo or standard care alone for individuals with moderate to severe disease | |||
| Agarwal 2020 | Individuals with moderate illness with partial pressure of oxygen in arterial blood/fraction of inspired oxygen (PaO2/FiO2) ratio between 200 mmHg and 300 mmHg or a respiratory rate of more than 24/min with oxygen saturation 93% or less on room air) were included. (According to WHO clinical progression scale: level 4 and 5) Critically ill individuals with PaO2/FiO2 < 200 mmHg were excluded |
|
|
| AlQahtani 2020 | Individuals with moderate illness, with PaO2/FiO2 ratio of 300 or less, or an oxygen saturation of less than or equal 92% on air, or PaO2 < 60 mmHg in arterial blood gas, requiring oxygen therapy and having radiological evidence of pneumonia were included. (According to WHO clinical progression scale: level 5‐6, as 19/20 in CP group and 17/20 in control group received oxygen via nasal cannula or face mask at baseline and 1/20 in CP group and 3/20 in control group received oxygen via nonrebreather mask or high flow nasal cannula) Individuals not requiring oxygen therapy and individuals requiring ventilatory support (invasive or non‐invasive) were excluded. |
|
|
| Avendano-Sola 2020 | Individuals with moderate illness, requiring hospitalisation for COVID‐19 with either radiographic evidence of pulmonary infiltrates or clinical evidence plus SpO2 ≤ 94% on room air were included. (According to WHO clinical progression scale: level 4‐5) Individuals requiring mechanical ventilatory support (invasive or non‐invasive) or high‐flow oxygen devices were excluded. |
|
|
| Gharbharan 2020 | Individuals with moderate to severe disease, hospitalised with either no oxygen (CP 16% and SC 2%), or oxygen by mask or nasal prongs, or noninvasive ventilation or high‐flow oxygen, or invasive ventilation (CP 84% and SC 98%) were included (According to WHO clinical progression level: 4‐7, as CP arm had 16% ≤ 4 and 84% ≥ 5 and SC arm had 2% ≤ 4 and 98% ≥ 5) Individuals having had invasive ventilation for more than 96 h already were excluded. |
|
|
| Hamdy Salman 2020 | Individuals with moderate illness and/or individuals with severe illness, all receiving received oxygen therapy, but no invasive ventilation or ECMO and having two or more of a four‐category illness‐severity scale: 1. respiratory frequency ≥ 24/min. (23/30 patients at baseline); 2. blood oxygen saturation ≤ 93% on room air, (19/30 patients at baseline); 3. partial pressure of arterial oxygen to fraction of inspired oxygen ratio < 300 mmHg, (21/30 patients at baseline); 4. pulmonary infiltrates occupying more than 50% of both lungs (21/30 patients at baseline) were included. (According to WHO clinical progression level: 4‐6) Individuals with septic shock or multiple organ failure were excluded. |
|
|
| Horby 2021 | Individuals with moderate illness and/or individuals with severe illness were included. (According to WHO clinical progression level: 4‐7, as 5% receiving invasive mechanical ventilation, 87% receiving oxygen only (with or without non‐invasive respiratory support) and 8% were receiving no oxygen therapy) |
|
|
| Li 2020 | Individuals with severe illness with respiratory distress and/or hypoxemia or individuals with life‐threatening illness having shock, organ failure, or requiring mechanical ventilation were included. (According to WHO 10‐point scale: level 6‐9) Individuals with severe septic shock or with a partial pressure of arterial oxygen to fraction of inspired oxygen ratio below 100 were excluded. |
|
|
| Ray 2020 | Individuals with moderate illness and/or individuals with severe disease having fever or suspected respiratory infection, plus one of the following: respiratory rate > 30 breaths/min, severe respiratory distress, SpO2 < 90% at room air) with mild ARDS (patients having partial pressure of oxygen in the arterial blood (PaO2)/fraction of inspired oxygen (FiO2) ratio of 200‐300 mmHg) or moderate ARDS (PaO2/FiO2 100‐200 mmHg), not on mechanical ventilation were included. For patients unable to maintain O2 saturation in blood above 90% with face mask with reservoir, high flow nasal cannula or in some cases MV were deployed. (According to WHO 10‐point scale: level 5‐7) |
No other outcome than mortality or duration of hospital stay assessed | N/A |
| Simonovich 2020 | Individuals with moderate illness with at least one of the following severity criteria: oxygen saturation (SaO2) below 93% while they were at rest and breathing ambient air, a ratio of the partial pressure of oxygen (PaO2) to the fraction of inspired oxygen (FiO2) below 300 mm Hg (PaO2:FiO2), or a SOFA or mSOFA score of two or more points above baseline status (scores range from 0 to 24, with higher scores indicating more severe disease) were included. At baseline, more than 90% received receiving oxygen and glucocorticoids at the time of entry into the trial, 64% received low flow nasal cannula, 21.5% venturi or nonrebreather mask, 4.8% high‐flow nasal cannula and 0% noninvasive ventilatory support. (According to WHO 10‐point scale: level 4‐6) Individuals with requirement for mechanical ventilation or multiorgan failure were excluded. |
|
|
| Convalescent plasma versus standard plasma for individuals with moderate to severe disease | |||
| Bajpai 2020 | All participants received supplemental oxygen at five litre/min with target SpO2 being ≥ 94%. If saturation remained below 94%, either of high flow oxygen or NIV (via BiPAP) was given. Individuals presenting with multi‐organ failure or on mechanical ventilation or a PaO2/FiO2 ratio less than 150 were excluded from the study. |
|
|
| O’Donnell 2021 | Individuals with moderate illness and individuals with severe illness, with infiltrates on chest imaging, oxygen saturation ≤ 94% on room air or requirement for supplemental oxygen (including non‐invasive positive pressure ventilation or high flow supplemental oxygen), IMV, or extracorporeal membrane oxygenation (ECMO) at the time of screening, were included. (According to WHO 10‐point scale: level 5‐9, as 57% (126/223) of participants required supplemental oxygen, 25% (55/223) required high‐flow oxygen therapy or non‐invasive mechanical ventilation, and 13% (28/223) required IMV or ECMO) Individuals with a duration of IMV or ECMO ≥ 5 days at time of screening or severe multi‐organ failure were excluded. |
|
|
| Convalescent plasma versus placebo or standard care alone for individuals with asymptomatic or mild disease | |||
| Libster 2020 | Individuals of 75 years or older with at least one coexisting condition enrolled and admitted to hospital after screening and RT‐PCR testing via home visits. Individuals with severe respiratory disease were excluded. |
|
|
Abbreviations: BiPAP: bilevel positive airway pressure; CI: confidence interval; CP: convalescent plasma; ECMO: extracorporeal membrane oxygenation; FiO2: fraction of inspired oxygen; HR: hazard ratio; IMV: invasive mechanical ventilation; IQR: interquartile range; mSOFA: modified sequential organ failure assessment; MV: mechanical ventilation; NIV: non‐invasive ventilation; OR: odds ratio; O2: oxygen; PaO2: partial pressure of oxygen in the arterial blood; RR: risk ratio; RT‐PCR: reverse transcription polymerase chain reaction; SaO2: oxygen saturation; SOFA: sequential organ failure assessment; SC: standard care; SpO2: peripheral oxygen saturation; WHO: World Health Organization.
Improvement of clinical status
The reported evidence in all eight studies (12,682 participants) did not suggest any differences in the odds for clinical improvement or time to clinical improvement when assessed by liberation from respiratory support (high‐certainty evidence, see Table 10).
One study (Gharbharan 2020), reporting on 77 participants, provided sufficient data to calculate a relative effect estimate for our predefined outcome 'liberation from supplemental oxygen' for the subgroup of participants that received any supplemental oxygen or ventilator support (i.e WHO ≥ 5). Considering the reported event rates within the study, we estimated that 614 of 1000 participants had the chance to be liberated from supplemental oxygen when treated with placebo or standard care alone. Evidence suggests little to no difference in the chance of being liberated from supplemental oxygen when treated with convalescent plasma (RR 1.10, 95% CI 0.81 to 1.48; 675 per 1000; low‐certainty evidence, Analysis 1.4). Our main reasons for downgrading were very serious imprecision due to few participants, and very wide confidence intervals.
1.4. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 4: Clinical improvement: liberation from supplemental oxygen in surviving patients, for subgroup of participants requiring any supplemental oxygen or ventilator support at baseline
Two studies reported weaning or liberation from invasive mechanical ventilation in surviving patients for 630 participants; the subgroup of participants that were ventilated at baseline (i.e. WHO ≥ 7). Considering the reported event rates across studies, we estimated that 362 of 1000 participants had the chance to be weaned or liberated from invasive mechanical ventilation when treated without convalescent plasma. Evidence suggests that treatment with convalescent plasma may have little to no impact on being weaned or liberated from invasive mechanical ventilation (RR 1.04, 95% CI 0.57 to 1.93; 377 per 1000; I² = 75%; low‐certainty evidence, Analysis 1.5). Our main reasons for downgrading were serious inconsistency because direction of effect was not consistent in both studies, and serious imprecision due to few participants, and wide confidence intervals.
1.5. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 5: Clinical improvement: weaning or liberation from invasive mechanical ventilation in surviving patients, for subgroups of participants requiring invasive mechanical ventilation at baseline
Worsening of clinical status
Four studies reported the need for invasive mechanical ventilation for 11,765 participants. Considering the reported event rates within the study, we estimated that 126 of 1000 participants had a need for invasive mechanical ventilation when treated without convalescent plasma. Evidence suggests that treatment with convalescent plasma results in little to no difference in the need for invasive mechanical ventilation when compared to no convalescent plasma (RR 0.98, 95% CI 0.89 to 1.08; 123 per 1000; I² = 0%; high‐certainty evidence, Analysis 1.6). Need for non‐invasive mechanical ventilation or high‐flow oxygen and need for oxygen by mask or nasal prongs were not reported in any study, in the way that we had defined the outcomes.
1.6. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 6: Clinical worsening: need for invasive mechanical ventilation, for subgroup of participants not requiring invasive mechanical ventilation at baseline
Subgroup analyses
Severity of disease
We identified no evidence for a difference in the effectiveness of convalescent plasma with regard to the need for invasive mechanical ventilation for people with moderate disease (WHO score 4‐5), according to WHO Clinical Progression Scale (WHO 2020e), when compared to the main analysis that also included participants with more severe disease, receiving non‐invasive ventilation or high‐flow oxygen at baseline (WHO score 6); see Analysis 1.6.
Other subgroups
We could not perform any other of our planned subgroup analyses for the comparison of convalescent plasma versus standard plasma for any outcome summarised under 'clinical status'.
Sensitivity analyses
We summarised the effects of sensitivity analyses in Table 9. Reported effects of our main analysis for the outcome need for invasive mechanical ventilation were robust when removing studies preprint articles. We did not include any studies at high risk of bias, or studies that were stopped early in the main analysis of this outcome.
Convalescent plasma versus standard plasma
Reporting of the clinical status or progression of disease was heterogeneous across studies. We summarised reported data per study in Table 10.
Improvement of clinical status
Both studies assessed improvement of clinical status with the need for respiratory support or oxygenation indices (see Table 10). The reported evidence in both studies was of very low‐certainty and we do not know whether convalescent plasma has any effect on clinical improvement. Our main concerns were serious indirectness because definition of outcomes differed from the definitions used in our review, and very serious imprecision because of few participants and few events.
Worsening of clinical status
Ony study reported the need for invasive mechanical ventilation for 29 participants (Bajpai 2020). Considering the reported event rates within the study, we estimated that 67 of 1000 participants had a need for invasive mechanical ventilation when treated with standard plasma. Evidence is uncertain whether treatment with convalescent plasma increases the need for invasive mechanical ventilation when compared to standard plasma (RR 3.21, 95% CI 0.38 to 27.40; 214 per 1000; low‐certainty evidence; Analysis 2.2). Our main reasons for downgrading were very serious imprecision due to few participants, few events, and very wide confidence intervals. Need for non‐invasive mechanical ventilation or high‐flow oxygen and need for oxygen by mask or nasal prongs were not reported in any study.
2.2. Analysis.

Comparison 2: Convalescent plasma versus standard plasma for individuals with moderate to severe disease, Outcome 2: Clinical worsening: need for invasive mechanical ventilation
Subgroup analyses
We could not perform any of our planned subgroup analyses for the comparison of convalescent plasma versus standard plasma for any outcome summarised under 'clinical status'.
Sensitivity analyses
We could not perform any of our planned sensitivity analyses for the comparison of convalescent plasma versus standard plasma for any outcome summarised under 'clinical status'.
Quality of life
We had planned to assess quality of life, including fatigue and neurological functioning of participants, if assessed with standardised scales (e.g. WHOQOL‐100) for the following time points: at up to 7 days, up to 30 days, and longest follow‐up available. However we did not identify any studies reporting quality of life on a standardised scale.
Convalescent plasma versus placebo or standard care alone
None of the identified studies reported quality of life on a standardised scale. One study (Agarwal 2020, reporting on 309 participants) assessed resolution of fatigue on day 7 (RR 1.21, 95% CI 1.02 to 1.42, estimated absolute effect with convalescent plasma: 727 of 1000 with symptom resolution, very low‐certainty evidence). We do not know whether convalescent plasma has any impact on quality of life, and are very uncertain about the effect on resolution of fatigue. Our main reasons for downgrading were very serious indirectness and serious imprecision, because only one symptom impacting quality of life was assessed, the outcome was not measured on a standardised scale, after a short observation period, and only for few participants.
Convalescent plasma versus standard plasma
We did not identify any study reporting this outcome.
Additional outcomes (not included in the 'Summary of findings' table)
Duration of hospitalisation, or time to discharge from hospital
Convalescent plasma versus placebo or standard care alone
Mean duration of hospitalisation was not reported in any study. Five studies reported time to discharge from hospital for 683 participants. Evidence suggests that more people treated with convalescent plasma may be discharged earlier than people treated without convalescent plasma (HR 1.15, 95% CI 0.95 to 1.40; I² = 17.6%; Analysis 1.7).
1.7. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 7: Time to discharge from hospital
Convalescent plasma versus standard plasma
One study reported duration of hospitalisation for 29 participants (Bajpai 2020). The study reported a mean duration of hospitalisation of 12.1 days (SD 4.1) in the convalescent plasma group and 16.1 days (SD 5.6) in the standard plasma group (MD ‐4.00, 95% CI ‐7.56 to ‐0.44; Analysis 2.3). Time to discharge from hospital was not reported in any study.
2.3. Analysis.

Comparison 2: Convalescent plasma versus standard plasma for individuals with moderate to severe disease, Outcome 3: Duration of hospitalisation
Admission to the ICU
Convalescent plasma versus placebo or standard care alone
One study reported admission to the ICU for 333 participants (Simonovich 2020). Evidence suggests that fewer people treated with convalescent plasma may have to be admitted to the ICU (RR 0.90, 95% CI 0.74 to 1.09; Analysis 1.8).
1.8. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 8: Admission to the intensive care unit (ICU)
Convalescent plasma versus standard plasma
We did not identify any study reporting this outcome.
Length of stay on the ICU, or time to discharge from ICU
Convalescent plasma versus placebo or standard care alone
We did not identify any study reporting this outcome.
Convalescent plasma versus standard plasma
We did not identify any study reporting this outcome.
Viral clearance
We included data of viral clearance if assessed with RT‐PCR test for SARS‐CoV‐2 for the following time points: at baseline, up to 3, 7, and 15 days.
Convalescent plasma versus placebo or standard care alone
Four studies reported viral clearance for 552 participants. Evidence suggests that more people treated with convalescent plasma may achieve viral clearance at up to day 3 (RR 1.73, 95% CI 0.98 to 3.04; 4 studies, 552 participants; I² = 76%; Analysis 1.9) day 7 (RR 1.55, 95% CI 0.99 to 2.43; 3 studies, 485 participants; I² = 75%; Analysis 1.10), and day 15 (RR 1.59, 95% CI 0.74 to 3.43; 2 studies, 129 participants; I² = 83%; Analysis 1.11).
1.9. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 9: Viral clearance at up to day 3
1.10. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 10: Viral clearance at up to day 7
1.11. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 11: Viral clearance at up to day 15
Convalescent plasma versus standard plasma
We did not identify any study reporting this outcome.
Need for dialysis
Convalescent plasma versus placebo or standard care alone
One study reported the need for dialysis for 11,442 participants (Horby 2021). Evidence suggests little to no difference between participants receiving convalescent plasma or not (RR 1.03, 95% CI 0.87 to 1.22; Analysis 1.12).
1.12. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 12: Need for dialysis
Convalescent plasma versus standard plasma
We did not identify any study reporting this outcome.
Safety of convalescent plasma
In addition to RCT data, we included data from controlled NRSIs, and non‐controlled NRSIs for safety outcomes, if the study was prospectively registered and reported on 500 or more participants receiving convalescent plasma.
Adverse events
We defined the outcome as the number of participants with any event and were interested in events of any grade, grade 1‐2, and grade 3‐4. We summarised data, including the potential relationship between intervention and adverse reaction, as reported in the primary studies in Table 11.
8. Adverse events of any grade.
| Study | Number of participants | Any grade adverse events | |
| CP group | Control group | ||
| Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease | |||
| Agarwal 2020 | 227 in CP group and 224 in control group |
|
|
| AlQahtani 2020 | 20 in CP group and 20 in control group |
|
|
| Avendano-Sola 2020 | 38 in CP group and 43 in control group |
|
|
| Gharbharan 2020 | 43 in CP group and 43 in control group | NR | NR |
| Hamdy Salman 2020 | 15 in CP group and 15 in control group |
|
NR |
| Horby 2021 | in CP group and in control group |
|
|
| Li 2020 | 52 in CP group and 51 in control group |
|
NR |
| Ray 2020 | 40 in CP group 43 in control group | Reported any grade events in the CP group:
|
NR |
| Simonovich 2020 | 228 in CP group and 105 in control group |
|
|
| Convalescent plasma versus standard plasma for individuals with moderate to severe disease | |||
| Bajpai 2020 | 14 in CP group and 15 in control group |
|
|
| O’Donnell 2021 | 147 in CP group and 72 in control group |
|
|
| Convalescent plasma (no comparison) for individuals with moderate to severe disease | |||
| Joyner 2020 | 35,322 (20,000 included in safety analysis) | NR | N/A (single‐arm study) |
| Convalescent plasma versus placebo or standard of care alone for individuals with asymptomatic or mild disease | |||
| Libster 2020 | 80 in CP group and 80 in control group |
|
|
Abbreviations: CP: convalescent plasma; NR: not reported; TACO: transfusion‐associated circulatory overload; TRALI: transfusion‐related acute lung injury.
Convalescent plasma versus placebo or standard care alone
One study reported any grade adverse events for both groups and a total of 333 participants (Simonovich 2020). Considering the reported event rates across studies, we estimated that 629 of 1000 participants experience any adverse event when treated without convalescent plasma. Evidence suggests little to no difference in the occurrence of any adverse events when treated with convalescent plasma (RR 1.06, 95% CI 0.89 to 1.26; 667 per 1000; Analysis 1.13).
1.13. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 13: Any grade adverse events
Four RCTs reported grade 3 or 4 events for both groups and a total of 905 participants. Considering the reported event rates across studies, we estimated that 64 of 1000 participants experience a grade 3 or 4 adverse event when treated without convalescent plasma. We are uncertain whether convalescent plasma reduces or increases the risk of grade 3 and 4 adverse events (RR 0.90, 95% CI 0.58 to 1.41; 57 per 1000; I² = 0%; low‐certainty evidence, Analysis 1.14). Our main reasons for downgrading were serious imprecision due to few participants, wide confidence intervals, and suspected publication bias, because most studies assessed and reported transfusion‐related events only; i.e. reported safety data only for the intervention group.
1.14. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 14: Grade 3 and 4 adverse events
Grade 1‐2 adverse events were not reported in any study in a way that we could pool data. We summarised any reported adverse reactions in Table 11, including potential relationships between events and transfusion.
Subgroup analyses
Severity of disease
All studies included in the main analysis for the outcome grade 3 or 4 adverse events, included participants with moderate disease (WHO score 4‐5), according to WHO Clinical Progression Scale (WHO 2020e); see Analysis 1.14. We could therefore not investigate subgroup differences between participants with moderate and severe disease.
Other subgroups
We could not perform any other of our planned subgroup analyses for the comparison of convalescent plasma versus standard plasma for any outcome summarised adverse events.
Sensitivity analyses
We summarised the effects of sensitivity analyses in Table 9. Reported effects of our main analysis for the outcome grade 3 or 4 adverse events were robust when removing studies preprint articles. We did not include any studies at high risk of bias, or studies that were stopped early in the main analysis of this outcome.
Convalescent plasma versus standard plasma
One study reported any grade adverse events for both groups and a total of 219 participants (O’Donnell 2021). Considering the reported event rates across studies, we estimated that 556 of 1000 participants experience any adverse event when treated without convalescent plasma. Evidence suggests an increase of any adverse events when treated with convalescent plasma (RR 1.18, 95% CI 0.93 to 1.49; 656 per 1000; very low‐certainty evidence; Analysis 2.4). None of the studies reported the number of participants experiencing any grade 1 or 2, or grade 3 or 4 adverse events. However, O’Donnell 2021 reported the number of participants experiencing grade 1, 2, 3, and 4 events, respectively (see Table 11). We are very uncertain whether or whether not convalescent plasma increases the risk of grade 3 or 4 adverse events, when compared to standard plasma. Our main concerns were serious indirectness because definition of outcomes differed from the definitions used in our review, and very serious imprecision because of few participants and few events.
2.4. Analysis.

Comparison 2: Convalescent plasma versus standard plasma for individuals with moderate to severe disease, Outcome 4: Any grade adverse events
Both studies further reported incidence of transfusion‐related events. Bajpai 2020 reported that one participant per group (14 participants in convalescent plasma group, and 15 participants in standard plasma group) experienced a mild transfusion reaction; O’Donnell 2021 reported that four of 147 participants in the convalescent plasma group, and two of 72 participants in the standard plasma group, experienced events that were probably or definitely transfusion‐related (see Table 11).
Convalescent plasma (no comparison)
We did not identify any study that matched our inclusion criteria and reported this outcome.
Serious adverse events
We defined the outcome as the number of participants with any event. Two RCTs (414 participants) reported serious adverse events for both groups. Further, four RCTs (6125 participants in convalescent plasma groups) reported on serious adverse events only in participants who received convalescent plasma, with no reporting in the control group. One of the included studies was a single‐arm expanded access study, reporting safety data for 20,000 of 35,322 transfused participants (Joyner 2020). A detailed report of the observed events of the five RCTs and the expanded access study is provided in Table 12, including potential relationships between events and transfusion.
9. Serious adverse events (SAEs).
| Study | Number of participants | Serious adverse events | |
| CP group | Control group | ||
| Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease | |||
| Agarwal 2020 | 227 in CP group and 224 in control group |
|
NR |
| AlQahtani 2020 | 20 in CP group and 20 in control group | NR | NR |
| Avendano-Sola 2020 | 38 in CP group and 43 in control group |
|
|
| Gharbharan 2020 | 43 in group and 43 in control group |
|
NR |
| Hamdy Salman 2020 | 15 in group and 15 in control group | NR | NR |
| Horby 2021 | 5795 in group and 5763 in control group |
|
NR |
| Li 2020 | 52 in CP group and 51 in control group | NR |
NR |
| Ray 2020 | 40 in CP group and 40 in control group | NR | NR |
| Simonovich 2020 | 228 in CP group and 105 in control group |
|
|
| Convalescent plasma versus standard plasma for individuals with moderate to severe disease | |||
| Bajpai 2020 | 14 in CP group and 15 in control group | NR | NR |
| O’Donnell 2021 | 147 in CP group an 72 in control group |
|
|
| Convalescent plasma (no comparison) for individuals with moderate to severe disease | |||
| Joyner 2020 | 35,322 (20,000 included in safety analysis) | Total events within 7 days after transfusion: 1282 Within 4 h after transfusion: 146 events reported
Within 7 days after transfusion: additional 1136 events reported
|
N/A (single‐arm study) |
| Convalescent plasma versus placebo or standard of care alone for individuals with mild disease | |||
| Libster 2020 | 80 in CP group and 80 in control group |
|
|
Abbreviations: CP: convalescent plasma; NR: not reported; TACO: transfusion‐associated circulatory overload; TRALI: transfusion‐related acute lung injury; SAEs: serious adverse events; SHOT: serious hazards of transfusion.
Convalescent plasma versus placebo or standard care alone
Considering the reported event rates reported across the control groups of the two RCTs, we estimated that 176 of 1000 participants experience a serious adverse event when treated without convalescent plasma. Convalescent plasma may or may not result in an increase of serious adverse events (RR 1.24, 95% CI 0.81 to 1.90; 218 per 1000; 2 studies, 414 participants; I² = 0%; low‐certainty evidence, Analysis 1.15). Our main reasons for downgrading were serious imprecision due to few participants, and wide confidence intervals, and suspected publication bias because most studies assessed and reported transfusion‐related events only; i.e. reported safety data only for the intervention group.
1.15. Analysis.

Comparison 1: Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease, Outcome 15: Serious adverse events
Subgroup analyses
Severity of disease
Both studies included in the main analysis for the outcome serious adverse events, included participants with moderate disease (WHO score 4‐5), according to WHO Clinical Progression Scale (WHO 2020e); see Analysis 1.15. We could therefore not investigate subgroup differences between participants with moderate and severe disease.
Other subgroups
We could not perform any other of our planned subgroup analyses for the comparison of convalescent plasma versus standard plasma for any outcome summarised adverse events.
Sensitivity analyses
We summarised the effects of sensitivity analyses in Table 9. Reported effects of our main analysis for the outcome grade 3 or 4 adverse events were robust when removing studies preprint articles. We did not include any studies at high risk of bias, or studies that were stopped early in the main analysis of this outcome.
Convalescent plasma versus standard plasma
One study reported serious adverse events for both groups and a total of 219 participants (O’Donnell 2021). Considering the reported event rates across studies, we estimated that 316 of 1000 participants experience any serious adverse event when treated without convalescent plasma. Treatment with convalescent plasma may decrease the risk of serious adverse events when compared to standard plasma (RR 0.73, 95% CI 0.49 to 1.11; 246 per 1000, see Analysis 2.5), but the evidence is uncertain. Our main reasons for downgrading were very serious imprecision because of few participants and few events.
2.5. Analysis.

Comparison 2: Convalescent plasma versus standard plasma for individuals with moderate to severe disease, Outcome 5: Serious adverse events
Convalescent plasma without a comparison
Agarwal 2020 (227 participants, intervention arm from the included RCT) reported that three observed deaths in the convalescent plasma group could be related to the transfusion. Gharbharan 2020 (43 participants, intervention arm from the included RCT) did not observe any serious transfusion‐related adverse events. Horby 2021 (5795 participants, intervention arm from the included RCT) assessed serious adverse events according to SHOT (serious hazards of transfusion), and reported that 13 transfused individuals experienced at least one event. Joyner 2020 reported safety data for 20,000 of 35,322 transfused participants from an ongoing USA FDA 'Expanded Access Program'. The study authors evaluated the incidence of serious adverse events in the first four hours after convalescent plasma transfusion, and additionally, within seven days after transfusion. Overall, 1282 events were reported, 146 of which occurred during the first four hours of observation, and 1136 additional events occurred within seven days after transfusion of convalescent plasma. Li 2020 (52 participants, intervention arm from the included RCT) mentioned that one participant suffered from shortness of breath, cyanosis, and severe dyspnoea within six hours of convalescent plasma transfusion, which they classified as possible severe transfusion‐associated dyspnoea (TAD). After medical treatment, the symptoms gradually improved over two hours.
Reporting of serious adverse events was variable across the included studies. The duration of follow‐up for observation of serious adverse events varied across all studies. Some, but not all, studies included death as a serious adverse event. In addition, it was difficult to ascertain whether some of the adverse events were related to convalescent plasma transfusion, or due to underlying disease or other treatments, or both. There was insufficient evidence to determine whether convalescent plasma therapy results in a clinically relevant increased risk of serious adverse events and our certainty in the evidence is low.
Individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease
In Table 3 we present certainty of the evidence for our prioritised outcomes (please see 'Summary of findings and assessment of the certainty of the evidence' in Data synthesis) for individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease, and the comparison of convalescent plasma versus placebo or standard care alone.
We did not identify any completed studies on hyperimmune immunoglobulins, yet, but monitor ongoing studies closely (see Table 7).
Effectiveness of convalescent plasma
One study included and analysed individuals with asymptomatic or mild disease (Libster 2020). The included study did not provide all the data on the effectiveness of the intervention in accordance with our pre‐specified outcomes.
Prioritised outcomes (included in the 'Summary of findings' table)
All‐cause mortality
We assessed all‐cause mortality at day 28, day 60, time‐to‐event, and at longest follow‐up available. One RCT reported mortality at an undefined time point, including 160 participants, from which 2 out of 80 participants had died due to COVID‐19 in the convalescent plasma group and 4 out of 80 participants in the comparison group (Libster 2020). We are very uncertain about the effect of convalescent plasma on all‐cause mortality when compared to placebo or standard care alone (RR 0.50, 95% CI 0.09 to 2.65; very low‐certainty evidence, Analysis 3.1). Our main reasons for downgrading were serious indirectness and very serious imprecision, because the study investigators defined the outcome as deaths associated with COVID‐19, and may not have reported other causes of mortality; and because we only identified one study with few participants and few events.
3.1. Analysis.

Comparison 3: Convalescent plasma versus placebo or standard care alone for individuals with mild disease, Outcome 1: All‐cause mortality
Development of moderate to severe clinical COVID‐19 symptoms
We defined moderate and severe clinical COVID‐19 symptoms according to the WHO clinical progression scale (WHO 2020e).
Development of severe disease
We assessed this outcome by the need for invasive mechanical ventilation, non‐invasive mechanical ventilation or high flow (i.e. score 6 or higher on WHO clinical progression scale, WHO 2020e). Need for invasive mechanical ventilation was reported by one RCT (Libster 2020), including 160 participants, from which 2 out of 80 participants developed the need for invasive mechanical ventilation in the convalescent plasma group compared to 4 out of 80 participants in the comparison group (RR 0.50, 95% CI 0.09 to 2.65; Analysis 3.2). The study authors further reported the risk for developing severe respiratory disease (RR 0.52, 95% CI 0.29 to 0.94), or critical illness with life‐threatening disease (RR 0.83, 95% CI 0.27 to 2.62), see Table 10. We are uncertain about the effect of convalescent plasma on the development of severe clinical symptoms when compared to placebo or standard care alone (very‐low certainty). Our main reason for downgrading was very serious imprecision due to few events and the small information size.
3.2. Analysis.

Comparison 3: Convalescent plasma versus placebo or standard care alone for individuals with mild disease, Outcome 2: Development of severe symptoms: need for invasive mechanical ventilation
Development of moderate disease
We assessed this outcome by the need for hospitalisation with supplemental oxygen by mask or nasal prongs or without oxygen therapy (i.e. scores 4 and 5 on WHO clinical progression scale, WHO 2020e). We did not identify any study reporting this outcome.
Quality of life
We did not identify any study reporting this outcome.
Additional outcomes (not included in the 'Summary of findings' table)
Admission to hospital
Libster 2020 recruited all participants via home visits but admitted all for observational purposes at study enrolment to the hospital. We did not identify any other study reporting this outcome.
Time to symptom onset
We did not identify any study reporting this outcome.
Length of hospital stay (for hospitalised patients)
We did not identify any study reporting this outcome.
Admission to the intensive care unit (ICU)
This outcome was reported by one RCT (Libster 2020), including 160 participants, from which 2 out of 80 participants were admitted to the intensive care unit in the convalescent plasma group and 6 out of 80 participants were admitted to ICU in the comparison group. Convalescent plasma therapy may decrease the rate of ICU admissions when compared to placebo treatment or standard care alone (RR 0.33, 95% CI 0.07 to 1.60; see Analysis 3.3), but the evidence is very uncertain.
3.3. Analysis.

Comparison 3: Convalescent plasma versus placebo or standard care alone for individuals with mild disease, Outcome 3: Admission to the intensive care unit (ICU)
Viral clearance (assessed with RT‐PCR)
We did not identify any study reporting this outcome.
Safety of convalescent plasma
In addition to RCT data, we had planned to include data from controlled NRSIs, and non‐controlled NRSIs for safety outcomes, if the study was prospectively registered and reported on 500 or more participants receiving convalescent plasma. However, we did not identify any non‐randomised study matching our inclusion criteria for individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease.
Adverse events
We defined the outcome as the number of participants with any event and were interested in events of any grade, grade 1‐2, and grade 3‐4. We summarised data, including the potential relationship between intervention and adverse reaction, as reported in the primary study, in Table 11.
One study reported that convalescent plasma was not associated with any solicited adverse events (Libster 2020). Because the definition was unclear whether only drug‐related adverse events were assessed, and we did not receive additional information from the study investigators, we did not include this outcome in analysis. We do not know whether convalescent plasma is associated with a higher risk for adverse events (very low‐certainty evidence).
We identified no study reporting adverse events of any grade, grade 1‐2, or grade 3‐4.
Serious adverse events
One study reported that convalescent plasma was not associated with any solicited serious adverse events (Libster 2020). Because the definition was unclear whether only drug‐related serious adverse events were assessed, and we did not receive additional information from the study investigators, we did not include this outcome in analysis. We do not know whether convalescent plasma is associated with a higher risk for serious adverse events (very low‐certainty evidence).
Discussion
Summary of main results
The aim of this review was to assess the effectiveness and safety of convalescent plasma and hyperimmune immunoglobulin in the treatment of COVID‐19. This is the fourth version of our living systematic review.
We identified 12 RCTs (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Bajpai 2020; O’Donnell 2021; Gharbharan 2020; Hamdy Salman 2020; Horby 2021; Li 2020; Libster 2020; Ray 2020; Simonovich 2020), and one non‐controlled NRSI (Joyner 2020). The studies evaluated 48,509 participants, of whom 41,880 received convalescent plasma. We did not identify any completed studies that evaluated hyperimmune immunoglobulin. We identified a further 100 ongoing studies evaluating convalescent plasma or hyperimmune immunoglobulin. We also identified 24 completed but not yet published studies, one study not yet completed but terminated early for futility, two studies where expanded access is no longer available but without results published yet and four completed studies with full texts in our weekly searches after the submission of the current review version, that we categorised as 'Awaiting classification', as well as two platform trials that we have placed in that category.
Risk of bias
Among those studies reporting a mortality outcome, we judged the risk of bias to be of some concern for three RCTs (Agarwal 2020; AlQahtani 2020; Gharbharan 2020) and high for one RCT (Ray 2020). Among those studies reporting at least one of the outcomes addressing clinical status and progression of disease, the risk of bias was of some concern for two RCTs (Agarwal 2020; Gharbharan 2020). As none of the studies reported quality of life, we could not assess the risk of bias for this outcome. The risk of bias of safety outcomes was of some concern for two RCTs (Agarwal 2020; AlQahtani 2020).
For safety outcomes, we also included and assessed non‐controlled NRSIs. In addition to the high risk of bias due to the non‐randomised and non‐controlled study design, we rated the overall risk of bias within the study to be low (Joyner 2020).
Effects of interventions
Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease
Eleven RCTs and one NRSI investigated the use of convalescent plasma for 48,349 participants with moderate to severe disease, of which nine RCTs compared convalescent plasma to placebo treatment or standard care alone, and two compared convalescent plasma to standard plasma.
Effectiveness of convalescent plasma
We included data on all RCTs (13,127 participants) to assess effectiveness of convalescent plasma. Nine RCTs (12,875 participants) compared convalescent plasma to placebo or standard care alone, and two RCTs (252 participants) compared convalescent plasma to standard plasma.
Convalescent plasma versus placebo or standard care alone
Convalescent plasma does not reduce all‐cause mortality at up to day 28 (RR 0.98, 95% CI 0.92 to 1.05; 7 RCTs, 12,646 participants; high‐certainty evidence), it has little to no impact on clinical improvement when assessed by liberation from respiratory support (RR not estimable; 8 RCTs, 12,682 participants; high‐certainty evidence) for all participants, or on the chance of being weaned or liberated from invasive mechanical ventilation for the subgroup of participants requiring invasive mechanical ventilation at baseline (RR 1.04, 95% CI 0.57 to 1.93; 2 RCTs, 630 participants; low‐certainty evidence), and does not reduce the need for invasive mechanical ventilation (RR 0.98, 95% CI 0.89 to 1.08; 4 RCTs, 11,765 participants; high‐certainty evidence). We identified no subgroup differences.
We did not identify any studies reporting quality of life, and therefore, do not know whether convalescent plasma has any impact on quality of life. One RCT (309 participants) assessed resolution of fatigue on day 7, but we are very uncertain about the effect (RR 1.21, 95% CI 1.02 to 1.42; very low‐certainty evidence).
Convalescent plasma versus standard plasma
We do not know whether convalescent plasma has any effect on all‐cause mortality at up to day 28 (RR 0.95, 95% CI 0.17 to 5.29; 2 RCTs, 252 participants; very low‐certainty evidence), and clinical improvement (RR not estimable; 2 RCTs, 252 participants; very low‐certainty evidence). Convalescent plasma may increase the need for invasive mechanical ventilation (RR 3.21, 95% CI 0.38 to 27.40; 1 RCT, 29 participants; low‐certainty evidence).
No study reported quality of life.
Safety of convalescent plasma
We included results from eight RCTs, and one NRSIs assessing safety of convalescent plasma. Reporting of safety data and duration of follow‐up was variable. Some of the RCTs reported on adverse events and serious adverse events only in participants receiving convalescent plasma. Some, but not all, studies included death as a serious adverse event.
Convalescent plasma versus placebo or standard care alone
We are uncertain whether convalescent plasma increases or reduces the risk of grade 3 and 4 adverse events (RR 0.90, 95% CI 0.58 to 1.41; 4 RCTs, 905 participants; low‐certainty evidence), and serious adverse events (RR 1.24, 95% CI 0.81 to 1.90; 2 RCTs, 414 participants; low‐certainty evidence), when compared to placebo treatment or standard care alone.
Convalescent plasma versus standard plasma
We are very uncertain whether or not convalescent plasma increases the risk for grade 3 or 4 adverse events (very low‐certainty evidence), because the identified studies did not report the number of participants experiencing any grade 3 or 4 adverse event. One study (219 participants) reported the number of participants experiencing any event of grade 3 ( 27/147 in convalescent plasma group versus 17/72 in standard plasma group), or grade 4 (26/147 in convalescent plasma group versus 15/72 in standard plasma group). The study also reported the number of participants experiencing at least one event of any grade (96/147 in convalescent plasma group versus 40/72 in standard plasma group, RR 1.18, 95% CI 0.93 to 1.49). Both identified studies reported on observed transfusion‐related events, with no severe side effects observed in one study (reporting on 29 participants); 4/147 in convalescent plasma group versus 3/72 in standard plasma group in the other study).
Convalescent plasma may decrease the risk of serious adverse events (RR 0.73, 95% CI 0.49 to 1.11; 1 RCT, 219 participants; low‐certainty evidence).
Convalescent plasma (no comparison)
We included data of one NRSI (reporting safety data for 20,000 of 35,322 transfused participants), and four RCTs reporting safety data only for participants that received convalescent plasma (6125 participants). The NRSI reported on SAEs within the first four hours and within an additional seven days after transfusion. There were 63 deaths, 12 were possibly related and one was probably related to transfusion. There were 146 SAEs within four hours and 1136 SAEs within seven days post‐transfusion. These were predominantly allergic or respiratory, thrombotic or thromboembolic and cardiac events. The four RCTs observed severe or serious transfusion‐related adverse events in 0 to 1.3% of participants receiving convalescent plasma, including severe transfusion‐associated dyspnoea, and probably‐transfusion‐related deaths.
Individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease
We identified one RCT reporting on 160 participants, and comparing convalescent plasma to placebo treatment (saline).
Effectiveness of convalescent plasma
We are very uncertain about the effect of convalescent plasma on all‐cause mortality (RR 0.50, 95% CI 0.09 to 2.65; very low‐certainty evidence). Convalescent plasma may decrease the risk for developing severe clinical COVID‐19 symptoms (RR not estimable; low‐certainty evidence), but the evidence is uncertain.
We identified no study reporting quality of life.
Safety of convalescent plasma
The identified study reported solicited adverse events only and reported that none have been observed in either group. We do not know whether convalescent plasma is associated with a higher risk of grade 3 or 4 adverse events (very low‐certainty evidence), or serious adverse events (very low‐certainty evidence).
Overall completeness and applicability of evidence
We identified 12 RCTs, and one NRSI (for safety outcomes only), evaluating convalescent plasma in adults. These studies included 48,509 participants, of whom 41,880 received convalescent plasma. Most of the participants had also received different treatment options, including antivirals, antimicrobials, corticosteroids, hydroxychloroquine, respiratory support (extracorporeal membrane oxygenation, mechanical ventilation or oxygen), or a combination of those.
Eleven RCTs and the NRSI investigated the use of convalescent plasma for moderately to severely ill individuals, of which nine RCTs compared convalescent plasma to placebo treatment or standard care alone, and two compared convalescent plasma to standard plasma. We included data of all RCTs (13,127 participants) to assess effectiveness of convalescent plasma therapy for individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease. We have high certainty in the evidence that treatment with convalescent plasma does not reduce all‐cause mortality at up to 28 days, and has little to no impact on clinical improvement when compared to placebo treatment or standard care alone. We have low certainty about the effect of convalescent plasma on all‐cause mortality at up to 28 days, and clinical improvement when compared to treatment with standard plasma. Not all of the included RCTs reported adverse events for the control arm. One large, non‐controlled NRSI provided serious adverse events data (reported data for 20,000 of 35,322 transfused participants) within seven days after convalescent plasma transfusion. Convalescent plasma therapy may result in a clinically relevant increased risk of serious adverse events, when compared to treatment with placebo or standard care alone (low‐certainty evidence). When compared to standard plasma, convalescent plasma therapy may results in a decreased risk of serious adverse events. The evidence for grade 3 and 4 adverse events is uncertain, but treatment with convalescent plasma therapy may result in a slight reduction of grade 3 and 4 adverse events (low‐certainty evidence). None of the studies comparing convalescent and standard plasma therapy reported other than transfusion‐related grade 3 or 4 adverse events.
The remaining RCT (160 participants) investigated the efficacy and safety of convalescent plasma compared to placebo treatment for individuals with pre‐existing co‐morbidities and mild symptoms. We are uncertain about the effect of convalescent plasma therapy on all‐cause mortality (very‐low certainty), the risk for developing severe clinical COVID‐19 symptoms (low‐certainty evidence), or the risk for experiencing severe or serious adverse events (very low‐certainty evidence).
We identified 31 studies (28 RCTs, two expanded assess studies and one NRSI with a planned enrolment of 600 participants) that were completed, terminated or no longer available as expanded access according to the trials registry entry. Of these, we identified the full texts of four studies in our weekly searches, after we submitted this version of the review. We have categorised these studies as 'Awaiting classification'. We will include them in the next version of this living systematic review. For the remaining studies, no outcome data were yet available; study investigators did not reply to our requests, or contact details of principal investigators were not reported in the trials registry. In addition, we identified two platform trials, which do not include our intended interventions. However, we wish to keep track of them, in the event that they may add arms on convalescent plasma or hyperimmune immunoglobulins. Therefore, we categorised these as 'Awaiting classification'. We will consider including them in an update of this review, once results are available.
We identified 100 ongoing studies, of which 85 are RCTs, seven are expanded access studies from the USA, two are single‐arm studies, two are non‐randomised controlled studies and four are pre‐registered observational studies. Of the ongoing studies, 90 are assessing the benefits and safety of convalescent plasma therapy for the treatment of COVID‐19 and ten studies are assessing the benefits and safety of hyperimmune immunoglobulins for the treatment of COVID‐19.
Certainty of the evidence
We included data of nine RCTs to assess effectiveness and safety of convalescent plasma for individuals with a confirmed diagnosis of COVID‐19 disease and moderate to severe symptoms when compared to treatment with placebo or standard care alone. We had high certainty in the identified evidence for effectiveness outcomes, and very low‐ to low certainty in the identified evidence for safety outcomes. Our main concerns were that safety outcomes were assessed and reported in most studies for the convalescent plasma group only, indicating selective reporting and publication bias and serious imprecision due to the small information size with comparative evidence.
We included data of two RCTs to assess effectiveness and safety of convalescent plasma for individuals with a confirmed diagnosis of COVID‐19 disease and moderate to severe symptoms when compared to treatment with standard plasma. We had very low‐ to low certainty in the identified evidence. Our main concerns were serious inconsistency for mortality outcomes, and very serious imprecision due to the small information size, and wide confidence intervals for mortality, and safety outcomes, as well as clinical worsening.
We included data of one RCT to assess effectiveness and safety of convalescent plasma for individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease when compared to treatment with placebo or standard care alone. We had very‐low to low certainty in the identified evidence. Our main concerns were very serious imprecision due to the small information size and serious indirectness for mortality and safety outcomes, because reported outcomes did not precisely match our outcome definition.
Potential biases in the review process
To avoid potential biases in the review process, we had planned to include the best available evidence and adhered to the guidance provided in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2019a) in any step of the review. However, as COVID‐19 is a novel disease, high quality evidence is still rare, although more RCTs became available since the last update (Chai 2020). For this update, we were able to include 12 RCTs and one non‐controlled NRSI. To increase the informative value of our review, we are tracking all registered trials and will continually update this review as more evidence becomes available. There are currently still many new trials being registered in trials registries, as can be seen from the additional 86 RCTs added to the list of ongoing studies in this update of the review.
Two experienced information specialists developed a sensitive search strategy, to identify all ongoing and completed studies. We searched all relevant databases and trials registries, and two review authors conducted all review steps independently and in duplicate.
Another consideration for this rapidly evolving field is the availability of preprint articles that have not yet undergone peer review. In this review, we also included such preprints. However, we are aware of the potentially lower quality of these publications, and investigated robustness of our analysis results in sensitivity analyses.
The necessary adaptation of review methods to the development of research output, as described in Table 4, is in general a potential source of bias in the review process. Since the available evidence changed rapidly in a comparably short period of time in the COVID‐19 pandemic, we needed to take this approach to give a comprehensive answer to the review question. Before starting with an update process, our interdisciplinary team of review authors meets to review the methods and to discuss necessary amendments. We follow the methods we agree upon, before starting with the update, and adhere to these decisions throughout each update process.
For this review update, we introduced separate analyses for ambulatory and hospitalised participants because of clinical heterogeneity between people with asymptomatic to mild symptoms, and people with moderate to severe symptoms. Furthermore, we added in the first version of this review (Valk 2020) that studies with the intervention 'standard immunoglobulin' will be excluded and 'standard immunoglobulin' and 'standard plasma' were added as eligible control treatments because of substantial biological differences of these treatment options, compared to the eligible interventions. We do not think that the bias arising from these adaptations was substantial, since these changes were driven by objective reasons.
For changes in outcomes and outcome measurement, we specified and redefined the outcome 'clinical status (assessed by need for respiratory support)' from self‐set cut‐offs until version 2 of this review (Piechotta 2020b) to standardised scales (WHO 2020e; WHO 2020f) from version 3 (Chai 2020) onward, as they became available later in the course of the COVID‐19 pandemic. We do not think that this change has led to any bias in the review process. Instead, we think that changing the inclusion criteria for outcome measurement to a standardised scale can facilitate identifying studies with objective and higher quality results and, additionally, can contribute to a lower heterogeneity among included studies. We also added a new secondary outcome 'quality of life' from version 2 of this review (Piechotta 2020b) onward, but do not suspect that this had an impact on bias since the outcome was suggested by an external patient representative.
For inclusion criteria regarding different study designs, we tried to anticipate possible changes of the evidence landscape already at protocol stage and therefore excluded study designs of lower level evidence as more RCTs were published. Nonetheless, for this update we decided that we needed to deviate from our previous specification by not excluding prospectively registered single‐arm studies with inclusion of 500 or more participants, even if upcoming RCTs report safety data for both groups. That was because we decided that data of large single‐arm studies like Joyner 2020 could still provide valuable information for safety outcomes and their results could not be neglected in our analyses. To mitigate risk of bias arising from the changes in inclusion criteria, we planned to only use safety data of such studies and to only include prospectively registered studies.
Regarding other changes in methods, it must also be noted that in a previous versions of this review (Piechotta 2020b), we used the former 'Risk of bias' tool to assess risk of bias for RCTs (Higgins 2011). Since the last update (Chai 2020), we assessed RCTs using 'Risk of Bias 2' (Sterne 2019). This led to changes in the risk of bias rating and the GRADE assessment for the outcomes mortality, clinical status and safety outcomes of one included study (Li 2020). We think using the revised 'Risk of Bias 2' tool (Sterne 2019) corrected our judgement from potential personal biases, since it is less sensitive to subjective interpretations.
Agreements and disagreements with other studies or reviews
This update of our systematic review identified moderate‐ to high‐certainty evidence that treatment with convalescent plasma is not effective for the treatment of COVID‐19, in individuals with moderate to severe disease, when compared to placebo treatment or standard care alone. Further, we identified low‐certainty evidence about the effects of convalescent plasma when compared to standard plasma in individuals with moderate to severe COVID‐19 and very low to low‐certainty evidence about the effects of convalescent plasma when compared to placebo or standard care alone in individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease.
To date, there have been published several systematic reviews on effectiveness and safety of convalescent plasma in the treatment of COVID‐19. For example, Janiaud 2021 analysed data of four peer‐reviewed RCTs, including 1,060 participants, and six other publicly available RCTs, including 10,722 participants. All but one included trial (Libster 2020) included participants with moderate to severe disease and they meta‐analysed trials that compared convalescent plasma to placebo and/or standard care. In contrast to our review, they did not differentiate populations by disease severity. However, they already included preliminary published data of Horby 2021, which had a big impact on overall meta‐analysis results, since the study contributed data from 10,406 participants. A meta‐analysis including all 10 RCTs indicated that there is moderate‐certainty evidence for no association of convalescent plasma on all‐cause mortality (RR 1.02, 95% CI 0.92 to 1.12). Furthermore, the analyses including all 10 RCTs showed that convalescent plasma use was not associated with length of hospital stay (HR 1.07, 95% CI 0.79 to 1.45) or the relative risk for the initiation of mechanical ventilation (RR 0.81 95% CI 0.42 to 1.58). Certainty of evidence for both outcomes were rated as low due to imprecision. The authors conducted no meta‐analysis for clinical improvement, clinical deterioration, or serious adverse events, due to inconsistent reporting. In concordance with our results, Janiaud 2021 concluded that convalescent plasma compared to standard care or placebo is not associated with all‐cause mortality or other clinical outcomes that were considered in their analysis. Janiaud 2021 included data from the same studies as we did, but we included additional safety data from one non‐RCT (Joyner 2020).
Klassen 2021 aggregated patient outcome data of 35,055 participants for the outcome mortality with varying length of follow‐up (2‐118 days). They included 10 RCTs, 20 matched‐control studies, two dose‐response studies, and 96 case‐reports or case‐series. They found a 42% reduction in mortality rate of participants treated with convalescent plasma (20%) compared to participants receiving standard treatment (28%), when combining results of RCTs and matched‐control studies (OR 0.58, 95% CI 0.47 to 0.71). When they synthesised results of RCTs only, they found no association between convalescent plasma therapy and mortality (OR 0.76, 95% CI 0.54 to 1.09). In a sensitivity analysis, the authors excluded Agarwal 2020 because approximately 70 percent of participants in the convalescent plasma arm received plasma with an antibody level of less than 1:80. Based on that, they found a reduction of 35% in mortality odds ratio of the plasma arm (11%) compared to the control group (16%) (OR 0.65, 95% CI 0.43 to 0.98). In contrast to our results, Klassen 2021 concluded that there is moderate to high certainty evidence favouring convalescent plasma over standard care.
Klassen 2021 did not include data of Horby 2021, since it was not available in the format of a preprint or peer‐reviewed publication at the time of their publication; this is likely to be one reason for the differences between their results and ours. Further reasons may be the post‐hoc exclusion of Agarwal 2020 , and the inclusion of Rasheed 2020, since they classified the study as a RCT. Rasheed 2020 reported that controls were matched to participants according to the disease stage, age, and sex, and assigned participants to convalescent plasma based on ABO compatibility and limited availability of plasma. In our opinion, this does not fit the criteria for a randomised allocation method and we classified it as a controlled NRSI. We also wrote to the authors of Rasheed 2020 to clarify their methods of randomisation, but their answer did not give any new insights.
The large‐scale clinical administration of convalescent plasma in the USA was regulated under an expanded access programme by the FDA with individual patient authorisation and collection of data (Joyner 2020). The initial purpose of the analysis was to provide data to establish the safety of administration of convalescent plasma (Joyner 2020). However, the data were thought to contain signals of efficacy and therefore re‐analysed, and although the data set did not include any data from a control cohort, the FDA and the USA government considered that there was sufficient evidence of efficacy to widen access to convalescent plasma under the 'Emergency Use Authorization' (EUA) issued on 23 August 2020 (FDA 2020). On 11 February 2021, the FDA revised the EUA of convalescent plasma. The authorization is now limited to the use of high titre convalescent plasma for hospitalised individuals at an early stage of disease (FDA 2021).
On 2 March 2021, the USA National Institutes of Health announced that a large trial of COVID‐19 convalescent plasma for participants with mild symptoms in the emergency department (NCT04355767) was halted because no strong evidence for a benefit of convalescent plasma was found (NIH 2021). They stated that they do not expect a change in results, even if enrolment was continued.
The adverse events associated with plasma transfusions are well characterised. Critically ill participants receiving plasma transfusions have an especially high risk of TACO, which is the leading cause of transfusion‐related mortality (Pandey 2012). Many countries have now introduced risk mitigation strategies to decrease the risk of TRALI. In the UK in 2018, there was only one confirmed case of TRALI. To date, there is no sufficient comparative safety data available for adverse events.
In this systematic review of the literature, which mainly identified studies that included people with COVID‐19 with severe or critical illness, we identified a small proportion of participants experiencing any grade 3 or 4 adverse events, or serious adverse events. With the information available at this moment from published trials registry entries, it is apparent that the majority of clinical trials are enrolling people with COVID‐19 who have progressed to moderate or severe disease. Despite there being some evidence from other infectious diseases that early therapy might be more effective (Mair‐Jenkins 2015), targeting this population is justifiable given the evident lack of effective interventions for COVID‐19. Recent subgroup analysis in RECOVERY (Horby 2021) did not identify strong evidence for a difference between receipt of intervention within seven days of symptom onset or after seven days, and results of our review show that evidence concerning safety and effectiveness of convalescent plasma for individuals with mild disease still is not sufficient.
Authors' conclusions
Implications for practice.
We currently have high certainty in the evidence that convalescent plasma for the treatment of individuals with moderate to severe disease does not reduce mortality and has little to no impact on measures of clinical improvement. We are uncertain about the safety of convalescent plasma in such patients, again when compared to placebo or standard care. Further, we identified very low‐ to low‐certainty evidence about the effects of convalescent plasma when compared to standard plasma in individuals with moderate to severe COVID‐19; very low‐ to low‐certainty evidence about the effects of convalescent plasma when compared to placebo or standard care alone in individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease; and no evidence on effectiveness or safety of hyperimmune immunoglobulins.
Implications for research.
For the fourth version of this living systematic review investigating the use of convalescent plasma or hyperimmune immunoglobulin for people with COVID‐19, we included data from 12 randomised controlled trials (RCTs) reporting on the effectiveness and safety of convalescent plasma, and in total considered the experience of almost 50,000 participants. We did not identify any completed studies that evaluated hyperimmune immunoglobulin, but there are currently still new studies being registered in trials registries. Studies should report outcomes in the same way, and should consider the importance of maintaining comparability in terms of co‐interventions administered in all study arms. There are 100 ongoing studies evaluating convalescent plasma and hyperimmune immunoglobulin and 27 studies reporting in a study registry as being completed, terminated or no longer available as expanded assess. After submitting this version of the review, our weekly searches identified four completed studies with full texts. We will also keep track of two platform trials. Of the 100 ongoing studies, 10 are investigating the effect of hyperimmune immunoglobulins. Publication of the results might dissolve some of the uncertainties around hyperimmune immunoglobulin therapy for people with any severity of disease, and convalescent plasma therapy for people with asymptomatic or mild disease.
What's new
| Date | Event | Description |
|---|---|---|
| 19 March 2021 | New citation required and conclusions have changed | High certainty in the evidence for some of the prioritised outcomes |
| 17 March 2021 | New search has been performed | 12 RCTs and one NRSI included |
History
Review first published: Issue 5, 2020
| Date | Event | Description |
|---|---|---|
| 30 August 2020 | New citation required and conclusions have changed | Additional safety data included (more than 20,000 participants) |
| 30 August 2020 | New search has been performed | Two RCTs, eight controlled NRSIs and nine non‐controlled NRSIs included |
| 3 June 2020 | New citation required and conclusions have changed | We included results from one RCT and three controlled NRSIs and added further safety data from non‐controlled NRSIs. |
| 31 May 2020 | New search has been performed | We included eight new studies. |
Risk of bias
Risk of bias for analysis 1.7 Time to discharge from hospital.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Avendano‐Sola 2020 | Low risk of bias | Participants were randomized through a web‐based eCRF system (ORACLE clinical) in a 1:1 ratio to receive either convalescent plasma in addition to the standard therapy or standard of care alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 81 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Low risk of bias | For the outcome "time to discharge from hospital", there is a low risk of bias for all the domains. |
| Gharbharan 2020 | Low risk of bias | Participants were randomized via a web‐based system in a 1:1 ratio to the current standard of care at each hospital with or without the addition of convalescent plasma. There are no baseline differences that would suggest a problem with randomisation. | Some concerns | Both participants and those delivering the intervention were aware of intervention received, but there was no information on deviations from intended intervention, as insufficient information was provided on whether treatments were balanced across arms. The analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 86 participants randomized. | Low risk of bias | The outcome assessors aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement or that the measurement differed between intervention groups. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Some concerns | For this outcome, there is a low risk of bias from the randomization process, due to missing outcome data, in measurement of the outcome and in selection of the reported result. However, there are some concerns for bias due to deviations from intended interventions. |
| Li 2020 | Low risk of bias | Participants were block randomized via computer‐generated random numbering in a 1:1 ratio to receive standard treatment coupled with convalescent plasma transfusion or standard treatment alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 103 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "time to discharge from hospital", all the domains have a low risk of bias. |
| Ray 2020 | Some concerns | Participants were probably allocated randomly to either the standard of care group alone or standard of care with convalescent plasma group and there were no further information given on the randomization process. There is no information on the allocation concealment, the trial registry only indicates concealment through "Case Record Numbers". There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 80 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Some concerns | For the outcome "time to discharge from hospital" in this study, there is a low risk of bias due to deviations from intended interventions, due to missing outcome data, in measurement of the outcome and in selection of the reported result. There are some concerns for bias from the randomization process. |
| Simonovich 2020 | Low risk of bias | Participants were randomized through a randomization program (REDCap) in a 2:1 ratio to receive either convalescent plasma or a placebo. The allocation sequence was random and concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and carers and people delivering the intervention were unaware of the assigned intervention received and the analysis was appropriate. | Low risk of bias | From the 334 participants randomized data was available for 228 participants in the convalescent plasma group and 105 participants in the placebo group. The data of one participant in control group was missing, due to withdrawal of consent after randomisation and therefore the participant was excluded. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "time to discharge from hospital ", all the domains have a low risk of bias. |
Risk of bias for analysis 1.9 Viral clearance at up to day 3.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Agarwal 2020 | Low risk of bias | The allocation sequence was random and concealed and there were no differences between intervention groups suggesting a problem with the randomization process. | Some concerns | Five participants in convalescent plasma arm and four in control arm did not receive the allocated intervention, which could be due to awareness of the intervention, as this trial was open‐label. According to the study, a per protocol analysis was performed for this secondary outcome, but there was no potential for a substantial impact (on the result) of the failure to analyse randomized participants. | Some concerns | Data for this outcome was available for 367 out of 451 participants included in the per protocol analysis. Data of 51 participants in the convalescent plasma arm and of 46 participants in the control arm were missing and there is no evidence that the result was not biased by missing outcome data. However, there it is not likely that missingness in the outcome depended on its true value. | High risk of bias | There was no information on whether the method of measuring the outcome was inappropriate, as it was unclear how the outcome was exactly measured and the measurement could have differed between intervention groups. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | High risk of bias | For this outcome "viral clearance", there is a low risk of bias from the randomization process and in selection of the reported result, there are some concerns for bias due to deviations from intended interventions and due to missing outcome data. However, there is a high risk in measurement of the outcome. |
| Avendano‐Sola 2020 | Low risk of bias | Participants were randomized through a web‐based eCRF system (ORACLE clinical) in a 1:1 ratio to receive either convalescent plasma in addition to the standard therapy or standard of care alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was not available for all participants randomized, but only for 68 out of 81 participants. 79.7% of serial scheduled samples in the plasma group and 66.54% in the control group were obtained, but it is unlikely that the missingness of outcome data depended on its true value. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Low risk of bias | For the outcome "viral clearance", there is a low risk of bias for all the domains. |
| Hamdy Salman 2020 | Low risk of bias | Participants were block randomized using website software in a 2:1 ratio to receive either convalescent plasma in addition to the standard therapy or standard of care alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and carers and people delivering the intervention were unaware of the assigned intervention received and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 30 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Some concerns | The data that produced this result was not analysed in accordance with the predefined outcomes stated in the trial registration. | Some concerns | For this outcome " viral clearance at day 3", there is a low risk of bias from the randomization process, due to deviations from intended interventions, due to missing outcome data and in measurement of the outcome. However, there are some concerns for bias in selection of the reported result. |
| Li 2020 | Low risk of bias | Participants were block randomized via computer‐generated random numbering in a 1:1 ratio to receive standard treatment coupled with convalescent plasma transfusion or standard treatment alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was not available for all, or nearly all, participants randomized (47/52 participants in plasma group and 40/51 participants in control group) and there is no evidence that the result was not biased by missing outcome data. However, it is unlikely that the missingness in the outcome could depend on its true value. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "viral clearance", there is a low risk of bias in all the domains. |
Risk of bias for analysis 1.10 Viral clearance at up to day 7.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Agarwal 2020 | Low risk of bias | The allocation sequence was random and concealed and there were no differences between intervention groups suggesting a problem with the randomization process. | Some concerns | Five participants in convalescent plasma arm and four in control arm did not receive the allocated intervention, which could be due to awareness of the intervention, as this trial was open‐label. According to the study, a per protocol analysis was performed for this secondary outcome, but there was no potential for a substantial impact (on the result) of the failure to analyse randomized participants. | Some concerns | Data for this outcome was available for 342 out of 451 participants included in the per protocol analysis and there is no evidence that the result was not biased by missing outcome data. However, there it is not likely that missingness in the outcome depended on its true value. | High risk of bias | There was no information on whether the method of measuring the outcome was inappropriate, as it was unclear how the outcome was exactly measured and the measurement could have differed between intervention groups. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | High risk of bias | For this outcome "viral clearance", there is a low risk of bias from the randomization process and in selection of the reported result, there are some concerns for bias due to deviations from intended interventions and due to missing outcome data. However, there is a high risk in measurement of the outcome. |
| Avendano‐Sola 2020 | Low risk of bias | Participants were randomized through a web‐based eCRF system (ORACLE clinical) in a 1:1 ratio to receive either convalescent plasma in addition to the standard therapy or standard of care alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was not available for all participants randomized, but only for 56 out of 81 participants. 79.7% of serial scheduled samples in the plasma group and 66.54% in the control group were obtained, but it is unlikely that the missingness of outcome data depended on its true value. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Low risk of bias | For the "viral clearance" in this study, there is a low risk of bias for all the domains. |
| Li 2020 | Low risk of bias | Participants were block randomized via computer‐generated random numbering in a 1:1 ratio to receive standard treatment coupled with convalescent plasma transfusion or standard treatment alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was not available for all, or nearly all, participants randomized (47/52 participants in plasma group and 40/51 participants in control group) and there is no evidence that the result was not biased by missing outcome data. However, it is unlikely that the missingness in the outcome could depend on its true value. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "viral clearance", there is a low risk of bias in all the domains. |
Risk of bias for analysis 1.11 Viral clearance at up to day 15.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Avendano‐Sola 2020 | Low risk of bias | Participants were randomized through a web‐based eCRF system (ORACLE clinical) in a 1:1 ratio to receive either convalescent plasma in addition to the standard therapy or standard of care alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was not available for all participants randomized, but only for 62 out of 81 participants. 79.7% of serial scheduled samples in the plasma group and 66.54% in the control group were obtained, but it is unlikely that the missingness of outcome data depended on its true value. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Low risk of bias | For the "viral clearance", there is a low risk of bias for all the domains. |
| Li 2020 | Low risk of bias | Participants were block randomized via computer‐generated random numbering in a 1:1 ratio to receive standard treatment coupled with convalescent plasma transfusion or standard treatment alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was not available for all, or nearly all, participants randomized (47/52 participants in plasma group and 40/51 participants in control group) and there is no evidence that the result was not biased by missing outcome data. However, it is unlikely that the missingness in the outcome could depend on its true value. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For the outcome "viral clearance", there is a low risk of bias in all the domains. |
Risk of bias for analysis 1.13 Any grade adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.13.1 Individuals with moderate disease | ||||||||||||
| Simonovich 2020 | Low risk of bias | Participants were randomized through a randomization program (REDCap) in a 2:1 ratio to receive either convalescent plasma or a placebo. The allocation sequence was random and concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and carers and people delivering the intervention were unaware of the assigned intervention received and the analysis was appropriate. | Low risk of bias | From the 334 participants randomized, data was available for 228 participants in the convalescent plasma group and 104 participants in the placebo group. The data of two participants in control group was missing, of which one missing participants was due to withdrawal of consent after randomisation and therefore that participant was excluded from the analysis. | Low risk of bias | The outcome measurement is an appropriate measure and it is unlikely that the measurement differed between intervention groups. The outcome assessors were not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For this outcome "Any grade adverse events", there is a low risk of bias for all the domains. |
Risk of bias for analysis 1.15 Serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.15.1 Individuals with moderate disease | ||||||||||||
| Avendano‐Sola 2020 | Low risk of bias | Participants were randomized through a web‐based eCRF system (ORACLE clinical) in a 1:1 ratio to receive either convalescent plasma in addition to the standard therapy or standard of care alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 81 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Low risk of bias | For this outcome "Serious adverse events", there is a low risk of bias for all the domains. |
| Simonovich 2020 | Low risk of bias | Participants were randomized through a randomization program (REDCap) in a 2:1 ratio to receive either convalescent plasma or a placebo. The allocation sequence was random and concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and carers and people delivering the intervention were unaware of the assigned intervention received and the analysis was appropriate. | Low risk of bias | From the 334 participants randomized, data was available for 228 participants in the convalescent plasma group and 105 participants in the placebo group. The data of one participant in control group was missing, due to withdrawal of consent after randomisation and therefore the participant was excluded from the analysis. | Low risk of bias | The outcome measurement is an appropriate measure and it is unlikely that the measurement differed between intervention groups. The outcome assessors were not aware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For this outcome "Serious adverse events", there is a low risk of bias for all the domains. |
Risk of bias for analysis 2.1 All‐cause mortality at up to day 28.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Bajpai 2020 | Low risk of bias | Participants were block randomized with concealed allocation using the Sequentially Numbered Opaque Sealed Envelopes (SNOSE) method, to receive either convalescent plasma with standard of care or standard plasma (fresh frozen plasma) with standard of care. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for nearly all participants randomized, in total 29 out of 31 participants (2 participants became PCR negative on the day of plasma transfusion). | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Low risk of bias | For this outcome "mortality at day 28", there is a low risk of bias for all the domains. |
| O’Donnell 2021 | Low risk of bias | Participants were block randomized using a web‐based randomization platform in a 2:1 ratio to receive either convalescent plasma or standard plasma and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and people delivering the intervention were unaware of the assigned intervention received and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 223 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Low risk of bias | For the outcome "28‐day mortality", there is a low risk of bias for all the domains. |
Risk of bias for analysis 2.2 Clinical worsening: need for invasive mechanical ventilation.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Bajpai 2020 | Low risk of bias | Participants were block randomized with concealed allocation using the Sequentially Numbered Opaque Sealed Envelopes (SNOSE) method, to receive either convalescent plasma with standard of care or standard plasma (fresh frozen plasma) with standard of care. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were aware of intervention received, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for nearly all participants randomized, in total 29 out of 31 participants (2 participants became PCR negative on the day of plasma transfusion). | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Low risk of bias | For this outcome "Clinical worsening: Need for invasive mechanical ventilation", there is a low risk of bias for all the domains. |
Risk of bias for analysis 2.3 Duration of hospitalisation.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Bajpai 2020 | Low risk of bias | Participants were block randomized with concealed allocation using the Sequentially Numbered Opaque Sealed Envelopes (SNOSE) method, to receive either convalescent plasma with standard of care or standard plasma (fresh frozen plasma) with standard of care. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | ‐ | Low risk of bias | ‐ | Low risk of bias | ‐ | Low risk of bias | ‐ | Low risk of bias | ‐ |
Risk of bias for analysis 2.4 Any grade adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| O’Donnell 2021 | Low risk of bias | Participants were block randomized using a web‐based randomization platform in a 2:1 ratio to receive either convalescent plasma or standard plasma and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and people delivering the intervention were unaware of the assigned intervention received and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for nearly all (219 out of 223) participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Low risk of bias | For the outcome "any adverse events", there is a low risk of bias for all the domains. |
Risk of bias for analysis 2.5 Serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| O’Donnell 2021 | Low risk of bias | Participants were block randomized using a web‐based randomization platform in a 2:1 ratio to receive either convalescent plasma or standard plasma and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and people delivering the intervention were unaware of the assigned intervention received and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for nearly all (219 out of 223) participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the predefined outcomes stated in the trial registration. | Low risk of bias | For the outcome "serious adverse events", there is a low risk of bias for all the domains. |
Risk of bias for analysis 3.1 All‐cause mortality.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Libster 2020 | Low risk of bias | Participants were randomized using an electronic system numbering to receive either convalescent plasma in addition to the standard therapy or standard of care alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | The participants were unaware of their assigned intervention and those delivering the intervention were aware of intervention assigned during the trial, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 160 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were probably unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For this outcome "all‐cause mortality", there is a low risk of bias for all the domains. |
Risk of bias for analysis 3.2 Development of severe symptoms: need for invasive mechanical ventilation.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Libster 2020 | Low risk of bias | Participants were randomized using an electronic system numbering to receive either convalescent plasma in addition to the standard therapy or standard of care alone and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | The participants were unaware of their assigned intervention and those delivering the intervention were aware of intervention assigned during the trial, but there were no deviations from intended interventions and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all 160 participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were probably unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For this outcome "Development of severe symptoms", there is a low risk of bias for all the domains. |
Acknowledgements
This review was published in collaboration with the Cochrane Editorial and Methods Department. We particularly thank Sarah Hodgkinson and Liz Bickerdike (Associate Editors, Cochrane Editorial and Methods Department), Mike Brown (Network Senior Editor, Cochrane Acute and Emergency Care), Clare Dooley (Managing Editor), Hacsi Horvath (Copy Editor) and Denise Mitchell (Plain Language Summary writer) for their excellent support. Thanks also to the Cochrane Editorial and Methods Department team, for their valuable comments on the review and timely management of the editorial process.
We thank Theresa Moore (Methodology Editor, Editorial and Methods Department) for reviewing our risk of bias assessments and the implementation of RoB 2.0, Robin Featherstone (Information Specialist, Cochrane Editorial and Methods Department) for commenting on the search strategy, and Gerald Gartlehner and Adrienne Stevens for their advice on rapid review methodology for previous versions of this review. We thank Carolyn Dorée (Information Specialist, Systematic Review Initiative, NHS Blood and Transplant Oxford) for developing the original search strategy for the first published review version. We thank Analysis of Review Group Output (ARGO) for their comments on the Abstract.
We thank the investigators of eight studies (Agarwal 2020; AlQahtani 2020; Avendano‐Sola 2020; Balcells 2020; Gharbharan 2020; Horby 2021; Li 2020; and Rasheed 2020) for providing us with additional information and data.
We thank all external peer reviewers who read and commented on this review. We thank Miquel Lozano (MD, PhD University Clinic Hospital, University of Barcelona, Spain), and Dr Michael James Ankcorn (Department of Virology, Northern General Hospital, Sheffield Teaching Hospitals NHS Foundation Trust, UK), who greatly helped to improve this review.
We thank Rujan Shrestha and Ya‐Ying Wang for translating and assessing articles in Chinese language, and Lev E. Korobchenko for translating and assessing articles in Russian language for us via Cochrane TaskExchange.
The research was supported by NHS Blood and Transplant and the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 101015756. The contents of this document reflect only the author's view and the Commission is not responsible for any use that may be made of the information it contains.
Appendices
Appendix 1. Planned methodology for study designs that are no longer included in this systematic review
Criteria for considering studies for this review
Types of studies
In case of insufficient evidence available from RCTs, we had planned to include prospective controlled non‐randomised studies of interventions (NRSIs), including quasi‐randomised controlled trials (e.g. assignment to treatment by alternation or by date of birth), controlled before‐and‐after (CBA) studies, and interrupted time series (ITS) studies. We had planned to use the methods proposed in the Cochrane Handbook for Systematic Reviews of Interventions for the inclusion of controlled NRSIs in systematic reviews (Reeves 2019).
We had further planned to include retrospective controlled NRSIs, in case of insufficient evidence (very low‐certainty evidence or no evidence) available from RCTs and prospective controlled NRSIs and to adapt the methods for the inclusion of controlled NRSIs in systematic reviews as specified by the Cochrane Handbook for Systematic Reviews of Interventions (Reeves 2019).
In case the evidence that we found from RCTs was at high risk of bias and at critical risk of bias for the controlled NRSIs for safety outcomes, we had planned to also included safety data from prospectively and retrospectively registered non‐controlled NRSIs, for example, case series, and followed the methodology as specified in the protocol (Piechotta 2020a).
Data collection and analysis
Assessment of risk of bias in included studies
Controlled non‐randomised studies of interventions
As reported above, we had planned to include controlled non‐randomised studies of intervention (NRSI) trials if there was insufficient evidence from RCTs.
Two review authors (VP, NS) would have independently assessed eligible studies for methodological quality and risk of bias (using the Risk Of Bias in Non‐randomised Studies ‐ of Interventions (ROBINS‐I) tool; Sterne 2016). The quality assessment strongly depends upon information on the design, conduct and analysis of the trial. The two review authors would have resolved any disagreements regarding quality assessments by discussion, and in case of discrepancies among their judgements, or inability to reach consensus, we had planned to consult a third review author until consensus could be reached. We asked the Cochrane Editorial and Methods Department (Theresa Moore) to review our judgements for reasonability for previous versions of this review. The categories for 'Risk of bias' judgements for controlled NRSIs using ROBINS‐I are 'low risk', 'moderate risk', 'serious risk' and 'critical risk' of bias.
We had planned to assess the following domains of bias.
Bias due to confounding
Bias in selection of participants into the study
Bias in classification of interventions
Bias due to deviations from intended interventions
Bias due to missing data
Bias in measurement of outcomes
Bias in selection of the reported result
For every criterion we had planned to make a judgement using one of five response options.
Yes
Probably yes
Probably no
No
No information
Measures of treatment effect
Controlled non‐randomised studies of interventions
For dichotomous outcomes, if available, we had planned to extract and report the RR with a 95% CI from statistical analyses adjusting for baseline differences (such as Poisson regressions or logistic regressions) or the ratio of RRs (i.e. the RR post‐intervention/RR pre‐intervention).
For continuous variables, if available, we had planned to extract and report the absolute change from a statistical analysis adjusting for baseline differences (such as regression models, mixed models or hierarchical models), or the relative change adjusted for baseline differences in the outcome measures (i.e. the absolute post‐intervention difference between the intervention and control groups, as well as the absolute pre‐intervention difference between the intervention and control groups/the post‐intervention level in the control group; EPOC 2017).
Data synthesis
We had planned to not synthesise efficacy data from controlled NRSIs if they were at critical risk of bias. If a meta‐analysis had been feasible for controlled NRSIs we had planned to analyse the different types of studies separately. We had planned to only analyse outcomes with adjusted effect estimates if these were adjusted for the same factors using the inverse‐variance method as recommended in Chapter 24 of the Cochrane Handbook for Systematic Reviews of Interventions (Reeves 2019).
Summary of findings and assessment of the certainty of the evidence
As we had planned to use the ROBINS‐I tool to assess risk of bias for controlled NRSIs, we had planned to follow GRADE guidance 18 to rate the certainty in the evidence for controlled NRSIs; starting from high‐certainty evidence with the opportunity to downgrade by three points for critical risk of bias (Schünemann 2019).
Appendix 2. Search strategy MEDLINE
# Searches
1 Coronavirus Infections/ or Coronavirus/
2 SARS‐CoV‐2/ or COVID‐19/
3 ("2019 nCoV" or 2019nCoV or coronavir* or coronovir* or COVID or COVID19 or HCoV* or "nCov 2019" or "SARS CoV2" or "SARS CoV 2" or SARSCoV2 or "SARSCoV 2").tw,kf.
4 ((corona* or corono*) adj1 (virus* or viral* or virinae*)).tw,kf.
5 "severe acute respiratory syndrome coronavirus 2".tw,kf,nm.
6 (anti‐flu* or anti‐influenza* or antiflu* or antinfluenza*).tw,kf.
7 or/1‐6
8 Plasma/
9 Immunoglobulins/
10 Immunoglobulins, Intravenous/
11 Immune Sera/
12 ((convalesc* or recovered or cured or rehabilitat* or survivor* or survived or virus‐positive or virus neutrali* or virus inactivated or antibod* or high‐titre* or high‐titer*) adj6 (plasma or blood or serum or sera)).mp.
13 ((plasma adj1 therap*) or gamma‐globulin* or "γ‐Globulin" or hyper‐Ig).tw,kf.
14 (hyperimmune* or hyper‐immune*).mp.
15 (high‐dos* adj3 (plasma or immunoglobulin* or IVIG* or immune globulin* or globulin* or IgG)).tw,kf.
16 (plasma adj5 (immun* or antibod* or exchange* or donor* or donat* or transfus* or infus*)).mp.
17 ((convalesc* or recovered or cured or rehabilitat* or survivor* or survived or virus‐positive or virus inactivated or antibody‐positive) adj5 (donor* or donat*)).mp.
18 (serum or sera or serotherap* or sero‐therap*).tw,kf.
19 exp Immunization, Passive/
20 (passiv* adj3 (antibod* transfer* or immunization* or immunotherap* or immuno‐therap* or vaccin*)).tw,kf.
21 ((immunoglobulin* or immune globulin*) adj2 (therap* or treatment* or neutrali?ing or prevent* or protect* or prophylax*)).tw,kf.
22 hIVIG.tw,kf.
23 (XAV‐19 or SAB‐185 or equine or INM005 or CSL760).tw,kf.
24 (IGY‐110 or IGY110 or GIGA‐2050 or GIGA2050).tw,kf.
25 (GC5131 or 5131A).tw,kf.
26 (((anti‐coronavirus or anticoronavirus) adj1 immunoglobulin*) or ITAC).tw,kf.
27 ("Hyperimmune anti‐COVID‐19 IVIG" or C‐IVIG or CIVIG).tw,kf.
28 (equine polyclonal antibod* or EpAbs).tw,kf.
29 Flebogamma.tw,kf.
30 or/8‐29
31 7 and 30
32 "Covid‐19 Serotherapy".px.
33 (Flu‐IVIG or ((anti‐flu* or anti‐influenza* or antiflu* or antinfluenza*) adj5 plasma)).mp.
34 or/31‐33
35 exp cohort studies/ or exp epidemiologic studies/ or exp clinical trial/ or exp evaluation studies as topic/ or exp statistics as topic/
36 ((control and (group* or study)) or (time and factors) or program or survey* or ci or cohort or comparative stud* or evaluation studies or follow‐up*).mp.
37 or/35‐36
38 (animals/ not humans/) or comment/ or editorial/ or exp review/ or meta analysis/ or consensus/ or exp guideline/
39 hi.fs. or case report.mp.
40 or/38‐39
41 37 not 40
42 randomized controlled trial.pt.
43 controlled clinical trial.pt.
44 randomi?ed.ab.
45 placebo.ab.
46 drug therapy.fs.
47 randomly.ab.
48 trial.ab.
49 groups.ab.
50 or/42‐49
51 exp animals/ not humans/
52 50 not 51
53 clinical trial, phase iii/
54 ("Phase 3" or "phase3" or "phase III" or P3 or "PIII").ti,ab,kw.
55 (53 or 54) not 51
56 52 or 55
57 34 and (41 or 56)
58 limit 57 to yr="2019 ‐Current"
Appendix 3. Search strategy Embase
# Searches
1 coronavirinae/ or coronaviridae/ or coronaviridae infection/
2 coronavirus disease 2019/
3 Coronavirus infection/
4 sars‐related coronavirus/
5 "Severe acute respiratory syndrome coronavirus 2"/
6 ((corona* or corono*) adj1 (virus* or viral* or virinae*)).tw,kw.
7 ("2019 nCoV" or 2019nCoV or coronavir* or coronovir* or COVID or COVID19 or HCoV* or "nCov 2019" or "SARS CoV2" or "SARS CoV 2" or SARSCoV2 or "SARSCoV 2").tw,kw.
8 "Severe acute respiratory syndrome coronavirus 2".mp.
9 (anti‐flu* or anti‐influenza* or antiflu* or antifluenza*).tw,kw.
10 or/1‐9
11 Plasma Transfusion/
12 exp Immunoglobulin/
13 ((convalesc* or recovered or cured or survivor* or survived or rehabilitat* or virus‐positive or virus‐neutrali* or virus inactived or antibody‐rich or high‐tire* or high‐titer*) adj6 (plasma or blood or serum or sera)).mp.
14 ((plasma adj1 therap*) or gamma‐globulin or "y‐Globulin" or hyper‐lg).tw,kw.
15 (plasma adj5 (immun* or antibod* or exchange* or donor* or donat* or transfus* or infus*)).mp.
16 (hyperimmune* or hyper‐immune*).mp.
17 (high‐dos* adj3 (plasma or immunoglobulin* or IVIG* or immune globulin* or globulin* or IgG)).tw,kw.
18 ((convalesc* or recovered or cured or rehabilitat* or survivor* or survived or virus‐positive or virus inactivated or antibody‐positive) adj5 (donor* or donat*)).mp.
19 (serum or sera or serotherap* or sero‐therap*).tw,kw.
20 passive immunization/
21 (passiv* adj3 (antibod* transfer* or immuni?ation* or immunotherap* or immuno‐therap* or immunit* transfer* or vaccin*)).tw,kw.
22 passive immunit*.tw,kw.
23 ((immunoglobulin* or immune globulin*) adj2 (therap* or treatment* or neutrali?ing or prevent* or protect* or prophylax*)).tw,kw.
24 hIVIG.tw,kw.
25 (CSL760 or INM005 or XAV‐19 or SAB‐185 or equine).tw,kw.
26 (IgY‐110 or IgY110 or GIGA‐2050 or GIGA2050).tw,kw.
27 (GC5131 or 5131A).tw,kw.
28 (((anti‐coronavirus or anticoronavirus) adj1 immunoglobulin*) or ITAC).tw,kw.
29 ("Hyperimmune anti‐COVID‐19 IVIG" or C‐IVIG or CIVIG).tw,kw.
30 (equine polyclonal antibod* or EpAbs).tw,kw.
31 flebogamma.tw,kw.
32 or/11‐31
33 (Flu‐IVIG or ((anti‐flu* or anti‐influenza* or antiflu* or antinfluenza*) adj5 plasma)).mp.
34 Clinical study/
35 (cross sectional adj (study or studies)).tw.
36 exp longitudinal study/
37 exp prospective study/
38 exp follow up/
39 cohort*.tw.
40 exp case control study/
41 (case* and control*).tw.
42 or/34‐41
43 Randomized controlled trial/
44 Controlled clinical study/
45 random*.ti,ab.
46 randomization/
47 intermethod comparison/
48 placebo.ti,ab.
49 (compare or compared or comparison).ti.
50 (open adj label).ti,ab.
51 ((double or single or doubly or singly) adj (blind or blinded or blindly)).ti,ab.
52 double blind procedure/
53 parallel group$1.ti,ab.
54 (crossover or cross over).ti,ab.
55 ((assign$ or match or matched or allocation) adj5 (alternate or group$1 or intervention$1 or patient$1 or subject$1 or participant$1)).ti,ab.
56 (controlled adj7 (study or design or trial)).ti,ab.
57 (volunteer or volunteers).ti,ab.
58 trial.ti.
59 or/43‐58
60 phase 3 clinical trial/
61 ("Phase 3" or "phase3" or "phase III" or P3 or "PIII").tw,kw.
62 or/60‐61
63 (10 and 32) or 33
64 63 and (42 or 59 or 62)
65 limit 64 to yr="2019 ‐Current"
Appendix 4. Search strategy Cochrane COVID‐19 Study Register
plasma OR convalesc* OR serum OR sera OR donor* OR donation* OR serotherapy OR "sero‐therapy" OR "flu‐IVIG" OR antiflu* OR "anti‐flu" OR hyperimmune* OR hyper‐immune* OR IVIG OR immunoglobulin OR "immune‐globulin" OR globulin OR "gamma‐globulin" OR "γ‐Globulin" OR "hyper‐Ig" OR immunization OR immunisation OR immunotherap* OR CSL760 OR INM005 OR equine OR "XAV‐19" OR "SAB‐185" OR hIVIG OR equine OR INOSARS OR "GIGA‐2050" or GIGA2050 OR "IGY‐110" OR IGY1109 OR "GC5131" OR "5131A" OR ITAC OR "C‐IVIG" OR CIVIG OR flebogamma OR EpAbs
Limits: treatment and management
Appendix 5. Search strategy PubMed
#1 "2019 ncov"[Title/Abstract] OR "2019nCoV"[Title/Abstract] OR "corona virus"[Title/Abstract] OR "corona viruses"[Title/Abstract] OR "Coronavirus"[Title/Abstract] OR "coronaviruses"[Title/Abstract] OR "COVID"[Title/Abstract] OR "COVID19"[Title/Abstract] OR "ncov 2019"[Title/Abstract] OR "SARS‐CoV2"[Title/Abstract] OR "SARS‐CoV‐2"[Title/Abstract] OR "SARSCoV2"[Title/Abstract] OR "SARSCoV‐2"[Title/Abstract] OR "COVID‐19"[MeSH Terms] OR "Coronavirus"[MeSH Terms:noexp] OR "SARS‐CoV‐2"[MeSH Terms] OR "COVID‐19"[Supplementary Concept] OR "severe acute respiratory syndrome coronavirus 2"[Supplementary Concept]
#2 ("convalesc*"[Title/Abstract] OR "recovered"[Title/Abstract] OR "cured"[Title/Abstract] OR "rehabilitat*"[Title/Abstract] OR "survivor*"[Title/Abstract] OR "survived"[Title/Abstract] OR "virus‐positive"[Title/Abstract] OR "virus neutrali*"[Title/Abstract] OR "virus inactivated"[Title/Abstract] OR "antibod*"[Title/Abstract] OR "high titre*"[Title/Abstract] OR "high titer*"[Title/Abstract]) AND ("plasma"[Title/Abstract] OR "blood"[Title/Abstract] OR "donor*"[Title/Abstract] OR "donat*"[Title/Abstract])
#3 ("plasma"[Title] AND ("immun*"[Title/Abstract] OR "transfus*"[Title/Abstract] OR "infus*"[Title/Abstract]))
#4 ("high dos*"[Title/Abstract] AND ("plasma"[Title/Abstract] OR "immunoglobulin*"[Title/Abstract] OR "ivig*"[Title/Abstract] OR "immune globulin*"[Title/Abstract] OR "globulin*"[Title/Abstract] OR "IgG"[Title/Abstract])
#5 "serum"[Title] OR "sera"[Title] OR "immunization, passive"[MeSH Terms] OR "hyperimmune"[Title/Abstract] OR "hyperimmunity"[Title/Abstract] OR "serotherap*"[Title/Abstract] OR "sero therap*"[Title/Abstract] OR "therapeutic plasma"[Title/Abstract] OR "plasma therapy"[Title/Abstract] OR "immune plasma"[Title/Abstract] OR "plasma exchange"[Title/Abstract] OR "gamma globulin*"[Title/Abstract] OR "gamma‐Globulin"[Title/Abstract] OR "hyper‐Ig"[Title/Abstract]
#6 ("passiv*"[Title/Abstract] AND (("antibod*"[All Fields] AND "transfer*"[All Fields]) OR "immunisation*"[All Fields] OR "vaccin*"[Title/Abstract] OR "immunization*"[All Fields] OR "immunotherap*"[All Fields] OR "immuno therap*"[All Fields] OR "vaccin*"[All Fields])
#7 ("immunoglobulin*"[Title] OR "immune globulin*"[Title]) AND ("therap*"[Title/Abstract] OR "treat*"[Title/Abstract] OR "prevent*"[Title/Abstract] OR "protect*"[Title/Abstract] OR "prophylax*"[Title/Abstract]))
#8 ("equine*"[Title/Abstract] OR "hivig*"[Title/Abstract] OR "c ivig*"[Title/Abstract] OR "XAV‐19"[Title/Abstract] OR "5131A"[Title/Abstract] OR "equine polyclonal antibod*"[Title/Abstract] OR "EpAbs"[Title/Abstract] OR "flebogamma*"[Title/Abstract] OR (("anti‐coronavirus"[Title/Abstract] OR "anticoronavirus"[Title/Abstract]) AND "immunoglobulin*"[Title/Abstract]))
#9 (("anti‐coronavirus"[Title/Abstract] OR "anticoronavirus"[Title/Abstract]) AND "immunoglobulin*"[Title/Abstract])
#10 #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9
#11 (("anti flu*"[Title/Abstract] OR "anti influenza*"[Title/Abstract] OR "antiflu*"[Title/Abstract] OR "antinfluenza*"[Title/Abstract]) AND ("plasma*"[Title/Abstract])) OR ("flu ivig*"[Title/Abstract])
#12 #1 AND #10
#13 #11 OR #12
#14 ("animals"[MeSH Terms] NOT "humans"[MeSH Terms])
#15 (publisher[sb] OR inprocess[sb] OR pubmednotmedline[sb])
#16 #13 NOT #14
#17 #15 AND #16 Filters: from 2020/1/1 ‐ 3000/12/12
Appendix 6. Search strategy ‐ World Health Organization COVID‐19 Global literature on coronavirus disease
Advanced search; search fileds: title, abstract, subject
Search 1: (plasma OR convalesc* OR serum OR sera OR donor* OR donation* OR serotherapy OR "sero‐therapy" OR "flu‐IVIG" OR antiflu* OR "anti‐flu" OR hyperimmune* OR hyper‐immune* OR IVIG) AND (random* OR placebo OR RCT)
Search 2: (immunoglobulin OR immune‐globulin OR globulin OR gamma‐globulin OR γ‐Globulin OR hyper‐Ig) AND (random* OR placebo OR RCT)
Search 3: ( immunization OR immunisation OR immunotherap*) AND (random* OR placebo OR RCT)
Search 4: ( CSL760 OR INM005 OR equine OR "XAV‐19" OR "SAB‐185" OR hIVIG) AND (random* OR placebo OR RCT)
Search 5: (INOSARS OR "GIGA‐2050" OR "GIGA2050" OR "IGY‐110" OR "IGY1109" OR GC5131 OR 5131A OR ITAC OR C‐IVIG OR CIVIG OR flebogamma OR EpAbs) AND (random* OR placebo OR RCT)
Appendix 7. Search strategy ‐ Epistemonikos, L*OVE List Coronavirus disease (COVID‐19)
by pico search: population: Covid‐19: intervention: passive immunization
Prevention or treatment: passive immunization: convalescent plasma: primary studies: RCTs
Prevention or treatment: passive immunization: Immunoglobulin therapy: primary studies: RCTs
Prevention or treatment: passive immunization: heterologous antibodies: primary studies: RCTs
Appendix 8. Cochrane Childhood Cancer assessment tool for observational studies 'Risk of bias' assessment for: convalescent plasma for people with COVID‐19 (safety)
We assessed methodological quality and risk of bias using the 'Risk of bias' assessment criteria for observational studies tool provided by Cochrane Childhood Cancer (see Table 6; Mulder 2019).
| 'Risk of bias' assessment of Joyner 2020 | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Representative study group (selection bias) | Adverse events | Low | Large population size, prospective study, interim analysis of safety data for 20,000 participants (out of 35,322 participants) |
| Outcome detectors blinded to intervention (detection bias) | Adverse events | Low | Assessment of outcome probably not biased through awareness of intervention |
| Complete outcome assessment/follow‐up (attrition bias) | Adverse events | Low | Interim results; serious adverse events assessed over 7 days after transfusion |
| Well‐defined study group (reporting bias) | Adverse events | Low | Study population and intervention well described |
| Well‐defined outcome (reporting bias) | Adverse events | Low | Interim results for 20,000 participants; assessed and reported for all participants |
| Well‐defined risk estimates (analyses) | Adverse events | Low | Quote: "cumulative incidence of each of a series of SAEs [serious adverse events] was summarised using a point estimate and 95% score confidence interval (CI)" |
| Important prognostic factors or follow‐up taken adequately into account (confounding) | Adverse events | Low | Not adjusted, but confounding unlikely |
Data and analyses
Comparison 1. Convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 All‐cause mortality at up to day 28 | 7 | 12646 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.92, 1.05] |
| 1.1.1 Individuals with moderate disease | 4 | 907 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.68, 1.41] |
| 1.1.2 Individuals with severe disease | 1 | 101 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.29, 1.46] |
| 1.1.3 Individuals with moderate or severe disease | 2 | 11638 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.92, 1.05] |
| 1.2 Mortality (time to event) | 5 | 12160 | Hazard Ratio (IV, Random, 95% CI) | 0.99 [0.92, 1.07] |
| 1.2.1 Individuals with moderate disease | 1 | 333 | Hazard Ratio (IV, Random, 95% CI) | 0.93 [0.47, 1.85] |
| 1.2.2 Individuals with severe disease | 2 | 189 | Hazard Ratio (IV, Random, 95% CI) | 0.64 [0.33, 1.25] |
| 1.2.3 Individuals with moderate or severe disease | 2 | 11638 | Hazard Ratio (IV, Random, 95% CI) | 1.00 [0.93, 1.07] |
| 1.3 All‐cause mortality at hospital discharge | 3 | 577 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.53, 1.53] |
| 1.3.1 Individuals with moderate disease | 2 | 491 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.71, 1.76] |
| 1.3.2 Individuals with severe disease | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.55 [0.22, 1.34] |
| 1.4 Clinical improvement: liberation from supplemental oxygen in surviving patients, for subgroup of participants requiring any supplemental oxygen or ventilator support at baseline | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.81, 1.48] |
| 1.5 Clinical improvement: weaning or liberation from invasive mechanical ventilation in surviving patients, for subgroups of participants requiring invasive mechanical ventilation at baseline | 2 | 630 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.57, 1.93] |
| 1.6 Clinical worsening: need for invasive mechanical ventilation, for subgroup of participants not requiring invasive mechanical ventilation at baseline | 4 | 11765 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.89, 1.08] |
| 1.7 Time to discharge from hospital | 5 | 683 | Hazard Ratio (IV, Random, 95% CI) | 1.15 [0.95, 1.40] |
| 1.8 Admission to the intensive care unit (ICU) | 1 | 333 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.74, 1.09] |
| 1.9 Viral clearance at up to day 3 | 4 | 552 | Risk Ratio (M‐H, Random, 95% CI) | 1.73 [0.98, 3.04] |
| 1.10 Viral clearance at up to day 7 | 3 | 485 | Risk Ratio (M‐H, Random, 95% CI) | 1.55 [0.99, 2.43] |
| 1.11 Viral clearance at up to day 15 | 2 | 149 | Risk Ratio (M‐H, Random, 95% CI) | 1.59 [0.74, 3.43] |
| 1.12 Need for dialysis | 1 | 11442 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.87, 1.22] |
| 1.13 Any grade adverse events | 1 | 332 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.89, 1.26] |
| 1.13.1 Individuals with moderate disease | 1 | 332 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.89, 1.26] |
| 1.14 Grade 3 and 4 adverse events | 4 | 905 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.58, 1.41] |
| 1.14.1 Individuals with moderate disease | 4 | 905 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.58, 1.41] |
| 1.15 Serious adverse events | 2 | 414 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.81, 1.90] |
| 1.15.1 Individuals with moderate disease | 2 | 414 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.81, 1.90] |
Comparison 2. Convalescent plasma versus standard plasma for individuals with moderate to severe disease.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 All‐cause mortality at up to day 28 | 2 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.17, 5.29] |
| 2.2 Clinical worsening: need for invasive mechanical ventilation | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 3.21 [0.38, 27.40] |
| 2.3 Duration of hospitalisation | 1 | 29 | Mean Difference (IV, Random, 95% CI) | ‐4.00 [‐7.56, ‐0.44] |
| 2.4 Any grade adverse events | 1 | 219 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.93, 1.49] |
| 2.5 Serious adverse events | 1 | 219 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.49, 1.11] |
Comparison 3. Convalescent plasma versus placebo or standard care alone for individuals with mild disease.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 All‐cause mortality | 1 | 160 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.09, 2.65] |
| 3.2 Development of severe symptoms: need for invasive mechanical ventilation | 1 | 160 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.09, 2.65] |
| 3.3 Admission to the intensive care unit (ICU) | 1 | 160 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.07, 1.60] |
Comparison 4. Subgroup analysis: duration since symptom onset for the comparison of convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 All‐cause mortality at up to day 28 | 1 | 11552 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.88, 1.10] |
| 4.1.1 Duration of symptom onset up to 7 days | 1 | 4466 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.84, 1.02] |
| 4.1.2 Duration of symptom onset more than 7 days | 1 | 7086 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.95, 1.14] |
Comparison 5. Subgroup analysis: antibodies in recipients detected at baseline for the comparison of convalescent plasma versus placebo or standard care alone for individuals with moderate to severe disease.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 All‐cause mortality at up to day 28 | 1 | 9385 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.89, 1.09] |
| 5.1.1 Antibodies detected at baseline | 1 | 5774 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.93, 1.16] |
| 5.1.2 No antibodies detected at baseline | 1 | 3611 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.85, 1.03] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Agarwal 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
AlQahtani 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Avendano‐Sola 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Bajpai 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Gharbharan 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Hamdy Salman 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Horby 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Joyner 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Li 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Libster 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
O’Donnell 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Ray 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Simonovich 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Abbreviations: AE: adverse event; ALT: alanine transaminase; ARDS: acute respiratory distress syndrome; AST: aspartate transaminase; BAL: bronchoalveolar lavage; BAT: best available therapy; BMI: body mass index; C: celsius; CDC: US Centers for Disease Control and Prevention; COI: conflict of interest; COPD: chronic obstructive pulmonary disease; CP: convalescent plasma; CPAP: continuous positive airway pressure; CPK: creatine phosphokinase; CRP: c‐reactive protein; CT: computed tomography; DFPP: double‐filtration plasmapheresis; DSMB: Data and Safety Monitoring Board; DVT: deep vein thrombosis; ECMO: extracorporeal membrane oxygenation; ED: emergency department; ELISA: enzyme‐linked immunosorbent assay; FDA: US Food and Drug Administration; FiO2: fractional inspired oxygen; GFR: glomerular filtration rate; HBV/HCV: hepatitis B/C; HCPOA: healthcare power of attorney; HLA: human leukocyte antigen; HNA: human neutrophil antigens; ICU: intensive care unit; IgA (B/G/M): immunoglobulin A (B/G/M); IL‐6: interleukin‐6; IMV: invasive mechanical ventilation; IQR: interquartile range; IV: intravenous; IVIG: intravenous immunoglobulin; LAR: legal authorised representative; LDH: lactate dehydrogenase; NR: not reported; NYHA: New York Heart Association; PaO2: arterial blood oxygen partial pressure; PCR: polymerase chain reaction; PE: pulmonary embolism; PRNT: plaque reduction neutralisation test; QoL: quality of life; RCT: randomised controlled trial; RCU: respiratory care unit; RNA: ribonucleic acid; RT‐PCR: reverse transcription polymerase chain reaction; SAE: serious adverse event; SARS: severe acute respiratory syndrome; SC: subcutaneous; SD: standard deviation; SOC: standard of care; SOFA: sequential organ failure assessment; SpO2: peripheral capillary oxygen saturation; TACO: transfusion‐associated circulatory overload; TAD: transfusion‐associated dyspnoea; TB: tuberculosis; TRALI: transfusion‐related acute lung injury; TTP: thrombotic thrombocytopenic purpura; UIP: usual interstitial pneumonia; ULN: upper limit of normal; VMNT: viral microneutralisation test; WBC: white blood count; WHO: World Health Organization.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Abdullah 2020 | Single arm study; fewer than 500 participants received convalescent plasma |
| Abolghasemi 2020 | Single arm study; fewer than 500 participants received convalescent plasma |
| Ahn 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Anderson 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Baklaushev 2020 | Controlled study, probably not truly randomised |
| Balcells 2020 | Performed another intervention comparison (early vs deferred plasma) |
| Bao 2020b | Single‐arm study; not pre‐registered in a clinical study registry |
| Bobek 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Bradfute 2020 | Single arm study; fewer than 500 participants received convalescent plasma |
| Brasil Ministerio 2020 | Standard operating procedure |
| Budhai 2020 | Feasibility of plasma collection only |
| Cantore 2020 | Single‐arm study compared to published cases; not pre‐registered in a clinical study registry |
| Cao 2020a | Ineligible intervention |
| Chen 2020b | Ineligible intervention |
| Chen 2020c | Ineligible intervention |
| ChiCTR2000029850 | Single arm study; fewer than 500 participants will receive convalescent plasma |
| ChiCTR2000030039 | Single arm study; fewer than 500 participants will receive convalescent plasma |
| ChiCTR2000030312 | Study cancelled before starting recruitment |
| ChiCTR2000030381 | Study cancelled before starting recruitment |
| ChiCTR2000030442 | Study cancelled before starting recruitment |
| ChiCTR2000031501 | Single arm study; fewer than 500 participants will receive convalescent plasma |
| ChiCTR2000033798 | Single arm study; fewer than 500 participants will receive convalescent plasma |
| Clark 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| CTRI/2020/04/024804 | Single arm study; fewer than 500 participants will receive convalescent plasma |
| CTRI/2020/08/027285 | Single arm study; fewer than 500 participants will receive convalescent plasma |
| CTRI/2020/10/028547 | Ineligible intervention |
| Çınar 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| de Assis 2020 | Ineligible indication |
| Díez 2020 | Ineligible intervention |
| Donato 2020 | Single arm study; fewer than 500 participants received convalescent plasma |
| Duan 2020 | Single arm study; fewer than 500 participants received convalescent plasma |
| Dulipsingh 2020 | Single‐arm study; fewer than 500 participants received convalescent plasma |
| Enzmann 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Erkurt 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Fan 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Figlerowicz 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Franchini 2020 | Standard operating procedure |
| Grisolia 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Hashim 2020 | Feasibility of plasma collection only |
| Hu 2020 | Ineligible intervention |
| Ibrahim 2020 | Single‐arm study; fewer than 500 participants received convalescent plasma |
| Im 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| IRCT20151228025732N53 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| IRCT20200406046968N2 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| IRCT20200414047072N1 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| IRCT20200416047099N1 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| ISRCTN86534580 | Ineligible intervention |
| Jamous 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Jiang 2020a | Single‐arm study; not pre‐registered in a clinical study registry |
| Jiang 2020b | Ineligible intervention |
| Jin 2020 | Single‐arm study; fewer than 500 participants received convalescent plasma |
| Karatas 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Kong 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Lin 2020 | Ineligible intervention |
| Liu 2020 | Single‐arm study; fewer than 500 participants received convalescent plasma |
| Liu 2020a | Single‐arm study; not pre‐registered in a clinical study registry |
| Madariaga 2020 | Single‐arm study; fewer than 500 participants received convalescent plasma |
| Martinez‐Resendez 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| McCuddy 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Ministerio de Salud 2020 | Standard operating procedure |
| Mira 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| NCT04261426 | Ineligible intervention |
| NCT04264858 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04292340 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04321421 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04323800 | Ineligible participant population (participants exposed to COVID‐19) |
| NCT04325672 | Study cancelled before starting recruitment |
| NCT04327349 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04332380 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04333355 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04344015 | Feasibility of plasma collection only |
| NCT04344379 | Ineligible intervention |
| NCT04344977 | Feasibility of plasma collection only |
| NCT04345679 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04346589 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04347681 | Controlled, non‐randomised studies with less than 500 participants receiving convalescent plasma or hyperimmune immunoglobulin |
| NCT04348877 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04350580 | Ineligible intervention |
| NCT04353206 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04354831 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04355897 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04356482 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04360278 | Feasibility of plasma collection only |
| NCT04365439 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04368013 | Ineligible intervention |
| NCT04374565 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04376034 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04377672 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04383548 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04384497 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04384588 | Controlled, non‐randomised studies with less than 500 participants receiving convalescent plasma or hyperimmune immunoglobulin |
| NCT04388527 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04389710 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04389944 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04390178 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04392232 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04393727 | Terminated in November 2020 (Study was stopped because the sponsor was changed and a new study on convalescent plasma sponsored by the Italian Medicines Agency (AIFA) was started in Italy) |
| NCT04397523 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04407208 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04408209 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04411602 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04412486 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04418531 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04432103 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04438694 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04458363 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04462848 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04467151 | Study withdrawn |
| NCT04471051 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04474340 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04476888 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04502472 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04513158 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04516954 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04535063 | Single‐arm study; fewer than 500 participants received convalescent plasma |
| NCT04554992 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04555109 | Study of plasma donors |
| NCT04565197 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04569188 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04570982 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04614012 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04616976 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04622826 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04638634 | Study on pharmacokinetics |
| NCT04644198 | Single‐arm study; fewer than 500 participants will receive convalescent plasma |
| NCT04661839 | Pharmacokinetics study |
| NCT04721236 | Single‐arm hyperimmune immunoglobulin study with fewer than 500 participants |
| Niu 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Olivares‐Gazca 2020 | single arm study; less than 500 participants received convalescent plasma |
| Pei 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Peng 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| PER‐031‐20 | Single arm study; less than 500 participants will receive convalescent plasma |
| Perotti 2020 | Single arm study; less than 500 participants received convalescent plasma |
| Qiu 2020 | No use of convalescent plasma. Reporting on generalised collection of information about COVID‐19 infection relating to aetiology, pathology, symptoms, clinical presentation and some generalised pharmacological treatment methods. The papers do not contain patient data that we required. Article translated by Rujan Shrestha and Ya‐Ying Wang via Cochrane TaskExchange |
| Rasheed 2020 | Controlled study, probably not truly randomised |
| RBR‐4vm3yy | Single arm study; less than 500 participants will receive convalescent plasma |
| Robbiani 2020 | Ineligible intervention |
| RPCEC00000323 | Single arm study; less than 500 participants will receive convalescent plasma |
| Salazar 2020a | single arm study; less than 500 participants received convalescent plasma |
| Salazar 2020b | Single‐arm study; not pre‐registered in a clinical study registry |
| Shen 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Shi 2020 | Ineligible intervention |
| Soleimani 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Taher 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Tan 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Tu 2020 | No use of convalescent plasma. Reporting on generalised collection of information about COVID‐19 infection relating to aetiology, pathology, symptoms, clinical presentation and some generalised pharmacological treatment methods. The papers do not contain patient data that we required. Article translated by Rujan Shrestha and Ya‐Ying Wang via Cochrane TaskExchange |
| Wang 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Wright 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Xia 2020 | Single arm study; less than 500 participants received convalescent plasma |
| Xie 2020 | Ineligible intervention |
| Xu 2020b | Single‐arm study; not pre‐registered in a clinical study registry |
| Yang 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Ye 2020 | Single‐arm study; not pre‐registered in a clinical study registry |
| Zeng 2020 | Single arm study; less than 500 participants received convalescent plasma |
| Zhang 2020a | Single‐arm study; not pre‐registered in a clinical study registry |
| Zhang 2020b | Single‐arm study; not pre‐registered in a clinical study registry |
| Zhang 2020c | Single‐arm study; not pre‐registered in a clinical study registry |
Characteristics of studies awaiting classification [ordered by study ID]
Beltran 2021.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
Bennett‐Guerrero 2021.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
CTRI/2020/05/025299.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
CTRI/2020/05/025328.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
CTRI/2020/09/027903.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
IRCT20120215009014N353.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
IRCT20150808023559N21.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
IRCT20200404046948N1.
| Methods |
|
| Participants | Participants
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
IRCT20200409047007N1.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
IRCT20200413047056N1.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
IRCT20200503047281N1.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
IRCT20200525047562N1.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
IRCT20201004048922N1.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
ISRCTN85216856.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
jRCT2031200174.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
Lopardo 2021.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT04315948.
| Methods |
|
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
|
| Notes | Recruitment status: active, not recruiting Prospective completion date: March 2023 Sponsor/funding: Institut National de la Santé et de la Recherche Médicale, France |
NCT04332835.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT04355767.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT04358211.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT04372368.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT04392414.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT04405310.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes | Recruitment status: completed Prospective completion date: 20 June 2020 Sponsor/funding: Grupo Mexicano para el Estudio de la Medicina Intensiva Hospital General Naval de Alta Especialidad ‐ Escuela Medico Naval National Institute of Pediatrics, Mexico Instituto Nacional de Enfermedades Respiratorias |
NCT04433910.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT04442958.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT04492501.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT04501978.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT04521309.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT04542941.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT04547127.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT04547660.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
NCT04610502.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
Pouladzadeh 2021.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
AE: adverse event; ARDS: acute respiratory distress syndrome; CAP: community‐acquired pneumonia; COPD: chronic obstructive pulmonary disease; CP: convalescent plasma; CRP: C‐reactive protein; CT: computed tomography; ECMO: extracorporeal membrane oxygenation; ELISA: enzyme‐linked immunosorbent assay; FiO2: fractional inspired oxygen; GFR: glomerular filtration rate; HLA: human leukocyte antigen; HNA: human neutrophil antigens; ICU: intensive care unit; IgA (B/G/M): immunoglobulin A (B/G/M); IL‐6: interleukin‐6; IQR: interquartile range; IV: intravenous; LDH: lactate dehydrogenase; NR: not reported; PaO2: arterial blood oxygen partial pressure; PCR: polymerase chain reaction; QoL: quality of life; RCT: randomised controlled trial; RT‐PCR: reverse transcription polymerase chain reaction; SAE: serious adverse event; SD: standard deviation; SOFA: sequential organ failure assessment; TACO: transfusion‐associated circulatory overload; TAD: transfusion‐associated dyspnoea; TRALI: transfusion‐related acute lung injury
Characteristics of ongoing studies [ordered by study ID]
ChiCTR2000030010.
| Study name | A randomized, double‐blind, parallel‐controlled, trial to evaluate the efficacy and safety of anti‐SARS‐CoV‐2 virus inactivated plasma in the treatment of severe novel coronavirus pneumonia patients (COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 19 February 2020 |
| Contact information | Liu Ying Wuhan Jinyintan Hospital (Wuhan Infectious Diseases Hospital) , 1 Yintan Road, Dongxihu District, Wuhan, Hubei, China , 430023, whsjytyy_gcp@163.com Zhang Dingyu 1 Yintan Road, Dongxihu District, Wuhan, Hubei, China, 430023, 1813886398@qq.com |
| Notes |
|
ChiCTR2000030179.
| Study name | Experimental study of novel coronavirus pneumonia rehabilitation plasma therapy severe novel coronavirus pneumonia (COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 24 February 2020 |
| Contact information | Liu Wei The First Affiliated Hospital of Nanchang University, 17 Yongwai Main Street, Nanchang, Jiangxi, China, 330006, cdyfyliuwei@163.com Le Aiping 17 Yongwai Main Street, Nanchang, Jiangxi, China, 330006, leaiping@126.com |
| Notes |
|
ChiCTR2000030627.
| Study name | Study on the application of convalescent plasma therapy in severe COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 February 2020 |
| Contact information | Guojun Zhang The First Affiliated Hospital of Zhengzhou University, 1 Jianshe Road East, Zhengzhou, He'nan, China, zlgj‐001@126.com Guojun Zhang 1 Jianshe Road East, Zhengzhou, He'nan, China, zlgj‐001@126.com |
| Notes |
|
ChiCTR2000030702.
| Study name | Convalescent plasma for the treatment of common COVID‐19: a prospective RCT |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 15 February 2020 |
| Contact information | Liu Zhong Institute of Blood Transfusion, Chinese Academy of Medical Sciences, 26 Huacai Road, Chenghua District, Chengdu, Sichuan, China, 610000, Liuz@ibt.pumc.edu.cn Cao Bin 2 Yinghua Street East, Chaoyang District, Beijing, China, 100029, caobin_ben@163.com |
| Notes |
|
ChiCTR2000030929.
| Study name | A randomized, double‐blind, parallel‐controlled trial to evaluate the efficacy and safety of anti‐SARS‐CoV‐2 virus inactivated plasma in the treatment of severe novel coronavirus pneumonia (COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 17 March 2020 |
| Contact information | Lianghao Zhang 11443556@qq.com Sinopharm Wuhan Blood Products Co., Ltd. 1 Golden Industrial Park Road, Zhengdian, Jiangxia District, Wuhan, Hubei, China |
| Notes |
|
CTRI/2020/04/024915.
| Study name | A phase II, open label, randomized controlled trial to assess the safety and efficacy of convalescent plasma to limit COVID‐19 associated complications |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 09/05/2020 |
| Contact information | Corresponding Author: Name: Dr Sangeeta Pathak Affiliation: Blood Bank, Max Super Speciality hospital, Saket (A unit of DevkiDevi Foundation) Full Address: Max Super Speciality Hospital (Devki Devi Foundation), East Block,Blood Bank, 2, Press enclave Road, Saket New DelhiNew DelhiDELHI110017India Email: sangeeta.pathak@maxhealthcare.com |
| Notes |
|
CTRI/2020/05/025346.
| Study name | A phase II, open label, randomized controlled trial to assess the safety and efficacy of convalescent plasma in severe COVID‐19 patients. |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 01/06/2020 |
| Contact information | Corresponding Author Name: Dr S Anbuselvi Mattuvar Kuzhali Affiliation: Madras Medical College Full Address: Institute of Physiology and Experimental Medicine, Madras MedicalCollege, Chennai 4B, third floor, KGEYES HYACINTH, LB road, Kamaraj Nagar, Thiruvanmiyur, Chennai, Chennai, TAMIL NADU 600003 India Email: dranbuselvimk@gmail.com |
| Notes |
|
CTRI/2020/06/025803.
| Study name | Efficacy of convalescent plasma therapy in patients with COVID‐19: a randomized control trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 18/06/2020 |
| Contact information | Corresponding Author Name: Dr Meenu Bajpai Affiliation: Institute of Liver and Biliary Sciences Full Address: Room No:16056, Department of Transfusion Medicine, PhaseI, Upper Basement, D‐1, Vasant Kunj New Delhi‐110070 South West DELHI 110070 India Email: meenubajpai@hotmail.com |
| Notes |
|
CTRI/2020/06/026123.
| Study name | A phase II, open label, randomized controlled trial to assess the safety and efficacy of convalescent plasma in severe COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 25/06/2020 |
| Contact information | Corresponding Author Name: Dr Sushant Meshram Affiliation: Government Medical College and Hospital, Nagpur Full Address: Dept of Pulmonary Medicine, 4th floor, Super Specialty Hospital, Government Medical College Hospital, Nagpur. Nagpur, MAHARASHTRA 440009 India Email: drsushant.in@gmail.com |
| Notes |
|
EUCTR2020‐001632‐10.
| Study name | A randomized open label phase‐II clinical trial with or without infusion of plasma from subjects after convalescence of SARS‐CoV‐2 infection in high‐risk patients with confirmed severe SARS‐CoV‐2 disease |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 04 May 2020 |
| Contact information | Corresponding Author Name: Prof. Dr. Carsten Müller‐Tidow Affiliation: University Hospital Heidelberg; Dpt. of Internal Medicine V Hematology, Oncology and Rheumatology Full Address: Im Neuenheimer Feld 410, 69120 Heidelberg, Germany Email: Carsten.Mueller‐Tidow@med.uni‐heidelberg.de |
| Notes |
|
EUCTR2020‐001936‐86.
| Study name | A prospective, randomized, open label phase 2 clinical trial to evaluate superiority of anti‐SARS‐CoV‐2 convalescent plasma versus standard‐of‐care in hospitalized patients with mild COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 19 October 2020 |
| Contact information | Corresponding Author/Contact Name: Institute of Transfusion Medicine, Hannover Medical School Affiliation: ‐ Full Address: Carl‐Neuberg‐Str. 1, Hannover, 30625, Germany Email: NR |
| Notes |
|
EUCTR2020‐002122‐82.
| Study name | Prospective open‐label randomized controlled phase 2b clinical study in parallel groups for the assessment of efficacy and safety of immune therapy with COVID‐19 convalescent plasma plus standard treatment vs. standard treatment alone of subjects with severe COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 12 June 2020 |
| Contact information | Corresponding Author Name: Holger Hackstein Affiliation: Universitätsklinikum Erlangen Full Address: Universitätsklinikum Erlangen, Transfusionsmedizinische Abteilung, Krankenhausstr. 12, Erlangen 91054, Germany Email: holger.hackstein@uk‐erlangen.de |
| Notes |
|
EUCTR2020‐005410‐18.
| Study name | Multicentre, randomized, double‐blind, placebo‐controlled, non‐commercial clinical trial to evaluate the efficacy and safety of specific anti‐SARS‐CoV‐2 immunoglobulin in the treatment of COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 16/11/2020 |
| Contact information | Samodzielny Publiczny Szpital Kliniczny Nr 1 w Lublinie Samodzielny Publiczny Zakład Opieki Zdrowotnej Krzysztof Tomasiewicz ul. Stanisława Staszica 16 Lublin20‐081 Poland 00488153 49 414564 00488153 49 410 krzysztoftomasiewicz@umlub.pl |
| Notes |
|
IRCT20200501047258N1.
| Study name | Investigation of the effects of COVID‐19 convalescent plasma in acute respiratory distress syndrome due to COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 04 May 2020 |
| Contact information | Corresponding Author Name: Rahim Asghari Affiliation: Oroumia University of Medical Sciences Full Address: Resalat st, UMSU, Urmia, West Azarbaijan Email: rahimasghari@gmail.com |
| Notes |
|
IRCT20200508047346N1.
| Study name | Evaluation of the effectiveness of rabbit antibody against coronavirus in patients. |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 21 May 2020 |
| Contact information | Corresponding Author Name: Prof. Mostafa Ghanei Affiliation: Bagheiat‐allah University of Medical Sciences Full Address: Mulla Sadra, Sheikh Baha'i, Baqiyatallah Al‐Azam Hospital, Teheran, 1435915371 Email: mghaneister@gmail.com |
| Notes |
|
NCT02735707.
| Study name | Randomized, embedded, multifactorial adaptive platform trial for community‐acquired pneumonia (REMAP‐CAP) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 11 April 2016 |
| Contact information | Cameron Green: info@remapcap.org |
| Notes |
|
NCT04333251.
| Study name | Evaluating convalescent plasma to decrease coronavirus associated complications. A phase I study comparing the efficacy and safety of high‐titer anti‐SARS‐CoV‐2 plasma versus best supportive care in hospitalized patients with interstitial pneumonia due to COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 April 2020 |
| Contact information | NR |
| Notes |
|
NCT04338360.
| Study name | Expanded access to convalescent plasma for the treatment of patients with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | NR |
| Contact information | Michael Joyner, MD; 507‐255‐4288; USCOVIDplasma@mayo.edu |
| Notes |
|
NCT04345289.
| Study name | Efficacy and safety of novel treatment options for adults with COVID‐19 pneumonia (CCAP) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 20 April 2020 |
| Contact information | Thomas Benfield, MD, DMSc: thomas.lars.benfield@regionh.dk |
| Notes |
|
NCT04345991.
| Study name | Efficacy of convalescent plasma to treat COVID‐19 patients, a nested trial in the CORIMUNO‐19 cohort |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 14 April 2020 |
| Contact information | Karine Lacombe: karine.lacombe2@aphp.fr |
| Notes |
|
NCT04348656.
| Study name | Convalescent plasma for hospitalized adults with COVID‐19 respiratory illness (CONCOR‐1) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 27 April 2020 |
| Contact information | Donald M Arnold, MD, McMaster University, Hamilton, Canada: arnold@mcmaster.ca |
| Notes |
|
NCT04352751.
| Study name | Experimental use of convalescent plasma for passive immunization in current COVID‐19 pandemic in Pakistan in 2020 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | April 2020 |
| Contact information | Dr. Arshi Naz, PhD: labarshi@yahoo.com |
| Notes |
|
NCT04358783.
| Study name | Phase II, randomized, double‐blind, controlled clinical trial evaluating the efficacy and safety of plasma from patients cured of COVID‐19 compared to the best available therapy in subjects with SARS‐CoV‐2 pneumonia |
| Methods |
Clinical trial comparing convalescent plasma to BAT for the treatment of severely ill and critically ill patient with COVID‐19 Patients with SARS‐CoV‐2 PCR‐confirmed infection with pulmonary infiltrates and hypoxaemia will be screened and invited to participate |
| Participants |
Patients with SARS‐CoV‐2 PCR‐confirmed infection with pulmonary infiltrates and hypoxaemia will be screened and invited to participate |
| Interventions |
|
| Outcomes |
|
| Starting date | 27 April 2020 |
| Contact information | Eduardo Pérez Alba, MD: +52 8117998705; md.eduardo.perez@gmail.com |
| Notes |
|
NCT04360486.
| Study name | Treatment of COVID‐19 with anti‐SARS‐CoV‐2 convalescent plasma (ASCoV2CP) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 24 April 2020 |
| Contact information | Andrew P Cap, MS, MD, PhD, FACP: andrew.p.cap.mil@mail.mil |
| Notes |
|
NCT04361253.
| Study name | A prospective, randomized, double‐masked, placebo‐controlled trial of high‐titer COVID‐19 convalescent plasma (HT‐CCP) for the treatment of hospitalized patients with COVID‐19 of moderate severity |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 30 April 2020 |
| Contact information | Richard Kaufman, MD: +1‐617‐732‐5232; rmkaufman@bwh.harvard.edu |
| Notes |
|
NCT04362176.
| Study name | A randomized, controlled clinical trial to test the safety and efficacy of convalescent donor plasma to treat COVID‐19 in hospitalized adults |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 24 April 2020 |
| Contact information | Amanda J Bistran‐Hall +1‐615‐875‐8531; amanda.j.bistran‐hall@vumc.org |
| Notes |
|
NCT04363034.
| Study name | Arkansas expanded access COVID‐19 convalescent plasma treatment program |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 27 April 2020 |
| Contact information | Danielle Evans: +1‐501‐526‐7906; DEvans@uams.edu |
| Notes |
|
NCT04364737.
| Study name | Convalescent plasma to limit coronavirus associated complications: a randomized blinded phase 2 study comparing the efficacy and safety of anti‐SARS‐CoV2 plasma to placebo in COVID‐19 hospitalized patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 17 April 2020 |
| Contact information | Mila B Ortigoza, MD, PhD: Mila.Ortigoza@nyulangone.org |
| Notes |
|
NCT04366245.
| Study name | Phase I/II multicentre, randomized and controlled clinical trial to evaluate the efficacy of treatment with hyperimmune plasma obtained from convalescent antibodies of COVID‐19 infection |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 23 April 2020 |
| Contact information | Ana Cardesa Gil: 697 95 69 41 ext 0034; ana.cardesa@juntadeandalucia.es |
| Notes |
|
NCT04372979.
| Study name | Evaluation of efficacy of COVID‐19 convalescent plasma versus standard plasma in the early care of COVID‐19 patients hospitalized outside intensive care units |
| Methods |
|
| Participants | Inclusion criteria:
OR 1 of the biological criteria :
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | May 2020 |
| Contact information |
|
| Notes |
|
NCT04373460.
| Study name | Comparison of the efficacy and safety of human coronavirus immune plasma (HCIP) vs. control (SARS‐CoV‐2 non‐immune) plasma among outpatients with symptomatic COVID‐19 |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 4 May 2020 |
| Contact information |
|
| Notes |
|
NCT04374370.
| Study name | Severe acute respiratory syndrome coronavirus 2 of the genus betacoronavirus (SARS‐CoV‐2) convalescent plasma (CP) expanded access protocol (EAP) |
| Methods |
|
| Participants | Inclusion criteria:
Severe disease is defined as:
Life‐threatening disease is defined as:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | NR |
| Contact information | Chris Ensor, Pharm D: 413.519.7056; Chris.Ensor@AdventHealth.com |
| Notes | Recruitment status: available Prospective completion date: NR Sponsor/funding: AdventHealth Orlando, Available: Orlando, Florida, United States, 32803, Principal Investigator: Eduardo Oliveira, MD AdventHealth |
NCT04374487.
| Study name | A phase II, open label, randomized controlled trial to assess the safety and efficacy of convalescent plasma to limit COVID‐19 associated complications |
| Methods |
|
| Participants | Inclusion criteria
Or in case of severe or immediately life‐threatening COVID‐19, for example:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 9 May 2020 |
| Contact information |
|
| Notes |
|
NCT04374526.
| Study name | Early transfusIon of COVID‐19 convalescent plasma in elderly COVID‐19 patients to prevent disease progression |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 27 May 2020 |
| Contact information | Raffaele Landolfi, Prof: 06 30154435 ext +39; raffaele.landolfi@unicatt.it Luciana Teofili, Prof: 06 30154180 ext +39; luciana.teofili@unicatt.it |
| Notes |
|
NCT04376788.
| Study name | Exchange transfusion versus plasma from convalescent patients with methylene blue in patients with COVID‐19 |
| Methods |
|
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
|
| Starting date | 6 May 2020 |
| Contact information |
|
| Notes |
|
NCT04377568.
| Study name | CONCOR‐KIDS: a randomized, multicentered, open‐label phase 2 clinical trial of the safety and efficacy of human coronavirus‐immune convalescent plasma for the treatment of COVID‐19 disease in hospitalized children |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 6 May 2020 |
| Contact information | Julia Upton: 416 813 7654 ext 208634, julia.upton@sickkids.ca Christoph Licht, christoph.licht@sickkids.ca |
| Notes |
|
NCT04380935.
| Study name | Effectiveness and safety of convalescent plasma therapy on COVID‐19 patients with acute respiratory distress syndrome |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 8 May 2020 |
| Contact information | Robert Sinto, MD: +628158835432, rsinto@yahoo.com |
| Notes |
|
NCT04385043.
| Study name | Phase 2b/3 trial to evaluate the safety and efficacy of plasma transfusion from convalescent patients with SARS‐CoV‐2 infection on severity and mortality of COVID‐19 in hospitalized patients |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 May 2020 |
| Contact information | Gabriella Talarico, MD0961883111, trasfusionale@aocz.it |
| Notes |
|
NCT04385186.
| Study name | Inactivated convalescent plasma as a therapeutic alternative in hospitalized patients COVID‐19 |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 20 June 2020 |
| Contact information |
|
| Notes |
|
NCT04385199.
| Study name | The use of convalescent plasma for patients hospitalized with COVID‐19 disease |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 4 May 2020 |
| Contact information |
|
| Notes |
|
NCT04388410.
| Study name | Phase 2b/3 trial to evaluate the safety and efficacy of plasma transfusion from convalescent patients with SARS‐CoV‐2 infection on severity and mortality of COVID‐19 in hospitalized patients. |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 June 2020 |
| Contact information |
|
| Notes |
|
NCT04390503.
| Study name | A phase 2 randomized, double‐blinded trial to evaluate the efficacy and safety of human anti‐SARS‐CoV‐2 plasma in close contacts of COVID‐19 cases |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | March 2021 (estimated) |
| Contact information |
|
| Notes |
|
NCT04391101.
| Study name | Efficacy of convalescent plasma for the treatment of severe SARS‐CoV‐2 infection: a randomized, open label clinical trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | June 2020 |
| Contact information |
|
| Notes |
|
NCT04395170.
| Study name | A multicenter randomized clinical trial to evaluate the efficacy and safety of the use of convalescent plasma (PC) compared to anti‐COVID‐19 human immunoglobulin and standard treatment in hospitalized patients |
| Methods |
|
| Participants |
|
| Interventions |
CP:
hyperimmune immunoglobulin:
|
| Outcomes |
|
| Starting date | June 2020 |
| Contact information |
|
| Notes |
|
NCT04397757.
| Study name | COVID‐19 convalescent plasma for the treatment of hospitalized patients with pneumonia caused by SARS‐CoV‐2 |
| Methods |
|
| Participants |
Note ‐ An exception must be requested to the Sponsor if ≥ 72 h since positive test
|
| Interventions |
|
| Outcomes |
|
| Starting date | 13 March 2020 |
| Contact information |
|
| Notes |
|
NCT04403477.
| Study name | Convalescent plasma transfusion therapy in severe COVID‐19 patients ‐ a tolerability, efficacy and dose‐response phase II RCT |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 20 May 2020 |
| Contact information |
|
| Notes |
|
NCT04408040.
| Study name | Use of convalescent plasma collected from donors recovered from COVID‐19 virus disease for transfusion, as an empirical and preemptive treatment during viral pandemic outbreak |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | June 2020 |
| Contact information | Stacey Brown 404‐780‐7965 stacey.brown@northside.com |
| Notes |
|
NCT04415086.
| Study name | Treatment of patients with COVID‐19 with convalescent plasma transfusion: a multicenter, open‐labeled, randomized and controlled study |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 June 2020 |
| Contact information |
|
| Notes |
|
NCT04418518.
| Study name | CONCOR‐1: a randomized open‐label trial of convalescent plasma for hospitalized adults with acute COVID‐19 respiratory illness |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 24 June 2020 |
| Contact information | Celine Arar: 212‐746‐4177; cea4002@med.cornell.edu |
| Notes |
|
NCT04420988.
| Study name | Investigational COVID‐19 convalescent plasma infusion for severely or life‐threateningly ill COVID‐19 patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | NR |
| Contact information |
|
| Notes |
|
NCT04421404.
| Study name | A randomized controlled adaptive study comparing COVID‐19 convalescent plasma (CCP) to non‐immune plasma to limit coronavirus‐associated complications in hospitalized patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 9 June 2020 |
| Contact information |
|
| Notes |
|
NCT04425837.
| Study name | Effectiveness and safety of convalescent plasma in patients with high‐risk COVID‐19: a randomized, controlled study CRI‐CP (Coronavirus Investigation ‐ Convalescent Plasma) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | July 2020 |
| Contact information |
|
| Notes |
|
NCT04425915.
| Study name | Efficacy of convalescent plasma therapy in patients with COVID‐19: a randomized control trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 9 June 2020 |
| Contact information |
|
| Notes |
|
NCT04428021.
| Study name | Effectiveness of adding standard plasma or COVID‐19 convalescent plasma to standard treatment, versus standard treatment alone, in patients with recent onset of COVID‐19 respiratory failure. A randomized, three‐arms, phase 2 trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 15 June 2020 |
| Contact information | Paola Maria Manzini, Principal Investigator, Azienda Ospedaliera Città della Salute e della Scienza di Torino |
| Notes |
|
NCT04429854.
| Study name | A randomized, open‐label, adaptive, proof‐of‐concept clinical trial of donated antibodies working against with COVID‐19: DAWN‐PLASMA |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 2 May 2020 |
| Contact information | Geert Meyfroidt, MD, PhD: geert.meyfroidt@uzleuven.be |
| Notes |
|
NCT04432272.
| Study name | Antibody‐level based analysis of COVID‐19 convalescent serum (ABACCuS) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 14 July 2020 |
| Contact information | Maureen Cooney, RN, BSN: Maureen.Cooney@beaumont.org |
| Notes |
|
NCT04438057.
| Study name | Evaluating the efficacy of convalescent plasma in symptomatic outpatients infected with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 6 July 2020 |
| Contact information |
|
| Notes |
|
NCT04442191.
| Study name | Infusion of convalescent plasma for the treatment of patients infected with severe acute respiratory syndrome‐coronavirus‐2 (COVID‐19): a double‐blinded, placebo‐controlled, proof‐of‐concept study |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 5 May 2020 |
| Contact information | Jessica Herrick, Assistant Professor of Clinical Medicine, University of Illinois at Chicago |
| Notes |
|
NCT04445207.
| Study name | Experimental expanded access treatment with convalescent plasma for the treatment of patients with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | NR |
| Contact information |
|
| Notes |
|
NCT04452812.
| Study name | Pilot clinical, statistical and epidemiological study on efficacy and safety of convalescent plasma for the management of patients with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 6 July 2020 |
| Contact information |
|
| Notes |
|
NCT04453384.
| Study name | Study to evaluate the safety and efficacy of XAV‐19 in patients with COVID‐19 induced moderate pneumonia (POLYCOR) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | September 1, 2020 |
| Contact information |
|
| Notes |
|
NCT04456413.
| Study name | Phase II randomized study of convalescent plasma from recovered COVID‐19 donors collected by plasmapheresis as treatment for subjects with early COVID‐19 infection |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | November 6, 2020 |
| Contact information |
|
| Notes |
|
NCT04463823.
| Study name | "NORPLASMA" COVID‐19 convalescent plasma treatment monitoring study |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 6 July 2020 |
| Contact information |
|
| Notes |
|
NCT04468009.
| Study name | Treatment of critically ill patients with COVID‐19 with convalescent plasma |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 25 June 2020 |
| Contact information |
|
| Notes |
|
NCT04468958.
| Study name | Safety, tolerability, and pharmacokinetics of SAB‐185 in healthy participants |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | August 5, 2020 |
| Contact information |
|
| Notes |
|
NCT04469179.
| Study name | Safety, tolerability, and pharmacokinetics of SAB‐185 in ambulatory participants with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 20 August 2020 |
| Contact information |
|
| Notes |
|
NCT04472572.
| Study name | Expanded access to convalescent plasma for the treatment of patients with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 15 July 2020 |
| Contact information | Principal Investigator: Michele Donato, Hackensack Meridian Health |
| Notes |
|
NCT04483960.
| Study name | An international multi‐centre randomised clinical trial to assess the clinical, virological and immunological outcomes in patients diagnosed with SARS‐CoV‐2 infection (COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 21 July 2020 |
| Contact information |
|
| Notes |
|
NCT04497324.
| Study name | PERUCONPLASMA: randomized clinical trial to evaluate safety and efficacy of the use of convalescent plasma in hospitalized patients with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 21 September 2020 |
| Contact information |
|
| Notes |
|
NCT04497779.
| Study name | Evaluation of coronavirus disease 19 (COVID‐19) convalescent plasma |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 21 August 2020 |
| Contact information | Contact: John Zaia 626‐218‐1817 jzaia@coh.org |
| Notes |
|
NCT04514302.
| Study name | Safety and efficacy of anti‐SARS‐CoV‐2 equine antibody fragments (INOSARS) for hospitalized patients with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 20 October 2020 (estimated start date) |
| Contact information |
|
| Notes |
|
NCT04516811.
| Study name | A prospective, randomized, placebo‐controlled, double‐blinded, phase III clinical trial of the therapeutic use of convalescent plasma in the treatment of patients with moderate to severe COVID‐19 |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 3 September 2022 |
| Contact information |
|
| Notes |
|
NCT04521036.
| Study name | Convalescent plasma for COVID‐19 patients (CPCP) (CPCP) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | December 1, 2020 |
| Contact information |
|
| Notes |
|
NCT04524507.
| Study name | COVID‐19 antibody plasma research study in hospitalized patients (UNC CCP RCT) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 27 August 2020 |
| Contact information | Corresponding Author Name: JoAnn Kuruc, RN, MSN Affiliation: NR Full Address: NR Email: joann_kuruc@med.unc.edu |
| Notes |
|
NCT04528368.
| Study name | Convalescent plasma for treating patients with COVID‐19 pneumonia without indication of ventilatory support |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | August 18, 2020 |
| Contact information | Contact: Eduardo M Rego, MD, PhD: edumrego@hotmail.com |
| Notes |
|
NCT04539275.
| Study name | VA coronavirus research and efficacy studies‐1 (VA CURES‐1) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 16/11/2020 |
| Contact information | Edward N Janoff, MD: (730) 723‐6255; Edward.Janoff@va.gov |
| Notes |
|
NCT04542967.
| Study name | Convalescent plasma as a treatment for patients with severe COVID‐19 disease |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 23 June 2020 |
| Contact information | Contact: Carmen G Torres, MD: dragabytorresalarcon@icloud.com |
| Notes |
|
NCT04545047.
| Study name | CSP #2030 ‐ Observational study of convalescent plasma for treatment of veterans with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 5 May 2020 |
| Contact information | Sponsors and Collaborators
Investigators
|
| Notes |
|
NCT04546581.
| Study name | Inpatient treatment with anti‐coronavirus immunoglobulin (ITAC) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 8 October 2020 |
| Contact information | Corresponding author Name: Jacqueline Nordwall Affiliation: NR Full Address: NR Email: jacquie@ccbr.umn.edu |
| Notes |
|
NCT04555148.
| Study name | A prospective, open‐label, randomized, multi‐center, Phase 2a study to evaluate the dose response, efficacy and safety of hyper‐Ig (hyper‐immunoglobulin) GC5131 in patients with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 20 September 2020 |
| Contact information |
|
| Notes |
|
NCT04558476.
| Study name | A multicenter randomized trial to assess the efficacy of convalescent plasma therapy in patients with invasive COVID‐19 and acute respiratory failure treated with mechanical ventilation: the CONFIDENT trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 September 2020 |
| Contact information | Benoit Misset, MD,PhD: benoit.misset@chuliege.be |
| Notes |
|
NCT04567173.
| Study name | A randomized, open‐label, single center clinical trial to assess the efficacy and safety of convalescent plasma to hospitalized adult COVID‐19 patients as adjunctive therapy to reduce the need for ICU admission: Co‐CLARITY trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 28 September 2020 |
| Contact information | Deonne Thaddeus V Gauiran, MD: +639088150248.; dvgauiran@up.edu.ph |
| Notes |
|
NCT04573855.
| Study name | Treatment with anti‐SARS‐CoV‐2 immunoglobulin in patients with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 December 2020 |
| Contact information |
|
| Notes |
|
NCT04589949.
| Study name | Early convalescent plasma therapy for high‐risk patients with COVID‐19 in primary care (the CoV‐Early Study) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 12 October 2020 |
| Contact information | Corresponding Author Name: Bart Rijnders, MD, PhD Affiliation: Erasmus Medical Center Full Address: Erasmus Medical Center, Rotterdam, Zuid‐Holland, Netherlands, 3000 CA Email: b.rijnders@erasmusmc.nl |
| Notes |
|
NCT04600440.
| Study name | Convalescent plasma in the treatment of COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 23 October 2020 |
| Contact information | Magnus Rasmussen, MD, Prof: +4646171000; magnus.rasmussen@med.lu.se |
| Notes |
|
NCT04621123.
| Study name | Plasma for early treatment in non‐hospitalised mild or moderate COVID‐19 patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 30 October 2020 |
| Contact information | Corresponding Author Name: Oriol Mitjà Villar, PhD, MD Affiliation: Germans Trias i Pujol Hospital Full Address: NR Email: omitja@flsida.org |
| Notes |
|
NCT04634422.
| Study name | Plasma exchange (PLEX) and convalescent plasma (CCP) in COVID‐19 patients with multiorgan failure ‐ the COVID PLEX+CCP Trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 18/11/2020 |
| Contact information | Wladimir M Szpirt, MD: 4535451767; mail@covid‐plex.com |
| Notes |
|
NCT04642014.
| Study name | A multi‐centre, 18‐months, single‐group study of application of convalescent plasma in the treatment of SARS CoV‐2 disease (COVID‐19) with metabolomic and laboratory evaluation of plasma therapy effectiveness |
| Methods | Methods
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 24 November 2020 |
| Contact information | Siddarth Agrawal, PhD: 0048717364000; siddarth.agrawal@umed.wroc.pl |
| Notes |
|
NCT04649879.
| Study name | Convalescent plasma for treatment of COVID‐19: an open randomised controlled trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 12 February 2020 |
| Contact information | Contact: Joakim Dillner, MD, PhD+46 (0) 72‐468 24 60; joakim.dillner@ki.se |
| Notes |
|
NCT04669990.
| Study name | Remdesivir and convalescent plasma therapy for treatment of COVID‐19 infection in Nepal: a registry study |
| Methods |
|
| Participants | Inclusion criteria
Severe COVID‐19 infection is defined by one or more of the following criteria:
Exlusion criteria
|
| Interventions |
|
| Outcomes |
|
| Starting date | 17 December 2020 |
| Contact information | Janak Koirala, MD,MPH: 9818762117 ext +977; clinicaltrialsnepal@gmail.com |
| Notes |
|
NCT04681430.
| Study name | Reconvalescent plasma/camostat mesylate early in SARS‐CoV‐2 Q‐PCR (COVID‐19) positive high‐risk individuals (RES‐Q‐HR) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 8 January 2021 |
| Contact information | Corresponding Author Name: Verena Keitel‐Anselmino, Prof.Dr.med. Affiliation: Universitätsklinikum Düsseldorf Klinik für Hepatologie und Infektiologie Full Address: Universitätsklinikum Düsseldorf Klinik für Hepatologie und Infektiologie, Duesseldorf, Germany, 40225 Email: keitelan@uni‐duesseldorf.de |
| Notes |
|
NCT04712344.
| Study name | Assessment of efficacy and safety of therapy with COVID‐19 convalescent plasma in subjects with severe COVID‐19 (IPCO) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 18 January 2021 |
| Contact information | Contact: Mario Schiffer, MD: +49913185 ext 39002; mario.schiffer@uk‐erlangen.de |
| Notes |
|
NCT04716556.
| Study name | Tranfusion of convalescent plasma for the early treatment of pneumonIa in COVID‐19 patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 16 July 2020 |
| Contact information | Corresponding Author Name: Elena Toschi Affiliation: Istituto Superiore di Sanità Full Address: INR Email: elena.toschi@iss.it |
| Notes |
|
NCT04730401.
| Study name | Convalescent plasma in the treatment of COVID‐19 (CP_COVID‐19) |
| Methods |
|
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
|
| Starting date | 27 January 2021 |
| Contact information |
|
| Notes |
|
NL8633.
| Study name | A randomized, double blinded clinical trial of convalescent plasma compared to standard plasma for treatment of hospitalized non‐ICU patients with COVID‐19 infections (COV‐PLAS) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 13 May 2020 |
| Contact information | Name: Jaap Jan Zwaginga Email: j.j.zwaginga@lumc.nl Phone: 0715264006 |
| Notes |
|
PACTR202006760881890.
| Study name | Lagos COVID‐19 convalescent plasma trial (LACCPT) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 24 September 2020 |
| Contact information |
|
| Notes |
|
PACTR202007653923168.
| Study name | A clinical trial comparing use of convalescent plasma therapy plus standard treatment to standard treatment alone in patients with severe COVID‐19 infection |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 August 2020 |
| Contact information |
|
| Notes |
|
PER‐013‐20.
| Study name | Convalescent plasma as treatment for COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 19th September 2020 |
| Contact information |
|
| Notes |
|
PER‐060‐20.
| Study name | Randomized phase 2 clinical trial to evaluate safety and efficacy of the use of plasma from convalescent patients with the new coronavirus disease (COVID‐19) for the experimental treatment of patients hospitalized in the Centro Médico Naval 'Cirujano Mayor Santiago Távara' |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 19 October 2020 |
| Contact information |
|
| Notes |
|
RBR‐7jqpnw.
| Study name | Therapeutic effectiveness of COVID‐19 convalescent plasma produced by HEMOPE: a multicenter, randomized and controlled clinical trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 July 2020 |
| Contact information |
|
| Notes |
|
AE: adverse event; ALT: alanine transaminase; ANC: absolute neutrophil count; ARDS: acute respiratory distress syndrome; AST: aspartate transaminase; BAL: bronchoalveolar lavage; BAT: best available therapy; B(i)PAP: bi‐level positive airway pressure; BMI: body mass index; CDC: Centers for Disease Control and Prevention; COI: conflict of interest; COPD: chronic obstructive pulmonary disease; CAP: community‐acquired pneumonia; CP: convalescent plasma; CPAP: continuous positive airway pressure; CPK: creatine phosphokinase; CRP: C‐reactive protein; CT: computed tomography; DBP: diastolic blood pressure; DFPP: double‐filtration plasmapheresis; DIC: disseminated intravascular coagulation; DMARD: disease‐modifying anti‐rheumatic drug; DVT: deep vein thrombosis; ECG: electrocardiogram; ECMO: extracorporeal membrane oxygenation; ED: emergency department; FDA: US Food and Drug Administration; FFP: fresh frozen plasma; FiO2: fractional inspired oxygen; GFR: glomerular filtration rate; HBV/HCV: hepatitis B/C; HCPOA: healthcare power of attorney; HLA: human leukocyte antigen; ICU: intensive care unit; IgA (B/G/M): immunoglobulin A (B/G/M); IL‐6: interleukin‐6; IV: intravenous; IVIG: intravenous immunoglobulin; LAR: legal authorised representative; LDH: lactate dehydrogenase; NR: not reported; NYHA: New York Heart Association; PaO2: arterial blood oxygen partial pressure; PCR: polymerase chain reaction; PE: pulmonary embolism; QoL: quality of life; RCT: randomised controlled trial; RF: respiratory failure; RNA: ribonucleic acid; RRT: renal replacement therapy; RT‐PCR: reverse transcription polymerase chain reaction; SAE: serious adverse event; SARS: severe acute respiratory syndrome; SBP: systolic blood pressure; SC: subcutaneous; SOFA: Sequential Organ Failure Assessment; SpO2: peripheral capillary oxygen saturation; SRD: severe respiratory disease; TACO: transfusion‐associated circulatory overload; TAD: transfusion‐associated dyspnoea; TB: tuberculosis; TRALI: transfusion‐related acute lung injury; TTP: thrombotic thrombocytopenic purpura; UIP: usual interstitial pneumonia; ULN: upper limit of normal; WBC: white blood count; WHO: World Health Organization
Differences between protocol and review
In this section, we do not only report the differences between the protocol and the current review version but the changes between each published version of the review. The summary of amendments are also provided in Table 4.
Types of studies
Differences between first and second published review version
(Valk 2020 to Piechotta 2020b)
As the evidence we found from the RCT was at unclear or high risk of bias and at serious risk of bias for the controlled NRSIs, and as none of these studies reported safety data for the control arm, we also included safety data from prospective and retrospective non‐comparative study designs (e.g. case series) and followed the methodology as specified in the protocol Piechotta 2020a). Because of the missing comparator, efficacy data of non‐controlled studies cannot be placed in context and therefore do not provide any useful evidence. In contrast to the protocol, we therefore decided to only include safety data of non‐controlled studies.
Differences between second and third published review version
(Piechotta 2020b to Chai 2020)
We decided to include registered non‐controlled NRSIs only to minimise selection bias.
Differences between third published review version and current version
(Chai 2020 to current version)
Originally we had planned to include different study designs in a top down approach: randomised controlled trials, prospective and retrospective controlled non‐randomised studies of interventions (NRSIs), and prospective and retrospective registered non‐controlled NRSIs. We had planned to include the next lower level in case we had low or very low certainty in the evidence of higher‐quality studies.
However, because large‐scale or expanded access studies could still provide valuable information on the safety of convalescent plasma or hyperimmune immunoglobulins, we decided to include prospectively registered single‐arm studies, even if upcoming RCTs report safety data for both groups. We decided to consider prospectively registered single‐arm studies only for safety data, and if 500 or more participants were included.
Types of participants
Differences between third published review version and current version
(Chai 2020 to current version)
After discussion with different attending physicians and clinical experts, we decided to perform separate analyses for populations with ambulatory mild disease and for hospitalised participants with moderate to severe disease, according to the latest WHO clinical progression score (https://revman.cochrane.org/#/660020041013463556/dashboard/htmlCompare/3.7.55/3.6#REF‐WHO‐2020e). We discussed that patient and study characteristics were not homogenous enough to be combined and outcomes of interest differ.
Types of intervention
Differences between first and second published review version
(Valk 2020 to Piechotta 2020b)
We added standard immunoglobulin as an eligible control treatment.
Types of outcome measures
Differences between protocol and first published review version
(Piechotta 2020a to Valk 2020)
We revised the secondary outcome 'Improvement of clinical symptoms, assessed through need for respiratory support at up to 7 days; 8 to 15 days; 16 to 30 days' and added the fourth bullet point: 'plus high‐flow oxygen', to differentiate from the third bullet point. After revision, it read:
oxygen by mask or nasal prongs
oxygen by NIV (non‐invasive ventilation) or high flow
intubation and mechanical ventilation
extracorporeal membrane oxygenation (ECMO)
Differences between first and second published review version
(Valk 2020 to Piechotta 2020b)
We added the outcome 'quality of life' after discussion with a patient representative.
Differences between second and third published review version
(Piechotta 2020b to Chai 2020)
We renamed the outcome 'time to death' as 'mortality (time to event)'. This did not change the outcome measurement we are interested in.
We revised the secondary outcome 'Improvement of clinical symptoms' according to the revised outcome measure set for COVID‐19 clinical research (COMET 2020). Instead of defining cut‐offs ourselves, we refer to the recommended standardised scales. It read:
Improvement of clinical symptoms, assessed by need for respiratory support with standardised scales (e.g. WHO Clinical Progression Scale (WHO 2020e), WHO Ordinal Scale for Clinical Improvement (WHO 2020f)) at up to 7 days, 8 to 15 days, 16 to 30 days.
We added the outcome 'virological response assessed with reverse transcription polymerase chain reaction (RT‐PCR) test for SARS‐CoV‐2 at baseline, up to 3, 7, and 15 days' because this was suggested during the peer review of the last version of this review.
Differences between third published review version and current version
(Chai 2020 to current version)
We divided efficacy outcomes for hospitalised individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease and ambulatory managed individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease, according to WHO clinical progression scale (WHO 2020e).
For individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease, we added the outcomes admission to hospital, development of moderate to severe clinical COVID‐19 symptoms, time to symptom onset, and any grade adverse events; and length of hospital stay, for subgroup of participants being hospitalised in the course of disease.
We revised and redefined outcomes for individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease after discussion with intensive care specialists and the German guideline panel for inpatient therapy of people with COVID‐19. We summarised different outcome measures for all‐cause mortality below one outcome, added sub‐outcomes for clinical improvement, and added clinical worsening to better reflect the course of disease and to detect group differences. We also added the outcome need for dialysis, and extended the definition of quality of life to also include fatigue and functional independence;
We renamed the outcome 'time to discharge from hospital' to 'Duration of hospitalisation, or time to discharge from hospital' to clarify that we are interested in both, continuous and time‐to‐event data. We renamed the outcome 'virological response' to 'viral clearance' to clarify that we are interested in test‐negativity and not in changes of viral load.
Electronic searches
Differences between protocol and first published review version
(Piechotta 2020a to Valk 2020)
As publication bias might influence all subsequent analyses and conclusions, we searched all potential relevant trials registries in detail to detect ongoing as well as completed studies, but not yet published studies. Nowadays, it is mandatory to provide results at least in the trials registry. In case results were not published elsewhere, we had planned to extract and analyse these data. However, no outcome data had yet been added to the trials registries.
Differences between first and second published review version
(Valk 2020 to Piechotta 2020b)
We decided to exclude individual study registries from the search strategy, because they are already included in the Cochrane COVID‐19 Study Register, which is updated Monday to Friday and to also exclude the WHO COVID‐19 Global Research Database. The WHO COVID‐19 Global Research Database and LitCov are included in the collection of Center for Disease Control and Prevention COVID‐19 Research Article Database. The search part for COVID‐19 was updated for the search strategies from IM and CD peer reviewed it.
Differences between third published review version and current version
(Chai 2020 to current version)
In the list of databases The Living Overview of Evidence (L*OVE) Covid‐19 provided from Epistemonikos is included due its variety of sources it contains (since September 2020) and the WHO COVID‐19 global literature on coronavirus disease was added because it integrates the CDC database (since October 2020).
New identified search terms like the MeSH term Immunization, Passive exploded and these search strings (passiv* adj3 (antibod* transfer* or immunization* or immunotherap* or immuno‐therap*)).tw,kf.; ((immunoglobulin* or immune globulin*) adj2 (therap* or treat*)).tw,kf.; (INM005 or CSL760).tw,kf.; (XAV‐19 or SAB‐185 or hIVIG or equine).tw,kf. were searched from November 2020. Due to the availability of more studies, the searches were focused on nonRCTs and RCTs with adequate study filters (since December 2020). At the beginning of 2021, new MeSH or EMTREE terms were inserted in Medline and Embase, so the whole search strategies were revised and new search terms like IGY‐110 or GIGA‐2050 or GC5131 or 5131A or INOSARS were added. The search string (passive adj2 vaccin*).tw,kf. and new terms for hyperimmune like equine polyclonal antibodies (EpAbs), hyperimmune anti‐COVID‐19 IVIG (C‐IVIG), anti‐coronavirus immunoglobulin (ITAC), flebogamma were included in February 2021.
Data extraction and management
Differences between protocol and first published review version
(Piechotta 2020a to Valk 2020)
We had planned to extract data using a standardised data extraction form developed in Covidence. However, we could not adapt the standardised form to our needs. Therefore we generated a customised data extraction form in Microsoft Excel (Microsoft Corporation 2018).
Differences between first and second published review version
(Valk 2020 to Piechotta 2020b)
Assessment of risk of bias in included studies
Randomised controlled trials
We had planned to use the Risk of Bias 2.0 (RoB 2) tool to analyse the risk of bias in the underlying study results (Sterne 2019). However, RoB 2 is not yet available in RevMan Web (Review Manager Web), and the Cochrane Editorial and Methods Department recommended for us to use the former 'Risk of bias' tool for this version of the review (Higgins 2011), instead.
Differences between second and third published review version
(Piechotta 2020b to Chai 2020)
Measures of treatment effect
We had planned to use the Excel tool of the purpose‐built method based on the Parmar and Tierney approach (https://revman.cochrane.org/#/660020041013463556/dashboard/htmlCompare/3.6/2.11#REF‐Parmar‐1998; https://revman.cochrane.org/#/660020041013463556/dashboard/htmlCompare/3.6/2.11#REF‐Tierney‐2007), to estimate hazard ratios (HRs) with the reported data, if HRs were not available. We were able to read off mortality data from the Kaplan‐Meier curve provided by https://revman.cochrane.org/#/660020041013463556/dashboard/htmlCompare/3.6/2.11#STD‐Gharbharan‐2020 per day. Because we did not have the rights to edit the Excel tool to add a greater number of time intervals, we could not use the Excel tool. We therefore used a digitising software (https://revman.cochrane.org/#/660020041013463556/dashboard/htmlCompare/3.6/2.11#REF‐GetData‐Graph‐Digitizer) to estimate the HR for https://revman.cochrane.org/#/660020041013463556/dashboard/htmlCompare/3.6/2.11#STD‐Gharbharan‐2020.
Assessment of risk of bias in included studies
Randomised controlled trials
The Risk of Bias 2.0 (RoB 2) tool is meanwhile available in RevMan web. We therefore decided to revert to our originally planned methodology for risk of bias assessment in randomised controlled trials, and used RoB 2.0 for any assessments.
Differences between third published review version and current version
(Chai 2020 to current version)
Subgroup analysis and investigation of heterogeneity
We had planned to add subgroup analyses for the following characteristics in this update of the review.
Duration since symptom onset
Level of antibody titre in donors
Level of antibody titre in recipients at baseline
SARS‐CoV‐2 variants
Considering the currently available evidence, we decided to add these subgroups, because their role in the effectiveness of convalescent plasma is currently discussed and needs to be further investigated.
Summary of findings and assessment of the certainty of the evidence
Differences between protocol and first published review version
(Piechotta 2020a to Valk 2020)
At protocol stage we had planned to assess the certainty of the evidence for our primary outcomes (all‐cause mortality at hospital discharge and time to death), only. However, as none of the included studies reported any deaths during their study periods, we decided to assess the certainty of the evidence also for prioritised secondary outcomes (clinical improvement, grade 3 and 4 adverse events, and serious adverse events) to increase the informative value on effectiveness and safety of convalescent plasma therapy.
Differences between first and second published review version
(Valk 2020 to Piechotta 2020b)
For the living systematic review we also prioritised patient quality of life as an important patient outcome and added this outcome to the 'Summary of findings' table. We specified in the methods how we graded the certainty of the evidence, especially for non‐randomised controlled trials using ROBINS‐I for 'Risk of bias' assessment, for calculation of absolute effects for time‐to‐event outcomes and for writing informative statements for the findings and certainty of the evidence.
Differences between third published review version and current version
(Chai 2020 to current version)
We decided to include two 'Summary of findings' tables, one for each population.
We amended the outcomes for inclusion into the summary of findings table, in accordance with redefining the types of outcome measures. We summarised the outcome all‐cause mortality and provide a hierachy of outcome measures that we would consider for inclusion in the summary of findings table. We added clinical worsening, in addition to clinical improvement, to better reflect the course of disease, and also provide a hierachy for sub‐outcomes of all‐cause mortality.
Contributions of authors
VP: methodological expertise, study selection, data extraction and assessment, conception and writing of the manuscript
CI: methodological expertise, study selection, data extraction and assessment, writing of the manuscript
KLC: clinical expertise, study selection, data extraction and assessment, conception and writing of the manuscript
SJV: clinical expertise, study selection, data extraction and assessment, conception and writing of the manuscript
CK: clinical expertise, study selection, and advice
ED: study selection, data extraction and assessment, writing of the manuscript
IM: development of the search strategy
EMW: clinical expertise and advice
AL: clinical expertise and advice
DJR: clinical expertise and advice
ZM: clinical expertise and advice
CS‐O: clinical expertise and advice
LJE: clinical expertise, and conception and writing of the manuscript
NS: methodological expertise, study selection, data extraction and assessment, conception and writing of the manuscript
Sources of support
Internal sources
-
Sanquin Blood Supply, Netherlands
Center for Clinical Transfusion Research
-
University Hospital of Cologne, Germany
Cochrane Cancer, Department I of Internal Medicine
-
Monash University, Australia
Transfusion Research Unit, Department of Epidemiology and Preventive Medicine
-
NHS Blood and Transplant, UK
NHS Blood and Transplant
-
Leukaemia Foundation and HSANZ, Australia
Haematology Society of Australia and New Zealand (HSANZ)
External sources
-
European Union's Horizon 2020 research and innovation programme, Belgium
SUPPORTing high‐quality evaluation of covid‐19 convalescent plasma throughout EUROPE (Support‐E)
Declarations of interest
VP: none known
CI: none known
KLC: HSANZ Leukaemia Foundation PhD scholarship to support studies at Monash University. This is not related to the work in this review.
SJV: is receiving a PhD scholarship from the not‐for‐profit Sanquin blood bank.
CK: none known
ED: none known
IM: none known
EMW: I have received funding support from Australian Medical Research Future Fund for a trial of convalescent plasma. I was not involved in bias assessment, data extraction or interpretation, but served as a content expert.
AL: none known
DJR: investigator on the REMAP‐CAP and RECOVERY trial. I was not involved in bias assessment, data extraction or interpretation, but served as a content expert.
ZM: I have received funding support from Australian Medical Research Future Fund for a trial of convalescent plasma. I was not involved in bias assessment, data extraction or interpretation, but served as a content expert.
CS‐O: is a member of the BEST Collaborative Clinical Study Group and Associate Editor for Transfusion Medicine Journal. I was not involved in bias assessment, data extraction or interpretation, but served as a content expert.
LJE: co‐lead of the COVID‐19 immunoglobulin domain of the REMAP‐CAP trial and investigator on the RECOVERY trial. I was not involved in bias assessment, data extraction or interpretation, but served as a content expert.
NS: none known
contributed equally
contributed equally
contributed equally
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Agarwal 2020 {published data only}
- Agarwal A , Mukherjee A, Kumar G, Chatterjee P, Bhatnagar T, Malhotra P, et al. Convalescent plasma in the management of moderate COVID-19 in India: an open-label parallel-arm phase II multicentre randomized controlled trial (PLACID Trial) (preprint). medRxiv 10 September 2020. [DOI: 10.1101/2020.09.03.20187252] [DOI]
- Agarwal A, Mukherjee A, Kumar G, Chatterjee P, Bhatnagar T, Malhotra P, et al. Convalescent plasma in the management of moderate Covid-19 in adults in India: open label phase II multicentre randomised controlled trial (PLACID trial). BMJ 2020;371:m3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
AlQahtani 2020 {published data only}
- AlQahtani M, Abdulrahman A, Almadani A, Alali SY, Al Zamrooni AM, Hejab A, et al. Randomized controlled trial of convalescent plasma therapy against standard therapy in patients with severe COVID-19 disease (preprint). Medrxiv 2020:2020.11.02.20224303. [DOI] [PMC free article] [PubMed]
Avendano‐Sola 2020 {published data only}
- Avendano-Sola C, Ramos-Martinez A, Munez-Rubio E, Ruiz-Antoran B, Malo de Molina R, Torres F, et al. Convalescent plasma for COVID-19: a multicenter, randomized clinical trial (preprint). medRxiv 2020:2020.08.26.20182444. [DOI: 10.1101/2020.08.26.20182444] [DOI]
- Diago-Sempere E, Bueno JL, Sancho-Lopez A, Munez-Rubio E, Torres F, Malo de Molina R, et al. Evaluation of convalescent plasma versus standard of care for the treatment of COVID-19 in hospitalized patients: study protocol for a phase 2 randomized, open-label, controlled, multicenter trial. Trials 2021;22(1):70. [DOI] [PMC free article] [PubMed]
Bajpai 2020 {published data only}
- Bajpai M, Kumar S, Maheshwari A, Chabra K, Kale P, Gupta A, et al. Efficacy of convalescent plasma therapy compared to fresh frozen plasma in severely ill COVID-19 patients: a pilot randomized controlled trial (preprint). Medrxiv 2020:2020.10.25.20219337. [DOI: 10.1101/2020.10.25.20219337] [DOI]
Gharbharan 2020 {published and unpublished data}
- Gharbharan A, Jordans C, GeurtsvanKessel C, Hollander J, Karim F, Mollema F, et al. Effects of potent neutralizing antibodies from convalescent plasma in patients hospitalized for severe SARS-CoV-2 infection (preprint). ResearchSquare 2020. [DOI] [PMC free article] [PubMed]
- Gharbharan A, Jordans CC, GeurtsvanKessel C, den Hollander JG, Karim F, Mollema FP, et al. Convalescent plasma for COVID-19. A randomized clinical trial (preprint). medRxiv 2020. [DOI: 10.1101/2020.07.01.20139857] [DOI]
Hamdy Salman 2020 {published data only}
- Hamdy Salman O, Ail Mohamed HS. Efficacy and safety of transfusing plasma from COVID-19 survivors to COVID-19 victims with severe illness. a double-blinded controlled preliminary study. Egyptian Journal of Anaesthesia 2020;36(1):264-272. [Google Scholar]
Horby 2021 {published data only}
- Horby PW, Estcourt L, Peto L, Emberson JR, Staplin N, Spata E, et al. Convalescent plasma in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial (preprint). medRxiv 2021:2021.03.09.21252736.
Joyner 2020 {published data only}
- Diago-Sempere E, Bueno LJ , Sancho-López A , Múñez Rubio E , Torres F, Malo de Molina R, et al. Evaluation of convalescent plasma versus standard of care for the treatment of COVID-19 in hospitalized patients: study protocol for a phase 2 randomized, open-label, controlled, multicenter trial. Trials 20 January 2021;22(1):70. [DOI: 10.1186/s13063-020-05011-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman W, Hess AS, Connor JP. Hospitalized COVID-19 patients treated with convalescent plasma in a mid-size city in the midwest (preprint). medRxiv 2020. [DOI: 10.1101/2020.06.19.20135830] [DOI] [PMC free article] [PubMed]
- Hegerova L, Gooley T, Sweerus KA, Maree CL, Bailey N, Bailey M, et al. Use of convalescent plasma in hospitalized patients with Covid-19 - case series. Blood 2020;136:759-762. [DOI: 10.1182/blood.2020006964] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner MJ, Bruno KA, Klassen SA, Kunze KL, Johnson PW, Lesser ER, et al. Safety update: COVID-19 convalescent plasma in 20,000 hospitalized patients. Mayo Clinical Proceedings 2020;95(9):1888-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner MJ , Senefeld JW , Klassen SA , Mills JR , Johnson PW , Theel ES, et al. Effect of convalescent plasma on mortality among hospitalized patients with COVID-19: initial three-month experience (preprint). medRxiv 12 August 2020. [DOI: ]
- Joyner MJ, Wright RS, Fairweather D, Senefeld JW, Bruno KA, Klassen SA, et al. Early safety indicators of COVID-19 convalescent plasma in 5,000 patients. Journal of Clinical Investigation 2020;130(9):4791-7. [DOI: 10.1172/JCI140200] [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2020 {published data only}
- Li L, Zhang W, Hu Y, Tong X, Zheng S, Yang J, et al. Effect of convalescent plasma therapy on time to clinical improvement in patients with severe and life-threatening COVID-19: a randomized clinical trial. JAMA 2020 Jun 3 [Epub ahead of print];324:460-470. [DOI: 10.1001/jama.2020.10044] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhang W, Hu Y. Correction to data in trial of convalescent plasma treatment for COVID-19. Jama 4 August 2020;324(5):519-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. Errors in trial of effect of convalescent plasma therapy on time to clinical improvement in patients with severe and life-threatening COVID-19. JAMA 2020;324(5):518-9. [DOI] [PubMed] [Google Scholar]
Libster 2020 {published data only}
- Libster R, Marc GP, Wappner D, Coviello S, Bianchi A, Braem V, et al. Early high-titer plasma therapy to prevent severe Covid-19 in older adults. New England Journal of Medicine 2021;384(7):610-618. [DOI: 10.1056/NEJMoa2033700] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libster R , Marc GP , Wappner D , Coviello S , Bianchi A , Braem V, et al. Prevention of severe COVID-19 in the elderly by early high-titer plasma (preprint). medRxiv 21 November 2020. [DOI: 10.1101/2020.11.20.20234013] [DOI]
O’Donnell 2021 {published data only}
- Eckhardt CM, Cummings MJ, Rajagopalan KN, Borden S, Bitan ZC, Wolf A, et al. Correction to: evaluating the efficacy and safety of human anti-SARS-CoV-2 convalescent plasma in severely ill adults with COVID-19: a structured summary of a study protocol for a randomized controlled trial. Trials 17 November 2020;21(1):927. [DOI: 10.1186/s13063-020-04877-z] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt CM, Cummings MJ, Rajagopalan KN, Borden S, Bitan ZC, Wolf A, et al. Evaluating the efficacy and safety of human anti-SARS-CoV-2 convalescent plasma in severely ill adults with COVID-19: a structured summary of a study protocol for a randomized controlled trial. Trials 2020;21(1):499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell M, Grinsztejn B, Cummings MJ, Justman J, Lamb MR, Eckhardt CM, et al. A randomized, double-blind, controlled trial of convalescent plasma in adults with severe COVID-19 (preprint). Medrxiv 2021. [DOI: 10.1101/2021.03.12.21253373] [DOI] [PMC free article] [PubMed]
Ray 2020 {published data only}
- Bandopadhyay P, Rozario R, Lahiri A, Sarif J, Ray Y, Paul SR, et al. Nature and dimensions of the cytokine storm and its attenuation by convalescent plasma in severe COVID-19 (preprint). medRxiv 2020:2020.09.21.20199109.
- Ray Y, Paul SR, Bandopadhyay P, D'Rozario R, Sarif J, Lahiri A, et al. Clinical and immunological benefits of convalescent plasma therapy in severe COVID-19: insights from a single center open label randomised control trial (preprint). medRxiv 2020. [DOI: 10.1101/2020.11.25.20237883] [DOI]
Simonovich 2020 {published data only}
- Simonovich VA, Burgos PLD, Scibona P, Beruto MV, Vallone MG, Vázquez C, et al. A randomized trial of convalescent plasma in Covid-19 severe pneumonia. New England Journal of Medicine 2020;384(7):619-629. [DOI: 10.1056/NEJMoa2031304] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Abdullah 2020 {published data only}
- Abdullah HM, Hama-Ali HH, Ahmed SN, Ali KM, Karadakhy KA, Mahmood SO, et al. A severe refractory COVID-19 patient responding to convalescent plasma; a case series. Annals of Medicine and Surgery 2020;56:125-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Abolghasemi 2020 {published data only}
- Abolghasemi H, Eshghi P, Cheraghali AM, Imani Fooladi AA, Bolouki Moghaddam F, Imanizadeh S, et al. Clinical efficacy of convalescent plasma for treatment of COVID-19 infections: results of a multicenter clinical study. Transfusion and Apheresis Science 2020 July 15 [Epub ahead of print];59(5):102875. [DOI: 10.1016/j.transci.2020.102875] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ahn 2020 {published data only}
- Ahn JY, Sohn Y, Lee SH, Cho Y, Hyun JH, Baek YJ, et al. Use of convalescent plasma therapy in two COVID-19 patients with acute respiratory distress syndrome in Korea. Journal of Korean Medical Science 2020;35(14):e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Anderson 2020 {published data only}
- Anderson J, Schauer J, Bryant S, Graves CR. The use of convalescent plasma therapy and remdesivir in the successful management of a critically ill obstetric patient with novel coronavirus 2019 infection: a case report. Case Reports in Women's Health 2020;27:e00221. [DOI: 10.1016/j.crwh.2020.e00221] [DOI] [PMC free article] [PubMed] [Google Scholar]
Baklaushev 2020 {published data only}
- Baklaushev VP, Averyanov AV, Sotnikova AG, Perkina AS, Ivanov AV, Yusubalieva GM, et al. Safety and efficacy of convalescent plasma for COVID-19: the preliminary results of a clinical trial. Journal of Clinical Practice 2020;11(2):38-50 %J J Clin Pract. [Google Scholar]
Balcells 2020 {published data only}
- Balcells ME, Rojas L, Le Corre N, Martínez-Valdebenito C, Ceballos ME, Ferrés M, et al. Early anti-SARS-CoV-2 convalescent plasma in patients admitted for COVID-19: a randomized phase II clinical trial (preprint). medRxiv 2020:2020.09.17.20196212. [DOI] [PMC free article] [PubMed]
Bao 2020b {published data only}
- Bao Y, Lin SY, Cheng ZH, Xia J, Sun YP, Zhao Q, et al. Clinical features of COVID-19 in a young man with massive cerebral hemorrhage—case report. SN Comprehensive Clinical Medicine 2020 May 23 [Epub ahead of print];2:703–709. [DOI: 10.1007/s42399-020-00315-y] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bobek 2020 {published data only}
- Bobek I, Gopcsa L, Reti M, Beko G, Hancz L, Lakatos B, et al. Successful administration of convalescent plasma in critically ill COVID-19 patients in Hungary: the first two cases. Orvosi Hetilap 2020;161(27):1111-21. [DOI] [PubMed] [Google Scholar]
Bradfute 2020 {published data only}
- Bradfute SB, Hurwitz I, Yingling AV, Ye C, Cheng Q, Noonan TP, et al. SARS-CoV-2 neutralizing antibody titers in convalescent plasma and recipients in New Mexico: an open treatment study in COVID-19 patients. Journal of Infectious Diseases 2020;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Brasil Ministerio 2020 {published data only}
- Brasil Ministério da Saúde, Secretaria de Ciência. (no translation) [Tratamento farmacológico para casos internados com SARS-COV-2, do Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo]. Available from fi-admin.bvsalud.org/document/view/27t7v (accessed 25 June 2020).
Budhai 2020 {published data only}
- Budhai A, Wu AA, Hall L, Strauss D, Paradiso S, Alberigo J, et al. How did we rapidly implement a convalescent plasma program? Transfusion 2020 May 25 [Epub ahead of print];60:1348-1355. [DOI: 10.1111/trf.15910] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cantore 2020 {published data only}
- Cantore I, Valente P. Convalescent plasma from COVID 19 patients enhances intensive care unit survival rate. A preliminary report. Transfusion and Apheresis Science 2020 Jun 10 [Epub ahead of print];59(5):102848. [DOI: 10.1016/j.transci.2020.102848] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cao 2020a {published data only}
- Cao W, Liu X, Bai T, Fan H, Hong K, Song H, et al. High-dose intravenous immunoglobulin as a therapeutic option for deteriorating patients with coronavirus disease 2019. Open Forum Infectious Diseases 2020;7(3):ofaa102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2020b {published data only}
- Chen X, Li Y, Wang J, Cai H, Cao H, Sheng J. Pregnant women complicated with COVID-19: a clinical analysis of 3 cases. Zhejiang da Xue Xue Bao. Yi Xue Ban = Journal of Zhejiang University. Medical Sciences 2020;49(2):240-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2020c {published data only}
- Chen Q, Quan B, Li X, Gao G, Zheng W, Zhang J, et al. A report of clinical diagnosis and treatment of nine cases of coronavirus disease 2019. Journal of Medical Virology 2020;92(6):683-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
ChiCTR2000029850 {published data only}
- ChiCTR2000029850. Efficacy and safety of convalescent plasma treatment for severe patients with novel coronavirus pneumonia (COVID-19): a prospective cohort study. Available from www.chictr.org.cn/showproj.aspx?proj=49533 (first received 15 February 2020).
ChiCTR2000030039 {published data only}
- ChiCTR2000030039. Clinical study for infusing convalescent plasma to treat patients with new coronavirus pneumonia (COVID-19). Available from www.chictr.org.cn/showproj.aspx?proj=49544 (first received 21 February 2020).
ChiCTR2000030312 {published data only}
- ChiCTR2000030312. Cancelled, due to modify the protocol A single-center, open-label and single arm trial to evaluate the efficacy and safety of anti-SARS-CoV-2 inactivated convalescent plasma in the treatment& [A single-center, open-label and single arm trial to evaluate the efficacy and safety of anti-SARS-CoV-2 inactivated convalescent plasma in the treatment of novel coronavirus pneumonia (COVID-19) patient]. Available from www.chictr.org.cn/showproj.aspx?proj=50258 (first received 23 April 2020).
ChiCTR2000030381 {published data only}
- ChiCTR2000030381. Cancelled by investigator. A randomized, open-label, controlled and single-center trial to evaluate the efficacy and safety of anti-SARS-CoV-2 inactivated convalescent plasma in the treatment of novel coronavirus pneumonia (COVID-19) patient [A randomized, open-label, controlled and single-center trial to evaluate the efficacy and safety of anti-SARS-CoV-2 inactivated convalescent plasma in the treatment of novel coronavirus pneumonia (COVID-19) patient]. Available from www.chictr.org.cn/showproj.aspx?proj=50290 (first received 23 April 2020).
ChiCTR2000030442 {published data only}
- ChiCTR2000030442. Combination of tocilizumab, IVIG and CRRT in severe patients with novel coronavirus pneumonia (COVID-19). Available from www.chictr.org.cn/showproj.aspx?proj=50380 (first received 23 April 2020).
ChiCTR2000031501 {published data only}
- ChiCTR2000031501. The efficacy of convalescent plasma in patients with critical novel coronavirus pneumonia (COVID-19): a pragmatic, prospective cohort study. Available from www.chictr.org.cn/showproj.aspx?proj=50254 (first received 2 April 2020).
ChiCTR2000033798 {published data only}
- ChiCTR2000033798. The efficacy and safety of convalescent plasma therapy in novel coronavirus pneumonia (COVID-19): a medical records based retrospective cohort study. Available from www.chictr.org.cn/showproj.aspx?proj=55194 (first received 12 June 2020).
Clark 2020 {published data only}
- Clark E, Guilpain P, Filip L, Pansu N, Le Bihan C, Cartron G, et al. Convalescent plasma for persisting COVID-19 following therapeutic lymphocyte depletion: a report of rapid recovery. British Journal of Haematology 2020;27:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
CTRI/2020/04/024804 {published data only}
- CTRI/2020/04/024804. Evaluation of safety and efficacy of convalescent plasma in COVID-19 patients. Available from ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=42849 (first received 22 April 2020).
CTRI/2020/08/027285 {published data only}
- CTRI/2020/08/027285. Safety of convalescent plasma (CVP) drawn from mild symptomatic COVID-19 patients. Available from www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=45050 (first received 21 August 2020).
CTRI/2020/10/028547 {published data only}
- CTRI/2020/10/028547. IND02 for prevention against SARS-CoV-2 infection: a randomized controlled study in moderate to high risk population. Available from ctri.nic.in/Clinicaltrials/showallp.php?mid1=48708&EncHid=&userName=2020/10/028547.
Çınar 2020 {published data only}
- Çınar OE, Sayınalp B, Karakulak EA, Karataş AA, Velet M, İnkaya AÇ, et al. Convalescent (immune) plasma treatment in a myelodysplastic COVID-19 patient with disseminated tuberculosis. Transfusion and Apheresis Science 2020;59(5):102821. [DOI: 10.1016/j.transci.2020.102821] [DOI] [PMC free article] [PubMed] [Google Scholar]
de Assis 2020 {published data only}
- Assis RR, Jain A, Nakajima R, Jasinskas A, Felgner J, Obiero JM, et al. Analysis of SARS-CoV-2 antibodies in COVID-19 convalescent plasma using a coronavirus antigen microarray (preprint). bioRxiv 2020. [DOI: 10.1101/2020.04.15.043364] [DOI] [PMC free article] [PubMed]
Díez 2020 {published data only}
- Díez JM, Romero C, Gajardo R. Currently available intravenous immunoglobulin contains antibodies reacting against severe acute respiratory syndrome coronavirus 2 antigens. Immunotherapy 2020 (Epub 2020 May );12:571-576. [DOI: 10.2217/imt-2020-0095] [DOI] [PMC free article] [PubMed] [Google Scholar]
Donato 2020 {published data only}
- Donato M, Park S, Baker M, Korngold R, Morawski A, Geng X, et al. Clinical and laboratory evaluation of patients with SARS-CoV-2 pneumonia treated with high-titer convalescent plasma: a prospective study (preprint). medRxiv 2020. [DOI: 10.1101/2020.07.20.20156398] [DOI] [PMC free article] [PubMed]
Duan 2020 {published data only}
- Duan K, Liu B, Li C, Zhang H, Yu T, Qu J, et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proceedings of the National Academy of Sciences of the United States of America 2020;117:9490-6. [DOI: 10.1073/pnas.2004168117] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan K, Liu B, Li C, Zhang H, Yu T, Qu J, et al. The feasibility of convalescent plasma therapy in severe COVID-19 patients: a pilot study (preprint). medRxiv 2020. [DOI: 10.1101/2020.03.16.20036145] [DOI]
Dulipsingh 2020 {published data only}
- Dulipsingh L, Danyal I, Crowell R, Diffenderfer M, Williams K, Lima C, et al. SARS-CoV-2 serology and virology trends in donors and recipients of convalescent plasma (preprint). SSRN [Preprint] 2020. [ssrn.com/abstract=3669324] [DOI] [PMC free article] [PubMed]
Enzmann 2020 {published data only}
- Enzmann MO, Erickson MP, Grindeland CJ, Lopez SM, Hoover SE, Leedahl DD. Treatment and preliminary outcomes of 150 acute care patients with COVID-19 in a rural health system in the Dakotas. Epidemiology and Infection 2020;148:e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Erkurt 2020 {published data only}
- Erkurt MA, Sarici A, Berber I, Kuku I, Kaya E, Ozgul M. Life-saving effect of convalescent plasma treatment in COVID-19 disease: clinical trial from eastern Anatolia. Transfusion and Apheresis Science 2020 Jun 27 [Epub ahead of print];59(5):102867. [DOI: 10.1016/j.transci.2020.102867] [DOI] [PMC free article] [PubMed] [Google Scholar]
Fan 2020 {published data only}
- Fan O, Qiang F, Shuhong G, Haibing Y, Xiangyang L, Min T, et al. Recovery from critical COVID-19 despite delays in diagnosis and respiratory treatment: a cautionary tale. Signa Vitae 2020;16(1):193-8. [Google Scholar]
Figlerowicz 2020 {published data only}
- Figlerowicz M, Mania A, Lubarski K, Lewandowska Z, Sluzewski W, Derwich K, et al. First case of convalescent plasma transfusion in a child with COVID-19-associated severe aplastic anemia. Transfusion and Apheresis Science 2020 July 1 [Epub ahead of print];59(5):102866. [DOI: 10.1016/j.transci.2020.102866] [DOI] [PMC free article] [PubMed] [Google Scholar]
Franchini 2020 {published data only}
- Franchini M, Marano G, Velati C, Pati I, Pupella S, Liumbruno GM. Operational protocol for donation of anti-COVID-19 convalescent plasma in Italy. Vox Sanguinis 2021;116(1):136-7. [DOI: 10.1111/vox.12940] [DOI] [PMC free article] [PubMed] [Google Scholar]
Grisolia 2020 {published data only}
- Grisolia G, Franchini M, Glingani C, Inglese F, Garuti M, Beccaria M, et al. Convalescent plasma for coronavirus disease 2019 in pregnancy: a case report and review. American Journal of Obstetrics and Gynecology 2020;2(3):100174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hashim 2020 {published data only}
- Hashim HT. Convalescent plasma to treat COVID-19: its challenges in Iraq situation. Ethics, Medicine and Public Health 2020 Jul 16 [Epub ahead of print];15:100564. [DOI: 10.1016/j.jemep.2020.100564] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hu 2020 {published data only}
- Hu H, Ma F, Wei X, Fang Y. Coronavirus fulminant myocarditis saved with glucocorticoid and human immunoglobulin. European Heart Journal 2020;16:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ibrahim 2020 {published data only}
- Ibrahim D, Dulipsingh L, Zapatka L, Eadie R, Crowell R, Williams K, et al. Factors associated with good patient outcomes following convalescent plasma in COVID-19: a prospective phase II clinical trial. Infectious Diseases and Therapy 2020 [Epub ahead of print];9:913–926. [DOI: 10.1007/s40121-020-00341-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Im 2020 {published data only}
- Im JH, Nahm CH, Baek JH, Kwon HY, Lee JS. Convalescent plasma therapy in coronavirus disease 2019: a case report and suggestions to overcome obstacles. Journal of Korean Medical Science 2020;35(26):e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
IRCT20151228025732N53 {published data only}
- IRCT20151228025732N53. Therapeutic effects of plasma of recovered people from COVID-19 on hospitalized patients with this disease. Available from en.irct.ir/trial/46931 (first received 10 April 2020).
IRCT20200406046968N2 {published data only}
- IRCT20200406046968N2. Efficacy of convalescent plasma transfusion of COVID-19 survivors on the treatment of respiratory failure of these patients. Available from www.irct.ir/trial/47115 (first received 22 April 2020).
IRCT20200414047072N1 {published data only}
- IRCT20200414047072N1. Use of convalescent plasma in the treatment of COVID-19. Available from en.irct.ir/trial/47163 (first received 28 April 2020).
IRCT20200416047099N1 {published data only}
- IRCT20200416047099N1. Plasma therapy in patient with COVID-19. Available from en.irct.ir/trial/47266 (first received 21 April 2020).
ISRCTN86534580 {published data only}
- ISRCTN86534580. A trial evaluating treatments for suspected coronavirus infection in people aged 50 years and above with pre-existing conditions and those aged 65 years and above. Available from www.isrctn.com/ISRCTN86534580 (first received 20 March 2020).
Jamous 2020 {published data only}
- Jamous F, Meyer N, Buus D, Ateeli H, Taggart K, Hanson T, et al. Critical illness due to COVID-19: a description of the surge in a single center in Sioux Falls. South Dakota Medicine 2020;73(7):312-7. [PubMed] [Google Scholar]
Jiang 2020a {published data only}
- Jiang J, Miao Y, Zhao Y, Lu X, Zhou P, Zhou X, et al. Convalescent plasma therapy: helpful treatment of COVID-19 in a kidney transplant recipient presenting with severe clinical manifestation and complex complications. Clinical Transplantation 2020 Jun 30 [Epub ahead of print];34:e14025. [DOI: 10.1111/ctr.14025] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jiang 2020b {published data only}
- Jiang Y, He S, Zhang C, Wang X, Chen X, Jin Y, et al. Clinical characteristics of 60 discharged cases of 2019 novel coronavirus-infected pneumonia in Taizhou, China. Annals of Translational Medicine 2020;8(8):547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jin 2020 {published data only}
- Jin C, Gu J, Yuan Y, Long Q, Zhang Q, Zhou H, et al. Treatment of 6 COVID-19 patients with convalescent plasma (preprint). medRxiv 2020. [DOI: 10.1101/2020.05.21.20109512] [DOI]
Karatas 2020 {published data only}
- Karatas A, Inkaya AC, Demiroglu H, Aksu S, Haziyev T, Cinar OE, et al. Prolonged viral shedding in a lymphoma patient with COVID-19 infection receiving convalescent plasma. Transfusion and Apheresis Science 2020 October [Epub ahead of print ];59(5):102871. [DOI: 10.1016/j.transci.2020.102871] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kong 2020 {published data only}
- Kong Y, Cai C, Ling L, Zeng L, Wu M, Wu Y, et al. Successful treatment of a centenarian with coronavirus disease 2019 (COVID-19) using convalescent plasma. Transfusion and Apheresis Science 2020 May 21 [Epub ahead of print];59:102820. [DOI: 10.1016/j.transci.2020.102820] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lin 2020 {published data only}
- Lin JH, Chen YC, Lu CL, Hsu YN, Wang WJ. Application of plasma exchange in association with higher dose CVVH in cytokine storm complicating COVID-19. Journal of the Formosan Medical Association 2020;119(6):1116-8. [DOI: 10.1016/j.jfma.2020.04.023] [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 2020 {published data only}
- Liu ST, Lin HM, Baine I, Wajnberg A, Gumprecht JP, Rahman F, et al. Convalescent plasma treatment of severe COVID-19: a propensity score–matched control study. Nature Medicine 2020 September 15;26:1708–1713. [DOI: ] [DOI] [PubMed] [Google Scholar]
Liu 2020a {published data only}
- Liu M, Chen Z, Dai MY, Yang JH, Chen XB, Chen D, et al. Lessons learned from early compassionate use of convalescent plasma on critically ill patients with COVID-19. Transfusion 2020 Aug 8 [Epub ahead of print];60:2210-2216. [DOI: 10.1111/trf.15975] [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Madariaga 2020 {published data only}
- Madariaga ML, Guthmiller J, Schrantz S, Jansen M, Christenson C, Kumar M, et al. Clinical predictors of donor antibody titer and correlation with recipient antibody response in a COVID-19 convalescent plasma clinical trial. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.06.21.20132944] [DOI] [PMC free article] [PubMed]
- Madariaga ML, Schrantz S, Jansen MO, Christensen C, Kumar M, Prochaska M, et al. Integrated COVID-19 convalescent plasma treatment and antibody research program at a single academic medical center. SSRN 2020. [DOI: 10.2139/ssrn.3605131] [DOI]
Martinez‐Resendez 2020 {published data only}
- Martinez-Resendez MF, Castilleja-Leal F, Torres-Quintanilla A, Rojas-Martinez A, Garcia-Rivas G, Ortiz-Lopez R, et al. Initial experience in Mexico with convalescent plasma in COVID-19 patients with severe respiratory failure, a retrospective case series (preprint). medRxiv 2020. [DOI: 10.1101/2020.07.14.20144469] [DOI]
McCuddy 2020 {published data only}
Ministerio de Salud 2020 {published data only}
- Ministerio de salud - Instituto Nacional de Salud. (no translation) [Lineamientos técnicos para uso de plasma convaleciente en pacientes con COVID-19]. Available from fi-admin.bvsalud.org/document/view/nruba 2020;1:20.
Mira 2020 {published data only}
- Mira E, Yarce OA, Ortega C, Fernández S, Pascual N, Gómez C, et al. Rapid recovery of a SARS-CoV-2 infected X-linked agammaglobulinemia patient after infusion of COVID-19 convalescent plasma. Journal of Allergy and Clinical Immunology: In Practice 2020;8(8):2793-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT04261426 {published data only}
- NCT04261426. The efficacy of intravenous immunoglobulin therapy for severe 2019-nCoV infected pneumonia. Available from clinicaltrials.gov/ct2/show/NCT04261426 (first received 23 April 2020).
NCT04264858 {published data only}
- ChiCTR2000030841. Treatment of acute severe COVID-19 with immunoglobulin from cured COVID-19 patients. Available from www.chictr.org.cn/showproj.aspx?proj=51072 (first received 15 March 2020).
- NCT04264858. An exploratory clinical study on the treatment of acute severe 2019-nCoV pneumonia with immunoglobulin from cured 2019-nCoV pneumonia patients. Available from clinicaltrials.gov/show/NCT04264858 (first received 11 February 2020).
NCT04292340 {published data only}
- NCT04292340. The efficacy and safety of anti-SARS-CoV-2 inactivated convalescent plasma in the treatment of novel coronavirus pneumonia patient (COVID-19): an observational study. Available from clinicaltrials.gov/show/NCT04292340 (first received 3 March 2020).
NCT04321421 {published data only}
- NCT04321421. Hyperimmune plasma for critical patients with COVID-19. Available from clinicaltrials.gov/ct2/show/NCT04321421 (first received 28 May 2020).
NCT04323800 {published data only}
- NCT04323800. Convalescent plasma to stem coronavirus: a randomized, blinded phase 2 study comparing the efficacy and safety human coronavirus immune plasma (HCIP) vs. control (SARS-CoV-2 non-immune plasma) among adults exposed to COVID-19. Available from clinicaltrials.gov/show/NCT04323800 (first received 23 April 2020).
NCT04325672 {published data only}
- NCT04325672. Convalescent plasma to limit coronavirus associated complications: an open label, phase 2A study of high-titer anti-SARS-CoV-2 plasma in hospitalized patients with COVID-19. Available from clinicaltrials.gov/show/NCT04325672 (first received 23 April 2020).
NCT04327349 {published data only}
- NCT04327349. Investigating effect of convalescent plasma on COVID-19 patients outcome: a clinical trial. Available from clinicaltrials.gov/show/NCT04327349 (first received 31 March 2020).
NCT04332380 {published data only}
- NCT04332380. Convalescent plasma for patients with COVID-19: a pilot study. Available from clinicaltrials.gov/show/NCT04332380 (first received 2 April 2020).
NCT04333355 {published data only}
- NCT04333355. Phase 1 study to evaluate the safety of convalescent plasma as an adjuvant therapy in patients with SARS-CoV-2 infection. Available from clinicaltrials.gov/show/NCT04333355 (first received 3 April 2020).
NCT04344015 {published data only}
- NCT04344015. COVID-19 plasma collection. Available from clinicaltrials.gov/show/NCT04344015 (first received 23 April 2020).
NCT04344379 {published data only}
- NCT04344379. Prevention of SARS-CoV-2 in hospital workers exposed to the virus. Available from clinicaltrials.gov/show/NCT04344379 (first received 14 April 2020).
NCT04344977 {published data only}
- NCT04344977. COVID-19 plasma collection. Available from clinicaltrials.gov/ct2/show/NCT04344977 (first received 14 April 2020).
NCT04345679 {published data only}
- NCT04345679. Anti COVID-19 convalescent plasma therapy. Available from clinicaltrials.gov/show/NCT04345679 (first received 14 April 2020).
NCT04346589 {published data only}
- NCT04346589. Convalescent antibodies infusion in critically ill COVID 19 patients. Available from clinicaltrials.gov/ct2/show/NCT04346589 (first received 15 April 2020).
NCT04347681 {published data only}
- Albalawi M, Zaidi SZA, AlShehry N, AlAskar A, Zaidi ARZ, Abdallah RNM, et al. Safety and efficacy of convalescent plasma to treat severe COVID-19: protocol for the Saudi collaborative multi-center phase II study. JMIR Research Protocol 2020;9(10):e23543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04347681. Potential efficacy of convalescent plasma to treat severe COVID-19 and patients at high risk of developing severe COVID-19. Available from clinicaltrials.gov/show/NCT04347681 (first received 15 April 2020).
NCT04348877 {published data only}
- NCT04348877. Plasma rich antibodies from recovered patients from COVID19. Available from clinicaltrials.gov/show/NCT04348877 (first received 16 April 2020).
NCT04350580 {published data only}
- NCT04350580. Polyvalent immunoglobulin in COVID-19 related ARDS. Available from clinicaltrials.gov/show/NCT04350580 (first received 17 April 2020).
NCT04353206 {published data only}
- NCT04353206. Convalescent plasma in ICU patients with COVID-19-induced respiratory failure. Available from clinicaltrials.gov/show/NCT04353206 (first received 20 April 2020).
NCT04354831 {published data only}
- NCT04354831. A study evaluating the efficacy and safety of high-titer anti-SARS-CoV-2 plasma in hospitalized patients with COVID-19 infection. Available from clinicaltrials.gov/ct2/show/NCT04354831 (first received 21 April 2020).
NCT04355897 {published data only}
- NCT04355897. COVID-19 plasma in treatment of COVID-19 patients. Available from clinicaltrials.gov/ct2/show/NCT04355897 (first received 21 April 2020).
NCT04356482 {published data only}
- NCT04356482. Convalescent plasma for ill patients by COVID-19. Available from clinicaltrials.gov/show/NCT04356482 (first received 22 April 2020).
NCT04360278 {published data only}
- NCT04360278. Plasma collection from convalescent and/or immunized donors for the treatment of COVID-19. Available from clinicaltrials.gov/show/NCT04360278 (first received 24 April 2020).
NCT04365439 {published data only}
- NCT04365439. Convalescent plasma for COVID-19. Available from clinicaltrials.gov/show/NCT04365439 (first received 28 April 2020).
NCT04368013 {published data only}
- NCT04368013. Host-pathogen interactions, immune response, and clinical prognosis at COVID-19 - the CoVUm trial. Available from clinicaltrials.gov/show/NCT04368013 (first received 20 April 2020).
NCT04374565 {published data only}
- NCT04374565. Convalescent plasma for treatment of COVID-19 patients with pneumonia. Available from clinicaltrials.gov/show/NCT04374565 (first received 5 May 2020).
NCT04376034 {published data only}
- NCT04376034. Convalescent plasma collection and treatment in pediatrics and adults. Available from clinicaltrials.gov/show/NCT04376034 (first received 6 May 2020).
NCT04377672 {published data only}
- NCT04377672. Human convalescent plasma for high-risk children exposed or infected with SARS-CoV-2. Available from clinicaltrials.gov/show/NCT04377672 (first received 6 May 2020).
NCT04383548 {published data only}
- NCT04383548. Clinical study for efficacy of anti-corona VS2 immunoglobulins prepared from COVID19 convalescent plasma prepared by VIPS mini-pool IVIG medical devices in prevention of SARS-CoV-2 infection in high risk groups as well as treatment of early cases of COVID. Available from clinicaltrials.gov/show/NCT04383548 (first received 12 May 2020).
NCT04384497 {published data only}
- NCT04384497. Convalescent plasma for treatment of COVID-19: an exploratory dose identifying study. Available from clinicaltrials.gov/ct2/show/NCT04384497 (first received 12 May 2020).
NCT04384588 {published data only}
- NCT04384588. COVID19-convalescent plasma for treating patients with active symptomatic COVID 19 infection (FALP-COVID). Available from clinicaltrials.gov/show/NCT04384588 (first received 12 May 2020).
NCT04388527 {published data only}
- NCT04388527. COVID-19 convalescent plasma for mechanically ventilated population. Available from clinicaltrials.gov/show/NCT04388527 (first received 14 May 2020).
NCT04389710 {published data only}
- NCT04389710. Convalescent plasma for the treatment of COVID-19. Available from clinicaltrials.gov/show/NCT04389710 (first received 15 May 2020).
NCT04389944 {published data only}
- NCT04389944. Amotosalen-ultraviolet a pathogen-inactivated convalescent plasma in addition to best supportive care and antiviral therapy on clinical deterioration in adults presenting with moderate to severe COVID-19. Available from clinicaltrials.gov/show/NCT04389944 (first received 15 May 2020).
NCT04390178 {published data only}
- NCT04390178. Convalescent plasma as treatment for acute coronavirus disease (COVID-19). Available from clinicaltrials.gov/show/NCT04390178 (first received 15 May 2020).
NCT04392232 {published data only}
- NCT04392232. A phase 2 study of COVID 19 convalescent plasma in high risk patients with COVID 19 infection. Available from clinicaltrials.gov/show/NCT04392232 (first received 18 May 2020).
NCT04393727 {published data only}
- NCT04393727. Transfusion of convalescent plasma for the early treatment of pneumonIa due to SARSCoV2. Available from clinicaltrials.gov/show/NCT04393727 (first received 19 May 2020).
NCT04397523 {published data only}
- NCT04397523. Efficacy and safety of COVID-19 convalescent plasma. Available from clinicaltrials.gov/show/NCT04397523 (first received 21 May 2020).
NCT04407208 {published data only}
- NCT04407208. Convalescent plasma therapy in patients with COVID-19. Available from clinicaltrials.gov/show/NCT04407208 (first received 29 May 2020).
NCT04408209 {published data only}
- NCT04408209. Convalescent plasma for the treatment of patients with severe COVID-19 infection. Available from clinicaltrials.gov/show/NCT04408209 (first received 29 May 2020).
NCT04411602 {published data only}
- NCT04411602. Feasibility study of anti-SARS-CoV-2 plasma transfusions in COVID-19 patients with SRD. Available from clinicaltrials.gov/show/NCT04411602 (first received 2 June 2020).
NCT04412486 {published data only}
- NCT04412486. COVID-19 convalescent plasma (CCP) transfusion. Available from clinicaltrials.gov/show/NCT04412486 (first received 2 June 2020).
NCT04418531 {published data only}
- NCT04418531. Convalescent antibodies infusion in COVID 19 patients. Available from clinicaltrials.gov/show/NCT04418531 (first received 5 June 2020).
NCT04432103 {published data only}
- NCT04432103. Treatment of severe and critical COVID-19 pneumonia with convalescent plasma. Available from clinicaltrials.gov/show/NCT04432103 (first received 16 June 2020).
NCT04438694 {published data only}
- NCT04438694. Use of convalescent plasma for treatment of patients with COVID-19 infection. Available from clinicaltrials.gov/show/NCT04438694 (first received 19 June 2020).
NCT04458363 {published data only}
- NCT04458363. Convalescent plasma in pediatric COVID-19. Available from clinicaltrials.gov/ct2/show/NCT04458363 (first received 07 July 2020).
NCT04462848 {published data only}
- NCT04462848. COVID-19 convalescent plasma as prevention and treatment for children with underlying medical conditions. Available from clinicaltrials.gov/show/NCT04462848 (first received 8 July 2020).
NCT04467151 {published data only}
- NCT04467151. Administration of anti-SARS-CoV-2 convalescent plasma in hospitalized, non-ICU patients with COVID-19. Available from clinicaltrials.gov/show/NCT04467151 (first received 10 July 2020).
NCT04471051 {published data only}
- NCT04471051. An observational cohort trial of outcomes and antibody responses following treatment with COVID19 convalescent plasma in hospitalized COVID-19 patients. Available from clinicaltrials.gov/show/NCT04471051 (first received 14 July 2020).
NCT04474340 {published data only}
- NCT04474340. COVID-19 convalescent plasma treatment in SARS-CoV-2 infected patients: multicenter interventional study. Available from clinicaltrials.gov/show/NCT04474340 (first received 16 July 2020).
NCT04476888 {published data only}
- NCT04476888. Convalescent plasma treatment in COVID-19. Available from clinicaltrials.gov/show/NCT04476888 (first received 20 July 2020).
NCT04502472 {published data only}
- NCT04502472. Open-label treatment of severe coronavirus disease 2019 (COVID-19) with convalescent plasma. Available from clinicaltrials.gov/show/NCT04502472 (first received 06 August 2020).
NCT04513158 {published data only}
- NCT04513158. Convalescent plasma in the early treatment of high-risk patients with SARS-CoV-2 (COVID-19) infection. Available from clinicaltrials.gov/show/NCT04513158 (first received 14 August 2020).
NCT04516954 {published data only}
- NCT04516954. Convalescent plasma for COVID-19 patients. Available from clinicaltrials.gov/show/NCT04516954 (first received 18 August 2020).
NCT04535063 {published data only}
- NCT04535063. Convalescent plasma as potential therapy for severe COVID-19 pneumonia. Available from clinicaltrials.gov/show/NCT04535063 (first received 01 September 2020).
NCT04554992 {published data only}
- NCT04554992. Convalescent plasma for the treatment of COVID-19. Available from clinicaltrials.gov/show/NCT04554992 (first received 18 September 2020).
NCT04555109 {published data only}
- NCT04555109. Convalescent plasma for COVID-19 research donor study. Available from clinicaltrials.gov/show/NCT04555109 (first received 18 September 2020).
NCT04565197 {published data only}
- NCT04565197. Convalescent plasma therapy for COVID-19 patients. Available from clinicaltrials.gov/show/NCT04565197 (first received 25 September 20209.
NCT04569188 {published data only}
- NCT04569188. Convalescent plasma in COVID-19 elderly patients. Available from clinicaltrials.gov/show/NCT04569188 (first received 29 September 2020).
NCT04570982 {published data only}
- NCT04570982. Clinical protocol for convalescent plasma and remdesivir therapy in Nepal. Available from clinicaltrials.gov/show/NCT04570982 (first received 30 September 2020).
NCT04614012 {published data only}
- NCT04614012. Hyperimmune plasma for patients with COVID-19. Available from clinicaltrials.gov/show/NCT04614012 (first received 03 November 2020).
NCT04616976 {published data only}
- NCT04616976. COVID-19 with convalescent plasma. Available from clinicaltrials.gov/show/NCT04616976 (first received 05 November 2020).
NCT04622826 {published data only}
- NCT04622826. plasmApuane CoV-2: efficacy and safety of immune Covid-19 plasma in Covid-19 pneumonia in non ITU patients. Available from clinicaltrials.gov/show/NCT04622826 (first received 10 November 2020).
NCT04638634 {published data only}
- NCT04638634. Pharmacokinetics, safety, and tolerability of CSL760, an anti- COVID-19 hyperimmune intravenous immunoglobulin, in healthy adult subjects. Available from clinicaltrials.gov/ct2/show/NCT04638634 (first received 20 November 2020).
NCT04644198 {published data only}
- NCT04644198. Convalescent plasma transfusion in severe COVID-19 patients in Jamaica. Available from clinicaltrials.gov/ct2/show/NCT04644198 (first received 25 November 2020).
NCT04661839 {published data only}
- NCT04661839. Phase 1 study to evaluate safety and pharmacokinetics of COVID-HIGIV administered as a single dose or a repeat dose in healthy adults. Available from clinicaltrials.gov/show/NCT04661839 (first received 10 December 2020).
NCT04721236 {published data only}
- NCT04721236. Early use of hyperimmune plasma in COVID-19 (COV-II-PLA). Available from clinicaltrials.gov/ct2/show/NCT04721236 (first received 22 January 2021).
Niu 2020 {published data only}
- Niu A, McDougal A, Ning B, Safa F, Luk A, Mushatt DM, et al. COVID-19 in allogeneic stem cell transplant: high false-negative probability and role of CRISPR and convalescent plasma. Bone Marrow Transplantation 2020;15:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Olivares‐Gazca 2020 {published data only}
- Olivares-Gazca JC, Priesca-Marin JM, Ojeda-Laguna M, Garces-Eisele J, Soto-Olvera S, Palacios-Alonso A, et al. Infusion of convalescent plasma is associated with clinical improvement in critically ill patients with COVID-19: a pilot study. Revista de Investigation Clinica 2020;72(3):159-64. [DOI] [PubMed] [Google Scholar]
Pei 2020 {published data only}
- Pei S, Yuan X, Zhimin ZZ, Run YR, Xie Y, Minxue SM, et al. Convalescent plasma to treat COVID-19: Chinese strategy and experiences (preprint). medRxiv 2020. [DOI: 10.1101/2020.04.07.20056440] [DOI]
Peng 2020 {published data only}
- Peng H, Gong T, Huang X, Sun X, Luo H, Wang W, et al. A synergistic role of convalescent plasma and mesenchymal stem cells in the treatment of severely ill COVID-19 patients: a clinical case report. Stem Cell Research and Therapy 2020;11(1):291. [DOI] [PMC free article] [PubMed] [Google Scholar]
PER‐031‐20 {published data only}
- PER-031-20. Phase 2 Study of efficacy and safety of plasma from convalescent patients with COVID-19 in patients with moderate disease (AUNA 20-01). https://www.ins.gob.pe/ensayosclinicos/rpec/recuperarECPBNuevoEN.asp?numec=031-20 (first received 06 July 2020).
Perotti 2020 {published data only}
- Perotti C, Baldanti F, Bruno R, Delfante C, Seminari E, Casari S, et al. Mortality reduction in 46 severe COVID-19 patients treated with hyperimmune plasma. A proof of concept single arm multicenter interventional trial. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.05.26.20113373] [DOI] [PMC free article] [PubMed]
- Perotti C, Baldanti F, Bruno R, Del Fante C, Seminari E, Casari S, et al. Mortality reduction in 46 severe COVID-19 patients treated with hyperimmune plasma. A proof of concept single arm multicenter trial. Haematologica 2020 July 23 [Epub ahead of print];105:2834-2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perotti C, Del Fante C, Baldanti F, Franchini M, Percivalle E, Vecchio Nepita E, et al. Plasma from donors recovered from the new coronavirus 2019 as therapy for critical patients with COVID-19 (COVID-19 plasma study): a multicentre study protocol. Internal and Emergency Medicine 2020;15(5):819-24. [DOI: 10.1007/s11739-020-02384-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Qiu 2020 {published data only}
- Qiu T, Wang J, Zhou J, Zou J, Chen Z, Ma X, et al. The report of two cases infection with novel coronavirus (2019-NCcoV) after kidney transplantation and the association literature analyzation. Chinese Journal of Organ Transplantation 2020;41(0):E004. [Google Scholar]
Rasheed 2020 {published data only}
- Rasheed AM, Fatak DF, Hashim HA, Maulood MF, Kabah KK, Almusawi YA, et al. The therapeutic effectiveness of convalescent plasma therapy on treating COVID-19 patients residing in respiratory care units in hospitals in Baghdad, Iraq (preprint). medRxiv 2020. [DOI: 10.1101/2020.06.24.20121905] [DOI] [PubMed]
RBR‐4vm3yy {published data only}
- RBR-4vm3yy. Effect of convalescent plasma in patients with severe COVID-19. www.ensaiosclinicos.gov.br/rg/RBR-4vm3yy/ (first received 11 May 2020).
Robbiani 2020 {published data only}
- Robbiani DF, Gaebler C, Muecksch F, Cetrulo LJ, Wang Z, Cho A, et al. Convergent antibody responses to SARS-CoV-2 infection in convalescent individuals (preprint). bioRxiv 2020. [DOI: 10.1101/2020.05.13.092619] [DOI] [PMC free article] [PubMed]
RPCEC00000323 {published data only}
- RPCEC00000323. Plasma treatment to asymptomatic patient with COVID-19 infection. rpcec.sld.cu/en/trials/RPCEC00000323-En (first received 03 July 2020).
Salazar 2020a {published data only}
- Salazar E, Christensen PA, Graviss EA, Nguyen DT, Castillo B, Chen J, et al. Treatment of COVID-19 patients with convalescent plasma reveals a signal of significantly decreased mortality. American Journal of Pathology 2020;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Salazar 2020b {published data only}
- Salazar E, Perez KK, Ashraf M, Chen J, Castillo B, Christensen PA, et al. Treatment of COVID-19 patients with convalescent plasma in Houston, Texas. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.05.08.20095471] [DOI] [PMC free article] [PubMed]
- Salazar E, Perez KK, Ashraf M, Chen J, Castillo B, Christensen PA, et al. Treatment of COVID-19 patients with convalescent plasma. American Journal of Pathology 2020 May 27 [Epub ahead of print];190:1680-1690. [DOI: 10.1016/j.ajpath.2020.05.014] [DOI] [PMC free article] [PubMed] [Google Scholar]
Shen 2020 {published data only}
- Shen C, Wang Z, Zhao F, Yang Y, Li J, Yuan J, et al. Treatment of 5 critically ill patients with COVID-19 with convalescent plasma. JAMA 2020;323(16):1582-9. [DOI: 10.1001/jama.2020.4783] [DOI] [PMC free article] [PubMed] [Google Scholar]
Shi 2020 {published data only}
- Shi H, Zhou C, He P, Huang S, Duan Y, Wang X, et al. Successful treatment of plasma exchange followed by intravenous immunoglobulin in a critically ill patient with 2019 novel coronavirus infection. International Journal of Antimicrobial Agents 2020;56(2):105974. [DOI: 10.1016/j.ijantimicag.2020.105974] [DOI] [PMC free article] [PubMed] [Google Scholar]
Soleimani 2020 {published data only}
- Soleimani Z, Soleimani A. ADRS due to COVID-19 in midterm pregnancy: successful management with plasma transfusion and corticosteroids. Journal of Maternal-Fetal & Neonatal Medicine 2020 Jul 26 [Epub ahead of print];26:1-4. [DOI: 10.1080/14767058.2020.1797669] [DOI] [PubMed] [Google Scholar]
Taher 2020 {published data only}
- Taher A, Alalwan AA, Naser N, Alsegai O, Alaradi A. Acute kidney injury in COVID-19 pneumonia: a single-center experience in Bahrain. Cureus 2020;12(8):e9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tan 2020 {published data only}
- Tan L, Kang X, Zhang B, Zheng S, Liu B, Yu T, et al. A special case of COVID-19 with long duration of viral shedding for 49 days (preprint). medRxiv 2020. [DOI: 10.1101/2020.03.22.20040071] [DOI] [PMC free article] [PubMed]
Tu 2020 {published data only}
- Tu Y, Wu X, Liu F, Wang J, Luo Y, Cai Z, et al. Two clinical cases of novel coronavirus pneumonia (NCP) in renal transplant recipients. Chinese Journal of Organ Transplantation 2020;41(0):E005. [Google Scholar]
Wang 2020 {published data only}
- Wang Y, Zhang Y, Yu Q, Zhu K. Convalescent plasma coupled with medications for the treatment of a severe COVID-19 patient: drugs analysis and pharmaceutical care based on the newly established guidelines for COVID-19 remedy. Frontiers in Pharmacology 2020;11:966. [DOI: 10.3389/fphar.2020.00966] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wright 2020 {published data only}
- Wright Z, Bersabe A, Eden R, Cap A. Successful use of COVID-19 convalescent plasma in a patient recently treated for follicular lymphoma. Clinical Lymphoma, Myeloma and Leukemia 2020 Jun 25 [Epub ahead of print];21(1):66-68. [DOI: 10.1016/j.clml.2020.06.012] [DOI] [PMC free article] [PubMed] [Google Scholar]
Xia 2020 {published data only}
- Xia X, Li K, Wu L, Wang Z, Zhu M, Huang B, et al. Improved clinical symptoms and mortality on severe/critical COVID-19 patients utilizing convalescent plasma transfusion. Blood 2020;23:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Xie 2020 {published data only}
- Xie Y, Cao S, Li Q, Chen E, Dong H, Zhang W, et al. Effect of regular intravenous immunoglobulin therapy on prognosis of severe pneumonia in patients with COVID-19. Journal of Infection 2020;81(2):318-56. [DOI: 10.1016/j.jinf.2020.03.044] [DOI] [PMC free article] [PubMed] [Google Scholar]
Xu 2020b {published data only}
- Xu T M, Lin B, Chen C, Liu LG, Xue Y. Non-optimal effectiveness of convalescent plasma transfusion and hydroxychloroquine in treating COVID-19: a case report. Virology Journal 2020;17(1):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yang 2020 {published data only}
- Yang X, Sui Y, Liu F, Kang Z, Wu S, Zhao J, et al. Clinical characteristics and convalescent plasma therapy in severe and critically ill COVID-19 patients (preprint). Social Science Research Network 2020 May 5. [DOI: 10.2139/ssrn.3576894] [DOI]
Ye 2020 {published data only}
- Ye M, Fu D, Ren Y, Wang F, Wang D, Zhang F, et al. Treatment with convalescent plasma for COVID-19 patients in Wuhan, China. Journal of Medical Virology 2020 April 15 [Epub ahead of print];92:1890-1901. [DOI: 10.1002/jmv.25882] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zeng 2020 {published data only}
- Zeng QL, Yu ZJ, Gou JJ, Li GM, Ma SH, Zhang GF, et al. Effect of convalescent plasma therapy on viral shedding and survival in COVID-19 patients. Journal of Infectious Diseases 2020;222(1):38-43. [DOI: 10.1093/infdis/jiaa228] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2020a {published data only}
- Zhang B, Liu S, Tan T, Huang W, Dong Y, Chen L, et al. Treatment with convalescent plasma for critically ill patients with severe acute respiratory syndrome coronavirus 2 infection. Chest 2020;158(1):e9-e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2020b {published data only}
- Zhang B, Liu S, Tan T, Huang W, Dong Y, Chen L, et al. Treatment with convalescent plasma for critically ill patients with SARS-CoV-2 infection. Chest 2020 Mar 31 [Epub ahead of print];158:e9-e13. [DOI: 10.1016/j.chest.2020.03.039] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2020c {published data only}
- Zhang L, Pang R, Xue X, Bao J, Ye S, Dai Y, et al. Anti-SARS-CoV-2 virus antibody levels in convalescent plasma of six donors who have recovered from COVID-19. Aging 2020 Apr 22 [Epub ahead of print];12:6536–6542. [DOI: 10.18632/aging.103102] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies awaiting assessment
Beltran 2021 {published data only}
- Beltran Gonzalez JL, Gonzalez Gamez M, Mendoza Enciso EA, Esparza Maldonado RJ, Hernandez Palacios D, Duenas Campos S, et al. Efficacy and safety of convalescent plasma and intravenous immunoglobulin in critically ill COVID-19 patients. A controlled clinical trial (preprint). Medrxiv 2021:2021.03.28.21254507. [DOI: ]
- NCT04381858. Convalescent plasma vs human immunoglobulin to treat COVID-19 pneumonia. Available from clinicaltrials.gov/show/NCT04381858 (first received 11 May 2020).
Bennett‐Guerrero 2021 {published data only}
- Bennett-Guerrero E, Romeiser JL, Talbot LR, Ahmed T, Mamone LJ, et al. Severe acute respiratory syndrome coronavirus 2 convalescent plasma versus standard plasma in coronavirus disease 2019 infected hospitalized patients in New York. Critical Care Medicine 2021 April 16 (Epub ahead of print). [DOI: 10.1097/CCM.0000000000005066] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04344535. Convalescent plasma vs. standard plasma for COVID-19. Available from clinicaltrials.gov/show/NCT04344535 (first received 14 April 2020).
CTRI/2020/05/025299 {published data only}
- Convalescent plasma to limit Coronavirus associated complications: an open label clinical study of anti-SARS-CoV-2 plasma in hospitalized patients with COVID-19. Available from www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=43752 (first received 21 May 2020).
CTRI/2020/05/025328 {published data only}
- CTRI/2020/05/025328. Study to assess the safety and efficacy of convalescent plasma on outcome of COVID-19 associated complications. Available from ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=43703 (first received 23 May 2020).
CTRI/2020/09/027903 {published data only}
- CTRI/2020/09/027903. Testing the efficacy and safety of a blood product COVID-19 hyper-immuneglobulin (human) solution in participants with active COVID-19. Available from ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=47147 (first received 18 September 2020).
IRCT20120215009014N353 {published data only}
- IRCT20120215009014N353. Effect of plasma of patients recovered from COVID-19 versus control group on treatment of COVID-19: a randomized clinical trial. Available from www.irct.ir/trial/47501 (first received 27 April 2020).
IRCT20150808023559N21 {published data only}
- IRCT20150808023559N21. The effect of convalescent plasma therapy on patients with 19-COVID. Available from en.irct.ir/trial/47594 (first received 9 May 2020).
IRCT20200404046948N1 {published data only}
- IRCT20200404046948N1. Efficacy and safety of convalescent plasma in the treatment of COVID-19. Available from en.irct.ir/trial/46973 (first received 15 April 2020).
IRCT20200409047007N1 {published data only}
- IRCT20200409047007N1. Effect of COVID 19 survivors plasma in COVID 19 patients with ARDS. Available from en.irct.ir/trial/47058 (first received 12 April 2020).
IRCT20200413047056N1 {published data only}
- IRCT20200413047056N1. Comparison between the efficacy of intravenous immunoglobulin and convalescent plasma in COVID-19. Available from en.irct.ir/trial/47212 (first received 17 April 2020).
IRCT20200503047281N1 {published data only}
- IRCT20200503047281N1. Evaluation of convalescent plasma therapy for COVID-19 patients. Available from en.irct.ir/trial/47632 (first received 25 July 2020).
IRCT20200525047562N1 {published data only}
- IRCT20200525047562N1. Treatment of COVID-19 patients with convalescent plasma. Available from en.irct.ir/trial/48493 (first received 14 June 2020).
IRCT20201004048922N1 {published data only}
- IRCT20201004048922N1. Evaluation of the effectiveness of intravenous infusion of human COVID-19 hyperimmune plasma with specific antibody titer in hospitalized patients with Covid-19: a randomized clinical trial. Available from www.irct.ir/trial/51443 (first received 19 July 2020).
ISRCTN85216856 {published data only}
- ISRCTN85216856. Using blood plasma to develop passive immunity to coronavirus in Ecuador. Available from www.isrctn.com/ISRCTN85216856 (first received 06 May 2020).
jRCT2031200174 {published data only}
- jRCT2031200174. An international multicenter, adaptive, randomized double-blind, placebo-controlled trial of the safety, tolerability and efficacy of anti-coronavirus hyperimmune intravenous immunoglobulin for the treatment of adult hospitalized patients at onset of clinical progression of COVID-19. Available from who.int/trialsearch/Trial2.aspx?TrialID=JPRN-jRCT2031200174 (first received 26 October 2020).
Lopardo 2021 {published data only}
- Lopardo G, Belloso W H, Nannini E, Colonna M, Sanguineti S, Zylberman V, et al. RBD-specific polyclonal F(ab´)2 fragments of equine antibodies in patients with moderate to severe COVID-19 disease: a randomized, multicenter, double-blind, placebo-controlled, adaptive phase 2/3 clinical trial. EClinicalMedicine 2021:100843. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04494984. Study to investigate the pharmacokinetics, efficacy and safety of INM005 in patients with SARS-CoV2 disease. Available from clinicaltrials.gov/show/NCT04494984 (first received 31 July 2020).
NCT04315948 {published data only}
- NCT04315948. Trial of treatments for COVID-19 in hospitalized adults (DisCoVeRy). Available from clinicaltrials.gov/ct2/show/NCT04315948 (first received March 20, 2020).
NCT04332835 {published data only}
- NCT04332835. Convalescent plasma for patients with COVID-19: a randomized, open label, parallel, controlled clinical study. Available from clinicaltrials.gov/show/NCT04332835 (first received 3 April 2020).
NCT04355767 {published data only}
- NCT04355767. Convalescent plasma vs. placebo in emergency room patients with COVID-19. Available from clinicaltrials.gov/ct2/show/NCT04355767 (first received 21 April 2020).
NCT04358211 {published data only}
- NCT04358211. Expanded access to convalescent plasma to treat and prevent pulmonary complications associated with COVID-19. Available from clinicaltrials.gov/show/NCT04358211 (first received 24 April 2020).
NCT04372368 {published data only}
- NCT04372368. Convalescent plasma for the treatment of patients with COVID-19. Available from clinicaltrials.gov/show/NCT04372368 (first received 04 May 2020).
NCT04392414 {published data only}
- NCT04392414. Hyperimmune convalescent plasma in moderate and severe COVID-19 disease. Available from clinicaltrials.gov/show/NCT04392414 (first received 18 May 2020).
NCT04405310 {published data only}
- NCT04405310. Convalescent plasma of COVID-19 to treat SARS-COV-2 a randomized double blind 2 center trial (CPC-SARS). Available from clinicaltrials.gov/show/NCT04405310 (first received 28 May 2020).
NCT04433910 {published data only}
- EUCTR2020-001310-38. A randomized, prospective, open label clinical trial on the use of convalescent plasma compared to best supportive care in patients with severe COVID-19. Available from clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2020-001310-38 (first received 23 April 2020).
- Koerper S, Jahrsdoerfer B, Appl T. Convalescent plasma for treatment of severe COVID-19: rationale and designing of a randomized, open-label clinical trial of convalescent plasma compared to best supportive care (CAPSID Trial). Transfusionsmedizin 2020;10(3):143-9. [Google Scholar]
- NCT04433910. A clinical trial of convalescent plasma compared to best supportive care for treatment of patients with severe COVID-19. Available from clinicaltrials.gov/show/NCT04433910 (first received 16 June 2020).
NCT04442958 {published data only}
- NCT04442958. Effectiveness of convalescent immune plasma therapy. Available from clinicaltrials.gov/show/NCT04442958 (first received 23 June 2020).
NCT04492501 {published data only}
- Investigational treatments for COVID-19 in tertiary care hospital of Pakistan. Available from clinicaltrials.gov/ct2/show/NCT04492501 (first received 30 July 2020).
NCT04501978 {published data only}
- NCT04501978. ACTIV-3: Therapeutics for Inpatients With COVID-19 (TICO). Available from clinicaltrials.gov/ct2/show/NCT04501978 (first received August 6, 2020).
NCT04521309 {published data only}
- Ali S, Luxmi S, Anjum F, Muhaymin SM, Uddin SM, Ali A, et al. Hyperimmune anti-COVID-19 IVIG (C-IVIG) therapy for passive immunization of severe and critically ill COVID-19 patients: A structured summary of a study protocol for a randomised controlled trial. Trials 2020;21(905):(no pagination). [DOI] [PMC free article] [PubMed] [Google Scholar]
- SARS-CoV-2 antibodies based IVIG therapy for COVID-19 patients. Available from clinicaltrials.gov/ct2/show/NCT04521309 (first received 20 August 2020).
NCT04542941 {published data only}
- NCT04542941. Assessment of safety and efficacy of CCP. Available from clinicaltrials.gov/show/NCT04542941 (first received 09 September 2020).
NCT04547127 {published data only}
- Ferrer Roca R, Llamas P, Manez R, Galban C, Quintana M, Sanchez-Garcia M, et al. Design of a study to evaluate the safety and efficacy of convalescent plasma to treat COVID-19 in critically ill patients. Intensive Care Medicine Experimental. Conference: 33rd European Society of Intensive Care Medicine Annual Congress, ESICM 2020;8, SUPPL 2.
- NCT04547127. A study to evaluate safety and efficacy of convalescent methylene blue treated (MBT) plasma from donors recovered from coronavirus disease 2019 (COVID-19). Available from clinicaltrials.gov/show/NCT04547127 (first received 14 September 2020).
NCT04547660 {published data only}
- NCT04547660. Convalescent plasma for severe COVID-19 patients. Available from clinicaltrials.gov/show/NCT04547660 (first received 14 September 2020).
NCT04610502 {published data only}
- NCT04610502. Efficacy and safety of two hyperimmune equine anti Sars-CoV-2 in COVID-19 patients. Available from clinicaltrials.gov/show/NCT04610502 (first received 30 October 2020).
Pouladzadeh 2021 {published data only}
- IRCT20200310046736N1. Comparison of the therapeutic effect of convalescent plasma and plasma-derived immunoglobulin-enriched solution on COVID-19 patients. Available from en.irct.ir/trial/46424 (first received 1 April 2020).
- Pouladzadeh M, Safdarian M, Eshghi P, Abolghasemi H, Bavani AG, Sheibani B, et al. A randomized clinical trial evaluating the immunomodulatory effect of convalescent plasma on COVID-19-related cytokine storm. Internal and Emergency Medicine 2021 April 10 [Epub ahead of print]:1–11. [DOI: 10.1007/s11739-021-02734-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
ChiCTR2000030010 {published data only}
- ChiCTR2000030010. A randomized, double-blind, parallel-controlled, trial to evaluate the efficacy and safety of anti-SARS-CoV-2 virus inactivated plasma in the treatment of severe novel coronavirus pneumonia patients (COVID-19). Available from www.chictr.org.cn/showproj.aspx?proj=49777 (first received 19 February 2020).
ChiCTR2000030179 {published data only}
- ChiCTR2000030179. Experimental study of novel coronavirus pneumonia rehabilitation plasma therapy severe novel coronavirus pneumonia (COVID-19). Available from www.chictr.org.cn/showproj.aspx?proj=50059 (first received 24 February 2020).
ChiCTR2000030627 {published data only}
- ChiCTR2000030627. Study on the application of convalescent plasma therapy in severe COVID-19. Available from www.chictr.org.cn/showproj.aspx?proj=50727 (first received 8 March 2020).
ChiCTR2000030702 {published data only}
- ChiCTR2000030702. Convalescent plasma for the treatment of common COVID-19: a prospective randomized controlled trial. Available from www.chictr.org.cn/showproj.aspx?proj=50537 (first received 10 March 2020).
ChiCTR2000030929 {published data only}
- ChiCTR2000030929. A randomized, double-blind, parallel-controlled trial to evaluate the efficacy and safety of anti-SARS-CoV-2 virus inactivated plasma in the treatment of severe novel coronavirus pneumonia (COVID-19). Available from www.chictr.org.cn/showproj.aspx?proj=50696 (first received 17 March 2020).
CTRI/2020/04/024915 {published data only}
- CTRI/2020/04/024915. A phase II, open label, randomized controlled trial to assess the safety and efficacy of convalescent plasma to limit COVID-19 associated complications. Available from ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=43332 (first received 29 April 2020).
CTRI/2020/05/025346 {published data only}
- CTRI/2020/05/025346. A phase II, open label, randomized controlled trial to assess the safety and efficacy of convalescent plasma in severe COVID-19 patients. Available from ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=43005 (first received 5 May 2020).
CTRI/2020/06/025803 {published data only}
- CTRI/2020/06/025803. Effect of convalescent plasma in COVID-19 patients. Available from ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=44478 (first received 11 June 2020).
CTRI/2020/06/026123 {published data only}
- CTRI/2020/06/026123. Plasma therapy in corona patients(Severe COVID-19). Available from ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=44667 (first received 24 June 2020).
EUCTR2020‐001632‐10 {published data only}
- EUCTR2020-001632-10. A randomized open label phase-II clinical trial with or without infusion of plasma from subjects after convalescence of SARS-CoV-2 infection in high-risk patients with confirmed severe SARS-CoV-2 disease. Available from www.clinicaltrialsregister.eu/ctr-search/trial/2020-001632-10/DE (first received 20 April 2020). [DOI] [PMC free article] [PubMed]
- Janssen M, Schakel U, Djuka Fokou U, Krisam J, Stermann J, Kriegsmann K, et al. A randomized open label phase-II clinical trial with or without infusion of plasma from subjects after convalescence of SARS-CoV-2 infection in high-risk patients with confirmed severe SARS-CoV-2 disease (RECOVER): a structured summary of a study protocol for a randomised controlled trial. Trials 2020;21(1):828. [DOI] [PMC free article] [PubMed] [Google Scholar]
EUCTR2020‐001936‐86 {published data only}
- EUCTR2020-001936-86. A prospective, randomized, open label Phase 2 clinical trial to evaluate superiority of anti-SARS-CoV-2 convalescent plasma versus standard-of-care in hospitalized patients with mild COVID-19. Available from www.clinicaltrialsregister.eu/ctr-search/trial/2020-001936-86/DE (first received 20 August 2020).
EUCTR2020‐002122‐82 {published data only}
- 2020-002122-82. Prospective open-label randomized controlled phase 2b clinical study in parallel groups for the assessment of efficacy and safety of immune therapy with COVID-19 convalescent plasma plus standard treatment alone of subjects with severe COVID-19. Available from www.clinicaltrialsregister.eu/ctr-search/trial/2020-002122-82/DE (first received 07 May 2020).
EUCTR2020‐005410‐18 {published data only}
- EUCTR2020-005410-18. Multicentre, randomized, double-blind, placebo-controlled, non-commercial clinical trial to evaluate the efficacy and safety of specific anti-SARS-CoV-2 immunoglobulin in the treatment of COVID-19. Available from www.clinicaltrialsregister.eu/ctr-search/trial/2020-005410-18/PL.
IRCT20200501047258N1 {published data only}
- IRCT20200501047258N1, U07. Effects of convalescent plasma in COVID-19. Available from en.irct.ir/trial/47629 (first received 04 May 2020).
IRCT20200508047346N1 {published data only}
- IRCT20200508047346N1. Evaluation of the effectiveness of rabbit antibody against coronavirus in patients. Available from www.irct.ir/trial/47953 (first received 13 May 2020).
NCT02735707 {published data only}
- NCT02735707. Randomized, embedded, multifactorial adaptive platform trial for community- acquired pneumonia (REMAP-CAP). NCT02735707 (first received 13 April 2016).
NCT04333251 {published data only}
- NCT04333251. Evaluating convalescent plasma to decrease coronavirus associated complications. A phase I study comparing the efficacy and safety of high-titer anti-Sars-CoV-2 plasma vs best supportive care in hospitalized patients with interstitial pneumonia due to COVID-19. Available from clinicaltrials.gov/show/NCT04333251 (first received 3 April 2020).
NCT04338360 {published data only}
- NCT04338360. Expanded access to convalescent plasma for the treatment of patients with COVID-19. Available from clinicaltrials.gov/show/NCT04338360 (first received 8 April 2020).
NCT04345289 {published data only}
- EUCTR2020-001367-88-DK. Efficacy and safety of novel treatment options for adults with COVID-19 pneumonia. Available from apps.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2020-001367-88-DK (first received 14 April 2020).
- NCT04345289. Efficacy and safety of novel treatment options for adults with COVID-19 pneumonia (CCAP). Available from clinicaltrials.gov/show/NCT04345289 (first received 14 April 2020).
NCT04345991 {published data only}
- NCT04345991. Efficacy of convalescent plasma to treat COVID-19 patients, a nested trial in the CORIMUNO-19 cohort. Available from clinicaltrials.gov/show/NCT04345991 (first received 15 April 2020).
NCT04348656 {published data only}
- Begin P, Callum J, Heddle N, Cook R, Zeller MP, Timmouth A, et al. Convalescent plasma for adults with acute COVID-19 respiratory illness (CONCOR-1): study protocol for an international, multicenter, randomized, open-label trial (preprint). Available from www.researchsquare.com/article/rs-268937/v1 04 March 2021. [DOI: 10.21203/rs.3.rs-268937/v1] [DOI] [PMC free article] [PubMed]
- NCT04348656. Convalescent plasma for hospitalized adults with COVID-19 respiratory illness (CONCOR-1). Available from clinicaltrials.gov/show/NCT04348656 (first received 16 April 2020).
NCT04352751 {published data only}
- NCT04352751. Experimental use of convalescent plasma for passive immunization in current COVID-19 pandemic in Pakistan in 2020. Available from clinicaltrials.gov/show/NCT04352751 (first received 20 April 2020).
NCT04358783 {published data only}
- NCT04358783. Convalescent plasma compared to the best available therapy for the treatment of SARS-CoV-2 pneumonia. Available from clinicaltrials.gov/show/NCT04358783 (first received 24 April 2020).
NCT04360486 {published data only}
- NCT04360486. Treatment of COVID-19 with anti-SARS-CoV-2 convalescent plasma (ASCoV2CP). Available from clinicaltrials.gov/show/NCT04360486 (first received 24 April 2020).
NCT04361253 {published data only}
- NCT04361253. Evaluation of SARS-CoV-2 (COVID-19) antibody-containing plasma therapy. Available from clinicaltrials.gov/show/NCT04361253 (first received 24 April 2020).
NCT04362176 {published data only}
- NCT04362176. Passive immunity trial of Nashville II. Available from clinicaltrials.gov/show/NCT04362176 (first received 24 April 2020).
- Self WH, Stewart TG, Wheeler AP, Atrouni WE, Bistran-Hall AJ, Casey JD et al. Passive immunity trial for our nation (PassITON): study protocol for a randomized placebo-control clinical trial evaluating COVID-19 convalescent plasma in hospitalized adults (preprint). Available from www.researchsquare.com/article/rs-227796/v1. [DOI] [PMC free article] [PubMed]
NCT04363034 {published data only}
- NCT04363034. Arkansas expanded access COVID-19 convalescent plasma treatment program. Available from clinicaltrials.gov/ct2/show/NCT04363034 (first received 27 April 2020).
NCT04364737 {published data only}
- NCT04364737. Convalescent plasma to limit COVID-19 complications in hospitalized patients. Available from clinicaltrials.gov/show/NCT04364737 (first received 28 April 2020).
- NCT04404634. Convalescent plasma to limit coronavirus associated complications. Available from clinicaltrials.gov/show/NCT04404634 (first received 28 May 2020).
NCT04366245 {published data only}
- NCT04366245. Clinical trial to evaluate the efficacy of treatment with hyperimmune plasma obtained from convalescent antibodies of COVID-19 infection. Available from clinicaltrials.gov/show/NCT04366245 (first received 28 April 2020).
NCT04372979 {published data only}
- NCT04372979. Efficacy of convalescent plasma therapy in the early care of COVID-19 patients. Available from clinicaltrials.gov/show/NCT04372979 (first received 04 May 2020).
NCT04373460 {published data only}
- NCT04373460. Convalescent plasma to limit SARS-CoV-2 associated complications. Available from clinicaltrials.gov/show/NCT04373460 (first received 04 May 2020).
NCT04374370 {published data only}
- NCT04374370. SARSCoV2 (COVID-19) convalescent plasma (CP) expanded access protocol (EAP). Available from clinicaltrials.gov/show/NCT04374370 (first received 5 May 2020).
NCT04374487 {published data only}
- NCT04374487. A phase II, open label, randomized controlled trial to assess the safety and efficacy of convalescent plasma to limit COVID-19 associated complications. Available from clinicaltrials.gov/show/NCT04374487 (first received 5 May 2020).
NCT04374526 {published data only}
- NCT04374526. Early transfusIon of convalescent plasma in elderly COVID-19 patients to prevent disease progression. Available from clinicaltrials.gov/show/NCT04374526 (first received 5 May 2020).
- Teofili L, Landolfi R, Cingolani A, Antinori A, Vecchiet J, Sanguinetti M, et al. Early transfusion of convalescent plasma in older patients with COVID-19 to prevent disease progression: A structured summary of a study protocol for a randomised controlled trial. Trials 2020;21(1):875. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT04376788 {published data only}
- NCT04376788. Exchange transfusion versus plasma from convalescent patients with methylene blue in patients with COVID-19. Available from clinicaltrials.gov/show/NCT04376788 (first received 6 May 2020).
NCT04377568 {published data only}
- NCT04377568. Efficacy of human coronavirus-immune convalescent plasma for the treatment of COVID-19 disease in hospitalized children. Available from clinicaltrials.gov/show/NCT04377568 (first received 6 May 2020).
NCT04380935 {published data only}
- NCT04380935. Effectiveness and safety of convalescent plasma therapy on COVID-19 patients with acute respiratory distress syndrome. Available from clinicaltrials.gov/show/NCT04380935 (first received 6 May 2020).
NCT04385043 {published data only}
- NCT04385043. Hyperimmune plasma in patients with COVID-19 severe infection. Available from clinicaltrials.gov/show/NCT04385043 (first received 12 May 2020).
NCT04385186 {published data only}
- NCT04385186. Inactivated convalescent plasma as a therapeutic alternative in patients COVID-19. Available from clinicaltrials.gov/show/NCT04385186 (first received 12 May 2020).
NCT04385199 {published data only}
- NCT04385199. Convalescent plasma for patients with COVID-19. Available from clinicaltrials.gov/show/NCT04385199 (first received 12 May 2020).
NCT04388410 {published data only}
- NCT04388410. Safety and efficacy of convalescent plasma transfusion for patients with SARS-CoV-2 infection. Available from clinicaltrials.gov/show/NCT04388410 (first received 14 May 2020).
NCT04390503 {published data only}
- NCT04390503. Convalescent plasma for COVID-19 close contacts. Available from clinicaltrials.gov/ct2/show/NCT04390503 (first received 15 May 2020).
NCT04391101 {published data only}
- NCT04391101. Convalescent plasma for the treatment of severe SARS-CoV-2 (COVID-19). Available from clinicaltrials.gov/show/NCT04391101 (first received 18 May 2020).
NCT04395170 {published data only}
- NCT04395170. Convalescent plasma compared to anti-COVID-19 human immunoglobulin and standard treatment (TE) in hospitalized patients. Available from clinicaltrials.gov/show/NCT04395170 (first received 20 May 2020).
NCT04397757 {published data only}
- NCT04397757. COVID-19 convalescent plasma for the treatment of hospitalized patients with pneumonia caused by SARS-CoV-2. Available from clinicaltrials.gov/show/NCT04397757 (first received 21 May 2020).
NCT04403477 {published data only}
- Chowdhury FR, Hoque A, Chowdhury FUH, Amin MR, Rahim A, Rahman MM, et al. Convalescent plasma transfusion therapy in severe COVID-19 patients- a safety, efficacy and dose response study: A structured summary of a study protocol of a phase II randomized controlled trial. Trials 2020;21(1):883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04403477. Convalescent plasma therapy in severe COVID-19 infection. Available from clinicaltrials.gov/show/NCT04403477 (first received 27 May 2020).
NCT04408040 {published data only}
- Use of convalescent plasma for COVID-19. Available from clinicaltrials.gov/ct2/show/NCT04408040 (first received 29 May 2020).
NCT04415086 {published data only}
- NCT04415086. Treatment of patients with COVID-19 with convalescent plasma. Available from clinicaltrials.gov/show/NCT04415086 (first received 4 June 2020).
NCT04418518 {published data only}
- NCT04418518. A trial of convalescent plasma for hospitalized adults with acute COVID-19 respiratory illness. Available from clinicaltrials.gov/show/NCT04418518 (first received 5 June 2020).
NCT04420988 {published data only}
- Investigational COVID-19 convalescent plasma infusion for severely or life-threateningly ill COVID-19 patients. Available from clinicaltrials.gov/ct2/show/NCT04420988 (first received 09 June 2020).
NCT04421404 {published data only}
- NCT04421404. Effects of COVID-19 convalescent plasma (CCP) on coronavirus-associated complications in hospitalized patients. Available from clinicaltrials.gov/show/NCT04421404 (first received 9 June 2020).
NCT04425837 {published data only}
- NCT04425837. Effectiveness and safety of convalescent plasma in patients with high-risk COVID-19. Available from clinicaltrials.gov/show/NCT04425837 (first received 11 June 2020).
NCT04425915 {published data only}
- NCT04425915. Efficacy of convalescent plasma therapy in patients with COVID-19. Available from clinicaltrials.gov/show/NCT04425915 (first received 11 June 2020).
NCT04428021 {published data only}
- NCT04428021. Standard or convalescent plasma in patients with recent onset of COVID-19 respiratory failure. Available from clinicaltrials.gov/show/NCT04428021 (first received 11 June 2020).
NCT04429854 {published data only}
- Devos T, Geukens T, Schauvlieghe A, Ariën K, Barbezange C, Cleeren M, et al. A randomized, multicentre, open-label phase II proof-of-concept trial investigating the clinical efficacy and safety of the addition of convalescent plasma to the standard of care in patients hospitalized with COVID-19: the Donated Antibodies Working against nCoV (DAWn-Plasma) trial. Trials 27 November 2020;21:981. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos T, Geukens T, Schauvlieghe A, Ariën K, Barbezange C, Cleeren M, et al. Correction to: a randomized, multicentre, open-label phase II proof-of-concept trial investigating the clinical efficacy and safety of the addition of convalescent plasma to the standard of care in patients hospitalized with COVID-19: the Donated Antibodies Working against nCoV (DAWn-Plasma) trial. Trials 14 December 2020;21:981. [DOI: 10.1186/s13063-020-04876-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos T, Geukens T, Schauwvlieghe A, Ariën K, Barbezange C, Cleeren M, et al. A randomized, multicentre, open-label phase II proof-of-concept trial investigating the clinical efficacy and safety of the addition of convalescent plasma to the standard of care in patients hospitalized with COVID-19: the Donated Antibodies Working against nCoV (DAWn-Plasma) trial. ResearchSquare 2020. [DOI] [PMC free article] [PubMed]
- NCT04429854. Donated antibodies working against nCoV. Available from clinicaltrials.gov/show/NCT04429854 (first received 12 June 2020).
NCT04432272 {published data only}
- NCT04432272. Antibody-level based analysis of COVID convalescent serum (ABACCuS). Available from clinicaltrials.gov/ct2/show/NCT04432272 (first received 16 June 2020).
NCT04438057 {published data only}
- NCT04438057. Evaluating the efficacy of convalescent plasma in symptomatic outpatients infected with COVID-19. Available from clinicaltrials.gov/show/NCT04438057 (first received 18 June 2020).
NCT04442191 {published data only}
- NCT04442191. Convalescent plasma as a possible treatment for COVID-19. Available from clinicaltrials.gov/show/NCT04442191 (first received 22 June 2020).
NCT04445207 {published data only}
- Experimental expanded access treatment with convalescent plasma for the treatment of patients with COVID-19. Available from clinicaltrials.gov/ct2/show/NCT04445207 (first received 24 June 2020).
NCT04452812 {published data only}
- NCT04452812. Statistical and epidemiological study based on the use of convalescent plasma for the management of patients with COVID-19. Available from clinicaltrials.gov/show/NCT04452812 (first received 30 June 2020).
NCT04453384 {published data only}
- EUCTR202000257427. A randomized, double-blind, placebo-controlled phase 2a and 2b study to evaluate the safety and efficacy of XAV-19 in patients with COVID-19 induced moderate pneumonia. Available from www.clinicaltrialsregister.eu/ctr-search/trial/2020-002574-27/FR.
- Gaborit B, Vanhove B, Vibet M A, Le Thuaut A, Lacombe K, Dubee V, et al. Evaluation of the safety and efficacy of XAV-19 in patients with COVID-19-induced moderate pneumonia: study protocol for a randomized, double-blinded, placebo-controlled phase 2 (2a and 2b) trial. Trials 2021;22(199):(no pagination). [DOI: 10.1186/s13063-021-05132-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04453384. Study to evaluate the safety and efficacy of XAV-19 in patients with COVID-19 induced moderate pneumonia. Available from clinicaltrials.gov/show/NCT04453384 (first received 01 July 2020).
NCT04456413 {published data only}
- NCT04456413. Convalescent plasma as treatment for subjects with early COVID-19 infection. Available from clinicaltrials.gov/show/NCT04456413 (first received 2 July 2020).
NCT04463823 {published data only}
- "NORPLASMA" COVID-19 convalescent plasma treatment monitoring study. Available from clinicaltrials.gov/ct2/show/NCT04463823 (first received 09 July 2020).
NCT04468009 {published data only}
- NCT04468009. Treatment of critically ill patients with COVID-19 with convalescent plasma. Available from clinicaltrials.gov/show/NCT04468009 (first received 13 July 2020).
NCT04468958 {published data only}
- NCT04468958. Safety, tolerability, and pharmacokinetics of SAB-185 in healthy participants. Available from clinicaltrials.gov/ct2/show/NCT04468958 (first received 13 July 2020).
NCT04469179 {published data only}
- NCT04469179. Safety, tolerability, and pharmacokinetics of SAB-185 in ambulatory participants with COVID-19. Available from clinicaltrials.gov/show/NCT04469179 (first received 13 July 2020).
NCT04472572 {published data only}
- NCT04472572. Expanded access to convalescent plasma for treatment of COVID-19. Available from clinicaltrials.gov/show/NCT04472572 (first received 15 July 2020).
NCT04483960 {published data only}
- NCT04483960. Australasian COVID-19 trial (ASCOT). Available from clinicaltrials.gov/show/NCT04483960 (first received 23 July 2020).
NCT04497324 {published data only}
- NCT04497324. Peruconplasma: evaluating the use of convalescent plasma as management of COVID-19. Available from clinicaltrials.gov/ct2/show/NCT04497324 (first received 04 August 2020).
- PER-016-20. PERUCONPLASMA: Evaluating the use of convalescent plasma as managment of COVID-19. Available from www.ins.gob.pe/ensayosclinicos/rpec/recuperarECPBNuevoEN.asp?numec=016-20 (first receibed 01 June 2020).
NCT04497779 {published data only}
- Evaluation of coronavirus disease 19 (COVID-19) convalescent plasma. Available from clinicaltrials.gov/ct2/show/NCT04497779 (first received 04 August 2020).
NCT04514302 {published data only}
- NCT04514302. Safety and efficacy of anti-SARS-CoV-2 equine antibody fragments (INOSARS) for hospitalized patients with COVID-19. Available from clinicaltrials.gov/show/NCT04514302 (first received 14 August 2020).
NCT04516811 {published data only}
- NCT04516811. Therapeutic use of convalescent plasma in the treatment of patients with moderate to severe COVID-19. Available from clinicaltrials.gov/show/NCT04516811 (first received 18 August 2020).
NCT04521036 {published data only}
- NCT04521036. Convalescent plasma for COVID-19 patients (CPCP). Available from clinicaltrials.gov/show/NCT04521036 (first received 20 August 2020).
NCT04524507 {published data only}
- NCT04524507. COVID-19 antibody plasma research study in hospitalized patients (UNC CCP RCT). Available from clinicaltrials.gov/show/NCT04524507 (first received 24 August 2020).
NCT04528368 {published data only}
- NCT04528368. Convalescent plasma for treating patients with COVID-19 pneumonia without indication of ventilatory support. Available from clinicaltrials.gov/show/NCT04528368 (first received 27 August 2020).
NCT04539275 {published data only}
- NCT04539275. COVID-19 (VA CURES-1). Available from clinicaltrials.gov/show/NCT04539275 (first received 04 September 2020).
NCT04542967 {published data only}
- NCT04542967. Study on the safety and efficacy of convalescent plasma in patients with severe COVID-19 disease. Available from clinicaltrials.gov/show/NCT04542967 (first received 09 September 2020).
NCT04545047 {published data only}
- NCT04545047. Observational study of convalescent plasma for treatment of veterans with COVID-19. Available from clinicaltrials.gov/show/NCT04545047 (first received 10 September 2020).
NCT04546581 {published data only}
- EUCTR2020-002542-16-GR. Treatment of patients with coronavirus infection with immunoglobulin. Available from who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2020-002542-16-GR (first received 14 September 2020).
- NCT04546581. Inpatient treatment with anti-coronavirus immunoglobulin (ITAC). Available from clinicaltrials.gov/show/NCT04546581 (first received 14 September 2020).
NCT04555148 {published data only}
- NCT04555148. COVIDIG (COVID-19 Hyper-ImmunoGlobulin). Available from clinicaltrials.gov/show/NCT04555148 (first received 18 September 2020).
NCT04558476 {published data only}
- Misset B, Hoste E, Donneau AF, Grimaldi D, Meyfroidt G, Moutschen M, et al. A multicenter randomized trial to assess the efficacy of CONvalescent plasma therapy in patients with Invasive COVID-19 and acute respiratory failure treated with mechanical ventilation: the CONFIDENT trial protocol. BMC Pulmonary Medicine 2020;20(317):(no pagination). [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04558476. Efficacy of convalescent plasma in patients with COVID-19 treated with mechanical ventilation. Available from clinicaltrials.gov/show/NCT04558476 (first received 22 September 2020).
NCT04567173 {published data only}
- NCT04567173. Convalescent plasma as adjunctive therapy for hospitalized patients with COVID-19. Available from clinicaltrials.gov/show/NCT04567173 (first received 28 September 2020).
NCT04573855 {published data only}
- NCT04573855 - unclear. Treatment with anti-SARS-CoV-2 immunoglobulin in patients with COVID-19. Available from clinicaltrials.gov/show/NCT04573855 (first received 05 October 2020).
NCT04589949 {published data only}
- NCT04589949. Early convalescent plasma therapy for high-risk patients with COVID-19 in primary care (the CoV-Early Study). Available from clinicaltrials.gov/show/NCT04589949 (first received 19 October 2020).
NCT04600440 {published data only}
- NCT04600440. Convalescent plasma in the treatment of COVID-19. Available from clinicaltrials.gov/show/NCT04600440 (first received 23 October 2020).
NCT04621123 {published data only}
- NCT04621123. Plasma for early treatment in non-hospitalised mild or moderate COVID-19 patients. Available from clinicaltrials.gov/show/NCT04621123 (first received 09 November 2020).
NCT04634422 {published data only}
- NCT04634422. Plasma exchange (PLEX) and convalescent plasma (CCP) in COVID-19 patients with multiorgan failure. Available from clinicaltrials.gov/show/NCT04634422 (first received 18 November 2020).
NCT04642014 {published data only}
- NCT04642014. Application of convalescent plasma in the treatment of SARS CoV-2 disease (COVID-19) with evaluation of therapy effectiveness. Available from clinicaltrials.gov/show/NCT04642014 (first received 24 November 2020).
NCT04649879 {published data only}
- NCT04649879. Convalescent plasma for treatment of COVID-19: an open randomised controlled trial. Available from clinicaltrials.gov/show/NCT04649879 (first received 02 December 2020).
NCT04669990 {published data only}
- NCT04669990. Remdesivir and convalescent plasma therapy for treatment of COVID-19 infection in Nepal: a registry study. Available from clinicaltrials.gov/ct2/show/NCT04669990 (first received 17 December 2020).
NCT04681430 {published data only}
- NCT04681430. Reconvalescent plasma/camostat mesylate early in SARS-CoV-2 Q-PCR (COVID-19) positive high-risk individuals (RES-Q-HR). Available from clinicaltrials.gov/ct2/show/NCT04681430 (first received 23 December 2020).
NCT04712344 {published data only}
- NCT04712344. Assessment of efficacy and safety of therapy with COVID-19 convalescent plasma in subjects with severe COVID-19 (IPCO). Available from clinicaltrials.gov/ct2/show/NCT04712344 (first received 15 January 2021).
NCT04716556 {published data only}
- NCT04716556. Tranfusion of convalescent plasma for the early treatment of pneumonIa in COVID-19 patients. Available from clinicaltrials.gov/show/NCT04716556 (first received 20 January 2021).
NCT04730401 {published data only}
- NCT04730401. Convalescent plasma in the treatment of COVID-19 (CP_COVID-19). Available from clinicaltrials.gov/ct2/show/NCT04730401 (first received January 27, 2021).
NL8633 {published data only}
- NL8633. A randomized, double-blinded clinical trial of convalescent plasma compared to standard plasma for treatment of hospitalized non-ICU patients with COVID-19 infections. Available from trialregister.nl/trial/8633 (first received 13 May 2020).
PACTR202006760881890 {published data only}
- PACTR202006760881890. Lagos COVID-19 convalescent plasma trial (LACCPT). Available from pactr.samrc.ac.za/TrialDisplay.aspx?TrialID=12168.
PACTR202007653923168 {published data only}
- PACTR202007653923168. A clinical trial comparing use of convalescent plasma therapy plus standard treatment to standard treatment alone in patients with severe COVID-19 infection. Available from pactr.samrc.ac.za/TrialDisplay.aspx?TrialID=11047 (first received 16 July 2020).
PER‐013‐20 {published data only}
- PER-013-20. Convalescent plasma as treatment for COVID-19. Available from ins.gob.pe/ensayosclinicos/rpec/recuperarECPBNuevoEN.asp?numec=013-20 (first received 25 June 2020).
PER‐060‐20 {published data only}
- PER-060-20. Randomized phase 2 clinical trial to evaluate the safety and efficacy of the use of plasma from convalescent patients with the new coronavirus disease (COVID-19). Available from who.int/trialsearch/Trial2.aspx?TrialID=PER-060-20 (first received 21 September 2020).
RBR‐7jqpnw {published data only}
- RBR-7jqpnw. Effect of COVID-19 convalescent plasma produced by HEMOPE: a randomized study, with a comparative group in several centers. Available from ensaiosclinicos.gov.br/rg/RBR-7jqpnw/ (first received 11 May 2020).
Additional references
Balshem 2011
- Balshem H, Helfand M, Schünemann HJ, Oxman AD, Kunz R, Brozek J, et al. GRADE guidelines: 3. Rating the quality of evidence. Journal of Clinical Epidemiology 2011;64(4):401-6. [DOI] [PubMed] [Google Scholar]
Bao 2020a
- Bao L, Deng W, Gao H, Xiao C, Liu J, Xue J, et al. Reinfection could not occur in SARS-CoV-2 infected rhesus macaques. bioRxiv [Preprint] 2020. [DOI: 10.1101/2020.03.13.990226] [DOI]
Baudel 2020
- Baudel JL, Vigneron C, Pras-Landre V, Joffre J, Marjot F, Ait-Oufella H, et al. Transfusion-related acute lung injury (TRALI) after intravenous immunoglobulins: French multicentre study and literature review. Clinical Rheumatology 2020;39(2):541-6. [DOI] [PubMed] [Google Scholar]
Beigel 2017
- Beigel JH, Tebas P, Elie-Turenne MC, Bajwa E, Bell TE, Cairns B, et al. Immune plasma for the treatment of severe influenza: an open-label, multicentre, phase 2 randomised study. Lancet Respiratory Medicine 2017;5(6):500-11. [DOI: 10.1016/S2213-2600(17)30174-1] [DOI] [PMC free article] [PubMed] [Google Scholar]
Beigel 2019
- Beigel JH, Aga E, Elie-Turenne M-C, Cho J, Tebas P, Clark CL, et al. Anti-influenza immune plasma for the treatment of patients with severe influenza A: a randomised, double-blind, phase 3 trial. Lancet Respiratory Medicine 2019;7(11):941-50. [DOI: 10.1016/S2213-2600(19)30199-7] [DOI] [PMC free article] [PubMed] [Google Scholar]
Beigel 2020
- Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of COVID-19 — preliminary report. New England Journal of Medicine 2020;383(10):992-4. [DOI: 10.1056/NEJMoa2007764] [DOI] [PubMed] [Google Scholar]
Bloch 2020
- Bloch EM, Shoham S, Casadevall A, Sachais BS, Shaz B, Winters JL, et al. Deployment of convalescent plasma for the prevention and treatment of COVID-19. Journal of Clinical Investigation 2020;130(6):2757-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Brennan 2003
- Brennan VM, Salomé-Bentley NJ, Chapel HM, Immunology Nurses Study. Prospective audit of adverse reactions occurring in 459 primary antibody-deficient patients receiving intravenous immunoglobulin. Clinical and Experimental Immunology 2003;133(2):247-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Casadevall 2020
- Casadevall A, Pirofski L. The convalescent sera option for containing COVID-19. Journal of Clinical Investigation 2020;130(4):1545-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
CDC 2020b
- Centers for Disease Control and Prevention (CDC). Interim clinical guidance for management of patients with confirmed coronavirus disease (COVID-19). Available from www.cdc.gov/coronavirus/2019-ncov/hcp/clinical-guidance-management-patients.html (accessed 22 March 2021).
Chen 2020a
- Chen L, Xiong J, Bao L, Shi Y. Convalescent plasma as a potential therapy for COVID-19. Lancet Infectious Diseases 2020;20(4):398-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chun 2016
- Chun S, Chung CR, Ha YE, Han TH, Ki CS, Kang ES, et al. Possible transfusion-related acute lung injury following convalescent plasma transfusion in a patient with Middle East respiratory syndrome. Annals of Laboratory Medicine 2016;36(4):393-5. [DOI: 10.3343/alm.2016.36.4.393] [DOI] [PMC free article] [PubMed] [Google Scholar]
COMET 2020
- Core outcome set developers’ response to COVID-19 (2nd April 2020). Available from www.comet-initiative.org/Studies/Details/1538 (accessed 9 April 2020).
Covidence [Computer program]
- Veritas Health Innovation Covidence. Version accessed 20 April 2020. Melbourne, Australia: Veritas Health Innovation, 2020. Available at covidence.org.
D'Antiga 2020
- D’Antiga L. Coronaviruses and immunosuppressed patients: the facts during the third epidemic. Liver Transplantation 2020;26(6):832-4. [DOI] [PubMed] [Google Scholar]
Davey 2019
- Davey RT Jr, Fernández-Cruz E, Markowitz N, Pett S, Babiker AG, Wentworth D, et al. Anti-influenza hyperimmune intravenous immunoglobulin for adults with influenza A or B infection (FLU-IVIG): a double-blind, randomised, placebo-controlled trial. Lancet Respiratory Medicine 2019;7(11):951-63. [DOI: 10.1016/S2213-2600(19)30253-X] [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2019
- Deeks JJ, Higgins JP, Altman DG, editor(s). Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Driggin 2020
- Driggin E, Madhavan MV, Bikdeli B, Chuich T, Laracy J, Biondi-Zoccai G, et al. Cardiovascular considerations for patients, health care workers, and health systems during the COVID-19 pandemic. Journal of the American College of Cardiology 2020;75(18):2352-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Eibl 2008
- Eibl MM. History of immunoglobulin replacement. Immunology and Allergy Clinics of North America 2008;28(4):737-64. [DOI: 10.1016/j.iac.2008.06.004] [DOI] [PubMed] [Google Scholar]
Eldridge 2016
- Eldridge S, Campbell M, Campbell M, Dahota A, Giraudeau B, Higgins JT, et al. Revised Cochrane risk of bias tool for randomized trials (RoB 2.0) additional considerations for cluster-randomized trials. Available from www.riskofbias.info/welcome/rob-2-0-tool/archive-rob-2-0-cluster-randomized-trials-2016 (accessed 22 March 2021).
EPOC 2017
- Cochrane Effective Practice and Organisation of Care (EPOC). What study designs can be considered for inclusion in an EPOC review and what should they be called? EPOC Resources for review authors. Available from epoc.cochrane.org/resources/epoc-resources-review-authors (accessed 22 March 2021).
FDA 2020
- US Food and Drug Administration. Recommendations for investigational COVID-19 convalescent plasma. Available from www.fda.gov/vaccines-blood-biologics/investigational-new-drug-ind-or-device-exemption-ide-process-cber/recommendations-investigational-covid-19-convalescent-plasma (accessed 22 March 2021).
FDA 2021
- US Food and Drug Administration. Recommendations for investigational COVID-19 convalescent plasma. Available from www.fda.gov/vaccines-blood-biologics/investigational-new-drug-ind-or-device-exemption-ide-process-cber/recommendations-investigational-covid-19-convalescent-plasma (accessed 22 March 2021).
Fung 2020
- Fung M, Babik JM. COVID-19 in immunocompromised hosts: what we know so far. Clinical Infectious Diseases 2020;72:340-350. [DOI: 10.1093/cid/ciaa863] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro GDT [Computer program]
- McMaster University (developed by Evidence Prime) GRADEpro GDT. Version accessed 20 April 2020. Hamilton (ON): McMaster University (developed by Evidence Prime), 2020. Available at gradepro.org.
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327:557-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Altman DG, Sterne JA, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Higgins 2019a
- Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook. [DOI] [PMC free article] [PubMed]
Higgins 2019b
- Higgins JPT, Eldridge S, Li T (editors). Chapter 23: Including variants on randomized trials. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook..
Higgins 2019c
- Higgins JP, Savović J, Page M, Elbers RG, Sterne JA. Chapter 8: Assessing risk of bias in a randomized trial. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Higgins 2019d
- Higgins JP, Deeks JJ, editor(s). Chapter 6: Choosing effect measures and computing estimates of effect. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Ho 2005
- Ho MS, Chen WJ, Chen HY, Lin SF, Wang MC, Di J, et al. Neutralizing antibody response and SARS severity. Emerging Infectious Diseases 2005;11(11):1730-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Horby 2020
- Horby P, Lim WS, Emberson J, Mafham M, Bell J, Linsell L, et al. Effect of dexamethasone in hospitalized patients with COVID-19: preliminary report. medRxiv [Preprint] 2020. [DOI: ]
Horby 2021
- Horby PW, Pessoa-Amorim G, Peto L, Brightling CE, Sarkar R, Thomas K, et al. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): preliminary results of a randomised, controlled, open-label, platform trial. medRxiv 11 February 2021. [DOI: ]
Huang 2020
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020;395(10223):497-506. [DOI: 10.1016/S0140-6736(20)30183-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hung 2013
- Hung IF, To KK, Lee CK, Lee KL, Yan WW, Chan K, et al. Hyperimmune IV immunoglobulin treatment: a multicenter double-blind randomized controlled trial for patients with severe 2009 influenza A(H1N1) infection. Chest 2013;144(2):464-73. [DOI: 10.1378/chest.12-2907] [DOI] [PubMed] [Google Scholar]
Iwasaki 2021
- Iwasaki Akiko. What reinfections mean for COVID-19. The Lancet Infectious Diseases 2021;21(1):3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Izquierdo‐Dominguez 2020
- Izquierdo-Dominguez A, Rojas-Lechuga MJ, Mullol J, Alobid I. Olfactory dysfunction in the COVID-19 outbreak. Journal of Investigational Allergology & Clinical Immunology 2020;30(5):317-326. [DOI: 10.18176/jiaci.0567] [DOI] [PubMed] [Google Scholar]
Janiaud 2021
- Janiaud P, Axfors C, Schmitt A M, Gloy V, Ebrahimi F, Hepprich M, et al. Association of convalescent plasma treatment with clinical outcomes in patients with COVID-19: a systematic review and meta-analysis. JAMA 2021;(no indication):e212747. [DOI: 10.1001/jama.2021.2747] [DOI] [PMC free article] [PubMed] [Google Scholar]
Johns Hopkins 2021
- Johns Hopkins University and Medicine. Mortality analyses. Available from www.coronavirus.jhu.edu/data/mortality (last accessed 12 May 2021).
Joyner 2021
- Joyner MJ, Carter RE, Senefeld JW, Klassen SA, Mills JR, Johnson PW, et al. Convalescent plasma antibody levels and the risk of death from Covid-19. New England Journal of Medicine 13 January 2021;384:1015-1027. [DOI: 10.1056/NEJMoa2031893] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kim 2020
- Kim D-H, Choe YJ, Jeong J-Y. Understanding and interpretation of case fatality rate of coronavirus disease 2019. Journal of Korean Medical Science 2020;35(12):e137. [DOI: 10.3346/jkms.2020.35.e137] [DOI] [PMC free article] [PubMed] [Google Scholar]
Klassen 2021
- Klassen SA, Senefeld JW, Johnson PW, Carter RE, Wiggins CC, Shoham S, et al. Evidence favoring the efficacy of convalescent plasma for COVID-19 therapy (Preprint). Available from https://www.medrxiv.org/content/10.1101/2020.07.29.20162917v4 2021. [DOI: 10.1101/2020.07.29.20162917] [DOI]
Lauer 2020
- Lauer SA, Grantz KH, Bi Q, Jones FK, Zheng Q, Meredith HR, et al. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Annals of Internal Medicine 2020;172(9):577-582. [DOI: 10.7326/M20-0504] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lefebvre 2019
- Lefebvre C, Glanville J, Briscoe S, Littlewood A, Marshall C, Metzendorf MI, et al. Chapter 4: Searching for and selecting studies. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Li 2019
- Li T, Higgins JP, Deeks JJ, editor(s). Chapter 5: Collecting data. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Liang 2020
- Liang W, Guan W, Chen R, Wang W, Li J, Xu K, et al. Cancer patients in SARS-CoV-2 infection: a nationwide analysis in China. Lancet Oncology 2020;21(3):335-7. [DOI: 10.1016/S1470-2045(20)30096-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
Luke 2006
- Luke TC, Kilbane EM, Jackson JL, Hoffman SL. Meta-analysis: convalescent blood products for Spanish influenza pneumonia: a future H5N1 treatment? Annals of Internal Medicine 2006;145(8):599-609. [DOI] [PubMed] [Google Scholar]
Mair‐Jenkins 2015
- Mair-Jenkins J, Saavedra-Campos M, Baillie JK, Cleary P, Khaw FM, Lim WS, et al. The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: a systematic review and exploratory meta-analysis. Journal of Infectious Diseases 2015;211(1):80-90. [DOI: 10.1093/infdis/jiu396] [DOI] [PMC free article] [PubMed] [Google Scholar]
McGuinness 2020
- McGuinness LA, Higgins JP. Risk-of-bias VISualization (robvis): An R package and Shiny web app for visualizing risk-of-bias assessments. Research Synthesis Methods 2020;12:55– 61. [DOI: 10.1002/jrsm.1411] [DOI] [PubMed] [Google Scholar]
McKenzie 2019
- McKenzie JE, Brennan SE, Ryan RE, Thomson HJ, Johnston RV, Thomas J. Chapter 3: Defining the criteria for including studies and how they will be grouped for the synthesis. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Microsoft Corporation 2018 [Computer program]
- Microsoft Excel. Microsoft Corporation, 2018. Available from office.microsoft.com/excel.
Mo 2006
- Mo H, Zeng G, Ren X, Li H, Ke C, Tan Y, et al. Longitudinal profile of antibodies against SARS-coronavirus in SARS patients and their clinical significance. Respirology 2006;11(1):49-53. [DOI: 10.1111/j.1440-1843.2006.00783.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Journal of Clinical Epidemiology 2009;62(10):1006-12. [DOI] [PubMed] [Google Scholar]
Morens 1994
- Morens DM. Antibody-dependent enhancement of infection and the pathogenesis of viral disease. Clinical Infectious Diseases 1994;19(3):500-12. [DOI] [PubMed] [Google Scholar]
Mulder 2019
- Mulder RL, Bresters D, Van den Hof M, Koot BG, Castellino SM, Loke YK, et al. Hepatic late adverse effects after antineoplastic treatment for childhood cancer. Cochrane Database of Systematic Reviews 2019, Issue 4. Art. No: CD008205. [DOI: 10.1002/14651858.CD008205.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
NIH 2021
- National Institute of Health. NIH halts trial of COVID-19 convalescent plasma in emergency department patients with mild symptoms. Available from www.nih.gov/news-events/news-releases/nih-halts-trial-covid-19-convalescent-plasma-emergency-department-patients-mild-symptoms (accessed 22 March 2021).
Otrock 2017
- Otrock ZK, Liu C, Grossman BJ. Transfusion‐related acute lung injury risk mitigation: an update. Vox Sanguinis 2017;112(8):694-703. [DOI] [PubMed] [Google Scholar]
Pandey 2012
- Pandey S, Vyas GN. Adverse effects of plasma transfusion. Transfusion 2012;52 Suppl 1:65S-79S. [DOI] [PMC free article] [PubMed] [Google Scholar]
Park 2020
- Park JJ, Harari O, Dron L, Lester RT, Thorlund K, Mills EJ. An overview of platform trials with a checklist for clinical readers. Journal of Clinical Epidemiology 2020;125:1-8. [DOI] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta-analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815-34. [DOI] [PubMed] [Google Scholar]
Payne 2016
- Payne DC, Iblan I, Rha B, Algasrawi S, Hin A, Al Nsour M, et al. Persistence of antibodies against Middle East respiratory syndrome coronavirus. Emerging Infectious Disease Journal 2016;22(10):1824-6. [DOI: 10.3201/eid2210.160706] [DOI] [PMC free article] [PubMed] [Google Scholar]
Reeves 2019
- Reeves BC, Deeks JJ, Higgins JP, Shea B, Tugwell P, Wells GA. Chapter 24: Including non-randomized studies on intervention effects. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Review Manager Web [Computer program]
- The Cochrane Collaboration Review Manager Web (RevMan Web). The Cochrane Collaboration, 2019. Available from revman.cochrane.org.
Ricke 2020
- Ricke D, Malone R. Medical countermeasures analysis of 2019-nCoV and vaccine risks for antibody-dependent enhancement (ADE). Preprints [Preprint] 2020. [DOI: 10.20944/preprints202003.0138.v1] [DOI]
Robbins 1995
- Robbins JB, Schneerson R, Szu SC. Perspective: hypothesis: serum IgG antibody is sufficient to confer protection against infectious diseases by inactivating the inoculum. Journal of Infectious Diseases 1995;171(6):1387-98. [DOI] [PubMed] [Google Scholar]
Rock 2011
- Rock G. A comparison of methods of pathogen inactivation of FFP. Vox Sanguinis 2011;100(2):169-78. [DOI] [PubMed] [Google Scholar]
Santesso 2020
- Santesso N, Glenton C, Dahm P, Garner P, Akl A, Alper B, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. Journal of Clinical Epidemiology 2020;119:126-35. [DOI] [PubMed] [Google Scholar]
Schünemann 2019
- Schünemann HJ, Cuello C, Akl EA, Mustafa RA, Meerpohl JJ, Thayer K, et al. GRADE guidelines: 18. How ROBINS-I and other tools to assess risk of bias in nonrandomized studies should be used to rate the certainty of a body of evidence. Journal of Clinical Epidemiology 2019;111:105-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2020
- Schünemann HJ, Higgins JP, Vist GE, Glasziou P, Akl EA, Skoetz N, et al. Chapter 14: Completing ‘Summary of findings’ tables and grading the certainty of the evidence. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). Cochrane, 2020. Available from www.training.cochrane.org/handbook.
Sekul 1994
- Sekul EA, Cupler EJ, Dalakas MC. Aseptic meningitis associated with high-dose intravenous immunoglobulin therapy: frequency and risk factors. Annals of Internal Medicine 1994;121(4):259-62. [DOI] [PubMed] [Google Scholar]
Skoetz 2020
- Skoetz N, Goldkuhle M, Van Dalen EC, Akl EA, Trivella M, Mustafa RA, et al. GRADE guidelines 27: how to calculate absolute effects for time-to-event outcomes in summary of findings tables and evidence profiles. Journal of Clinical Epidemiology 2020;118:124-31. [DOI] [PubMed] [Google Scholar]
Sterne 2016
- Sterne JA, Hernán MA, Reeves BC, Savović J, Berkman ND, Viswanathan M, et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions. BMJ 2016;355:i4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2019
- Sterne JA, Savovic J, Page MJ, Elbers RG, Blencowe NS, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ 2019;366:l4898. [DOI] [PubMed] [Google Scholar]
Stiehm 2013
- Stiehm ER. Adverse effects of human immunoglobulin therapy. Transfusion Medicine Reviews 2013;27(3):171-8. [DOI] [PubMed] [Google Scholar]
Team 2020
- Team NCPERE . Vital surveillances: the epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) – China. China CDC Weekly 2020;2(8):113-22. [DOI: 10.3760/cma.j.issn.0254-6450.2020.02.003] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time-to-event data into meta-analysis. Trials 2007;8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tolouian 2020
- Tolouian R, Vahed SZ, Ghiyasvand S, Tolouian A, Ardalan M. COVID-19 interactions with angiotensin-converting enzyme 2 (ACE2) and the kinin system; looking at a potential treatment. Journal of Renal Injury Prevention 2020;9(2):e19. [DOI: 10.34172/jrip.2020.19] [DOI] [Google Scholar]
US Covid Plasma 2020
- US Covid Plasma. COVID-19 expanded access program. Available from www.uscovidplasma.org (accessed 22 March 2021).
Van de Veerdonk 2020
- Van de Veerdonk F, Netea MG, Van Deuren M, Van der Meer JW, De Mast Q, Bruggemann RJ, et al. Kinins and cytokines in COVID-19: a comprehensive pathophysiological approach. Preprints [Preprint] 2020. [DOI: 10.20944/preprints202004.0023.v1] [DOI]
Wan 2020
- Wan Y, Shang J, Sun S, Tai W, Chen J, Geng Q, et al. Molecular mechanism for antibody-dependent enhancement of coronavirus entry. Journal of Virology 2020;94(5):e02015-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2014
- Wang S-F, Tseng S-P, Yen C-H, Yang J-Y, Tsao C-H, Shen C-W, et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochemical and Biophysical Research Communications 2014;451(2):208-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2007
- World Health Organization (WHO). Cumulative number of reported probable cases of SARS. Available from www.who.int/csr/sars/country/2003_07_11/en/ (accessed 22 March 2021).
WHO 2019
- World Health Organization (WHO). Middle East respiratory syndrome coronavirus (MERS-CoV). Available from www.who.int/emergencies/mers-cov/en/ (accessed 22 March 2021).
WHO 2020a
- World Health Organization (WHO). Report of the WHO‐China Joint Mission on coronavirus disease 2019 (COVID‐19); February 2020. Available from www.who.int/docs/default-source/coronaviruse/who-china-joint-mission-on-covid-19-final-report (accessed 22 March 2021).
WHO 2020b
- World Health Organization (WHO). Rolling updates on coronavirus diseases (COVID‐19). Available from www.who.int/emergencies/diseases/novel-coronavirus-2019/events-as-they-happen (accessed 22 March 2021).
WHO 2020c
- World Health Organization (WHO). Coronavirus disease (COVID-19) situation report-209; 16 August 2020. Available from https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200816-covid-19-sitrep-209.pdf?sfvrsn=5dde1ca2_2 (accessed 22 March 2021).
WHO 2020d
- World Health Organization (WHO). Clinical management of severe acute respiratory infection (SARI) when COVID19 disease is suspected: interim guidance. World Health Organization 2020;WHO/2019-nCoV/clinical/2020.4.
WHO 2020e
- WHO Working Group on the Clinical Characterisation and Management of COVID-19 infection. A minimal common outcome measure set for COVID-19 clinical research. Lancet Infectious Diseases 2020;20(8):e192-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2020f
- World Health Organization. COVID-19 Therapeutic Trial Synopsis. Available from www.who.int/blueprint/priority-diseases/key-action/COVID-19_Treatment_Trial_Design_Master_Protocol_synopsis_Final_18022020.pdf (accessed 22 March 2021).
WHO 2020g
- World Health Organization. Draft landscape of COVID-19 candidate vaccines. Available from www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (accessed 22 March 2021).
Williamson 2020
- Williamson E, Walker AJ, Bhaskaran KJ, Bacon S, Bates C, Morton CE, et al. OpenSAFELY: factors associated with COVID-19-related hospital death in the linked electronic health records of 17 million adult NHS patients. medRxiv [Preprint] 2020. [DOI: ]
Wu 2020a
- Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Internal Medicine 2020;180:934-943. [DOI: 10.1001/jamainternmed.2020.0994] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wu 2020b
- Wu F, Wang A, Liu M, Wang Q, Chen J, Xia S, et al. Neutralizing antibody responses to SARS-CoV-2 in a COVID-19 recovered patient cohort and their implications. medRxiv [Preprint] 2020. [DOI: ]
Xu 2020a
- Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respiratory Medicine 2020;8(4):420-2. [DOI: 10.1016/S2213-2600(20)30076-X] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhao 2020a
- Zhao J, Yuan Q, Wang H, Liu W, Liao X, Su Y, et al. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.03.02.20030189] [DOI] [PMC free article] [PubMed]
References to other published versions of this review
Chai 2020
- Chai KL, Valk SJ, Piechotta V, Kimber C, Monsef I, Doree C, et al. Convalescent plasma or hyperimmune immunoglobulin for people with COVID‐19: a living systematic review. Cochrane Database of Systematic Reviews 2020, Issue 10. Art. No: CD013600. [DOI: 10.1002/14651858.CD013600.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Piechotta 2020a
- Piechotta V, Valk SJ, Chai KL, Wood EM, Lamikanra A, Kimber C, et al. Safety and effectiveness of convalescent plasma or hyperimmune globulin for people with COVID-19: a rapid review. available at: doi.org/10.17605/OSF.IO/DWF53 2020. [DOI: 10.17605/OSF.IO/DWF53] [DOI] [PMC free article] [PubMed]
Piechotta 2020b
- Piechotta V, Chai KL, Valk SJ, Doree C, Monsef I, Wood EM, et al. Convalescent plasma or hyperimmune immunoglobulin for people with COVID-19: a living systematic review. Cochrane Database of Systematic Reviews 2020, Issue 7. Art. No: CD013600. [DOI: 10.1002/14651858.CD013600.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Piechotta 2021
- Piechotta V, Iannizzi C, Chai KL, Valk SJ, Kimber C, Dorando E, et al. Risk of bias assessments for version 4 of the Cochrane review 'Convalescent plasma or hyperimmune immunoglobulin for people with COVID-19: a living systematic review'. https://zenodo.org/record/4715089#.YIKvBOj7RaQ 2021. [DOI: 10.5281/zenodo.4715089] [DOI] [PMC free article] [PubMed]
Valk 2020
- Valk SJ, Piechotta V, Chai KL, Doree C, Monsef I, Wood EM, et al. Convalescent plasma or hyperimmune immunoglobulin for people with COVID-19: a rapid review. Cochrane Database of Systematic Reviews 2020, Issue 5. Art. No: CD013600. [DOI: 10.1002/14651858.CD013600] [DOI] [PMC free article] [PubMed] [Google Scholar]