

Abstract
Impressive progress has been made over the last several years toward understanding how almost every aspect of the immune system contributes to the expression of systemic autoimmunity. In parallel, studies have shed light on the mechanisms that contribute to organ inflammation and damage. New approaches that address the complicated interaction between genetic variants, epigenetic processes, sex and the environment promise to enlighten the multitude of pathways that lead to what is clinically defined as systemic lupus erythematosus. It is expected that each patient owns a unique ‘interactome’, which will dictate specific treatment.
It took almost 100 years to realize that lupus erythematosus, which was initially thought to be a skin entity, is a systemic disease that spares no organ and that an aberrant autoimmune response is involved in its pathogenesis. The involvement of vital organs and tissues such as the brain, blood and the kidney in most patients, the vast majority of whom are women of childbearing age, impels efforts to develop diagnostic tools and effective therapeutics. The prevalence ranges from 20 to 150 cases per 100,000 people and appears to be increasing as the disease is recognized more readily and survival rates improve. In the United States, people of African, Hispanic or Asian ancestry, as compared to those of other racial or ethnic groups, tend to have an increased prevalence of systemic lupus erythematosus (SLE) and greater involvement of vital organs. The 10-year survival rate has increased significantly over the last 50 years to more than 70%, mostly because of greater awareness of the disease, the extensive and wiser use of immunosuppressive drugs and a more efficient treatment of infections, the major cause of death1–3.
Although low levels of autoreactivity and autoimmunity are necessary for lymphocyte selection and, in general, for the regulation of the immune system, in certain individuals, autoimmunity advances through multiple pathways (reviewed in ref. 4) and leads to organ inflammation and damage. The diverse mechanisms do not contribute equally to the expression of disease in all patients with SLE, as will be discussed below. It appears that the clinical heterogeneity of the disease is matched by the multiple pathogenic processes, which justifies the call for the development of personalized medicine (Fig. 1).
Fig. 1 |. The pathogenetic landscape of SLE.
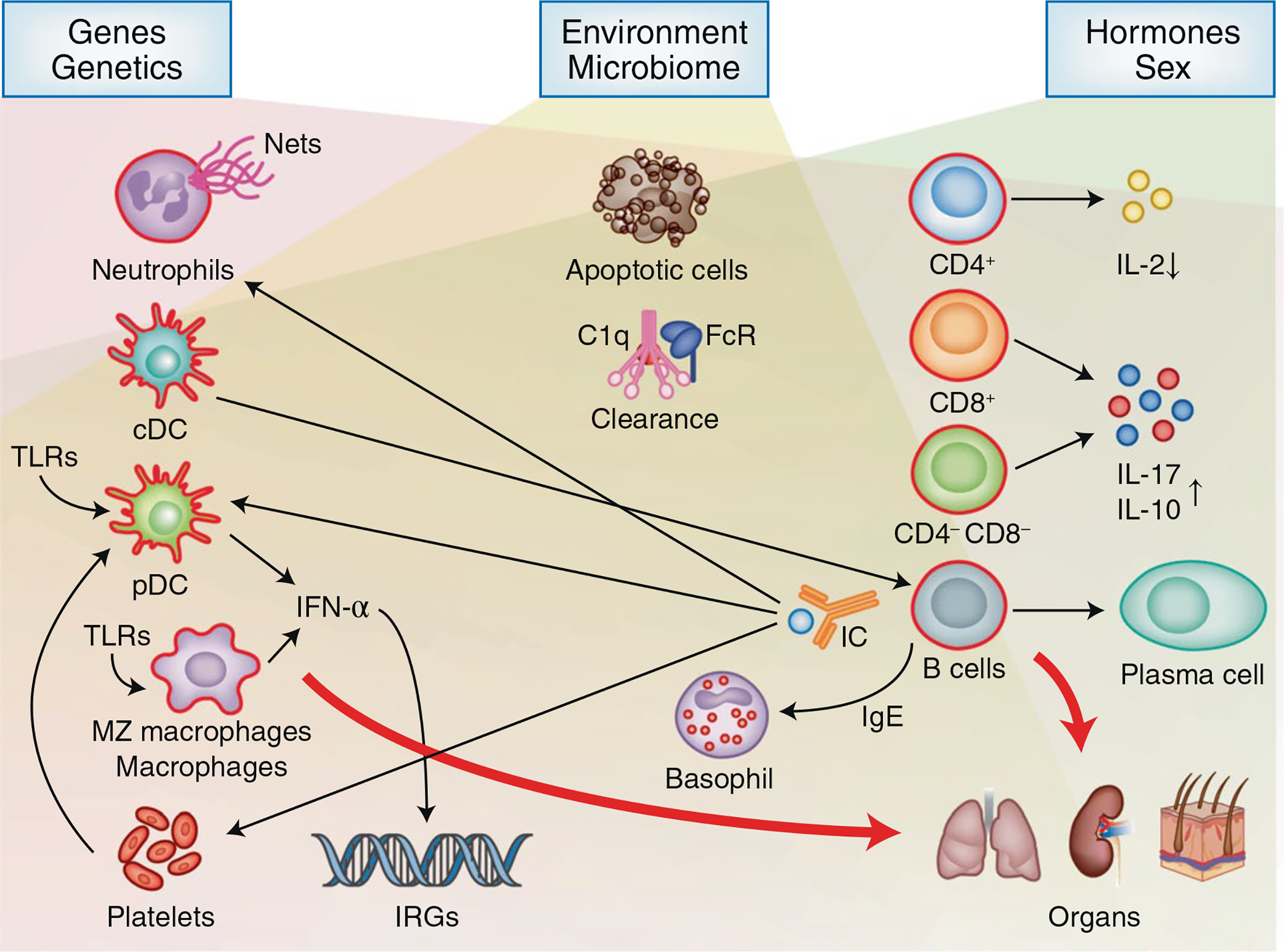
Genetic, environmental and hormonal factors act on various elements of the innate and adaptive immune responses. Gene copy variants (for example, C4 and FCR) and SNPs influence the expression of many genes involved in the immune response as well as in the control of organ damage (for example, APOL1 and KLK). Environmental factors, including UV light, drugs and products of the microbiome, alter T and B cell responses and the functions of innate cells by stimulating TLRs. Hormones and genes defined by the X chromosome contribute to disease expression by altering the function of lymphocytes and of cells of the innate immune response. The involved factors lead eventually to loss of tolerance of B and T cells to autoantigens, which are present in abundance because of both increased rates of apoptosis and defects in mechanisms responsible for their clearance. The T cell response to antigen is aberrant in and late signaling events (Fig. 2) and results in misbalanced levels of cytokines, including decreased IL-2 and increased IL-17 production. T cells, through distinct pathways, also acquire a greater ability to invade tissues and contribute to the inflammatory response. B cells, in response to cognate and non-cognate (cytokines) interactions with T cells, produce antibodies. Antibodies enter tissues directly or in the form of immune complexes (IC), which contribute to tissue inflammation. Cells of the innate immune response, under the influence of the involved pathogenic factors, produce cytokines (including IFN-α) or, through the direct interaction with lymphocytes, contribute significantly to the inflammatory organ-damaging response. The clinical heterogeneity of the disease is highly correlated with the multitude of the pathways that lead to organ injury. Although several pathways operate in each individual, the relative contribution of each pathway varies from person to person. Finally, local factors dictate which organ will be afflicted by the autoinflammatory response.
Genes and genetics
Over the past two decades, extensive genome-wide association studies and meta-analyses have identified close to 150 new SLE risk loci across multiple ancestries5,6. Extensive use of exome sequencing has revealed an increasing number of monogenic cases of SLE, listed in ref. 7. Interestingly, several of the risk loci span multiple autoimmune diseases8,9, stipulating disease commonality. Functional studies of shared loci may eventually help reclassify autoimmune diseases according to shared pathways, along with clinical characteristics. Detailed lists of gene variants have been published, and a limited number is listed in Table 1, along with evidence supporting involvement in disease pathogenesis. Several variants have been linked genetically, some with supporting biology, to pathogenic mechanisms and specific clinical manifestations, pointing strongly to the heterogeneity of the disease. Several genes linked to the immune response are regulated through long-distance chromatin interactions10,11. Studies addressing long-distance interactions between gene variants in SLE are still missing, but, with the advent of new technologies, such studies will emerge.
Table 1 |.
Select gene variants linked to SLE and suggested function
| Gene | Function | Ref. |
|---|---|---|
| IRF5 (promoter variants) | Increased expression, which leads to induction of proinflammatory cytokines and the definition of an inflammatory macrophage phenotype | 126 |
| IRF7 (single SNP) | Increased production of type I IFN by DCs | 127 |
| STAT4 | Binds the transcription factor HMGA1; shared with rheumatoid arthritis | 128 |
| STAT1 | Increased binding to a variant of the transcription factor ETS1 | 129 |
| PRDM1 | Linked to increased production of IL-6 and cathepsin S (needed for loading peptides into MHC-II) | 16,17 |
| IKZF1 | Involved in the expression of C1QB and five type I IFN–response genes | 130 |
| Fc receptor genes | Clearance of apoptotic cells and immune complexes; antibody-mediated cell cytotoxicity | 131 |
| C1q, C4 | Clearance of apoptotic cells; negative immune selection | 132 |
| C4 | Decreased number of copy variants is linked to SLE | 133 |
| FCGR3B | Decreased number of copy variants is linked to SLE | 134 |
| NCF1 | Decreased number of copies predispose to, whereas increased protects against, SLE | 135 |
| ITGAM | Impaired phagocytosis of complement-opsonized targets in monocytes, neutrophils and macrophages | 136 |
| NCF2 | Essential to LC3-associated phagocytosis | 137 |
| ATG5, PRKCD, NCF1 | Essential for autophagy | 137 |
| TLR7 | Increased IFN-α production | |
| TNFAIP3 | Encodes the deubiquitinating enzyme A20, which is involved in the termination of NF-κB signaling; A20-deficient mice develop severe autoinflammatory disease | 138,139 |
| PTPN22 | Expansion of effector and memory T cells and increased production of IFN-γ, TNF and GM-CSF | 140 |
| Major histocompatibility antigens | ||
| HLA-DR2 and HLA-DR3 | Contributes to SLE in Europeans | 134 |
| HLA-DRB1*15:01 and HLA-DQB1*6:02 | Contributes to SLE in Asians | 141 |
| HLA-DR3 and HLA-DR15 | Contributes to SLE across all ancestries | 6 |
| BANK1 and BLK | Interaction between the SLE-associated variants | 142 |
| IgG1 | A knockin mouse with a patient-identified IgG1 SNP developed lupus | 143 |
| APOL1 | Linked to lupus nephritis | 144 |
| VGLL3 | Sex-biased transcription factor | 27 |
Better understanding of the epigenome is needed to understand how it supplements the genetic contribution to the disease. Decreased DNA methylation of certain genes in SLE T cells has been recognized and has been attributed to poor function of the CpG remethylating enzyme DNMT1. For example, hypomethylation of TNFSF5, located on the X chromosome, results in increased CD40-ligand (CD40L) expression in T cells from women, and hypomethylation of Il10 increases interleukin (IL)-10 production. Yet methylation of genes in SLE is more complicated and does not follow a unidirectional pattern. For example, the CD8 locus is methylated in T cells from patients with SLE, resulting in the generation of CD3+CD4−CD8− T cells, whereas the Il2 locus is hypermethylated, resulting in low production of IL-2 (reviewed in ref. 12). Delivery of a demethylating agent specifically to either CD4+ or CD8+ cells in lupus-prone mice suppresses disease expression by enhancing the expression of FoxP3 and sustaining the expression of CD813.
In SLE T cells, the cyclic AMP response element modifier (CREMα) binds to various regulatory elements within the CD8 cluster and recruits histone modifiers, including DNMT3a and the histone methyltransferase G9a, causing stable silencing of CD8A and CD8B14. A similar process takes place in the Il2 locus, resulting in decreased IL-2 production15. In contrast, STAT3 recruitment to Il10 regulatory regions mediates the recruitment of histone acetyltransferase p300, resulting in enhanced gene expression16.
A large number of microRNAs are suspected to control at least one-third of human mRNA stability and translation, and, reasonably, they have been studied in SLE. A limited list is presented in Table 2. MicroRNA serum concentrations may serve as disease biomarkers17, and the development of antagomirs, used to silence endogenous micoRNAs, may help to control the disease.
Table 2 |.
MicroRNAs control aspects of the immune response and organ damage in SLE
| MicroRNA | Function | Ref. |
|---|---|---|
| miR-146a | Regulates the innate immune response | 145 |
| miR-125a | Regulates the anti-inflammatory response | 146 |
| miR-155 | Controls the expression of SH2-domain-containing inositol 5′-phosphatase 1 (SHIP1), which is important in B cell activation; miR155−/−Faslpr mice have decreased autoimmunity and nephritis | 147 |
| miR-148a | Impairs B cell tolerance by suppressing the expression of the autoimmune suppressor Gadd45a, the tumor suppressor PTEN and the proapoptotic protein Bim, which promotes the survival of immature B cells after engagement of the B cell antigen receptor | 148 |
| miR-23b | Suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKKα mRNA | 149 |
| miR-17[sim]92 | Promotes TFH cell differentiation; promotes DC activation through the microRNA let-7c and BLIMP-1 |
150 151 |
| miR-150 | Promotes renal fibrosis in lupus nephritis by downregulating expression of SOCS1 | 152 |
The contribution of epigenetic modifications to the expression of the disease complements the genetic susceptibility. Better understanding of the biochemical processes involved should offer unique opportunities to develop new therapeutics, and in a personalized manner.
Environment
Approximately a third of monozygotic twins are clinically concordant for SLE, which signifies the importance of environmental factors in the expression of the disease. The environment, including ultraviolet (UV) light, and cross-reactivity between self-antigens and molecules defined by viruses and other pathogens is important in the pathogenesis of SLE. Microbes, including innocuous commensal organisms that colonize the gut, skin, nasal cavities and the vagina, may trigger and sustain autoimmune inflammation in genetically susceptible hosts. Microbiota have been amply shown to shape the immune response, including the development of T helper type 1 (TH1), TH2 and regulatory T (Treg) cells and have been implicated in several autoimmune diseases18.
Cross-reactivity between bacterial species and autoantigens has been long claimed to contribute to the expression of disease in susceptible individuals. The bacterium Propionibacterium propionicum, which encodes an ortholog of the RNA-binding protein Ro60, was found in cutaneous lesions of patients with subcutaneous lupus erythematosus and was shown to stimulate memory T cells from patients with SLE19, suggesting a direct involvement of pathogens in T cell proliferation and the production of autoantibodies.
Segmented filamentous bacteria induce intestinal IL-17-producing TH17 cells20, and gut microbiota drive autoimmune arthritis by promoting the generation of follicular helper T (TFH) cells21. Microbiota apparently use Toll-like receptor (TLR) signaling because TNFAIP3 (A20)-deficient mice, which develop autoimmunity, fail to do so when MyD88 (a central TLR signaling molecule) is genetically deleted, or the mice are treated with antibiotics22. Microbiota translocate from the gut to the mesenteric lymph nodes, spleen23 and the liver and induce TH17 and TFH cells as well as innate immune pathways, including the plasmacytoid dendritic cell (pDC)–type I interferon (IFNα/β) axis. Interestingly, certain microbiota can be found in the liver of patients with SLE or autoimmune hepatitis24. Microbiota contribute to disease expression through a number of mechanisms, including molecular mimicry, engagement of the innate immune response and the propagation of proinflammatory TH17 cells. Accordingly, better understanding of the role of microbiota in the expression of SLE should reveal simple approaches for controlling autoimmunity though dietary changes or changing the distribution of microbiota in the gut and elsewhere.
Sex
Despite the fact that more than 90% of the people affected with SLE are women, we still do not have a clear understanding of the causative mechanisms. It is well known that people with XXX (Klinefelter syndrome) are prone to SLE and that epigenetic changes in certain pathogenic genes (for example, TNFSF5, encoding CD40L) contribute to disease expression. Also, six SLE susceptibility loci map to the X chromosome, four of which (TLR7, TMEM187, IRAK1 (MyD88-interacting kinase) and the IFN-α-inducible CXorf21) can escape X-chromosome inactivation25.
Estrogen alters the thresholds for B cell apoptosis and activation, and estrogen receptor α contributes to T cell–mediated autoimmune inflammation by promoting T cell activation, and it also promotes lupus in NZB × NZW F1 mice. At the molecular level, estrogen upregulates CREMα expression, which is known to control the expression of Il2 and Il17 (reviewed in ref. 26). Gene expression analysis revealed a female-biased autoimmunity-related network driven by the transcription factor VGLL3 that is linked with autoimmune diseases, including SLE, Sjogren’s syndrome and scleroderma27. While we still do not understand why women represent the vast majority of people with SLE, new evidence points to distinct molecular processes and the probable activation of X-chromosome-defined genes that are genetically linked to SLE.
Innate immune cell disturbances
Genetic and epigenetic factors contribute directly to alter cells of both the innate and adaptive immune responses. It is probable that certain immune aberrations may elicit others. Studies in mice and humans are still limited because of the reductionist approaches that are needed to understand the contribution of each abnormality to the expression of the disease. It will require artificial intelligence approaches to understand the sequence of events in any given patient. Such knowledge will be necessary for the application of precision treatment protocols.
Neutrophils in patients with SLE display an increased capacity to form neutrophil extracellular traps (NETosis) that harbor autoantigens, including chromatin, dsDNA and granular proteins. In patients with SLE, NETs are poorly cleared and stimulate pDCs to produce type I IFN through TLR9 stimulation28. Endothelin-1 and hypoxia-inducible factor-1α appear to mediate the expression of the stress-response protein REDD1, which drives the formation of NETs in SLE. NETs are decorated with tissue factor and IL-17 and are abundant in discoid skin lesions and in the kidneys of patients with SLE29. Further, splenic neutrophils localized in the perimarginal zone can induce immunoglobulin (Ig) class switching, somatic hypermutation and antibody production by activating marginal zone B cells. Interestingly, patients who are neutropenic have fewer and less mutated marginal zone B cells and less preimmune Ig specific for T-independent antigens, suggesting neutrophils generate an additonal level of innate immunity in the antibacterial defense30.
DCs link innate and adaptive immune responses and have been identified in the expression of SLE, as their uncontrolled activation may drive autoimmunity31. Although the numbers are decreased in the periphery, they are found activated in the inflamed tissues, producing inflammatory cytokines and helping T and B cells. Immune complexes containing RNA induce OX40-ligand expression by conventional SLE DCs. Subsequently, they drive the differentiation of naive and memory CD4+ T cells into TFH cells, which are able to help B cells32 and impair Treg function33. Conventional DCs in SLE instruct the differentiation of IgG and IgA plasmablasts and contribute to the formation of ectopic lymphoid structures34.
pDCs are distinguished from conventional DCs by morphology and cell surface markers and are similarly low in the periphery, probably because they lodge inflamed areas. Triggered by TLR7/9 agonists, they produce IFN type I35 to contribute to disease expression, and duplication of TLR7 promotes disease36. Specific depletion of pDCs in mice reduces disease manifestations such as autoantibody production, glomerulonephritis and expression of IFN-inducible genes37. At the clinical level, targeting of pDCs with a BDCA2 antibody ameliorates skin disease in patients with SLE38, while chronic triggering of pDCs through TLR7 and TLR9 renders pDCs resistant to inhibition of the NF-κB pathway and leads to steroid resistance39.
Marginal zone macrophages surrounding the splenic follicles are crucial for the efficient clearance of apoptotic cells and for the induction of tolerance to autoantigens. Phagocytosis of apoptotic cells by splenic marginal zone macrophages requires the megakaryoblastic leukemia 1 transcriptional coactivator–mediated mechanosensing pathway40. The production of type I IFN by macrophages in response to TLR7 engagement is enabled by the TREML4 receptor expressed on myeloid cells, and macrophages from Treml4−/− mice are hyporesponsive to TLR7 agonists, while TREML4-deficient MRL-lpr lupus-prone mice display decreased autoimmunity and nephritis41. It should also be noted that IFN type I and tumor necrosis factor (TNF) cooperate to promote an inflammatory signature in monocytes, and such cooperation also occurs in monocytes from patients with SLE42.
Type I IFN, which affects multiple components of the immune system, has been demonstrated to contribute to the pathogenesis of adult and pediatric SLE43 and to reflect disease activity (reviewed in detail in ref. 44). Yet type I IFN alone may not be sufficient to cause disease expression45 and, in some murine strains, may even be beneficial46.
Proper processing of apoptotic material involves the activation of the transcription factor aryl hydrocarbon receptor (AhR) following engagement of TLR9, which leads to a series of events that suppress inflammation, including the production of IL-10. Deletion of AhR in myeloid cells causes autoimmunity, and its transcription signature correlates with disease activity in mice and humans47. This observation may explain why TLR9 has a protective effect against autoimmunity.
Platelets are activated in patients and mice with SLE through a number of mechanisms, including the action of immune complexes and contact with injured endothelial cells, and they display a type I IFN signature48. Once activated, platelets express and release CD40L and modulate adaptive immunity by activating antigen-presenting cells, including DCs. Platelets interact with pDCs in patients to increase the secretion of type I IFN by triggering TLR9 and TLR749. Understanding of the contribution of platelets may reveal adjuvant tools for the treatment of SLE.
Autoreactive IgE causes basophils to home to lymph nodes, promote TH2 cell differentiation and enhance the production of self-reactive antibodies that cause lupus-like nephritis in mice lacking the protein tyrosine kinase Lyn. Patients with SLE with elevated concentrations of self-reactive IgEs and activated basophils have increased disease activity and active lupus nephritis50.
Studies in mice have shown, in a definitive manner, the roles of neutrophils, basophils, pDCs, TLR activation and IFN type I production in the expression of SLE. Several of these contribute directly to organ damage, whereas others instruct, directly or indirectly, the aberrations of the adaptive immune response. The diversity of the involved pathways underlines the wide clinical spectrum of the disease, and it is quite possible that each cellular element contributes to the expression of disease, to varying degrees.
Lymphocyte disturbances in SLE
B cells in SLE have been reviewed extensively51–53. Loss of B cell tolerance at distinct check points has explained the production of autoantibodies. B cell antigen receptor (BCR)–sequencing studies in children with SLE suggested that defects at distinct checkpoints in early B cell development accounted for autoantibody production54. Although autoimmunity results from the failure of tolerance checkpoints, there is evidence that it may arise from the expansion of existing autoreactive cells55.
Age-associated B cells (which include IgD−CD27− and CD21lo B cells) have been detected in human autoimmune disorders, including SLE. Their expansion is controlled by the transcription factor IRF5, variants of which are linked to SLE, through IL-21 expression and unique landscape remodeling56. In mice, age-associated B cells are expanded in the absence of GTPase regulatory proteins (DEF6 and SWAP70), and DEF6 variants have been identified as conferring increased susceptibility to SLE57.
The SLE molecular signature in B cells appears to become established during the resting naive phase and is dominated by the enrichment of accessible chromatin motifs for the transcription factors AP-1 and EGR58, which is facilitated by, probably among other factors, IFN-γ59. B cells that lack IgD and CD27 are known to be expanded in patients with SLE and to produce autoantibodies. These cells are hyperresponsive to TLR7 agonists and IL-21, lack the TLR regulator TRAF560 and have features of freshly activated naive B cells61.
Although allergic reactions are not more frequent in people with SLE as compared to normal subjects, IgE antibodies specific for dsDNA are present in the sera of patients with SLE. These IgE antibodies bind to the high-affinity FcγRI receptor for IgE, can activate pDCs and transfer DNA to TLR9 in phagosomes. This activation results in the secretion of substantial amounts of IFN-α62.
T cells are key players in promoting the autoimmune response by providing help to B cells and by activating antigen-presenting cells through cytokine release and direct cellular contact. Additionally, they infiltrate tissues and promote local inflammation. Autoreactive CD4+ T cells are presumed to respond to nucleosomal antigens and, in particular, to peptides derived from histones63. An interesting T cell subset of unknown pathogenic importance was recorded in patients with SLE and multiple sclerosis (CXCR3+CD38+CD39+PD-1+HLA-DR+CD161+KLRG1−CD28+OX40+), which differs from TFH cells and was first recognized in the gut of patients with celiac disease by virtue of binding gluten64.
TFH cells promote B cell function and evolve from CD4+ T cells in the presence of IL-6, IL-21 and inducible T cell costimulatory (ICOS)65. ICOS deficiency protects MRL-lpr mice from disease66. A CD4+ cell subset that resembles TFH cells is expanded in the peripheral blood of patients with active SLE65. The ATP-gated ionotropic P2X7 receptor restricts the expansion of aberrant TFH cells, but TFH cells from patients with SLE are resistant to P2X7-mediated inhibition of cytokine-driven expansion, pointing to a signaling defect67. CXCR5−CXCR3+PD-1+ helper T cells, different from TFH cells, are present in the periphery and in kidney tissues of people with SLE, and provide help to B cells by producing IL-10 and succinate68.
CD8+ T cell cytotoxic responses are decreased in SLE and contribute to increased rates of infection7. A CD8+CD38+ T cell population is expanded in the peripheral blood of patients with SLE. CD8+CD38+ T cells display decreased production of granzymes and perforin and reduced cytotoxic capacity, and patients with SLE, for whom this population is expanded, experience infections more frequently. CD38, a marker of T cell exhaustion, is an ectonucleotidase that degrades NAD and, through the histone methyltransferase EZH2, suppresses the expression of cytotoxicity-related molecules69. Specific inhibitors of CD38-mediated NAD degradation ameliorate age-related metabolic dysfunction and may be of use in restoring CD8+ T cell cytotoxic activity in people with SLE70. Although exhaustion, defined by the levels of expression of molecules, has been argued to be desirable in autoimmunity71, it is important to understand the metabolic processes involved in further detail.
Treg cells are characterized by constitutive expression of the transcription factor FoxP3 and high expression of the high-avidity IL-2 receptor α chain (CD25); in humans, some activated T effector (Teff) cells also transiently express this molecule. The number of Treg cells is reduced during the early phases of the disease, whereas the CD45RA−FoxP3lo non-Treg cell population is increased in active SLE72. The realization that Treg cells have higher affinity receptors for IL-2 and, therefore, stronger IL-2 receptor (IL-2R)–mediated signaling than Teff cells, suggested that administration of IL-2 at a lower dose than that used for Teff cells should promote Treg cell expansion and function. Administration of low-dose IL-2 to lupus-prone mice expanded the population of Treg cells and shrank the pool of CD3+CD4−CD8− IL-17-producing T cells73, which are known to contribute to the development of lupus nephritis74. Low-dose IL-2 administered to people with SLE has been reported75 to produce clinical benefit. A caveat to the apparent success of low-dose IL-2 is evidence that the IL-2–IL-2R–p-STAT5 signaling pathway in SLE T cells is compromised76. IL-2 has the potential to reverse several pathogenic processes involved in the development of SLE, including poor Treg cell function, increased IL-17 production, increased TFH cell activity and the expansion of the population of CD4−CD8− T cells77. The demonstration that Treg cells contribute to tissue repair78 and the possibility that Treg cells are limited in the kidneys of patients with lupus nephritis79 encourage the consideration of approaches that enrich Treg cells in the kidneys or other tissues.
The phenotype and function of T cells isolated from patients with SLE has been extensively studied in the search for clues to explain the pathogenesis of the disease and in an attempt to identify molecules that can serve as biomarkers and/or therapeutic targets80. These studies have revealed that, in the context of SLE, T cell function is severely compromised as a result of a large number of signaling aberrations that distort gene expression profiles and skew the cellular immune response toward a proinflammatory type (reviewed in refs. 80,81). In brief, CD3-mediated T cell signaling is abnormal in people with SLE, and this is followed by aberrant expression of kinases, phosphatases, transcription factors, chemokine receptors and adhesion molecules and the production of chemokines and proinflammatory cytokines (Fig. 2).
Fig. 2 |. T cell early and late signaling aberrations in T cells from patients with SLE.
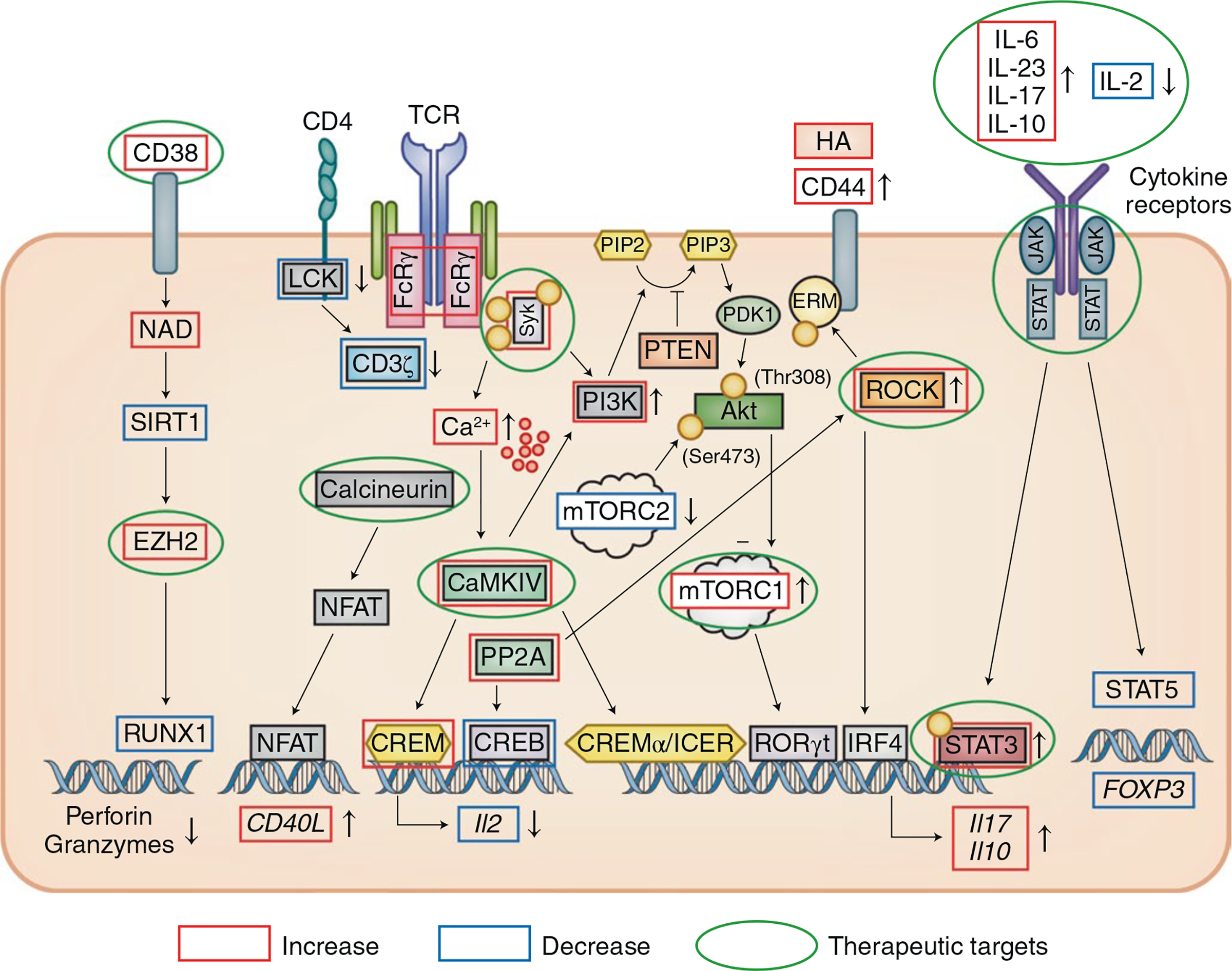
Engagement of the CD3 complex either by autoantigen or circulating CD3/TCR antibodies results in an increased intracytoplasmic calcium response. The CD3 complex is rewired, with the CD3ζ chain replaced with the FcRγ chain, which recruits the kinase Syk. The calcium-requiring kinase CaMKIV enhances the binding of CREMα/ICER to the Il2 and Il17 or Il10 promoters to suppress and enhance their expression, respectively. Calcineurin also dephosphorylates the transcription factor NFAT, which binds to the promoter of CD40L and increases its expression. Phosphatase PP2A is increased in T cells with diverse effects: in Treg cells, it dephosphorylates mTORC1 and promotes Treg cell function and, in effector T cells, it enhances the binding of IRF4 to the Il17 promoter and it dephosphorylates p-CREB. PP2A also promotes ROCK activity, which phosphorylates ERM, which in turn enhances the ability of CD44 to bind its ligand hyaluronic acid (HA, expressed in the kidney and other tissues). IL-2 signaling is defective with decreased amounts of p-STAT5, whereas IL-6 signaling is increased with increased binding of p-STAT3 to the Il17 promoter. An increased proportion of CD8+ T cells express the ectonucleotidase CD38, which suppresses the level of NAD and the activity of the deacetylase SIRT1, which in turn enhances the activity of the histone methyltransferase EZH2. As indicated by the green circles, a number of the molecules have been considered as therapeutic targets: Syk, ROCK, calcineurin, EZH2, IL-17, IL-23, JAK and mTOR as well as IL-2, which can be replenished with low doses.
Metabolic abnormalities have been recognized in people with SLE and in lupus-prone mice and have been linked to abnormal T cell function. SLE T cells display increased oxidative stress, as indicated by the depletion of glutathione (via NADPH loss), metabolic checkpoint kinase complex mTORC1, glycolysis and glutaminolysis. Inhibition of increased mTORC1, glycolysis or glutaminolysis mitigates disease in lupus-prone mice (reviewed in ref. 82).
Lupus nephritis
The kidney is involved in more than half of patients with SLE and contributes significantly to morbidity. The contribution of autoantibodies with a number of reactivities and of immune complexes in the expression of kidney inflammation has been reviewed extensively over the years. Their role in instigating injury is considered to be mediated through the activation of the complement system, which accounts for the inflammatory response83. Podocytes express increased amounts of the serine/threonine kinase CaMK4, which, through a distinct series of biochemical events, causes injury, and cell-targeted inhibition of CaMK4 in podocytes averts the deposition of immune complexes and nephritis84. These findings signify the importance of resident cells in the initiation and propagation of kidney inflammation.
T cell migration to the kidney is important in the development of lupus-like nephritis85. MRL-lpr mice that lack TCRαβ do not develop lupus nephritis86. Although kidney-infiltrating cells were thought to be exhausted87, clonally expanded CD4+ and CD8+ T cells with memory effector cell markers are present in the kidneys of lupus nephritis. CD8+ T cells are present in all biopsy samples and were found to adhere to the Bowman’s capsule and to infiltrate the tubular epithelium88 and contribute to kidney damage. IL-17-producing T cells have been found within kidney cell infiltrates of patients with lupus nephritis74, and IL-17 is important for the development of lupus nephritis89. TFH cells are present in the kidneys in close association with B cells in people with lupus nephritis, suggesting that they may provide help to them90. Intrarenal B cells form germinal center–like structures produce antibodies to vimentin, which is a dominant target in human lupus tubulointerstitial nephritis91. Application of deep convolutional neural network methodology in specimens from patients with lupus nephritis enabled cell-distance mapping, which confirmed that DCs present antigen to CD4+ T cells92. Understanding the cellular architectures of in situ immunity in lupus nephritis should expand our understanding of the involved pathogenic processes.
Intrarenal macrophages have been considered important in the development of lupus nephritis (reviewed in ref. 93). Analyses of macrophage and DC infiltrates in murine lupus nephritis have shown considerable heterogeneity. Monocytes are located around glomeruli and adjacent to tubules and peritubular capillaries in the renal interstitium and derive from the expanded circulating Gr1lo monocyte population94. Brief ischemia accelerates the infiltration of Ly6Chi inflammatory macrophages into the kidneys of MRL-lpr mice95. DCs also infiltrate the kidneys in people with lupus nephritis, probably propagating local adaptive immune responses. Myeloid DC infiltration is associated with the accumulation of lymphoid aggregates in the kidneys94. The identification of anti-inflammatory monocytes in the kidneys of patients with lupus nephritis96 is especially important in considering prohealing rather than immunosuppressive therapeutic approaches. Lastly, CD43hiCD11c+F4/80loMHC-II− patrolling monocytes, which are known to orchestrate experimental kidney inflammation97, are present in the kidneys of patients with lupus nephritis and lupus-prone mice. Their function depends on TNFAIP3-interacting protein 1 (also referred to as ABIN1) and its absence promotes lupus nephritis in a TLR-dependent manner98.
Single-cell RNA sequencing of kidney and skin biopsy material from patients with lupus nephritis revealed type I IFN−response signatures in tubular cells and keratinocytes. Also, high IFN response and fibrotic signatures in tubular cells were associated with failure to respond to treatment99. Recent single-cell RNA sequencing of kidney samples from people with lupus nephritis revealed 21 subsets of disease-active leukocytes, including multiple populations of myeloid cells, T cells, natural killer cells and B cells, that demonstrated both pro-inflammatory responses and inflammation-resolving responses. Also, evidence of activated B cells and of progressive stages of monocyte differentiation were detected in the kidney. A clear IFN type I response was observed in most cells. Two chemokine receptors, CXCR4 and CX3CR1, were broadly expressed, implying they may have central roles in cell trafficking100. Similar studies that investigate resident kidney cells in parallel may reveal how the invasion of inflammatory cells alters the gene expression landscape and the function of kidney cells.
Nephritis can develop independently of systemic autoimmunity. Mice lacking ABIN1 develop glomerulonephritis and autoimmunity, both of which depend on TLR signaling, but ABIN1-deficient Rag1−/− and C3−/− mice develop glomerulonephritis without autoimmunity98. Similarly, B6.Nr4a1.Sle1.yaa mice, which have a duplication of the Tlr7 locus, lack patrolling monocytes and are prone to developing autoimmunity, do not develop glomerulonephritis but display ample evidence of systemic autoimmunity98. A lack of association between autoimmunity and kidney damage has been previously suggested by studies of congenic lupus-prone NZM2328 strains101. The notion that the two processes are independent explains why certain patients with lupus nephritis develop end-stage renal disease despite heavy treatment with immunosuppressive drugs, while many people with systemic autoimmunity never develop clinical renal disease.
There are a multitude of mechanisms that are involved in the expression of lupus nephritis, including immune complexes, complement, infiltrating proinflammatory T cells and antibody-producing B cells, and monocytes and the inherent processes of resident cells account for the majority of the clinical heterogeneity of lupus nephritis and for the variable response to drugs and biologics. Understanding the dominant operative mechanism in each patient is the only way to develop personalized medicine.
Central nervous system (CNS) SLE
The central nervous system is involved frequently in people and mice with SLE. The clinical manifestations are quite diverse, apparently reflecting numerous immune and local pathogenic processes102. So far, antibody-mediated neuronal injury, microglial cell activation and infiltrating T cells are involved in the expression of brain injury. Antibodies that recognize double-stranded DNA can cross-react with the NR2A and NR2B subunits of the N-methyl-D-aspartate receptor and cause neuronal death, primarily via increased neuronal calcium influx, which mimics glutamate excitotoxicity103. Transfer of these antibodies to normal mice104 or immunization with the NMDAR-derived DWEYS pentapeptide105 causes neuropsychiatric disease.
Within the brain, resident microglia are the predominant immune cells of the CNS and are potent cytokine producers. It appears that neuronal injury is followed by microglial activation, which involves the activation of the angiotensin-converting enzyme. Blockade of microglial activation with an angiotensin-converting enzyme inhibitor limits neuronal injury106, offering another treatment option.
Lymphocytes and other immune cells are probably important in the expression of CNS disease. Tertiary lymphoid structures are present in the choroid plexus of lupus-prone mice and people with SLE106, and lymphocytes apparently enter the brain parenchyma, but their nature and function have not been studied. The recent reports on the presence of T cells in the brains of people with autism107 and Alzheimer’s disease108 call for more attention on the role of T cells in brain tissue injury.
Cutaneous lupus
Four of the eleven American College of Rheumatology–established criteria for the classification of the disease involve skin manifestations that exposure to sun may elicit or worsen. UV light–induced skin inflammation depends on the production of the cytokine CSF-1 by keratinocytes, which in turn recruits and activates monocytes, which enhance apoptosis of keratinocytes109. Dying keratinocytes release autoantigens, including Ro60 (Ro antibodies have long been linked to cutaneous lupus), which may propagate the autoimmune response. Mice and humans deficient in the complement protein C1q are known to have a defect in the clearance of apoptotic material and have lupus-like skin manifestations110. C1q-coated apoptotic cells are engulfed by DCs, macrophages and endothelial cells through binding the receptor SCARF1 on the surface of these cells. SCARF1-deficient mice develop lupus-like disease111.
TH17 cells, which are present in skin biopsy material, may contribute to the inflammatory process112. pDCs have the unique capacity to rapidly produce huge amounts of IFN-α upon recognition of viral RNA and DNA through TLR7 and TLR9 or through other pathogen recognition receptors that are present in skin lesions113. The development of skin lesions depends on FasL expressed on infiltrating TH1 cells recognizing cognate antigen and on TLR7 in the absence of TLR9, revealing a complex regulation of skin inflammation in lupus. pDCs and IFN-α have been shown to be abundant in skin lesions114.
The importance of TNF in the expression of skin lesions has been shown in experiments in which lupus IgG was injected into the skin of various genetically modified mice. In brief, monocytes but not lymphocytes or Ig were required for the induction of skin inflammation (reviewed in ref. 115). Although TNF was required, only TNF receptor type I and not II trimerization was needed for the induction of inflammation. While blockade of IL-17 may have value in the treatment of patients with skin lupus, it is unclear whether TNF receptor trimerization inhibitors may have any value, particularly given that TNF blockade may lead to autoimmune manifestations.
Cardiovascular disease in SLE
Patients with SLE, and particularly those with oxidized LDL and β2-glycoprotein I, have a 2-fold increased risk for cardiovascular disease116. Multiple mechanisms have been found to contribute to the expression of vascular damage. Type I IFN has been shown to inhibit the production of endothelial nitric oxide synthase and to cause endothelial damage117. Low-density granulocytes damage the vasculature because of their increased propensity for NETosis and promote vascular leakage and endothelial-to-mesenchymal transition through the degradation of vascular endothelial cadherin118. CCR5+T-bet+FoxP3+ CD4+ effector T cells are present in atherosclerotic plaques119. Invariant natural killer T cells, however, were recently claimed to interact with monocytes and promote an atheroprotective effect120.
Whereas inflammation promotes atherosclerosis, it has been demonstrated that atherogenic hyperlipidemia promotes autoimmune TH17 cell responses in vivo121. An atherogenic environment induces the production of IL-27 by DCs in a TLR4-dependent manner, which in turn triggers the differentiation of CXCR3+ TFH cells, which are increased in mice66 and patients with SLE122, while inhibiting the differentiation of follicular regulatory T cells123.
Prospects and needs
Amazing progress has been made over the last few years due to increased funding from various sources and the recruitment of skilled researchers from various sections of immunology and other fields of medicine, including nephrology, dermatology and cardiology, and other areas of science, including advanced molecular biology and bioinformatics. As this Review briefly summarizes, every cellular, molecular and biochemical aspect of the immune system contributes somehow to the expression of the disease. In humans it is obviously difficult to assign the time of entry for each recorded abnormality in the pathogenesis of the disease and classify it as either primary or secondary. Lupus-prone mice and engineered mice, in which one suspected molecule is deleted, overexpressed or structurally altered, invariably demonstrate that each molecule is able and sufficient to cause autoimmunity, reflecting a ‘house of cards’ effect, rather than what actually happens in people with SLE. While the complete understanding of each pathway, be it cellular or molecular, is of enormous significance, more important is the need to master technology to identify which pathway is the driver in each individual person with SLE.
The list of failing clinical trials keeps expanding and has been regularly updated, but there has been no effort to change the approach to clinical trials in a radical manner1. It is unfortunate, although unavoidable, that we define the disease using diverse classification criteria. There is no doubt that each biologic helps some people with SLE, mostly because that pathway is central to the expression of the disease in the responding group of patients, but this is not sufficient. There are two solutions: administer to each patient with SLE multiple drugs/biologics in the hope that a greater number will experience clinical benefit, or identify the driving pathway in each patient and treat her accordingly. A corollary to the first approach is to administer biologics that correct multiple pathways.
The expanding use of whole-exome sequencing along with genome-wide association studies has increased the number of patients with monogenic lupus, which will most probably continue to increase. For these patients, besides treatment with drugs to control manifestations, there is the possibility, with advancing technologies, of talking about a cure. Although still early, it is important to identify patients in whom two or a few gene variants contribute to the expression of the disease (oligogenic patients). New technologies should be adapted by investigators in the field to study how individual single nucleotide polymorphisms (SNPs) interact with remote genes or their variants to enable autoimmune pathology124.
The expanding use of single-cell sequencing will help to identify previously unknown functional immune cell subsets that may be upstream in immune system dysregulation and could offer new targets for treatment. Single-cell sequencing of involved tissues may reveal more features of the invading immune cells and, more importantly, that local resident cells have both previously unrecognized functions100 and the ability to produce molecules that cause organ destruction.
It is unfathomable that, although practically all patients are women (9 or more in 10), we know very little of what lies behind this. The X chromosome packs numerous genes involved in the immune response, and it appears that improper inactivation may perturb the immune balance in favor of autoimmunity125. Are there master regulators encoded by genes of the X chromosome that commandeer all that has been reported for SLE? Do these genes need to be expressed in a homozygous fashion through unleashing the inactive allele? In the same line of thinking, the study of the epigenome is still nascent in SLE.
Studies of the involved organs, including skin, the kidney and the brain, have revealed local processes that are responsible for tissue damage. Without ignoring the role of the autoimmune response in instigating organ damage, it is possible that the local inflammatory processes proceed independently with little or no outside input. Better understanding of these processes should open new approaches to disease treatment.
Acknowledgements
I thank my colleagues current and past who have helped me acquire a better understanding of this formidable disease known as lupus. I want to thank M. Tsokos for her support and feedback during the preparation of this report and N. Plummer for helping with the assembly of references. The work in my laboratory has been supported by the NIH.
Footnotes
Competing interests
The author declares no competing interests.
Reprints and permissions information is available at www.nature.com/reprints.
Editor recognition statement L. A. Dempsey was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
References
- 1.Durcan L, O’Dwyer T & Petri M Management strategies and future directions for systemic lupus erythematosus in adults. Lancet 393, 2332–2343 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Dörner T & Furie R Novel paradigms in systemic lupus erythematosus. Lancet 393, 2344–2358 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Tsokos GC Systemic lupus erythematosus. N. Engl. J. Med. 365, 2110–2121 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Theofilopoulos AN, Kono DH & Baccala R The multiple pathways to autoimmunity. Nat. Immunol. 18, 716–724 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deng Y & Tsao BP Updates in lupus genetics. Curr. Rheumatol. Rep. 19, 68 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Langefeld CD et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat. Commun. 8, 16021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsokos GC, Lo MS, Costa Reis P & Sullivan KE New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 12, 716–730 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Teruel M & Alarcon-Riquelme ME Genetics of systemic lupus erythematosus and Sjögren’s syndrome: an update. Curr. Opin. Rheumatol. 28, 506–514 (2016).27227345 [Google Scholar]
- 9.Acosta-Herrera M et al. Genome-wide meta-analysis reveals shared new loci in systemic seropositive rheumatic diseases. Ann. Rheum. Dis. 78, 311–319 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deligianni C & Spilianakis CG Long-range genomic interactions epigenetically regulate the expression of a cytokine receptor. EMBO Rep. 13, 819–826 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jhunjhunwala S et al. The 3D structure of the immunoglobulin heavy-chain locus: implications for long-range genomic interactions. Cell 133, 265–279 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hedrich CM, Mäbert K, Rauen T & Tsokos GC DNA methylation in systemic lupus erythematosus. Epigenomics 9, 505–525 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H et al. Precision DNA demethylation ameliorates disease in lupus-prone mice. JCI Insight 3, 120880 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hedrich CM et al. cAMP-responsive element modulator α (CREM α) trans-represses the transmembrane glycoprotein CD8 and contributes to the generation of CD3+CD4−CD8− T cells in health and disease. J. Biol. Chem. 288, 31880–31887 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hedrich CM, Rauen T & Tsokos GC cAMP-responsive element modulator (CREM)α protein signaling mediates epigenetic remodeling of the human interleukin-2 gene: implications in systemic lupus erythematosus. J. Biol. Chem. 286, 43429–43436 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hedrich CM et al. Stat3 promotes IL-10 expression in lupus T cells through trans-activation and chromatin remodeling. Proc. Natl Acad. Sci. USA 111, 13457–13462 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin F et al. Serum microRNA profiles serve as novel biomarkers for autoimmune diseases. Front. Immunol. 9, 2381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coit P & Sawalha AH The human microbiome in rheumatic autoimmune diseases: a comprehensive review. Clin. Immunol. 170, 70–79 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Greiling TM et al. Commensal orthologs of the human autoantigen Ro60 as triggers of autoimmunity in lupus. Sci. Transl. Med. 10, eaan2306 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivanov II et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teng F et al. Gut microbiota drive autoimmune arthritis by promoting differentiation and migration of Peyer’s patch T follicular helper cells. Immunity 44, 875–888 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turer EE et al. Homeostatic MyD88-dependent signals cause lethal inflammation in the absence of A20. J. Exp. Med. 205, 451–464 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zegarra-Ruiz DF et al. A diet-sensitive commensal Lactobacillus strain mediates TLR7-dependent systemic autoimmunity. Cell Host Microbe 25, 113–127.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manfredo Vieira S et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 359, 1156–1161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Odhams CA et al. Interferon inducible X-linked gene CXorf21 may contribute to sexual dimorphism in Systemic Lupus Erythematosus. Nat. Commun. 10, 2164 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moulton VR & Tsokos GC Why do women get lupus? Clin. Immunol. 144, 53–56 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Liang Y et al. A gene network regulated by the transcription factor VGLL3 as a promoter of sex-biased autoimmune diseases. Nat. Immunol. 18, 152–160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia-Romo GS et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 3, 73ra20 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frangou E et al. REDD1/autophagy pathway promotes thromboinflammation and fibrosis in human systemic lupus erythematosus (SLE) through NETs decorated with tissue factor (TF) and interleukin-17A (IL-17A). Ann. Rheum. Dis. 78, 238–248 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puga I et al. B cell–helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat. Immunol. 13, 170–180 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blanco P, Palucka AK, Gill M, Pascual V & Banchereau J Induction of dendritic cell differentiation by IFN-α in systemic lupus erythematosus. Science 294, 1540–1543 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Jacquemin C et al. OX40 ligand contributes to human lupus pathogenesis by promoting T follicular helper response. Immunity 42, 1159–1170 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacquemin C et al. OX40L/OX40 axis impairs follicular and natural Treg function in human SLE. JCI Insight 3, e122167 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joo H et al. Serum from patients with SLE instructs monocytes to promote IgG and IgA plasmablast differentiation. J. Exp. Med. 209, 1335–1348 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barrat FJ et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 202, 1131–1139 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pisitkun P et al. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 312, 1669–1672 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Rowland SL et al. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J. Exp. Med. 211, 1977–1991 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Furie R et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J. Clin. Invest. 129, 1359–1371 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guiducci C et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 465, 937–941 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H et al. Interferon-induced mechanosensing defects impede apoptotic cell clearance in lupus. J. Clin. Invest. 125, 2877–2890 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramirez-Ortiz ZG et al. The receptor TREML4 amplifies TLR7-mediated signaling during antiviral responses and autoimmunity. Nat. Immunol. 16, 495–504 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park SH et al. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat. Immunol. 18, 1104–1116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banchereau R et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell 165, 551–565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barrat FJ, Crow MK & Ivashkiv LB Interferon target-gene expression and epigenomic signatures in health and disease. Nat. Immunol. 20, 1574–1583 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhuang H, Szeto C, Han S, Yang L & Reeves WH Animal models of interferon signature positive lupus. Front. Immunol. 6, 291 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwarting A et al. Interferon-β: a therapeutic for autoimmune lupus in MRL-Faslpr mice. J. Am. Soc. Nephrol. 16, 3264–3272 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Shinde R et al. Apoptotic cell-induced AhR activity is required for immunological tolerance and suppression of systemic lupus erythematosus in mice and humans. Nat. Immunol. 19, 571–582 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lood C et al. Platelet transcriptional profile and protein expression in patients with systemic lupus erythematosus: up-regulation of the type I interferon system is strongly associated with vascular disease. Blood 116, 1951–1957 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Duffau P et al. Platelet CD154 potentiates interferon-α secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Sci. Transl. Med. Sci. Transl. Med. 2, 47ra63 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Charles N, Hardwick D, Daugas E, Illei GG & Rivera J Basophils and the T helper 2 environment can promote the development of lupus nephritis. Nat. Med. 16, 701–707 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dörner T & Lipsky PE Beyond pan-B-cell-directed therapy — new avenues and insights into the pathogenesis of SLE. Nat. Rev. Rheumatol. 12, 645–657 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Tipton CM, Hom JR, Fucile CF, Rosenberg AF & Sanz I Understanding B-cell activation and autoantibody repertoire selection in systemic lupus erythematosus: a B-cell immunomics approach. Immunol. Rev. 284, 120–131 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dörner T, Giesecke C & Lipsky PE Mechanisms of B cell autoimmunity in SLE. Arthritis Res. Ther. 13, 243 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yurasov S et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J. Exp. Med. 201, 703–711 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suurmond J et al. Loss of an IgG plasma cell checkpoint in patients with lupus. J. Allergy Clin. Immunol. 143, 1586–1597 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manni M et al. Regulation of age-associated B cells by IRF5 in systemic autoimmunity. Nat. Immunol. 19, 407–419 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun C et al. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat. Genet. 48, 323–330 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scharer CD et al. Epigenetic programming underpins B cell dysfunction in human SLE. Nat. Immunol. 20, 1071–1082 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zumaquero E et al. IFNγ induces epigenetic programming of human T-bethi B cells and promotes TLR7/8 and IL-21 induced differentiation. Elife 8, e41641 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jenks SA et al. Distinct effector B cells induced by unregulated Toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity 49, 725–739.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tipton CM et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat. Immunol. 16, 755–765 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Henault J et al. Self-reactive IgE exacerbates interferon responses associated with autoimmunity. Nat. Immunol. 17, 196–203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mohan C, Adams S, Stanik V & Datta SK Nucleosome: a major immunogen for pathogenic autoantibody-inducing T cells of lupus. J. Exp. Med. 177, 1367–1381 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Christophersen A et al. Distinct phenotype of CD4+ T cells driving celiac disease identified in multiple autoimmune conditions. Nat. Med. 25, 734–737 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim SJ, Lee K & Diamond B Follicular helper T cells in systemic lupus erythematosus. Front. Immunol. 9, 1793 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Odegard JM et al. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J. Exp. Med. 205, 2873–2886 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Faliti CE et al. P2X7 receptor restrains pathogenic Tfh cell generation in systemic lupus erythematosus. J. Exp. Med. 216, 317–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caielli S et al. A CD4+ T cell population expanded in lupus blood provides B cell help through interleukin-10 and succinate. Nat. Med. 25, 75–81 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Katsuyama E et al. The CD38/NAD/SIRTUIN1/EZH2 axis mitigates cytotoxic CD8 T cell function and identifies patients with SLE prone to infections. Cell Rep. 30, 112–123.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tarrago MG et al. A potent and specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD+ decline. Cell Metab. 27, 1081–1095.e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McKinney EF, Lee JC, Jayne DR, Lyons PA & Smith KG T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 523, 612–616 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharabi A et al. Regulatory T cells in the treatment of disease. Nat. Rev. Drug Discov. 17, 823–844 (2018). [DOI] [PubMed] [Google Scholar]
- 73.Mizui M et al. IL-2 protects lupus-prone mice from multiple end-organ damage by limiting CD4−CD8− IL-17–producing T cells. J. Immunol. 193, 2168–2177 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Crispin JC et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J. Immunol. 181, 8761–8766 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.He J et al. Low-dose interleukin-2 treatment selectively modulates CD4+ T cell subsets in patients with systemic lupus erythematosus. Nat. Med. 22, 991–993 (2016). [DOI] [PubMed] [Google Scholar]
- 76.Comte D et al. Brief report: CD4+ T cells from patients with systemic lupus erythematosus respond poorly to exogenous interleukin-2. Arthritis Rheumatol. 69, 808–813 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moulton VR et al. Pathogenesis of human systemic lupus erythematosus: a cellular perspective. Trends Mol. Med. 23, 615–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Burzyn D et al. A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Afeltra A et al. The involvement of T regulatory lymphocytes in a cohort of lupus nephritis patients: a pilot study. Intern. Emerg. Med. 10, 677–683 (2015). [DOI] [PubMed] [Google Scholar]
- 80.Crispin JC, Hedrich CM, Suárez-Fueyo A, Comte D & Tsokos GC SLE-associated defects promote altered T cell function. Crit. Rev. Immunol. 37, 39–58 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moulton VR & Tsokos GC T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J. Clin. Invest. 125, 2220–2227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sharabi A & Tsokos GC T cell metabolism: new insights in systemic lupus erythematosus pathogenesis and therapy. Nat. Rev. Rheumatol. 16, 100–112 (2020). [DOI] [PubMed] [Google Scholar]
- 83.Flores-Mendoza G, Sansón SP, Rodriguez-Castro S, Crispin JC & Rosetti F Mechanisms of tissue injury in lupus nephritis. Trends Mol. Med. 24, 364–378 (2018). [DOI] [PubMed] [Google Scholar]
- 84.Maeda K et al. CaMK4 compromises podocyte function in autoimmune and nonautoimmune kidney disease. J. Clin. Invest. 128, 3445–3459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Steinmetz OM et al. CXCR3 mediates renal Th1 and Th17 immune response in murine lupus nephritis. J. Immunol. 183, 4693–4704 (2009). [DOI] [PubMed] [Google Scholar]
- 86.Peng SL et al. Murine lupus in the absence of alpha beta T cells. J. Immunol. 156, 4041–4049 (1996). [PubMed] [Google Scholar]
- 87.Tilstra JS et al. Kidney-infiltrating T cells in murine lupus nephritis are metabolically and functionally exhausted. J. Clin. Invest. 128, 4884–4897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Winchester R et al. Immunologic characteristics of intrarenal T cells: trafficking of expanded CD8+ T cell β-chain clonotypes in progressive lupus nephritis. Arthritis Rheum. 64, 1589–1600 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Amarilyo G, Lourenço EV, Shi FD & La Cava A IL-17 promotes murine lupus. J. Immunol. 193, 540–543 (2014). [DOI] [PubMed] [Google Scholar]
- 90.Liarski VM et al. Cell distance mapping identifies functional T follicular helper cells in inflamed human renal tissue. Sci. Transl. Med. 6, 230ra246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kinloch AJ et al. Vimentin is a dominant target of in situ humoral immunity in human lupus tubulointerstitial nephritis. Arthritis Rheumatol. 66, 3359–3370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liarski VM et al. Quantifying in situ adaptive immune cell cognate interactions in humans. Nat. Immunol. 20, 503–513 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Davidson A, Aranow C & Mackay M Lupus nephritis: challenges and progress. Curr. Opin. Rheumatol. 31, 682–688 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sahu R, Bethunaickan R, Singh S & Davidson A Structure and function of renal macrophages and dendritic cells from lupus-prone mice. Arthritis Rheumatol. 66, 1596–1607 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Iwata Y et al. Aberrant macrophages mediate defective kidney repair that triggers nephritis in lupus-susceptible mice. J. Immunol. 188, 4568–4580 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Berthier CC et al. Cross-species transcriptional network analysis defines shared inflammatory responses in murine and human lupus nephritis. J. Immunol. 189, 988–1001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Finsterbusch M et al. Patrolling monocytes promote intravascular neutrophil activation and glomerular injury in the acutely inflamed glomerulus. Proc. Natl Acad. Sci. USA 113, E5172–E5181 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kuriakose J et al. Patrolling monocytes promote the pathogenesis of early lupus-like glomerulonephritis. J. Clin. Invest. 129, 2251–2265 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Der E et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat. Immunol. 20, 915–927 (2019); erratum 20, 1556 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Arazi A et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat. Immunol. 20, 902–914 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ge Y et al. Cgnz1 allele confers kidney resistance to damage preventing progression of immune complex-mediated acute lupus glomerulonephritis. J. Exp. Med. 210, 2387–2401 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schwartz N, Stock AD & Putterman C Neuropsychiatric lupus: new mechanistic insights and future treatment directions. Nat. Rev. Rheumatol. 15, 137–152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.DeGiorgio LA et al. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat. Med. 7, 1189–1193 (2001). [DOI] [PubMed] [Google Scholar]
- 104.Kowal C et al. Human lupus autoantibodies against NMDA receptors mediate cognitive impairment. Proc. Natl Acad. Sci. USA 103, 19854–19859 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kowal C et al. Cognition and immunity; antibody impairs memory. Immunity 21, 179–188 (2004). [DOI] [PubMed] [Google Scholar]
- 106.Stock AD et al. Tertiary lymphoid structures in the choroid plexus in neuropsychiatric lupus. JCI Insight 4, 124203 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.DiStasio MM, Nagakura I, Nadler MJ & Anderson MP T lymphocytes and cytotoxic astrocyte blebs correlate across autism brains. Ann. Neurol. 86, 885–898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Heneka MT An immune-cell signature marks the brain in Alzheimer’s disease. Nature 577, 322–323 (2020). [DOI] [PubMed] [Google Scholar]
- 109.Menke J et al. Sunlight triggers cutaneous lupus through a CSF-1-dependent mechanism in MRL-Faslpr mice. J. Immunol. 181, 7367–7379 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Martens HA et al. Analysis of C1q polymorphisms suggests association with systemic lupus erythematosus, serum C1q and CH50 levels and disease severity. Ann. Rheum. Dis. 68, 715–720 (2009). [DOI] [PubMed] [Google Scholar]
- 111.Ramirez-Ortiz ZG et al. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat. Immunol. 14, 917–926 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Oh SH et al. Expression of interleukin-17 is correlated with interferon-α expression in cutaneous lesions of lupus erythematosus. Clin. Exp. Dermatol. 36, 512–520 (2011). [DOI] [PubMed] [Google Scholar]
- 113.Guiducci C et al. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J. Exp. Med. 207, 2931–2942 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mande P et al. Fas ligand promotes an inducible TLR-dependent model of cutaneous lupus-like inflammation. J. Clin. Invest. 128, 2966–2978 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Deng GM & Tsokos GC Pathogenesis and targeted treatment of skin injury in SLE. Nat. Rev. Rheumatol. 11, 663–669 (2015). [DOI] [PubMed] [Google Scholar]
- 116.Liu Y & Kaplan MJ Cardiovascular disease in systemic lupus erythematosus: an update. Curr. Opin. Rheumatol. 30, 441–448 (2018). [DOI] [PubMed] [Google Scholar]
- 117.Buie JJ, Renaud LL, Muise-Helmericks R & Oates JC IFN-α negatively regulates the expression of endothelial nitric oxide synthase and nitric oxide production: implications for systemic lupus erythematosus. J. Immunol. 199, 1979–1988 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Carlucci PM et al. Neutrophil subsets and their gene signature associate with vascular inflammation and coronary atherosclerosis in lupus. JCI Insight 3, 99276 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li J et al. CCR5+T-bet+FoxP3+ effector CD4 T cells drive atherosclerosis. Circ. Res. 118, 1540–1552 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Smith E et al. Cross-talk between iNKT cells and monocytes triggers an atheroprotective immune response in SLE patients with asymptomatic plaque. Sci. Immunol. 1, eaah4081 (2016). [DOI] [PubMed] [Google Scholar]
- 121.Lim H et al. Proatherogenic conditions promote autoimmune T helper 17 cell responses in vivo. Immunity 40, 153–165 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Choi JY et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol. 67, 988–999 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ryu H et al. Atherogenic dyslipidemia promotes autoimmune follicular helper T cell responses via IL-27. Nat. Immunol. 19, 583–593 (2018); erratum 19, 1036 (2018). [DOI] [PubMed] [Google Scholar]
- 124.Hedrich CM & Tsokos G SNPs talk to genes using landlines: long-range chromatin interactions link genetic risk with epigenetic patterns in Takayasu arteritis. Ann. Rheum. Dis. 78, 1293–1295 (2019). [DOI] [PubMed] [Google Scholar]
- 125.Libert C, Dejager L & Pinheiro I The X chromosome in immune functions: when a chromosome makes the difference. Nat. Rev. Immunol. 10, 594–604 (2010). [DOI] [PubMed] [Google Scholar]
- 126.Kottyan LC et al. The IRF5–TNPO3 association with systemic lupus erythematosus has two components that other autoimmune disorders variably share. Hum. Mol. Genet. 24, 582–596 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lee MN et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343, 1246980 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pelikan RC et al. Enhancer histone-QTLs are enriched on autoimmune risk haplotypes and influence gene expression within chromatin networks. Nat. Commun. 9, 2905 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lu X et al. Lupus risk variant increases pSTAT1 binding and decreases ETS1 expression. Am. J. Hum. Genet. 96, 731–739 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Westra HJ et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mayadas TN, Tsokos GC & Tsuboi N Mechanisms of immune complex–mediated neutrophil recruitment and tissue injury. Circulation 120, 2012–2024 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liszewski MK, Java A, Schramm EC & Atkinson JP Complement dysregulation and disease: insights from contemporary genetics. Annu. Rev. Pathol. 12, 25–52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yang Y et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am. J. Hum. Genet. 80, 1037–1054 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Deng Y & Tsao BP Advances in lupus genetics and epigenetics. Curr. Opin. Rheumatol. 26, 482–492 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhao J et al. A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat. Genet. 49, 433–437 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kim K et al. Variation in the ICAM1–ICAM4–ICAM5 locus is associated with systemic lupus erythematosus susceptibility in multiple ancestries. Ann. Rheum. Dis. 71, 1809–1814 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Levine B & Kroemer G Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang S, Wen F, Wiley GB, Kinter MT & Gaffney PM An enhancer element harboring variants associated with systemic lupus erythematosus engages the TNFAIP3 promoter to influence A20 expression. PLoS Genet. 9, e1003750 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Vereecke L, Beyaert R & van Loo G Genetic relationships between A20/TNFAIP3, chronic inflammation and autoimmune disease. Biochem. Soc. Trans. 39, 1086–1091 (2011). [DOI] [PubMed] [Google Scholar]
- 140.Salmond RJ, Brownlie RJ, Morrison VL & Zamoyska R The tyrosine phosphatase PTPN22 discriminates weak self peptides from strong agonist TCR signals. Nat. Immunol. 15, 875–883 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Molineros JE et al. Amino acid signatures of HLA Class-I and II molecules are strongly associated with SLE susceptibility and autoantibody production in Eastern Asians. PLoS Genet. 15, e1008092 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Castillejo-López C et al. Genetic and physical interaction of the B-cell systemic lupus erythematosus-associated genes BANK1 and BLK. Ann. Rheum. Dis. 71, 136–142 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chen X et al. An autoimmune disease variant of IgG1 modulates B cell activation and differentiation. Science 362, 700–705 (2018). [DOI] [PubMed] [Google Scholar]
- 144.Freedman BI et al. End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol. 66, 390–396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tang Y et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 60, 1065–1075 (2009). [DOI] [PubMed] [Google Scholar]
- 146.Zhao X et al. MicroRNA-125a contributes to elevated inflammatory chemokine RANTES levels via targeting KLF13 in systemic lupus erythematosus. Arthritis Rheum. 62, 3425–3435 (2010). [DOI] [PubMed] [Google Scholar]
- 147.Thai TH et al. Deletion of microRNA-155 reduces autoantibody responses and alleviates lupus-like disease in the Faslpr mouse. Proc. Natl Acad. Sci. USA 110, 20194–20199 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gonzalez-Martin A et al. The microRNA miR-148a functions as a critical regulator of B cell tolerance and autoimmunity. Nat. Immunol. 17, 433–440 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhu S et al. The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-α. Nat. Med. 18, 1077–1086 (2012). [DOI] [PubMed] [Google Scholar]
- 150.Baumjohann D et al. The microRNA cluster miR-17~92 promotes TFH cell differentiation and represses subset-inappropriate gene expression. Nat. Immunol. 14, 840–848 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kim SJ, Gregersen PK & Diamond B Regulation of dendritic cell activation by microRNA let-7c and BLIMP1. J. Clin. Invest. 123, 823–833 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhou H et al. miR-150 promotes renal fibrosis in lupus nephritis by downregulating SOCS1. J. Am. Soc. Nephrol. 24, 1073–1087 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]