

Abstract
The systematic identification of host genetic risk factors is essential for the understanding and treatment of coronavirus disease 2019 (COVID-19). By performing a meta-analysis of two independent genome-wide association summary datasets (N = 680 128), a novel locus at 21q22.11 was identified to be associated with COVID-19 infection (rs9976829 in IFNAR2-IL10RB, odds ratio = 1.16, 95% confidence interval = 1.09–1.23, P = 2.57 × 10−6). The rs9976829 represents a strong splicing quantitative trait locus for both IFNAR2 and IL10RB genes, especially in lung tissue (P = 1.8 × 10−24). Integrative genomics analysis of combining genome-wide association study with expression quantitative trait locus data showed the expression variations of IFNAR2 and IL10RB have prominent effects on COVID-19 in various types of tissues, especially in lung tissue. The majority of IFNAR2-expressing cells were dendritic cells (40%) and plasmacytoid dendritic cells (38.5%), and IL10RB-expressing cells were mainly nonclassical monocytes (29.6%). IFNAR2 and IL10RB are targeted by several interferons-related drugs. Together, our results uncover 21q22.11 as a novel susceptibility locus for COVID-19, in which individuals with G alleles of rs9976829 have a higher probability of COVID-19 susceptibility than those with non-G alleles.
Introduction
Coronavirus disease 2019 (COVID-19) has rapidly evolved into a global pandemic (1). The health and economy systems of most nations worldwide are suffering from severe disruptions (2). As of July 13th, 2020, there were >12.9 million confirmed patients worldwide with >550 000 deaths (3). The clinical manifestations of COVID-19 range from asymptomatic to severe respiratory failure (4). Early studies on COVID-19 infection have concentrated on epidemiology (5,6), clinical characteristics (7,8) and genomic features of virus (9,10). Understanding host genetic factors contributing to COVID-19 susceptibility is essential for the precise management in the community.
Recently, a growing number of researchers have concentrated on the involvement of host genetic factors in COVID-19. Through performing a genome-wide association study (GWAS) with 1610 severe COVID-19 patients and 2205 controls, Ellinghaus et al. (11) reported two important gene clusters of 3p21.31 and 9q34.2 as genetic susceptibility loci for severe COVID-19, and confirmed a potential involvement of the ABO blood-group system. From a population perspective, the COVID-19 Host Genetic Consortium launched the ‘COVID-19 Host Genetics Initiative’ to collect data from the genetics community to uncover the genetic determinants of COVID-19 susceptibility, severity, and outcomes (2). However, identification of more host genetic risk factors is limited by the sample size of a single study.
Here, we performed a meta-analysis by combining two independent GWAS summary statistics with a large-scale sample size to identify novel variants for COVID-19 susceptibility. The systematic bioinformatics analyses were performed, including gene-based association analysis, S-PrediXcan and S-MultiXcan analysis, Sherlock-based integrative genomics analysis, functional enrichment analysis, gene-property analysis, in silico permutation analysis, single-cell RNA analysis and drug–gene interaction analysis, to uncover risk genes and biological pathways implicated in COVID-19 infection and give a clue of the potential effective drugs for treating COVID-19.
Results
SNP-level association analysis reveals a novel susceptible locus 21q22.11 for COVID-19
By conducting a meta-analysis of GWAS summary data from Ellinghaus et al. (11) (COVID_I: 1610 COVID-19 patients and 2205 controls) and the COVID-19 Host Genetic Consortium (2) (ANA5: 1678 COVID-19 patients and 674 635 controls), we confirmed two reported loci of 3p21.31 and 9q34.2 to be associated with COVID-19 infection (rs11385942 in SLC6A20, P = 2.87 × 10−16, and rs8176719 in ABO, P = 4 × 10−7; Fig. 1, Table 1, and Supplementary Material, Figs S1 and S2). The reported rs657152 in ABO, which is high linkage disequilibrium with rs8176719, remains suggestively significant (P = 5.53 × 10−6). Notably, we identified a novel locus at 21q22.11 to be associated with COVID-19 infection (rs9976829 in IFNAR2-IL10RB, odds ratio (OR) = 1.16, 95% confidence interval (CI) = 1.09–1.23, P = 2.57 × 10−6; Table 1 and Fig. 1). The rs9976829 represents a splicing quantitative trait locus (sQTL) for both IFNAR2 and IL10RB genes with significant associations across multiple tissues, especially the strongest significant sQTL for IFNAR2 in the lung tissue (P = 1.8 × 10−24; Supplementary Material, Fig. S3A and B). Meanwhile, rs9976829 shows significant expression quantitative trait locus (eQTL) associations for both IFNAR2 and IL10RB across multiple tissues (Supplementary Material, Fig. S3C and D).
Figure 1 .
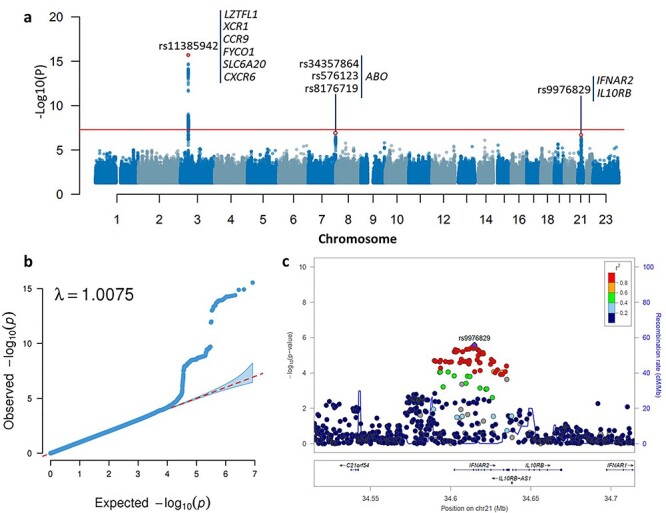
Meta-analysis of GWAS summary data highlighting susceptibility loci for COVID-19. a) Manhattan plot of the meta-analysis GWAS summary statistics highlighting three susceptibility loci for COVID-19. The Manhattan plot is shown of the meta-GWAS summary statistics of meta-analyzing the COVID_I GWAS data (controlled for potential population stratification) with ANA5 GWAS data. The red horizontal line marks the genome-wide significance threshold of a P value < 5 × 10−8. b) Quantile–quantile (QQ) plot of the meta-analysis GWAS summary statistics. All 8 424 883 high-quality SNPs with a MAF ≥ 1% and imputation R2 ≥ 0.6 were used for plotting. In QQ plot the 2.5th and 97.5th centiles of the distribution under random sampling and the null hypothesis form the 95% concentration band. The genomic inflation factor lambda (λ) is 1.0075. c) Regional association plot for 21q22.11 locus of meta-GWAS summary statistics. Regional association plot is shown for 21q22.11 locus of the meta-GWAS summary statistics of meta-analyzing the COVID_I GWAS data (controlled for potential population stratification) with ANA5 GWAS data. The purple diamond marks the most strongly associated SNP of rs9976829 with COVID-19. The color illustrates LD information with rs9976829, as shown in the color legend.
Table 1.
Susceptibility loci associated with COVID-19 identified by meta-analysis of GWAS summary data
| SNP | CHR | Position | Loci | ALT | REF | COVID_I | ANA5 | Meta-analysis | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | P-value | OR | 95% CI | P-value | OR | 95% CI | P-value | ||||||
| rs11385942 | 3 | 45 876 459 | 3p21.31 | GA | G | 1.77 | 1.49–2.11 | 1.15 × 10−10 | 1.50 | 1.29–1.74 | 1.10 × 10−7 | 1.61 | 1.43–1.80 | 2.87 × 10−16 |
| rs8176719 | 9 | 136 132 908 | 9q34.2 | TC | T | 1.32 | 1.19–1.46 | 9.93 × 10−8 | 1.10 | 1.01–1.19 | 2.10 × 10−2 | 1.17 | 1.10–1.25 | 4.76 × 10−7 |
| rs657152 | 9 | 136 139 265 | 9q34.2 | A | C | 1.33 | 1.20–1.47 | 4.95 × 10−8 | 1.07 | 0.99–1.15 | 7.86 × 10−2 | 1.15 | 1.08–1.21 | 5.53 × 10−6 |
| rs9976829 | 21 | 34 614 834 | 21q22.11 | G | A | 1.18 | 1.07–1.32 | 1.77 × 10−3 | 1.15 | 1.06–1.24 | 3.58 × 10−4 | 1.16 | 1.09–1.23 | 2.57 × 10−6 |
Note: CHR = chromosome, OR = odds ratio, 95% CI = 95% confidence interval, ALT = Altered allele, REF = Reference allele, COVID_I = COVID_I GWAS summary statistics (Dataset #1), ANA5 = ANA5 GWAS summary statistics (Dataset #2). The meta-analysis data were based on the combination of COVID_I GWAS summary data (controlled for potential population stratification) with ANA5 GWAS summary data.
Gene-based association analysis identifies nine risk genes for COVID-19
We performed a MAGMA gene-based association analysis by using the meta-GWAS results and found that nine genes in three loci of 3p21.31 (LZTFL1, XCR1, CCR9, FYCO1, SLC6A20 and CXCR6), 9q21.32 (HNRNPK and RMI1) and 21q22.11 (IFNAR2) were significantly associated with COVID-19 infection (false discovery rate (FDR) < 0.05; Fig. 2 and Table 2). As expected, the gene of ABO showed a nominally significant association with COVID-19 (P = 6.55 × 10−4) consistent with the previous report (11). Of note, IFNAR2 (P = 2.58 × 10−7), HNRNPK (P = 1.46 × 10−5), and RMI1 (P = 1.86 × 10−5) were identified for the first time to be associated with COVID-19 infection. There were other 30 genes showing suggestive associations with COVID-19 (P < 1 × 10−3, Supplementary Material, Table S1). Meanwhile, we performed two MAGMA gene-based association analyses for COVID_I GWAS summary data from Ellinghaus et al. (11) and the COVID-19 Host Genetic Consortium (ANA5) (2), respectively. We found that these nine identified genes showed significant or suggestive associations with COVID-19 in both COVID-I and ANA5 datasets (Table 2), indicating that our meta-analysis based on larger samples enhance the statistical power to uncover risk genes for COVID-19.
Figure 2 .
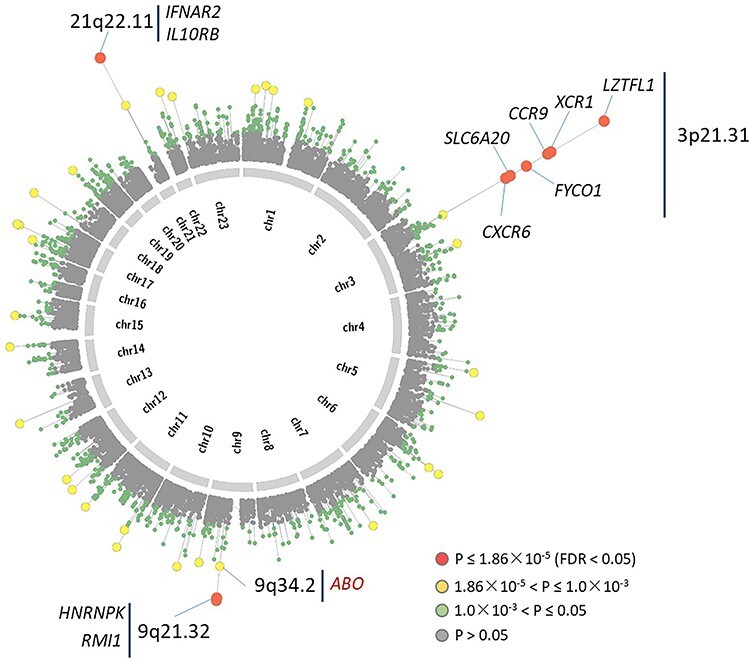
Circus plot showed the results of gene-based association analysis. Note: The inner ring shows the 22 autosomal human chromosomes (Chr1–22) and X chromosome (Chr23). A circular symbol in the outer ring represents a gene. Color indicates the statistical significance of genes (red marks genes significantly associated with COVID-19 with FDR < 0.05, yellow indicates genes with 1.86 × 10−5 < P ≤ 1 × 10−3, green marks genes with 1 × 10−3 < P ≤ 0.05, and gray represents genes with P > 0.05).
Table 2.
Significant genes associated with COVID-19 identified by MAGMA gene-based association analysis
| Gene | CHR | Start position | Stop position | Loci | MAGMA on COVID_I | MAGMA on ANA5 | MAGMA on meta-analysis data | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Z score | P-value | Z score | P-value | Z score | P-value | FDR | |||||
| LZTFL1 | 3 | 45 844 808 | 45 977 216 | 3p21.31 | 5.71 | 5.61 × 10−9 | 4.51 | 3.29 × 10−6 | 7.05 | 8.83 × 10−13 | 1.69 × 10−8 |
| XCR1 | 3 | 46 042 291 | 46 088 979 | 3p21.31 | 4.82 | 7.13 × 10−7 | 4.14 | 1.71 × 10−5 | 6.03 | 8.41 × 10−10 | 8.03 × 10−6 |
| CCR9 | 3 | 45 907 996 | 45 964 667 | 3p21.31 | 4.81 | 7.70 × 10−7 | 3.61 | 1.54 × 10−4 | 5.96 | 1.28 × 10−9 | 8.14 × 10−6 |
| FYCO1 | 3 | 45 939 391 | 46 057 316 | 3p21.31 | 4.28 | 9.55 × 10−6 | 3.32 | 4.49 × 10−4 | 5.50 | 1.95 × 10−8 | 9.31 × 10−5 |
| SLC6A20 | 3 | 45 776 941 | 45 858 039 | 3p21.31 | 4.05 | 2.60 × 10−5 | 2.72 | 3.26 × 10−3 | 5.11 | 1.58 × 10−7 | 6.03 × 10−4 |
| IFNAR2 | 21 | 34 582 231 | 34 656 831 | 21q22.11 | 3.53 | 2.10 × 10−4 | 3.50 | 2.31 × 10−4 | 5.02 | 2.58 × 10−7 | 7.58 × 10−4 |
| CXCR6 | 3 | 45 964 973 | 46 009 845 | 3p21.31 | 3.85 | 5.99 × 10−5 | 3.13 | 8.83 × 10−4 | 5.01 | 2.78 × 10−7 | 7.58 × 10−4 |
| HNRNPK | 9 | 86 562 998 | 86 615 692 | 9q21.32 | 2.02 | 2.16 × 10−2 | 3.18 | 7.42 × 10−4 | 4.18 | 1.46 × 10−5 | 3.48 × 10−2 |
| RMI1 | 9 | 86 575 321 | 86 638 989 | 9q21.32 | 2.09 | 1.84 × 10−2 | 3.15 | 8.29 × 10−4 | 4.12 | 1.86 × 10−5 | 3.94 × 10−2 |
| ABO | 9 | 136 110 563 | 136 170 630 | 9q34.2 | 4.85 | 6.28 × 10−7 | 0.10 | 0.46 | 3.21 | 6.55 × 10−4 | 0.43 |
Note: CHR = chromosome, FDR = False discovery rate, COVID_I = COVID_I GWAS summary statistics (Dataset #1), ANA5 = ANA5 GWAS summary statistics (Dataset #2). The meta-analysis data were based on the combination of COVID_I GWAS summary data (controlled for potential population stratification) with ANA5 GWAS summary data.
Cytokine-related pathways enriched by risk genes for COVID-19
As mentioned above, there were 41 genes showing significant or suggestive associations with COVID-19 (P < 1 × 10−3, Supplementary Material, Table S1). We performed a pathway enrichment analysis of these 41 identified genes for COVID-19 susceptibility (Methods), and found they were significantly enriched in two pathways of cytokine-cytokine receptor interaction (FDR = 0.009) and chemokine signaling pathway (FDR = 0.009) (Fig. 3a). Additionally, there were seven pathways showing suggestive associations (P < 0.05; Supplementary Material, Table S2), including Kaposi sarcoma-associated herpesvirus infection (P = 0.0086), human cytomegalovirus infection (P = 0.014), and human papilloma virus infection (P = 0.027). Meanwhile, we conducted a Gene Ontology (GO) enrichment analysis and found 3 GO terms were significantly overrepresented (FDR < 0.05; Fig. 3b and Supplementary Material, Table S3). That is, cytokine receptor activity (FDR = 1.08 × 10−5), cytokine binding (FDR = 0.022), and peptide receptor activity (FDR = 0.027). Previous studies demonstrated that soluble cytokines activate an anti-viral and anti-proliferate state by inducing the expression of many interferon-stimulated genes to prevent viral replication (12). Network-based enrichment analysis showed that these 41 genes are at least partially biologically connected (P = 0.013, Supplementary Material, Fig. S8). These results suggest cytokine-related pathways or functional terms may play important roles in the process of COVID-19 infection.
Figure 3 .
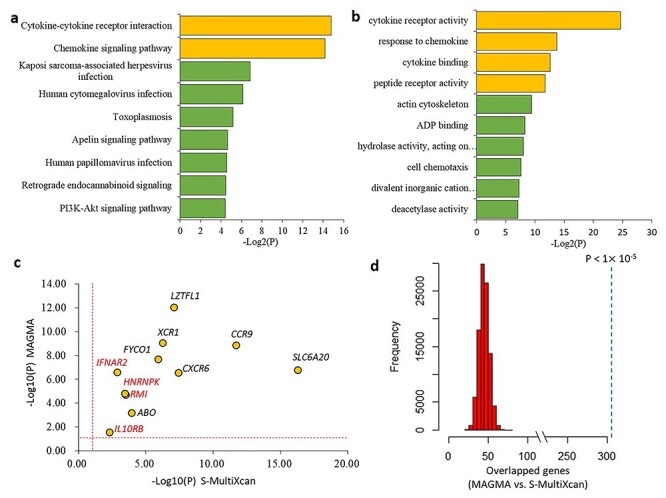
Functional enrichment analysis of genes associated with COVID-19. a) Pathway enrichment analysis identified 9 significant or suggestive KEGG pathways enriched by COVID-19-associated genes. b) GO enrichment analysis identified 10 significant or suggestive GO-terms enriched by COVID-19-associated genes. a and b) The green bar represents a suggestive enrichment (P < 0.05), and the orange bar represents a significant enrichment (FDR < 0.05). c) Scatter plot show the consistency of 11 risk genes identified from both MAGMA and S-MultiXcan analysis. The vertical and horizontal dotted lines represent -log10 (P = 0.05). d) In silico permutation analysis of 100 000 times of random selections. This permutation analysis was used to compare the overlapped genes between MAGMA and S-MultiXcan (see Methods). The empirical P value is less than 1 × 10−5.
Expression of IFNAR2 and IL10RB associated with COVID-19 across multiple tissues
To highlight the functional association of these 11 identified genes with COVID-19, we conducted two independent integrative genomics analyses by incorporating meta-GWAS summary data with eQTL data across 49 GTEx tissues. Using S-PrediXcan analysis, we found that the expression variations of these 11 genes have prominent effects on COVID-19 in various types of tissues (Supplementary Material, Table S5). Consistently, Sherlock-based integrative genomics analysis showed seven genes including IFNAR2 and IL10RB whose genetically regulated expression were significantly associated with COVID-19 across multiple tissues (Supplementary Material, Table S6). For example, the IFNAR2 gene was associated with COVID-19 across six tissues including lung (P = 2.44 × 10−4). The IL10RB gene showed associations with COVID-19 across 24 tissues with the most significant tissue of cells-transformed fibroblasts (P = 2.80 × 10−5).
We further used S-MultiXcan to meta-analyze the tissue-specific associations from S-PrediXcan across 49 GTEx tissues, and found these 11 genes whose expression were significantly associated with COVID-19, which support our results from the MAGMA analysis (Fig. 3c and Supplementary Material, Table S7). Additionally, our in silico permutation analysis also showed that genes identified from MAGMA analysis had a significantly higher overlap with S-MultiXcan-identified genes than random events (permuted P < 1 × 10−5; Fig. 3d).
IFNAR2 and IL10RB specially expressed in immunity associated cell types in lung tissue
To examine the links between tissue-specific gene expression profiles and COVID-19 gene associations, we conducted a MAGMA gene-property analysis in 53 specific tissue types and 30 general tissue types. We found that the association signals were enriched in lung, thyroid and esophagus tissue in 30 general tissue types (Supplementary Material, Fig. S4a). In the analysis of 53 specific tissues, COVID-19 gene associations were also enriched in lung, cultured fibroblasts and thyroid (Supplementary Material, Figure S4b). These results suggest that these identified genes for COVID-19 may have important functions in lung tissue.
Using 50 cell populations across four compartments (epithelial, endothelial, stromal and immune) of lung tissue, we identified the primarily expressed cells of these 11 risk genes associated with COVID-19 (Fig. 4, Supplementary Material, Table S4 and Fig. S5). The HNRNPK gene was widely expressed in various cell types across all four compartments (Supplementary Material, Fig. S6). However, the majority of IFNAR2-expressing cells were dendritic cells (40%) and plasmacytoid dendritic cells (38.5%) (Fig. 4). The IFNAR2 was also expressed within lipofibroblast (20%), ciliated (19.1%) and nonclassical monocyte (18.5%), albeit at diminished abundance compared with dendritic cells; IL10RB was primarily expressed within nonclassical monocyte (29.6%). Plasmacytoid dendritic cells produce large amounts of type I interferons-proteins that are important for immunity to viruses (13,14). To further validate the cell-specific expression of IFNAR2 and IL10RB, we re-performed the scRNA-seq analysis in two reported large-scale datasets based on peripheral immune cells and lung tissue (15,16). Based on the scRNA-seq data on lung tissue, we found these two genes were highly expressed in monocyte-like cells (30.21% for IFNAR2 and 32.52% for IL10RB, Supplementary Material, Table S8). Consistently, these two genes remained to be highly expressed in monocyte-like cells (36.27% for IFNAR2 and 35.02% for IL10RB) in the peripheral scRNA-seq dataset. These two genes also showed their expressions in dendritic cells in both scRNA-seq datasets (Supplementary Material, Tables S8, S9 and Fig. S10). Our findings indicated that IFNAR2 and IL10RB could play regulatory roles in the pulmonary immune response.
Figure 4 .
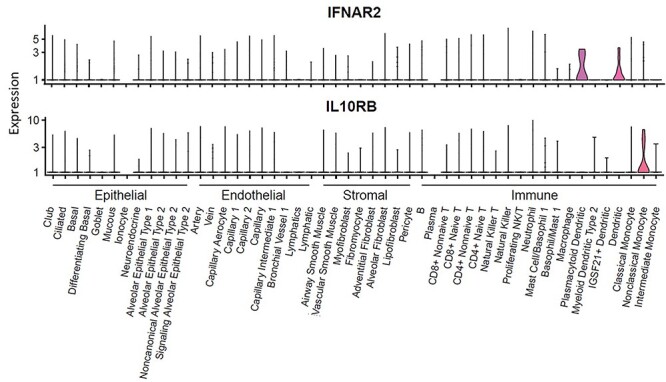
Expression of IFNAR2 and IL10RB among 50 cellular populations from lung tissue. This plot is based on a data set as a part of the Human Lung Atlas, consisting of 50 cell populations across 4 compartments (epithelial, endothelial, stromal, and immune) of lung tissue (x axis). y axis represents the expression level with log transformed count.
Potential drugs targeted with IFNAR2 and IL10RB
By performing a drug-gene interaction analysis and literature mining, we found that seven of 11 COVID-19-associated genes (63.6%) were enriched in five potential ‘druggable’ gene categories (Supplementary Material, Table S10 and Fig. S7). The gene of CCR9 is targeted by two FDA-approved drugs including hydroxyurea and hydralazine (Supplementary Material, Table S11). There are nine FDA-approved drugs showing agonist-receptor interactions with IFNAR2 (Supplementary Material, Table S11), including interferon alfa-2a, interferon alfacon-1, and interferon beta-1a, which could be useful alone or in combination with other antiviral drugs for treating SARS-CoV infection (17). Loutfy et al. (18) reported that interferon alfacon-1 plus corticosteroids showed association with improved oxygen saturation and more rapid resolution of radiographic lung opacities than systemic corticosteroid alone in severe acute respiratory syndrome (SARS). Treatment with interferon has shown preliminary benefits for patients with COVID-19 (19) and is being evaluated by numerous ongoing clinical trials. The IFN-beta treatment could effectively block SARS-CoV-2 replication (20). The IL10RB gene is targeted by peginterferon lambda-1a (Supplementary Material, Table S11), which has been designed to treat mild COVID-19 in a clinical trial (identifier: NCT04331899). These results suggest that interferons associated with IFNAR2 and IL10RB exert potential effects on the treatment of COVID-19.
Discussion
Using a meta-analytic method to combine two existing GWAS summary datasets with a large-scale sample size, we validated two reported genetic loci on chromosome 3p21.31 and 9q34.2 to be significantly associated with COVID-19 infection, and found a novel locus (rs9976829 in IFNAR2- IL10RB) at a chromosome 21q22.11 gene cluster implicated in COVID-19 susceptibility. The rs9976829 shows significant sQTL and eQTL associations with both IFNAR2 and IL10RB in multiple tissues including lung tissue. Recently, Pairo-Castineira et al. (21) performed a GWAS study of 2244 critically ill COVID-19 patients from 208 UK intensive care units (ICUs), and found a polymorphism of rs2236757 (P = 4.99 × 10−8) in IFNAR2 show an association with severe COVID-19 at genome-wide significant level and low expression of IFNAR2 is associated with life-threating disease, providing supportive evidence of the important role of IFNAR2 in COVID-19 risk.
On chromosome 21q22.11, the peak association signal covered two genes of IFNAR2 and IL10RB, which have biological functions that probably related to COVID-19. Subsequently, we used two widely-used tools (i.e. S-PrediXcan and Sherlock) to integrate GWAS summary data with eQTL data across 49 GTEx tissues independently for determining whether these two identified risk genes (IFNAR2 and IL10RB) with genetic variants affect COVID-19 through dysregulation of gene expression traits. In light of multi-tissue data can be leveraged to identify genes and pathways influenced by human disease-associated variation, it is plausible to combine information available across multiple tissues in GTEx for mechanistically explain gene regulation and the genetic basis of disease (22). Thus, we further leveraged the S-MultiXcan tool to examine the mediating role of gene expression variation on COVID-19 by combining information across all 49 tissues collectively with GWAS summary statistics. Based on these analyses, we found that genetically predicted gene expression of both IFNAR2 and IL10RB are significantly associated with COVID-19 infection among multiple tissues, such as lung, artery coronary, liver, blood and heart, of which tissue-specific comorbidities including hypertension, asthma, diabetes, and liver diseases have been reported to be associated with COVID-19 infection (23–25). These results provided supportive evidence of both IFNAR2 and IL10RB genes could be good candidates for uncovering the etiology of COVID-19.
IFNAR2 encodes a type I membrane protein, which forms one of the two chains of a receptor for interferons alpha and beta. The deficiency of IFNAR2 supports an essential role for interferons alpha and beta in human antiviral immunity (26). Notably, dysregulation of type I interferon response has been observed in COVID-19 patients (27). Impaired type I interferon activity in the blood could be a hallmark of severe COVID-19 (28). Furthermore, IFNAR2 is required for anti-influenza immunity and related to the risk of post-influenza bacterial superinfections (29). As for IL10RB, its encoded protein belongs to the cytokine receptor family and is an accessory chain essential for the active interleukin 10 receptor complex. Variants in IFNAR2 and IL10RB gene were associated with the susceptibility to hepatitis B virus (HBV) infection (30). Most recently, the interferon pathway is identified to be targeted by the COVID-19 viral protein of Nsp13 (31). Consistently, several recent studies (28,32,33) have provided supportive evidence of the important roles of type I interferon deficiency in severe COVID-19. For example, Zhang et al. (32) reported inborn errors of type I interferon immunity can underlie life-threatening COVID-19 pneumonia in patients with no prior severe infection.
Functional enrichment analysis showed that cytokine-related pathways including cytokine receptor activity, cytokine–cytokine receptor interaction, and chemokine signaling pathway were significantly enriched by genes associated with COVID-19. Seven risk genes of IFNAR2, IL10RB, XCR1, CXCR6, CCR9, CCR1 and GNG12 have implicated in these pathways. There were four genes encoding chemokine receptors, including the X-C motif chemokine receptor 1 (XCR1), the C-X-C motif chemokine receptor 6 (CXCR6), the CC motif chemokine receptor 9 (CCR9) and the CC motif chemokine receptor 1 (CCR1). Vaccine molecules targeting XCR1 on cross-presenting dendritic cells enhance a protective cluster of differentiation 8 (CD8+) T-cell responses against influenza virus (34). CXCR6 modulates the localization of lung tissue-resident memory CD8+ T-cells throughout the sustained immune response to respiratory pathogens, including influenza viruses (35). Both CCR9 and CCR1 also have related functions on immune response to respiratory influenza infection (36–38). Varied manifestations in COVID-19 infection may result from different host genetic factors, which are probably related to immune response (39). Through inducing the expression of many interferon-stimulated genes, soluble cytokines have anti-viral, anti-proliferate and immunomodulatory effects on obstructing viral replication (12). Together, cytokine-related pathways potentially play important roles in the pathogenesis of COVID-19 infection, and more relevant studies should be performed to explore the underlying biological mechanisms.
With regard to the novel gene of HNRNPK, it belongs to the subfamily of ubiquitously expressed heterogeneous nuclear ribonucleoproteins (hnRNP), of which proteins have important roles in cell cycle progression. HNRNPK acts as a central hub in the replication cycle of multiple viruses including HCV (40). An interaction of HNRNPK and HNRNPA2B1 with hepatitis E virus (HEV) promoters has important roles in HEV replication (41). Two cellular RNA binding proteins of hnRNPK and NS1-BP have important roles in regulating influenza A virus RNA splicing (42). As for the RMI1 gene, its protein is an essential component of the RMI complex, which has a crucial role in DNA repair and maintaining genome stability (43). Since there existed important functions of these identified genes, molecular studies are warrant to illustrate the functional consequences of detected association signals.
In light of the ongoing COVID-19 spread worldwide, there exist accumulating interests in understanding host genetic factors associated with COVID-19 susceptibility. Many candidate gene-based association studies and GWASs have been conducted/reported to uncover risk genes implicated in COVID-19, and numerous genes, such as SCL6A20, CXCR6, CCR9, FYCO1, XCR1, LZTFL1, ACE2, TMPRSS2, OAS1, TYK2, DPP9, IFNAR2 and IL10RB, have been identified to be of significance (2,11,21,44–51). For example, Hou et al. (51) investigated two key host genes of ACE2 and TMPRSS2 for COVID-19 susceptibility by testing genetic variants in these two genes from approximately 81 000 human genomes, and indicated that genetic variants in ACE2 and TMPRSS2 were likely associated with genetic susceptibility of COVID-19, which were replicated by other studies (52). Together, these reported COVID-19-related genes could be treated as good candidate genes for future studies.
The power of our study is limited by the difference in study design between the two datasets we analyzed: the COVID-19 Host Genetic Consortium included patients with mild or severe COVID-19 but the Ellinghaus et al. (11) study only included severe COVID-19. Because most of the COVID-19 infected individuals are asymptomatic, population-based controls used in the original studies may contain a substantial proportion of asymptomatic patients, which further reduced the power of our meta-analysis. Due to data from the two included GWASs were based on summary statistics, the population stratification was not assessed in the current meta-analysis. For the Ellinghaus et al. (11) study, to examine for population stratification within and across Italian and Spanish panels, a principal component analysis (PCA) was performed by using the FlashPCA (53). Covariates from 10 PCA were conducted to control for potential population stratification (COVID_I). As for the ANA5 GWAS summary data from the COVID-19 Host Genetic Consortium constructed by 10 contributing studies, the strategy of population stratification is unknown. There should be cautious that only a small sample size of COVID-19 patients (N = 3288) in the current meta-GWAS analysis, which may lead to the reduced power of uncovering risk variants. In light of the association of IFNAR2 gene with severe COVID-19 has been reported by a previous study (21), this limited the novelty of IFNAR2 gene for COVID-19 susceptibility in the current investigation. More replicated studies for the effects of IFNAR2 and IL10RB on COVID-19 are needed in the near future.
In summary, our findings uncover 21q22.11 as a novel risk locus for COVID-19 susceptibility and implicate the potential role of interferons targeting IFNAR2 and IL10RB in the treatment of COVID-19. Individuals with the G alleles of rs9976829 have a 16% greater chance of COVID-19 infection compared with these carrying no such allele. The efficacy and the safety of interferon products are still being evaluated by numerous ongoing clinical trials, which may be strengthened by subgrouping the patients according to their genotypes of the IFNAR2 and the IL10RB loci. Further studies are needed to delineate current findings and understand the underlying pathophysiology of COVID-19.
Materials and Methods
GWAS summary data from Ellinghaus et al. (Dataset #1)
For this GWAS recently reported by Ellinghaus et al. (11), there were 1980 patients with severe COVID-19 enrolled from seven hospitals in the Italian and Spanish epicenters of the SARS-CoV-2 pandemic in the Europe. A total of 2381 control participants were enrolled from Italy and Spain. After stringent quality control and excluding population outliers, 1610 patients with COVID-19 with respiratory failure (835 Italian and 775 Spanish COVID-19 cases) and 2205 control participants (1255 Italian and 950 Spanish controls) were included in the final GWAS. In total, 8 965 091 high-quality single-nucleotide polymorphisms (SNPs, post imputation R2 ≥ 0.6 and minor allele frequency (MAF) ≥ 1%) were included in the Italian cohort and 9 140 716 high-quality SNPs in the Spanish cohort. The GWAS summary statistics (COVID_I) are publicly available in the website (www.c19-genetics.eu). For more detailed information, please refer to the original article (11).
GWAS summary data from the COVID-19 Host Genetic Consortium (Dataset #2)
This GWAS summary statistics of the publicly available COVID-19 HGI GWAS meta-analyses round 2 (ANA5, susceptibility [affected vs. population]) was downloaded from the official website of the COVID-19 Host Genetic Consortium (2) (www.covid19hg.org/results; analysis named ‘20 200 508-results-ANA5_ALL_inv_var_meta’; file named ‘COVID19_HGI_ANA5_20200513.txt.gz’; release date of May 152 020). There were 1678 COVID-19 patients and 674 635 control participants from 10 contributing studies. For the GWAS summary statistics, there were a total of 34 010 457 genetic variants included with a MAF threshold of 0.0001 and an imputation score filter of 0.6. For more detailed information, please refer to the original article (2).
Meta-analysis of GWAS summary data
By using the meta-analysis tool of METAL (54), a fixed-effects meta-analysis was performed to identify risk genes for COVID-19 across two GWAS summary datasets (Dataset #1: COVID_I GWAS summary statistics, and Dataset #2: ANA5 GWAS summary statistics). After removing low-quality and non-matched SNPs, there were 8 424 883 high-quality SNPs with a MAF ≥ 1% and imputation R2 ≥ 0.6 that were common to both datasets with the use of effect-size estimates (BETA) and their standard errors (SE) for the meta-analysis. With regard to the genome-wide meta-analysis, we adopted the widely-used threshold of 5 × 10−8 for combined P values to determine statistical significance. As reported in Ellinghaus et al. (11), we used the combined P value and combined effect (E) with its SE generated by the METAL to compute the OR and its 95% CI: 1) OR = exp (E); 2) the upper confidence limit (OR_95 U) = exp (E + 1.96*SE); 3) the lower confidence limit (OR_95L) = exp (E—1.96*SE). In the current meta-GWAS analysis, we performed the heterogeneity analyses for whole genome-wide SNPs (n = 8 424 883), the vast majority of SNPs (8 008 729/8424882 = 95.1%) showed non-significant heterogeneity between two GWAS summary datasets (PHeterogeneity > 0.05, Supplementary Material, Fig. S9A). Furthermore, all the top-ranked SNPs identified from our meta-GWAS analysis obtained non-significant heterogeneous results (Supplementary Material, Fig. S9B). Thus, the fixed-effects model applied in the current analysis is reasonable. The qqman package in R platform was used to generate Manhattan plot and quantile-quantile (QQ) plot. The web-access tool of LocusZoom (55) was used to visualize regional association plots (http://locuszoom.sph.umich.edu/).
Cis-sQTL and cis-eQTL analysis
To further explore the regulatory function of identified risk SNPs, we performed cis-sQTL and cis-eQTL analysis based on the Genotype-Tissue Expression (GTEx) project data (version 8, dbGaP Accession phs000424.v8.p2). RNA-seq expression outliers were excluded by using a multidimensional extension of the statistics (56). Samples with <10 million mapped reads were excluded. For each gene in given tissue, expression value was normalized across samples using TMM as implemented in edgeR (57). Using LeafCutter (58) to compute the intron excision phenotypes for quantifying splicing values. The genotype data used for analysis were based on genome-wide sequencing from 838 donors, which all had RNA-seq data available in GTEx version 8. The tool of FastQTL (59) was used to perform cis-sQTL and cis-eQTL analyses. Data visualization was performed by using the web-access tool of GTEx portal (www.gtexportal.org) (60).
Gene-based association analysis
We conducted a gene-based association analysis of our meta-GWAS summary data for COVID-19 by using the Multi-marker Analysis of GenoMic Annotation (MAGMA) (61), which utilizes a multiple regression method to identify multi-marker aggregated effects that account for SNP P values and linkage disequilibrium (LD) between SNPs. The analyzed SNP set of each gene was based on whether the SNP located in the gene body region or within extended +/− 20 kb downstream or upstream of the gene. The LDs among SNPs were calculated based on the 1000 Genomes Phases 3 European Panel (62). The Benjamini–Hochberg FDR method was used to correct the association results for multiple testing. The P value threshold of 1.86 × 10−5 was applied.
Functional enrichment analysis
To annotate the molecular functions and biological pathways of these COVID-19-associated genes (Supplementary Material, Table S2), we performed a functional enrichment analysis by using the WebGestalt tool (63) based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and GO terms. Using the overrepresentation analysis, the WebGestalt could identify functional association between COVID-19-associated genes and KEGG pathways. Furthermore, GO enrichment analysis was performed by using 3 categories of GO terms: biological process, cellular component and molecular function. The redundancies of GO-terms were removed. The hypergenometric test was applied to assess the statistical significance. P values were corrected for multiple testing using the FDR, and a P value threshold of 2.9 × 10−4 was applied.
Gene-property analysis
We performed a MAGMA gene-property analysis (61), which is implemented in FUMA (64). The gene expression data from 83 tissues GTEx RNA-seq data (version 8) were used to parse the gene expression profiles. Expression values (TPM) were log2 transformed and average expression values were adopted per tissue. The gene-property analysis was conducted for 53 specific tissue types and 30 general tissue types, respectively. Bonferroni correction for multiple testing was used for the examined tissue types.
Single-cell RNA-seq analysis for lung tissue
We performed single-cell data analysis from normal lung tissue sequenced by using Smart-Seq2. This data set is a part of the Human Lung Atlas (65), consisting of 50 cell populations across 4 compartments (epithelial, endothelial, stromal, and immune) of lung tissue. All 9404 cells with distinct cellular identities were download from the Human Lung Atlas. Cells from 3 donors with FACS-sorted strategy were available at the Synapse (accession numbers: syn22168639, syn22168625 and syn22168622).
S-PrediXcan and S-MultiXcan analysis
We applied S-PrediXcan (66) to integrate expression quantitative trait loci (eQTL) data with genetic associations from GWAS summary statistics to identify genes, which genetically predicted expression levels are associated with COVID-19. S-PrediXcan firstly estimates gene expression weights by training a linear prediction model (MASHR model) in samples with both SNP genotype and gene expression data. These estimated weights are processed with beta values and standard errors from meta-GWAS summary data on COVID-19 to predict gene expression from GWAS summary statistics, while combining the variance and co-variance of SNPs from an LD reference panel based on the 1000 Genomes Project Phase 3 genotypes (62). The eQTL data for 49 tissues from the GTEx Project (version 8) were used in the current analysis. S-PrediXcan was performed for each of 49 tissues for a total of 659 158 gene-tissue pairs. To increase power to identify genes whose expression is similarly differentially regulated across tissues, we meta-analyzed the S-PrediXcan results with the use of the S-MultiXcan method (67), which employs multivariate regression to integrate evidence across 49 GTEx tissues with a total of 22 327 genes. Significant associations were determined by using Bonferroni correction.
Sherlock-based integrative genomics analysis
To further validate these identified host genes for COVID-19, we also applied an independent approach of Sherlock-based integrative genomics analysis based on a Bayesian inference algorithm (68). The Sherlock-based integrative analysis incorporates genetic information from meta-GWAS summary statistics on COVID-19 with eQTL data across 49 tissues from the GTEx Project (version 7) to prioritize important risk genes. It should be noted that Sherlock employs different algorithm and strategies to perform the statistical inference compared with S-PrediXcan. Briefly, Sherlock first searches expression-associated SNPs (named as eSNPs) across different GTEx tissues. Then, Sherlock estimates the possible association of eSNPs with COVID-19 using our meta-GWAS summary data. Sherlock computes individual Logarithm of the Bayes Factor (LBF) for each SNP pair, and the sum of these constitutes the final LBF score for each gene. There are three potential scenarios: 1) if an eSNP in a given gene showed a significant association with COVID-19, a positive score would be assigned; 2) if an eSNP in a given gene showed a non-significant association with COVID-19, a negative score would be assigned; 3) no score was assigned if an SNP was not eSNP but showed a significant association with COVID-19. The Sherlock applied the simulation analysis to compute the P value of the Bayes factor for each gene, as reference of a method of Bayes/non-Bayes compromise (69). We used the Bonferroni correction method to correct the significance for multiple testing. Due to we tested the Sherlock analysis in 49 GTEx tissues, there existed different thresholds across tissues. For example, the P value threshold was 6.64 × 10−6 (0.05/7529) for lung tissue.
In silico permutation analysis
To determine whether there exist a higher overlapped rate of genes identified from MAGMA analysis (Gene set #1: N = 1005, P < 0.05) with genes from S-MultiXcan analysis (Gene set #2: N = 1141, P < 0.05) than that from random selections, we conducted a computer-based permutation analysis of 100 000 times of random selections (70,71). First, we counted the overlapped genes between Gen sets #1 and #2 (N observation). Secondly, we used all tested genes from S-MultiXcan analysis as background genes (N background = 22 326 genes). Then, through randomly selecting the same number of genes as Gene set #2 from background genes to compare with Gene set #1 for 100 000 times, we counted the number of overlapped genes in each time (N random). In the third step, we summed the counts of N observation ≤ N random and divided by 100 000 to calculate empirically permuted P value. P < 0.05 is considered to be significant.
Drug-gene interaction analysis
We submitted these 11 COVID-19-associated genes into the widely-used Drug Gene Interaction Database (DGIdb v.3.0.2; http://www.dgidb.org/) to identify drug–gene interactions with Food and Drug Administration (FDA)-approved pharmaceutical compounds as well as antineoplastic and immunotherapies drugs depended on 20 databases with 51 known interaction types, and search 10 databases to find genes with potential drug abilities.
Supplementary Material
Acknowledgements
We thanks to the helpful suggestions of Dr Guijun Zhang. We appreciate all the authors from the COVID-19 Host Genetic Consortium, as well as Ellinghaus and his colleagues who have deposited and shared GWAS summary data on public databases.
Conflict of Interest Statement. The authors declare no conflict of interest.
Contributor Information
Yunlong Ma, Institute of Biomedical Big Data, School of Ophthalmology & Optometry and Eye Hospital, School of Biomedical Engineering, Wenzhou Medical University, Wenzhou 325027, China.
Yukuan Huang, Institute of Biomedical Big Data, School of Ophthalmology & Optometry and Eye Hospital, School of Biomedical Engineering, Wenzhou Medical University, Wenzhou 325027, China.
Sen Zhao, Beijing Key Laboratory for Genetic Research of Skeletal Deformity, Key laboratory of big data for spinal deformities, Department of Orthopedic Surgery, Peking Union Medical College Hospital, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing 100730, China.
Yinghao Yao, Wenzhou Institute, University of Chinese Academy of Sciences, Wenzhou 325011, China.
Yaru Zhang, Institute of Biomedical Big Data, School of Ophthalmology & Optometry and Eye Hospital, School of Biomedical Engineering, Wenzhou Medical University, Wenzhou 325027, China.
Jia Qu, Institute of Biomedical Big Data, School of Ophthalmology & Optometry and Eye Hospital, School of Biomedical Engineering, Wenzhou Medical University, Wenzhou 325027, China.
Nan Wu, Beijing Key Laboratory for Genetic Research of Skeletal Deformity, Key laboratory of big data for spinal deformities, Department of Orthopedic Surgery, Peking Union Medical College Hospital, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing 100730, China.
Jianzhong Su, Institute of Biomedical Big Data, School of Ophthalmology & Optometry and Eye Hospital, School of Biomedical Engineering, Wenzhou Medical University, Wenzhou 325027, China; Wenzhou Institute, University of Chinese Academy of Sciences, Wenzhou 325011, China.
Data Availability
All the GWAS summary statistics used in this study can be accessed in the official websites (www.covid19hg.org/results and www.c19-genetics.eu). The GTEx eQTL data (version 8) were downloaded from Zenodo repository (https://zenodo.org/record/3518299#.Xv6Z6igzbgl). All analysis code in the Methods is available in a publicly available GitHub repository at https://github.com/mayunlong89/Host_genetics_for_COVID-19.
Funding
The National Natural Science Foundation of China (61871294 to J.S., 81501852 to N.W.); the Scientific Research Foundation for Talents of Wenzhou Medical University (KYQD20201001 to Y.M.); and Science Foundation of Zhejiang Province (LR19C060001 to J.S).
Author Contributions
J.S., N.W., Y.M. and J.Q. conceived and designed the study. Y.M., Y.H., S.Z., G.Q. and J.Z. contributed to management of data collection. Y.M., Y.H., G.Q., J.Z., J.Q., Z.M., J.Y., J.S., N.W. and S.Z. conducted bioinformatics analysis and data interpretation. Y.M., J.S., N.W. and S.Z. wrote the manuscripts. All authors reviewed and approved the final manuscript.
References
- 1. Li, Q., Guan, X., Wu, P., Wang, X., Zhou, L., Tong, Y., Ren, R., Leung, K.S.M., Lau, E.H.Y., Wong, J.Y. et al. (2020) Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med., 382, 1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. The COVID-19 Host Genetics Initiative (2020) et al. Eur. J. Human Genetics, 28, 715–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dong, E., Du, H. and Gardner, L. (2020) An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis., 20, 533–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu, Z. and McGoogan, J.M. (2020) Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA, 323(13), 1239–1242. [DOI] [PubMed] [Google Scholar]
- 5. Chan, J.F., Yuan, S., Kok, K.H., To, K.K., Chu, H., Yang, J., Xing, F., Liu, J., Yip, C.C., Poon, R.W. et al. (2020) A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet (London, England), 395, 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Onder, G., Rezza, G. and Brusaferro, S. (2020) Case-fatality rate and characteristics of patients dying in relation to COVID-19 in Italy. JAMA, 323(18), 1775–1776. [DOI] [PubMed] [Google Scholar]
- 7. Zhou, F., Yu, T., Du, R., Fan, G., Liu, Y., Liu, Z., Xiang, J., Wang, Y., Song, B., Gu, X. et al. (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet (London, England), 395, 1054–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., Zhang, L., Fan, G., Xu, J., Gu, X. et al. (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (London, England), 395, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou, P., Yang, X.L., Wang, X.G., Hu, B., Zhang, L., Zhang, W., Si, H.R., Zhu, Y., Li, B., Huang, C.L. et al. (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu, R., Zhao, X., Li, J., Niu, P., Yang, B., Wu, H., Wang, W., Song, H., Huang, B., Zhu, N. et al. (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet (London, England), 395, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ellinghaus, D., Degenhardt, F., Bujanda, L., Buti, M., Albillos, A., Invernizzi, P., Fernández, J., Prati, D., Baselli, G., Asselta, R. et al. (2020) Genomewide association study of severe Covid-19 with respiratory failure. N. Engl. J. Med., 383(16), 1522–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Platanias, L.C. (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol., 5, 375–386. [DOI] [PubMed] [Google Scholar]
- 13. Barrat, F.J. and Su, L. (2019) A pathogenic role of plasmacytoid dendritic cells in autoimmunity and chronic viral infection. J. Exp. Med., 216, 1974–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Macal, M., Jo, Y., Dallari, S., Chang, A.Y., Dai, J., Swaminathan, S., Wehrens, E.J., Fitzgerald-Bocarsly, P. and Zúñiga, E.I. (2018) Self-renewal and toll-like receptor signaling sustain exhausted plasmacytoid dendritic cells during chronic viral infection. Immunity, 48, e735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ren, X., Wen, W., Fan, X., Hou, W., Su, B., Cai, P., Li, J., Liu, Y., Tang, F., Zhang, F. et al. (2021) COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell, 184, 1895–1913.e1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Su, Y., Chen, D., Yuan, D., Lausted, C., Choi, J., Dai, C.L., Voillet, V., Duvvuri, V.R., Scherler, K., Troisch, P. et al. (2020) Multi-omics resolves a sharp disease-state shift between mild and moderate COVID-19. Cell, 183, 1479–1495.e1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cinatl, J., Morgenstern, B., Bauer, G., Chandra, P., Rabenau, H. and Doerr, H.W. (2003) Treatment of SARS with human interferons. Lancet (London, England), 362, 293–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Loutfy, M.R., Blatt, L.M., Siminovitch, K.A., Ward, S., Wolff, B., Lho, H., Pham, D.H., Deif, H., LaMere, E.A., Chang, M. et al. (2003) Interferon alfacon-1 plus corticosteroids in severe acute respiratory syndrome: a preliminary study. JAMA, 290, 3222–3228. [DOI] [PubMed] [Google Scholar]
- 19. Hung, I.F., Lung, K.C., Tso, E.Y., Liu, R., Chung, T.W., Chu, M.Y., Ng, Y.Y., Lo, J., Chan, J., Tam, A.R. et al. (2020) Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet, 395, 1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lei, X., Dong, X., Ma, R., Wang, W., Xiao, X., Tian, Z., Wang, C., Wang, Y., Li, L., Ren, L. et al. (2020) Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun., 11, 3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pairo-Castineira, E., Clohisey, S., Klaric, L., Bretherick, A.D., Rawlik, K., Pasko, D., Walker, S., Parkinson, N., Fourman, M.H., Russell, C.D. et al. (2020) Genetic mechanisms of critical illness in Covid-19. Nature, 591(7848), 92–98. [DOI] [PubMed] [Google Scholar]
- 22. Battle, A., Brown, C.D., Engelhardt, B.E. and Montgomery, S.B. (2017) Genetic effects on gene expression across human tissues. Nature, 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cai, Q., Huang, D., Ou, P., Yu, H., Zhu, Z., Xia, Z., Su, Y., Ma, Z., Zhang, Y., Li, Z. et al. (2020) COVID-19 in a designated infectious diseases hospital outside Hubei Province, China. Allergy, 75, 1742–1752. [DOI] [PubMed] [Google Scholar]
- 24. Ejaz, H., Alsrhani, A., Zafar, A., Javed, H., Junaid, K., Abdalla, A.E., Abosalif, K.O.A., Ahmed, Z. and Younas, S. (2020) COVID-19 and comorbidities: deleterious impact on infected patients. J. Infect. Public Health, 13, 1833–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Feldman, E.L., Savelieff, M.G., Hayek, S.S., Pennathur, S., Kretzler, M. and Pop-Busui, R. (2020) COVID-19 and diabetes: a collision and collusion of two diseases. Diabetes, 69, 2549–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Duncan, C.J., Mohamad, S.M., Young, D.F., Skelton, A.J., Leahy, T.R., Munday, D.C., Butler, K.M., Morfopoulou, S., Brown, J.R., Hubank, M. et al. (2015) Human IFNAR2 deficiency: lessons for antiviral immunity. Sci. Transl. Med., 7(307), 307ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Blanco-Melo, D., Nilsson-Payant, B.E., Liu, W.C., Uhl, S., Hoagland, D., Moller, R., Jordan, T.X., Oishi, K., Panis, M., Sachs, D. et al. (2020) Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell, 181, 1036–1045 e 1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hadjadj, J., Yatim, N., Barnabei, L., Corneau, A., Boussier, J., Smith, N., Péré, H., Charbit, B., Bondet, V., Chenevier-Gobeaux, C. et al. (2020) Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science, 369, 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shepardson, K.M., Larson, K., Johns, L.L., Stanek, K., Cho, H., Wellham, J., Henderson, H. and Rynda-Apple, A. (2018) IFNAR2 is required for anti-influenza immunity and alters susceptibility to post-influenza bacterial superinfections. Front. Immunol., 9, 2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gong, Q.M., Kong, X.F., Yang, Z.T., Xu, J., Wang, L., Li, X.H., Jin, G.D., Gao, J., Zhang, D.H., Jiang, J.H. et al. (2009) Association study of IFNAR2 and IL10RB genes with the susceptibility and interferon response in HBV infection. J. Viral Hepat., 16, 674–680. [DOI] [PubMed] [Google Scholar]
- 31. Gordon, D.E., Jang, G.M., Bouhaddou, M., Xu, J., Obernier, K., White, K.M., O'Meara, M.J., Rezelj, V.V., Guo, J.Z., Swaney, D.L. et al. (2020) A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature, 583(7816), 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang, Q., Bastard, P., Liu, Z., Le Pen, J., Moncada-Velez, M., Chen, J., Ogishi, M., Sabli, I.K.D., Hodeib, S., Korol, C. et al. (2020) Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science, 370(6515), eabd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bastard, P., Rosen, L.B., Zhang, Q., Michailidis, E., Hoffmann, H.H., Zhang, Y., Dorgham, K., Philippot, Q., Rosain, J., Béziat, V. et al. (2020) Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science, 370(6515), eabd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fossum, E., Grødeland, G., Terhorst, D., Tveita, A.A., Vikse, E., Mjaaland, S., Henri, S., Malissen, B. and Bogen, B. (2015) Vaccine molecules targeting Xcr1 on cross-presenting DCs induce protective CD8+ T-cell responses against influenza virus. Eur. J. Immunol., 45, 624–635. [DOI] [PubMed] [Google Scholar]
- 35. Wein, A.N., McMaster, S.R., Takamura, S., Dunbar, P.R., Cartwright, E.K., Hayward, S.L., McManus, D.T., Shimaoka, T., Ueha, S., Tsukui, T. et al. (2019) CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J. Exp. Med., 216, 2748–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang, J., Li, F., Wei, H., Lian, Z.X., Sun, R. and Tian, Z. (2014) Respiratory influenza virus infection induces intestinal immune injury via microbiota-mediated Th17 cell-dependent inflammation. J. Exp. Med., 211, 2397–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Henson, S.M., Snelgrove, R., Hussell, T., Wells, D.J. and Aspinall, R. (2005) An IL-7 fusion protein that shows increased thymopoietic ability. J. Immunol., 175, 4112–4118. [DOI] [PubMed] [Google Scholar]
- 38. Zhou, J., Law, H.K., Cheung, C.Y., Ng, I.H., Peiris, J.S. and Lau, Y.L. (2006) Differential expression of chemokines and their receptors in adult and neonatal macrophages infected with human or avian influenza viruses. J Infect Dis, 194, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang, W., Xu, Y., Gao, R., Lu, R., Han, K., Wu, G. and Tan, W. (2020) Detection of SARS-CoV-2 in different types of clinical specimens. JAMA, 323, 1843–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Poenisch, M., Metz, P., Blankenburg, H., Ruggieri, A., Lee, J.Y., Rupp, D., Rebhan, I., Diederich, K., Kaderali, L., Domingues, F.S. et al. (2015) Identification of HNRNPK as regulator of hepatitis C virus particle production. PLoS Pathog., 11(1), e1004573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kanade, G.D., Pingale, K.D. and Karpe, Y.A. (2019) Protein interactions network of hepatitis E virus RNA and polymerase with host proteins. Front. Microbiol., 10, 2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tsai, P.L., Chiou, N.T., Kuss, S., García-Sastre, A., Lynch, K.W. and Fontoura, B.M. (2013) Cellular RNA binding proteins NS1-BP and hnRNP K regulate influenza a virus RNA splicing. PLoS Pathog., 9(6), e1003460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fang, L., Sun, X., Wang, Y., Du, L., Ji, K., Wang, J., He, N., Liu, Y., Wang, Q., Zhai, H. et al. (2019) RMI1 contributes to DNA repair and to the tolerance to camptothecin. FASEB J., 33, 5561–5570. [DOI] [PubMed] [Google Scholar]
- 44. Zhou, S., Butler-Laporte, G., Nakanishi, T., Morrison, D., Afilalo, J., Afilalo, M., Laurent, L., Pietzner, M., Kerrison, N., Zhao, K. et al. (2020) A Neanderthal OAS1 isoform protects against COVID-19 susceptibility and severity: results from Mendelian randomization and case-control studies. Nat. Med., 27(4), 659–667. [DOI] [PubMed] [Google Scholar]
- 45. Pathak, G.A., Singh, K., Miller-Fleming, T.W., Wendt, F.R., Ehsan, N., Hou, K., Johnson, R., Lu, Z., Gopalan, S., Yengo, L. et al. (2020) Integrative analyses identify susceptibility genes underlying COVID-19 hospitalization. medRxiv, in press., 2020.2012.2007.20245308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shelton, J.F., Shastri, A.J., Ye, C., Weldon, C.H., Filshtein-Somnez, T., Coker, D., Symons, A., Esparza-Gordillo, J., Aslibekyan, S. and Auton, A. (2020) Trans-ethnic analysis reveals genetic and non-genetic associations with COVID-19 susceptibility and severity. medRxiv in press., 2020.2009.2004.20188318. [DOI] [PubMed] [Google Scholar]
- 47. Roberts, G.H.L., Park, D.S., Coignet, M.V., McCurdy, S.R., Knight, S.C., Partha, R., Rhead, B., Zhang, M., Berkowitz, N., Haug Baltzell, A.K. et al. (2020) Ancestry DNA COVID-19 host genetic study identifies three novel loci. medRxiv, in press., 2020.2010.2006.20205864. [Google Scholar]
- 48. Di Maria, E., Latini, A., Borgiani, P. and Novelli, G. (2020) Genetic variants of the human host influencing the coronavirus-associated phenotypes (SARS, MERS and COVID-19): rapid systematic review and field synopsis. Hum. Genomics, 14, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ovsyannikova, I.G., Haralambieva, I.H., Crooke, S.N., Poland, G.A. and Kennedy, R.B. (2020) The role of host genetics in the immune response to SARS-CoV-2 and COVID-19 susceptibility and severity. Immunol. Rev., 296, 205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zeberg, H. and Pääbo, S. (2020) The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature, 587, 610–612. [DOI] [PubMed] [Google Scholar]
- 51. Hou, Y., Zhao, J., Martin, W., Kallianpur, A., Chung, M.K., Jehi, L., Sharifi, N., Erzurum, S., Eng, C. and Cheng, F. (2020) New insights into genetic susceptibility of COVID-19: an ACE2 and TMPRSS2 polymorphism analysis. BMC Med., 18, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., Schiergens, T.S., Herrler, G., Wu, N.H., Nitsche, A. et al. (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell, 181, e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abraham, G., Qiu, Y. and Inouye, M. (2017) Flash PCA2: principal component analysis of biobank-scale genotype datasets. Bioinformatics, 33, 2776–2778. [DOI] [PubMed] [Google Scholar]
- 54. Willer, C.J., Li, Y. and Abecasis, G.R. (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pruim, R.J., Welch, R.P., Sanna, S., Teslovich, T.M., Chines, P.S., Gliedt, T.P., Boehnke, M., Abecasis, G.R. and Willer, C.J. (2010) Locus zoom: regional visualization of genome-wide association scan results. Bioinformatics, 26, 2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wright, F.A., Sullivan, P.F., Brooks, A.I., Zou, F., Sun, W., Xia, K., Madar, V., Jansen, R., Chung, W., Zhou, Y.H. et al. (2014) Heritability and genomics of gene expression in peripheral blood. Nat. Genet., 46, 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Robinson, M.D. and Oshlack, A. (2010) A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol., 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li, Y.I., Knowles, D.A., Humphrey, J., Barbeira, A.N., Dickinson, S.P., Im, H.K. and Pritchard, J.K. (2018) Annotation-free quantification of RNA splicing using leaf cutter. Nat. Genet., 50, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ongen, H., Buil, A., Brown, A.A., Dermitzakis, E.T. and Delaneau, O. (2016) Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics, 32, 1479–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.GTEx Consortium. (2013) The genotype-tissue expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. de Leeuw, C.A., Mooij, J.M., Heskes, T. and Posthuma, D. (2015) MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol., 11(4), e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Auton, A., Brooks, L.D., Durbin, R.M., Garrison, E.P., Kang, H.M., Korbel, J.O., Marchini, J.L., McCarthy, S., McVean, G.A. and Abecasis, G.R. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang, B., Kirov, S. and Snoddy, J. (2005) Web gestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res., 33, W741–W748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Watanabe, K., Taskesen, E., van Bochoven, A. and Posthuma, D. (2017) Functional mapping and annotation of genetic associations with FUMA. Nat. Commun., 8, 1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Travaglini, K.J., Nabhan, A.N., Penland, L., Sinha, R., Gillich, A., Sit, R.V., Chang, S., Conley, S.D., Mori, Y., Seita, J. et al. (2020) A molecular cell atlas of the human lung from single cell RNA sequencing. bioRxiv, in press., 742320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Barbeira, A.N., Dickinson, S.P., Bonazzola, R., Zheng, J., Wheeler, H.E., Torres, J.M., Torstenson, E.S., Shah, K.P., Garcia, T., Edwards, T.L. et al. (2018) Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun., 9, 1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Barbeira, A.N., Pividori, M., Zheng, J., Wheeler, H.E., Nicolae, D.L. and Im, H.K. (2019) Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet., 15(1), e1007889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. He, X., Fuller, C.K., Song, Y., Meng, Q., Zhang, B., Yang, X. and Li, H. (2013) Sherlock: detecting gene-disease associations by matching patterns of expression QTL and GWAS. Am. J. Hum. Genet., 92, 667–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Servin, B. and Stephens, M. (2007) Imputation-based analysis of association studies: candidate regions and quantitative traits. PLoS Genet., 3, e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ma, X., Wang, P., Xu, G., Yu, F. and Ma, Y. (2020) Integrative genomics analysis of various omics data and networks identify risk genes and variants vulnerable to childhood-onset asthma. BMC Med. Genet., 13, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dong, Z., Ma, Y., Zhou, H., Shi, L., Ye, G., Yang, L., Liu, P. and Zhou, L. (2020) Integrated genomics analysis highlights important SNPs and genes implicated in moderate-to-severe asthma based on GWAS and eQTL datasets. BMC Pulm. Med., 20, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the GWAS summary statistics used in this study can be accessed in the official websites (www.covid19hg.org/results and www.c19-genetics.eu). The GTEx eQTL data (version 8) were downloaded from Zenodo repository (https://zenodo.org/record/3518299#.Xv6Z6igzbgl). All analysis code in the Methods is available in a publicly available GitHub repository at https://github.com/mayunlong89/Host_genetics_for_COVID-19.