

Abstract
Overproduction of reactive oxygen species and compromised antioxidant defenses perturb intracellular redox homeostasis and is associated with a myriad of human diseases as well as with the natural process of aging. Hydrogen sulfide (H2S), which is biosynthesized by organisms ranging from bacteria to man, influences a broad range of physiological functions. A highly touted molecular mechanism by which H2S exerts its cellular effects is via post-translational modification of the thiol redox proteome, converting cysteine thiols to persulfides, in a process referred to as protein persulfidation. The physiological relevance of this modification in the context of specific signal transmission pathways remains to be rigorously established, while a general protective role for protein persulfidation against hyper-oxidation of the cysteine proteome is better supported. A second mechanism by which H2S modulates redox homeostasis is via remodeling the redox metabolome, targeting the electron transfer chain and perturbing the major redox nodes i.e., CoQ/CoQH2, NAD+/NADH and FAD/FADH2. The metabolic changes that result from H2S-induced redox changes fan out from the mitochondrion to other compartments. In this review, we discuss recent developments in elucidating the roles of H2S and its oxidation products on redox homeostasis and its role in protecting the thiol proteome.
Keywords: Sulfide oxidation pathway, sulfide quinone oxidoreductase, reactive sulfur species, mitochondrial bioenergetics, redox proteome, redox metabolome, persulfidation
A necessary fallout of life on oxygen is the production of reactive oxygen species (ROS) including superoxide anion, hydrogen peroxide, and hydroxyl radical. Oxidative metabolism concomitantly generates reducing power, which is captured by redox cofactors such as NAD+ and FAD that are critical for powering cellular functions and, together with antioxidant systems such as glutathione (GSH) and thioredoxin, are important for establishing redox homeostasis. The reduced cofactors, NADH and FADH2 are recycled via the electron transport chain (ETC), which is a central hub in the cellular redox network and is susceptible to regulation by endogenous modulators such as H2S and NO. The regulated generation of ROS such as superoxide and peroxide is important in redox signaling and influences processes such as cell growth, differentiation, and death (Holmstrom and Finkel, 2014). The wanton reactivity of hydroxyl radicals on the other hand, spells danger and is associated with ferroptosis, characterized by iron-dependent accumulation of lipid peroxides (Dixon et al., 2012). The proteome is a structurally diverse class of cellular macromolecules that represents a potentially large sink for oxidants (Hansen, Roth, and Winther, 2009). Cellular proteostasis is maintained via a carefully coordinated response to oxidative stress (Reichmann, Voth, and Jakob, 2018).
The thiol redox proteome collectively refers to proteins that undergo reversible or irreversible modification of cysteines that can serve as functional switches at an individual protein level (van Montfort et al., 2003; Suzuki and Yamamoto, 2017) while conferring protection against oxidation-induced unfolding and aggregation at a global level. Cysteine is underrepresented in many proteomes and its prevalence ranges from ~0.4% in Archaea to 2.26% in mammals (Miseta and Csutora, 2000). It is estimated that under conditions of oxidative stress, the thiol redox proteome can shift from a basal state of 5-12% to >40% of cysteines being oxidized (Go and Jones, 2013). A range of reversible thiol modifications has been described including sulfenylation, glutathionylation, persulfidation and nitrosylation and hundreds of proteins with oxidatively modified cysteines are typically identified by mass spectrometry methods in response to different oxidant triggers (Yang, Carroll, and Liebler, 2016; Gao et al., 2015). There is some but certainly far from complete overlap in targets that are modified by different oxidants and the basis for target selectivity is not understood.
H2S is a recently recognized addition to the cellular repertoire of redox regulators and modulates both the redox proteome and metabolome (Kabil and Banerjee, 2010; Kabil, Vitvitsky, and Banerjee, 2014; Kabil and Banerjee, 2014; Filipovic et al., 2018). Its discovery as an endogenously synthesized cellular mediator in eukaryotes (Abe and Kimura, 1996) emerged decades after its notoriety as a toxic gas was widely known. The toxicity of H2S derives from its ability to poison complex IV in the respiratory chain (Nicholls and Kim, 1982). Some organisms are adapted to life in environments with remarkably high H2S exposure and partner with sulfide-metabolizing endosymbionts to achieve tolerance (Stewart and Cavanaugh, 2006). The lower gut in humans similarly represents a high sulfide environment that is shaped by microbial metabolism (Macfarlane, Gibson, and Cummings, 1992). The adaptive responses of colonocytes to this exposure have only just begun to be understood (Libiad et al., 2019). In this review, we examine the impacts of H2S on the redox metabolome via its effects on mitochondrial bioenergetics, and on the thiol proteome via persulfidation, which could be important in a multipronged cellular strategy for countering stress.
H2S and reactive sulfur species biogenesis
The capacity for cellular H2S biogenesis in mammals resides in at least three enzymes of which two, cystathionine β-synthase (CBS) and γ-cystathionase (CSE) produce H2S in secondary reactions resulting from relaxed substrate specificities (Banerjee, 2017; Singh and Banerjee, 2011; Chiku et al., 2009). The canonical reactions catalyzed by CBS and CSE lead to the conversion of homocysteine to cysteine in the cytoplasmic transsulfuration pathway (Figure 1a), while the same amino acids can serve as a source of H2S in γ-replacement or β-replacement reactions, respectively. Metabolic track switching from cysteine to H2S synthesis by the transsulfuration pathway enzymes has been ascribed to heme-dependent regulation of CBS by CO and NO (Kabil, Yadav, and Banerjee, 2016; Taoka, West, and Banerjee, 1999; Taoka and Banerjee, 2001; Puranik et al., 2006). The third enzyme that can catalyze H2S synthesis is mercaptopyruvate sulfurtransferase (MPST). It is expressed as two isoforms that are found in the cytoplasm and mitochondrion, respectively (Yadav et al., 2013; Yadav et al., 2020). MPST is involved in cysteine catabolism and catalyzes sulfur transfer from 3-mercaptopyruvate to an active site cysteine, and from there, to a thiophilic acceptor, which can be a small molecule (e.g., cysteine) or a protein (e.g., thioredoxin) (Figure 1b). H2S is eliminated from the resulting persulfide product in the presence of reductant or via intramolecular disulfide bond formation as in thioredoxin.
Figure 1. Biogenesis and clearance of H2S and reactive sulfur species.
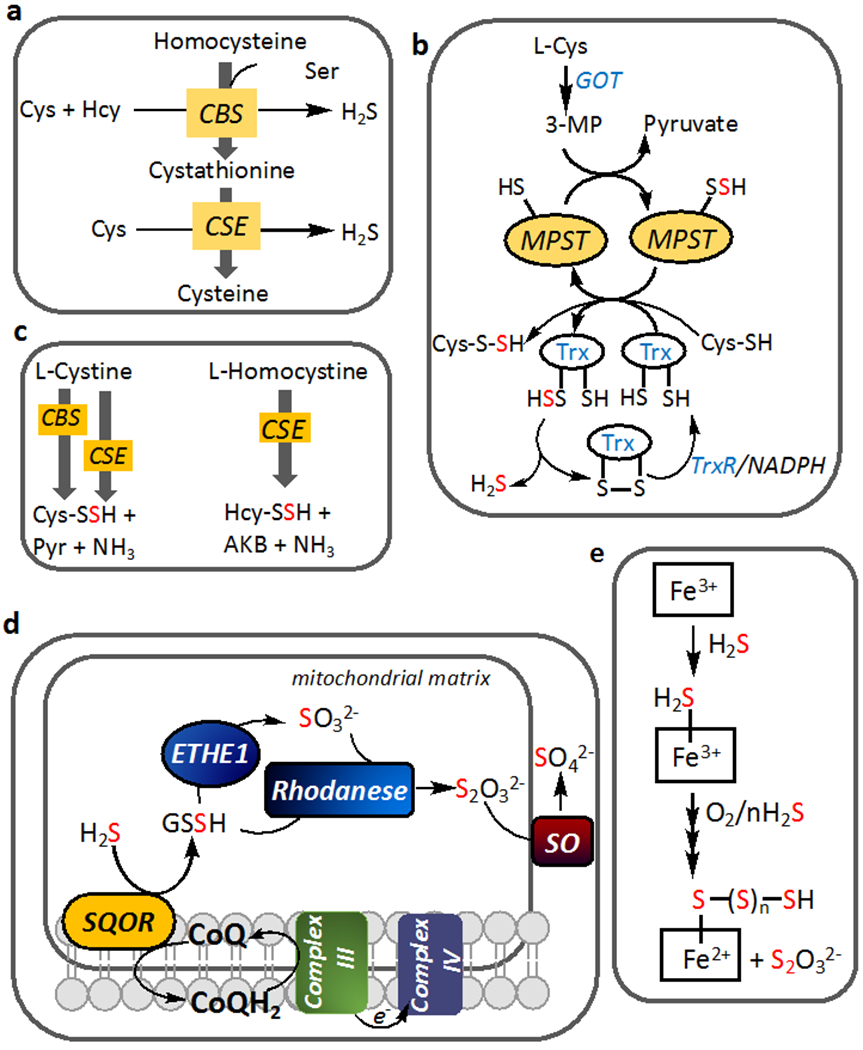
(a) Reactions showing the major routes for H2S synthesis by CBS and CSE at cellular concentrations of substrates and their canonical reactions in the transsulfuration pathway. (b) H2S generation by MPST from 3-mercaptopyruvate (3-MP), which is derived from cysteine via the action of glutamate-oxaloacetate transaminase (GOT), which is also referred to as cysteine aminotransferase. A persulfide intermediate is formed on MPST and transferred to a low molecular weight (LMW) acceptor like cysteine or to thioredoxin (Trx). TrxR represents thioredoxin reductase. (c) LMW persulfides can be synthesized by CBS and CSE, with the latter being the major source under cellular conditions. Pyr and AKB denote pyruvate and α-ketobutyrate. (d) The mitochondrial sulfide oxidation pathway enzymes lead to oxidation of H2S to thiosulfate and sulfate. Electrons from the first step in the pathway, which is catalyzed by SQOR (sulfide quinone oxidoreductase), enter the ETC at the level of complex III. ETHE1 is a persulfide dioxygenase and SO denotes sulfite oxidase (e) Sulfide can be oxidized at ferric heme centers in globins leading to formation of thiosulfate and iron bound polysulfides.
All three enzymes can also generate persulfide products in addition to H2S (Figure 1b,c). Cysteine persulfide (CBS, CSE) and homocysteine persulfide (CSE) can be synthesized from cystine and homocystine, respectively (Yadav et al., 2016; Ida et al., 2014) while MPST can synthesize cysteine persulfide (Yadav et al., 2020). Simulations based on the kinetics of CBS and CSE, their protein levels and physiological substrate concentrations, predict that CSE accounts for ~97% of liver H2S synthesis by the transsulfuration pathway enzymes (Kabil et al., 2011). Mathematical modeling also predicts that at physiologically relevant substrate concentrations in the liver, persulfide synthesis by CBS and CSE is negligible compared to H2S (Yadav et al., 2016). Thus, the reducing intracellular milieu results in the dominance of cysteine over the cystine pool leading to substrate level regulation of CBS and CSE, which influences the competing flux of sulfur to H2S versus persulfide synthesis. However, if intracellular cystine concentrations increase from the steady state levels of ~0.2 μM to >5 μM, such as under oxidative stress conditions, then, CSE-catalyzed cysteine-persulfide synthesis could become more substantial (Yadav et al., 2016). Low molecular weight persulfides are also products of the mitochondrial sulfide oxidation pathway as discussed below (Figure 1d) (Mishanina, Libiad, and Banerjee, 2015).
Reactive sulfur species (RSS) other than persulfides can also be generated via metal-mediated sulfide oxidation. Ferric heme proteins including hemoglobin (Vitvitsky et al., 2017; Vitvitsky et al., 2015), myoglobin (Bostelaar et al., 2016), neuroglobin (Ruetz et al., 2017) and cytochrome c (Vitvitsky et al., 2018) can bind sulfide. Under aerobic conditions, ferric sulfide is oxidized to a mixture of thiosulfate and metal bound polysulfides (Figure 1e). Sulfide inhibits myeloperoxidase and also reacts with its product, hypochlorous acid, forming polysulfides (Palinkas et al., 2014; Nagy and Winterbourn, 2010). On the other hand, binding of polysulfides to ferric heme activates indoleamine-2,3-dioxygenase, which is involved in immune regulation via the kynurenine pathway (Nelp et al., 2019). The dielectric constant in the active site of heme proteins modulates the fate of the ferric sulfide complex, with a polar environment favoring heme iron reduction and simultaneous sulfide oxidation (Pietri et al., 2009).
RSS can also be formed in solution via reaction of H2S with one- and two-electron oxidants (Carballal et al., 2011; Das et al., 1999). The rate constants for the reaction of sulfide with one-electron oxidants vary in magnitude from being relatively low as with superoxide (200 M−1s−1), to almost diffusion controlled, as with the carbonate (2 ×108 M−1s−1) or hydroxyl (1.5×1010 M−1s−1) radicals. The rate constants for its reaction with two electron oxidants also exhibit a wide range, with H2O2 at the low end (0.73 M−1s−1), peroxynitrite (4.8 × 103 M−1s−1) and sulfenic acid (1 × 105 M−1s−1) in the middle, and hypochlorite at the high end (8 × 107 M−1s−1). The products of these reactions are described in Table 1 (equations 1–7).
Table 1.
Rate constants for reactions of H2S with one- and two-electron oxidants.
| O2•− + H2S + H+ → H2O2 + HS• | 200 M−1s−1 | [1] |
| •OH + H2S → H2O + HS• | 1.5 × 1010 M−1s−1 | [2] |
| CO3•− + H2S → HS• +CO32− + H+ | 2 × 108 M−1s−1 | [3] |
| H2O2+ H2S → HSOH + H2O | 0.73 M−1s−1 | [4] |
| ONOOH + H2S → HSOH + HNO2 | 4.8 × 103 M−1s−1 | [5] |
| HSOH + H2S → HSSH + H2O | 1 × 105 M−1s−1 | [6] |
| HOCl + H2S → HSCl + H2O | 8 × 107 M−1s−1 | [7] |
The thiyl radical produced from the one-electron oxidation of sulfide (equations 1–3) can dimerize with a second HS• forming hydrodisulfide (HSSH, 4 × 109 M−1s−1), or react with O2 forming a highly reactive sulfur dioxide radical anion (SO2•−, 5 × 109 M−1s−1), or react with sulfide forming the disulfanyl radical anion (HSS•2−, 4 × 109 M−1s−1) (Das et al., 1999). Sulfenic acid is produced from the two-electron oxidation of sulfide (equations 4, 5) and can itself react with H2S forming hydrodisulfide (equation 6). The products of H2S oxidation are reactive species and can generate a complex mixture of products particularly under aerobic conditions, including elemental sulfur (S8) (Carballal et al., 2011).
The mitochondrial sulfide oxidation pathway
The committing step in the sulfide oxidation pathway, which converts H2S to thiosulfate or sulfate as end products, is catalyzed by sulfide quinone oxidoreductase (SQOR) (Figure 1d). The latter transfers electrons to coenzyme Q (CoQ) via an FAD cofactor as it oxidizes sulfide to sulfane sulfur that is transferred to a thiophilic acceptor (Hildebrandt and Grieshaber, 2008; Landry, Ballou, and Banerjee, 2017; Jackson, Melideo, and Jorns, 2012). Human SQOR has a unique redox site comprising a cysteine trisulfide, which is estimated to confer an ~105-fold rate enhancement for sulfide addition compared to a cysteine disulfide. The cysteine trisulfide thus accounts for much of the 2×107-fold rate acceleration for sulfide addition to SQOR versus a cysteine disulfide in solution (Landry et al., 2019; Landry et al., 2020; Cuevasanta et al., 2015). SQOR, like CBS and CSE, exhibits considerable substrate promiscuity (Banerjee, 2017) and can transfer sulfane sulfur from an active site bound persulfide to a variety of acceptors including sulfite, methanethiol, CoA and GSH (Jackson, Melideo, and Jorns, 2012; Mishanina et al., 2015; Landry, Ballou, and Banerjee, 2018). GSH is predicted to be the physiologically relevant acceptor and forms glutathione persulfide (GSSH) (Landry, Ballou, and Banerjee, 2017; Libiad et al., 2014). SQOR-derived CoA persulfide inhibits butyrate oxidation, which could be important for prioritizing sulfide clearance over fuel utilization in colonocytes during an acute H2S exposure (Landry et al., 2019).
Since CoQ is the electron acceptor for the SQOR reaction, the electrons released from H2S oxidation enter the ETC at the level of complex III (Figure 1d). The remainder of the sulfide oxidation pathway involves dioxygenation of GSSH to sulfite catalyzed by ETHE1, a sulfur transfer reaction catalyzed by rhodanese (or TST), forming thiosulfate, and sulfite oxidation to sulfate catalyzed by sulfite oxidase. Electrons from sulfite oxidation enter the ETC at the level of complex IV via cytochrome c. The sulfide oxidation pathway enzymes, SQOR, ETHE1 and TST in addition to the sulfur transferase TSTD1 (discussed later), exhibit a marked apical localization in colonic crypts (Libiad et al., 2019; Libiad et al., 2018). This location at the host-microbiome interface is likely to be functionally important for protecting colonocytes from high H2S exposure derived from microbial metabolism.
H2S targets mitochondrial bioenergetics and remodels the redox metabolome
Of the various purported mechanisms by which H2S modulates physiology, ETC inhibition at the level of complex IV, which is lethal if sustained, is the most rigorously established mechanism of its action (Nicholls and Kim, 1982). In evaluating the cellular rationale for H2S biogenesis, it is therefore instructive to consider the potential metabolic and signaling consequences of inhibiting the ETC, a central nerve in the cellular bioenergetic network. In quiescent differentiated cells, oxidative phosphorylation is important for maximizing ATP yield from limiting nutrients (Ward and Thompson, 2012). In contrast, proliferating cells cued by growth factors to increase nutrient uptake, decrease flux through oxidative phosphorylation, divert TCA cycle intermediates for biomass synthesis, and utilize aerobic glycolysis to meet ATP needs. These metabolic alterations are also germane to proliferation of malignant cells with the exception that they are independent of growth factor instruction. The shift in flux through the ETC is correlated with metabolic remodeling that balances the cellular demands for ATP, reduced carbon, reduced nitrogen and NADPH (Ward and Thompson, 2012). The metabolic vulnerability and response to H2S will therefore be influenced by whether cells are quiescent versus proliferating as well as whether the exposure is acute versus chronic.
Since H2S oxidation requires CoQ, it has the potential to perturb the major redox nodes that converge at the ETC including NAD+/NADH, FAD/FADH2 and CoQ/COQH2 causing an insufficiency of electron acceptors, which are needed for nutrient oxidation to support cell proliferation (Figure 2). Additionally, through its action on the ETC, H2S can affect the proton motive force, which can be monitored by the membrane potential (ΔΨ) and ATP levels. How these H2S-induced perturbations in mitochondrial bioenergetics might fan out through the metabolic network is discussed next.
Figure 2. H2S interactions with the ETC.
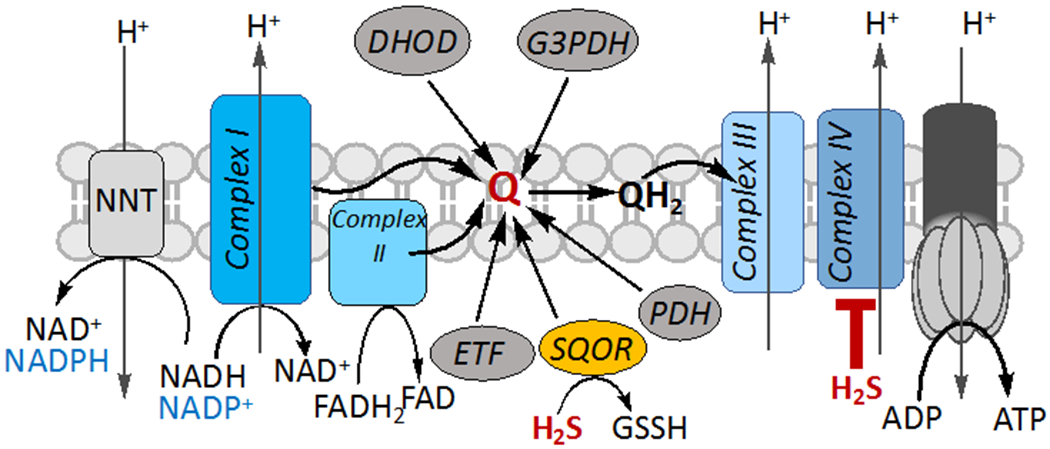
Oxidation of H2S by SQOR requires CoQ (Q), which is also used as an electron acceptor by other enzymes and by complexes I and II. At higher concentrations, H2S inhibits the ETC by targeting complex IV. H2S oxidation limits the oxidized CoQ pool which together with complex IV inhibition is predicted to lead to a reductive shift in the NAD+/NADH, FAD/FADH2 and CoQ/CoQH2 redox couples, affect the proton motive force and decrease ATP. Under these conditions, the NADPH concentration could also increase via the activity of the nucleotide transhydrogenase (NNT). DHOD, G3PDH, ETF and PDH denote dihydroorotate dehydrogenase, glycerol 3-phosphate dehydrogenase, electron transfer flavoprotein and proline dehydrogenase, respectively.
The free NAD+/NADH ratio is estimated to range from ~500-1000 in the cytosol and 5-10 in the mitochondrion (Williamson, Lund, and Krebs, 1967). The reductive shift in the NAD+/NADH ratio induced by H2S is exacerbated in SQOR knockdown cells due to their greater sensitivity to complex IV inhibition (Libiad et al., 2019). The free NADP+/NADPH ratio is estimated to be ~0.01 in the cytosol, i.e. ~105-fold lower than the corresponding value for free NAD+/NADH (Veech, Eggleston, and Krebs, 1969). Since the mitochondrial NADH and NADPH pools are linked via the proton-translocating nucleotide transhydrogenase (NNT) (Figure 2), an H2S-mediated increase in the NADH pool could potentially increase the NADPH pool and inhibit or activate enzymes that use the oxidized or reduced form, respectively of either pyridine nucleotide cofactor.
The malate aspartate shuttle represents a metabolic avenue for maintaining the steep difference in the cytoplasmic versus mitochondrial NAD+/NADH ratio. This shuttle uses malate to piggyback electron equivalents across the mitochondrial inner membrane using the electroneutral and reversible α-ketoglutarate/malate carrier (Figure 3a). The electrogenic aspartate/glutamate carrier couples the efflux of aspartate from the mitochondrion to the influx of glutamate and is driven by the membrane potential. While the role of this shuttle in communicating redox changes elicited by H2S in the mitochondrion to the cytoplasm is not known, the total malate, aspartate, glutamate and α-ketoglutarate concentrations are lower in H2S-treated versus control cells (Libiad et al., 2019).
Figure 3. Metabolic pathways that are potentially affected by H2S.
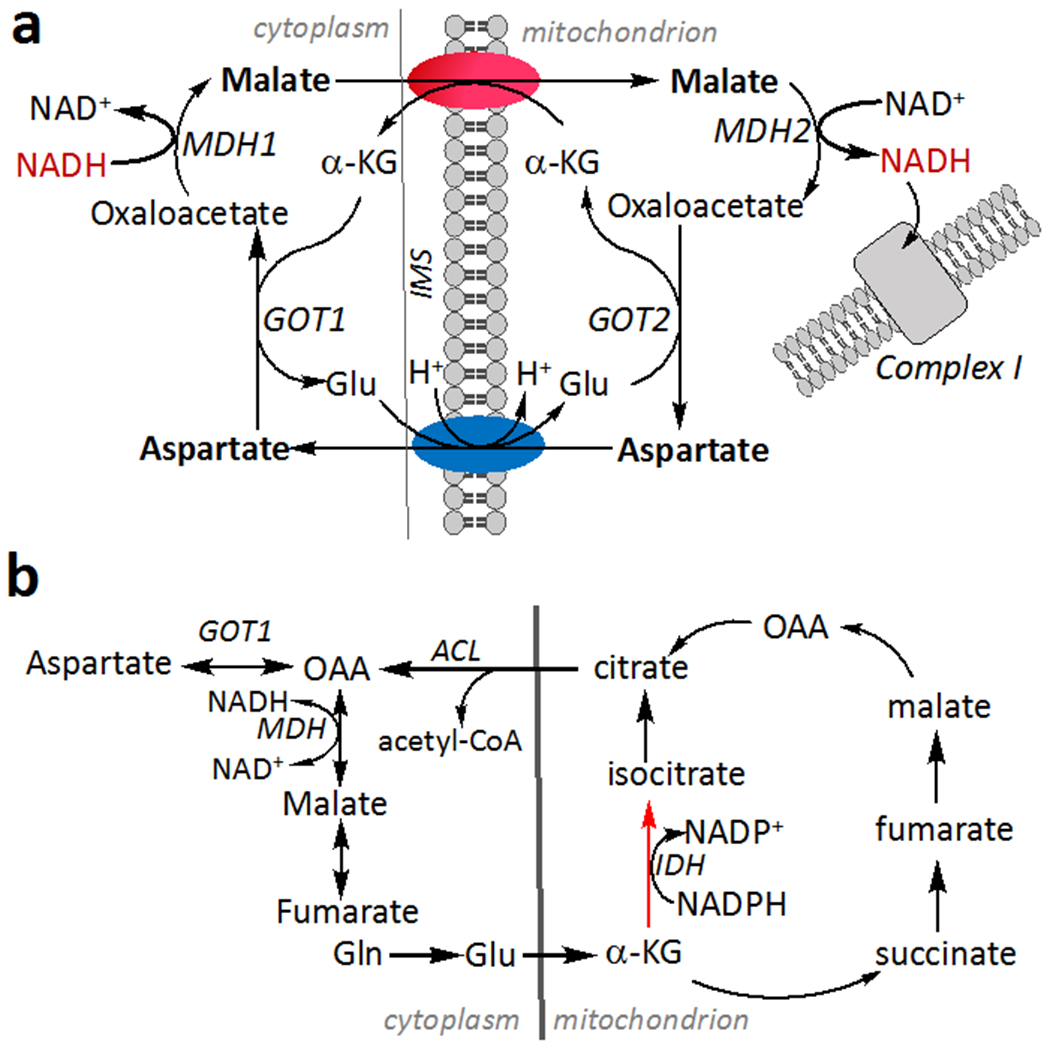
(a) The malate aspartate shuttle transfers NADH equivalents as malate, which crosses the mitochondrial inner membrane via a reversible α-ketoglutarate/malate carrier. Aspartate generated in the mitochondrion is exchanged for glutamate by an electrogenic carrier and converted to malate via the activity of cytosolic glutamate oxoglutarate transaminase (GOT1). (b) H2S stimulates reductive carboxylation (red arrow), which is catalyzed by isocitrate dehydrogenase (IDH). Cleavage of citrate by acetyl-CoA lyase (ACL) provides an alternative source of cytosolic oxaloacetate (OAA), which is converted by GOT1 to aspartate. IMS is inter mitochondrial membrane space.
The NAD(P)ome, which refers to the collection of gene products that utilize NAD(P)+ as a substrate/cofactor or are involved in its biogenesis, is sizeable (Goodman, Calvo, and Mootha, 2018). It is estimated to number 426 proteins of which less than half are localized in the cytosol and mitochondria (Goodman, Calvo, and Mootha, 2018). In addition to enzymes that use pyridine nucleotides as cofactors, NAD+ also serves as a substrate for protein and nucleic acid modifications that are catalyzed by sirtuins and poly(ADP-ribose) polymerases, respectively (Bai and Canto, 2012; Imai and Guarente, 2014). An additional consequence of an H2S-induced reductive shift could therefore be on enzymes that use pyridine nucleotides as substrates with the magnitude of change in the NAD(P)+ or NAD(P)H concentration relative to the KM/KD values of individual enzymes, determining the impact on enzymatic function.
Glutamine-dependent reductive metabolism becomes a major route for generating macromolecular precursors when mitochondrial oxidative metabolism is impaired (Mullen et al., 2012). H2S induced mitochondrial reprogramming stimulates reductive carboxylation of α-ketoglutarate (Figure 3b), which was tracked by mass isotopomer analysis of citrate labeled from [U-13C]-glutamine (Libiad et al., 2019). Metabolomic changes in central carbon metabolism induced by H2S largely map to enzymes that utilize redox cofactors. For example, metabolites that accumulate in H2S-treated HT29 cells are substrates (glyceraldehyde 3-phosphate) of NAD+-or products of NAD(P)H-dependent (lactate, isocitrate/citrate) or FADH2-dependent (succinate) enzymes (Libiad et al., 2019). Chronic H2S exposure restricts cell proliferation, which is partially rescued by uridine or pyruvate supplementation and fully rescued when the metabolites are added together (Libiad et al., 2019). Overreduction of the CoQ pool in the presence of H2S is expected to decrease the activity of dihydrouridine dehydrogenase, limiting uridine availability (Figure 2). Pyruvate, on the other hand serves as an electron acceptor and is reduced to lactate, regenerating NAD+ in a reaction catalyzed by lactate dehydrogenase. Interestingly, uridine and aspartate also alleviate the antiproliferative effect of H2S, which is explained by aspartate becoming growth limiting in some cells facing an electron acceptor insufficiency (Birsoy et al., 2015; Sullivan et al., 2015). Aspartate is more oxidized than the nutrients from which it is derived (i.e., glucose or glutamine) and is needed for nucleotide and protein synthesis. Under conditions of electron acceptor insufficiency, aspartate is primarily derived from glutamine rather than glucose (Figure 3a,b). The flux through the reverse isocitrate dehydrogenase and the malate aspartate shuttle is apparently insufficient however, and limits proliferation (Birsoy et al., 2015; Sullivan et al., 2015).
In cardiomyocytes, H2S suppresses β-adrenergic stimulation-induced hypertrophy and is associated with transcriptomic changes that converge on NAD(P)H homeostasis, affecting the steroid/isoprenoid, pentose phosphate and glutathione pathways (Chhabra et al., 2018). The decreased NADP+/NADPH ratio induced by H2S was connected to decreased expression of p53, which inhibits formation of active glucose 6-phosphate dehydrogenase dimers. Based on these results, the authors concluded that H2S plays an important role in regulating the NADPH-linked redox metabolic state (Chhabra et al., 2018). A connection between the reductive shift in the NADP+/NADPH ratio and the mitochondrial NAD+/NADH ratio due to ETC dysfunction was not explored in this study. It is likely that the specific pathways impacted by H2S-dependent metabolic reprogramming vary to some extent in a cell type and tissue specific manner.
Dual effects of H2S on mitochondrial oxygen consumption
In numerous studies, the effects of H2S on the kinetics of oxygen consumption by isolated mitochondria or in intact cells have been characterized using the Seahorse Analyzer. Due to the volatility of H2S and the open well format of this experimental setup as well as the time needed for injections of ETC inhibitors into wells, this system does not provide reliable data. In contrast, the traditional Clark electrode or the Oroboros respirometer in which the chamber can be filled with sample and is largely sealed from the atmosphere, provide reproducible data. The dual effects of H2S on electron flux through the ETC, which is increased via the action of SQOR but decreased via inhibition of complex IV, are differentially sensitive to H2S concentration. Thus, at lower concentrations, sulfide stimulates the oxygen consumption rate (OCR) but inhibits the same at higher concentrations (Lagoutte et al., 2010), indicating that SQOR has a higher affinity than complex IV for H2S. The relative concentrations of complex IV and SQOR, which is likely to be cell-type dependent, should impact the threshold concentration at which H2S switches from activating to inhibiting OCR. SQOR knockdown renders cells very sensitive to ETC poisoning (Libiad et al., 2019), while its overexpression increases resistance to sulfide toxicity (Lagoutte et al., 2010), highlighting the important function of this enzyme as a sulfide shield.
Low concentrations of sulfide in the presence of oligomycin, which inhibits the FI/Fo ATP synthase, transiently increase OCR and lead to membrane energization, as expected (Goubern et al., 2007). At concentrations of H2S that inhibit complex IV, an increase in succinate concomitant with a decrease in malate is observed, which led to the proposal that complex II reversal occurs under these conditions (Goubern et al., 2007). In a later study, the same authors proposed that H2S induces reverse electron transfer through complex I, albeit only in colonic (HT29) but not in other (CHO, THP1) cell lines (Lagoutte et al., 2010). However, neither model was experimentally evaluated.
In a more recent study, the effects of an H2S donor (i.e. increased OCR and decreased ATP in oligomycin-treated murine microglia) were interpreted as evidence of H2S-induced mitochondrial uncoupling and reverse electron transfer (Jia et al., 2020). An over-reduced CoQ pool and a high mitochondrial membrane potential (ΔΨ) are known drivers of reverse electron transfer through complex I and lead to a transient increase in ROS. The study on murine microglia (Jia et al., 2020) reported variable changes in ΔΨ in response to the H2S donor, i.e. a relatively short-lived increase (10 min) followed by a sustained decrease (30 min) while ROS production was prolonged (24 h). These discrepancies between the kinetics of membrane potential changes and ROS generation raise questions about the validity of the proposed model. Furthermore, the reported magnitude of changes elicited by the H2S donor ADT-OH ([5-(4-hydroxyphenyl)-3H-1, 2-dithiole-3-thione]) in the CoQ/CoQH2, NAD+/NADH and ATP pools, were significantly greater than is typically seen during metabolic reprogramming. It is unclear whether the donor scaffold itself contributed to the observed effects. Finally, the prolonged stimulation of ROS, which is deleterious, is inconsistent with the cytoprotective effects of H2S when administered concomitantly with reperfusion (Elrod et al., 2007), and the absence of H2S-induced changes in ROS levels reported in other studies (Chhabra et al., 2018).
Persulfidation protects the thiol proteome against hyperoxidation
The beneficial effects of H2S have been described in an array of physiological processes including angiogenesis, attenuation of myocardial ischemia reperfusion injury and anti-inflammatory responses (Elrod et al., 2007; Papapetropoulos et al., 2009; Whiteman et al., 2010). While targeted cysteine persulfidation is commonly invoked to explain the physiological effects of H2S (Mustafa et al., 2009), the lack of known mechanisms for achieving target specificity is superficially dealt with in the literature despite the obvious questions that it raises about whether it can function in specific signaling pathways. It is critical to weigh factors such as H2S concentration, reactivity and reaction specificity to assess the possible relevance of persulfidation in specific signaling pathways versus the more plausible role for persulfidation as a general strategy for conferring global protection of the thiol proteome when it is under siege due to oxidative stress.
The versatility of sulfur in biology derives from the availability of its full valence range, extending from −2 in H2S to +6 in sulfate. This, in turn, translates into a range of sulfur chemotypes that can oxidize, reduce or otherwise react with other biomolecules (Mishanina, Libiad, and Banerjee, 2015). For uncatalyzed protein persulfidation to occur, either sulfide or the cysteine thiol must first be oxidized (Figure 4a). The intrinsic reactivity (i.e. the pH independent rate) of H2S with H2O2 is 25-fold lower than of free cysteine, due to the twin effects of the absence of an inductive effect in sulfide and the lower Brønsted basicity of sulfide versus cysteine thiol (Carballal et al., 2011), a difference that could be further exacerbated in a protein environment. Cysteine disulfide or mixed disulfides represent other forms of oxidized protein thiols but their concentrations are generally low in the reducing cytoplasmic milieu. This is however, not the case in the endoplasmic reticulum and the mitochondrial intermembrane space, where an oxidizing microenvironment is maintained to support oxidative protein folding (Herrmann and Kohl, 2007; Hwang, Sinskey, and Lodish, 1992). In these compartments, protein persulfidation could potentially occur in the absence of oxidative stress (Figure 4b). However, the rate constant for the addition of sulfide into mixed disulfides is low, on the order of 1-10 M−1 s−1 (Cuevasanta et al., 2015).
Figure 4. Mechanisms of protein persulfidation.
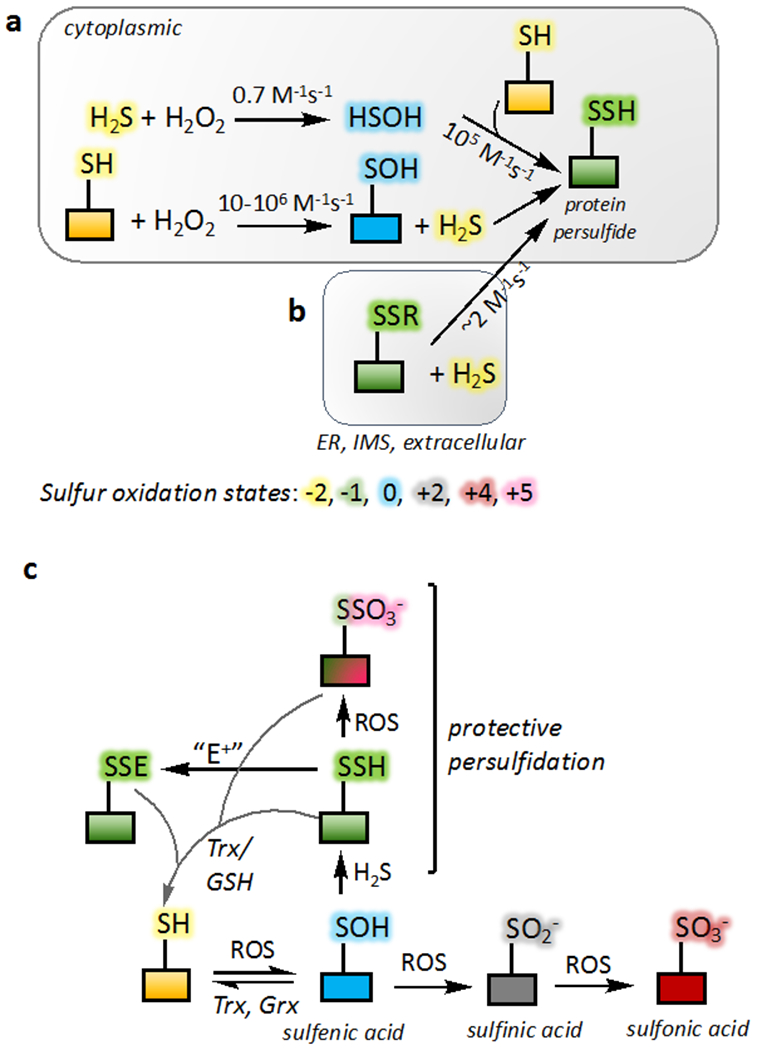
(a) Protein persulfidation requires an initial oxidation of the protein thiol or H2S. The bimolecular rate constant for the reaction of HSOH with free (105 M−1s−1) versus protein thiols is noted. (b) In oxidizing compartments within the cell such as the endoplasmic reticulum (ER) and the intermitochondrial membrane space (IMS), H2S can add into protein disulfides. The rate constant shown is for the reaction of H2S with a mixed disulfide between human serum albumin and GSH (Cuevasanta et al., 2015). (c) The irreversibility of some oxidized thiol modifications (sulfinic and sulfonic acid) can be averted via protein persulfidation at the level of sulfenic acid. Similarly, persulfide modification by an electrophile (E+) is reversible unlike modification of a thiol. The products can be reduced back to the thiol form by a disulfide reductases like thioredoxin (Trx) and glutaredoxin (Grx) or by GSH.
The thiol proteome is a quantitatively significant redox active pool and, like GSH, functions as a redox buffer (Hansen, Roth, and Winther, 2009; Thomas, Poland, and Honzatko, 1995). The concentration of accessible thiols in the proteome is estimated to be of the same order of magnitude as the concentration of GSH in mammalian cells (i.e. ~10 mM) (Hansen, Roth, and Winther, 2009) and vastly greater than of H2S, which is estimated to be ~10-30 nM (Furne, Saeed, and Levitt, 2008; Vitvitsky, Kabil, and Banerjee, 2012). Given this steep difference in concentrations and the low reactivity of H2S with H2O2, the oxidation of protein thiols rather than H2S is a more likely mechanism for priming proteins for persulfidation (Figure 4a).
Persulfide proteomics in basal cell culture conditions have revealed a large number of targets, which further increase upon exposure to exogenous H2S (Gao et al., 2015; Libiad et al., 2019; Fu et al., 2019). While these approaches furnish data on fold changes in persulfidation, technical challenges have precluded the quantitative assessment of how much of a given protein is modified in a cellular setting, thus limiting assessment of its physiological relevance. The sheer number of proteins that are persulfidated (~800 in MIN6 cells (Gao et al., 2015)) suggests that this modification represents a stochastic marking of the proteome that is determined by a combination of factors such as protein concentration, cysteine reactivity and proximity to locations where H2S is generated.
Oxidative cysteine modification, i.e., protein S-thiolation is an early stress response to oxidative challenge. This is a dynamic process in which the oxidative markings on the thiol proteome are reversed by repair systems such as thioredoxin, GSH and glutaredoxin (Figure 4c). The reactivity of protein thiols span a wide range (1-107 M−1s−1), which could underlie selectivity and explain the greater susceptibility of some targets versus others (D’Autreaux and Toledano, 2007). The initial product of cysteine thiol oxidation by ROS is sulfenic acid, which can be reduced back to the thiol state or further oxidized to sulfinic and sulfonic acid (Figure 4c). The latter two cysteine oxoforms are not reduced by the major cellular reductants, GSH and thioredoxin, and in fact, the sulfonic acid modification is irreversible. Sulfinylated peroxiredoxin can be repaired by a dedicated enzyme, sulfiredoxin (Biteau, Labarre, and Toledano, 2003).
Protein persulfides, like protein thiols, can also undergo ROS-mediated oxidation forming the corresponding perthiosulfenic, perthiosulfinic and perthiosulfonic acid, respectively (Heppner et al., 2018). Furthermore, both thiols and persulfides can add to electrophiles, with persulfides being more reactive. However, unlike thiols, chemical modifications on cysteine persulfides are reversible by virtue of the intervening disulfide linkage (Cys-S-S-R), which can be reduced (Figure 4c). This access to reversible modification supports the purported potential for persulfidation being a cellular strategy for protecting against hyperoxidation. Protein perthiosulfonic acid and persulfide levels are reportedly comparable in murine liver samples (Doka et al., 2020).
Decreased expression of CSE or the pharmacological inhibition of CSE, CBS or the cystine transporter system xc-decrease cellular persulfidation levels while overexpression of CSE or activating MPST-dependent H2S production, increase global persulfidation (Zivanovic et al., 2020). Growth factor (epidermal growth factor or vascular endothelium-derived growth factor) and insulin-induced increases in protein persulfidation are preceded by increased protein sulfenylation, consistent with cysteine sulfenylation priming proteins for subsequent persulfidation (Figure 4c) (Zivanovic et al., 2020). It is unclear however, how H2S synthesis increased under these conditions, especially in the short (30 min) treatment durations. Global persulfidation appears to decrease with age and is loosely correlated with decreases in H2S producing enzymes in some but not all tissues where this phenomenon was examined (Zivanovic et al., 2020).
If protective persulfidation does indeed serve an antioxidant role, it raises the question as to how H2S availability via increased production and/or decreased clearance is enhanced under oxidative stress conditions to promote this posttranslational modification. The mechanisms underlying coordination of H2O2 and H2S biogenesis in response to growth factors or other inducers of signaling pathways also remain to be systematically described in addition to the kinetics of cysteine sulfenate persulfidation versus further oxidation to sulfinic acid. Additionally, the role of RSS, i.e., a downstream product of H2S oxidation, rather than H2S per se, in persulfidation needs to be evaluated since it can occur independently of a priming sulfenylation reaction (equations 8 versus 9 and 10). The outer sulfur of persulfides have dual electrophilic and nucleophilic character (Benchoam et al., 2020). The pKa value of the H2S oxidation product GSSH (5.45) (Benchoam et al., 2020), predicts that it is largely present in the anionic (GSS−) form, which would disfavor electrophilic addition.
| [8] |
| [9] |
| [10] |
The inherent reactivity of persulfides would potentially increase the vulnerability of persulfidated proteins to crosslinking and lead to aggregation in the crowded intracellular milieu. This complication needs to be evaluated when invoking protein persulfidation as a signaling mechanism. Cellular strategies to protect against such an unregulated increase in proteomic complexity are needed and a general mechanism would be to maintain low steady-state H2S levels, and consequently, reduced global persulfidation levels. High turnover of H2S is maintained by its efficient removal by the mitochondrial sulfide oxidation pathway (Vitvitsky, Kabil, and Banerjee, 2012).
Sulfur transferases as potential mediators of protein persulfidation
Sulfur transferases are ubiquitous enzymes that catalyze sulfane sulfur production, although their specific physiological roles are largely unknown. Some sulfur transferases are involved in sulfide (Libiad et al., 2014; Libiad et al., 2018) and selenium (Lima, 2002) metabolism, iron-sulfur cluster biogenesis (Braymer and Lill, 2017), and cyanide detoxification (Leininger and Westley, 1968). Many sulfur transferases like rhodanese/TST (Ploegman et al., 1978), MPST (Yadav et al., 2013) and the bacterial persulfide dioxygenase-rhodanese fusion protein (Motl et al., 2017) stabilize cysteine persulfides in their active sites, which has led to their crystallization in this form (Figure 5a,b).
Figure 5. Structures and reactions catalyzed by three rhodanese domain containing sulfur transferases found in humans.
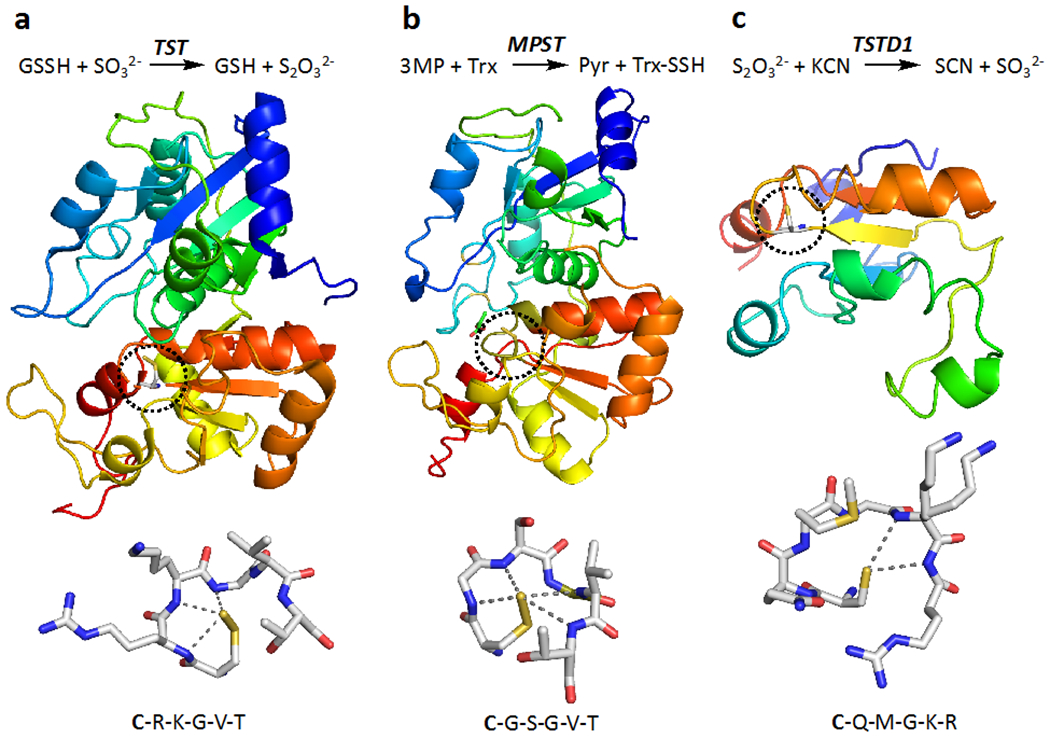
(a, b, c) While each of the sulfur transferases catalyzes multiple reactions, the reaction reported to exhibit the highest kcat/KM is shown in the top panel for rhodanese (or TST), MPST and TSTD1. In the middle panel, the structures of bovine TST (PDB: 1RHD), human MPST (PDB: 4JGT) and human TSTD1 (PDB: 6BEV) are shown and reveal that the first two have a tandem repeat of rhodanese domains while TSTD1 has a single rhodanese domain. The dotted circles highlight the active site cysteine and each protein is shown in rainbow hues ranging from blue (N-terminus) to red (C-terminus). The blue-green and yellow-orange colors depict the two domains in TST and MPST. In the lower panel, close-ups of the active site cysteine are shown. In TST and MPST, the cysteine was captured in the persufide form. The cysteine in TSTD1 is surface exposed and might serve as a persulfide donor to larger (protein) acceptor. The sequence adjacent to the active site cysteine (bold lettering), which is believed to confer substrate specificity to each protein, is shown below.
In humans, there are three sulfur transferases which belong to the rhodanese superfamily (Cipollone, Ascenzi, and Visca, 2007). They include the mitochondrial rhodanese, two isoforms of MPST found in the cytoplasm (MPST1) and mitochondrion (MPST2), respectively and three isoforms of the cytoplasmic thiosulfate sulfur transferase-like domain containing protein (TSTD-1, −2 and −3). Of these, rhodanese/TST and MPST harbor active sites that are located between a tandem repeat of rhodanese domains (Figure 5a,b). The preferred sulfur donors in the rhodanese and MPST-catalyzed reactions are GSSH (Libiad, Sriraman, and Banerjee, 2015) and 3-mercaptopyruvate (Yadav et al., 2013), respectively. TSTD1 is a single rhodanese domain protein with a relatively exposed active site (Figure 5c), which does not stabilize a persulfide intermediate (Libiad et al., 2018). TSTD3, lacks an active site cysteine, is not expected to be active as a sulfur transferase, while the function of TSTD2 is unknown. Due to its relatively accessible active site, it has been speculated that TSTD1 might play a role in persulfidation of proteins although its cellular sulfur donor(s) and acceptor(s) remain to be identified (Libiad et al., 2018). The residues immediately downstream of the active site cysteine, which is persulfidated in the catalytic cycle, reside on a loop and confer substrate specificity to respective sulfur transferases (Figure 5).
Clinical data place important constraints on interpreting the physiological roles of enzymes in an organismal context. MPST deficiency is inherited as an autosomal recessive disorder and associated with mercaptolactate-cysteine disulfiduria (Crawhall et al., 1969) that is also seen in mpst−/− mice, which are not otherwise overtly different from wild-type mice (Akahoshi et al., 2020). Inherited disorders of rhodanese and TSTD1/2 on the other hand, have not been reported. Interestingly in mice, elevated expression of rhodanese in adipocytes is correlated with leanness and indices of metabolic health (Morton et al., 2016). In humans, rhodanese expression correlates negatively with fat mass and positively with insulin sensitivity in adipose tissue (Morton et al., 2016).
Although not generally thought of as a sulfur transferase, SQOR catalyzes a sulfur transfer reaction from H2S forming a persulfide product as it reduces CoQ to CoQH2 (Figure 6a) (Landry, Ballou, and Banerjee, 2020). Unlike the sulfur transferases discussed above, SQOR belongs to the flavin disulfide reductase rather than the rhodanese superfamily and is structurally distinct (Figure 6b). Addition of sulfide to the active site trisulfide in SQOR results in the formation of a pair of persulfides at Cys-201 and at Cys-379. The latter serves as the sulfur donor to thiophilic acceptors such as GSH forming GSSH (Figure 6a).
Figure 6. Structure and mechanism of human SQOR, a sulfurtransferase (PDB: 6OI6).
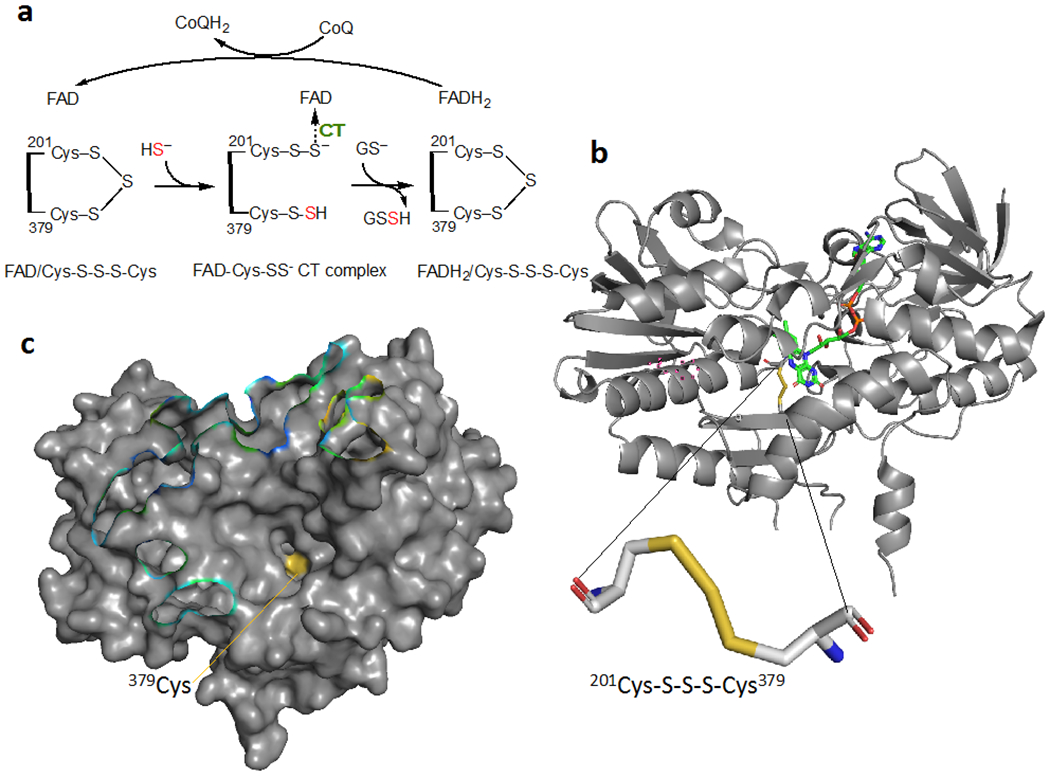
(a) Reaction mechanism of SQR. CT denotes charge transfer. (b) A unique trisulfide between Cys-201 and Cys-379 represents the redox-active motif in the active site of human SQOR. The FAD cofactor is shown in green. (c) Surface representation of SQOR reveals a large cavity leading to the active site in which Cys-379 (yellow) that transfers the sulfane sulfur to an external acceptor, is relatively surface exposed.
The surface accessibility of Cys-379, which is located at the bottom of a large solvent-exposed cavity (Figure 6c), raises the possibility that SQOR transfers sulfane sulfur to proteins in addition to the small molecule acceptors (e.g. GSH, CoA, methanethiol) that have been characterized (Landry et al., 2019; Landry, Ballou, and Banerjee, 2018). To our knowledge, thioredoxin is the only protein acceptor that has been tested with human SQOR, but did not show activity (Libiad et al., 2014).
Conclusions and Future Perspectives
Despite the profusion of literature on the many cellular and physiological effects of H2S, there is a paucity of rigorous studies to illuminate our understanding of these phenomena. Structural enzymology studies on H2S biogenesis, and on H2S oxidation by enzymes in the mitochondrial sulfide oxidation pathway and by globins are providing the kinetic parameters and mechanistic insights needed to assess the contributions of these reactions in physiological contexts. In contrast, insights into how these reactions are regulated and respond to cellular queues for dialing H2S levels up or down are poorly understood but are critical to understanding the cellular settings that tap into H2S for mediating a response. The challenges to clarity and consistency in the field are manifold. They begin with the basic problems with handling a volatile and redox active metabolite and are amplified by the plethora of H2S “donors”, persulfide derivatives and other RSS that are introduced in cell culture medium in various laboratories. The stability of these compounds, their intracellular and/or extracellular (i.e., in the culture medium) H2S release kinetics, and their metabolic fates are seldom rigorously characterized, contributing to the variability in and irreproducibility of some of the published data. Studies in which highly reactive and unstable compounds are added to culture medium at nonphysiological concentrations raise additional questions about relevance. Compounding these challenges, are the technical limitations of methodologies that are widely used to detect H2S including the growing use of sensors that are often not well characterized. These limitations, however, also represent opportunities for overcoming the technical challenges and making significant contributions. Thus, the application of chemically rigorous and quantitative approaches in cellular and organismal settings hold the promise for answering fundamentally important questions like why cells generate H2S and under what conditions, how H2S affects the redox proteome and metabolome and to what end. Coupled with biophysical and biochemical approaches to furnish molecular and mechanistic insights, such information could hold translational promise.
Acknowledgements
This work was supported in part by a grant from the National Institutes of Health (GM130183).
Footnotes
Conflict of Interest
RB is a paid member of the scientific advisory board of Apneo Therapeutics and owns equity in the company.
References
- 1.Holmstrom KM, Finkel T. (2014).Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol, 15, 411–21 [DOI] [PubMed] [Google Scholar]
- 2.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR. (2012).Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell, 149, 1060–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansen RE, Roth D, Winther JR. (2009).Quantifying the global cellular thiol-disulfide status. Proc Natl Acad Sci U S A, 106, 422–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reichmann D, Voth W, Jakob U. (2018).Maintaining a Healthy Proteome during Oxidative Stress. Mol Cell, 69, 203–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. (2003).Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature, 423, 773–7 [DOI] [PubMed] [Google Scholar]
- 6.Suzuki T, Yamamoto M. (2017).Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J Biol Chem, 292, 16817–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miseta A, Csutora P. (2000).Relationship Between the Occurrence of Cysteine in Proteins and the Complexity of Organisms. Mol Biol Evol, 17, 1232–39 [DOI] [PubMed] [Google Scholar]
- 8.Go YM, Jones DP. (2013).The redox proteome. J Biol Chem, 288, 26512–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J, Carroll KS, Liebler DC. (2016).The Expanding Landscape of the Thiol Redox Proteome. Mol Cell Proteomics, 15, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao XH, Krokowski D, Guan BJ, Bederman I, Majumder M, Parisien M, Diatchenko L, Kabil O, Willard B, Banerjee R, Wang B, Bebek G, Evans CR, Fox PL, Gerson SL, Hoppel C, Liu M, Arvan P, Hatzoglou M. (2015).Quantitative H2S-mediated protein sulfhydration reveals metabolic reprogramming during the Integrated Stress Response. Elife, 4, e10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kabil O, Banerjee R. (2010).The redox biochemistry of hydrogen sulfide. J Biol Chem, 285, 21903–07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kabil O, Vitvitsky V, Banerjee R. (2014).Sulfur as a signaling nutrient through hydrogen sulfide. Ann Rev Nutr, 34, 171–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kabil O, Banerjee R. (2014).Enzymology of H2S Biogenesis, Decay and Signaling. Antioxid Redox Signal, 20, 770–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filipovic MR, Zivanovic J, Alvarez B, Banerjee R. (2018).Chemical Biology of H2S Signaling through Persulfidation. Chem Rev, 118, 1253–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abe K, Kimura H. (1996).The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci, 16, 1066–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicholls P, Kim JK. (1982).Sulphide as an inhibitor and electron donor for the cytochrome c oxidase system. Can J Biochem, 60, 613–23 [DOI] [PubMed] [Google Scholar]
- 17.Stewart FJ, Cavanaugh CM. (2006).Symbiosis of thioautotrophic bacteria with Riftia pachyptila. Prog Mol Subcell Biol, 41, 197–225 [DOI] [PubMed] [Google Scholar]
- 18.Macfarlane GT, Gibson GR, Cummings JH. (1992).Comparison of fermentation reactions in different regions of the human colon. J Appl Bacteriol, 72, 57–64 [DOI] [PubMed] [Google Scholar]
- 19.Libiad M, Vitvitsky V, Bostelaar T, Bak DW, Lee HJ, Sakamoto N, Fearon E, Lyssiotis CA, Weerapana E, Banerjee R. (2019).Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J Biol Chem, 294, 12077–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banerjee R (2017).Catalytic promiscuity and heme-dependent redox regulation of H2S synthesis. Curr Opin Chem Biol, 37, 115–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh S, Banerjee R. (2011).PLP-dependent H2S biogenesis. Biochim Biophys Acta, 1814, 1518–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiku T, Padovani D, Zhu W, Singh S, Vitvitsky V, Banerjee R. (2009).H2S Biogenesis by Human Cystathionine γ-Lyase Leads to the Novel Sulfur Metabolites Lanthionine and Homolanthionine and Is Responsive to the Grade of Hyperhomocysteinemia. J Biol Chem, 284, 11601–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kabil O, Yadav V, Banerjee R. (2016).Heme-dependent metabolite switching regulates H2S synthesis in response to ER stress. J Biol Chem, 291, 16418–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taoka S, West M, Banerjee R. (1999).Characterization of the heme and pyridoxal phosphate cofactors of human cystathionine beta-synthase reveals nonequivalent active sites. Biochemistry, 38, 2738–44 [DOI] [PubMed] [Google Scholar]
- 25.Taoka S, Banerjee R. (2001).Characterization of NO binding to human cystathionine [beta]-synthase:Possible implications of the effects of CO and NO binding to the human enzyme. J Inorg Biochem, 87, 245–51 [DOI] [PubMed] [Google Scholar]
- 26.Puranik M, Weeks CL, Lahaye D, Kabil O, Taoka S, Nielsen SB, Groves JT, Banerjee R, Spiro TG. (2006).Dynamics of Carbon Monoxide Binding to Cystathionine beta-Synthase. J Biol Chem, 281, 13433–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yadav PK, Yamada K, Chiku T, Koutmos M, Banerjee R. (2013).Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J Biol Chem, 288, 20002–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yadav PK, Vitvitsky V, Carballal S, Seravalli J, Banerjee R. (2020).Thioredoxin regulates human mercaptopyruvate sulfurtransferase at physiologically-relevant concentrations. J Biol Chem, 295, 6299–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yadav PK, Martinov M, Vitvitsky V, Seravalli J, Wedmann R, Filipovic MR, Banerjee R. (2016).Biosynthesis and Reactivity of Cysteine Persulfides in Signaling. J Am Chem Soc, 138, 289–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T, Yamamoto M, Ono K, Devarie-Baez NO, Xian M, Fukuto JM, Akaike T. (2014).Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc Natl Acad Sci U S A, 111, 7606–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kabil O, Vitvitsky V, Xie P, Banerjee R. (2011).The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid Redox Signal, 15, 363–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mishanina TV, Libiad M, Banerjee R. (2015).Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat Chem Biol, 11, 457–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vitvitsky V, Yadav PK, An S, Seravalli J, Cho US, Banerjee R. (2017).Structural and Mechanistic Insights into Hemoglobin-Catalyzed Hydrogen Sulfide Oxidation and the Fate of Polysulfide Products. J Biol Chem, 292, 5584–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vitvitsky V, Yadav PK, Kurthen A, Banerjee R. (2015).Sulfide oxidation by a noncanonical pathway in red blood cells generates thiosulfate and polysulfides. J Biol Chem, 290, 8310–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bostelaar T, Vitvitsky V, Kumutima J, Lewis BE, Yadav PK, Brunold TC, Filipovic M, Lehnert N, Stemmler TL, Banerjee R. (2016).Hydrogen Sulfide Oxidation by Myoglobin. J Am Chem Soc, 138, 8476–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruetz M, Kumutima J, Lewis BE, Filipovic MR, Lehnert N, Stemmler TL, Banerjee R. (2017).A Distal Ligand Mutes the Interaction of Hydrogen Sulfide with Human Neuroglobin. J Biol Chem, 292, 6512–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vitvitsky V, Miljkovic JL, Bostelaar T, Adhikari B, Yadav PK, Steiger AK, Torregrossa R, Pluth MD, Whiteman M, Banerjee R, Filipovic MR. (2018).Cytochrome c Reduction by H2S Potentiates Sulfide Signaling. ACS Chem Biol, 13, 2300–07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palinkas Z, Furtmuller PG, Nagy A, Jakopitsch C, Pirker KF, Magierowski M, Jasnos K, Wallace JL, Obinger C, Nagy P. (2014).Interactions of hydrogen sulfide with myeloperoxidase. Br J Pharmacol, 172, 1516–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagy P, Winterbourn CC. (2010).Rapid Reaction of Hydrogen Sulfide with the Neutrophil Oxidant Hypochlorous Acid to Generate Polysulfides. Chem Res Toxicol, 23, 1541–3 [DOI] [PubMed] [Google Scholar]
- 40.Nelp MT, Zheng V, Davis KM, Stiefel KJE, Groves JT. (2019).Potent Activation of Indoleamine 2,3-Dioxygenase by Polysulfides. J Am Chem Soc, 141, 15288–300 [DOI] [PubMed] [Google Scholar]
- 41.Pietri R, Lewis A, Leon RG, Casabona G, Kiger L, Yeh SR, Fernandez-Alberti S, Marden MC, Cadilla CL, Lopez-Garriga J. (2009).Factors controlling the reactivity of hydrogen sulfide with hemeproteins. Biochemistry, 48, 4881–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carballal S, Trujillo M, Cuevasanta E, Bartesaghi S, Moller MN, Folkes LK, Garcia-Bereguiain MA, Gutierrez-Merino C, Wardman P, Denicola A, Radi R, Alvarez B. (2011).Reactivity of hydrogen sulfide with peroxynitrite and other oxidants of biological interest. Free Radic Biol Med, 50, 196–205 [DOI] [PubMed] [Google Scholar]
- 43.Das TN, Huie RE, Neta P, Padmaja S. (1999).Reduction potential of the sulfhydryl radical: pulse radiolysis and laser flash photolysis studies of the formation andreactions of •SH and HSSH•− in aqueous solutions. J Phys Chem, A 103, 5221–26 [Google Scholar]
- 44.Hildebrandt TM, Grieshaber MK. (2008).Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J, 275, 3352–61 [DOI] [PubMed] [Google Scholar]
- 45.Landry AP, Ballou DP, Banerjee R. (2017).H2S oxidation by nanodisc-embedded human sulfide quinone oxidoreductase. J Biol Chem, 292, 11641–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jackson MR, Melideo SL, Jorns MS. (2012).Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry, 51, 6804–15 [DOI] [PubMed] [Google Scholar]
- 47.Landry AP, Moon S, Kim H, Yadav PK, Guha A, Cho US, Banerjee R. (2019).A Catalytic Trisulfide in Human Sulfide Quinone Oxidoreductase Catalyzes Coenzyme A Persulfide Synthesis and Inhibits Butyrate Oxidation. Cell Chem Biol, 26, 1515–25 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Landry AP, Moon S, Bonanata J, Cho US, Coitino EL, Banerjee R. (2020).Dismantling and Rebuilding the Trisulfide Cofactor Demonstrates Its Essential Role in Human Sulfide Quinone Oxidoreductase. J Am Chem Soc, 142, 14295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cuevasanta E, Lange M, Bonanata J, Coitino EL, Ferrer-Sueta G, Filipovic MR, Alvarez B. (2015).Reaction of hydrogen sulfide with disulfide and sulfenic acid to form the strongly nucleophilic persulfide. J Biol Chem, 290, 26866–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mishanina TV, Yadav PK, Ballou DP, Banerjee R. (2015).Transient Kinetic Analysis of Hydrogen Sulfide Oxidation Catalyzed by Human Sulfide Quinone Oxidoreductase. J Biol Chem, 290, 25072–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Landry AP, Ballou DP, Banerjee R. (2018).Modulation of Catalytic Promiscuity during Hydrogen Sulfide Oxidation. ACS Chem Biol, 13, 1651–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Libiad M, Yadav PK, Vitvitsky V, Martinov M, Banerjee R. (2014).Organization of the human mitochondrial sulfide oxidation pathway. J Biol Chem, 289, 30901–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Libiad M, Motl N, Akey DL, Sakamoto N, Fearon ER, Smith JL, Banerjee R. (2018).Thiosulfate sulfurtransferase-like domain-containing 1 protein interacts with thioredoxin. J Biol Chem, 293, 2675–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ward PS, Thompson CB. (2012).Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell, 21, 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Williamson DH, Lund P, Krebs HA. (1967).The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J, 103, 514–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Veech RL, Eggleston LV, Krebs HA. (1969).The redox state of free nicotinamide-adenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem J, 115, 609–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goodman RP, Calvo SE, Mootha VK. (2018).Spatiotemporal compartmentalization of hepatic NADH and NADPH metabolism. J Biol Chem, 293, 7508–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bai P, Canto C. (2012).The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab, 16, 290–5 [DOI] [PubMed] [Google Scholar]
- 59.Imai S, Guarente L. (2014).NAD+ and sirtuins in aging and disease. Trends Cell Biol, 24, 464–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. (2012).Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature, 481, 385–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. (2015).An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell, 162, 540–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. (2015).Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell, 162, 552–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chhabra A, Mishra S, Kumar G, Gupta A, Keshri GK, Bharti B, Meena RN, Prabhakar AK, Singh DK, Bhargava K, Sharma M. (2018).Glucose-6-phosphate dehydrogenase is critical for suppression of cardiac hypertrophy by H2S. Cell Death Discov, 4, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lagoutte E, Mimoun S, Andriamihaja M, Chaumontet C, Blachier F, Bouillaud F. (2010).Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta, 1797, 1500–11 [DOI] [PubMed] [Google Scholar]
- 65.Goubern M, Andriamihaja M, Nubel T, Blachier F, Bouillaud F. (2007).Sulfide, the first inorganic substrate for human cells. FASEB J, 21, 1699–706 [DOI] [PubMed] [Google Scholar]
- 66.Jia J, Wang Z, Zhang M, Huang C, Song Y, Xu F, Zhang J, Li J, He M, Li Y, Ao G, Hong C, Cao Y, Chin YE, Hua ZC, Cheng J. (2020).SQR mediates therapeutic effects of H2S by targeting mitochondrial electron transport to induce mitochondrial uncoupling. Sci Adv, 6, eaaz5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, Kimura H, Chow CW, Lefer DJ. (2007).Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A, 104, 15560–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, Jeschke MG, Branski LK, Herndon DN, Wang R, Szabo C. (2009).Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci U S A, 106, 21972–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Whiteman M, Li L, Rose P, Tan CH, Parkinson DB, Moore PK. (2010).The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid Redox Signal, 12, 1147–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. (2009).H2S signals through protein S-sulfhydration. Sci Signal, 2, ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hersrmann JM, Kohl R. (2007).Catch me if you can! Oxidative protein trapping in the intermembrane space of mitochondria. J Cell Biol, 176, 559–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hwang C, Sinskey AJ, Lodish HF. (1992).Oxidized redox state of glutathione in the endoplasmic reticulum. Science, 257, 1496–502 [DOI] [PubMed] [Google Scholar]
- 73.Thomas JA, Poland B, Honzatko R. (1995).Protein sulfhydryls and their role in the antioxidant function of protein S-thiolation. Arch Biochem Biophys, 319, 1–9 [DOI] [PubMed] [Google Scholar]
- 74.Furne J, Saeed A, Levitt MD. (2008).Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am J Physiol Regul Integr Comp Physiol, 295, R1479–85 [DOI] [PubMed] [Google Scholar]
- 75.Vitvitsky V, Kabil O, Banerjee R. (2012).High turnover rates for hydrogen sulfide allow for rapid regulation of its tissue concentrations. Antioxid Red Signal, 17, 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fu L, Liu K, He J, Tian C, Yu X, Yang J. (2019).Direct Proteomic Mapping of Cysteine Persulfidation. Antioxid Redox Signal, 33, 1061–76 [DOI] [PubMed] [Google Scholar]
- 77.D’Autreaux B, Toledano MB. (2007).ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol, 8, 813–24 [DOI] [PubMed] [Google Scholar]
- 78.Biteau B, Labarre J, Toledano MB. (2003).ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature, 425, 980–4 [DOI] [PubMed] [Google Scholar]
- 79.Heppner DE, Hristova M, Ida T, Mijuskovic A, Dustin CM, Bogdandi V, Fukuto JM, Dick TP, Nagy P, Li J, Akaike T, van der Vliet A. (2018).Cysteine perthiosulfenic acid (Cys-SSOH): A novel intermediate in thiol-based redox signaling? Redox Biol, 14, 379–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Doka E, Ida T, Dagnell M, Abiko Y, Luong NC, Balog N, Takata T, Espinosa B, Nishimura A, Cheng Q, Funato Y, Miki H, Fukuto JM, Prigge JR, Schmidt EE, Arner ESJ, Kumagai Y, Akaike T, Nagy P. (2020).Control of protein function through oxidation and reduction of persulfidated states. Sci Adv, 6, eaax8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zivanovic J, Kourous E, Kohl JB, Adhikari B, Bursac B, Schott-Roux S, Petrovic D, Miljkovic JL, Thomas-Lopez D, Jung Y, Miler M, Mitchell S, Milosevic V, Gomes JE, Benhar M, Gonzalez-Zorn B, Ivanovic-Burmazovic I, Torregrossa R, Mitchell JR, Whiteman M, Schwarz G, Snyder SH, Paul BD, Carroll KS, Filipovic MR. (2020).Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab, 30, 1152–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Benchoam D, Cuevasanta E, Moller MN, Alvarez B. (2020).Persulfides, at the crossroads between hydrogen sulfide and thiols. Essays Biochem, 64, 155–68 [DOI] [PubMed] [Google Scholar]
- 83.Benchoam D, Semelak JA, Cuevasanta E, Mastrogiovanni M, Grassano JS, Ferrer-Sueta G, Zeida A, Trujillo M, Moller MN, Estrin DA, Alvarez B. (2020).Acidity and nucleophilic reactivity of glutathione persulfide. J Biol Chem, 295, 15466–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lima CD. (2002).Analysis of the E. coli NifS CsdB protein at 2.0 A reveals the structural basis for perselenide and persulfide intermediate formation. J Mol Biol, 315, 1199–208 [DOI] [PubMed] [Google Scholar]
- 85.Braymer JJ, Lill R. (2017).Iron-sulfur cluster biogenesis and trafficking in mitochondria. J Biol Chem, 292, 12754–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leininger KR, Westley J. (1968).The mechanism of the rhodanese-catalyzed thiosulfate-cyanide reaction. Thermodynamic and activation parameters. J Biol Chem, 243, 1892–9 [PubMed] [Google Scholar]
- 87.Ploegman JH, Drent G, Kalk KH, Hol WG, Heinrikson RL, Keim P, Weng L, Russell J. (1978).The covalent and tertiary structure of bovine liver rhodanese. Nature, 273, 124–9 [DOI] [PubMed] [Google Scholar]
- 88.Motl N, Skiba MA, Kabil O, Smith JL, Banerjee R. (2017).Structural and biochemical analyses indicate that a bacterial persulfide dioxygenase-rhodanese fusion protein functions in sulfur assimilation. J Biol Chem, 292, 14026–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cipollone R, Ascenzi P, Visca P. (2007).Common themes and variations in the rhodanese superfamily. IUBMB Life, 59, 51–9 [DOI] [PubMed] [Google Scholar]
- 90.Libiad M, Sriraman A, Banerjee R. (2015).Polymorphic Variants of Human Rhodanese Exhibit Differences in Thermal Stability and Sulfur Transfer Kinetics. J Biol Chem, 290, 23579–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Crawhall JC, Parker R, Sneddon W, Young EP. (1969).Beta-mercaptolactate-cysteine disulfide in the urine of a mentally retarded patient. Am J Dis Child, 117, 71–82 [DOI] [PubMed] [Google Scholar]
- 92.Akahoshi N, Minakawa T, Miyashita M, Sugiyama U, Saito C, Takemoto R, Honda A, Kamichatani W, Kamata S, Anan Y, Ishii I. (2020).Increased Urinary 3-Mercaptolactate Excretion and Enhanced Passive Systemic Anaphylaxis in Mice Lacking Mercaptopyruvate Sulfurtransferase, a Model of Mercaptolactate-Cysteine Disulfiduria. Int J Mol Sci, 21, 818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morton NM, Beltram J, Carter RN, Michailidou Z, Gorjanc G, McFadden C, Barrios-Llerena ME, Rodriguez-Cuenca S, Gibbins MT, Aird RE, Moreno-Navarrete JM, Munger SC, Svenson KL, Gastaldello A, Ramage L, Naredo G, Zeyda M, Wang ZV, Howie AF, Saari A, Sipila P, Stulnig TM, Gudnason V, Kenyon CJ, Seckl JR, Walker BR, Webster SP, Dunbar DR, Churchill GA, Vidal-Puig A, Fernandez-Real JM, Emilsson V, Horvat S. (2016).Genetic identification of thiosulfate sulfurtransferase as an adipocyte-expressed antidiabetic target in mice selected for leanness. Nat Med, 22, 771–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Landry AP, Ballou DP, Banerjee R. (2020).Hydrogen Sulfide Oxidation by Sulfide Quinone Oxidoreductase. Chembiochem [DOI] [PMC free article] [PubMed] [Google Scholar]