


Abstract
Novel tools for in silico design of RNA constructs such as riboregulators are required in order to reduce time and cost to production for the development of diagnostic and therapeutic advances. Here, we present MoiRNAiFold, a versatile and user-friendly tool for de novo synthetic RNA design. MoiRNAiFold is based on Constraint Programming and it includes novel variable types, heuristics and restart strategies for Large Neighborhood Search. Moreover, this software can handle dozens of design constraints and quality measures and improves features for RNA regulation control of gene expression, such as Translation Efficiency calculation. We demonstrate that MoiRNAiFold outperforms any previous software in benchmarking structural RNA puzzles from EteRNA. Importantly, with regard to biologically relevant RNA designs, we focus on RNA riboregulators, demonstrating that the designed RNA sequences are functional both in vitro and in vivo. Overall, we have generated a powerful tool for de novo complex RNA design that we make freely available as a web server (https://moiraibiodesign.com/design/).
INTRODUCTION
RNA synthetic biology has experienced tremendous growth in recent years, with substantial contributions in novel approaches in the biomedical and bioengineering fields. A large amount of RNA-based scientific publications (1–3), patents and commercial applications (related but not limited to vaccines, therapeutics and diagnostics) have arisen and show enormous potential or outright success. For instance, in the current pandemic context, Moderna and Pfizer/BioNtech (4,5) have developed synthetic mRNA vaccines, winning the race to produce the first COVID-19 vaccines. Part of the success of synthetic RNA approaches is the capability of rapidly adapting to new pathogens and/or diseases. Diagnostic approaches such as SHERLOCK (6) have been rapidly adapted to the detection of SARS-CoV-2 (7). RNA vaccines and other mRNA-based therapeutics, as they require low hydrolysis rates, can be substantially improved by using structure design algorithms for RNA stability predictions (8). Also, mRNA secondary structure has an impact on translation rates (9). Importantly, most RNA designs rely on a specific structure or structure switch to become functional, and designed sequences do not always fold into the desired structure either in vitro or in vivo (or both). Therefore, in order to speed up the time to production of these synthetic RNA tools, better and more versatile de novo RNA design approaches are needed.
MoiRNAiFold is a Constraint Programming (CP) based tool for RNA design in the same spirit as RNAiFold (10–13). In fact, it inherits its design constraints and philosophy, while introducing novel modeling concepts that impact its overall efficiency. A large number of tools for RNA design have appeared over the years (for a review see (14)). RNAinverse (15), from the the ViennaRNA Package, was the first in 1994, followed by others such as RNA-SSD (16), INFO-RNA (17), Modena (18), NUPACK (19), antaRNA (20), incaRNAfbinv (21), etc. All of them are based on some form of heuristic local search to solve the RNA design problem (or RNA inverse folding problem). More recently, more sophisticated algorithms have been developed, some based on Reinforcement Learning (22), Deep Learning (23), MultiObjective Optimization (24) and others in collaborative approaches (25). The main goal of these approaches is to outperform each other in solving certain established benchmarks. Even though NUPACK (19) seems to be the preferred method when attempting to design functional RNA sequences, it does not perform extremely well in benchmarks (24).
To date, many interesting functional RNA constructs have been described: Small Transcriptional Activating RNAs (STARs) (26), hammerhead ribozyme switches (27) or hairpin ribozyme switches (28), to name a few. For experimental validation purposes here, we chose to focus on the most well-known and established RNA constructs for gene expression control through translation regulation: toehold switches (29,30). Noteworthy, toehold switches have been successfully applied to Zika virus diagnostics (31). We also show a STARs example to demonstrate that transcriptional control can be equally effective by RNA structures designed by our novel algorithm and web server.
MoiRNAiFold introduces new modeling concepts that aim at outperforming any other approaches in solving complex benchmarking sets, while at the same time including novel dozens of design constraints and quality measures necessary for the design of complex functional RNAs, specifically with a focus on control of gene expression (translation) via RNA–RNA interactions. Our goal is to be able to design RNA sequences for complex RNA target structures, while at the same time and more importantly, producing functional RNA sequences when validated both in vitro and in vivo.
MATERIALS AND METHODS
Computational methods
MoiRNAiFold is an extension and partial rewrite of RNAiFold (10–13), which has been updated to work with the new versions of the ViennaRNA (2.4.12) and OR-Tools (7.3) libraries. Supplementary Table S1 lists all its features, highlighting new ones from inherited ones and Supplementary Table S2 highlights the algorithmic novelties with respect to RNAiFold (10,11,13). As is the case with RNAiFold, MoiRNAiFold is based on CP and it solves the RNA inverse folding problem, i.e. the problem of given a target structure (#RNAscdstr) find the sequence or sequences whose Minimum Free Energy (MFE) structure is the target structure. It is worth noting that CP provides a complete search, meaning that, given enough time, the algorithm will either find all possible solutions or prove that none exists. This is why the result of MoiRNAiFold for a given target structure and with a time limit can be one of the following: (i) No solution found—meaning it was not able to find sequences for which the MFE structure is the target structure in the allotted time; (ii) no possible solution—meaning the algorithm has proven that no sequences exist whose MFE is the target structure; (iii) a list of solutions, i.e., a list of sequences whose MFE structure is the target structure; also, in this case, if the algorithm can prove that all the sequences that exist have been found, it will produce the message All solutions found at the end of the list.
CP is a powerful framework for solving combinatorial optimization problems, providing both a rich modeling language and an efficient branch and prune search strategy. It follows that, to solve a problem with CP, one needs to define both the model (variables, domains and constraints) and the search (variable and value heuristics).
Modeling novelties
Recall that RNAiFold (10) maintains a set of variables X = {x1, x2, …, xn} for each nucleotide position in the target structure, each with the domain dom(X) = {1, 3, 7, 12} corresponding to {G, A, C, U}. This is based on the marks in an optimal Golomb Ruler (32) of size 5, and it facilitates keeping track of the possible values of the base pairs (10). MoiRNAiFold maintains also a set of variables Y = {y1, y2, …, yn} for each nucleotide position in the target structure, each with the domain dom(Y) = {0, 1, 2, 3} corresponding to {A, C, G, U}. Channeling constraints ensure the values of both sets of variables are consistent with one another. Moreover, RNAiFold maintains a set of variables BP = {bp1, bp2, …, bpm}, corresponding to the m base pairs in the target structure, where if the ith base pair corresponds to the positions k and l, we have bpi = (xl – xk)2. Given this, MoiRNAiFold introduces a novel type of variable of Groups of Unpaired Positions (GUP) that correspond to unpaired positions that have an impact in the Free Energy of the secondary structure. These types involve from one to four unpaired variables as it can be seen in Figure 1A. As such the values for each GUP are calculated with the general formula: GUPi = yi1*40 + yi2*41 + yi3*42 + yi4*43, where yik are the variables corresponding to the GUPi positions. Unpaired Positions (UP) are still maintained, and are all unpaired positions (all not BPs) that are not GUPs. This seemingly small addition has great impact in solving complex RNA structures, as we show in the benchmarking section. All new constraints are also part of the modeling, but for the sake of flow are explained in Supplementary Information (‘MoiRNAiFold: New Features’; section A). Their impact is mostly on the ability to design functional RNAs although they also have an impact on the speed in which a design is solved, due to the reduction of the search space they imply.
Figure 1.

Three key features of MoiRNAiFold. (A) Twelve of the most relevant types of GUPs (in red). These positions are assigned their values as a unique variable. This new concept impacts both heuristics and LNS restarts. (B) Key difference between Salis et al. and our approach to TE calculation. In Salis et al. (top panel) the relevant sequence is calculated selecting 75 nucleotides (in red) around the AUG regardless of the actual overall secondary structure. Instead, we calculate the actual secondary structure and extend the selected region to the start and end of complete helices (bottom panel). This yields a significant difference in TE calculation, especially in the context of small riboregulators. (C) Screenshot of the input form (left) and the results page (right) of MoiRNAiFold web server for a particular structure. For a detailed description of specific MoiRNAiFold novel constraints see Supplementary Information (‘MoiRNAiFold: New Features’; section A).
Search novelties
CP performs an exhaustive exploration of the search space in a depth first manner, assigning variables and values in a given order. These are called variable and value heuristics. Our variable heuristic is divided into several steps, but the first step, as in RNAiFold, is to break down the target structure into a tree of helices. The value heuristics are based on the Free Energy contribution modulated by a random component. This random component is now different for each set of variables (BP, UP and GUP) and it is now a percentage value signifying how accurately the algorithm has to follow the ordering defined by the actual Free Energies of each value. We have added a new option named Competition that automatically fixes all heuristic related aspects (i.e. it fixes the value of the random components to 100, 90 and 90, respectively). It also activates Large Neighborhood Search (see below) and defines the following variable ordering: (i) all external loop (UPs) positions of root helix; (ii) all GUPs from the root helix; (iii) then, for each helix by levels from bottom to top it assigns the variables in the order: GUPs, UPs of the parent helix, UPs and BPs.
Without the competition option, the variable heuristic is as in RNAiFold with the addition of the new GUP variables that are assigned last within the helix. Supplementary Figure SF1 shows a toy comparison of the variable heuristics for RNAiFold and MoiRNAiFold both with the Competition option and without it. Moreover, we have added an option (#LocalCstrsOrder) to set the variable heuristic via local constraints (Supplementary Information, ‘MoiRNAiFold: New Features’; section A). In this case, all variables involved in the local constraint are assigned first in the order in which they appear in the input file.
Large neighborhood search
LNS is a meta-heuristic that iteratively changes a candidate solution until it becomes a valid solution. It is not complete, as opposed to CP, meaning it is not guaranteed to find a solution or prove that none exists. As in RNAiFold, MoiRNAiFold introduces this option seemingly by repeating the process of performing CP search, stopping after a certain amount of time, fixing the value of some variables and restarting the CP search. Also, after a few of such restarts, a hard restart occurs (controlled by #LNSunchangedRestarts), i.e., no variables are fixed and the CP search restarts from scratch. As in RNAiFold, at each restart, positions that present a correct status (paired or unpaired) with respect to the target structure in the partial solution are fixed with a certain probability. MoiRNAiFold introduces a new fixing strategy at each restart as it relates to GUP positions. Briefly, as GUP positions are unpaired, it is often the case that they are unpaired in a partial solution, however, their values might be jeopardizing the correct overall structure of the target structure (or parent helix in the tree decomposition). Therefore, MoiRNAiFold checks whether this is the case before fixing their values with a certain probability. Moreover, MoiRNAiFold introduces other small changes in the restarting heuristic such as: the probability of a position being fixed decreases at each restart as long as the number of fixed variables is not the highest found so far (exploration); the time to a restart increases when the number of fixed variables is the highest found so far (exploitation); also, the random component for each GUP and BP position decreases each time the restart fixes more positions than in the previous restart; and, both the random component of each variable and the time to the next restart are reset at each hard restart.
Translation efficiency calculation
Among the many quality measures of the designed sequences, we introduce a novel measure of Translation Efficiency (TE). This measure calculates ribosomal TE as in Salis et al. (33) with modifications to make it more suitable for the design of small riboregulators, such as toehold switches (29,30). In particular, it maintains the same features but changes the manner in which such features are calculated. In Salis et al. (33), the structure relevant for such features is what can be found from 75 nucleotides upstream of the Ribosome Binding Site (RBS) to 75 nucleotides downstream of the RBS. However, for most of the RNA constructs related to gene expression regulation (Figure 1B), structural changes occur further away from the RBS and as such, this approach cannot be used. Instead, we calculate the structure of the whole RNA sequence in consideration and, starting 75 nucleotides away from the RBS, extend the structure for feature calculations to the start and end of complete helices. We find that this simple modification extends Salis et al. (33) approach in a fashion that allows us to use it in the gene expression regulation context without jeopardizing its efficiency.
Web server
We have developed a completely new web server that incorporates all new and inherited design constraints with an intuitive and user-friendly input form, and a novel sequence visualization tool (Figure 1C). The complete set of constraints and quality measures is shown in Supplementary Table S1 and the MoiRNAiFold web server manual is shown in Supplementary Information (section B).
RNA sequences selection for experimental validation
MoiRNAiFold was used to design a few thousand RNA sequences for each RNA riboregulator design. Afterwards, the resulting sequence list was ordered based on the Ensemble Defect and TE in a few different ways. From this order list, three top ranked sequences were selected for experimental validation. Supplementary Data 1 shows the ranked RNA sequences (along with the ordering criteria) for toehold activators, classic and 3WJ repressors and the riboregulators designed for in vivo assays.
Bacterial strains and growth conditions
Escherichia coli strain K-12 substrain MG1655 (F− λ− ilvG− rfb−50 rph−1) were used for in vivo assays. DH5α (Thermo Scientific) were used for transformation and expansion of plasmid DNA. All strains were grown in LB medium with appropriate antibiotics (i.e. ampicillin) when needed at 37°C and 200 rpm.
Toehold switch activators
To test our generated toehold switch activator constructs in our in vitro system we picked the top ranked construct described by Green et al. (29). In brief, the toehold switch activator positive control sequence preceded by a T7 promoter in ssDNA form (Toehold Activator Control, Supplementary Table S3) obtained from Integrated DNA Technologies (IDT, Coralville, IA, USA) was used in a PCR reaction to introduce the full gfpmut3b-ASV gene sequence (29) in frame (downstream of the toehold sequence) to obtain a dsDNA template. This DNA was purified by GeneJET purification kit (Thermo Scientific), quantified by NanoDrop (Thermo Fisher) and used with PURExpress™ (New England Biolabs) for in vitro transcription and translation assay. A total quantity of 150 ng of the purified DNA were added in the presence or absence of 8 or 16 μM synthetic RNA target (sRNA, Supplementary Table S3) and tested it in a low volume assay (6.4 μl, black bottom 384-well plates, Nunc, Thermo Fisher). In parallel, we tested our MoiRNAiFold-designed toehold activator constructs (Moirai BD Act1–3; Supplementary Data 2), following the same protocol. The GFP raw fluorescence unit (RFU) readings were performed in a BioTek plate reader (Synergy H1 Hybrid Multi-Mode Reader) at 475/510 nm (excitation/emission wavelengths, respectively) at 1–3 h reaction time (Supplementary Figure SF2A).
Toehold classic and three-way junction (3WJ) repressors
As for the toehold switch activators, to compare the toehold classic and 3WJ repressors we picked the top ranked constructs described in Kim et al. (30) and our MoiRNAiFold-generated constructs and tested them in our in vitro system. In brief, the toehold switch positive control sequences (Supplementary Table S3) and Moirai BD Repr1–3 and Moirai BD 3WJ1–3 (Supplementary Data 2) preceded by a T7 promoter in ssDNA form, obtained from IDT (Coralville, CA, USA), were run in a PCR to introduce the full gfpmut3b-ASV gene sequence (29) in frame (downstream of the toehold sequence) to obtain a dsDNA template. Again, this was purified by GeneJET purification kit (Thermo Scientific), quantified by NanoDrop (Thermo Fisher) and used with PURExpress™ (New England Biolabs) for in vitro transcription and translation assay. As previously, 150 ng of the purified DNA were added in the presence or absence of 8 or 16 μM synthetic RNA target (sRNA, Supplementary Table S3 and Supplementary Data 2) and tested it in a low volume assay (6.4 μl, black bottom 384-well plates, Nunc, Thermo Fisher). The GFP RFU readings were performed in a BioTek plate reader (Synergy H1 Hybrid Multi-Mode Reader) at 475/510 nm (excitation/ emission wavelengths, respectively) at 1–3 h reaction time (Supplementary Figure SF2B and C). All in vitro toehold riboregulator data graphs (including activators, classic repressors and 3WJ) and statistical analysis shown in Figure 3 were done with Graph Pad Software 6.0.
Figure 3.
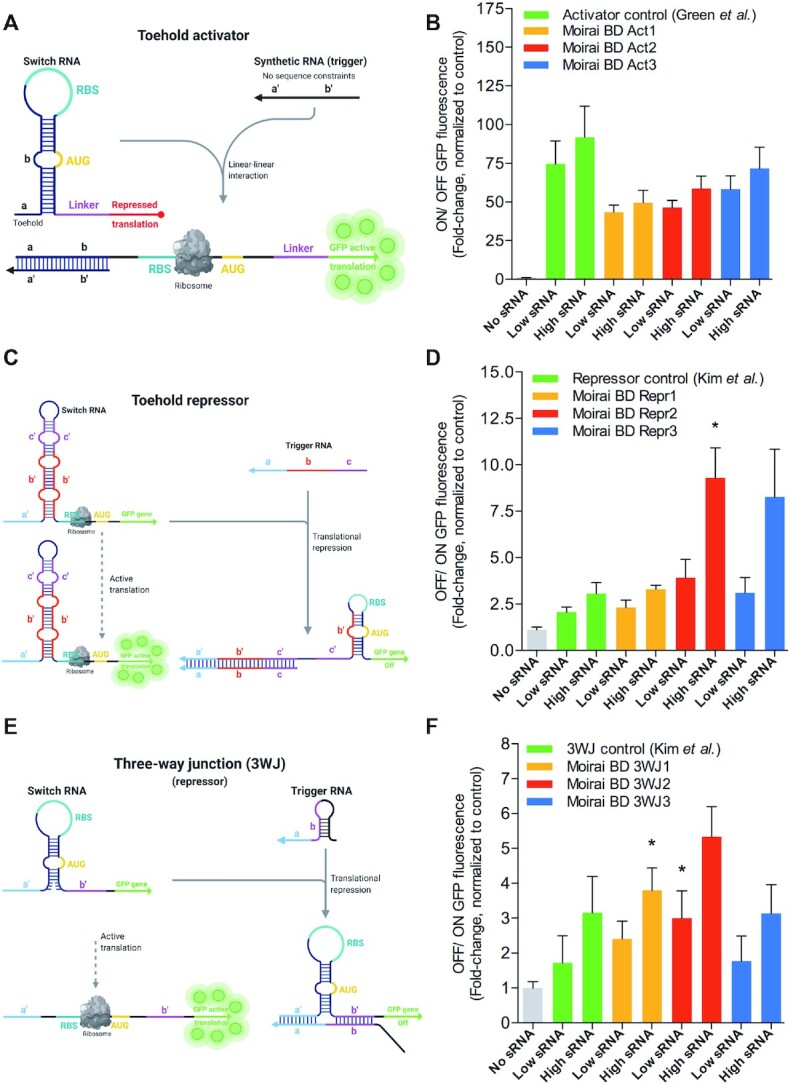
De novo MoiRNAiFold-generated toehold activators and repressor switches perform optimally in vitro. (A) Schematic mechanism of toehold switches regulating activation of translation of GFPmut3b-ASV (GFP) as described in Green et al. (29). (B) In vitro cell-free assay comparison of ON/OFF GFP fluorescence ratio toehold activator Control (Green et al. (29)) compared to three MoiRNAiFold-generated riboregulators (Moirai BD Act1–3, orange, red and blue, respectively) at low (8 μM) and high (16 μM) concentrations of synthetic RNA target (sRNA). Results represent the average (AVG) ± standard error of the mean (SEM) of at least five experiments. A Student t-test analysis shows no significant difference between sRNA treated positive control samples and sRNA treated Moirai BD Act1–3. (C) Schematic mechanism of toehold switches regulating repression of translation of GFP as described in Kim et al. (30). (D) In vitro cell-free assay comparison of GFP translation fold-change ratio (OFF/ ON) in classic repressor control (Kim et al. (24)) compared to three MoiRNAiFold-generated toeholds (Moirai BD Repr1–3, orange, red and blue, respectively) at low and high concentrations of sRNA. Results represent the AVG ± SEM of at least five experiments. A Student t-test analysis shows a significant difference between sRNA treated control and high concentration sRNA treated Moirai BD Repr2 (*P < 0.05). No other significant differences were found. (E) Schematic mechanism of 3WJ RNAs regulating repression of translation of GFP as described in Kim et al. (30); (F) In vitro cell-free assay comparison of GFP translation fold-change ratio (OFF/ ON) in 3WJ control from Kim et al. (24) compared to three MoiRNAiFold-generated 3WJ (Moirai BD 3WJ 1–3, orange, red and blue, respectively) at low and high sRNA. Results represent the AVG ± SEM of at least five experiments. A Student's t-test analysis shows significant differences between sRNA treated control and sRNA treated Moirai BD 3WJ1 and 3WJ2 for high and low sRNA, respectively (*P < 0.05). Raw data, average and deviation plus statistical analysis for panels B, D and F are shown in Supplementary Data 5.
RyhB in vivo assays
A synthetic gene (ryhB positive control) containing the nucleotide sequence of a ProD promoter, ryhB gene sensor, a DNA linker, gfpmut3b-ASV and a T7 terminator (Supplementary Data 3) based on Green et al. (29) plus BglII and NdeI restriction sites was obtained from IDT (Coralville, CA, USA) and used as a template for plasmid construction. The dsDNA was subcloned into the bacterial expression vector pET15b (kindly gifted by Antoni Planas, Laboratory of Biochemistry, Institut Químic de Sarrià, University Ramon Llull, Barcelona, Spain) using BglII and NdeI restriction and sequence verified (Nahum Meller, Custom DNA Constructs Plasmid Cloning Company, NY). Similarly, RyhB Moirai BD1–BD3 constructs were cloned into pET15b using the same strategy (Nahum Meller, Custom DNA Constructs Plasmid Cloning Company, NY). The final bacterial expression vectors containing ryhB positive control, ryhB Moirai BD1, 2 and 3 (Supplementary Data 3) were transformed into E. coli strain K-12 substrain MG1655 and single colonies were grown overnight at 37°C in the presence of 100 μM ampicillin. Before running the experiments, cultures were diluted 1/100 in LB plus 100 μM FeSO4 and were grown during 1.5 h. To induce RyhB expression, 2,2-bipyridyl was added at different concentrations (0–0.6 mM) and the GFP expression was followed after 1 h by fluorescence reading on FL1 channel by flow cytometry (Gallios, Beckman Coulter). Gating strategy used is shown in Supplementary Figure SF3. GFP expression data is represented as fold-induction with respect to normalized data to GFP background levels at 0 mM 2,2-bipyridyl samples as traces of Fe2+ or Fe3+ present in the LB media were partially activating (inducing GFP expression) in all RyhB-transformed cultures. In vivo toehold switches data graphs and statistical analysis shown in Figure 4 was done with Graph Pad Software 6.0.
Figure 4.

De novo MoiRNAiFold-generated toehold switch endogenous RNA sensors in vivo assays. (A) Schematic mechanism of toehold switches (ryhB sensor) regulating GFPmut3b-ASV translation upon endogenous ryhB small RNA (sRNA) as described in Green et al. (29). (B) In vivo assay in E.coli (strain K-12 substrain MG1655) grown in LB plus 100 μM FeSO4 was started by inducing GFP expression by 2,2-bipyridyl at different concentrations (0 mM -control- to 0.6 mM) during 1h and fluorescence in FL1 followed by flow cytometry. Data represent the AVG ± SEM of four experiments. (C) Bar graph representing the GFP fold-induction of ryhB sensor positive control and Moirai BD1–3 in the presence of 0.3 mM 2,2-bipyridyl. Data represent the AVG ± SEM of four experiments normalized to no 2,2-bipyridyl (0 mM) control. A paired t-test analysis shows significant differences between ryhB sensor control and ryhB Moirai BD1–3 at 0.3 mM 2,2-bipyridyl (**P < 0.01 and *P < 0.05, for Moirai BD1–2 and Moirai BD3, respectively). Raw data and complete statistical analysis and P values for each 2,2-bipyridyl concentration are shown in Supplementary Data 6.
RESULTS
Benchmarking
Our first aim was to outperform all previous approaches in solving complex and well established challenges such as the EteRNA benchmark puzzles (34). This is a set of 100 complex RNA structures, most of which cannot be solved under the current RNA Energy Model Turner2004 (35), mainly due to single base pairs, large internal loops and complex Multiloops. Therefore, as is the case for other algorithms (including RNAiFold), we use the Turner1999 (36) energy model for all benchmarking purposes. We run each puzzle 10 times with different time limits and the results show that MoiRNAiFold solves most of them and many of them in seconds (Figure 2A; resulting sequences in Supplementary Information, ‘EteRNA sequences’; section C). The best performing algorithm in the literature (24) solves 73 puzzles with a time limit of one day (although 61 of them are solved in less than a minute), while MoiRNAiFold solves 91 puzzles within the same time limit, 84 of them within a time limit of 1 minute (Figure 2A) and, to the best of our knowledge, 12 of them for the first time (Figure 2B and Supplementary Figure SF4). A broad comparison of solved puzzles for several other algorithms (adapted from (24,34)) is depicted in Supplementary Figure SF4. A puzzle is considered solved when the algorithm returns a solution within the time limit. Note that this accomplishment is carried on in a very hard benchmark, where other approaches that have appeared over the years were only able to solve a few more puzzles than the previous best solver. Of note, RNAiFold was only able to solve 43 puzzles, demonstrating the impact of the novel concepts introduced in MoiRNAiFold, especially the GUPs, the new variable heuristic and the new fixing strategy in the LNS restarts (Supplementary Figure SF5 shows a comparison of RNAiFold and MoiRNAiFold at solving one of the EteRNA puzzles). For completion, we show that MoiRNAiFold performs extremely well in simpler benchmarks, such as the Rfam dataset (18) (structures selected from Rfam database (37)), where most of the structures are solved in less than one second in median time (Supplementary Table S4 and Supplementary Information, ‘Rfam sequences’ section D) with MoiRNAiFold being the only program solving all puzzles with a possible design by current thermodynamic models and algorithms (Supplementary Figure SF6). All benchmarks were run with the Competition heuristic setup.
Figure 2.

MoiRNAiFold performance in common EteRNA benchmark RNA structures. (A) Solved structures (Y axis) versus number (#) of solutions obtained (X axis) of EteRNA benchmark RNA puzzles by MoiRNAiFold algorithms in less than 1 min (light blue), 2 h (orange) and 24 h (red). Structures that were not solved (i.e. #96, #97 or #100) show no bars (0 solutions obtained after a 24 h test). (B) RNA secondary structures only solved by MoiRNAiFold represented by Pseudoviewer web server (http://pseudoviewer.inha.ac.kr/). Structures of EteRNA benchmark are shown in Supplementary Information (‘EteRNA sequences’; section C).
Experimental validation
Once the performance of MoiRNAiFold has been benchmarked, we turn to the experimental validation of designed RNA molecules, by focusing on RNA regulation of gene expression via RNA–RNA interactions. Given the expressive range of our set of constraints and the multitude of RNA intrinsic quality measures, we can set our goal to generate several highly constrained sequences, select very few in terms of these intrinsic quality measures, and validate them experimentally to confirm their efficiency. In contrast to the benchmarking approach, all sequences designed in the remainder of the paper use the functional setup, and the key issues for their success are, specially, the expressiveness of the novel constraints (Supplementary Information) and the new TE measure for selecting final candidates. Here, we show how MoiRNAiFold is capable of designing functional, biologically relevant RNA structures, bridging the gap between theoretical complex puzzles and functional designs.
In vitro riboregulators
First, we approached the design of toehold activators that switch from a secondary structure that prevents translation to a structure that allows it upon binding of a trans-acting synthetic RNA (sRNA) target as reported in Green et al. (29). The structure and mechanism of toehold activators is depicted in Figure 3A (the input file for MoiRNAiFold is detailed in Supplementary Data 4). We used the top ranked forward-engineered toehold switch from Green et al. (29) (generated by NUPACK (19)) (Supplementary Table S3) as activator RNA switch positive control and compared the ON/OFF ratio (sRNA versus non-sRNA added) of GFP fluorescence with our de novo generated toehold switches selected based on measures such as Ensemble Defect (10) and ribosomal TE difference between the ON and OFF states (RNA sequences, their corresponding sRNA targets and quality measures are shown in Supplementary Data 2). In our assays in vitro, the ON/OFF ratio of toehold activator positive control ranged between ∼75 to 90 in the presence of low and high concentration of sRNA, respectively, while our three de novo designed toehold switches showed an optimal activation of GFP levels but slightly lower ratio (∼45–70) (Figure 3B and Supplementary Data 5). Of note, despite the lower activation ratio, our three activators also showed a lower standard deviation (∼30–45 for positive control versus 10–30 on average for our RNAs, Supplementary Data 5) suggesting a higher reproducibility performance in vitro and corresponding to greater statistical significance of sRNA added versus non-sRNA controls (P < 0.001, Moirai BD Act1–2 and P < 0.01 for Moirai BD Act3 versus P < 0.05, Toehold Activator Ctrl, Supplementary Data 5).
Second, we tested whether we could design actively translating GFP RNA constructs that are repressed upon sRNA hybridization by secondary structure conformational switch (Figure 3C), as previously shown by Kim et al. (30). MoiRNAiFold input file is shown in Supplementary Data 4 and selected RNA sequences in Supplementary Data 2. For comparison, we considered the top de novo-designed translation-repressing riboregulator in the aforementioned study (30) as positive control (Supplementary Table S3). The OFF/ON ratios achieved in the control repressor construct in vitro ranged from ∼2.0 to 3.6 fold-change (equivalent to 50–70% reduction), whereas our three newly designed RNAs achieved a translation repression of ∼2.3 to 9.3 fold-change (∼50–87% reduction) at low and high concentrations of sRNA, respectively (Figure 3D). Thus, our generated toehold repressors were as potent or slightly better (at high sRNA concentrations) as Kim et al. (30) top-ranked one (Supplementary Data 5) with Moirai BD Repressor 2 showing significantly higher translational repression at high sRNA concentrations with respect to the repressor positive control from Kim et al. (P < 0.05, Figure 3D and Supplementary Data 3).
Third, a different translation-repressing RNA design known as three-way junction (3WJ) riboregulators were also described by Kim et al. (30). By using MoiRNAiFold we designed (Figure 3E and input file detailed in Supplementary Data 4) and ranked the resulting 3WJ constructs based on their potential to perform in vitro (Supplementary Data 2). 3WJ control from Kim et al. showed a fold-change of ∼1.7 to 2.9 (corresponding to an inhibition of ∼31% and 62%) in the presence of low and high sRNA concentration, respectively. Of note, our MoiRNAiFold-generated 3WJ showed similar or better potency of inhibition (ranging from ∼1.8 to 5.3 fold-change (corresponding to ∼38 to 80%, respectively), with two constructs showing significantly higher repression at low or high concentrations of sRNA (P < 0.05, Figure 3F and Supplementary Data 5).
Of note, for toehold activators and repressors constructs we decided to use in vitro cell-free assays as it represented the easiest and fastest way of building and testing the constructs (containing our designed RNA sequences and a gene encoding a fluorescent protein—i.e. GFP—for the outcome) and due to the advantage of not having to manipulate living organisms in the laboratory. Therefore, any difference in fold-changes found in our study in comparison to previous ones (29,30) (with top-ranked riboregulator RNA sequences used as positive controls) are due to this distinct assay performance. While previous studies mostly tested the toehold switches in cells in vivo, we used the cell-free transcription and translation in vitro assays and thus raw fluorescent values as well as fold-changes calculated from them can vary significantly.
Overall, we can conclude that our de novo designed toehold activators and repressors were significantly active and optimally performing in vitro, similar to those resulting from hundreds of tested RNA molecules in previous studies (29,30).
In vivo riboregulators
Finally, we aimed to design RNAs for in vivo performance comparison. In this case, we used endogenous RNA regulation via a toehold switch sensor to detect the E. coli small RNA RyhB (Figure 4A) (29), a small transcript that downregulates a series of iron-associated genes when iron endogenous levels are low (38). Using the input file depicted in Supplementary Data 4, we generated many sequences and selected the three top ranked as explained for in vitro constructs (Supplementary Data 6).
Escherichia coli cells constitutively expressing RyhB-responsive toehold switches (RyhB control (29) and RyhB Moirai BD1–3) were induced by chelating iron with 2,2-bipyridyl at different concentrations (0–0.6 mM) during 1 h and the GFP expression was followed by flow cytometry. All RyhB constructs showed GFP levels increasing up to 0.3 mM 2,2-bipyridyl, beyond which concentration levels plateau (Figure 4B). At 0.3 mM 2,2-bipyridyl, RyhB control (29) reached a peak of ∼1.5-fold-induction with respect to non-chelated control, whereas our de novo designed RyhB constructs showed a 2.0- to 4.3-fold-induction (Figure 4C), demonstrating a significantly greater response to endogenous RyhB small RNA (P < 0.005 for Moirai BD1 and BD2 and P < 0.05 for Moirai BD3 construct, Figure 4C and Supplementary Data 6).
DISCUSSION
We have generated a novel RNA design tool that allows for a fast generation of efficient complex structured RNAs with a specific biological function without the need of testing thousands of designs by high-throughput assays. Overall, our RNA design software outperforms all previous approaches (both in speed and number of solved structures) in established EteRNA benchmarks (34). This paves the way towards the possibility of efficiently generating highly structured RNA molecules that cannot be designed by any other software. More importantly, MoiRNAiFold is capable of generating, in a user-friendly, intuitive and versatile manner, de novo RNA constructs that perform similarly or as well as the top ranked constructs obtained from a screening of a battery of hundreds of designs (29,30). Note that we were able to generate functional in vitro performing translation-activating, classic and 3WJ repressing riboregulators without the need of synthesizing and testing a large collection of constructs. More remarkably, in our in vivo approach, the three constructs generated performed significantly better than the one previously reported in Green et al. (29), confirming the capability of our MoiRNAiFold software to generate RNA designs with great potential to work in in vivo settings. Moreover, to show that MoiRNAiFold is capable of designing functional RNAs that regulate not only translation, we also designed and tested STARs, proving to regulate gfpmut3b-ASV gene transcription (Supplementary Methods, ‘STARs’; section E and Supplementary Table S5). Both STARs designed showed >2-fold activation with most conditions showing an average of ∼5-fold increase in GFP levels with respect to no STAR control (Supplemental Figure SF7).
Very recently, two publications reported Deep Learning approaches applied to thousands of toehold switches in order to infer characteristics that render some of them extremely efficient in vitro (39,40). This approach is orthogonal to our approach, however, they require extensive experimental validations that are costly and time consuming, and would have to be carried out for any new RNA constructs that can be devised in the future. Moreover, these inferred characteristics can be applied via constraints in MoiRNAiFold to improve even further the designed sequences. Also, recent promising tools for the design of specific RNA constructs have been developed (41,42) (https://yiplab.cse.cuhk.edu.hk/toehold and https://ribosoft2.fungalgenomics.ca/). However, we believe our approach can be applied at any time with the expectation that the number of RNA molecules to test will be dramatically reduced without jeopardizing functionality and, therefore, reducing the time and cost to production in any scenario.
DATA AVAILABILITY
MoiRNAiFold is freely available as a web server at https://moiraibiodesign.com/design/.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Judit Domingo, Dani Poglayen and Amadís Pagès from Moirai Biodesign for their critical reading of the manuscript and fruitful discussions and suggestions throughout the whole process of the study. We would also like to thank Michelle Meyer and Sonia Hernandez-Diaz for their useful comments. All the illustrations in the figures and supplementary data figures were created using BioRender.com, unless otherwise indicated in the figure captions.
Contributor Information
Gerard Minuesa, Moirai Biodesign, c/ Baldiri Reixach s/n, Parc Científic de Barcelona (PCB), 08028 Barcelona, Spain.
Cristina Alsina, Moirai Biodesign, c/ Baldiri Reixach s/n, Parc Científic de Barcelona (PCB), 08028 Barcelona, Spain.
Juan Antonio Garcia-Martin, Bioinformatics for Genomics and Proteomics. National Centre for Biotechnology (CNB-CSIC). c/ Darwin 3, 28049 Madrid, Spain; Grupo Interdisciplinar de Sistemas Complejos (GISC), Universidad Carlos III de Madrid, 28911 Madrid, Spain.
Juan Carlos Oliveros, Bioinformatics for Genomics and Proteomics. National Centre for Biotechnology (CNB-CSIC). c/ Darwin 3, 28049 Madrid, Spain.
Ivan Dotu, Moirai Biodesign, c/ Baldiri Reixach s/n, Parc Científic de Barcelona (PCB), 08028 Barcelona, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Gerard Minuesa leading to these results has received funding from the European Union's Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie grant agreement No 712949 (TECNIOspring PLUS) and from the Agency for Business and Competitiveness (ACCIÓ) of the Government of Catalonia. Funding for open access charge: European Union's Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie grant agreement No 712949 (TECNIOspring PLUS) and from the Agency for Business and Competitiveness (ACCIÓ) of the Government of Catalonia.
Conflict of interest statement. None declared.
REFERENCES
- 1. Chappell J., Watters K.E., Takahashi M.K., Lucks J.B.. A renaissance in RNA synthetic biology: new mechanisms, applications and tools for the future. Curr. Opin. Chem. Biol. 2015; 28:47–56. [DOI] [PubMed] [Google Scholar]
- 2. Schmidt C.M., Smolke C.D.. RNA switches for synthetic biology. Cold Spring Harb. Perspect. Biol. 2019; 11:a032532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim J., Franco E.. RNA nanotechnology in synthetic biology. Curr. Opin. Biotechnol. 2020; 63:135–141. [DOI] [PubMed] [Google Scholar]
- 4. Corbett K.S., Edwards D.K., Leist S.R., Abiona O.M., Boyoglu-Barnum S., Gillespie R.A., Himansu S., Schäfer A., Ziwawo C.T., DiPiazza A.T.et al.. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature. 2020; 586:567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vogel A.B., Kanevsky I., Che Y., Swanson K.A., Muik A., Vormehr M., Kranz L.M., Walzer K.C., Hein S., Güler A.et al.. A prefusion SARS-CoV-2 spike RNA vaccine is highly immunogenic and prevents lung infection in non-human primates. 2020; bioRxiv doi:08 September 2020, preprint: not peer reviewed 10.1101/2020.09.08.280818. [DOI]
- 6. Gootenberg J.S., Abudayyeh O.O., Lee J.W., Essletzbichler P., Dy A.J., Joung J., Verdine V., Donghia N., Daringer N.M., Freije C.A.et al.. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017; 356:438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Broughton J.P., Deng X., Yu G., Fasching C.L., Servellita V., Singh J., Miao X., Streithorst J.A., Granados A., Sotomayor-Gonzalez A.et al.. CRISPR–Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020; 38:870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wayment-Steele H.K., Kim D.S., Choe C.A., Nicol J.J., Wellington-Oguri R., Sperberg R.A.P., Huang P.-S., Das R.. Theoretical basis for stabilizing messenger RNA through secondary structure design. 2020; bioRxiv doi:19 February 2021, preprint: not peer reviewed 10.1101/2020.08.22.262931. [DOI] [PMC free article] [PubMed]
- 9. Mauger D.M., Joseph Cabral B., Presnyak V., Su S.V., Reid D.W., Goodman B., Link K., Khatwani N., Reynders J., Moore M.J.et al.. mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:24075–24083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garcia-Martin J.A., Clote P., Dotu I.. RNAiFOLD: A constraint programming algorithm for RNA inverse folding and molecular design. J. Bioinform. Comput. Biol. 2013; 11:1350001. [DOI] [PubMed] [Google Scholar]
- 11. Garcia-Martin J.A., Clote P., Dotu I.. RNAiFold: a web server for RNA inverse folding and molecular design. Nucleic Acids Res. 2013; 41:W465–W470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dotu I., Garcia-Martin J.A., Slinger B.L., Mechery V., Meyer M.M., Clote P.. Complete RNA inverse folding: computational design of functional hammerhead ribozymes. Nucleic Acids Res. 2014; 42:11752–11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garcia-Martin J.A., Dotu I., Clote P.. RNAiFold 2.0: a web server and software to design custom and Rfam-based RNA molecules. Nucleic Acids Res. 2015; 43:W513–W521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Churkin A., Retwitzer M.D., Reinharz V., Ponty Y., Waldispühl J., Barash D.. Design of RNAs: comparing programs for inverse RNA folding. Brief. Bioinform. 2018; 19:350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hofacker I.L., Fontana W., Stadler P.F., Bonhoeffer L.S., Tacker M., Schuster P.. Fast folding and comparison of RNA secondary structures. Monatsh. Chem. Chem. Mon. 1994; 125:167–188. [Google Scholar]
- 16. Andronescu M., Fejes A.P., Hutter F., Hoos H.H., Condon A.. A new algorithm for RNA secondary structure design. J. Mol. Biol. 2004; 336:607–624. [DOI] [PubMed] [Google Scholar]
- 17. Busch A., Backofen R.. INFO-RNA - a fast approach to inverse RNA folding. Bioinformatics. 2006; 22:1823–1831. [DOI] [PubMed] [Google Scholar]
- 18. Taneda A. MODENA: a multi-objective RNA inverse folding. Adv. Appl. Bioinforma. Chem. 2011; 4:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zadeh J.N., Wolfe B.R., Pierce N.A.. Nucleic acid sequence design via efficient ensemble defect optimization. J. Comput. Chem. 2011; 32:439–452. [DOI] [PubMed] [Google Scholar]
- 20. Kleinkauf R., Mann M., Backofen R.. AntaRNA: ant colony-based RNA sequence design. Bioinformatics. 2015; 31:3114–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Drory Retwitzer M., Reinharz V., Ponty Y., Waldispühl J., Barash D.. incaRNAfbinv: a web server for the fragment-based design of RNA sequences. Nucleic Acids Res. 2016; 44:W308–W314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eastman P., Shi J., Ramsundar B., Pande V.S.. Solving the RNA design problem with reinforcement learning. PLoS Comput. Biol. 2018; 14:e1006176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Runge F., Stoll D., Falkner S., Hutter F.. Learning to design RNA. ICLR 2019 Conference Paper. 2019; 28 Sept 2018 (modified: 12 Apr 2019). [Google Scholar]
- 24. Rubio-Largo Á., Vanneschi L., Castelli M., Vega-Rodríguez M.A.. Multiobjective metaheuristic to design RNA sequences. IEEE Trans. Evol. Comput. 2019; 23:156–169. [Google Scholar]
- 25. Lee J., Kladwang W., Lee M., Cantu D., Azizyan M., Kim H., Limpaecher A., Yoon S., Treuille A., Das R.. RNA design rules from a massive open laboratory. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:2122–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chappell J., Westbrook A., Verosloff M., Lucks J.B.. Computational design of small transcription activating RNAs for versatile and dynamic gene regulation. Nat. Commun. 2017; 8:1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sack M., Stifel J., Kreft S.G., Deuerling E., Hartig J.S.. Neomycin-dependent hammerhead ribozymes for the direct control of gene expression in Saccharomyces cerevisiae. Methods. 2019; 161:35–40. [DOI] [PubMed] [Google Scholar]
- 28. Vauléon S., Müller S.. External regulation of hairpin ribozyme activity by an oligonucleotide effector. Chem. Bio. Chem. 2003; 4:220–224. [DOI] [PubMed] [Google Scholar]
- 29. Green A.A., Silver P.A., Collins J.J., Yin P.. Toehold switches: de-novo-designed regulators of gene expression. Cell. 2014; 159:925–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim J., Zhou Y., Carlson P.D., Teichmann M., Chaudhary S., Simmel F.C., Silver P.A., Collins J.J., Lucks J.B., Yin P.et al.. De novo-designed translation-repressing riboregulators for multi-input cellular logic. Nat. Chem. Biol. 2019; 15:1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pardee K., Green A.A., Takahashi M.K., Braff D., Lambert G., Lee J.W., Ferrante T., Ma D., Donghia N., Fan M.et al.. Rapid, low-cost detection of zika virus using programmable biomolecular components. Cell. 2016; 165:1255–1266. [DOI] [PubMed] [Google Scholar]
- 32. Babcock W.C. Intermodulation interference in radio systems. Frequency of occurrence and control by channel selection. Bell Syst. Tech. J. 1953; 32:63–73. [Google Scholar]
- 33. Salis H.M., Mirsky E.A., Voigt C.A.. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009; 27:946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anderson-Lee J., Fisker E., Kosaraju V., Wu M., Kong J., Lee J., Lee M., Zada M., Treuille A., Das R.. Principles for predicting RNA secondary structure design difficulty. J. Mol. Biol. 2016; 428:748–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mathews D.H., Disney M.D., Childs J.L., Schroeder S.J., Zuker M., Turner D.H.. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:7287–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xia T., SantaLucia J. Jr, Burkard M.E., Kierzek R., Schroeder S.J., Jiao X., Cox C., Turner D.H.. Thermodynamic parameters for an expanded nearest-neighbor model for the formation of RNA duplexes with Watson-Crick Base Pairs. Biochemistry. 1998; 37:14719–14735. [DOI] [PubMed] [Google Scholar]
- 37. Kalvari I., Nawrocki E.P., Ontiveros-Palacios N., Argasinska J., Lamkiewicz K., Marz M., Griffiths-Jones S., Toffano-Nioche C., Gautheret D., Weinberg Z.et al.. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2020; 49:D192–D200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Massé E., Salvail H., Desnoyers G., Arguin M.. Small RNAs controlling iron metabolism. Curr. Opin. Microbiol. 2007; 10:140–145. [DOI] [PubMed] [Google Scholar]
- 39. Valeri J.A., Collins K.M., Ramesh P., Alcantar M.A., Lepe B.A., Lu T.K., Camacho D.M.. Sequence-to-function deep learning frameworks for engineered riboregulators. Nat. Commun. 2020; 11:5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Angenent-Mari N.M., Garruss A.S., Soenksen L.R., Church G., Collins J.J.. A deep learning approach to programmable RNA switches. Nat. Commun. 2020; 11:5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kharma N., Varin L., Abu-Baker A., Ouellet J., Najeh S., Ehdaeivand M.R., Belmonte G., Ambri A., Rouleau G., Perreault J.. Automated design of hammerhead ribozymes and validation by targeting the PABPN1 gene transcript. Nucleic Acids Res. 2015; 44:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ching-Yuet To A., Ho-Ting Chu D., RuoningWang A., Cheuk-Yau Li F., Wai-On Chiu A., Gao D.Y., Choi C.H.J., Kong S.K., Chan T.F., Chan K.M.et al.. A comprehensive web tool for toehold switch design. Bioinformatics. 2018; 34:2862–2864. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MoiRNAiFold is freely available as a web server at https://moiraibiodesign.com/design/.