


Abstract
Long non-coding RNAs have emerged as critical regulators of cell homeostasis by modulating gene expression at chromatin level for instance. Here, we report that the lncRNA ANRIL, associated with several pathologies, binds to thousands of loci dispersed throughout the mammalian genome sharing a 21-bp motif enriched in G/A residues. By combining ANRIL genomic occupancy with transcriptomic analysis, we established a list of 65 and 123 genes potentially directly activated and silenced by ANRIL in trans, respectively. We also found that Exon8 of ANRIL, mainly made of transposable elements, contributes to ANRIL genomic association and consequently to its trans-activity. Furthermore, we showed that Exon8 favors ANRIL’s association with the FIRRE, TPD52L1 and IGFBP3 loci to modulate their expression through H3K27me3 deposition. We also investigated the mechanisms engaged by Exon8 to favor ANRIL’s association with the genome. Our data refine ANRIL’s trans-activity and highlight the functional importance of TEs on ANRIL’s activity.
INTRODUCTION
Over the last decade, next generation sequencing approaches highlighted the unexpected diversity of RNAs lacking obvious protein-coding capacity (ncRNAs). The ncRNAs longer than 200-nts are named long non-coding RNAs (lncRNAs). Nowadays, more than 167 000 lncRNAs have been identified in human, many of them being involved in various critical processes including cell proliferation (1). In the present state of knowledge, lncRNAs are already associated with >500 pathologies, strongly reinforcing the need to understand their mechanisms of action (1–4).
LncRNAs are key regulators of gene expression carrying both cytoplasmic and nuclear functions (5). Cytoplasmic lncRNAs mainly modulate gene expression by affecting mRNA stability or translation, while nuclear lncRNAs associate with the genome to regulate gene expression at the chromatin level. The latter implies the activities of epigenetic writers to targeted genomic loci, including for instance the Polycomb group proteins (PcG) composed by the Polycomb Repressive Complexes 1 and 2 (PRC1 and PRC2). These complexes are responsible for the conversion of euchromatin into heterochromatin by catalyzing the ubiquitylation of histone H2A on lysine 119 (H2AK119Ub) and the trimethylation of histone H3 on lysine 27 (H3K27me3), respectively. Interestingly, multiple lncRNAs have been shown to associate with the PcG (6–10) and 20% of them are specific PRC2-binders in human cells (11).
ANRIL (antisense noncoding RNA in the INK4 locus) is one of the lncRNAs associated with PcG activities and several pathologies (12,13). It is transcribed from the 9p21 locus in the opposite direction to the CDKN2A and CDKN2B (cyclin dependent kinase inhibitors 2A and 2B) genes. One crucial point is the possible production of at least 24 alternatively spliced isoforms of ANRIL, the overexpression of some of them being correlated with severe and highly frequent pathologies such as coronary artery disease, diabetes and cancers (14–20). This is especially the case for the three major ANRIL isoforms named DQ485454, EU741058 and NR_003529 (17,18). The longest isoform NR_003529 (3837 nts), hereafter called NR, contains exons 1–12 and 15–21. The shorter isoforms DQ485454 (2660 nts) and EU741058 (962 nts), hereafter called DQ and EU, contain the 13 first exons and the 5 exons (1,5-7,13), respectively.
ANRIL promotes in cis the transcriptional silencing of the CDKN2A and B genes by recruiting the PcG to the 9p21 locus resulting in an increased cell proliferation rate (12,13). In addition, ANRIL is expected to modulate in trans the expression of genes distant from the 9p21 locus. This is evidenced by the differential expression of >200 genes involved in the maintenance of chromatin architecture, cellular proliferation, growth and apoptosis upon the overexpression of ANRIL sub-fragments in HeLa or in HEK293 cells (21,22). Expression changes were also observed for 20 genes, implicated in proliferation and apoptosis, upon ANRIL siRNA knockdown in vascular smooth muscle cells (VSMCs) (23). Recently, by genome editing of induced pluripotent stem cells (iPSC)-derived VSMCs, a 60 kb region within the 9p21 locus overlapping the ANRIL gene was removed. The expression of >3000 genes involved in cell cycle regulation, cell adhesion, muscle development and contraction was dysregulated following this deletion (20). By RNA-FISH, ANRIL was also shown to have a nuclear localization not restricted to the 9p21 locus, but dispersed at several genomic loci (24,25). Even though previous studies demonstrated the trans-regulatory activity of ANRIL, the molecular mechanisms involved need to be refined. For instance, the coding and non-coding genes, which are directly contacted and regulated by ANRIL remain unknown. Also, the mechanisms engaged by ANRIL to specifically associate with the genome still have to be deciphered.
Transposable elements (TEs) are the major contributors to the bulk of the genomic DNA in mammals. They can provide novel regulatory sequences such as promoters and enhancers (26). Recently, several studies focused on the possible relationship between TEs and lncRNA functions. This revealed that nearly half of the lncRNA sequences (41%) are derived from TEs (27). Interestingly, lncRNA exons are strongly and non-randomly enriched in Endogenous RetroViruses (ERVLs) belonging to the LTR class, while other classes of TEs, like SINE (Alu) and LINE (LINE1 and LINE2) are under-represented (27). It was shown for several lncRNAs that the presence of TEs termed RIDLs (repeat insertion domains of long noncoding RNAs) impacts their localization and/or functions (27–34). Furthermore, Holdt and coll. identified Alu sequences within ANRIL and within 5 kb regions of gene promoters affected by the overexpression of ANRIL sub-fragments (21), suggesting that TEs within ANRIL sequence might be involved in its trans-regulatory activities.
In the present study, we investigated whether TEs participate in ANRIL’s chromatin recognition necessary for gene trans-regulation. We identified genome-wide the chromatin occupancy of ANRIL in HEK293 cells by applying the ChIRP-seq approach and found that ANRIL associates with 3227 binding sites mostly composed of G/A residues. By crossing the ChIRP-seq with transcriptomic data from ANRIL knocked-down cells, we established a list of 188 genes corresponding to the potential primary trans-targets of ANRIL, since they were both contacted by ANRIL and affected in terms of expression. Among them, 123 and 65 genes were negatively and positively regulated by ANRIL, respectively. Our data not only support dual-activities of ANRIL but importantly are in favor of mechanisms distinct from those previously described. In silico approaches highlighted the presence of multiple classes of TEs throughout ANRIL exons. In particular, 70% of the longest Exon8 is made up of ERVL elements. We investigated its putative role in ANRIL’s trans-activity. We showed that its presence is required for the efficient association of ANRIL to the chromatin, since Exon8 deletion resulted in a significant reduction of ANRIL’s genomic occupancy. By applying stringent criteria, we accurately identified 9 out of the 123 potential trans-silenced genes of ANRIL, which expression depends explicitly on the presence of Exon8. By further in silico, in cellulo and in vitro characterization, we showed that Exon8 is likely to contribute to the ANRIL’s recognition of the FIRRE, TPD52L1 and IGFBP3 genes and subsequently to their repression by modulating H3K27me3 deposition. We also brought data in favor of a recognition mode for the FIRRE and TPD52L1 genes involving direct DNA/DNA:RNA complex formation. Overall, our work improves the knowledge on ANRIL’s trans-activities and reinforces the emergent idea of a role of TEs in chromatin recognition by nuclear lncRNAs.
MATERIALS AND METHODS
Cell culture
Human embryonic kidney (HEK293) cells were grown in Dulbecco's modified Eagle's medium-high glucose (DMEM) (Sigma-Aldrich) supplemented with 10% Fetal Bovine Serum (FBS) (Dutscher), 1% penicillin/streptomycin (Sigma-Aldrich) and 1% l-glutamine (Sigma-Aldrich).
Generation of knocked-out cells by CRISPR/Cas9 approach
Two sgRNAs targeting the 5′ and 3′ extremities of Exon8 and Exon21 were designed using the CHOPCHOP website (https://chopchop.cbu.uib.no/) and inserted into the pSpCas9BB-2A-puro (35). The two vectors containing the sgRNAs were co-transfected into the HEK293 cells using lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations. Clonal selections were performed according to the manufacturer's recommendations. Clones were then isolated and DNA was extracted followed by end point PCR screening for homozygous deletions. Positive clones were verified by sequencing. The oligonucleotides used in this study are listed in the Supplementary Table S3.
LNA GapmeRs transfection
LNA GapmeRs either targeting unique regions of ANRIL isoforms (Figure 2A) or non-targeting any region (scrambled, used as a negative control) were designed by QIAGEN. Five hundred thousand HEK293 cells were seeded per well in six-well plates 12–16 h before transfection. Transfection was performed using Lipofectamine 2000 (Invitrogen). A mix of the four ANRIL LNA GapmeRs or scrambled LNA GapmeRs was used for transfection at a final concentration of 25 nM. All samples were collected 48h post-transfection in RLT lysis buffer (RNeasy mini kit QIAGEN) for total RNA extraction. The LNA GapmeR sequences are listed below:
Figure 2.
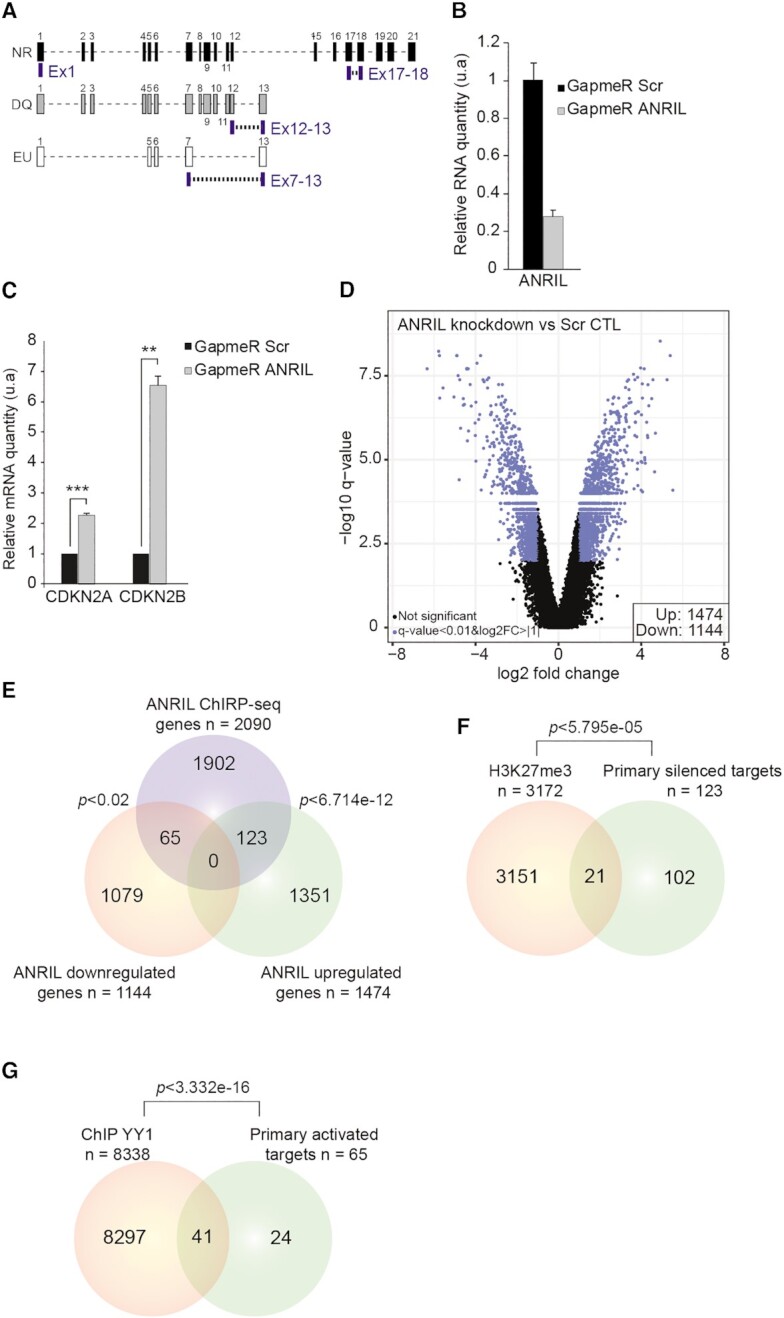
ANRIL is likely to regulate the expression of 188 genes in a direct manner. (A) Schematic representation of the hybridization position of the four different LNA GapmeRs used to silence ANRIL. (B) RTqPCR analysis after LNA GapmeR transfection revealed up to 75% reduction in ANRIL’s expression (n = 3). Values are normalized to the GAPDH housekeeping gene. (C) RTqPCR analysis of CDKN2A and CDKN2B expression following ANRIL knockdown by LNA GapmeRs (n = 3). Values are normalized to the GAPDH housekeeping gene. (D) Volcano plot representing the significance versus fold change for all differentially expressed (n = 2618, FDR < 0.01, log2FC > |1|) genes upon ANRIL knockdown (n = 5). (E) Venn diagram showing the intersection between the ANRIL upregulated genes, downregulated genes, and the ChIRP-seq dataset. A list of 188 genes has been identified as potential direct trans-regulated targets. (F) Venn diagram showing the intersection between H3K27me3 ChIP-seq data in HEK293 cells and the 123 silenced primary targets of ANRIL. List of 21 genes identified to be potentially repressed through PcG dependent mechanism. (G) Venn diagram showing the intersection between YY1 ChIP-seq data in HEK293 cells and the 65 primary activated targets of ANRIL. Forty-one genes were identified to be potentially activated through YY1. The P-values of Venn diagrams were obtained by hypergeometric test using the whole set of microarray genes as a background. Data are represented as mean ± SEM. P-values: moderated t-statistics, *P < 0.05, **P < 0.01, ***P < 0.001.
GapmeR Scrambled: GCTCCCTTCAATCCAA
GapmeR Exon1: TCAGAGGCGTGCAGCG
GapmeR Exon17–18: TAAGATCCAGTGGTGG
GapmeR Exon12–13: CGTAATCATCCATGCA
GapmeR Exon7–13: AATCATCCTGTCAAA
Total RNA extraction and RTqPCR
Total RNAs were collected using RNeasy mini kit (QIAGEN) and extracted following the manufacturer's recommendation. Quantification of the extracted RNAs was done using the nanodrop 2000. DNase step was performed on 1.25 μg of RNA for 1 h at 37°C using DNase I recombinant, RNase-free (Roche). Then RNAs were reverse transcribed using the Superscript III kit (Thermo Fisher Scientific) following the manufacturer's recommendation. cDNAs were diluted 2.5 times in water and RNA expression level was assessed by real time quantitative PCR (RTqPCR) using the iTaq™ Universal SYBR® Green Supermix (Bio-Rad) and ViiA-7 Real-Time PCR system (Applied Biosystems). Transcript RNA levels were normalized against GAPDH reference gene following the relative standard curve method. The RTqPCR primers were used at 1 μM final concentration. The RTqPCR primers used in this study are listed in the Supplementary Table S4.
Microarray expression profiling
The integrity of the RNA was first validated by pico-chip bioanalyzer 2100 (EPI-RNA seq platform from IBSLor, UMS2008, France). Then 5 ng of RNA samples were analyzed using the Clariom D Human Assay Microarrays (Applied Biosystems) which includes transcriptome wide gene- and exon-level expression probesets. Microarray hybridization and scanning was conducted in IMoPA, France according to the manufacturer's standard protocols. Briefly, each purified RNA sample was transcribed to double-strand cDNA, followed by cRNA synthesis and biotin-labeling. The labeled cRNAs were then hybridized onto the Clariom D microarray. After washing, the arrays were scanned using the GeneChip Scanner 3000 (Applied Biosystems). Data analysis was performed using the Transcriptome Analysis Console (TAC). The signal obtained was normalized using the SST-RMA method and the annotation of the probe sets was done using the ‘Clariom_D_Human.r1.na36.hg38.a1.transcript.csv’ annotation file obtained from Affymetrix. Differential expression was calculated using the ‘Limma’ package (takes into consideration the low sample numbers) and the P-value was adjusted using the eBayes correction. Differentially expressed RNAs between condition and control were identified based on fold change and FDR.
Chromatin preparation
Five millions of HEK293 cells were crosslinked in 1% methanol free formaldehyde (Thermo Fisher Scientific) for 10 min and then quenched with 0.125 mM glycine for 5 min. Samples were then lysed using the ChIRP lysis buffer (50 mM Tris–HCl pH 7.0, 10 mM EDTA, 1% SDS) supplemented with protease inhibitor cocktail 100× (Thermo Fisher Scientific) and Ribolock RNase inhibitor (Thermo Fisher Scientific). Samples were then sonicated using the Covaris M220 ultrasonicator and 25 μg of sheared chromatin was treated with 200 μg of proteinase K for 45 min at 50°C. DNA was then extracted using GeneJET Gel Extraction kit (Thermo Fisher Scientific) and quantified by the nanodrop 2000. 600 ng of the subsequent DNA were loaded on agarose gel 1.2% to verify the shearing efficiency (Supplementary Figure S1B). The sheared chromatin was then flash frozen in liquid nitrogen and stored at –80°C for later use.
RNA extraction from chromatin
Twenty-five μg of sheared chromatin was treated with 200 μg of proteinase K for 45 min at 50°C. RNA was extracted from the treated chromatin using the RNeasy MinElute Cleanup kit (QIAGEN) according to the manufacturer's recommendation. DNase and reverse transcription were then performed as described above. cDNAs were diluted 10 times in water and ANRIL enrichment level was assessed by RTqPCR using the iTaq™ Universal SYBR® Green Supermix (Bio-Rad) and ViiA-7 real-time PCR system (Applied Biosystems). Transcripts RNA levels were normalized against the Input.
ChIRP-seq and data analysis
ChIRP-seq adapted to ANRIL was performed as previously described (36). Briefly, ChIRP antisense biotinylated probes were designed using online designer at www.singlemoleculefish.com against the ANRIL full-length sequence. Twenty-three probes were generated tiling the whole lncRNA ANRIL and split into two independent even and odd probe pools based on their relative positions along ANRIL sequence. Similarly, 20 probes against LacZ mRNA were used as negative control. The ChIRP-seq probes used in this study are listed in the Supplementary Table S5. ChIRP-seq was performed on 30 μg of sheared chromatin followed by RNA elution using the RNeasy MinElute Cleanup kit (QIAGEN) and DNA elution using GeneJET Gel Extraction kit (Thermo Fisher Scientific) on two independent replicates. High-throughput sequencing libraries were constructed using the NEBNext Ultra II DNA Kit according to the manufacturer's recommendation (IBSLor Epitranscriptomics and Sequencing Core Facility, Nancy, France). Paired-end sequencing was done on the NextSeq 500 with a read length of 43 bp and with 45 million reads per sample (I2BC sequencing platform, Paris, France). Data analysis was adapted from the ChIRP-seq pipeline (36) (Supplementary Figure S1E). Briefly, the fastq files of replicates 1 and 2 were aligned to the hg19 genome using bowtie2 (37). Then the aligned reads of both even and odd bam files of each replicate were intersected and merged using bedtools (38). Peak calling was then performed against LacZ negative control using MACS2 peak caller (39). Peaks were further filtered based on the score ≥15, and FDR ≤0.05. Peaks located in blacklisted regions of the genome identified by ENCODE were discarded. Finally, only common peaks between both replicates were kept and considered as ‘True Peaks’. The true peaks were annotated using the ChIPseeker package in R (40). Peak distribution was calculated by normalizing the total length of peaks per chromosome by the size of their respective chromosome. Validation of several peaks was performed by quantitative PCR (qPCR) using the ViiA-7 real-time PCR system (Applied Biosystems). The qPCR primers were used at 1 μM final concentration. The qPCR primers used in this study can be found in Supplementary Table S6.
Chromatin Immunoprecipitation
ChIP experiments were performed in HEK293 cells according to the X-ChIP abcam protocol. Briefly, ∼25 μg of sheared DNA was used per IP and incubated overnight with 3 μg of H3K27me3 (PA5_31817, Thermo Fisher Scientific), 2 μg of H3K27ac (Ab4729, Abcam) or 2 μg of H3K4me3 (Ab213224, Abcam) antibodies/20 μl of Magna ChIP™ Protein A+G Magnetic Beads (Merck Millipore) complexes. The following day, the beads were subsequently washed in low salt wash (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8.0, 150 mM NaCl), high salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8.0, 500 mM NaCl) and LiCl wash buffer (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris–HCl pH 8.0). Samples were then treated with 200 μg of proteinase K in a total volume of 200 μl for 45 min at 50°C. DNA was prepared using the GeneJET Gel Extraction kit (Thermo Fisher Scientific) according to the manufacturer's recommendations, eluted in 15 μl of elution buffer and diluted two times with water. Primer list used can be found in Supplementary Table S6.
Motif analysis
The MEME package from MEME Suite was used to identify consensus DNA motifs enriched in the ANRIL ChIRP-seq peaks identified above (41,42). Default parameters were used as such:
The width of the expected motif was set between 6 and 50.
The expected occurrence per sequence was set to zero or one (zoops).
The maximum number of motifs to search for was 5.
FIMO package from MEME suite was also used to scan for the presence of the identified motif 1 within the ANRIL sequence using the default parameters.
Triple helix identification
Triplex Domain Finder (TDF) analysis was performed according to (43). Full length ANRIL sequence (FASTA format) and ChIRP-seq peaks (BED format) were used as inputs in the analysis. The genome used was the hg19 and the minimum length of triplex was set to 15.
Electrophoretic mobility shift assays
This protocol was adapted from (44). Briefly, purine rich strand DNA oligos were 5′-labeled with γ[32P]ATP (Perkin Elmer) and annealed in equimolar ratios to their complementary pyrimidine rich strand DNA oligos in an annealing buffer 1× (10 mM Tris-acetate pH 7.4, 50 mM NaCl, 5 mM Mg-acetate) for 2 min at 95°C and slowly cooled down to 20°C. For triplex formation, RNA was incubated with 100 fmol of radiolabeled duplex oligos for 16 h at 4°C in Triplex-buffer B [10 mM Tris–HCl pH 7.4, 50 mM KCl, 5 mM MgCl2, 10 μg salmon sperm DNA (Thermo Fisher Scientific), protease inhibitor cocktail 1× (Thermo Fisher Scientific), 20 U of Ribolock (Thermo Fisher Scientific)] in a final volume of 10 μl. Triplex formation was monitored by electrophoresis on 12% native polyacrylamide gels at 15 mA and revealed using a typhoon scanner. Sequences of DNA and RNA oligos can be found in Supplementary Table S7.
Transient transfection of ANRIL exons and isoforms
Calcium phosphate mediated transfection was used to overexpress separately ANRIL isoforms (NR, DQ and EU) and exons 3, 8 and 12 in the HEK293 cells according to the manufacturer's recommendations. Briefly, 360 000 HEK293 cells were seeded per well in six-well plates 12–16 h before transfection. One and half μg of pcDNA3.1 expression vectors were used for transfection in 2 ml final volume. Samples were collected 48 h post-transfection in RLT lysis buffer (RNeasy mini kit QIAGEN) for total RNA extraction.
Triplex capture assay
This protocol was adapted from (44). Briefly, RNA-free genomic DNA was sheared with Covaris M220 ultrasonicator to an average size of 200–500 bp and 75 μg of fragmented DNA were incubated with 40 pmol of in vitro transcribed Exon8 for 1 h at 30°C in 940 μl of Triplex buffer (10 mM Tris–HCl pH 7.4, 50 mM KCl, 5 mM MgCl2, 40 U of Ribolock RNase inhibitor – Thermo Fisher Scientific) for triplex formation. The formed DNA–RNA complexes were incubated with 100 pmol of biotinylated probe complemented with 40 U of Ribolock RNase inhibitor (Thermo Fisher Scientific) for 4 h at 30°C and isolated using the MyOne Streptavidin C1 Dynabeads (Thermo Fisher Scientific). After three washes with 700 μl of wash buffer (10 mM Tris–HCl pH 7.4, 50 mM KCl, 5 mM MgCl2, 0.05% Tween-20) DNA was eluted by incubation of the beads with 100 μl of elution buffer (150 mM NaCl, 12.5 mM EDTA, 100 mM Tris–HCl pH 7.5, 1% SDS) for 5 min at 75°C. DNA was then purified and concentrated using the GeneJET Gel Extraction kit (Thermo Fisher Scientific) according to the manufacturer's recommendations. For one replicate, two experiments were performed in parallel, eluted in 10 μl of elution buffer, diluted two times with water and pooled together. Input is prepared according to the same protocol and diluted 40 times. Samples were analyzed by qPCR. Primer list used can be found in Supplementary Table S6.
RESULTS
ANRIL binds 3227 loci across the genome of HEK293 cells
We first evaluated the ability of ANRIL to associate with the chromatin fraction in HEK293 cells. Chromatin was prepared by formaldehyde cross-linking followed by shearing (Supplementary Figures S1A and B). RNAs associated with cross-linked chromatin and cellular RNAs (INPUT) were extracted and analyzed by RTqPCR (Supplementary Figure S1A) (44). We observed a relative enrichment of ANRIL in the chromatin fraction compared to the INPUT (2.8×) and to the unrelated RplpO transcript encoding a ribosomal protein (14×) (Figure 1A). We then assessed the genome-wide occupancy of ANRIL at high resolution by applying the ChIRP-seq approach (36,45). Using tiling biotinylated antisense 20-mer oligos, we efficiently captured the endogenous ANRIL from chromatin (Supplementary Figure S1C and Supplementary Table S5). When compared to the unrelated GAPDH mRNA and the negative control LacZ mRNA, which is not expressed in eukaryotic cells, ANRIL enrichments of 532- and 375-fold were observed for the even and odd probe pools, respectively (Supplementary Figure S1D). The purified DNA was then analyzed by high-throughput sequencing. Data analysis was done from two independent experiments as previously described (36), followed by peak calling using MACS2 peak caller (39) (Supplementary Figure S1E, F). This allowed us to identify 3227 ANRIL-peaks corresponding to the genomic sites for ANRIL occupancy. Figure 1B shows a representative ANRIL-peak (score ≥ 15, and FDR ≤ 0.05) found on the X chromosome that we validated by ChIRP-qPCR (Figure 1B and C). Similar experiments validated the 9p21 locus used as a positive control of ANRIL binder, in addition to the MX1 and STAT1 peaks we identified by ANRIL ChIRP-seq. No enrichment was observed for the TERC locus used as a negative control (Supplementary Figure S1G). Peak distribution analysis showed that almost all the chromosomes were contacted by ANRIL. Few peaks (10,8,5,4) belonged to chromosomes 4, 8, 13 and 14 respectively, while 15% (176) and 23% (754) of them were on the chromosomes 19 and X, respectively (Figure 1D). Additionally, most of the ANRIL-peaks were located in intergenic and intronic regions (1608 and 1301 respectively) compared to UTR, promoter or exonic regions (Figure 1E). Based on human genome composition, no significant peak-enrichment was observed for any DNA sub-categories (Supplementary Figure S1H). We then compared the number of genomic binding sites to the number of ANRIL’s molecules estimated at 440 copies per HEK293 cells (Supplementary Figure S1I). Even though ANRIL’s genome occupancy may be slightly heterogeneous between each individual cell, the number of ANRIL molecules per cell (440) compared to its number of genomic binding sites (3227) suggests that one ANRIL molecule might interact in average with seven genomic sites. Altogether, these results indicate that ANRIL is a chromatin-associated lncRNA able to contact several loci dispersed throughout the genome of HEK293 cells.
Figure 1.

ANRIL binds 3227 loci across the genome of HEK293 cells. (A) RNA extraction experiments showing ANRIL enrichment in the chromatin fraction (n = 3). Values are normalized to the Input. (B) Representative example of one of the ANRIL ChIRP-seq peaks located on the X chromosome. (C) ChIRP-qPCR validation of the peak localized on the X chromosome (n = 4). (D) ANRIL does not coat all the chromosomes to the same extent. Approximately 20% of the ANRIL peaks are localized on the X chromosome. (E) ANRIL ChIRP-seq peaks are mostly distributed in distal intergenic and intronic regions. The percentages of each class were calculated by normalizing the sum of nucleotides in ANRIL ChIRP-seq dataset relative to the human genome. The number of peaks within each DNA category is indicated. (F) MEME motif analysis identifying a G/A rich motif in ANRIL ChIRP-seq peaks (n = 3167/3227). Data are represented as mean ± SEM. P-values: moderated t-statistics, *P < 0.05, **P < 0.01, ***P < 0.001, ns: not significant.
To further characterize the interaction between ANRIL and the genome, motif analysis was performed on the 3227 ANRIL ChIRP-seq peaks, using the MEME suite (http://meme-suite.org/). The most significant motif (E-value = 1.8e–048) corresponded to a highly predominant 21-bp long element present in 3167 out of the 3227 ANRIL ChIRP-seq peaks (Figure 1F). Interestingly, this motif, mainly composed of G and A residues, shows a high degree of similarity with those previously identified by ChIRP-seq experiments as genomic binding sites for the lncRNAs roXes and HOTAIR (36). The other less significant motifs are provided in Supplementary Figure S2A. We also looked for Alu motifs that were previously shown to be enriched within 5 kb fragments from promoters of multiple genes up- or down-regulated upon ANRIL overexpression (21). Interestingly, a similar Alu sequence was identified in motif 2 (41-bp long), that we detected for 48 genomic binding sites of ANRIL (Supplementary Figures S2A and B). Overall, our data suggest that purine-rich DNA regions and some TEs may be used by ANRIL as anchors for the recognition of specific genomic regions.
In HEK293 cells, ANRIL is likely to modulate the expression of 188 genes in a direct manner
To characterize in depth ANRIL’s trans-activity and to identify the genes potentially regulated by ANRIL in a direct manner, we silenced the expression of the main ANRIL isoforms expressed in HEK293 cells followed by genome-wide expression analysis. This was achieved by using a mix of four LNA GapmeRs (single stranded antisense oligos (ASO)) hybridizing to unique regions of the main ANRIL isoforms as such: GapmeR Exon1 (all isoforms), Exon17–18 (NR isoform), Exon12–13 (DQ isoform) and Exon7–13 (EU isoform) (Figure 2A). A reduction of 75% of ANRIL’s expression level upon treatment of HEK293 cells with this GapmeR mix was observed as compared to treatment with scrambled GapmeR (Figure 2B). This reduction was accompanied by a 2.2- and 6.5-fold increase of CDKN2A and CDKN2B mRNA levels, respectively (Figure 2C). These results were consistent with the ANRIL’s cis-activity previously described (12). Then, total RNAs were extracted and analyzed by next generation Clariom D microarrays from Affymetrix. Upon ANRIL knockdown, 2618 genes (1474 upregulated and 1144 downregulated with an FDR < 0.01, log2FC > |1|) experienced changes in RNA level (Figure 2D). The effects observed on some of the genes upon ANRIL knockdown were further validated by RTqPCR (Supplementary Figure S3A).
We next sought to identify the primary targets of ANRIL. Because one primary target can regulate the expression of many downstream genes, we hypothesized that the genes being both affected by ANRIL knockdown and in direct contact with ANRIL in the chromatin structure are likely to be primary targets of ANRIL. We therefore compared the two lists of the 1474 upregulated and 1144 downregulated genes with the ANRIL ChIRP-seq data. This identified 188 genes fulfilling conditions to be directly regulated (Figure 2E and Supplementary Tables S1 and S2). From the 188 potential direct targets, 123 (P < 6.714e–12) were silenced while 65 (P <0.02) were activated by ANRIL. Gene ontology analysis did not reveal any enriched pathways in the list of the 123 potential direct trans-silenced genes. In contrast, five pathways mainly involved in signal transduction were significantly enriched in the potential direct trans-activated genes dataset (Supplementary Figure S3B). Altogether, we concluded that ANRIL activities encompass repressive and activating functions on a small subset of genes compared to the number of ChIRP peaks.
Since it was documented that ANRIL associates with the PcG to silence genes, we postulated that it might repress a significant number out of the 123 potential direct trans-silenced targets through a similar mechanism (12,13,21). Crossing publicly available H3K27me3 ChIP-seq data performed in HEK293 cells (46) with the list of these genes showed an overlap of only 17% (P < 5.795e–05) (Figure 2F). This suggested that ANRIL is likely to silence genes by both PcG-dependent and independent mechanisms. In the same line, it was described that ANRIL may associate with YY1 to modulate positively gene expression in HUVEC cells (47). To determine the overlap between ANRIL and YY1 targets, we intersected ChIP-seq data of YY1 in HEK293 cells (GEO:GSE127598) with the list of the 65 potential ANRIL activated-primary targets (48). This identified 41 genes (P < 3.332e–16) indicating that YY1 is well suited to be involved in the activation of 63% of them (Figure 2G). In conclusion, our data support the dual-activities of ANRIL depending on PRC2 and YY1. Importantly they are also in favor of mechanisms implicating additional factors, not identified yet, important for ANRIL to modulate gene expression.
TEs in Exon8 are involved in ANRIL’s binding to the genome and gene regulation of 9 potential ANRIL direct trans-targets
Since the three most documented ANRIL isoforms are composed of different combinations of exons proposed to differentially affect gene expression (21), we postulated that each of them might contain unique functional domains (Figure 3A). In order to reveal those responsible for the binding of ANRIL to the genome, we first evaluated the ability of the NR, DQ and EU isoforms to associate with the chromatin fraction. RNA extraction from chromatin was performed after individual overexpression of these MS2-tagged isoforms by transient transfection in HEK293 cells (Figure 3B and Supplementary Figure S4A). Note that these experiments could not be performed on the endogenous isoforms because of their respective low expression levels (Supplementary Figure S4B). This identified both NR and DQ isoforms as DNA/Chromatin binders, but not the EU isoform when compared to the MS2-CTL. This suggested that the exons uniquely found in NR and DQ (exons 2, 3, 4, 8, 9, 10, 11 and 12) may contain RNA domains required for chromatin recognition by ANRIL (Figure 3A). Importantly, it has been shown that TEs in lncRNAs can serve for their chromatin occupancies (10,21,30,32,49). Thus, by using the RepeatMasker version 4.1.0 (http://www.repeatmasker.org/), we looked for such elements and found that only exons 3, 8 and 12 contained TEs as DNA element, LTR and SINE, respectively (Figure 3C and Supplementary Figure S4C). To determine which ones of these exons bind efficiently to the chromatin fraction, we performed RNA extraction from chromatin after individual overexpression of MS2-tagged exons 3, 8 and 12 in HEK293 cells (Supplementary Figure S4D). This identified only exons 3 and 8 as chromatin binders when compared to the MS2-CTL (Figure 3D). Interestingly, two LTRs belonging to the ERVL-MaLRs family were found to cover almost 70% of the 696 nts of Exon8, while repeat elements covered only 17% of the 313 nts of exon 3 (Figure 3C and Supplementary Figure S4E). Note that ERVs are enriched in lncRNAs compared to SINE and LINE classes and are thought to be critical for their genomic association (27,32,44). Thus, we decided to investigate whether Exon8 containing-ERVL could impact ANRIL’s genomic occupancy and subsequently its trans-activity. To test this, we engineered by the CRISPR–Cas9 approach ANRIL gene truncated for the Exon8 in HEK293 cells, hereafter called ΔExon8 HEK293 cells (Supplementary Figure S4F-H). The deletion did not affect the overall expression level of ANRIL nor the CDKN2A and 2B expression (Supplementary Figure S4I). However, RNA extraction from chromatin performed on ΔExon8 cells revealed a significant reduction by 60% in chromatin association of ANRIL, but not for RplpO which was used as a negative control (Figure 3E). These results strongly suggest that Exon8 containing-ERVL is involved, at least partially, in the genomic association of ANRIL.
Figure 3.

Exon8 is involved in the association of ANRIL with the chromatin. (A) Schematic representation of the three major ANRIL isoforms NR, DQ, and EU. Exons and introns are represented by numbered rectangles and dashed lines, respectively. (B) RNA extraction experiments after transient overexpression of the MS2-tagged NR, DQ, and EU isoforms (n = 2). This identified the NR and DQ isoforms as DNA/chromatin binders but not the EU compared to the MS2-CTL. Values are normalized to Input. (C) RepeatMasker analysis showing the distribution of TEs in ANRIL’s exons 3, 8, and 12. (D) RNA extraction experiments after transient overexpression of the MS2-tagged exons 3, 8 and 12 (n = 2). This identified the exons 3 and 8 of ANRIL as DNA/chromatin binders but not the exon 12 compared to the MS2-CTL. Values are normalized to Input. (E) RNA extraction experiments from ΔExon8 HEK293 cell lines which revealed a reduction in chromatin association of ANRIL by 60% but not for RplpO compared to the HEK293 WT cell lines (n = 3). Data are represented as mean ± SEM. P-values: moderated t-statistics, *P < 0.05, **P < 0.01, ***P < 0.001.
To evaluate the global impact of the absence of Exon8 on gene expression, transcriptome analysis was performed on ΔExon8 HEK293 cells using the Clariom D microarrays from Affymetrix. Interestingly, 450 genes showed changes in expression in mutated cells when compared to HEK293 WT (279 upregulated and 171 downregulated with an FDR <0.05, log2FC > |0.6|) (Figure 4A). By intersecting these ΔExon8 up- and down-regulated genes with the ANRIL LNA knockdown and ChIRP-seq datasets, we identified nine common genes (P < 5.053e–08) (TPD52L1, FIRRE, IGFBP3, RYR2, PRDM1, LSM14A, GPATCH1, KRTDAP, ST20-MTHFS) and 1 (UBQLN2) (P < 0.2, not significant) respectively, that could be considered as potential primary targets which expression depends on ANRIL Exon8 (Figure 4B and C). Note that the intersection of the ANRIL and ΔExon8 downregulated genes (n = 1144 and n = 171 respectively) with the ANRIL ChIRP-seq genes (n = 2090) reveals a very restricted overlap suggesting that Exon8 has only a minor contribution in the trans-activation mediated by ANRIL. Therefore, we decided to focus on the nine genes identified as potential direct silenced-targets which expression depends on ANRIL Exon8 called hereafter ΔExon8 trans-silenced targets (Figure 4D). We validated by RTqPCR the significant upregulation of these nine genes in the ΔExon8 cell line compared to the WT HEK293 cells. No pronounced changes were observed for the negative controls HYPM, STAT1 and MX1 (Figure 5A). To go further, we generated a CRISPRed-engineered HEK293 cell line truncated for ANRIL’s Exon21. RTqPCR analysis on this cell line did not reveal any effects on the expression of the ΔExon8 trans-silenced targets except for FIRRE (Supplementary Figure S5A–D). Altogether, our data show that the silencing of nine distal loci is at least in part dependent on the presence of ANRIL’s Exon8.
Figure 4.

The expression of 10 primary trans-targets depends on the presence of Exon8. (A) Volcano plot representing the significance versus fold change for all differentially expressed (n = 450, FDR < 0.05, log2FC > |0.6|) genes in ΔExon8 HEK293 cells (n = 5). (B) Venn diagram showing the intersection between the ANRIL ChIRP-seq genes and the upregulated genes in the ΔExon8 and ANRIL LNA knockdown datasets. A list of nine primary targets has been identified as being potentially silenced by the presence of Exon8 containing-ERVL in a direct manner. (C) Venn diagram showing the intersection between the ANRIL ChIRP-seq genes and the downregulated genes in the ΔExon8 and ANRIL LNA knockdown datasets. One target has been identified as being potentially activated by the Exon8 containing-ERVL in a direct manner. The P-values were obtained by hypergeometric test using the whole set of microarray genes as a background. (D) Table summarizing the 10 ΔExon8 trans-regulated genes. Gene symbol, chromosome, fold change, FDR, ANRIL ChIRP-seq peak number and peak position are represented.
Figure 5.

ANRIL directly contacts specific genes throughout the genome using its Exon8 to regulate their expression through the deposition of H3K27me3. (A) RTqPCR validation of the differentially expressed ΔExon8 trans-silenced targets in ΔExon8 HEK293 cells (n = 5). (B) ChIRP-qPCR on the ΔExon8 trans-silenced targets shows that in the absence of Exon8, ANRIL significantly dissociates from some of these loci (n = 5). Values are normalized to the Input then fold enrichment is calculated by normalizing to LacZ. (C) ChIP-qPCR using H3K27me3 and control IgG antibodies on promoter regions of the ΔExon8 trans-silenced targets (n = 5). Values are normalized to the Input. Data are represented as mean ± SEM. P-values: moderated t-statistics, *P < 0.05, **P < 0.01, ***P < 0.001, ns: not significant.
Exon8 favors ANRIL’s association with specific genomic loci to modulate their expression through H3K27me3 deposition
To gain further insight into the importance of Exon8 in the association of ANRIL with the ΔExon8 trans-silenced targets in cellulo, we performed ANRIL ChIRP-qPCR on the ΔExon8 cells. We verified that the removal of Exon8 did not affect the efficiency of RNA retrieval after capturing the endogenous ANRIL from the chromatin (Supplementary Figure S6A). ChIRP-qPCR analysis showed that ANRIL depleted for Exon8 tended to dissociate from the tested loci. Note that this dissociation appeared more pronounced for the FIRRE, TPD52L1, IGFBP3, RYR2 and PRDM1 loci as significantly evidenced by >3-fold reduction in ΔExon8 HEK293 when compared to the WT cells (Figure 5B). These results confirmed the importance of Exon8 in targeting and tethering ANRIL to at least five specific trans silenced-loci.
Since our data suggested that gene silencing of ANRIL’s primary targets may be mediated by the recruitment of the PcG in a range of 17% (Figure 2F), we sought that the loss of Exon8 might affect H3K27me3 levels at the promoters of some of the ΔExon8 trans-silenced targets. Thus, we performed ChIP-qPCR experiments using antibodies against H3K27me3 or control IgG. A reduction of 60% to 80% of H3K27me3 was observed at the promoters of the FIRRE, TPD52L1, IGFBP3 and KRTDAP genes in ΔExon8 cells compared to WT cells. No reduction in H3K27me3 level was observed at the remaining ΔExon8 trans-silenced targets nor at the HYPM and GAPDH loci used as negative controls (Figure 5C). Additionally, no global changes were observed for H3K4me3 and H3K27ac marks (Supplementary Figure S6B and C). This strongly suggested that their increased expression level observed upon ANRIL knockdown is likely to be independent of active euchromatin mark deposition. In conclusion, our results highlight the implication of Exon8 in the H3K27me3 deposition at the FIRRE, TPD52L1, IGFBP3 and KRTDAP promoters.
Exon8 is presumably involved in ANRIL’s association with the FIRRE and TPD52L1 loci through complex DNA/DNA:RNA structure
It remained to document the mechanism engaged by Exon8 to specifically associate ANRIL with its target genes. The recent development of computational approaches coupled to chromatin purification by RNA selection have provided evidences for a mechanism relying on the formation of DNA/DNA:lncRNA triple helix structures, hereafter called triplex (31,43,44,50–52). Triplex are formed when a single stranded RNA fragment accommodates the major groove of the double stranded DNA by Hoogsteen or reverse Hoogsteen hydrogen bonds in either parallel or anti-parallel orientation. The DNA and RNA regions involved in triplex formation are called Triplex Target Sites (TTS) and DNA Binding Domains (DBD), respectively (53). In order to test the hypothesis of ANRIL interaction with the chromatin via triplex formation, we used Triplex Domain Finder (TDF), a computational method which predicts triplex-forming potential between TTS and DBD based on Hoogsteen hydrogen bonds search (43,53). We submitted the genomic coordinates of the 3,227 ANRIL genomic binding sites against the full length ANRIL sequence. Strikingly, only the Exon8 was predicted to contain a significant DBD (P-value = 0.0013) (Figure 6A and Supplementary Figure S7A). The predicted Ex8-DBD had a length of 42-nts and was rich in purine residues. Interestingly, it was also located within the second LTR/ERVL-MaLR element reinforcing the idea of a role of the Exon8 containing-ERVL in ANRIL’s genome association (Figure 6B and Supplementary Figure S4E). The Ex8-DBD was predicted to form triplex with 422 potential DNA TTSs (13.07%) out of the 3227 ANRIL genomic binding sites (Supplementary Figure S7B). This set of TTSs was not significantly enriched for any particular DNA categories when compared to the human genome (Supplementary Figure S7C). Likewise, no enrichment was observed amongst the ANRIL trans-targets since 18.6% (n = 23) and 16.9% (n = 11) of the potential silenced (n = 123) and activated (n = 65) direct trans-targets contained predicted TTSs, respectively (Figure 2E, Supplementary Figure S7D). Then we intersected the list of the predicted TTSs (n = 422) with the list of the ΔExon8 trans-silenced targets (n = 9). This identified three genes FIRRE, TPD52L1 and LSM14A (P <3.999e–05), containing intronic TTSs, as being potentially targeted by ANRIL Exon8 via triplex formation (Figure 6C). To investigate the triplex forming potential of Exon8 with these three loci, we performed a triplex capture assay on genomic DNA using an adapted protocol (44) (Figure 6D). In this approach, an antisense biotinylated DNA oligo hybridizing to Exon8 was used to capture triplex formed with the full length in vitro transcribed Exon8 incubated with sheared genomic DNA (Figure 6D). After recovery of triplex on streptavidin beads, associated DNA was eluted and analyzed by qPCR. Upon Exon8 pulldown with streptavidin magnetic beads, we found a significant recovery of FIRRE (×2.44) and TPD52L1 (×1.91) TTSs containing-DNA regions compared to GAPDH where no triplex was expected to be formed and that was used as negative control (Figure 6E). Also, no significant enrichment was observed for most of the remaining ΔExon8 trans-silenced targets (except for GPATCH1; ×1.37), which are not predicted to associate with Exon8 by triplex formation. Thus, this was in favor of a potential triplex interaction between Exon8 and the TTSs of FIRRE and TPD52L1. To go further, we checked the chromatin accessibility at these loci by using publicly available ATAC-seq and DNase-seq performed in HEK293 cells (54). Indeed, it has been demonstrated that chromatin located adjacent to triplex forming regions is likely to be depleted in nucleosomes and consequently more accessible to tn5 and DNaseI activities (55,56). We found that ANRIL’s binding sites to TPD52L1 and FIRRE loci are located near DHS and ATAC-seq peaks (Figure 6F). This was not the case for LSM14A, neither for KRTDAP and MX1, which did not harbor predicted TTSs and were used as negative controls (Figure 6F). These data were in agreement with a potential association between ANRIL and the FIRRE and TPD52L1 genes through triplex formation. Next, using electrophoretic mobility shift assay (EMSA), we tested in vitro the triplex forming capacity of ANRIL’s Ex8-DBD (42 nts single-stranded RNA, ssRNA) with the DNA duplex (dsDNA) sequences containing the TTS associated with the selected ANRIL target genes (FIRRE and TPD52L1). As a positive control for our experiment, we used a DBD (40 nts) from the lncRNA NEAT1 which has been shown to form triplex with FLI1 dsDNA (44). Upon incubation of TPD52L1 radiolabeled DNA duplex with ANRIL Ex8-DBD, a decreased electrophoretic mobility was observed on gel indicating an interaction between the DNA and the RNA (Figure 6G). A similar result was obtained with NEAT1-DBD and FLI1 dsDNA while no reduced mobility was observed with FIRRE dsDNA (Supplementary Figure S7E–G). Importantly, the mobility of the formed complex was not affected upon treatment with RNase H, indicating that the observed gel shift was not due to Watson–Crick, but to Hoogsteen or reverse Hoogsteen interactions (Figure 6G and Supplementary Figure S7E–G). Overall, these data suggest that Exon8 contains elements required by ANRIL to contact and modulate the expression of TPD52L1 and FIRRE loci likely through triplex formation.
Figure 6.

Exon8 harbors a DBD potentially involved in triplex formation. (A) TDF prediction using ANRIL full-length against the ChIRP-seq dataset. This revealed the potential DBD on ANRIL’s sequence located in Exon8 (P-value = 0.0013) with its associated TTS (n = 422) hereafter called ChIRP-seq TTSs. (B) Schematic representation of the position and purine-rich sequence of Ex8-DBD. (C) Venn diagram showing the intersection between the predicted ChIRP-seq TTSs and the ΔExon8 trans-silenced targets. A list of three genes (FIRRE, TPD52L1 and LSM14A) was predicted to contain TTSs. The P-values were obtained by hypergeometric test using the whole set of microarray genes as a background. (D) Schematic representation of the RNA-based DNA capture assay adapted from (44) to validate triplex formation. An antisense biotinylated DNA oligo hybridizing to the Exon8 was used to capture triplex formed with full length in vitro transcribed Exon8 incubated with sheared genomic DNA. After recovery of triplex on streptavidin beads, associated DNA was eluted and analyzed by qPCR. (E) Enrichment of FIRRE, TPD52L1 and GPATCH1 peak sequences were obtained but not GAPDH nor the remaining ΔExon8 trans-silenced targets after capturing Exon8 using antisense biotinylated DNA oligo on streptavidin beads (n = 4). The enrichment is presented as the ratio between these loci and TERC which was used as a negative control. (F) ATAC-seq and DNase-seq data evaluating the chromatin accessibility in HEK293 cells at the TPD52L1, FIRRE, LSM14A, MX1 and KRTDAP loci. Signals observed represent open chromatin regions. (G) EMSA using 14 μM of synthetic Ex8-DBD (42 nts) with 100 fmol of double–stranded 32P-labeled double stranded oligonucleotides harboring a predicted TTS of TPD52L1 (Supplementary Table S7). Gel shift was resistant to RNaseH indicating a Hoogsteen base pairing. Potential Hoogsteen base pairing between Ex8-DBD represented in blue and TPD52L1 dsDNA sequences is shown; mismatches are marked *. Data are represented as mean ± SEM. P-values: moderated t-statistics, *P < 0.05, **P < 0.01, ***P < 0.001, ns: not significant.
DISCUSSION
The transcriptional complexity of the ANRIL locus is reflected by the production of several isoforms in a tissue specific manner. The expression of at least three of them positively correlate with severe pathologies such as coronary artery disease, diabetes and cancers (14–18,20,23,57–59). Therefore, they are believed to participate in disease development by inappropriate modulation of gene expression. However, the high variability in the number and identity of the regulated genes according to the model studied obscures our understanding of the mechanistic link between ANRIL and pathologies. In the present study, we provide novel information on how ANRIL trans-regulates some genes, through identification of its potential direct trans-target genes (20,21,47). To circumvent the fact that ANRIL is likely to modulate the expression of many gene regulators, we combined ChIRP-seq with transcriptomic analysis. We found 188 genes that we defined as potential direct trans-targets of ANRIL (Figure 7, Supplementary Tables S1 and S2).
Figure 7.

Summary of ANRIL’s functions. ANRIL interacts with 3227 of loci dispersed throughout the HEK293 cell genome sharing a 21-bp motif enriched in G/A residues. Combining ANRIL genomic occupancy with transcriptomic analysis identified 65 and 123 genes potentially trans-activated and silenced by ANRIL in a direct manner, respectively. Exon8 of ANRIL, mainly made of TEs, contributes to ANRIL genomic association and consequently to its trans-activity. ANRIL might also act as splicing regulator.
The overlap between the genes that were previously identified upon ANRIL knockout or overexpression was low likely due to the heterogeneity in the methods and cellular models used (20,21). For instance, when we compare our work with Holdt et al. (21), even though we both used HEK293 cells, the methods to characterize ANRIL’s trans-activity largely differ. First, Holdt and colleagues overexpressed fragments of ANRIL while we depleted ANRIL by LNA treatment. We preferred gene expression analysis upon ANRIL knockdown compared to overexpression since it may generate cellular stresses. Also, we considered that overexpressing ANRIL in HEK293 cells where ANRIL is expressed would not result in a drastic downregulation of the potential repressed primary targets since they are expected to be already silenced. The main difference compared to the other studies was the use of different cell lines such endothelial and vascular muscle cells (20,47).
Gene ontology analysis revealed in the list of the 65 trans-activated genes several pathways significantly enriched mainly involved in regulation of receptor internalization (for instance the GPC3 and GJA1 genes). This pathway ensures the proper delivery of internalized receptors to specific subcellular destinations in order to maintain cellular homeostasis (60). Interestingly, this pathway closely modulates cell proliferation and some genes such as GPC3 and GJA1 are linked to cancers reminiscent to diseases associated with ANRIL (61,62). In contrast, gene ontology analysis failed to identify common pathways shared by its potential 123 primary silenced-targets. However, we could identify several genes involved in cell cycle progression (CDC5L) and inflammation (IL16), pathways which are again reminiscent of cancer and cardiovascular diseases linked to ANRIL (63,64). Importantly, our list of potential ANRIL trans-target genes includes non-coding genes ignored so far (SNORA14B, SNORA33, TSIX, LINC01023, LINC00923 and FIRRE). As such ncRNAs may play critical functions in cellular homeostasis (65), this finding opens new avenues for future investigations of ANRIL’s functions, in particular in the view to better understand the connection between ANRIL and disease progression.
As mentioned above, we found that 123 genes out of the 188 direct trans-targets experienced a higher expression upon ANRIL depletion (Figure 7, Supplementary Table S1). We also found that only 17% of them are likely to be regulated by a PcG-dependent mechanism. This observation suggests the implication of PcG-independent mechanisms engaged by ANRIL to silence gene expression. Several studies have uncovered examples of lncRNAs that are able to silence gene expression by recruiting different repressive complexes. This is the case for HOTAIR which can associate with the PRC2 and CoREST complexes responsible for H3K27me3 deposition and H3K4me1–2 removal at the HOXD locus, respectively (66,67). We also identified 65 genes positively regulated by ANRIL arguing for its activating role. One study showed that ANRIL interacts with the transcription activator YY1 in HUVEC cells (47). Interestingly, we found that 63% of the potential trans-activated primary targets of ANRIL are likely to be bound by YY1 in HEK293 cells (Figure 7). This reinforced the potential implication of YY1 in ANRIL’s activating function and further studies are needed to discover the underlying mechanisms.
As we found that TEs cover 35% of the ANRIL sequence (Supplementary Figure S8), we evaluated their putative importance on ANRIL’s trans-silencing activity. We demonstrated that Exon8 which is 70% covered by the subcategory of LTR named ERVL-MaLR is involved in ANRIL’s genomic occupancy. Note that clinical investigation aiming to precisely correlate the presence of Exon8 in ANRIL isoforms to disease occurrence has never been done. This point should be examined in depth in the future.
Importantly, Exon8 deletion affected the expression of nine genes out of the 123 potential trans-silenced targets of ANRIL. Since CDKN2A and CDKN2B were not found among them, we concluded that Exon8 containing-ERVL does not function in cis but in trans on a limited number of genes. This limited number of potential ΔExon8 trans-silenced targets emphasizes the importance of other TEs which may help ANRIL to fully act in trans. They also indicate that ANRIL variants are likely constituted by functional blocks and that combination of these blocks may confer particular features for chromatin-linked activities (Figure 7). In this line of idea, we observed that the expression of FIRRE is affected in cells lacking Exon8 or Exon21 (Figure 5A and Supplementary Figure S5D), suggesting that both exons might cooperate to modulate its expression. In addition, our results suggest that Exon8 certainly contributes more by its role in helping ANRIL to recognize specific genomic loci rather than by directly recruiting the PcG since Exon8 deletion results in a significant eviction of ANRIL from the chromatin structure. Therefore, Exon8 containing-ERVL may serve essentially for specific chromatin association, while Alu sequences, for instance, would potentially favor protein recruitment. Accordingly, deletion of Alu sequences abolished the so-called pro-atherogenic functions of ANRIL (21) and have been shown to interact with a large repertoire of proteins such as chromatin modifiers, transcription factors, RNA binding proteins and RNA polymerase II (68–71).
Recent studies suggested a potential implication of repeat elements in DNA/DNA:RNA triplex formation. Thus, we used an in silico predictive approach to screen for possible direct ANRIL–DNA triplex formation (43,44,50,52,72). The ERVL-MaLR in Exon8 contained a DBD predicted to form triplex with TTSs identified in 3 of the 9 ΔExon8 trans-silenced targets (the non-coding gene FIRRE, and the protein coding genes TPD52L1 and LSM14A). Accordingly, a recently published article showed that ANRIL regulates in cis the CDKN2B gene by forming triple helices via a DBD located in ANRIL’s Exon1 (73).
We provide by alternative approaches pieces of evidence suggesting triplex formation between the Exon8 and FIRRE and TPD52L1 loci. Note that we only confirmed the Hoogsteen base-pairing formation by EMSA for the latter. This may be explained by the fact that conditions for triplex formation in vitro differ from those in cellulo where different factors may be involved, such as nucleosomes which were shown to stabilize triplex structures (55). Nevertheless, the examination of publicly available data of ATAC-seq and DNase-seq identified nucleosome free regions adjacent to triplex forming region of FIRRE and TPD52L1, which is in favor of triplex formation.
FIRRE and TPD52L1 are good candidates for a better understanding of how ANRIL impacts disease etiology. Indeed, TPD52L1 is a protein coding gene highly upregulated in breast cancer cell lines that was identified as a cell cycle regulator important for the completion of mitosis by interacting with 14–3–3, a negative regulator of the G2/M phase transition. Similarly, ANRIL also behaves as a cell cycle regulator by mediating the expression of tumor suppressor genes (12,13). In human, the lncRNA FIRRE, which is encoded from the X chromosome, is involved in post-transcriptional regulation of inflammatory genes, a pathway that is linked to ANRIL in the context of cardiovascular diseases (47,74). Upregulated in human cancer, FIRRE is considered as a marker for prognosis and diagnosis in human head and neck squamous cell carcinoma (HNSCC) (75). In mouse, Firre was shown to regulate the nuclear architecture through distinct interchromosomal interactions with five genomic regions (76). Additional functions have been attributed to Firre such as modulating adipogenesis, key pluripotency pathways and anchoring the mouse inactive X chromosome to maintain H3K27me3 status (76–78). Even though our results display coherent connections with ANRIL-associated pathways such as inflammation and cell proliferation (12,17,20,21,47), studies evaluating the link between ANRIL and FIRRE/TPD52L1 in pathological situations will likely yield further mechanistic insights on the role of ANRIL’s trans-regulatory activities in the establishment of diseases.
The pioneer ChIRP-seq experiment we performed revealed that besides its 188 potential trans-targets, ANRIL associates much widely with the genome by binding ∼3000 sites (Figure 7). This may reflect the fact that, our ChIRP-seq experiments were done using tiling probes hybridizing to all ANRIL exons. Therefore, they capture as a whole the genomic sites of the full set of ANRIL variants. Unfortunately, due to the limited abundance of some of the ANRIL isoforms, we could not evaluate their individual genomic occupancy using the dChIRP approach (data not shown) (79). We also observed that most of the ANRIL binding sites are located in non-coding areas such as introns and intergenic regions. This location is in agreement with the modulator roles of lncRNAs on enhancers activity, alternative splicing and chromatin organization (36,80–83). For instance, the contribution of lncRNAs on splicing was exemplified by the regulatory activity of the lncRNA asFGFR2 on the alternative splicing of the FGFR2 transcript (83). Remarkably, 40.3% of the ANRIL sites are intronic suggesting a possible role of ANRIL as a splicing regulator (Figure 7). Moreover, we found that 24% of the genes contacted by ANRIL are affected in terms of alternative splicing upon ANRIL knockdown (data not shown). More in-depth investigations are required to better understand the possible role of ANRIL as a splicing regulator.
Finally, we observed that the ANRIL binding sites within the genome are enriched in G/A nucleotides (motif 1) (Figure 7). This property was also observed for the HOTAIR, MEG3, TERRA and NEAT1 lncRNAs (36,44,52,82,84). This supports the emergent idea that G/A-rich sequences might serve as anchoring motifs to direct lncRNAs toward specific genomic loci (85,86). We can speculate that such composition may favor triplex formation since G/A residues promote the most stable Hoogsteen base-pairing formation (87). Note that 13% of the ChIRP-peaks have a TTS for which no significant enrichment was found neither for the up- and -down regulated primary trans-targets nor for any DNA classes (Supplementary Figure S7C). This suggests that triplex formation occurs independently of DNA sub-categories and ANRIL’s trans-functions. Importantly, this indicates that this genome recognition mode cannot solely explain ANRIL’s binding to the chromatin and that alternative modes engaged by ANRIL to associate with the genome have to be considered. LncRNA-chromatin recognition can take place through the interaction with specific protein partners that serve as bridge between the DNA and the lncRNA. One of the most characterized protein showing this property is the heterogeneous nuclear RiboNucleoProtein U (hnRNP U) matrix protein (76,88). The use of publicly available CLIP-seq data did not provide any evidences of direct hnRNP U binding to ANRIL (89), which suggests that ANRIL/chromatin association is most probably not dependent on hnRNP U. Another mechanism relies on the direct interaction of the lncRNA with the DNA molecule via RNA-DNA hybrid duplexes formed by canonical Watson-Crick base-pairing. The resulting hybrid is named R-loop (80,90). The FIMO analysis we performed to scan for the presence of motif 1 within the ANRIL sequence identified 6 sequences located in Exon1, 8 and 21 that might hybridize to the negative strand of the motif 1 (Supplementary Figure S9). This may indicate that ANRIL contains sequence elements compatible with R-loop formation with the motif 1. Even though this observation helps to better understand how ANRIL associates with thousands of genomic loci, further investigations are needed to demonstrate genome-wide the existence of R-loops formed between ANRIL and the motif 1.
DATA AVAILABILITY
All data generated or analyzed in this study are included in the published article and its supplementary information. The GEO accession number of the ChIRP-seq and microarray data is GSE162224.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the ‘RNA-RNP team’ for critical reading of the manuscript.
Author contributions: Conceptualization, S.M., I.B.-A., and C.A.; Methodology, S.M., I.B.-A., C.A., Y.M. and C.B.; Investigation, S.M., I.B.-A., C.A., A.S., R.R., Q.T., V.I.-B., V.M. and Y.M.; Writing – Original Draft, S.M., I.B.-A. and C.A.; Writing – Review & Editing, S.M., I.B.-A., C.A., C.B., Y.M. and V.M.; Funding Acquisition, S.M., I.B.-A. and C.B.; Supervision, S.M. and I.B.-A.
Contributor Information
Charbel Alfeghaly, Université de Lorraine, CNRS, IMoPA, F-54000 Nancy, France.
Aymeric Sanchez, Université de Lorraine, CNRS, IMoPA, F-54000 Nancy, France.
Raphael Rouget, Université de Lorraine, CNRS, IMoPA, F-54000 Nancy, France.
Quentin Thuillier, Université de Lorraine, CNRS, IMoPA, F-54000 Nancy, France.
Valérie Igel-Bourguignon, Université de Lorraine, CNRS, IMoPA, F-54000 Nancy, France; Université de Lorraine, CNRS, INSERM, UMS2008 IBSLor, Epitranscriptomics and RNA Sequencing (EpiRNA-Seq) Core Facility, F-54000 Nancy, France.
Virginie Marchand, Université de Lorraine, CNRS, INSERM, UMS2008 IBSLor, Epitranscriptomics and RNA Sequencing (EpiRNA-Seq) Core Facility, F-54000 Nancy, France.
Christiane Branlant, Université de Lorraine, CNRS, IMoPA, F-54000 Nancy, France.
Yuri Motorin, Université de Lorraine, CNRS, IMoPA, F-54000 Nancy, France.
Isabelle Behm-Ansmant, Université de Lorraine, CNRS, IMoPA, F-54000 Nancy, France.
Sylvain Maenner, Université de Lorraine, CNRS, IMoPA, F-54000 Nancy, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
French PIA project “Lorraine Université d’Excellence" [ANR-15-IDEX-04-LUE, in part]; PIA RHU program FIGHT-HF [ANR-15-RHU-004]; CNRS-UL and the FR3209 (renamed UMS2008-IBSLor) in the context of Action de Site Mirabelle and PEPS initiatives, respectively.
Conflict of interest statement. None declared.
REFERENCES
- 1. Zhao Y., Li H., Fang S., Kang Y., wu W., Hao Y., Li Z., Bu D., Sun N., Zhang M.Q.et al.. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016; 44:D203–D208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bao Z., Yang Z., Huang Z., Zhou Y., Cui Q., Dong D.. LncRNADisease 2.0: an updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019; 47:D1034–D1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mercer T.R., Dinger M.E., Mattick J.S.. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 2009; 10:155–159. [DOI] [PubMed] [Google Scholar]
- 4. Wapinski O., Chang H.Y.. Long noncoding RNAs and human disease. Trends Cell Biol. 2011; 21:354–361. [DOI] [PubMed] [Google Scholar]
- 5. Yao R.-W., Wang Y., Chen L.-L.. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019; 21:542–551. [DOI] [PubMed] [Google Scholar]
- 6. Achour C., Aguilo F.. Long non-coding RNA and Polycomb: an intricate partnership in cancer biology. Front. Biosci. Landmark Ed. 2018; 23:2106–2132. [DOI] [PubMed] [Google Scholar]
- 7. Davidovich C., Cech T.R.. The recruitment of chromatin modifiers by long noncoding RNAs: lessons from PRC2. RNA. 2015; 21:2007–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Long Y., Wang X., Youmans D.T., Cech T.R.. How do lncRNAs regulate transcription. Sci. Adv. 2017; 3:eaao2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maenner S., Blaud M., Fouillen L., Savoye A., Marchand V., Dubois A., Sanglier-Cianférani S., Van Dorsselaer A., Clerc P., Avner P.et al.. 2-D structure of the a region of Xist RNA and its implication for PRC2 association. PLoS Biol. 2010; 8:e1000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao J., Sun B.K., Erwin J.A., Song J.-J., Lee J.T.. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008; 322:750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khalil A.M., Guttman M., Huarte M., Garber M., Raj A., Rivea Morales D., Thomas K., Presser A., Bernstein B.E., van Oudenaarden A.et al.. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:11667–11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kotake Y., Nakagawa T., Kitagawa K., Suzuki S., Liu N., Kitagawa M., Xiong Y.. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15INK4B tumor suppressor gene. Oncogene. 2011; 30:1956–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yap K.L., Li S., Muñoz-Cabello A.M., Raguz S., Zeng L., Mujtaba S., Gil J., Walsh M.J., Zhou M.-M.. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell. 2010; 38:662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheng J., Cai M.-Y., Chen Y.-N., Li Z.-C., Tang S.-S., Yang X.-L., Chen C., Liu X., Xiong X.-D.. Variants in ANRIL gene correlated with its expression contribute to myocardial infarction risk. Oncotarget. 2017; 8:12607–12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cunnington M.S., Santibanez Koref M., Mayosi B.M., Burn J., Keavney B.. Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with ANRIL expression. PLos Genet. 2010; 6:e1000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Folkersen L., Kyriakou T., Goel A., Peden J., Mälarstig A., Paulsson-Berne G., Hamsten A., Franco-Cereceda A., Gabrielsen A., Eriksson P.. Relationship between CAD risk genotype in the chromosome 9p21 locus and gene expression. Identification of eight new ANRIL splice variants. PLoS One. 2009; 4:e7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Holdt L.M., Beutner F., Scholz M., Gielen S., Gaebel G., Bergert H., Schuler G., Thiery J., Teupser D.. ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler. Thromb. Vasc. Biol. 2010; 30:620–627. [DOI] [PubMed] [Google Scholar]
- 18. Olga J., Stewart Alexandre F.R., Robert R., George W., Paulina L., Thet N., Christine B., McLean Bradley W., Cook Richard C., Parker Joel S.et al.. Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler. Thromb. Vasc. Biol. 2009; 29:1671–1677. [DOI] [PubMed] [Google Scholar]
- 19. Kumar A., Thomas S.K., Wong K.C., Lo Sardo V., Cheah D.S., Hou Y.-H., Placone J.K., Tenerelli K.P., Ferguson W.C., Torkamani A.et al.. Mechanical activation of noncoding-RNA-mediated regulation of disease-associated phenotypes in human cardiomyocytes. Nat. Biomed. Eng. 2019; 3:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lo Sardo V., Chubukov P., Ferguson W., Kumar A., Teng E.L., Duran M., Zhang L., Cost G., Engler A.J., Urnov F.et al.. Unveiling the role of the most impactful cardiovascular risk locus through haplotype editing. Cell. 2018; 175:1796–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holdt L.M., Hoffmann S., Sass K., Langenberger D., Scholz M., Krohn K., Finstermeier K., Stahringer A., Wilfert W., Beutner F.et al.. Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLos Genet. 2013; 9:e1003588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sato K., Nakagawa H., Tajima A., Yoshida K., Inoue I.. ANRIL is implicated in the regulation of nucleus and potential transcriptional target of E2F1. Oncol. Rep. 2010; 24:701–707. [DOI] [PubMed] [Google Scholar]
- 23. Congrains A., Kamide K., Katsuya T., Yasuda O., Oguro R., Yamamoto K., Ohishi M., Rakugi H.. CVD-associated non-coding RNA, ANRIL, modulates expression of atherogenic pathways in VSMC. Biochem. Biophys. Res. Commun. 2012; 419:612–616. [DOI] [PubMed] [Google Scholar]
- 24. Cabili M.N., Dunagin M.C., McClanahan P.D., Biaesch A., Padovan-Merhar O., Regev A., Rinn J.L., Raj A.. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 2015; 16:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thomas A.A., Feng B., Chakrabarti S.. ANRIL: a regulator of VEGF in diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 2017; 58:470–480. [DOI] [PubMed] [Google Scholar]
- 26. Makałowski W. Genomic scrap yard: how genomes utilize all that junk. Gene. 2000; 259:61–67. [DOI] [PubMed] [Google Scholar]
- 27. Kelley D., Rinn J.. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012; 13:R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carlevaro-Fita J., Polidori T., Das M., Navarro C., Zoller T.I., Johnson R.. Ancient exapted transposable elements promote nuclear enrichment of human long noncoding RNAs. Genome Res. 2018; 208–222.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chishima T., Iwakiri J., Hamada M.. Identification of transposable elements contributing to tissue-specific expression of long non-coding RNAs. Genes. 2018; 9:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grote P., Wittler L., Hendrix D., Koch F., Währisch S., Beisaw A., Macura K., Bläss G., Kellis M., Werber M.et al.. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell. 2013; 24:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jalali S., Singh A., Maiti S., Scaria V.. Genome-wide computational analysis of potential long noncoding RNA mediated DNA:DNA:RNA triplexes in the human genome. J. Transl. Med. 2017; 15:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson R., Guigó R.. The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs. RNA. 2014; 20:959–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kannan S., Chernikova D., Rogozin I.B., Poliakov E., Managadze D., Koonin E.V., Milanesi L.. Transposable element insertions in long intergenic non-coding RNA genes. Front. Bioeng. Biotechnol. 2015; 3:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Loewer S., Cabili M.N., Guttman M., Loh Y.-H., Thomas K., Park I.H., Garber M., Curran M., Onder T., Agarwal S.et al.. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat. Genet. 2010; 42:1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chu C., Qu K., Zhong F., Artandi S.E., Chang H.Y.. Genomic maps of lincRNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell. 2011; 44:667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yu G., Wang L.-G., He Q.-Y.. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015; 31:2382–2383. [DOI] [PubMed] [Google Scholar]
- 41. Bailey T.L., Elkan C.. Fitting a mixture model by expectation maximization to discover motifs in biopolymer. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994; 2:28–36. [PubMed] [Google Scholar]
- 42. Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kuo C.-C., Hänzelmann S., Sentürk Cetin N., Frank S., Zajzon B., Derks J.-P., Akhade V.S., Ahuja G., Kanduri C., Grummt I.et al.. Detection of RNA–DNA binding sites in long noncoding RNAs. Nucleic Acids Res. 2019; 47:e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sentürk Cetin N., Kuo C.-C., Ribarska T., Li R., Costa I.G., Grummt I.. Isolation and genome-wide characterization of cellular DNA:RNA triplex structures. Nucleic Acids Res. 2019; 47:2306–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Engreitz J.M., Pandya-Jones A., McDonel P., Shishkin A., Sirokman K., Surka C., Kadri S., Xing J., Goren A., Lander E.S.et al.. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X-chromosome. Science. 2013; 341:1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oki S., Ohta T., Shioi G., Hatanaka H., Ogasawara O., Okuda Y., Kawaji H., Nakaki R., Sese J., Meno C.. ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018; 19:e46255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhou X., Han X., Wittfeldt A., Sun J., Liu C., Wang X., Gan L.-M., Cao H., Liang Z.. Long non-coding RNA ANRIL regulates inflammatory responses as a novel component of NF-κB pathway. RNA Biol. 2015; 13:98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R.et al.. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jeon Y., Lee J.T.. YY1 tethers Xist RNA to the inactive X nucleation center. Cell. 2011; 146:119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Buske F.A., Bauer D.C., Mattick J.S., Bailey T.L.. Triplexator: detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res. 2012; 22:1372–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kalwa M., Hänzelmann S., Otto S., Kuo C.-C., Franzen J., Joussen S., Fernandez-Rebollo E., Rath B., Koch C., Hofmann A.et al.. The lncRNA HOTAIR impacts on mesenchymal stem cells via triple helix formation. Nucleic Acids Res. 2016; 44:10631–10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mondal T., Subhash S., Vaid R., Enroth S., Uday S., Reinius B., Mitra S., Mohammed A., James A.R., Hoberg E.et al.. MEG3 long noncoding RNA regulates the TGF-β pathway genes through formation of RNA–DNA triplex structures. Nat. Commun. 2015; 6:7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li Y., Syed J., Sugiyama H.. RNA-DNA triplex formation by long noncoding RNAs. Cell Chem. Biol. 2016; 23:1325–1333. [DOI] [PubMed] [Google Scholar]
- 54. Karabacak Calviello A., Hirsekorn A., Wurmus R., Yusuf D., Ohler U.. Reproducible inference of transcription factor footprints in ATAC-seq and DNase-seq datasets using protocol-specific bias modeling. Genome Biol. 2019; 20:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Maldonado R., Schwartz U., Silberhorn E., Längst G.. Nucleosomes stabilize ssRNA-dsDNA triple helices in human cells. Mol. Cell. 2019; 73:1243–1254. [DOI] [PubMed] [Google Scholar]
- 56. Li Z., Schulz M.H., Look T., Begemann M., Zenke M., Costa I.G.. Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 2019; 20:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Burd C.E., Jeck W.R., Liu Y., Sanoff H.K., Wang Z., Sharpless N.E.. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010; 6:e1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hubberten M., Bochenek G., Chen H., Häsler R., Wiehe R., Rosenstiel P., Jepsen S., Dommisch H., Schaefer A.S.. Linear isoforms of the long noncoding RNA CDKN2B-AS1 regulate the c-myc-enhancer binding factor RBMS1. Eur. J. Hum. Genet. 2019; 27:80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sarkar D., Oghabian A., Bodiyabadu P.K., Joseph W.R., Leung E.Y., Finlay G.J., Baguley B.C., Askarian-Amiri M.E.. Multiple Isoforms of ANRIL in Melanoma Cells: Structural Complexity Suggests Variations in Processing. Int. J. Mol. Sci. 2017; 18:1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang J., Fedoseienko A., Chen B., Burstein E., Jia D., Billadeau D.D.. Endosomal receptor trafficking: Retromer and beyond. Traffic. 2018; 19:578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fernández D., Guereño M., Lago Huvelle M.A., Cercato M., Peters M.G.. Signaling network involved in the GPC3-induced inhibition of breast cancer progression: role of canonical Wnt pathway. J. Cancer Res. Clin. Oncol. 2018; 144:2399–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Busby M., Hallett M.T., Plante I.. The complex subtype-dependent role of Connexin 43 (GJA1) in breast cancer. Int. J. Mol. Sci. 2018; 19:693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mathy N.L., Scheuer W., Lanzendörfer M., Honold K., Ambrosius D., Norley S., Kurth R.. Interleukin-16 stimulates the expression and production of pro-inflammatory cytokines by human monocytes. Immunology. 2000; 100:63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mu R., Wang Y.-B., Wu M., Yang Y., Song W., Li T., Zhang W.-N., Tan B., Li A.-L., Wang N.et al.. Depletion of pre-mRNA splicing factor Cdc5L inhibits mitotic progression and triggers mitotic catastrophe. Cell Death. Dis. 2014; 5:e1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Salviano-Silva A., Lobo-Alves S.C., de Almeida R.C., Malheiros D., Petzl-Erler M.L.. Besides pathology: long non-coding RNA in cell and tissue homeostasis. Non-Coding RNA. 2018; 4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rinn J.L., Kertesz M., Wang J.K., Squazzo S.L., Xu X., Brugmann S.A., Goodnough L.H., Helms J.A., Farnham P.J., Segal E.et al.. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007; 129:1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tsai M.-C., Manor O., Wan Y., Mosammaparast N., Wang J.K., Lan F., Shi Y., Segal E., Chang H.Y.. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010; 329:689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Blackwell B.J., Lopez M.F., Wang J., Krastins B., Sarracino D., Tollervey J.R., Dobke M., Jordan I.K., Lunyak V.V.. Protein interactions with piALU RNA indicates putative participation of retroRNA in the cell cycle, DNA repair and chromatin assembly. Mob. Genet. Elem. 2012; 2:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Goodier J.L., Cheung L.E., Kazazian H.H.. Mapping the LINE1 ORF1 protein interactome reveals associated inhibitors of human retrotransposition. Nucleic Acids Res. 2013; 41:7401–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mariner P.D., Walters R.D., Espinoza C.A., Drullinger L.F., Wagner S.D., Kugel J.F., Goodrich J.A.. Human Alu RNA is a modular transacting repressor of mRNA transcription during heat shock. Mol. Cell. 2008; 29:499–509. [DOI] [PubMed] [Google Scholar]
- 71. Fasolo F., Patrucco L., Volpe M., Bon C., Peano C., Mignone F., Carninci P., Persichetti F., Santoro C., Zucchelli S.et al.. The RNA-binding protein ILF3 binds to transposable element sequences in SINEUP lncRNAs. FASEB J. 2019; 33:13572–13589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vance K.W., Ponting C.P.. Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends Genet. 2014; 30:348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ou M., Li X., Zhao S., Cui S., Tu J.. Long non-coding RNA CDKN2B-AS1 contributes to atherosclerotic plaque formation by forming RNA-DNA triplex in the CDKN2B promoter. EBioMedicine. 2020; 55:102694. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 74. Lu Y., Liu X., Xie M., Liu M., Ye M., Li M., Chen X.-M., Li X., Zhou R.. The NF-κB–responsive long noncoding RNA FIRRE regulates posttranscriptional regulation of inflammatory gene expression through interacting with hnRNPU. J. Immunol. Author Choice. 2017; 199:3571–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nohata N., Abba M.C., Gutkind J.S.. Unraveling the oral cancer lncRNAome: identification of novel lncRNAs associated with malignant progression and HPV infection. Oral Oncol. 2016; 59:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hacisuleyman E., Goff L.A., Trapnell C., Williams A., Henao-Mejia J., Sun L., McClanahan P., Hendrickson D.G., Sauvageau M., Kelley D.R.et al.. Topological organization of multi-chromosomal regions by Firre. Nat. Struct. Mol. Biol. 2014; 21:198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sun L., Goff L.A., Trapnell C., Alexander R., Lo K.A., Hacisuleyman E., Sauvageau M., Tazon-Vega B., Kelley D.R., Hendrickson D.G.et al.. Long noncoding RNAs regulate adipogenesis. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:3387–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yang F., Deng X., Ma W., Berletch J.B., Rabaia N., Wei G., Moore J.M., Filippova G.N., Xu J., Liu Y.et al.. The lncRNA Firre anchors the inactive X chromosome to the nucleolus by binding CTCF and maintains H3K27me3 methylation. Genome Biol. 2015; 16:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Quinn J.J., Ilik I.A., Qu K., Georgiev P., Chu C., Akhtar A., Chang H.Y.. Revealing long noncoding RNA architecture and functions using domain-specific chromatin isolation by RNA purification. Nat. Biotechnol. 2014; 32:933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Arab K., Karaulanov E., Musheev M., Trnka P., Schäfer A., Grummt I., Niehrs C.. GADD45A binds R-loops and recruits TET1 to CpG island promoters. Nat. Genet. 2019; 51:217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Blank-Giwojna A., Postepska-Igielska A., Grummt I.. lncRNA KHPS1 activates a poised enhancer by triplex-dependent recruitment of epigenomic regulators. Cell Rep. 2019; 26:2904–2915. [DOI] [PubMed] [Google Scholar]
- 82. Chu H.-P., Cifuentes-Rojas C., Kesner B., Aeby E., Lee H., Wei C., Oh H.J., Boukhali M., Haas W., Lee J.T.. TERRA RNA antagonizes ATRX and protects telomeres. Cell. 2017; 170:86–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gonzalez I., Munita R., Agirre E., Dittmer T.A., Gysling K., Misteli T., Luco R.F.. A lncRNA regulates alternative splicing via establishment of a splicing-specific chromatin signature. Nat. Struct. Mol. Biol. 2015; 22:370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. West J.A., Davis C.P., Sunwoo H., Simon M.D., Sadreyev R.I., Wang P.I., Tolstorukov M.Y., Kingston R.E.. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell. 2014; 55:791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kim M.J., Kim H., Shin J.S., Chung C.-H., Ohlrogge J.B., Suh M.C.. Seed-specific expression of sesame microsomal oleic acid desaturase is controlled by combinatorial properties between negative cis-regulatory elements in the SeFAD2 promoter and enhancers in the 5′-UTR intron. Mol. Genet. Genomics MGG. 2006; 276:351–368. [DOI] [PubMed] [Google Scholar]
- 86. Rose A.B. Introns as gene regulators: a brick on the accelerator. Front. Genet. 2019; 9:672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kunkler C.N., Hulewicz J.P., Hickman S.C., Wang M.C., McCown P.J., Brown J.A.. Stability of an RNA•DNA–DNA triple helix depends on base triplet composition and length of the RNA third strand. Nucleic Acids Res. 2019; 47:7213–7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hasegawa Y., Brockdorff N., Kawano S., Tsutui K., Tsutui K., Nakagawa S.. The matrix protein hnRNP U is required for chromosomal localization of Xist RNA. Dev. Cell. 2010; 19:469–476. [DOI] [PubMed] [Google Scholar]
- 89. Li J.-H., Liu S., Zhou H., Qu L.-H., Yang J.-H.. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014; 42:D92–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gibbons H.R., Shaginurova G., Kim L.C., Chapman N., Spurlock C.F., Aune T.M.. Divergent lncRNA GATA3-AS1 regulates GATA3 transcription in T-Helper 2 cells. Front. Immunol. 2018; 9:2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed in this study are included in the published article and its supplementary information. The GEO accession number of the ChIRP-seq and microarray data is GSE162224.