

Abstract
The broad host range of Fusarium virguliforme represents a unique comparative system to identify and define differentially induced responses between an asymptomatic monocot host, maize (Zea mays), and a symptomatic eudicot host, soybean (Glycine max). Using a temporal, comparative transcriptome-based approach, we observed that early gene expression profiles of root tissue from infected maize suggest that pathogen tolerance coincides with the rapid induction of senescence dampening transcriptional regulators, including ANACs (Arabidopsis thaliana NAM/ATAF/CUC protein) and Ethylene-Responsive Factors. In contrast, the expression of senescence-associated processes in soybean was coincident with the appearance of disease symptom development, suggesting pathogen-induced senescence as a key pathway driving pathogen susceptibility in soybean. Based on the analyses described herein, we posit that root senescence is a primary contributing factor underlying colonization and disease progression in symptomatic versus asymptomatic host–fungal interactions. This process also supports the lifestyle and virulence of F. virguliforme during biotrophy to necrotrophy transitions. Further support for this hypothesis lies in comprehensive co-expression and comparative transcriptome analyses, and in total, supports the emerging concept of necrotrophy-activated senescence. We propose that F. virguliforme conditions an environment within symptomatic hosts, which favors susceptibility through transcriptomic reprogramming, and as described herein, the induction of pathways associated with senescence during the necrotrophic stage of fungal development.
Fusarium-induced root senescence is differentially regulated in symptomatic hosts (e.g. soybean) versus asymptomatic hosts (e.g. maize).
Introduction
Analyses of contrasting phenes have illuminated our understanding of the complexity of processes in plants, examples of which include the regulation and interaction between plant development and the environment (York et al., 2013) and response to abiotic stress (Lynch et al., 2014). Indeed, “genotype × phenotype” interaction studies have aided in the identification of a multitude of genetic interactions underpinning host susceptibility and resistance to pathogens. For example, comparative studies evaluating phenotypically distinct responses within a single plant to a single pathogen have provided insight at physiological-to-genetic/genomic-scale resolution (O'Connell et al., 2012; Lorrain et al., 2018). Indeed, when coupled with functional analyses of gene networks, comparative transcriptome-based approaches have provided insight into a wide breadth of immune signaling processes. Key examples include processes governing (1) the activation of the hypersensitive response, a hallmark of effector-triggered immunity (Chisholm et al., 2006) and the activation of resistance (R) genes (Etalo et al., 2013), (2) pattern-triggered immunity (Bagnaresi et al., 2012; Zhang et al., 2018a), and (3) the role of antimicrobial signaling and metabolism (Dupont et al., 2015). Taken together, the array of these processes illustrates the temporal colonization dynamics between susceptible and resistant plant cultivars during their interaction(s) with a variety of pathogenic organisms (Kong et al., 2015; Burkhardt and Day, 2016).

While numerous processes required for the regulation of plant immunity have been described through the use of pairwise genomics- and transcriptomics-based approaches, most studies to date have focused on comparison(s) within cultivars from a single species, or species within a single genera (Lanubile et al., 2015; Burkhardt and Day, 2016; Chen et al., 2016; Chowdhury et al., 2017). Thus, knowledge gaps still remain in our understanding of how pathogens with broad host ranges modulate immune signaling within diverged hosts. This is particularly limiting in the case of fungal pathogens, many of which colonize a broad range of agronomically important crops, yet only a few are true pathogens (Gdanetz and Trail, 2017; Banerjee et al., 2019). For example, phytopathogenic Ascomycetes found within the species complexes of Fusarium solani and F. oxysporum colonize more than 200 plant species, including both monocots and eudicots (Michielse and Rep, 2009; Šišić et al., 2018). We posit that a comparison of host responses across highly diverged plant species colonized by the same fungus will provide insight(s) into immunity-induced pathways relevant to both resistance and tolerance. Moreover, such comparisons would be highly relevant and translatable to monoculture-based agroecosystems, particularly those comprising rotational crops, which are typically exposed to similar microbial community compositions each growing season (Gdanetz and Trail, 2017; Katan, 2017; Leandro et al., 2018). Relevant to the work described herein, rotation-based management practices are proving to be unsustainable as many field crops support the asymptomatic persistence of numerous pathogenic fungi (Kolander et al., 2012; Malcolm et al., 2013; Lofgren et al., 2018).
A mechanistic understanding of the molecular-genetic fundamentals of plant immunity has been largely built upon the framework of the model plant Arabidopsis thaliana, and from this, a robust understanding of eudicot immune signaling exists. However, immune signaling in monocots remains largely enigmatic (Balmer et al., 2012, 2013), and whilst defense gene homologs in monocots and eudicots have been described, there are many examples where well-defined eudicot pathways are not consistently conserved, nor do they function the same in monocots (Humphry et al., 2010; Lu et al., 2011; Balmer et al., 2013). Furthermore, attempting to infer the function and activity of below-ground host immune responses based on knowledge generated through the analysis of above-ground colonization processes and disease symptom development is unrealistic (De Coninck et al., 2015; Chuberre et al., 2018). In this regard, the work described herein offers insight into the contrasting signaling processes that exist between an asymptomatic monocot and a symptomatic eudicot following fungal pathogen colonization.
Differential host responses are prominent among crops colonized by Fusarium virguliforme, the causal agent of soybean (Glycine max) sudden death syndrome (SDS), a devastating disease yielding annual losses in the United States in excess of $274 million dollars (Koenning and Wrather, 2010; Allen et al., 2017). As one example of management strategies to reduce yield losses, soybean and maize (Zea mays) are typically grown in rotation (Katan, 2017). However, a recent study showed that maize can serve as an asymptomatic host for F. virguliforme, thereby providing a potential reservoir for this devastating soybean pathogen (Kolander et al., 2012). In the current study, we conducted a temporal transcriptome-based analysis to identify, and compare, genetic signatures underlying asymptomatic and symptomatic responses in maize and soybean roots during fungal pathogen colonization. Through this, we have defined the early temporal interactions between F. virguliforme and two hosts: maize (asymptomatic) and soybean (symptomatic) to generate a transcriptional atlas of pathogen-induced responses in each crop species. Using this approach, we asked if orthologous transcriptional responses are induced in a temporally independent manner between soybean and maize. Based on the output of the analysis presented herein, we suggest that pathogen-induced host senescence plays a predominant role in the establishment of fungal necrotroph colonization, a process we hypothesize distinguishes symptomatic (i.e. pathogen-induced) senescence in soybean from asymptomatic colonization and persistence of the pathogen in maize.
Results
Fusarium virguliforme colonizes soybean and maize
We recently reported a role for fungal transcriptome plasticity in the regulation and adaptation of F. virguliforme in association with asymptomatic (maize) and symptomatic (soybean) hosts (Baetsen-Young et al., 2020). To extend this analysis, and to identify induced host defense signaling responses associated with fungal pathogen colonization, we conducted a comprehensive analysis of fungal colonization of soybean and maize roots over a 2-week period of colonization. Following pathogen inoculation, soybean radical elongation was observed from 0 to 4 days after inoculation (DAI), with subsequent development of lateral and secondary lateral roots at 7 and 10 DAI, respectively (Figure 1A;Supplemental Figure 1A). By 14 DAI, lateral and tap root growth was abundant, with full expansion of cotyledons in soybean hosts following mock- and pathogen-inoculation. This represented a vegetative cotyledon (Fehr and Caviness, 1977) stage for soybean.
Figure 1.
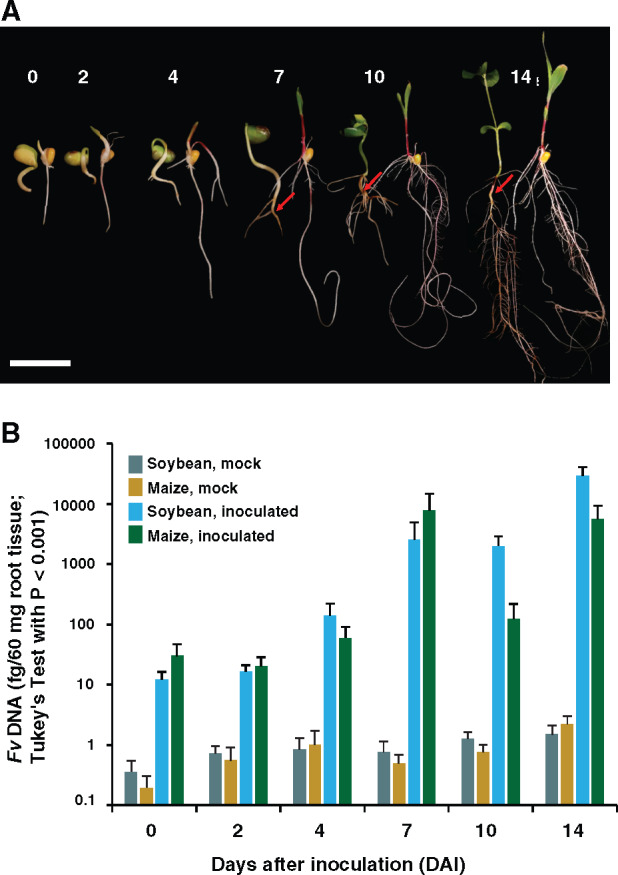
Fusarium virguliforme growth on maize and soybean. (A) Plant growth and development over 14 days (numbered) post inoculation with F. virguliforme. Soybean is on the left and maize is on the right in each pair. Bar = 4 cm. (B) F. virguliforme DNA in planta quantification from mock and inoculated soybean and maize roots. F. virguliforme levels were determined using a TaqMan-based quantitative PCR assay. Values shown are the average of three biological replicates, each of which contained two plants, ± sem (n = 6).
Consistent with the hemibiotrophic lifestyle of F. virguliforme, no symptoms were apparent on soybean roots from 0 to 4 DAI, illustrative of the biotrophic stage of the pathogen (Ngaki et al., 2016). At 7 DAI, discoloration of the lower tap root of F. virguliforme-inoculated samples was observed, which subsequently developed into necrotrophic streaking at 10 DAI. Beyond 10 DAI, complete necrosis of the lower hypocotyl was observed, with subsequent disease symptom spread to the lateral roots adjoining the tap root. Both mock- and pathogen-inoculated maize showed comparable growth and development patterns to those observed in soybean (Figure 1A;Supplemental Figure 1B). In brief, maize radical and seminal roots slowly emerged from 0 to 3 DAI, and by 4 DAI, had expanded in length. At 7 DAI, lateral roots initialized along the primary root, followed by crown root emergence (Supplemental Figure 1B). By 14 DAI, the seminal, crown, and primary roots had further elongated, with parallel expansion of leaves to the V1 growth stage (i.e. full extension of the first leaf).
Given that we did not observe visual symptoms on F. virguliforme-inoculated maize roots, we next employed quantitative real-time polymerase chain reaction (qPCR) to evaluate the in planta fungal growth in both maize and soybean. Using this approach, we observed that F. virguliforme DNA concentrations increased on both soybean and maize roots over the timecourse of analysis, with 0 and 2 DAI displaying significantly lower levels of F. virguliforme DNA quantities than 14 DAI (P < 0.0001; Figure 1B). Interestingly, both inoculated soybean and maize exhibited similar levels of F. virguliforme, as determined by qPCR at most timepoints, with notable increases between soybean and maize between 10 and 14 DAI. All sampled timepoints after fungal inoculation were significantly different from mock treatments (Tukey’s test with P < 0.001; Supplemental Table 1). As previously observed, we detected nominal background levels of DNA in tissue samples from mock-inoculated roots using F. virguliforme-specific DNA primers, an artifact that is associated with the non-specific nature of the qPCR assay at detection limits below 10 fg of DNA resulting in modest signals associated with nonspecific DNA (plant or fungal) binding (Wang et al., 2015).
Temporal expression of induced defense genes in soybean and maize
Having established that F. virguliforme colonizes both hosts, we conducted a comparative RNA-seq experiment using samples from six selected timepoints over a 0- to 14-day inoculation period. Timepoints were selected based on preliminary colonization timecourse experiments (Figure 1;Supplemental Figure 1). The impetus for this approach was to (1) determine if the timing of defense responses underpin symptomatic and asymptomatic phenotype development and (2) identify pathway rewiring in host plants by the pathogen which potentially function in modulating tolerance and susceptibility (Chen et al., 2016; Chowdhury et al., 2017).
Mock- and fungal-inoculated maize and soybean roots from 0, 2, 4, 7, 10, and 14 DAI were subjected to RNA-seq. Each of these biological samples were pooled from six independent plants, as well as repeated in three independent growth chamber runs. Sampling of plant tissues from both mock- and fungal-inoculated events at each timepoint allowed us to discover genes specifically related fungal colonization. In addition, all sampling and phenotypic observations were performed from the same inoculation site within the root, thereby enabling a comprehensive evaluation of host response to fungal inoculation, including fungal adhesion, development and colonization, and penetration and proliferation. Sequencing of each sample to an average of 70 million reads yielded uniquely mapped reads (after adapter trimming and quality thresholding) in the range of 76%–83% for soybean and 77%–81% for maize (Supplemental Figure 2 and Supplemental Data Set 1).
As a key step in demonstrating the relatedness of gene expression profiles across sampling as a function of host and time, we observed a high correlation (>96%) of gene expression patterns among biological replicates (Supplemental Figure 3), illustrating consistency among independent replicates and within treatments. To explore expression patterns between mock- and F. virguliforme-infected plants, samples were grouped using principal component analysis, the output of which revealed that gene expression patterns in maize were primarily separated from each other by time, with distinct groupings at 0, 2–7, 10, and 14 DAI (Supplemental Figure 2B). This is in contrast to groupings by treatment over time, indicating that root development had a greater impact on gene expression than did fungal colonization. Conversely, transcripts from soybean were primarily grouped by treatment (Supplemental Figure 2C). By 4 DAI, mock- and fungal-inoculated treatments had separated into distinct groups, and the remaining samples were separated by treatment as the time course proceeded. The large separation between treatments suggests large global expression changes within soybean roots, as the F. virguliforme induced disease symptoms.
To further catalog gene expression changes as those that were identified as differentially expressed following F. virguliforme inoculation, we first filtered the dataset for genes that were significantly up- or downregulated (multiple-testing adjusted P < 0.05, |log2(fold-change)| > 1) between fungal-inoculated samples and mock-inoculated treatments within each host species at each timepoint. Among the 28,956 expressed maize genes identified in at least one timepoint, 600 unique responsive [i.e. differentially expressed genes {DEGs}] genes were detected across all timepoints. Among the 600 significantly responsive genes identified in maize in response to F. virguliforme, 266 were upregulated and 336 were downregulated with two significantly up and downregulated at different timepoints, with the majority of the identified differential expression changes occurring at 0 and 14 DAI (Figure 2, A andTable 1; Supplemental Table S2, Figure S4 and Supplemental Data Set 2). In contrast, among 43,308 soybean genes expressed at one or more timepoints, 10,898 were significantly differentially expressed following F. virguliforme colonization (Supplemental Table 3 and Supplemental Data Set 3). Interestingly, there was an almost equal number of up- and downregulated genes from soybean (5,643 and 5,325, respectively), with a majority of DEGs identified at 7 DAI and beyond (Figure 2B andTable 1). This contrasts with the temporal pattern of response changes observed in maize, an observation we posit further highlights that differential host gene expression may underpin the divergence in symptomatic versus asymptomatic phenotypes in soybean and maize, respectively.
Figure 2.

Temporal pattern of defense gene expression in maize and soybean. (A and B) The number of significantly DEGs between mock and inoculated maize with log2(FC) > 1, respectively, of early differentially regulated genes in maize or soybean from six timepoints over the colonization timecourse. (C and D) Heatmap of log2-fold gene expression changes among transcripts cataloged in ontologies significantly enriched in upregulated genes across pooled timepoints for maize (n = 266) and soybean (n = 5,643). (E and F) Heatmap of significantly enriched gene ontologies from F. virguliforme-induced downregulated genes across pooled timepoints for maize (n = 336) and soybean (n = 5,325).
Table 1.
Transcriptomic landscape of soybean and maize during colonization by F. virguliforme over a 2-week timecourse of colonization
| Transcriptome | Soybean | Maize |
|---|---|---|
| Total expressed genes | 43,308 | 28,956 |
| DEGs | 10,898 | 600 |
| Orthologous genes | 23,274 | 14,542 |
| Orthologs, N (%) | ||
| Orthologous DEGs | 4,528 (41.5) | 221 (36.8) |
| Orthologous DEGs, up | 2,296 (50.7) | 86 (38.9) |
| Orthologous DEGs, down | 2,257 (49.8) | 137 (62) |
| Nonorthologs, N (%) | ||
| Nonorthologous DEGs | 6,370 (58.5) | 379 (63.2) |
| Nonorthologous DEGs, up | 3,347 (52.5) | 180 (47.5) |
| Nonorthologous DEGs, down | 3,068 (48.2) | 199 (52.5) |
The total percent of orthologous DEGs that were up- and downregulated in soybean reflects the percentage assignment(s) resulting from 25 genes in soybean being both up- and downregulated over the timecourse. The total percent of orthologous DEGs that were up- and downregulated in soybean reflects the percentage assignment(s) resulting from two genes in soybean being both up- and downregulated over the timecourse. The total percent of nonorthologous DEGs that were up- and downregulated in soybean reflects the percentage assignment(s) resulting from 45 genes in soybean being both up- and downregulated over the timecourse.
Previous studies have demonstrated that fungi elicit an array of pro-immune transcriptional responses in their host(s) roots, including the activation of metabolic processes such as the generation of reactive oxygen species (ROS) and the release of secondary metabolites (Pusztahelyi et al., 2016; Zhang et al., 2017). To gain insight into processes that underpin the observed phenotypic differences between symptomatic and asymptomatic host responses, we first conducted a gene ontology (GO) enrichment analysis of upregulated DEGs. We identified defense-related transcriptional response categories as the most enriched processes from those within upregulated genes over the timecourse of colonization of maize (Supplemental Table S4). Indeed, and as illustrated in Figure 2C, we observed an enrichment of genes from maize that were associated with defense response processes following F. virguliforme colonization, including those associated with oxidation–reduction and terpenoid biosynthesis (Boba et al., 2020). This suggests that maize, while phenotypically asymptomatic, recognized F. virguliforme and activated defense-associated processes following inoculation.
Based on the observation that F. virguliforme inoculation elicited a broader alteration of gene expression in soybean than in maize, we predicted that a greater diversity of enriched processes would be evident in soybean as compared to maize. Specifically, 12 significantly (multiple-testing adjusted P < 0.05) enriched biological process categories were identified in maize (Supplemental Table 4), including defense response signaling and the regulation of oxidation reduction processes. In contrast, 46 significantly (multiple-testing adjusted P < 0.05) enriched biological process categories were identified in soybean (Supplemental Table 5). Among these were numerous categories such as phosphate metabolism, protein ubiquitination, and cell-to-cell communication (Figure 2D). The increased number of identified categories not only points to broader disruption in soybean following pathogen colonization, but in the case of altered metabolic functions, is associated with the alteration of soybean root physiological processes and associated F. virguliforme colonization. When we compared the same enriched categories across hosts, we observed that defense-related ontologies were upregulated much earlier (i.e. 0–4 DAI) in samples from inoculated maize roots than in soybean (Figure 2, C and D). We hypothesize that this may underpin F. virguliforme tolerance in maize and is further supportive of a diverged temporal response between the two hosts. Indeed, we observed that the same gene categories that were also enriched early (i.e. induced) in maize were induced at later points in the colonization of soybean (7–14 DAI).
To identify biological processes that were repressed in each host over the duration of the colonization timecourse, we conducted an enrichment analysis of downregulated genes and their associated ontologies. As shown in Figure 2E, we identified 336 genes in maize, enriched among ∼13 biological process categories, that were significantly (multiple-testing adjusted P < 0.05) downregulated over the timecourse of F. virguliforme colonization (Table 1). Notably, among these were those associated with plant growth and development, as well as several key defense processes (Supplemental Data Set 2). In contrast, in soybean, 5,325 downregulated genes and their enriched categories—especially at later timepoints of the infection—were identified (Supplemental Data Set 3). Among these, many have previously been described as host processes essential for fungal growth and development, and nitrogen metabolism (Figure 2F;Fagard et al., 2014). For example, the downregulation of photosynthesis during pathogen infection has been demonstrated to not only coincide with the activation of robust defense signaling (Su et al., 2018), but is also associated with pathogen-induced downregulation of photosynthesis as a mechanism to induce ROS accumulation in chloroplasts, which leads to reduced levels of photosystems I and II (reviewed in Kretschmer et al., 2019).
As a final step in this stage of our analysis, we conducted a comparative co-expression analysis of each host’s transcriptome using a differential gene correlation analysis (DGCA). The impetus for this was to discover significant changes within gene pairs caused by the treatment, by employing the median z-score difference of the gene pair correlations in the first condition compared to the second condition, and then compared to all gene pairs (McKenzie et al., 2016). Using this approach, we identified 128 gene pairs (ca. 0.00003%) in maize that were significantly differentially correlated; that is, these gene pairs were observed to undergo a change in correlated expression in comparison of mock versus pathogen inoculation (Supplemental Data Set 4). Among these, two transcripts from maize with homologs to Arabidopsis R-genes were identified as having significant differential correlations. The first, RESISTANCE TO PSEUDOMONAS SYRINGAE pv. MACULICOLA (RPM1; Zm00001d014099), showed an expression profile that was positively correlated with XYLOGLUCAN GALACTOSYLTRANSFERASE (Zm00001d029862) in response to fungal inoculation, but negatively correlated in mock treatments. Specifically, expression of the putative maize RPM1 ortholog was downregulated at 0 DAI (ca. 19.5-fold change), but expression gradually increased over the timecourse to an approximate two-fold change. The second, RESISTANCE TO PERONOSPORA PARASITICA 13-LIKE-4 [RPP13L4; Zm00001d018786, containing a Rho-N domain {Zm0001d051967}], showed a negative expression correlation during colonization, yet was positively correlated under mock-inoculation conditions. The former suggests possible fungal modulation of R-gene expression during colonization, a scenario supported by previous work which identified this R-gene as a candidate for SDS resistance (Zhang et al., 2015). Additionally, the observed co-regulation of R-gene expression with genes associated with cell wall-associated remodeling processes (i.e. xyloglucan galactosyltransferase) further illustrates the connectivity of immunity with pathogen-induced changes in host cell architecture (Day and Graham, 2007).
In soybean, many more gene pairs (7,950,000) were identified as significantly differentially correlated when we compared samples from mock- and F. virguliforme-inoculated roots. To determine how these identified gene pairs were co-expressed, we constructed a planar filtered network (PFN), dissecting the resultant data into 1,160 modules (Supplemental Data Modules). Using this, we next explored the enrichment of co-regulated defense genes in the above modules and found that while they were significantly enriched in “metabolic process” and “primary metabolic process” (Supplemental Figure 5 and Supplemental Data Set 5), we did not identify enrichment in defense-related processes. This further supports our assertion that biotrophic fungal pathogens suppress plant cell death and manipulate plant metabolism in soybean (Doehlemann et al., 2017).
Orthologous host gene analysis reveals differential patterns of induced defense responses
Previous studies have described temporal changes in gene expression during compatible and/or incompatible pathogen interactions, an observation that associates with the onset of disease symptom development (Dupont et al., 2015; Kong et al., 2015). Here, we observed the enrichment of defense-related GOs at distinct timepoints in soybean and maize. To address whether defense-induced responses in soybean and maize occur in a temporally similar, or diverged, manner, we first identified the orthologous genes between soybean and maize. We reasoned that this would enable us to compare the temporal expression pattern changes between orthologs, which are descended from a single gene stemming from the last common ancestor. First, we clustered the predicted protein sequences from the maize and soybean genomes using OrthoFinder, which yielded 10,700 orthogroups, containing 23,273 and 14,542 expressed genes in soybean and maize, respectively (Supplemental Data Set 6). For each orthogroup, if orthologs contain more than a one-to-one relationship (e.g. two maize orthologs to one soybean ortholog), the median of the log2-fold changes value for each DEG was calculated at each timepoint. The variation in DE expression between soybean and maize orthologs was assessed to capture the greatest differential by the median transformation of gene expression values, as represented by significant correlations at several timepoints (Supplemental Figure 6)
To understand the significance of the soybean–maize orthologous gene set in light of the host response to F. virguliforme colonization, we next explored whether the DEGs were contained in the orthologs. Overall, a similar number of DEGs were identified as orthologous in both soybean and maize (41.5% versus 36.8%). However, of the orthologous genes cataloged from soybean and maize, an apparent difference was observed in the percentage of genes that were either up- or downregulated within each host. For example, in soybean, we identified a similar number of up- and downregulated genes from among the list of orthologous DEGs (50.7% up and 49.8% down), while in maize, 38.9% were upregulated and 62% were downregulated (Table 1; Supplemental Figure 4 and Supplemental Data Sets 7 and 8).
To determine if the host response to fungal colonization was unique to each host, we cataloged each orthogroup depending on whether significant DEGs, or groups of genes, were maize-specific, soybean-specific, or occurred in both hosts. As shown in Figure 3A, we identified 3,003 significantly upregulated orthogroups, whereby 93.3% of the orthogroups classified as significantly upregulated were uniquely upregulated in soybean. In contrast, only 3.3% of the orthogroups were uniquely upregulated in maize. In addition, 3.4% were upregulated in both hosts following F. virguliforme inoculation (Figure 3A;Supplemental Data Set 9), indicating that more than half of the upregulated genes in maize were also upregulated in soybean. To further explore the processes contained within the list of identified orthologous DEGs, we developed a merged gene ontologies annotation containing gene annotations from maize and soybean. This new GO annotation contained 4,685 orthogroups with GO annotations from maize, and an additional 6,015 orthogroups were supported by both soybean and maize annotations. Interestingly, no orthogroups were solely associated with soybean (Supplemental Data Set 10). Orthogroups that were uniquely upregulated in soybean were highly enriched for responses to oxygen containing compounds, defense, and hormone metabolism (Supplemental Data Set 11). Orthogroups that were upregulated in both hosts in response to F. virguliforme colonization were enriched for response to organic substance, stimulus, and defense (Supplemental Data Set 11). Orthogroups that were upregulated and unique to maize were enriched with cytoskeleton-associated processes and DNA methylation (Supplemental Data Set 12).
Figure 3.

An analysis of putative orthologous host processes reveals differential patterns of induced defense responses. (A and B) Heatmap of expression of significantly (log2(FC) > 1) upregulated (n = 3,003) and downregulated (n = 1,660) genes from mock- and fungal-inoculated samples. Gray indicates orthologous genes from soybean and maize are significant, blue indicates orthologous genes from only soybean are significant, and green illustrates orthologous genes from maize that were uniquely significantly DE. (C and D) Mean expression patterns of log2(FC) > 1 gene expression profiles from among significantly up- and downregulated orthologous genes, respectively, at a single timepoint in both hosts over the colonization timecourse. Error bars indicate one standard deviation of the mean.
In addition, we identified 1,660 significantly downregulated orthogroups; as shown in Figure 3B, 92.2% of the orthogroups classified as significantly downregulated were uniquely downregulated in soybean, while only 5.8% of the orthogroups were uniquely downregulated in maize. Approximately 2.0% were downregulated in both hosts (Supplemental Data 9). Specifically, orthogroups that were uniquely downregulated in soybean were enriched for cell wall organization, root development, and auxin transport (Supplemental Data Set 11). Surprising, we observed that the most significantly enriched process was photosynthesis, a finding that is agreement with previous studies demonstrating that the rate of photosynthesis decreases during leaf senescence (Wojciechowska et al., 2018). While roots and leaves utilize numerous distinct processes during growth, development, and environment interactions, the process of cellular and tissue senescence in each follow similar patterns of gene process expression (Liu et al., 2019). Leveraging this similarity, we exploited our root-specific transcriptome datasets as a guide to discover and further characterize the processes that are associated with pathogen-induced senescence in soybean. Using this approach, we observed that a small proportion of genes downregulated in both soybean and maize was highlighted by only 19 processes being significantly enriched (Supplemental Data Set 11). Not surprisingly, these included response to stimulus, root development, and response to external stimulus. Orthogroups that were uniquely downregulated in maize were enriched for cell cycle processes, the regulation of reproduction, and the negative regulation of metabolic process. Additionally, we evaluated F. virguliforme GO enrichments during colonization of both maize and soybean (Supplemental Figures 7 and 8). We did not observe an increase in the expression of F. virguliforme genes involved in chitin metabolic processes (a key indicator of fungal growth and development), yet we did identify a downregulation of F. virguliforme genes associated with ROS production from samples collected from colonized maize roots. Based on this, we surmised that these observations provide a basis from which we could proceed to compare and identify orthologous responses to colonization between the two hosts (Supplemental Figure 9, A and B, Supplemental Figure 10, A and B). Surprisingly, we did not observe an enrichment in soybean genes associated with defense response (e.g. “defense response to fungus”, “immune response”, and “root development”). Taken together, these data highlight the different host responses in soybean and maize in response to F. virguliforme.
To address how the temporal patterns of orthogroup induction differed between soybean and maize, we next focused on the defense-relevant GO categories that were enriched in maize during the early timepoints of the colonization (0–4 DAI; Supplemental Data Set 11). To do this, we first identified upregulated orthogroups in biological processes associated with host–pathogen interactions, including: (1) defense response; (2) defense response to fungus; (3) negative regulation of cellular protein; (4) oxidation reduction; and (5) terpenoid biosynthetic process (Figure 3C). As shown, we observed a shift in the induced patterns of expression between maize and soybean, with a notable upregulation of orthologous genes at 2 and 4 DAI in maize. Interestingly, and consistent with our hypothesis of a delayed response to pathogen colonization being one of the underlying factors driving susceptibility, these same responses were not observed as enriched/induced until 4 to 7 DAI in soybean. Genes within these upregulated orthogroups encoded PATHOGENESIS-RELATED PROTEIN 10 (PR10), STRESS-INDUCED PROTEIN 1, TERPENE SYNTHASE, and BROWMAN-BIRK TYPE TRYPSIN INHIBITOR. Based on this, it is tempting to hypothesize that while similar host defense-associated genes are activated during the course of F. virguliforme colonization in both soybean and maize, the induced expression of these genes at earlier time intervals in maize associates with the absence of disease symptom progression, as compared to soybean.
Using an approach similar to the above, we also identified downregulated defense-relevant process groups (Figure 3D;Supplemental Data Set 11). Among these, we observed that the expression of downregulated genes associated with response to chemical and endogenous stimulus were slightly diminished between maize and soybean; specifically, we observed an upregulation of orthologous genes in maize, but a downregulation in soybean and 7–10 DAI. We posit that this further supports the hypothesis that a delayed response to pathogen in soybean underpins, yet is not wholly responsible for, susceptibility to F. virguliforme colonization. Indeed, if one considers the in planta lifestyle transition (i.e. biotrophy to necrotrophy) of F. virguliforme, pathogen co-option of host processes at discrete stages of this transition in a host-specific manner (Supplemental Figures 7 and 8) likely serves as a critical virulence strategy. Thus, it is reasonable to hypothesize that the pathogen’s ability to alter the timing and activation sequence of host defense processes underpins host transitions from pathogen recognition, to defense, to either susceptibility, resistance, and/or tolerance (Supplemental Figure 9, A and B, Supplemental Figure 10, A and B; Doehlemann et al., 2017).
Nonorthologous defense processes within maize and soybean
While the identification of orthologous defense-related processes in soybean and maize identified temporal divergence in host response(s) to fungal colonization, an analysis of the novel host-specific defense signaling processes from each was not apparent. To resolve this, we surveyed the specific processes unique to each host associated with upregulated and downregulated DEGs as a comparison between maize and soybean. As shown in Figure 4, A and B, we identified non-orthologous genes that were significantly upregulated (180 and 3,347 in maize and soybean, respectively) and downregulated (199 and 3,068 in maize and soybean, respectively) following fungal colonization (Table 1; Supplemental Data Sets 12 and 13, Supplemental Tables 6 and 7).
Figure 4.

Analysis of nonorthologous host processes reveals host-specific patterns of induced defense responses that are consistent with disease and pathogen tolerance. (A and B) Heatmap of expression of log2(FC) > 1 genes from a list of significantly upregulated (n = 180) and downregulated (n = 199) genes at a single timepoint from maize, and significantly upregulated genes (n = 3,347) and downregulated genes (n = 3,068) from soybean at a single timepoint. Orange indicates significantly upregulated DE nonorthologous genes and blue indicates significantly downregulated DE nonorthologous genes. (C and D) Heatmap of significantly enriched gene ontologies from upregulated genes across pooled timepoints for maize and soybean. (E and F) Heatmap of significantly enriched gene ontologies from downregulated genes across pooled timepoints for maize and soybean, respectively, following F. virguliforme inoculation. (G) Mean expression patterns of log2(FC) > 1 of significantly upregulated and downregulated genes, respectively, at a single timepoint in both hosts, over the colonization timecourse. Error bars indicate one standard deviation of the mean.
We next asked if there were induced defense responses derived from defense gene categories that were unique to each host (i.e. genes that did not have an apparent ortholog). As shown in Figure 4C, upregulated DEGs in maize following F. virguliforme inoculation were associated with GO enrichment of processes including defense response to fungus, immune signaling, the hypersensitive response, and host programmed cell death (Supplemental Table 8 and Supplemental Data Set 14). Among these, and consistent with the initial biotic interaction lifestyle of this fungus, processes in maize were primarily associated with host programmed cell death, a mechanism likely associated with localized defenses preventing fungal proliferation and further invasion (Jones, 2001; Deshmukh et al., 2006; Schäfer et al., 2007; Diamond et al., 2013). Conversely, in soybean, we identified upregulated genes associated with processes such as glucose catabolic and response to carbohydrate catabolic process, previously identified as metabolic signatures of the onset of host–fungal interactions, host susceptibility, and disease (Figure 4D;Supplemental Table 8, Supplemental Data Set 15; Divon et al., 2007; Deveau et al., 2008; Askew et al., 2009). In addition, we explored the significantly upregulated nonorthologous genes pattern changes between maize and soybean within the same biological processes. As shown in Figure 5A, processes associated with “defense response”, “defense response to fungus”, “oxidation reduction”, and “terpenoid biosynthetic process” showed similar responses to fungal colonization in maize (ca. 2–7 DAI) than observed in soybean. In maize, the majority of downregulated genes identified were differentially regulated at later points in the colonization (i.e. 14 DAI; Figure 4E;Supplemental Data Set 16). In soybean, the host processes known to be associated with survival during fungal colonization were enriched at early timepoints in the colonization; specifically, as early as 2 DAI (Figure 4F;Supplemental Data Set 17).
Figure 5.

Processes unique to maize are associated with host immune responses following F. virguliforme inoculation. (A) Mean expression patterns of log2(FC) > 1 of significantly upregulated nonorthologous genes from at least a single timepoint in both hosts over the colonization timecourse. Genes for analysis were selected based on those identified within the same gene ontology categories, but themselves are nonorthologous. Error bars indicate one standard deviation of the mean. (B) Gene expression profile of maize root defense markers that do not share a soybean orthologous group across the colonization timecourse.
Next, we explored the nonorthologous response to F. virguliforme between maize and soybean within each timepoint. The impetus for this was to identify the various, and differentially induced, biological processes utilized by each host in their response to fungal colonization, including as a function of the timing of fungal lifestyle transitions (Supplemental Figure 9, C and D, Supplemental Figure 10, C and D). Additionally, we queried if defense processes identified using this approach were relevant to the observed host phenotypes (Supplemental Figure 1, Supplemental Tables 6 and 7), which identified the enrichment of several key biological processes that presented as potentially host-specific (Figure 4G;Supplemental Tables 8 and 9, Supplemental Data Sets 14–17). In soybean, enriched processes included “primary metabolite biosynthesis” and “signaling”, as well as “nucleotide catabolic processes”. In maize, this also included well-defined processes, such as those associated with the elicitation of the hypersensitive response, as well as signaling associated with response to chitin and fungus. Similar to orthologous defense gene comparisons described above, the GO process of “defense response” containing processes related to antifungal activity were upregulated earlier in maize than in soybean and include PATHOGENESIS-RELATED MAIZE SEED (PRms), TIFY10B (containing JASMONATE-ZIM DOMAIN repressors), and hevein-like (Figure 5A). Interestingly, both PRms and hevein were previously identified as being induced following fungal colonization (Majumdar et al., 2017; Wong et al., 2017). Similarly, TIFY10B was also upregulated in maize, and further analysis revealed that key marker genes within the jasmonic acid (JA) biosynthesis pathway were minimally impacted (mock versus treated).
Previous studies have demonstrated that the JA pathway plays a crucial role in protecting plants against both pathogen colonization and wounding (Zhang et al., 2018b). Based on the observation that JA precursor biosynthesis gene expression was not altered by F. virguliforme colonization of maize (Figure 5B;Supplemental Figure 11), we were curious as to how maize perceives and responds to F. virguliforme colonization. To address this, we next interrogated the expression patterns of several defense marker genes previously demonstrated as specific to maize root fungal colonization (Balmer et al., 2013; Chuberre et al., 2018). As shown in Figure 5B, pathogenesis-related (PR) genes were induced over the timecourse of colonization. At the same time, while we detected the induction of several PR genes, NONEXPRESSER OF PR GENES 1 (NPR1) was not upregulated in maize. While salicylic acid (SA)-dependent defense signaling does not specifically require the induced expression of NPR1, we hypothesize an additional bifurcation in SA-associated signaling, and that the observed upregulation of PR in response to F. virguliforme, herein, occurs in a SA-independent manner (Balmer et al., 2012). Additional hormone signaling processes, such as auxin and gibberellic acid biosynthesis, were also not significantly altered as a function of transcriptional expression throughout the timecourse of colonization. Interestingly, however, ethylene biosynthesis was rapidly upregulated (0 DAI) in maize roots (Supplemental Figure 11), as was the expression of ETHYLENE RESPONSIVE FACTOR 105-LIKE (ERF-105-like). This is significant, as ethylene biosynthesis was recently reported to be induced in SDS-resistant soybean cultivars (Abdelsamad et al., 2019), a broader defense mechanism hypothesized to be associated with resistance to necrotrophic pathogens (Laluk and Mengiste, 2010).
Constitutive expression of maize genes within orthologous defense
To date, numerous studies have focused on induced plant defense responses following fungal colonization (reviewed in van der Does and Rep, 2017). However, the role of constitutive defense-associated responses, particularly those in maize in response to F. virguliforme colonization, remains largely unaddressed. To determine if the duration and amplitude of constitutive orthologous gene expression underpins these responses, we next compared soybean and maize orthogroups following mock-inoculation, as opposed to fungal-treatment, detailed above. Using this approach, we successfully identified 182 orthogroups that were significantly upregulated in maize (compared to soybean) over the mock-inoculation timecourse (Figure 6; Supplemental Data Set 18). Based on the output of this analysis (i.e. 182 orthogroups), we hypothesize that the function of these genes might provide insight into host tolerance to F. virguliforme (i.e. maize) through a mechanism associated with constitutive, sustained, elevated defense gene expression. Additionally, we hypothesized that the expression of these orthogroups will decrease over the course of fungal colonization in maize (i.e. tolerant, asymptomatic host), in contrast to a robust induction of defense-associated processes in soybean (i.e. susceptible, symptomatic host). Surprisingly, we identified a similar pattern of expression of constitutively upregulated orthogroups at 2–14 DAI in both fungal-inoculated hosts. Indeed, constitutively induced genes in maize were on average four-fold higher in the F. virguliforme treatment when compared to the constitutive expression of the same genes following mock-inoculation at 0 DAI. In summary, these expression profiles support our hypothesis that the rapid upregulation of a core group of defense-associated transcripts during the initial interaction between maize and F. virguliforme is associated with the establishment of the asymptomatic interaction.
Figure 6.
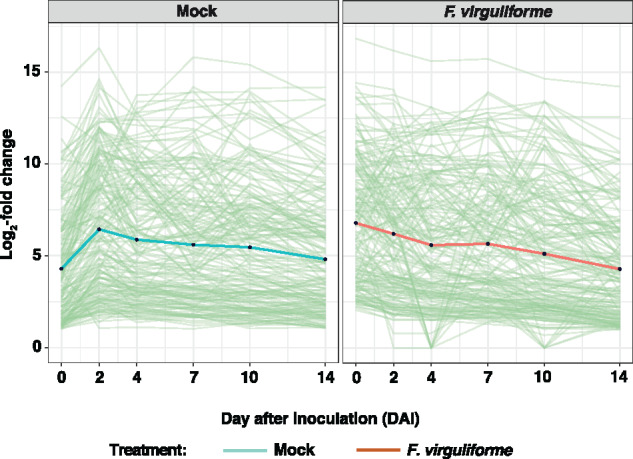
Conservation of defense gene expression patterns preceding inoculation with F. virguliforme. Line graph of expression patterns of orthogroups that are uniquely upregulated in maize when compared to soybean log2(FC) > 1, and corresponding regulation when maize and soybean are colonized by F. virguliforme. Individual green lines represent individual orthogroups. Solid green or brown lines represent the mean of orthogroups from mock- or F. virguliforme-inoculated.
Defense expression patterns of orthologous transcription factors is diverged across soybean and maize
The expression patterns of defense-induced orthologous genes in response to F. virguliforme inoculation varied by host, suggesting potential differences in host pathway activation, or repression. To define the regulation of these expression patterns within our orthologous soybean and maize datasets, we next compared the patterns of transcription factor (TF) expression (i.e. induced or upregulated) in each host following F. virguliforme inoculation. Similar to the orthologous genes identified as upregulated by F. virguliforme, the vast majority (93.3%) of the DE TFs were uniquely expressed within soybean (Supplemental Data Sets 19 and 20). Specifically, our analyses revealed that only 4 TF orthogroups were uniquely induced in maize (i.e. bHLH-157, MYB-41, Homeobox-transcription factor 29, and GRAS-transcription factor 11), while 11 were induced in both soybean and maize in response to F. virguliforme (Supplemental Data Set 19).
To narrow our focus to the analysis of those TFs whose expression patterns contained altered gene expression in both hosts, we next evaluated the expression patterns of TFs that were upregulated in both maize and soybean following fungal colonization. As shown in Figure 7A (dashed black boxes), among the selected TF orthogroups upregulated in both species, we identified three TFs that were expressed at least four-fold higher in maize than soybean at 0 DAI. Among these was a C2H2‐zinc finger TF, ZINC FINGER OF ARABIDOPSIS THALIANA12, previously identified as being induced in response to elevated hydrogen peroxide generation in maize (Mittler et al., 2006), and a NAC042 TF, which exhibited the largest expression change in comparison to mock versus fungal-inoculated maize. This is noteworthy, as NAC042 was also shown to be responsive to hydrogen peroxide generation (Wu et al., 2012), illustrating that ROS production in maize may underlie the more than four-fold difference in expression compared with soybean. The third TF that was identified as induced was DEHYDRATION-RESPONSIVE ELEMENT-BINDING PROTEIN 1A, a gene regulated by NAC042, and hypothesized to function in the activation of oxidative stress in tomato (Thirumalaikumar et al., 2018). Based on the sum of these analyses, we hypothesize that maize rapidly attenuates ROS production following defense activation to dampen cellular stress. Of significance to this process is the potential role of NAC042, which has been shown to function as a negative regulator of senescence (Wu et al., 2012). Taken together, we propose a model wherein F. virguliforme activates ethylene defense responses in maize, and that prolonged expression of NAC042 leads to the promotion of cell longevity via dampening of cell stress responses and the inhibition of cell death (Figure 7C). This hypothesis is consistent with the observation that NAC042 expression in soybean did not approach expression levels of more than four-fold change until 7 DAI, a timepoint that coincides with symptom development. Based on these data, we hypothesize that host senescence plays a key role in SDS development.
Figure 7.

Divergence of defense expression patterns of orthologous transcription factors. (A) Heatmap of log2(FC) > 1 of significantly upregulated genes at a single timepoint in at least one host between mock and inoculated (n = 215). Gray indicates orthologous genes from soybean and maize were significant, blue indicates orthologous genes from only soybean were significant, and green indicates that maize orthologous genes were uniquely significantly DE. Black dashed boxes highlight changes in transcription factor expression between hosts. (B) Heatmap of positive-regulatory TFs in maize (n = 9) and soybean (n = 4), and negative regulatory TFs in maize (n = 5) and soybean (n = 3). (C) Representation of reactive oxygen species induced NO APICAL MERISTEM (NAM), ATAF1/2, CUP-SHAPED COTYLEDON-2 (CUC2) pathways in soybean that exhibited differential expression patterns of log2(FTAbleC) > 1 between mock and pathogen-inoculated samples. The corresponding orthologs or homologs in maize associated with senescence and root vascular development. Each gene heatmap illustrates temporal changes from 0 to 14 DAI in soybean (blue) and maize (green). Green font indicates orthologous genes.
Fusarium virguliforme promotes susceptibility via regulatory biological processes that control senescence
Previous studies indicated that root senescence follows an intrinsically regulated developmental program that is regulated by multiple fundamental signaling pathways (Liu et al., 2019). As such, we hypothesized that pathogen-induced senescence is a key virulence strategy, which supports pathogen colonization and disease progression. To test this, we compared gene expression patterns in related GO processes between maize and soybean previously identified as required for senescence-associated signaling (e.g. fatty acid, chromatin, hydrolase process, response to water, JA, ethylene, pigment, carbohydrate, and ion relative biological process; Yang and Ohlrogge, 2009; Woo et al., 2013; Li et al., 2019). As shown in Figure 8, A and B, we observed that some of the orthologs assigned to these GO terms were DE in each host (Supplemental Data Set 21). For example, protein metabolic processes were generally unchanged in the early timepoints of colonization in maize yet were downregulated at later stages of the colonization. In contrast, in soybean, more than half of the DEGs were upregulated at the later stages of fungal colonization. In addition, genes involved in response to water deprivation were upregulated at early timepoints in maize. In soybean, transcripts assigned to this processes were generally not DE over the course of colonization. Indeed, and in further support of pathogen manipulation of senescence-associated signaling processes, we identified an increase in the expression of genes associated with pigment biosynthesis processes in maize (Zhao et al., 2018), yet in soybean, the DE of these processes was absent.
Figure 8.

Gene expression patterns of regulatory pathways that control senescence-associated processes. (A and B) Gene expression pattern of orthologous genes in maize (103) and soybean (376). (C and D) Gene expression pattern of nonorthologous genes in maize (159) and soybean (668).
Based on the analysis above detailing the differential patterns of expression of defense-associated orthologs from soybean and maize, we next queried the list of differentially regulated nonorthologous genes. As shown in Figure 8, C and D, processes associated with protein metabolism, ROS metabolism, response to water derivation, and JA biosynthesis were upregulated in maize, while no significant changes were observed in soybean. Interestingly, the expression pattern of nonorthologous genes associated with these biological processes was similar to the pattern of orthologous genes (Figure 8, A and B; Supplemental Data Set 21). Taken together, these results demonstrate that processes related to senescence were altered in maize, yet not affected in soybean during symptom development.
The data presented above highlight the importance of early, rapid, changes in host gene expression of F. virguliforme recognition and colonization, a process we hypothesize underscores the differential response, and outcome, between soybean and maize. To investigate the role of pathogen manipulation of host senescence-associated transcriptional reprogramming of growth and development, as well as the downregulation of host defenses, we first investigated the expression of TFs known to regulate senescence-associated processes, including those previously identified as positive and/or negative regulators of leaf senescence (Koyama, 2014; Koyama et al., 2017). Using this published dataset as a template to guide our discovery of root-associated senescence processes, we identified 9 and 5 previously characterized positive and negative regulators, respectively, of leaf senescence. As shown in Figure 7B, we observed a general trend of downregulation of maize gene expression profiles at early timepoints (i.e. 0–4 DAI) of TFs associated with the activation of senescence-associated processes (Supplemental Data Set 22). In support of our hypothesis that pathogen-induced host senescence is associated with disease symptom development in soybean, we observed that the same positive regulatory TFs previously identified (i.e. Koyama et al., 2017) were significantly upregulated in soybean, supporting the hypothesis for a mechanism of host transcriptome reprogramming in favor of induced host senescence. Specifically, among the identified induced TFs identified was the senescence-positive factor Teosinte Branched1/Cycloidea/Proliferating cell factors, which promotes the expression of JA biosynthetic enzyme genes (Schommer et al., 2008; Koyama, 2014). As predicted, TFs that negatively regulate senescence-associated processes, such as WRKY54 (Besseau et al., 2012), were significantly induced in maize, while in soybean, their induction was not observed. Taken together, these results suggest that the regulation of host senescence during the early stages of pathogen colonization is a key process associated with susceptibility and disease development.
As described above, we observed that the upregulation of positive-senescence-associated TFs decreased in maize compared to soybean at early timepoints of the colonization, while the pattern of expression of negative-senescence-associated TFs increased in comparison with soybean. To further test the hypothesis that host senescence plays a key role in SDS development, we interrogated the soybean transcriptome for significant expression changes in ANAC (ABSCISIC-ACID-RESPONSIVE NAC (NAM, ATAF1,2, and CUC2); Podzimska-Sroka et al., 2015) genes. Using this approach, we identified an additional seven ANACs that were DE in soybean but not in maize. Excitingly, and in further agreement with our hypothesis, previous work demonstrated that these ANACs play a role in both the negative and positive regulation of senescence, as well as in root development (reviewed in Majid and Abbas, 2019; Figure 7C;Supplemental Data Set 23). Specifically, three ANACs—ANAC047, ANAC055, and ANAC087—have established roles in senescence-associated signaling in Arabidopsis, and two, ANAC055 and ANAC087, have been demonstrated to be abscisic acid (ABA) responsive (Huysmans et al., 2018). In the context of the current work, we further observed that the expression of 9-CIS-EXPOSYCAROTENOID DIOXYGENASE, the product of which is required for the first step in ABA production (Xiong and Zhu, 2003), increased at 2 DAI in soybean, and was followed by a 16-fold increase in expression of ANAC055 and ANAC087 (Suppplemental Figure 10). Interestingly, these genes were not upregulated in maize. Along this same theme, several senescence-associated genes (SAGs) were also upregulated in soybean, but not in maize, as early as 4–7 DAI. This is noteworthy, as previous work showed that expression of SENESCENCE-ASSOCIATED GENE 13 (SAG13) is associated with the cell death response (Pell et al., 2004), with additional work revealing that SAG21 and SAG24 are upregulated during early senescence (Salleh et al., 2012). Interestingly, SAG20 expression was found to be upregulated by necrosis- and ethylene-inducing peptides from Fusarium spp., as well as by phytotoxins produced by F. virguliforme (Chang et al., 2016).
As a potential link to the development-associated gene networks in each of the hosts, we observed that NAC TFs (specifically those associated with cell development) were also induced in soybean roots infected with F. virguliforme. This is interesting, as previous studies showed that NAC022 plays a significant role in lateral root growth (Xie et al., 2000); herein, NAC022 was downregulated following F. virguliforme inoculation. In a similar manner, NAC074, which positively regulates xylem development (Xia et al., 2018), was observed as being downregulated in soybean in response to F. virguliforme colonization. Likewise, NAC083, which plays a role in vascular development (Yamaguchi et al., 2010), was upregulated over the timecourse of colonization. In total, the expression profiles of key development-associated regulators identified in the current study support a role for the alteration of root development and the concomitant acceleration of senescence by F. virguliforme as a potential mechanism of disease symptom development and susceptibility in soybean.
Discussion
Whole-transcriptome analyses of resistant and susceptible host responses to phytopathogens are a common approach to evaluate host–pathogen dynamics during colonization. In total, the sum of these studies provided a foundation to comparatively explore how immune pathways across diverged hosts respond to a single, common, pathogen. In the current study, we exploited the broad host range of F. virguliforme as a comparative system to probe differentially induced root responses between a monocot and a eudicot to uncover immune regulatory responses encoding tolerance and susceptibility. To do this, we generated 72 transcriptomes from mock and inoculated hosts to identify host responses specific to fungal root colonization.
Soybean and maize have distinct phenotypic responses to F. virguliforme colonization. For example, soybean shows marked signs of symptom development as early as 7 DAI, with chlorosis manifesting at 10 DAI, and tap root necrosis at 14 DAI. In maize, disease symptoms do not develop in response to F. virguliforme colonization, yet as noted above, maize supports similar levels of fungal colonization as observed in soybean. In total, this apparent dichotomy suggests a difference in the underlying genetic and/or physiological interactions between the host and pathogen that yields asymptomatic versus symptomatic colonization. For example, an analysis of maize revealed that root growth and development were unaltered in response to F. virguliforme colonization. However, and contrary to the lack of visible changes, we did identify significant changes in gene expression following colonization, most of which included immune response-associated genes. Conversely, an analysis of soybean revealed a massive transcriptomic reprogramming of defense signaling responses concomitant with symptom development. These observations are in agreement with a divergence in the magnitude of host responses as previously noted in the analysis of a single host independently colonized by both pathogenic and nonpathogenic isolates (Lanubile et al., 2015).
While the number of host genes induced by F. virguliforme varied substantially, both hosts exhibited gene expression changes associated with marked enrichments in defense-related ontologies, indicating that the temporal induction of immunity plays a predominant role in the tolerance of F. virguliforme by maize. Indeed, when we compared the expression of orthologous defense-related processes, we observed a strikingly different response between soybean and maize, with very few orthologous genes identified as uniquely DE in both hosts. This observation supports our hypothesis of a significant divergence in the transcriptome of orthologous genes. Indeed, orthologs encoding defense responses identified as induced by F. virguliforme exhibited disparate temporal patterns of induction. This is significant, as this timeframe coincides with the recognition of the fungus and the subsequent initiation of host defense responses. This also coincides with the developmental transition of the pathogen from a biotrophic phase to a necrotrophic phase in soybean. Similar shifts in host gene expression as a function of fungal lifestyle changes during colonization have been described previously in the case of hemibiotrophic interactions (Njiti et al., 1996; Chowdhury et al., 2017). Coincident with developmental transitions in pathogen lifestyle, defense-related processes typically upregulated in infected hosts are commonly associated with processes, for example, associated with hydrogen peroxide production and the release of anti-microbial compounds.
In the current study, our analyses reveal that as fungal colonization develops over time, numerous orthologous defense genes in maize were downregulated, indicating a dampening of host defense signaling by the fungus. While this observation is consistent with previous work evaluating gene expression change in resistant cultivars (Chen et al., 2016), there are limited examples of this occuring in an asymptomatic host. Not unexpectedly, we oberved that soybean defense responses increased to maximal induced levels at 10–14 DAI, further highlighting a delayed host response to F. virguliforme colonization. Taken together, these data point to the temporal activation of defense signaling as the underlying mechanism distinguishing symptomatic versus asymptomatic responses. For example, nonorthologous defense-induced responses exhibited equivalent trends between hosts for a majority of defense-associated processes. Again, these data support a role for transcriptional control (i.e. TF expression) as a critical process underpinning F. virguliforme tolerance in maize.
The analyses described herein point to a key role for the early recognition and activation of defenses, including both the timing and amplitude of expression. For example, our data suggest that the early activation of defense responses in maize may stem from the specific induction of ethylene and ROS production, a process that was previously identified as a process associated with reduced disease severity in soybean in response to F. virguliforme colonization (Abdelsamad et al., 2019). The analyses presented herein highlight a potential role for ERF-105 (Bolt et al., 2017) and NAC042 (Thirumalaikumar et al., 2018) as key regulators of symptomatic versus asymtompatic signaling during fungal colonization of soybean and maize, respectively. Indeed, as F. virguliforme transitions from a biotrophic to necrotrophic lifestyle on soybean, a substantial transcriptomic rewiring occurs. Necrotrophic plant pathogens promote plant cell death, which results in dramatic shifts in host gene expression and cellular metabolism. Through an analysis of host gene expression changes over the timecourse of colonization, we identified changes in the regulation of several senescence-related TFs in soybean following pathogen colonization. As a result of these changes in gene expression, downstream signaling through additional senescence-related genes within soybean occurred, the outcome of which is the inhibition of protein synthesis, the hydrolysis of macromolecules, and the degeneration of cells (Podzimska-Sroka et al., 2015). As a function of pathogen proliferation and host cell death, the observed onset of senescence in soybean is associated with the timing of fungal pathogen necrotrophy and the appearance of disease symptoms. Similar processes have previously been described during pathogen infection and defense and include the production of ROS—a defense mechanism that is concomitant with the induction of NAC-associated host senescence (reviewed in Häffner et al., 2015).
As a further link to the work described herein, senescence-associated processes which signal through NAC TFs are also induced by abscisic acid. This is noteworthy, as we also identified the induced expression of genes associated with abscisic acid biosynthesis from samples collected from soybean roots at 2 DAI. This also includes the observation of NAC055 and NAC087 induction at 4 and 7 DAI, respectively. We posit that F. virguliforme susceptibility in soybean is mediated in part by pathogen-induced senescence processes, similar to those previously described in the case of Sclerotinia sclerotium (Williams et al., 2011). Based on the data presented herein, we predict that future research in this area will help clarify previous observations suggesting that plant senescence and defense share many overlapping, critical, signaling pathways. These include, for example, the modulation of expression of key NAC TFs, most of which have demonstrable roles in immunity and susceptibility (Bu et al., 2008; Saga et al., 2012; Yuan et al., 2019). What remains unclear is if gene expression is an indicator of pathogen-triggered senescence or host-induced susceptibility, or both. However, the mounting evidence presented herein, and previously seems to suggest that necrotrophic pathogen manipulation of genetic pathways leading to a favorable host environment are evident (Häffner et al., 2010, 2015; Williams et al., 2011; Podzimska-Sroka et al., 2015; Chowdhury et al., 2017). In conclusion, the data presented in the current study demonstate that the intersection of pathogen lifestyle, host genetics, and plant development play a key role in the interaction of defense, virulence, and processes of pathogen-accelerated host senescence.
Materials and methods
Plant and F. virguliforme assay
Soybean (Glycine max cv. Sloan; gift from Martin Chilvers, Michigan State University) and maize (Zea mays) hybrid E13022S (Epley Brothers Hybrids Inc., gift from Martin Chilvers) were surface sterilized in 70% ethanol for 30 s, followed by 10% bleach for 20 min, and then rinsed three times with sterile distilled water for 1 min. Surface sterilized soybean seeds were placed between two sheets of sterile Whatman filter paper soaked with 5 mL of sterile water, which was then placed inside a sterile Petri dish. Surface sterilized maize seeds were incubated in the dark in sterile water for 24 h and were then placed between two sheets of sterile 100-mm Whatman filter paper with 5 mL of sterile water inside a Petri dish. All seeds were incubated in the dark for 5 days at 21°C before transplanting.
The F. virguliforme Mont-1 isolate was propagated for 7 weeks on potato dextrose agar (Difco, Thermo-Fisher). Asexual macroconidia spores were collected, diluted to 1 × 105 macroconidia mL−1, and sprayed onto 5-day-old maize or soybean seedlings using a 3-oz travel spray bottle. Twenty-five sprays were applied to the seedlings at angles of 0°, 90°, 180°, and 270° to ensure that seedlings were thoroughly inoculated. For mock-inoculated samples, sterile water was sprayed onto the seedlings. Seedlings were incubated for 30 min with the inoculum (including mock-inoculated), after which time the excess inoculum was removed, and seedlings were incubated for an additional 1 h. Three seedlings (maize and soybean) were placed into CYG germination pouches (Mega International; CYG-19LB) containing 25 mL of sterile distilled water, and the pouches were transferred to a BioChambers Bigfoot Series Model AC-60 growth chamber with 140 µE m−2 s−1 (T5 fluorescent bulbs) and a 14:10 h light/dark cycle at 12°C for 7 days and then 25°C for 7 days. Plants were watered as needed with sterile water. Tap roots from soybean, or radicals from maize, were collected at the same time of day (i.e. 16:00 h) from the original 4-cm inoculation site. The 2-week timecourse was repeated three times (independent biological replicates) in the same growth chamber, with sampling of six plants for RNA isolation and three plants for DNA isolation at 0, 2, 4, 7, 10, and 14 DAI for each biological repeat. Sampling of timepoint 0 was performed after completion of mock and fungal inoculations at 1-h post-fungal inoculum application. Plant growth and disease symptomology were recorded at each timepoint by photography with a D50 Nikon camera.
Analysis of fungal colonization: visual phenotypes and quantification
To visualize fungal growth on plant roots, microscopy analyses of maize and soybean were conducted at each timepoint for all treatments. Roots (n = 6) were cleared in 100% ethanol, followed by staining in a 0.05% trypan blue solution containing equal parts of water, glycerol, and lactic acid (Savory et al., 2012). Fungal structures were observed using a MZ16 dissecting microscope (Leica).
To determine the amount of fungal biomass present in inoculated samples over the duration of the colonization timecourse, genomic DNA was extracted from flash-frozen root tissue and used for qPCR analysis. A total of 60 mg of ground root tissue was extracted from individual maize or soybean plants from each timepoint. DNA was extracted using the NucleoSpin Plant II Kit (Macherey-Nagel) according to the manufacturer’s protocol, with an additional incubation of 1 h at 65°C for the first step in DNA isolation (i.e. cell lysis step). DNA samples for qPCR-based detection of F. virguliforme were prepared following the method of Wang et al. (2015). The 5′-end of the F. virguliforme TaqMan probe (prb) was labeled with 6-fluorescein (6-FAM; Life Technologies), whereas the 3′-end was modified with a minor groove binder nonfluorescent quencher (MGBNFQ; Thermo-Fisher). PrimeTime dual-labeled probes were labeled with 5′ 6-FAM, internal ZEN quencher and 3′ Iowa Black FQ quencher (3IABkFQ; IDT Technologies). The exogenous control HHIC (Haudenshield and Hartman Internal Control) assay primers and probe were purchased from IDT Technologies and are described in Haudenshield and Hartman (2011). All DNA primers for F. virguliforme quantification are listed in Supplemental Table 10. Real-time qPCR reactions were performed using the Applied Biosystems 7500 Fast Real-Time PCR System (Thermo-Fisher). At least three biological replicates containing three technical repeats were performed for each timepoint. Reaction mixtures consisted of 10 µL of TaqMan Universal real-time PCR master mix (2X) (Applied Biosystems), 2 µL of DNA, 0.5 µL of FvPrb-3 (Fv TaqMan probe; 10 µM), 0.5 µL of F6-3 and R6 primers (20 µM, each), 0.6 µL of HHIC-F (forward primer; 20 µM) primer, 0.2 µL of HHIC-R (reverse primer 20 µM), 0.4 µL of HHIC-prb (10 µM), 0.5 µL of linearized HHIC DNA plasmid (10 fg/µL; Haudenshield and Hartman (2011), 0.4 µL of bovine serum albumin (New England BioLabs, catalog # B9000S) at 10 mg/mL, and 4.4 µL of distilled water (Thermo-Fisher). Cycling conditions were as follows: 1 cycle at 50°C for 2 min, 1 cycle at 95°C for 10 min, and 40 cycles at 95°C for 15 s, and a final step at 60°C for 1 min. Fluorescence data were collected during the annealing and extension stages of the program.
Analysis of variance was calculated for DNA quantities using the lme4 (Bates et al., 2015) and Car (Fox and Weisberg, 2011) packages in R (v3.4.1; R Development Core Team, 2010; Supplemental Table 1). Fungal DNA means were separated by Tukey’s least significant difference test using the multicomp package (Hothorn et al., 2008) at P ≤ 0.05.
RNA extraction
Root tissue from mock and infected samples from 0, 2, 4, 7, 10, and 14 DAI was used for RNA-sequencing. Biological samples were pooled from six independent plants, and for RNA-sequencing, three independent replicates were included from three independent growth chamber experiments. Sampling of plant tissue from both mock and inoculated roots at each timepoint allowed us to discover genes specifically related to fungal colonization. Additionally, all tissue was sampled from the same inoculation site within the root, further enabling us to explore responses within the inoculation site stemming from fungal adhesion, colonization, and proliferation. mRNA was prepared using the miRNeasy Mini Kit (Qiagen) from a total RNA isolation derived from 200 mg of either ground flash frozen germinating macroconidia or plant root sample. Contaminating genomic DNA was removed using the TURBO DNase Free kit (Invitrogen). RNA quality was evaluated by gel electrophoresis using the 2100 Bioanalyzer (Agilent) in combination with the Agilent RNA 6000 Pico kit, according to the manufacturer’s instructions.
Library preparation and sequencing
mRNA libraries were prepared from three biological repeats of each timepoint of F. virguliforme or mock inoculated maize or soybean or germinating macroconidia sample using the Illumina TruSeq mRNA Library Preparation Kit by the Michigan State Research Technology and Support Facility. Samples were pooled and sequenced on the Illumina HiSeq 4000 (single end 50-bp mode). Base calling was performed using the Illumina real-time analysis (RTA; v2.7.7), and the output of RTA was demultiplexed and converted to FastQ format using Illumina Bcl2fastq (v2.19.1). Sequencing of each sample was performed to an average yield of 70 million reads.
Quantification of RNA-seq expression and differential analysis
Reads were adapter-trimmed and evaluated for read quality using Trimmomatic (v0.33; Bolger et al., 2014). After trimming for adapters and quality thresholding, uniquely mapped soybean reads ranged from 76% to 83%, while maize reads were in the range of 77%–81% (Supplemental Figure 2A and Supplemental Data Set 1). Resultant trimmed reads were mapped to the corresponding reference genomes of soybean (Wm82.a2.v1) and maize (B73 RefGen_v4, AGPv4) using HISAT2 (v 2.1.0; Kim et al., 2015). The following parameters were applied: –dta–rna-strandness F.
Hits from HISAT2 were converted from SAM to BAM format by Picard (v2.18.1; http://broadinstitute.github.io/picard/). Sequence alignments were counted using HTSeq (v0.6.1; Anders et al., 2014) with the following options: –minaqual 50-m intersection-strict-s reverse–idattr=gene_id. Gene counts were imported into DESeq2 (v1.22.2; Love et al., 2014), executed in R (R Development Core Team, 2010), normalized for library size, and log2-transformed to determine the correlation of biological replicates at each timepoint.
To determine differential gene expression, DESeq2 (v1.22.2), executed in R with raw HTSeq counts, was used. Gene counts of less than 10 were excluded. DESeq2 was used to identify DE transcripts which met the requirement of an adjusted P ≤ 0.05 and greater than |1-log2| fold difference in expression between mock- and F. virguliforme-inoculated samples. Pairwise comparisons were evaluated at each timepoint and within each host, including mock- and F. virguliforme-inoculated samples.
Differential gene co-expression network analysis
Gene counts were filtered for DGCA (implemented in R; McKenzie et al., 2016) for 90% of genes with less than 10 reads across all samples. The resultant 43,308 soybean and 28,956 maize genes were variance stabilized, transformed for importation, and the Pearson correlation of individual gene pairs within each treatment was calculated and compared to mock- or F. virguliforme-inoculated treatments. To classify differential correlation significance, DGCA transforms correlation coefficients to z-scores and uses differences in z-scores to calculate P-values of differential between gene pairs (e.g. ρ < 0, adjusted P < 0.05 for negative correlation, and ρ > 0, adjusted P < 0.05 for positive correlation), with a Bonferroni correction of P-value (i.e. McKenzie et al., 2016). Significant gene pairs from differentially induced classes (e.g. +/0, −/0, 0/−, +/0, +/−, and −/+) were weighted by the z-score difference between treatments to convert into a PFN assembled upon unique links between significantly differentially correlated gene pairs within a spherical surface to derive with the most relevant information from a similarity matrix based on a topological sphere. Gene pairs were imported into MEGENA and multiscale modules and hubs were identified through partitioning of the parent PFN by k-splitting via evaluation of network compactness through multi-scale embedded gene co-expression analysis, with a hub detection significance threshold of P < 0.05, module significance threshold of P < 0.05, network permutations of 100, and module size greater than 10 (Song and Zhang, 2015). Differential gene expression correlation analysis of samples from maize identified 128 significant gene pairs from 419,152,582 total gene pairs between mock- and pathogen-inoculated. Analysis of the correlation between soybean gene expression identified 5,526,057 significant gene pairs from 904,379,186 gene pairs between treatments. Soybean differential gene pairs were clustered into 1,161 modules using MEGENA (https://cran.r-project.org/web/packages/MEGENA/index.html). Modules were visualized in R using ggplot2 (v3.1.1; Wickham, 2016).
Identification of orthologous genes
Protein sequences were retrieved from Ensembl Plants (http://plants.ensembl.org/index.html). The longest protein sequences for genes from soybean and maize were analyzed by OrthoFinder using program default settings (v2.2.7; Emms and Kelly, 2019). A total of 23,274 soybean and 14,542 maize genes were discovered and subsequently classified into 10,700 orthogroups. This dataset was next analyzed to filter log2-fold changes between mock and inoculated maize or soybean at the orthogroup level. Because many orthogroups contained more than a one-to-one relationship, the median of log2-fold changes of all genes within one host orthogroup was evaluated. The median transformation of genes within an orthogroup captured the most variation between maize and soybean (Supplemental Data Set 9).
Differential expression analysis of orthogroups
To determine differential orthogroup expression, DESeq2 (v1.22.2; executed in R) was employed using the median transformed gene HTSeq counts within each orthogroup. Orthogroup counts with a value less than 10 were filtered from the analysis. Significant differential expression patterns were determined using an adjusted P ≤ 0.05. A greater than one-fold difference between mock treatments between maize and soybean, as well as inoculated treatments between maize and soybean, at each timepoint was used as a cutoff for analyses.
GO enrichment analysis
GO was retrieved from the GFF file of the soybean genome annotation (Wm82.a2.v1) and the revised GO annotation for maize (Wimalanathan et al., 2018). The GO annotation varies in completeness between soybean and maize. For example, the defense response term (GO:0006952) has 374 annotated genes; however, in soybean, only 86 genes are associated with this GO annotation. To generate an ontology that is similar for an ortholog comparison, we merged gene ontologies from maize and soybean within each orthogroup and removed redundant ontology terms. In total, all 10,700 orthogroups had GO assignments.
Orthogroup lists from differential analyses were analyzed by TopGO (2.34.0) conducted in R (Alexa and Rahnenfuhrer, 2018). Fisher’s exact test, set at an adjusted P ≤ 0.05, was conducted on each orthogroup to determine the relative significance of enrichments across all orthogroups. Additionally, lists of genes from differential analysis or gene differential clustering modules were analyzed for GO term enrichment by the singular enrichment analysis using the AgriGO (v2) website (Tian et al., 2017) with the Fisher statistical adjustment method set at a significance level of 0.05.
Orthologous TF analysis
TF sequences from maize were retrieved from: http://planttfdb.cbi.pku.edu.cn/.
Maize genome v3 gene IDs were converted to v4 IDs for the 2,290 TFs and orthologous genes in soybean; this included 508 orthogroups. Orthogroups were filtered for significant defense induction over the timecourse.
Positive and negative senescence TFs analysis
Positive and negative senescence TFs were selected from previous studies (Koyama, 2014), with a total of nine positive TFs and nine negative TFs chosen for further expression analysis herein. The gene expression value of each TFs was derived from mock treatments between maize and soybean, as well as inoculated treatments between maize and soybean, at each timepoint after quantification of differential gene expression.
Simplify GO enrichment analysis
GO enrichments from F. virguliforme (Baetsen-Young et al., 2020), maize, and soybean were analyzed by using a simplified enrichGo method in the clusterProfiler package of R (Yu et al., 2012) to reduce redundant GO terms from the generated output of the enrichGO function. All user-defined parameters were set to the program default mode.
GO analysis
Related gene ontologies of F. virguliforme (Baetsen-Young et al., 2020) were selected based on its biotrophy–necrotrophy switch in pathogen lifestyle (Doehlemann et al., 2017). Related gene ontologies of maize and soybean were selected based on the biological processes related to response to F. virguliforme colonization (Vargas et al., 2012). Gene count numbers were calculated based on gene numbers that sorted into each selected GO category and as a function of up- or downregulated expression changes.
Accession numbers
The RNA-seq data generated herein are contained within the National Center for Biotechnology Information (NCBI) Short Read Archive (SRA) in BioProject under project ID: PRJNA549915.
Supplementary Material
Acknowledgments
We would like to acknowledge the C.S. Mott Foundation for fellowship support to A.B.Y. Thanks to Martin Chilvers for providing the Fusarium virguliforme Mont-1 isolate, and to the support staff at the MSU Institute for Cyber-Enabled Research (High Performance Computing Cluster) for assistance in software optimization. Special thanks to Ching-Man Wai for advice during the development of the research, and to Noel Day for critical reading of the manuscript.
Funding
Research in the laboratory of B.D. was supported by the MSU Plant Resilience Institute and the National Institutes of General Medical Sciences (1R01GM125743). Research in the laboratory of S.-H.S. was supported by the National Science Foundation (IOS-1546617 and DEB-1655386), and the Department of Energy Great Lakes Bioenergy Research Center (BER DE-SC0018409).
Supplemental data
Supplemental Figure 1. Plant growth and development across the colonization timecourse.
Supplemental Figure 2. Read alignment and principal component analysis.
Supplemental Figure 3. Correlation of gene expression values in three biological replicates from 14 DAI samples of maize inoculated with F. virguliforme.
Supplemental Figure 4. Graphical representation of expressed and DEGs in soybean and maize following F. virguliforme inoculation.
Supplemental Figure 5. Module hierarchy of hubs containing defense-related genes.
Supplemental Figure 6. Correlation of median transformed log2-fold changes between F. virguliforme- and mock-inoculated orthogroups within soybean compared to maize.
Supplemental Figure 7. Gene ontology enrichment of DEGs maize and soybean.
Supplemental Figure 8 . Biological processes of DEGs from F. virguliforme at each DAI during growth on maize and soybean.
Supplemental Figure 9. Gene ontology enrichment of DEGs among orthologous and nonorthologous genes.
Supplemental Figure 10. Biological processes of different expression genes for maize and soybean at each DAI.
Supplemental Figure 11. Expression of orthologous genes in defense-relevant processes.
Supplemental Table 1 . ANOVA tables.
Supplemental Table 2. Significantly induced genes in maize.
Supplemental Table 3. Significantly induced genes in soybean.
Supplemental Table 4. Gene ontology enrichment of biological processes of genes from maize significantly upregulated between mock- and F. virguliforme-inoculated treatments over a timecourse of 2 weeks.
Supplemental Table 5. Gene ontology enrichment of biological processes of genes from soybean that were significantly upregulated between mock- and F. virguliforme-inoculated treatments over a timecourse of 2 weeks.
Supplemental Table 6. Significantly induced nonorthologous gene ontologies in maize.
Supplemental Table 7. Significantly induced nonorthologous gene ontologies in soybean.
Supplemental Table 8. Gene ontology enrichment of biological processes in nonorthologous genes from maize significantly upregulated between mock- and F. virguliforme-inoculated treatments over the 2-week timecourse.
Supplemental Table 9. Gene ontology enrichment of biological processes in nonorthologous genes from soybean significantly upregulated between mock- and F. virguliforme-inoculated treatments over the 2-week timecourse.
Supplemental Table 10. Primer sequences for qPCR to quantify F. virguliforme in planta levels.
Supplemental Data Set 1. Read mappings of maize and soybean libraries to either reference genomes of soybean (Wm82.a2.v1) and maize (B73 RefGen_v4, AGPv4).
Supplemental Data Set 2. DEGs at each timepoint of maize colonization between F. virguliforme- and mock-inoculated, with mock as a comparison base.
Supplemental Data Set 3. DEGs at each timepoint of soybean colonization between F. virguliforme- and mock-inoculated, with mock as a baseline for comparison.
Supplemental Data Set 4. Significant differential gene pairs between F. virguliforme-inoculated (corA) and mock (corB) treatments in maize.
Supplemental Data Set 5. Gene ontology enrichment of genes in modules for soybean.
Supplemental Data Set 6. Gene identities assigned to corresponding orthogroups for maize and soybean.
Supplemental Data Set 7. DE (log2-fold change) soybean genes have orthologs in maize.
Supplemental Data Set 8. DE (log2-fold change) maize genes that have orthologs in soybean.
Supplemental Data Set 9. DE orthogroup median of log2-fold changes between mock and inoculated treatments within each host.
Supplemental Data Set 10. Gene ontology annotation of orthogroups between maize and soybean.
Supplemental Data Set 11. Gene Ontology enrichment of DE orthogroups.
Supplemental Data Set 12. DE maize genes log2-fold changes without orthologues in soybean.
Supplemental Data Set 13. DE soybean genes log2-fold changes that are not orthologous with maize.
Supplemental Data Set 14. Gene ontology enrichment of DE upregulated nonorthologous genes in maize.
Supplemental Data Set 15. Gene ontology enrichment of DE upregulated nonorthologous soybean genes.
Supplemental Data Set 16. Gene ontology enrichment of DE downregulated nonorthologous maize genes.
Supplemental Data Set 17. Gene ontology enrichment of DE downregulated nonorthologous soybean genes.
Supplemental Data Set 18. Gene list of 182 orthologous gens that were significantly upregulated in maize.
Supplemental Data Set 19. DE transcription orthogroup median log2-fold changes between inoculated and mock in each host. Differential expression is grouped by shared or unique in each host.
Supplemental Data Set 20. Orthologous transcription factors.
Supplemental Data Set 21. Orthologous and nonorthologous genes are involved in a variety of regulatory pathways that control senescence.
Supplemental Data Set 22. TFs that positively and negatively regulate senescence-associated gene processes.
Supplemental Data Set 23. Gene expression of senescence-associated genes and NAC transcription factors.
Additional Supplemental Data Modules are deposited at Dryad: https://datadryad.org/stash/share/bl9xHgsTaIvsAZjYeInMc78Y1fNz8-Px3uGBsw7C99Q
A.B.Y., H.C., S.-H.S., and B.D. designed the framework; A.B.Y. conducted the experiments; A.B.Y., H.C., B.D. analyzed data; A.B.Y., H.C., S.-H.S., and B.D. wrote the article.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell) is: Brad Day (bday@msu.edu).
References
- Abdelsamad NA, MacIntosh GC, Leandro LFS (2019) Induction of ethylene inhibits development of soybean sudden death syndrome by inducing defense-related genes and reducing Fusarium virguliforme growth. PLoS One 14: e0215653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexa A, Rahnenfuhrer J (2018) topGO: Enrichment analysis for gene ontology. R package version 2.36.0
- Allardyce JA, Rookes JE, Hussain HI, Cahill DM (2013) Transcriptional profiling of Zea mays roots reveals roles for jasmonic acid and terpenoids in resistance against Phytophthora cinnamomi. Funct Integr Genom 13: 217–228 [DOI] [PubMed] [Google Scholar]
- Allen TW, Bradley CA, Sisson AJ, Byamukama E, Chilvers MI, Coker CM, Collins AA, Damicone JP, Dorrance AE, Dufault NS, et al. (2017) Soybean yield loss estimates due to diseases in the United States and Ontario, Canada, from 2010 to 2014. Plant Health Prog 18: 19–27 [Google Scholar]
- Anders S, Pyl PT, Huber W (2014) HTSeq—a python framework to work with high-throughput sequencing data. Bioinform 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew C, Sellam A, Epp E, Hoques H, Mullick A, Nantel A, Whiteway M (2009) Transcriptional regulation of carbohydrate metabolism in the human pathogen Candida albicans. PLoS Pathog 5: e1000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baetsen-Young A, Wei CM, VanBuren R, Day B (2020) Fusarium virguliforme transcriptional plasticity revealed by diverged host colonization. Plant Cell 32: 336–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnaresi P, Biselli C, Orrù L, Urso S, Crispino L, Abbruscato P, Piffanelli P, Lupotto E, Cattivelli L, Valè G (2012) Comparative transcriptome profiling of the early response to Magnaporthe oryzae in durable resistant vs susceptible rice (Oryza sativa L.) genotypes. PLoS One 7: e51609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer D, Planchamp C, Mauch-Mani B (2012) On the move: induced resistance in monocots. J Exp Bot 64: 1249–1261 [DOI] [PubMed] [Google Scholar]
- Balmer D, de Papajewski DV, Planchamp C, Glauser G, Mauch-Mani B (2013) Induced resistance in corn is based on organ-specific defence responses. Plant J 74: 213–225 [DOI] [PubMed] [Google Scholar]
- Banerjee S, Walder F, Büchi L, Meyer M, Held AY, Gattinger A, Keller T, Charles R, van der Heijden MGA (2019) Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. ISME J 13: 1722–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D, Machler M, Bolker B, Walker S (2015) Fitting linear mixed-effect models using lme4. J Stat Soft 67: 1–48 [Google Scholar]
- Besseau S, Li J, Palva ET (2012) WRKY54 and WRKY70 co-operate as negative regulators of leaf senescence in Arabidopsis thaliana. J Exp Bot 63: 2667–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boba A, Kostyn K, Zozak B, Wojtasik W, Preisner W, Prescha A, Gola EM, Lysh D, Dudek B, Szopa J, et al. (2020) Fusarium oxysporum infection activates the plastidial branch of the terpenoid biosynthesis pathway in flax, leading to increased ABA synthesis. Planta 251: 50. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolt S, Zuther E, Zintl S, Hincha DK, Schmülling T (2017) ERF105 is a transcription factor gene of Arabidopsis thaliana required for freezing tolerance and cold acclimation. Plant Cell Environ 40: 108–120 [DOI] [PubMed] [Google Scholar]
- Bu Q, Jiang H, Li C-B, Zhai Q, Zhang J, Wu X, Sun J, Xie Q, Li C (2008) Role of the Arabidopsis thaliana NAC transcription factors ANAC019 and ANAC055 in regulating jasmonic acid-signaled defense responses. Cell Res 18: 756–767 [DOI] [PubMed] [Google Scholar]
- Burkhardt A, Day B (2016) Transcriptome and small RNAome dynamics during a resistant and susceptible interaction between cucumber and downy mildew. Plant Genome 9(1), doi: 10.3835/plantgenome2015.08.0069 [DOI] [PubMed] [Google Scholar]
- Chang HX, Domier LL, Radwan O, Yendrek CR, Hudson ME, Hartman GL (2016) Identification of multiple phytotoxins produced by Fusarium virguliforme including a phytotoxic effector (FvNIS1) associated with sudden death syndrome foliar symptoms. Mol Plant-Microbe Interact 29: 96–108 [DOI] [PubMed] [Google Scholar]
- Chen Y, Yin H, Gao M, Zhu H, Zhang Q, Wang Y (2016) Comparative transcriptomics atlases reveals different gene expression pattern related to Fusarium wilt disease resistance and susceptibility in two Vernicia species. Front Plant Sci 7: 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm S, Coaker G, Day B, Staskawicz BJ (2006) Host-microbe interactions: shaping the evolution of the plant immune response. Cell 124: 1–12 [DOI] [PubMed] [Google Scholar]
- Chowdhury S, Basu A, Kundu S (2017) Biotrophy-necrotrophy switch in pathogen evoke differential response in resistant and susceptible sesame involving multiple signaling pathways at different phases. Sci Rep 7: 17251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuberre C, Plancot B, Driouich A, Moore JP, Bardor M, Gügi B, Vicré M (2018) Plant immunity is compartmentalized and specialized in roots. Front Plant Sci 9, doi: 10.3389/fpls.2018.01692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day B, Graham T (2007) The plant host pathogen interface: cell wall and membrane dynamics of pathogen-induced responses. Ann NY Acad Sci 1113: 123–134 [DOI] [PubMed] [Google Scholar]
- De Coninck B, Timmermans P, Vos C, Cammue BP, Kazan K (2015) What lies beneath: belowground defense strategies in plants. Trends Plant Sci 20: 91–101 [DOI] [PubMed] [Google Scholar]
- Deshmukh S, Hückelhoven R, Schäfer P, Imani J, Sharma M, Weiss M, Waller F, Kogel K-H (2006) The root endophytic fungus Piriformospora indica requires host cell death for proliferation during mutualistic symbiosis with barley. Proc Natl Acad Sci USA 103: 18450–18457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveau A, Kohler A, Frey-Klett P, Martin F (2008) The major pathways of carbohydrate metabolism in the ectomycorrhizal Basidiomycete laccaria bicolor S238N. New Phytol 180: 379–390 [DOI] [PubMed] [Google Scholar]
- Diamond M, Reape TJ, Rocha O, Doyle SM, Kacprzyk J, Doohan FM, McCabe PF (2013) The Fusarium mycotoxin deoxynivalenol can inhibit plant apoptosis-like Programmed cell death. PLoS One 8: e69542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divon HH, Fluhr R (2007) Nutrition acquisition strategies during fungal infection of plants. FEMS Microbiol Lett 266: 65–74 [DOI] [PubMed] [Google Scholar]
- Doehlemann G, Ökmen B, Zhu W, Sharon A (2017) Plant pathogenic fungi. Microbiol Spectr 5, doi: 10.1128/microbiolspec.FUNK-0023-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont P-Y, Eaton CJ, Wargent JJ, Fechtner S, Solomon P, Schmid J, Day RC, Emms DM, Kelly S (2019) OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etalo DW, Stulemeijer IJE, Peter van Esse H, de Vos RCH, Bouwmeester HJ, Joosten MHAJ (2013) System-wide hypersensitive response-associated transcriptome and metabolome reprogramming in tomato. Plant Physiol 162: 1599–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagard M, Launay A, Clément G, Courtial J, Dellagi A, Farjad M, Krapp A, Soulié M-C, Masclaux-Daubresse C (2014) Nitrogen metabolism meets phytopathology. J Exp Bot 65: 5643–5656 [DOI] [PubMed] [Google Scholar]
- Fehr WR, Caviness CE (1977) Stages of soybean development. Iowa State University Special Report. 87. http://lib.dr.iastate.edu/specialreports/87
- Fox J, Weisberg S (2011) An R Companion to Applied Regression. Sage, Thousand Oaks, CA [Google Scholar]
- Gdanetz K, Trail F (2017) The wheat microbiome under four management strategies, and potential for endophytes in disease protection. Phytobiomes J 1: 158–168 [Google Scholar]
- Häffner E, Karlovsky P, Diederichsen E (2010) Genetic and environmental control of the Verticillium syndrome in Arabidopsis thaliana. BMC Plant Biol 10: 235–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häffner E, Konietzki S, Diederichsen E (2015) Keeping control: the role of senescence and development in plant pathogenesis and defense. Plants 4: 449–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haudenshield JS, Hartman GL (2011) Exogenous controls increase negative call veracity in multiplexed, quantitative PCR assays for Phakopsora pachyrhizi. Plant Dis 95: 343–352 [DOI] [PubMed] [Google Scholar]
- Hothorn T, Bretz F, Westfall P (2008) Simultaneous inference in general parametric models. Biom J 50: 346–363 [DOI] [PubMed] [Google Scholar]
- Humphry M, Bednarek P, Kemmerling B, Koh S, Stein M, Göbel U, Stüber K, Piślewska-Bednarek M, Loraine A, Schulze-Lefert P, et al. (2010) A regulon conserved in monocot and dicot plants defines a functional module in antifungal plant immunity. Proc Natl Acad Sci USA 107: 21896–21901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huysmans M, Buono RA, Skorzinski N, Radio MC, De Winter F, Parizot B, Mertens J, Karimi M, Fendrych M, Nowack MK (2018) NAC transcription factors ANAC087 and ANAC046 control distinct aspects of programmed cell death in the Arabidopsis columella and lateral root cap. Plant Cell 30: 2197–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A (2001) Programmed cell death in development and disease. Plant Physiol 125: 94–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katan J (2017) Diseases caused by soilborne pathogens: biology, management and challenges. J Plant Pathol 99: 305–315 [Google Scholar]
- Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12: 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenning SR, Wrather JA (2010) Suppression of soybean yield potential in the continental United States by plant diseases from 2006 to 2009. Plant Health Prog 11. doi: 10.1094/php-2010-1122-1001-rs [Google Scholar]
- Kolander TM, Bienapfl JE, Kurle JE, Malvick DK (2012) Symptomatic and asymptomatic host range of Fusarium virguliforme, the causal agent of soybean sudden death syndrome. Plant Dis 96: 1148–1153 [DOI] [PubMed] [Google Scholar]
- Kong W, Chen N, Liu T, Zhu J, Wang J, He X, Jin Y (2015) Large-scale transcriptome analysis of cucumber and Botrytis cinerea during infection. PLoS One 10: e0142221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama T (2014) The roles of ethylene and transcription factors in the regulation of onset of leaf senescence. Front Plant Sci 5: 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama T, Sato F, Ohme-Takagi M (2017) Roles of miR319 and TCP transcription factors in leaf development. Plant Physiol 175: 874–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer M, Damoo D, Djamei A, Kronstad J (2019) Chloroplasts and plants: where are the fungal effectors? Pathogens 9: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laluk K, Mengiste T (2010) Necrotroph attacks on plants: wanton destruction or covert extortion? Arabidopsis Book 8: e0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanubile A, Muppirala UK, Severin AJ, Marocco A, Munkvold GP (2015) Transcriptome profiling of soybean (Glycine max) roots challenged with pathogenic and non-pathogenic isolates of Fusarium oxysporum. BMC Genom 16: 1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leandro LFS, Eggenberger S, Chen C, Williams J, Beattie GA, Liebman M (2018) Cropping system diversification reduces severity and incidence of soybean sudden death syndrome caused by Fusarium virguliforme. Plant Dis 102: 1748–1758 [DOI] [PubMed] [Google Scholar]
- Li Z, Pan X, Guo X, Fan K, Lin W (2019) Physiological and transcriptome analyses of early leaf senescence for ospls1 mutant rice (Oryza sativa L.) during the grain-filling stage. Int J Mol Sci 20: 1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lofgren LA, LeBlanc NR, Certano AK, Nachtigall J, LaBine KM, Riddle J, Broz K, Dong Y, Bethan B, Kafer CW, et al. (2018) Fusarium graminearum: pathogen or endophyte of North American grasses? New Phytol 217: 1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorrain C, Marchal C, Hacquard S, Delaruelle C, Pétrowski J, Petre B, Hecker A, Frey P, Duplessis S (2018) The rust fungus Melampsora larici-populina expresses a conserved genetic program and distinct sets of secreted protein genes during infection of its two host plants, larch and poplar. Mol Plant-Microbe Interact 31: 695–706 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genom Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Marella CBN, Hartmann A, Hajirezaei MR, von Wirén N (2019) An age-dependent sequence of physiological processes defines developmental root senescence. Plant Physiol 181: 993–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Friesen TL, Faris JD (2011) Molecular characterization and genomic mapping of the pathogenesis-related protein 1 (PR-1) gene family in hexaploid wheat (Triticum aestivum L.). Mol Genet Genom 285: 485–503 [DOI] [PubMed] [Google Scholar]
- Lynch JP, Chimungu JG, Brown KM (2014) Root anatomical phenes associated with water acquisition from drying soil: targets for crop improvement. J Exp Bot 65: 6155–6166 [DOI] [PubMed] [Google Scholar]
- Majid I, Abbas N (2019) Signal transduction in leaf senescence: an overview. InSarwat M, Tuteja M (eds), Senescence Signalling and Control in Plants. Academic Press, Cambridge, MA, pp 41–59 [Google Scholar]
- Majumdar R, Rajasekaran K, Sickler C, Lebar M, Musungu BM, Fakhoury AM, Payne GA, Geisler M, Carter-Wientjes C, Wei Q, et al. (2017) The pathogenesis-related corn seed (PRms) gene plays a role in resistance to Aspergillus flavus infection and aflatoxin contamination. Front Plant Sci 8: 1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcolm GM, Kuldau GA, Gugino BK, del Mar Jimenez-Gasco M (2013) Hidden host plant associations of soilborne fungal pathogens: an ecological perspective. Phytopathology 103: 538–544 [DOI] [PubMed] [Google Scholar]
- McKenzie AT, Katsyv I, Song WM, Wang M, Zhang B (2016) DGCA: a comprehensive R package for differential gene correlation analysis. BMC Syst Biol 10: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michielse CB, Rep M (2009) Pathogen profile update: Fusarium oxysporum. Mol Plant Pathol 10: 311–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittler R, Kim Y, Song L, Coutu J, Coutu A, Ciftci-Yilmaz S, Lee H, Stevenson B, Zhu J-K (2006) Gain- and loss-of-function mutations in Zat10 enhance the tolerance of plants to abiotic stress. FEBS Lett 580: 6537–6542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngaki MN, Wang B, Sahu BB, Srivastava SK, Farooqi MS, Kambakam S, Swaminathan S, Bhattacharyya MK (2016) Tanscriptomic study of the soybean-Fusarium virguliforme interaction revealed a novel ankyrin-repeat containing defense gene, expression of whose during infection led to enhanced resistance to the fungal pathogen in transgenic soybean plants. PLoS One 11: e0163106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njiti VN, Shenaut MA, Suttner RJ, Schmidt ME, Gibson PT (1996) Soybean response to sudden death syndrome: inheritance influenced by cyst nematode resistance in Pyramid × Douglas progenies. Crop Sci 36: 1165–1170 [Google Scholar]
- O'Connell RJ, Thon MR, Hacquard S, Amyotte SG, Kleemann J, Torres MF, Damm U, Buiate EA, Epstein L, Alkan N, et al. (2012) Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat Genet 44: 1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pell EJ, Miller JD, Bielenberg DG (2004) 20 effects of airborne pollutants. InNoodén LD, ed, Plant Cell Death Processes. Academic Press, San Diego, CA, pp 295–306 [Google Scholar]
- Podzimska-Sroka D, O'Shea C, Gregersen PL, Skriver K (2015) NAC transcription factors in senescence: from molecular structure to function in crops. Plants 4: 412–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusztahelyi T, Holb IJ, Pócsi I (2016) Plant-fungal interactions: special secondary metabolites of the biotrophic, necrotrophic, and other specific interactions. InMérillon JM, Ramawat K, eds, Fungal Metabolites. Reference Series in Phytochemistry. Springer, Cham, Switzerland, pp 1–58 [Google Scholar]
- R Development Core Team (2010) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Saga H, Ogawa T, Kai K, Suzuki H, Ogata Y, Sakurai N, Shibata D, Ohta D (2012) Identification and characterization of ANAC042, a transcription factor family gene involved in the regulation of camalexin biosynthesis in Arabidopsis. Mol Plant-Microbe Interact 25: 684–696 [DOI] [PubMed] [Google Scholar]
- Salleh FM, Evans K, Goodall B, Machin H, Mowla SB, Mur LAJ, Runions J, Theodoulou FL, Foyer CH, Rogers HJ (2012) A novel function for a redox-related LEA protein (SAG21/AtLEA5) in root development and biotic stress responses. Plant Cell Environ 35: 418–429 [DOI] [PubMed] [Google Scholar]
- Savory EA, Adhikari BN, Hamilton JP, Vaillancourt B, Buell CR, Day B (2012) mRNA-seq analysis of the Pseudoperonospora cubensis transcriptome during cucumber (Cucumis sativus l.) infection. PLoS One 7: e35796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer P, Khatabi B, Kogel K-H (2007) Root cell death and systemic effects of Piriformospora indica: a study on mutualism. FEMS Microbiol Lett 275: 1–7 [DOI] [PubMed] [Google Scholar]
- Šišić A, Baćanović-Šišić J, Al-Hatmi AMS, Karlovsky P, Ahmed SA, Maier W, de Hoog GS, Finckh MR (2018) The ‘forma specialis’ issue in Fusarium: a case study in Fusarium solani f. sp. pisi. Sci Rep 8: 1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schommer C, Palatnik JF, Aggarwal P,, Chételat A, Cubas P, Farmer EE, Nath U, Weigel D (2008) Control of jasmonate biosynthesis and senescence by miR319 targets. PLoS Biol 6: e230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WM, Zhang B (2015). Multiscale embedded gene co-expression network analysis. PLoS Comput Biol 11: e1004574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J, Yang L, Zhu Q, Wu H, He Y, Liu Y, Xu J, Jiang D, Zhang S (2018) Active photosynthetic inhibition mediated by MPK3/MPK6 is critical to effector-triggered immunity. PLoS Biol 16: e2004122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirumalaikumar VP, Devkar V, Mehterov N, Ali S, Ozgur R, Turkan I, Mueller-Roeber B, Balazadeh S (2018) NAC transcription factor JUNGBRUNNEN1 enhances drought tolerance in tomato. Plant Biotechnol J 16: 354–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian T, Liu Y, Yan H, You Q, Yi X, Du Z, Xu W, Su Z (2017) Agrigo v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res 45: W122–W129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Does HC, Rep M (2017) Adaptation to the host environment by plant pathogenic fungi. Ann Rev Phytopathol 55: 427–450 [DOI] [PubMed] [Google Scholar]
- Vargas WA, Martín JM, Rech GE, Rivera LP, Benito EP, Díaz-Mínguez JM, Thon MR, Sukno SA (2012) Plant defense mechanisms are activated during biotrophic and necrotrophic development of Colletotrichum graminicola in maize. Plant Physiol 158: 1342–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Jacobs JL, Byrne JM, Chilvers MI (2015) Improved diagnoses and quantification of Fusarium virguliforme, causal agent of soybean sudden death syndrome. Phytopathology 105: 378–387 [DOI] [PubMed] [Google Scholar]
- Wickham H (2016) ggplot2: Elegant Graphics for Data Analysis. Springer, New York [Google Scholar]
- Williams B, Kabbage M, Kim H-J, Britt R, Dickman MB (2011) Tipping the balance: Sclerotinia sclerotiorum secreted oxalic acid suppresses host defenses by manipulating the host redox environment. PLoS Pathog 7: e1002107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimalanathan K, Friedberg I, Andorf CM, Lawrence-Dill CJ (2018) Corn GO annotation—methods, evaluation, and review (Corn-GAMER). Plant Direct 2: e00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciechowska N, Sobieszczuk-Nowicka E, Bagniewska-Zadworna A (2018) Plant organ senescence – regulation by manifold pathways. Plant Biol 20:167–181 [DOI] [PubMed] [Google Scholar]
- Wong KH, Tan WL, Kini SG, Xiao T, Serra A, Sze SK, Tam JP (2017) Vaccatides: antifungal glutamine-rich hevein-like peptides from Vaccaria hispanica. Front Plant Sci 8:1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo HR, Kim HJ, Nam HG, Lim PO (2013) Plant leaf senescence and death – regulation by multiple layers of control and implications for aging in general. J Cell Sci 126: 4823–4833 [DOI] [PubMed] [Google Scholar]
- Wu A, Allu AD, Garapati P, Siddiqui H, Dortay H, Zanor MI, Asensi-Fabado MA, Munne-Bosch S, Antonio C, Tohge T, et al. (2012) JUNGBRUNNEN1, a reactive oxygen species-responsive NAC transcription factor, regulates longevity in Arabidopsis. Plant Cell 24: 482–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Zhao Y, Burks P, Pauly M, Brown PJ (2018) A sorghum NAC gene is associated with variation in biomass properties and yield potential. Plant Direct 2, doi: 10.1002/pld3.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Frugis G, Colgan D, Chua N-H (2000) Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Dev 14: 3024–3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L, Zhu J-K (2003) Regulation of abscisic acid biosynthesis. Plant Physiol 133: 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Ohtani M, Mitsuda N, Kubo M, Ohme-Takagi M, Fukuda H, Demura T (2010) VND-INTERACTING2, a NAC domain transcription factor, negatively regulates xylem vessel formation in Arabidopsis. Plant Cell 22: 1249–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Ohlrogge JB (2009) Turnover of fatty acids during natural senescence of Arabidopsis, Brachypodium, and switchgrass and in Arabidopsis beta-oxidation mutants. Plant Physiol 150: 1981–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- York LM, Nord EA, Lynch J (2013) Integration of root phenes for soil resource acquisition. Front Plant Sci 4: 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, He Q-Y (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS: J Int Biol 16: 284–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Wang H, Cai J, Li D, Song F (2019) NAC transcription factors in plant immunity. Phytopathol Res 1: 3 [Google Scholar]
- Zhang J, Singh A, Mueller DS, Singh AK (2015) Genome-wide association and epistasis studies unravel the genetic architecture of sudden death syndrome resistance in soybean. Plant J 84: 1124–1136 [DOI] [PubMed] [Google Scholar]
- Zhang W, Zhao F, Jiang L, Chen Cc, Wu L, Liu Z (2018a) Different pathogen defense strategies in Arabidopsis: more than pathogen recognition. Cells 7: 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tian L, Yan D-H, He W (2018b) Genome-wide transcriptome analysis reveals the comprehensive response of two susceptible poplar sections to Marssonina brunnea infection. Genes 9: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Valdes-Lopez O, Arellano C, Stacey G, Balint-Kurti P (2017) Genetic dissection of the corn (Zea mays L.) MAMP response. Theor Appl Genet 130: 1155–1168 [DOI] [PubMed] [Google Scholar]
- Zhao Z, Li Y, Zhao S, Zhang J, Zhang H, Fu B, He F, Zhao M, Liu P (2018) Transcriptome analysis of gene expression patterns potentially associated with premature senescence in Nicotiana tabacum L. Molecules 23: 2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.