

Abstract
This paper explores the pipeline of new and upcoming vaccines as it relates to monitoring their safety. Compared with most currently available vaccines, that are constituted of live attenuated organisms or inactive products, future vaccines will also be based on new technologies. Several products that include such technologies are either already licensed or at an advanced stage of clinical development. Those include viral vectors, genetically attenuated live organisms, nucleic acid vaccines, novel adjuvants, increased number of antigens present in a single vaccine, novel mode of vaccine administration and thermostabilisation. The Global Advisory Committee on Vaccine Safety (GACVS) monitors novel vaccines, from the time they become available for large scale use. GACVS maintains their safety profile as evidence emerges from post-licensure surveillance and observational studies. Vaccines and vaccine formulations produced with novel technologies will have different safety profiles that will require adapting pharmacovigilance approaches. For example, GACVS now considers viral vector templates developed on the model proposed by Brighton Collaboration. The characteristics of those novel products will also have implications for the risk management plans (RMPs). Questions related to the duration of active monitoring for genetic material, presence of adventitious agents more easily detected with enhanced biological screening, or physiological mechanisms of novel adjuvants are all considerations that will belong to the preparation of RMPs. In addition to assessing those novel products and advising experts, GACVS will also consider how to more broadly communicate about risk assessment, so vaccine users can also benefit from the committee’s advice.
Keywords: vaccines, immunisation
Summary box.
Novel vaccine technologies include genetic modifications of micro-organisms, viral vectors, use of nucleic acids or novel adjuvants, they also include increased valences and different routes of administration.
Those new characteristics will modify untoward reactions, specific and non-specific, and will also require new pharmaco-epidemiological approaches.
The risk management plans for those products will have to factor those new theoretical concerns and propose ways of monitoring them during the products life-cycle.
As several novel vaccines are developed against diseases prevalent in countries with weak pharmacovigilance systems, vaccine introductions will require establishing active monitoring to rule out serious untoward effects in early adopter populations.
Introduction
As compared with the 20th century many recent vaccines, and others currently under development, present several novel features that will also impact their safety profile. While older vaccines included live attenuated organisms or inactive substances (such as inactivated germs, subunits, modified toxins or recombinant proteins), it is now also possible to apply newer technologies. The recent COVID-19 pandemic has led to the largest and most diverse ever vaccine development effort. As at end July 2020, less than 7 months after identification and characterisation of the virus, close to 200 different vaccine products are at different stages of development, including at least five in phase III trials.1 Candidate COVID-19 vaccines encompass a broad spectrum of products, including classic products but also the use of several novel technologies such as viral vectors, genetically attenuated live organisms or nucleic acid vaccines. In addition, future vaccines may also include novel adjuvants, increased number of antigens, novel mode of vaccine administration and thermostabilisation. Those technological developments will also affect untoward reactions to vaccine administration, sometimes towards better tolerance, and will require additional monitoring approaches to account for the novel characteristics.
For over 20 years, the Global Advisory Committee on Vaccine Safety (GACVS) has monitored new vaccines from the time they reach market authorisation. GACVS does characterise and maintain the safety profile of WHO-recommended vaccines as evidence emerges from postlicensure surveillance and observational studies. GACVS also provides advice of priorities for vaccine safety research and normative guidance about preferred methodologies. In this review, we describe novel vaccine technologies, with a perspective over the next 10 years. We discuss known and theoretical implications with respect to monitoring their safety and also examine how this will impact the risk management of novel vaccine products.
Situation analysis
Vaccines and vaccine technologies pipeline
WHO supports vaccines and novel technologies from early product development through to the publication of global policy recommendations.2 WHO’s Product Development for Vaccines Advisory Committee (PDVAC) facilitates early assessment of pipeline products, typically in clinical phase I or II, for which there is the greatest unmet public health need.3 Since its inception in 2014, PDVAC has reviewed 36 pathogen areas and prioritised 10 urgently required vaccines (figure 1). A key component of PDVAC’s remit is horizon scanning of novel candidates, an increasing number of which are based on innovative manufacturing, product presentation or delivery technologies. These assessments provide a landscape analysis of the most advanced and applicable novel platforms. Since these candidates are typically 5–10 years away from licensure, they present a forward-looking perspective on innovations to come for WHO to proactively prepare guidance.
Figure 1.

Overview of the PDVAC pipeline by novel antigen presentation platform. Regimens involving heterologous prime boost approaches, or candidates incorporating more than one platform are shown as stripped bars. Ebola virus vaccines are overseen by the R&D blueprint, but are included in this PDVAC overview to reflect the pipeline status of novel platforms for this antigen. ETEC, enterotoxigenic Echerichia coli; GAS, group A streptococcus; GBS, group B streptococcus; HSV, herpes simplex virus; PDVAC, product development for vaccines advisory committee; R&D, research and development; RSV, respiratory syncytial virus; TB, tuberculosis.
As the number of WHO-recommended vaccines increases,4 there is a critical need for more effective products that are cheaper and easier to deliver. On the production side, healthy markets that provide affordable essential products in sufficient quantity, with the ability to rapidly manufacture and scale up in response to disease outbreaks, remain an elusive goal. These requirements prioritise technologies that offer one or more of the following advantages: (1) increased vaccine efficacy that enables dose sparing, or a reduction in the number of doses per regimen, such as with genetic attenuation of live organisms, novel adjuvants and potentially novel delivery routes; (2) ability to combine multiple antigens, either from the same pathogen or different pathogens within the same formulation, possibly with the inclusion of genetic adjuvants to reduce the number of vaccinations, such as through the use of viral vectors and nucleic acid vaccines, or combinations of individual antigens; (3) ability to rapidly manufacture low-cost vaccines with new antigen sequences in a matter of weeks rather than months, such as with nucleic acid-based platforms and (4) improved safety and acceptability of vaccines at the point of delivery, such as through the use of needle-free and integrated reconstitution technologies.
Figure 1 presents an overview of the vaccine candidates for the PDVAC priority pathogens by phase of clinical development. It also highlights those that are based on novel technologies, either not previously licensed or for which there is currently limited human safety data available. The most clinically advanced candidates are for respiratory syncytial virus (RSV), tuberculosis (TB) and HIV vaccines. For example, one HIV candidate is based on a novel technology, namely a viral vector, and is being administered through heterologous prime boost which is itself a novel delivery approach.5 Approximately half of the candidates in phase II clinical development for priority pathogens are based on novel technologies, including three based on nucleic acids (DNA, mRNA), eight based on viral vectors (modified Vaccinia Ankara and chimpanzee adenovirus), and several on live attenuated and on novel adjuvants (Matrix M, etc). The TB,6 RSV and Shigella pipelines are predominantly based on novel technologies.
Safety considerations
Viral vectors
Recombinant viral vectors are attractive platforms for developing novel vaccines. They provide an efficient means for heterologous antigen expression in vivo and induce robust cellular and humoral immunity, both without need for exogenous adjuvant.7 Targeted deletion of specific genes allows dampened or complete elimination of viral replication thereby increasing safety. Viral vectors are the most popular approach among the many new approaches emerging from the biotechnology revolution. About half (n=11) of the 20 awards for vaccine development by the Coalition for Epidemic Preparedness and Innovation (CEPI) to date support use of viral vectors and several of the others support use of recombinant nucleic acids. Several veterinary and human viral vector vaccines (Japanese encephalitis, dengue and Ebola virus vaccines) have been licensed with many others in the pipeline.7 There is an increasing but still limited clinical experience about the efficacy and safety of such vectors in humans, however.
Much has been learnt about viral vectors since the first studies with Vaccinia in 1984.8 9 Several issues of critical importance about this platform remained unknown and warranted investigation per a 2003 WHO informal consultation.10 These included issues such as recombination with wild-type pathogenic strains and public acceptance (box 1). With increasing numbers of viral vector vaccines entering human clinical trials, new regulatory measures to ensure their quality, safety and efficacy have been established.11 12
Box 1. Issues of critical importance for viral vector vaccines (adapted from reference17).
Potential recombination with wild-type pathogenic strains.
Impact of prior infections on the immunogenicity of vectored vaccines.
Genetic stability of replicating recombinant viruses in vivo.
Impact of addition of foreign genes on a viral vector compared with parent virus.
Reversion to virulence of attenuated vectors.
Demonstration of sustained absence of replication for replication incompetent vectors.
Public acceptance of vectored vaccines with specific safety concerns.
Possible induction of immunosuppressive window or alternatively immune activation.
Defining duration of monitoring for adverse events following immunisation after administration of vectored vaccine.
Archiving vectored vaccine samples to allow future assessment of adventitious agents.
Assessing possible secondary transmission of vectored vaccine virus.
On occasion, vector-based vaccines have been associated with unexpected higher rates of the disease they are designed to prevent. This has been the case with HIV infection acquisition among participants of the STEP and Phambili13 trials who had received a replication-defective Ad5 vector vaccine candidate. Despite these setbacks, the overall advantages of viral vector vaccines seem to outweigh the disadvantages.14 The first HIV vaccine candidate to show (modest) protection in large human trials consisted of a recombinant canary pox virus vector vaccine (ALVAC-HIV (vCP1521)) and a recombinant glycoprotein 120 subunit vaccine. Follow-on trials using multiple updated viral vector vaccines are underway in Southern Africa. A recombinant rhesus cytomegalovirus vaccine vector engineered to express simian immunodeficiency virus (SIV) proteins has resulted in progressive clearance of a pathogenic SIV infection in rhesus macaques in Africa.15 An Ebola vaccine based on the vesicular stomatitis virus (VSV) vector has proven to be highly efficacious16 and is now registered in some African countries.
Since 2008, the Brighton Collaboration has been working to improve our ability to anticipate safety issues and meaningfully assess and interpret safety data from clinical trials of new viral vector vaccines, thereby enhancing public confidence in their safety and efficacy, and in turn the vaccines’ acceptance and uptake.17 This work has provided guidance on several issues of critical importance needing further investigation. These include to date: (1) archiving samples of biological materials for retrospective analysis18; (2) potential for and theoretical consequences of recombination with wild type virus strains19 and (3) defining the interval for monitoring potential adverse events following immunisation after receipt of live viral vectored vaccines.20
In addition to extending the paradigm of standardised case definitions to this realm, another major activity has been to adapt and use a standardised template for collection of key information for benefit–risk assessment of a novel vaccine vector or vector-based vaccine. Since this domain of vaccine development is highly technical with many acronyms and since key stakeholders (eg, regulators, institutional review boards, public health practitioners, public at large) lack technical training, the risk for misunderstanding is high. By organising the key questions in understandable language, the standardised template will hopefully facilitate scientific discourse among key stakeholders and increase the transparency and comparability of vital information. The templates published to date include the yellow fever 17D vaccine vector, the recombinant VSV and its application to the Ebola virus.16 At its 6 June2019 meeting the GACVS recognised the value of the structured information conveyed Brighton Collaboration template to policy-makers and urged other new vaccine developers to also complete such template, starting with the most advanced Ebola vaccine candidates.21
As COVID-19 vaccine developers are using a broad range of technology platforms beyond viral vectors, the Brighton Collaboration is developing standardisation templates for nucleic acid, protein, inactivated viral and live attenuated viral vaccines as well. At ist May 27–28 2020 meeting, the GACVS recommended that any review of new vaccine safety be based on the appropriate Brighton Collaboration standardised templates for benefit–risk assessment of vaccines. GACVS advised that templates be pilot tested in a number of scenarios and then adapted accordingly.21
Genetic attenuation of live organisms
Most licensed viral vaccines and those in development are live, attenuated viruses derived from virulent clinical isolates through multiple passage in culture (eg, measles, mumps, yellow fever, polio). Attenuated viral vaccine strains have the potential to mutate and regain virulence during growth in cell substrates resulting in an unacceptable safety profile of the vaccine, potential reduction in potency and efficacy. State of the art technology allows genetic modification of wild-type viruses, bacteria and other micro-organisms to generate attenuated vaccine strains that can replicate the pattern of natural infection without causing disease or other untoward side effects. Examples include reassortant influenza vaccines combining RNA segments from different strains,22 chimeric flaviviruses,23 including a recently licensed dengue vaccine,24 as well as genetically modified enteric bacteria.25 New strains for oral polio vaccine (OPV) represent another prominent example of the use of targeted systematic genetic modifications to improve vaccine safety as described below.
Genetic mutations occurring in Sabin poliovirus from conventional OPV strains result in regaining of virulent properties and can cause cases of vaccine associated paralytic polio.26 Following eradication of wild-type 2 polioviruses paralytic cases were caused by virulent circulating type 2 vaccine derived polioviruses (cVDPV).27 Even though trivalent OPV was withdrawn and replaced with bivalent vaccine that no longer contained the type 2 polio component, outbreaks of type 2 cVDPV that occurred prior to the switch cannot be controlled by inactivated polio virus vaccine nor monovalent type 2 OPV. State of the art technology combined with knowledge of poliovirus biology and genetics were used to rationally design and generate two genetically modified strains of poliovirus predicted to be safe and immunogenic, but with increased genetic stability compared with conventional Sabin strains. The modifications included re-coding the RNA element in the 5’-untranslated region (5’-UTR) that is a critical determinant of attenuation and virulence, to decrease the potential for mutations to occur.28 Second, a critically important cis-acting replicative element29 was transplanted from the centre of the molecule into the 5’-UTR to prevent elimination of this stabilised element due to recombination. Third, mutations were introduced to the viral RNA-replicase to increase its fidelity to prevent the emergence of mutations and to limit the ability of these genetically engineered viruses to recombine with other enteroviruses.30 Finally, RNA sequences coding for the viral capsid proteins were recoded using different combination of codons, while preserving the same amino acid sequence.31 Phase 1 clinical trials32 that evaluated shed polioviruses using deep sequencing and the transgenic mouse neurovirulence test have demonstrated that these strains are indeed more genetically stable than the original Sabin virus. It is expected that after the phase 2 clinical immunogenicity evaluations are complete, these new OPV constructs will be used in final polio eradication efforts. This project represents the first example of targeted rational design of vaccine virus genomes, which may serve as a future paradigm for the development of viral vaccines against other diseases. It also illustrates that use of new technologies, such as de novo chemical synthesis of vaccine genomes and next-generation (deep) sequencing to evaluate the genetic composition of the entire genome of vaccines viruses, are important tools for monitoring the molecular consistency and genetic stability of polio vaccines and vaccine products in general.33 The use of vaccines derived by genetic engineering will require safety surveillance and environmental monitoring to confirm that these modifications increase product safety in large-scale public immunisation campaigns.
Adjuvants and novel formulations
Adjuvants have been added to vaccines for nearly a hundred years, initially with aluminium salts and more recently oil emulsions (notably squalene-containing oil-in-water emulsions such as MF59 and AS03), bacterial membrane extracts such as monophosphoryl lipid A (MPL) or synthetic derivatives, plant extracts such as the Quillaia saponins used in AS01 and MatrixM adjuvants, and synthetic oligonucleotides such as CpG. These adjuvants enhance the antibody titres to the antigen (such as aluminium in most childhood vaccines), and some also enhance the breadth of the response (such as the MPL in HPV vaccines) permitting antigen-dose reduction and enabling immunisation in older adults.
Numerous allegations of safety issues have been directed at vaccine adjuvants. Aluminium salts have the longest and largest safety track record of all vaccine adjuvants. Cases of macrophagic myofasciitis (MMF) in which aluminium was found within intracytoplasmic inclusions, has led some researchers to suggest that alum in vaccines was the cause of clinical manifestations.34 After a review of the presence of aluminium and clinical MMF GACVS did not find credible association and concluded that there was no basis for recommending a change in vaccination practices.35
Oil-in-water emulsions were added to two monovalent pandemic influenza vaccines that were used during the 2009 H1N1 pandemic (Pandemrix and Focetria). These emulsions contained squalene, an oil purified from sharks, and the emulsion in Pandemrix further contained alpha-tocopherol (vitamin E). The public acceptance and uptake of these vaccines was hampered by media claims of the dangers of squalene, a concern that derived from a 2002 claim that soldiers returning from the Gulf War with the so-called ‘gulf war syndrome’ had antisqualene antibodies induced through receiving vaccines allegedly containing squalene.36 Although vaccines used in the Gulf War did not apparently contain squalene, the GACVS reviewed all available data including data from clinical trials with the approved squalene-containing influenza vaccine Fluad and did not find evidence that squalene could induce pathological antisqualene antibodies.37 During the 2009 influenza ‘pandemic’, one of the adjuvant-containing pandemic influenza vaccines (Pandemrix, containing AS03) was associated with an observed increase in narcolepsy rates in several European countries. Proposed mechanisms whereby the adjuvant heightened and broadened the immune response to include induction of pathological mimicry responses38 are difficult to reconcile with the uneventful use of the same adjuvant in another H1N1 pandemic influenza vaccine (Arepanrix).39 A more recent hypothesis is that it could have occurred from an interaction among populations being vaccinated in Europe during wild-type virus circulation.40
Other adjuvant-associated concerns include the observed increase in solicited local symptoms with AS01 adjuvant.41 A nasally delivered inactivated influenza vaccine, containing a mutated heat-labile toxin from Escherichia coli as an adjuvant, was found to be associated with increased rates of Bell’s palsy resulting in the vaccine being withdrawn.42 Subsequent studies43 showed that the adjuvant can induce transient facial nerve paralysis.
Combination vaccines and increasing valency
Adding antigens to an existing vaccine formulation increases the protection against additional germs and simplifies vaccine administration. The number of antigens in individual vaccines has increased for several combinations including measles, mumps and rubella vaccines in various bivalent to tetravalent combinations and combined products with diphtheria, tetanus and pertussis (whole cell or acellular) vaccines with other non-live vaccines. The diphtheria-tetanus-acellular-pertussis-inactivated polio-hepatitis B-Haemophilus influenzae type b (DTaP-IPV-HepB-Hib) vaccine referred to as a hexavalent vaccine currently protects against the largest number of diseases (six different micro-organisms). One available presentation contains five different antigens against pertussis and three types of poliomyelitis vaccines and is therefore composed of 12 antigens in total.44
Multivalent vaccines are more complex to produce than monovalent ones. Each component must be manufactured, and quality tested separately and then in combination. Clinical development requires demonstrating non-inferiority of the combination for each component of the vaccine.45 In most cases, multivalent vaccines have been found to be equally immunogenic as the same components administered separately. This was, for example, essentially the case with measles, mumps and rubella.46 A quadrivalent live influenza vaccine was, however, found to have lower efficacy than non-live preparations.47 More antigen per dose administered is sometimes related with increased vaccine reactogenicity. For example, the measles-mumps-rubella-varicella vaccine causes more febrile seizures than measles-mumps-rubella and varicella vaccines administered separately at the same time.48 When compared with similar products with fewer antigens, DTaP-IPV-HepB-Hib safety profile has a slightly higher risk of fever.49
Multivalent vaccines are composed of different types of the same micro-organism. Trivalent live and inactivated poliovirus vaccines were the first example of such products. Today, pneumococcal polysaccharide vaccine (33 components) has the largest valence and a 20-valent pneumococcal conjugate is under clinical development.50
A larger antigen charge, the impact of higher carrier protein levels, and occasionally the need for higher adjuvant titres, can increase the frequency and intensity of non-specific local and systemic reactions. With HPV vaccines, a recent 9-valent product induces a higher frequency of adverse events than its 4-valent predecessor. Higher amounts of antigen and adjuvant are a likely explanation for this observation. On the other hand, polyvalent vaccines can replace older components with less reactogenic ones. The prime example being use of acellular pertussis instead of whole cell. This, however, appears to come at the expense of actual protective efficacy. GACVS did review the increasing number of vaccines administered and concluded that immune overload is not a real concern.51
Innovative vaccine delivery technologies
At present, most injectable human vaccines are presented as liquid or lyophilised formulations, in single or multidose vials, the latter of which often contain preservatives for non-live products. Doses are typically administered via the intramuscular or subcutaneous routes. These conventional presentations include potential safety hazards: vaccine reconstitution with an incorrect diluent, contamination of multidose vials52 and needle-stick injury and consequent disease transmission.53 These risks, as well as the need to improve ease of administration and ensure vaccine potency at the point of delivery, are driving the development of innovative vaccine delivery technologies with a potential to improve vaccine and vaccination safety.
One of the most compelling innovations in the development pipeline is the microarray patch (MAP). A MAP consists of hundreds or thousands of projections, coated with or composed of dry vaccine formulation, on a ‘patch’ backing. The most clinically advanced vaccine MAPs either have no applicator,54 or an integrated applicator55 and when pressed on the skin, deliver the vaccine to the dermis or epidermis over seconds or a few minutes. MAPs are ‘pre-filled’, needle-free, single use presentations that reduce the risks of contamination, use of incorrect diluent or needle stick injuries. In addition, novel antigen formulations that will need to be developed for the MAP presentation presents an opportunity to improve vaccine thermostability so that it can be stored outside a regular cold chain.56 57
Since the intradermal compartment is rich in antigen-presenting cells, it is conceivable that MAPs may offer the opportunity for dose sparing. This has been demonstrated in animal models for several antigens58 and in the clinic with a monovalent influenza antigen.56 However, this increase in immunogenicity may be associated with high and unacceptable levels of reactogenicity. Minor local reactions lasting several days following application have been observed in clinical studies54 59 60 and may be more pronounced with adjuvanted vaccines. To date, there have only been clinical studies with influenza vaccine and data with additional antigens are needed.
For liquid vaccines, next-generation compact prefilled autodisable devices are in the pipeline. Their presentation is similar to the commercially available Uniject58 but they can be manufactured by an automated, continuous, aseptic process known as Blow-Fill-Seal. This reduces the potential for contamination during manufacturing. This presentation is envisaged to be composed of a blister package containing a single dose of vaccine, a preassembled needle hub with an auto disable feature and a removeable vaccine shield. If all these elements are included in the final presentation, they offer the potential to reduce vaccine contamination, needle stick injuries and disease transmission.
While these innovations, and others in the pipeline, offer the potential of significant safety benefits, they are likely to incrementally increase the procurement cost per dose of the vaccine. Efforts are needed to evaluate these trade-offs, and to further rationalise the programmatic and public health benefit that can be offered.
Nucleic acid vaccines
In 1990, direct gene transfer into mouse muscle in vivo demonstrated that plasmid DNA (pDNA) containing a gene of interest when directly inoculated into host tissue resulted in in situ production of the corresponding protein.61 In the context of vaccination, this observation suggested that nucleic acid technology (NAT) could deliver and express target immunogens and genetic adjuvants as a simple and versatile platform for vaccination. A promising feature of NAT vaccines is the potential to induce both humoral and cellular immunity to the target immunogen. There is also an apparent absence of adaptive immune response (although not the absence of certain innate immune responses) against the vector. For a variety of technical reasons (eg, stability and ease of manufacturing), the focus of early NAT vaccine efforts was directed to pDNA rather than other DNA or RNA approaches and hundreds of clinical trials with pDNA ensued.62 Although no NAT vaccines have been licensed for use in humans, three have been licensed to date for veterinary use to prevent infectious diseases.
Not unexpectedly for a novel immunogen and vaccine delivery system, early on there was uncertainty about both efficacy and safety of pDNA in humans. Safety concerns included somatic chromosomal integration, germline alterations, autoimmunity and immunopathology. Initial guidance documents called for extensive nonclinical studies, including biodistribution, chromosomal integration, germline evaluation, in addition to more typical vaccine toxicology studies. There is abundant nonclinical evidence that pDNA does not biodistribute or persist throughout the host when delivered parenterally into muscle, subcutaneous tissue or various dermal layers. Published data from hundreds of clinical trials indicate that pDNA vaccine candidates have been generally safe, with acceptable reactogenicity profiles. Clinical reactogenicity relates more to delivery method than to pDNA (eg, electroporation, particle-mediated bombardment). As such, both the nonclinical and clinical sections of the WHO Guidelines for assuring the quality, safety and efficacy of DNA vaccines are being revised in light of existing non-clinical and clinical data.63
Clinical evaluation suggests that pDNA alone in most instances does not induce enough immune response in humans to warrant late-stage clinical development. To enhance the immune response optimisation of the primary and secondary structure of the pDNA, addition of excipients that enhance delivery or have an adjuvant effect, and changes in the mode or route of administration are being evaluated. More effective immunogenicity seems to be induced when pDNA is used to prime immune responses that are subsequently boosted by delivery of a heterologous vaccine candidate (protein antigen or gene-based viral vectors). In recent years, there has been a decline in publications on pDNA and a recent increase in publications and clinical evaluation of a variety of RNA-based NAT vaccine approaches (figure 2).
Figure 2.
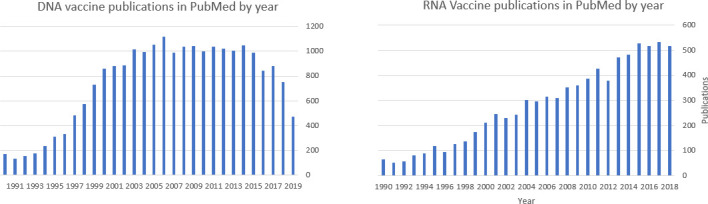
Publications on DNA and RNA vaccines 1990–2019.
RNA vaccines now being clinical tested incorporate the use of modified nucleosides and novel delivery excipients. Those appear to have overcome previous limitations of instability, transient production of encoded protein, and undesired innate immune responses.61 Like pDNA, the current uncertainty of both efficacy and safety of RNA-based vaccine candidates in humans have yielded to more streamlined evaluations, as evidenced by the unprecedented speed from sequence availability to first subject enrolled in phase 1 safety and immunogenicity study of a mRNA-based SARS-CoV-2 candidate vaccine.64
Evolving risk-management plans
Modern regulatory review of health products includes examination and continued monitoring of risk management plans (RMPs). RMPs have a product life cycle perspective. They typically include information on a product’s safety profile, based on data from clinical evaluation, possible safety signals and any other theoretical considerations. If by the time of marketing authorisation, no significant issues are identified, risks are primarily monitored using passive safety surveillance systems after products have reached the market. When safety issues have been identified or when safety data are limited, RMPs will include the conduct of pharmacoepidemiologic studies and other activities to gain more knowledge about the safety and efficacy of a product.65 Council for International Organizations of Medical Science recently published a guide specifically dedicated to such active vaccines safety surveillance.66 RMPs also discuss how risks will be prevented or minimised in patients and measure the effectiveness of risk-minimisation measures.
Novel vaccines, based on genetic modifications, open a whole new field of research. The Brighton Collaboration standardised viral vector templates provide an illustration of the many dimensions related to assessing stability of the new constructs and their potential for recombination and interaction that will have to be factored in and similar issues will have to be considered for nucleic acid vaccines. RMPs of the next decade will also include novel methods to characterise products. Questions related to the duration of active monitoring for genetic material, presence of adventitious agents as has been observed with several viral vaccines67 more easily detected with enhanced biological screening, or physiological mechanisms of novel adjuvants are all considerations that will belong to the preparation of RMPs.
Consideration for use in emergency settings and fragile states
The routine evaluation of a new candidate vaccine in low-income and middle-income country settings with limited resources, infrastructure, regulatory and clinical trial experience is already very challenging.68 69 Conduct such vaccine trials under emergency settings and in fragile states does compound these difficulties exponentially. While these challenges may have been most apparent during the recent Ebola outbreaks in West and Central Africa,69–71 they presumably were present historically with clinical trials of vaccines against any epidemic disease in such settings.72 73 Several papers documenting how interlocking challenges for conducting phase 2b trial from infrastructure, to staffing, participant communication and technology integration were designed, planned, tackled and solved during the constantly evolving West African Ebola outbreak from various perspectives are now available, including some key lessons learnt.69 71 74 75
In 2015, two additional emerging infections, Zika and Middle East Respiratory Syndrome (MERS) struck the globe.76 77 Plotkin78 argued that a ‘Global Vaccine Development Fund’ was urgently needed to provide the resources and momentum necessary to carry candidate vaccines against such pathogens from their conception through development and licensure—thereby averting future Ebola crisis. These ideas have gelled into the formation of the CEPI (www.cepi.net), a new initiative targeted at developing candidate vaccines against Lassa fever, MERS, SARS, Nipah virus, Rift Valley fever, chikungunya and now SARS-CoV-2 on list of deadly pathogens without a vaccine.74 79
CEPI has funded the Brighton Collaboration’ Safety Platform for Emergency vACcines (SPEAC) to help assess the safety of various CEPI-funded vaccine candidates undergoing clinical trials. SPEAC will help provide members with safety expertise to independent data safety monitoring boards (DSMB). It will also constitute a ‘meta-DSMB’ that will help oversee across vaccine platform against the same pathogen, as well as across pathogens using the same vaccine platform. The quality of safety data will be optimised by creating an online vaccine safety resource, which will include technical guidance, tools, a platform for information exchange and training modules. A Brighton Collaboration standardised viral vector template will be completed for each CEPI vaccine candidate.17
Conclusion
There is an exciting evolving vaccine pipeline that will display very different characteristics than those of currently available products. Changes in non-specific vaccine reactogenicity can be expected: stronger, as could be the case with more potent adjuvants; or milder, if doses can be reduced or new delivery methods are used. Use of genetically modified organisms and nucleic acids will require attention to any possibilities of mutation or recombination, an area for which the current vaccine experience is primarily around the evolution of oral polioviruses.
GACVS has started reviewing new vaccines using the Brighton Collaboration standardised template for benefit-risk assessment of vaccines, now available for the major technology platforms, as they offer a structured approach to evaluating safety.21 The challenges of monitoring genetic modifications will possibly require adding expertise on this area to the committee. The rapid development and urgent need for COVID-19 vaccines will likely require enhanced safety monitoring of vaccine products beyond phase III trials with close alignment of immunisation policies. Finally, as new vaccine combinations will likely be proposed, understanding the reactogenicity of each individual component will remain essential before assessing their effects when combined. In addition to assessing those novel products and advising experts, GACVS will also consider how to more broadly communicate about risk assessment, so vaccine users can also benefit from the committee’s advice.
Acknowledgments
This paper was initially prepared as a situation paper for a symposium on '20 years of GACVS, the Future of Vaccine Safety' held during the Global Vaccine Safety Summit on 2–3 December 2019. Comments received from meeting participants and the participation in the panel by WHO Chief, Dr Soumya Swaminathan, helped revise the manuscript.
Footnotes
Handling editor: Seye Abimbola
Contributors: Every author contributed the original draft of one or several sections of the manuscript and reviewed every version up to the final one. The lead author, in addition, assembled the manuscript and prepared its submission version.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study.
References
- 1. Vaccine Centre, London School of Hygiene & Tropical Medicine . COVID-19 vaccine 145 development pipeline, 2020. Available: https://vac-lshtm.shinyapps.io/ncov_vaccine_landscape/
- 2. From Vaccine Development to Policy . A brief review of who vaccine-related activities and advisory processes, 2017. Available: https://www.who.int/immunization/policy/WHO_vaccine_development_policy.pdf
- 3. WHO . Product development for vaccines advisory committee (established April 2014). meetings, terms of reference and composition, 2020. Available: https://www.who.int/immunization/research/committees/pdvac/en/
- 4. WHO . WHO recommendations for routine immunization - summary tables, 2020. Available: https://www.who.int/immunization/policy/immunization_tables/en/
- 5. Robinson HL. Hiv/Aids vaccines: 2018. Clin Pharmacol Ther 2018;104:1062–73. 10.1002/cpt.1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. TBVI . Tuberculosis vaccine initiative pipeline of vaccines, 2020. Available: https://www.tbvi.eu/what-we-do/pipeline-of-vaccines/
- 7. Rauch S, Jasny E, Schmidt KE, et al. New vaccine technologies to combat outbreak situations. Front Immunol 2018;9:9. 10.3389/fimmu.2018.01963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moss B, Smith GL, Gerin JL, et al. Live recombinant vaccinia virus protects chimpanzees against hepatitis B. Nature 1984;311:67–9. 10.1038/311067a0 [DOI] [PubMed] [Google Scholar]
- 9. Ramezanpour B, Haan I, Osterhaus A, et al. Vector-based genetically modified vaccines: exploiting Jenner's legacy. Vaccine 2016;34:6436–48. 10.1016/j.vaccine.2016.06.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. WHO . Who informal consultation on characterization and quality aspect of vaccines based on live viral vectors, 2003. Available: https://www.who.int/vaccine_research/documents/vvreport/en/
- 11. European Medicines Agency . Guideline on quality, non-clinical and clinical aspects of live recombinant viral vectored vaccines. In: EMA/CHMP/VWP/141697/2009. London, UK: E.M. Agency, 2010. https://www.ema.europa.eu/en/quality-non-clinical-clinical-aspects-live-recombinant-viral-vectored-vaccines [Google Scholar]
- 12. FDA CBER . Determining the need for and content of environmental assessments for gene therapies, vectored vaccines and related recombinant viral or microbial products: guidance for industry, 2015. Available: https://www.fda.gov/media/91425/download
- 13. Gray G, Buchbinder S, Duerr A. Overview of step and Phambili trial results: two phase IIb test-of-concept studies investigating the efficacy of MRK adenovirus type 5 gag/pol/nef subtype B HIV vaccine. Curr Opin HIV AIDS 2010;5:357–61. 10.1097/COH.0b013e32833d2d2b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Humphreys IR, Sebastian S. Novel viral vectors in infectious diseases. Immunology 2018;153:1–9. 10.1111/imm.12829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stephenson KE, D’Couto HT, Barouch DH. New concepts in HIV-1 vaccine development. Curr Opin Immunol 2016;41:39–46. 10.1016/j.coi.2016.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Monath TP, Fast PE, Modjarrad K, et al. rVSVΔG-ZEBOV-GP (also designated V920) recombinant vesicular stomatitis virus pseudotyped with Ebola Zaire glycoprotein: standardized template with key considerations for a risk/benefit assessment. Vaccine X 2019;1:100009. 10.1016/j.jvacx.2019.100009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen RT, Carbery B, Mac L, et al. The Brighton collaboration viral vector vaccines safety working group (V3SWG). Vaccine 2015;33:73–5. 10.1016/j.vaccine.2014.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klug B, Robertson JS, Condit RC, et al. Adventitious agents and live viral vectored vaccines: considerations for archiving samples of biological materials for retrospective analysis. Vaccine 2016;34:6617–25. 10.1016/j.vaccine.2016.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Condit RC, Williamson A-L, Sheets R, et al. Unique safety issues associated with virus-vectored vaccines: potential for and theoretical consequences of recombination with wild type virus strains. Vaccine 2016;34:6610–6. 10.1016/j.vaccine.2016.04.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kochhar S, Excler J-L, Bok K, et al. Defining the interval for monitoring potential adverse events following immunization (AEFIs) after receipt of live viral vectored vaccines. Vaccine 2019;37:5796–802. 10.1016/j.vaccine.2018.08.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. WHO . Global advisory committee on vaccine safety, 27-28 may 2020. Wkly Epi Rec 2020;95:331–6. [Google Scholar]
- 22. Houser K, Subbarao K. Influenza vaccines: challenges and solutions. Cell Host Microbe 2015;17:295–300. 10.1016/j.chom.2015.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lai C-J, Monath TP. Chimeric flaviviruses: novel vaccines against dengue fever, tick-borne encephalitis, and Japanese encephalitis. Adv Virus Res 2003;61:469–509. 10.1016/s0065-3527(03)61013-4 [DOI] [PubMed] [Google Scholar]
- 24. Malisheni M, Khaiboullina SF, Rizvanov AA, et al. Clinical efficacy, safety, and immunogenicity of a live attenuated tetravalent dengue vaccine (CYD-TDV) in children: a systematic review with meta-analysis. Front Immunol 2017;8:863. 10.3389/fimmu.2017.00863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Milligan R, Paul M, Richardson M, et al. Vaccines for preventing typhoid fever. Cochrane Database Syst Rev 2018;5:CD001261. 10.1002/14651858.CD001261.pub4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nkowane BM, et al. Vaccine-associated paralytic poliomyelitis. JAMA 1987;257:1335–40. 10.1001/jama.1987.03390100073029 [DOI] [PubMed] [Google Scholar]
- 27. Burns CC, Diop OM, Sutter RW, et al. Vaccine-derived polioviruses. J Infect Dis 2014;210 Suppl 1:S283–93. 10.1093/infdis/jiu295 [DOI] [PubMed] [Google Scholar]
- 28. Knowlson S, Burlison J, Giles E, et al. New strains intended for the production of inactivated polio vaccine at low-containment after eradication. PLoS Pathog 2015;11:e1005316. 10.1371/journal.ppat.1005316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yin J, Paul AV, Wimmer E, et al. Functional dissection of a poliovirus cis-acting replication element [PV-cre(2C)]: analysis of single- and dual-cre viral genomes and proteins that bind specifically to PV-cre RNA. J Virol 2003;77:5152–66. 10.1128/JVI.77.9.5152-5166.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pfeiffer JK, Kirkegaard K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc Natl Acad Sci U S A 2003;100:7289–94. 10.1073/pnas.1232294100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burns CC, Campagnoli R, Shaw J, et al. Genetic inactivation of poliovirus infectivity by increasing the frequencies of CpG and uPA dinucleotides within and across synonymous capsid region codons. J Virol 2009;83:9957–69. 10.1128/JVI.00508-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Van Damme P, De Coster I, Bandyopadhyay AS, et al. The safety and immunogenicity of two novel live attenuated monovalent (serotype 2) oral poliovirus vaccines in healthy adults: a double-blind, single-centre phase 1 study. The Lancet 2019;394:148–58. 10.1016/S0140-6736(19)31279-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Neverov A, Chumakov K. Massively parallel sequencing for monitoring genetic consistency and quality control of live viral vaccines. Proc Natl Acad Sci U S A 2010;107:20063–8. 10.1073/pnas.1012537107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brenner A. Macrophagic myofasciitis: a summary of Dr. Gherardi's presentations. Vaccine 2002;20 Suppl 3:S5–6. 10.1016/S0264-410X(02)00167-6 [DOI] [PubMed] [Google Scholar]
- 35. WHO . Global advisory committee on vaccine safety, 20-21 June 2002. Wkly Epidemiol Rec 2002;77:389–94. [PubMed] [Google Scholar]
- 36. Asa PB, Wilson RB, Garry RF. Antibodies to squalene in recipients of anthrax vaccine. Exp Mol Pathol 2002;73:19–27. 10.1006/exmp.2002.2429 [DOI] [PubMed] [Google Scholar]
- 37. WHO . Global Advisory Committee on vaccine safety, 6-7 June 2006. Wkly Epidemiol Rec 2006;81:273–8. [PubMed] [Google Scholar]
- 38. Cohet C, van der Most R, Bauchau V, et al. Safety of AS03-adjuvanted influenza vaccines: a review of the evidence. Vaccine 2019;37:3006–21. 10.1016/j.vaccine.2019.04.048 [DOI] [PubMed] [Google Scholar]
- 39. Weibel D, Sturkenboom M, Black S, et al. Narcolepsy and adjuvanted pandemic influenza A (H1N1) 2009 vaccines – multi-country assessment. Vaccine 2018;36:6202–11. 10.1016/j.vaccine.2018.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Edwards K, Lambert P-H, Black S. Narcolepsy and pandemic influenza vaccination: what we need to know to be ready for the next pandemic. Pediatr Infect Dis J 2019;38:873–6. 10.1097/INF.0000000000002398 [DOI] [PubMed] [Google Scholar]
- 41. Leroux-Roels G, Marchant A, Levy J, et al. Impact of adjuvants on CD4(+) T cell and B cell responses to a protein antigen vaccine: Results from a phase II, randomized, multicenter trial. Clin Immunol 2016;169:16–27. 10.1016/j.clim.2016.05.007 [DOI] [PubMed] [Google Scholar]
- 42. Mutsch M, Zhou W, Rhodes P, et al. Use of the Inactivated Intranasal Influenza Vaccine and the Risk of Bell’s Palsy in Switzerland. N Engl J Med 2004;350:896–903. 10.1056/NEJMoa030595 [DOI] [PubMed] [Google Scholar]
- 43. Lewis DJM, Huo Z, Barnett S, et al. Transient facial nerve paralysis (Bell’s palsy) following intranasal delivery of a genetically detoxified mutant of escherichia coli heat labile toxin. PLoS One 2009;4:e6999. 10.1371/journal.pone.0006999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Syed YY. DTaP5-HB-IPV-Hib vaccine (Vaxelis®): a review of its use in primary and booster vaccination. Pediatr Drugs 2017;19:69–80. 10.1007/s40272-016-0208-y [DOI] [PubMed] [Google Scholar]
- 45. Schlingmann B, Castiglia KR, Stobart CC, et al. Polyvalent vaccines: high-maintenance heroes. PLoS Pathog 2018;14:e1006904. 10.1371/journal.ppat.1006904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lerman SJ, Bollinger M, Brunken JM. Clinical and serologic evaluation of measles, mumps, and rubella (HPV-77:DE-5 and RA 27/3) virus vaccines, singly and in combination. Pediatrics 1981;68:18–22. [PubMed] [Google Scholar]
- 47. Gill MA, Schlaudecker EP. Perspectives from the society for pediatric research: decreased effectiveness of the live attenuated influenza vaccine. Pediatr Res 2018;83:31–40. 10.1038/pr.2017.239 [DOI] [PubMed] [Google Scholar]
- 48. Klein NP, Fireman B, Yih WK, et al. Measles-mumps-rubella-varicella combination vaccine and the risk of febrile seizures. Pediatrics 2010;126:e1–8. 10.1542/peds.2010-0665 [DOI] [PubMed] [Google Scholar]
- 49. Xu J, Stek JE, Ziani E, et al. Integrated safety profile of a new approved, fully liquid DTaP5-HB-IPV-Hib vaccine. Pediatr Infect Dis J 2019;38:439–43. 10.1097/INF.0000000000002257 [DOI] [PubMed] [Google Scholar]
- 50. Thompson A, Lamberth E, Severs J, et al. Phase 1 trial of a 20-valent pneumococcal conjugate vaccine in healthy adults. Vaccine 2019;37:6201–7. 10.1016/j.vaccine.2019.08.048 [DOI] [PubMed] [Google Scholar]
- 51. WHO . Global Advisory Committee on vaccine safety, 6–7 June 2006. Wkly Epid Rec 2006;81:273–8. [PubMed] [Google Scholar]
- 52. WHO . E-Learning course on vaccine safety basics, 2020. Available: https://www.who.int/vaccine_safety/initiative/tech_support/ebasic/en/
- 53. Riddell A, Kennedy I, Tong CYW. Management of sharps injuries in the healthcare setting. BMJ 2015;351:h3733. 10.1136/bmj.h3733 [DOI] [PubMed] [Google Scholar]
- 54. Rouphael NG, Paine M, Mosley R, et al. The safety, immunogenicity, and acceptability of inactivated influenza vaccine delivered by microneedle patch (TIV-MNP 2015): a randomised, partly blinded, placebo-controlled, phase 1 trial. The Lancet 2017;390:649–58. 10.1016/S0140-6736(17)30575-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Guillermet E, Alfa DA, Phuong Mai LT, et al. End-user acceptability study of the nanopatch™; a microarray patch (MAP) for child immunization in low and middle-income countries. Vaccine 2019;37:4435–43. 10.1016/j.vaccine.2019.02.079 [DOI] [PubMed] [Google Scholar]
- 56. Forster AH, Witham K, Depelsenaire ACI, et al. Safety, tolerability, and immunogenicity of influenza vaccination with a high-density microarray patch: results from a randomized, controlled phase I clinical trial. PLoS Med 2020;17:e1003024. 10.1371/journal.pmed.1003024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fernando GJP, Hickling J, Jayashi Flores CM, et al. Safety, tolerability, acceptability and immunogenicity of an influenza vaccine delivered to human skin by a novel high-density microprojection array patch (Nanopatch™). Vaccine 2018;36:3779–88. 10.1016/j.vaccine.2018.05.053 [DOI] [PubMed] [Google Scholar]
- 58. Arya J, Prausnitz MR. Microneedle patches for vaccination in developing countries. J Control Release 2016;240:135–41. 10.1016/j.jconrel.2015.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hirobe S, Azukizawa H, Matsuo K, et al. Development and clinical study of a self-dissolving microneedle patch for transcutaneous immunization device. Pharm Res 2013;30:2664–74. 10.1007/s11095-013-1092-6 [DOI] [PubMed] [Google Scholar]
- 60. Sutanto A, Suarnawa IM, Nelson CM, et al. Home delivery of heat-stable vaccines in Indonesia: outreach immunization with a prefilled, single-use injection device. Bull World Health Organ 1999;77:119–26. [PMC free article] [PubMed] [Google Scholar]
- 61. Wolff J, Malone R, Williams P, et al. Direct gene transfer into mouse muscle in vivo. Science 1990;247:1465–8. 10.1126/science.1690918 [DOI] [PubMed] [Google Scholar]
- 62. Liu MA . A comparison of plasmid DNA and mRNA as vaccine technologies. Vaccines 2019;7:37–57. 10.3390/vaccines7020037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sheets R, Kang H-N, Meyer H, et al. Who informal consultation on the guidelines for evaluation of the quality, safety, and efficacy of DNA vaccines, Geneva, Switzerland, December 2019. NPJ Vaccines 2020;5:52. 10.1038/s41541-020-0197-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jackson LA, Anderson EJ, Rouphael NG, et al. An mRNA vaccine against SARS-CoV-2 - preliminary report. N Engl J Med 2020. 10.1056/NEJMoa2022483. [Epub ahead of print: 14 Jul 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tsintis P, La Mache E. CIOMS and ICH initiatives in pharmacovigilance and risk management: overview and implications. Drug Saf 2004;27:509–17. 10.2165/00002018-200427080-00004 [DOI] [PubMed] [Google Scholar]
- 66. Council for International Organizations of Medical Science . CIOMS guide to active vaccine safety surveillance. Geneva: World Health Organization, 2017: 82. [Google Scholar]
- 67. Petricciani J, Sheets R, Griffiths E, et al. Adventitious agents in viral vaccines: lessons learned from 4 case studies. Biologicals 2014;42:223–36. 10.1016/j.biologicals.2014.07.003 [DOI] [PubMed] [Google Scholar]
- 68. Cutts FT, Enwere G, Zaman SMA, et al. Operational challenges in large clinical trials: examples and lessons learned from the Gambia pneumococcal vaccine trial. PLoS Clin Trials 2006;1:e16. 10.1371/journal.pctr.0010016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Grenham A, Villafana T. Vaccine development and trials in low and lower-middle income countries: key issues, advances and future opportunities. Hum Vaccin Immunother 2017;13:2192–9. 10.1080/21645515.2017.1356495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Henao-Restrepo AM, Preziosi M-P, Wood D, et al. On a path to accelerate access to ebola vaccines: the WHO’s research and development efforts during the 2014-2016 Ebola epidemic in West Africa. Curr Opin Virol 2016;17:138–44. 10.1016/j.coviro.2016.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Carter RJ, Idriss A, Widdowson M-A, et al. Implementing a multisite clinical trial in the midst of an Ebola outbreak: lessons learned from the Sierra Leone trial to introduce a vaccine against Ebola. J Infect Dis 2018;217:S16–23. 10.1093/infdis/jix657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rothman DJ. Ethical and social issues in the development of new drugs and vaccines. Bull N Y Acad Med 1987;63:557–68. [PMC free article] [PubMed] [Google Scholar]
- 73. Marchetti E, Mazarin-Diop V, Chaumont J, et al. Conducting vaccine clinical trials in sub-Saharan Africa: operational challenges and lessons learned from the meningitis vaccine project. Vaccine 2012;30:6859–63. 10.1016/j.vaccine.2012.09.008 [DOI] [PubMed] [Google Scholar]
- 74. Gupta SB, Coller B-A, Feinberg M. Unprecedented PACE and partnerships: the story of and lessons learned from one Ebola vaccine program. Expert Rev Vaccines 2018;17:913–23. 10.1080/14760584.2018.1527692 [DOI] [PubMed] [Google Scholar]
- 75. Higgs ES, Dubey SA, Coller BAG, et al. Accelerating vaccine development during the 2013-2016 West African Ebola virus disease outbreak. Curr Top Microbiol Immunol 2017;411:229–61. 10.1007/82_2017_53 [DOI] [PubMed] [Google Scholar]
- 76. Petersen LR, Jamieson DJ, Powers AM, et al. Zika virus. N Engl J Med 2016;374:1552–63. 10.1056/NEJMra1602113 [DOI] [PubMed] [Google Scholar]
- 77. Zumla A, Hui DS, Perlman S. Middle East respiratory syndrome. The Lancet 2015;386:995–1007. 10.1016/S0140-6736(15)60454-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Plotkin SA. Vaccines for epidemic infections and the role of CEPI. Hum Vaccin Immunother 2017;13:2755–62. 10.1080/21645515.2017.1306615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ghebreyesus TA, Swaminathan S. Scientists are sprinting to outpace the novel coronavirus. Lancet 2020;395:762–4. 10.1016/S0140-6736(20)30420-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable as no datasets generated and/or analysed for this study.