

Abstract
Comorbid chronic diseases affect cancer patients with an increasing frequency as populations get older. They negatively and disproportionately impact underserved populations and influence cancer diagnosis, tumor biology and metastasis, and choice of treatment. Many comorbidities are associated with a delayed cancer diagnosis. Although the relationship between comorbidities and cancer risk and survivorship has been studied extensively, we still lack knowledge on how they affect tumor biology and the metastatic process. Here, we will discuss our current understanding of mechanisms linking comorbidities to an adverse tumor biology and lethality and introduce thoughts of how we can close existing gaps in this knowledge. We argue that research into comorbidity-induced alterations in cancer metastasis, immunity, and metabolism should be prioritized.
Keywords: comorbidity, cancer, metabolism, immunity, metastasis, survival, obesity, diabetes
Chronic diseases modify cancer risk and survival
A comorbidity among cancer patients is generally defined as the coexistence of a disorder/chronic disease in addition to cancer. These disorders include chronic cardiovascular, liver and renal diseases, diabetes, metabolic syndrome, connective tissue diseases, chronic infectious diseases, dysbiosis (see glossary), neurological disorders such as dementia and chronic stress and depressive disorders, and autoimmune diseases such as rheumatoid arthritis, inflammatory bowel disease, systemic lupus erythematosus, or Sjogren’s disease. Although not a chronic disease, COVID-19 infections have recently been associated with an excessive mortality among cancer patients [1]. Many of these comorbidities share risk factors with cancer, thus commonly co-occur with cancer. It has been estimated that three-quarters of cancer patients have at least one comorbidity [2] while data from Medicare beneficiaries in the United States indicate a prevalence of 40% [3]. Comorbidities do not affect all segments of the US populations equally. Native Americans and African Americans have significantly elevated rates for obesity, diabetes, chronic kidney disease, and hypertension, when compared to other population groups [4].
Comorbidities can influence cancer outcomes. For example, epidemiology has shown that comorbid human immune deficiency virus (HIV) infections may increase cancer mortality [5]. They impede the participation of cancer patients in clinical trials and adversely affect the discussion and offer of trial participation [6]. The uptake of cancer screening is inversely associated with the severity of comorbidities, potentially delaying a cancer diagnosis in those most impacted by multiple comorbidities who also tend to be among the poor and underserved [2, 7]. In contrast, having just one comorbid condition may lead to increased screening participation because of increased contact with health services [8]. Yet, individual chronic diseases may have different effects on screening. Diabetes was found to associate with decreased cancer screening whereas an infection with HIV may not uniformly affect cancer screening decisions [9, 10]. Comorbidities have an adverse effect on cancer survival [3]. They are strong prognostic factors of poor survival in colorectal cancer patients independent of sociodemographic factors and tumor characteristics [11]. The negative impact of comorbidities on cancer outcomes tends to increase with increasing severity of the comorbidities and their impact is generally greater for cancers with otherwise better outcomes. The presence of a comorbidity will influence treatment selection and particularly the use of chemotherapy [12]. Cancer patients with a comorbidity are generally less likely to receive curative treatment for their cancer than those without the comorbidity [3].
In this opinion piece, we will summarize key evidence that links comorbidities to cancer development and an adverse tumor biology. We will then discuss the mechanisms that underly these relationships - to the extent they are known - and offer opportunities to close existing gaps in this knowledge. Much of the presented research investigated two comorbidities, obesity and diabetes, as modifiers of tumor biology and cancer outcomes because these conditions can readily be studied in experimental models of cancer. Other comorbidities still lack suitable experimental models or are studied with cancer models that may not fully recapitulate the human disease, such as the autoimmune component of inflammatory bowel disease.
Comorbid conditions and their treatment frequently impact tumor biology
Comorbidities may modulate cancer risks by affecting tumor biology (Figure 1). The hypothesis is robustly supported by clinical and epidemiological studies and laboratory investigations with animal models [2, 13]. Diabetes has been linked to a doubling of liver and pancreas cancer incidence and is associated with the risk of breast, cholangiocarcinoma, colorectal, endometrial and gallbladder cancer but may have a protective effect against prostate cancer [14, 15]. It is thought that diabetes and hyperinsulinemia promote cancer development and progression through insulin and insulin-like growth factor signaling, as indicated by human studies and further supported by experimental research [15–17]. Additional mechanisms, like chronic inflammation, are likely involved. Antidiabetic medications can interfere with tumor biology. Metformin inhibits breast, colorectal, and endometrial cancer development whereas other agents such as insulin (glargine), pioglitazone, and sulfonylureas have been associated with modestly increased cancer risks [18]. A systematic review and meta-analysis established diabetes as a significant risk factor for non-alcoholic fatty liver disease (NAFLD) with an estimated 50–60% global prevalence of NAFLD among patients with type 2 diabetes [19]. Many of these patients will develop hepatic fibrosis, conferring increased liver cancer risk [20]. Patients with rheumatoid arthritis have a heightened risk of developing Hodgkin and non-Hodgkin lymphoma and the clinical course of these lymphomas is often aggressive [21]. The correlation between rheumatoid arthritis disease activity and lymphoma risk suggest that both chronic inflammation and immune stimulation induced by a rheumatoid arthritis are possible drivers for this relationship although experimental data supporting the hypothesis are missing. Alternatively, immune suppression induced by anti-rheumatoid arthritis therapy may increase the risk of developing lymphoma. Lastly, hypertension has been associated with an elevated risk of developing kidney, colorectal, and breast cancer [22]. Changes in calcium metabolism in hypertensive patients may broadly increase cancer cell proliferation while hypertension-induced chronic renal hypoxia, together with a deregulated renin-angiotensin system, is a proposed mechanism that may increase the risk for kidney cancer.
Figure 1:
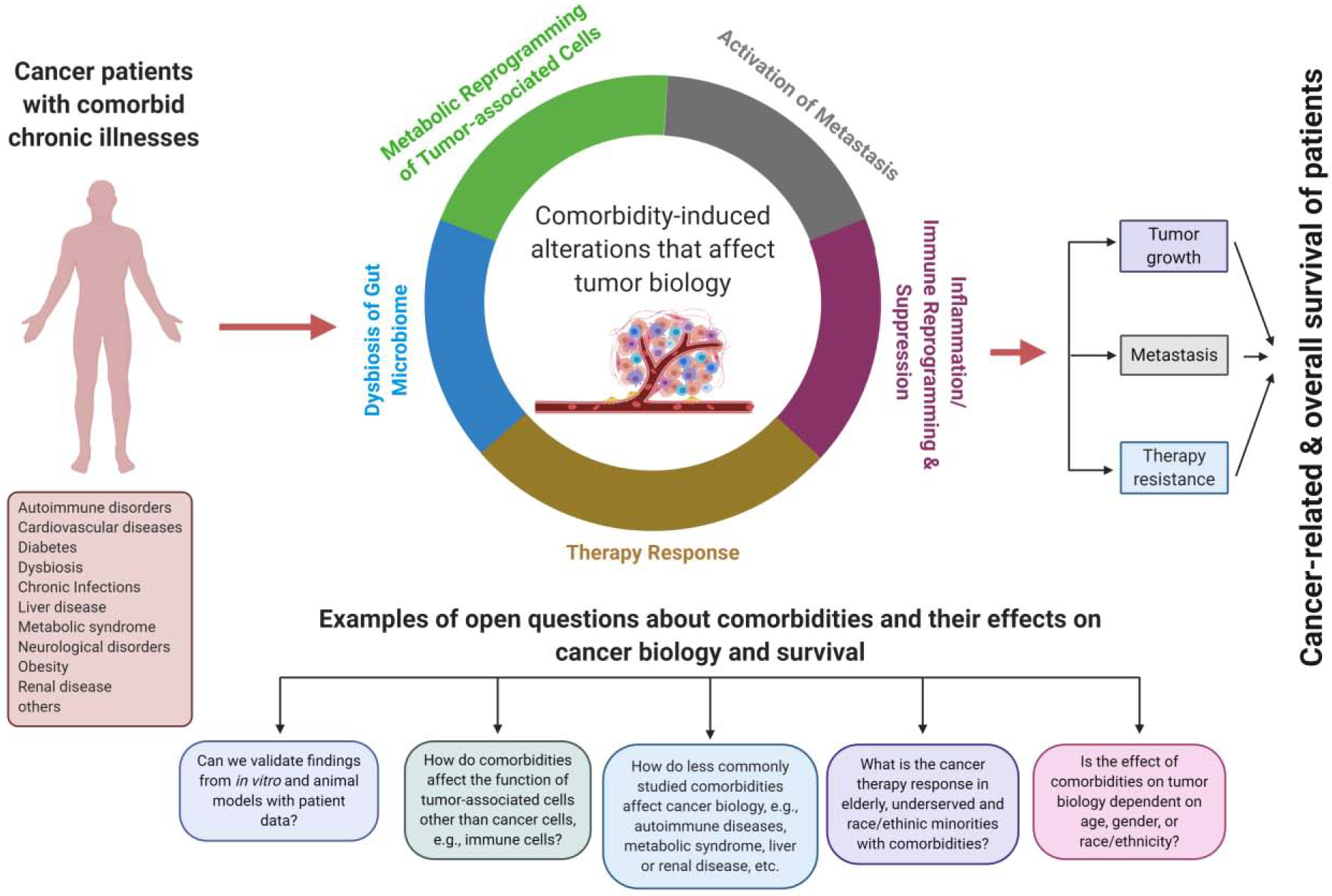
Current knowledge and open questions about cancer-related comorbidities and their effects on cancer biology and survival
Comorbidities promote cancer metastasis
Comorbidities are associated with an increased risk of cancer-specific mortality, indicating that comorbid conditions may enhance the metastatic spread of cancer, the primary cause of cancer deaths [2]. Yet little is known about the mechanisms by which comorbidities may impact metastasis. Obesity frequently associates with lethal cancer [15]. In mouse models of breast cancer, a high-fat diet and obesity promoted metastasis through impaired tumor vascularization and increased mesenchymal differentiation of cancer cells, altered chemokine signaling, changes to the tumor immune environment, and activation of the sphingolipid pathway [23–25]. A mesenchymal differentiation of cancer cells is also induced by diabetes, as shown for non-small cell lung cancer patients [26]. Hyperglycemia, a hallmark of diabetes, increases metastatic seeding of 4T1 breast cancer cells. In the 4T1 mouse model of breast cancer metastasis, hyperglycemia impaired tumor vascularization and secretion of granulocyte colony-stimulating factor and subsequent recruitment of neutrophils into metastatic sites [27]. Others reported that hyperglycemia promotes metastatic colonization of pancreatic ductal adenoma cells through activation of the pro-metastatic Runx3/Col6a1 pathway [28], whereas obesity-induced inflammation may lead to pancreatic cancer progression and resistance to therapy through stellate cell activation and increased desmoplasia [29]. Heart disease and cancer have shared risk factors. Still, there is evidence that heart failure per se is oncogenic and promotes cancer development and spread through release of soluble factors like serpins and signaling molecules from the renin-angiotensin-aldosterone pathway [30, 31]. Psychosocial factors such as chronic stress and depressive disorders are associated with cancer survival [32]. A pro-metastatic niche has been described for breast tumors from socially isolated women [33] and a decrease in chronic depression may slow metastasis in breast cancer patients [34]. Chronic stress and sympathetic nervous system signaling enhance breast cancer metastasis in animal models through increased secretion of colony stimulating factor 1, recruitment of M2 macrophages, and vascular endothelial factor C-induced lymphatic remodeling, enhancing the odds of metastasis [35, 36]. Thus, there is indication that co-morbidities are causatively linked to cancer metastasis although the existing literature remains sparse and needs to be expanded. There is, however, a lack of studies exploring the biology of primary tumors that give rise to metastasis in cancer patients with co-morbidities, or of studies that investigate the characteristics of metastatic lesions in these patients, and how those may relate to co-existing comorbidities.
Comorbidity-induced changes to the tumor immune environment that drive cancer progression and therapy resistance
Various malignancies are reproducibly cured in mouse models using immune therapy and chemotherapy. Yet, most of these therapies show objective responses in only a fraction of treated patients in the clinic. One reason for this disconnect might be the use of young, lean mice that lack immune-altering comorbidities present in elderly cancer patients. Obesity, diabetes, and viral infections like HIV/AIDS or chronic viral hepatitis have been shown to cause significant changes to the immune system and may reduce local and systemic immunity. One study showed that genetically and diet-induced obesity in the B16 melanoma and 4T1 breast cancer models results in PD-1-mediated T cell dysfunction, increased immune aging, and tumor progression which was partly driven by leptin [37]. In this study, obesity was also associated with increased efficacy of PD-1 blockade in both tumor-bearing mice and cancer patients. Another study examined the effect of obesity in two immunotherapeutic models, namely systemic anti–CTLA-4 monoclonal antibody therapy and delivery of a TRAIL-encoding adenovirus plus CpG. Here, both therapies were effective in lean mice but did not provide a survival benefit in mice with diet-induced obesity. Further analysis showed that leptin was a mediator of therapy resistance in this model, suggesting that leptin is a therapeutic target to improve tumor immunotherapy when immune-modulating comorbidities are present [38]. Chronic stress and depression are other comorbidities with good evidence that they can affect the systemic and intratumor immune environment, namely through activation of hypothalamic-pituitary-adrenal signaling as the central stress response system [39, 40].
Obesity and diabetes are cancer risk factors that induce changes to the gut microbiome and increase the risk of mucosal infections, establishing a link between dysbiosis and cancer (Box 1). There is good evidence that obesity, diabetes, certain chronic infections, and stress exposures can alter the tumor immune environment and gut microflora, but most other cancer-associated comorbidities have not been studied in this context, and we still lack patient data that would confirm the findings from animal studies.
Box 1. Comorbidities can alter the gut microbiome.
The gut microbiome affects human health. A dysbiosis can increase cancer risk. Comorbidities may confer their cancer risk through effects on the gut microbiome. There is evidence that a dysbiosis can be a cause of cancer [41, 42]. Diabetes can lead to depletion of beneficial butyrate-producing taxa in the human gut microbiota [43], whereas hyperglycemia can cause intestinal barrier dysfunction and enteric infections [44]. A high-fat diet can promote intestinal carcinogenesis in K-ras mutant mice by causing a gut microbiota dysbiosis, driven by a decrease in Paneth-cell-mediated antimicrobial host defense and compromised dendritic cell recruitment [45]. The gut microbiome also promotes obesity-associated liver cancer by inducing deoxycholic and lipoteichoic acid synthesis and a senescence-associated secretory phenotype in hepatic stellate cells with increases in inflammatory and tumor promoting factors and prostaglandin E2-mediated suppression of antitumor immunity [46, 47]. Recently, it was shown that the gastrointestinal microbiome influences efficacy of PD-1-based cancer immunotherapy in mice and patients, largely due to certain commensal species that define the clinical response [48]. The study indicated that an existing dysbiosis as a comorbidity may negatively affect immunotherapy efficacy. Furthermore, comorbidities may contribute to persistent gene expression alterations and epigenome remodeling that predispose to cancer or promote disease progression [49–51]. An obesity-associated microbiome was found to reprogram the intestinal epigenome as well, leading to persistent alterations that may promote carcinogenesis [52].
Chronic disease-induced inflammation is a cancer risk factor and modifier of tumor biology
Chronic infections commonly cause chronic inflammation which is a cancer risk factor. The link between chronic inflammatory diseases and cancer has been reviewed in the past [13]. Obesity causes macrophage accumulation in adipose tissue and systemic chronic inflammation, leading to obesity-related insulin resistance [53]. Both dietary and genetic obesity promote liver inflammation and tumorigenesis through upregulation of proinflammatory cytokines, including interleukin 6 and tumor necrosis factor α [54]. Recently, it was shown that interleukin 6-induced androgen receptor signaling leads to up-regulation of cell cycle-related kinase and mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular cancer [55]. In breast cancer, obesity upregulates adipose inflammation and estrogen synthesis by the aromatase pathway and activates the NLRC4 inflammasome pathway leading to increased interleukin 1β and VEGFA expression and enhanced tumor angiogenesis [56]. In pancreatic cancer, obesity-induced inflammation may lead to disease progression and resistance to therapy through stellate cell activation and increased desmoplasia [29]. Yet, many aspects of the inflammation-to-cancer axis remain incompletely understood for most obesity-associated cancer types. Furthermore, the role of inflammation in comorbidity-induced cancer progression has rarely been studied for comorbidities other than obesity and infectious diseases.
Impact of comorbidities on cancer metabolism
The metabolic health status of a person, rather than obesity per se, may confer cancer risk [57]. Comorbidity-induced changes in cancer metabolism have been studied for few conditions, namely, obesity, diabetes, metabolic syndrome, dysbiosis, and chronic stress exposure. Persistent metabolic alterations are hallmarks of cancer that maintain tumor growth and induce epithelial-to-mesenchymal transition and metastatic spread, but also provide vulnerabilities that make cancer metabolism a target for cancer therapy [58]. Stress-induced catecholamine signaling may promote cancer stem cell traits through a lactate dehydrogenase A-dependent mechanism that rewires cancer metabolism [59]. Reprogramming of cancer metabolism by aberrant Myc signaling or the acquisition of mutations in metabolic enzymes, such as isocitrate and succinate dehydrogenases, or fumarate hydratase, commonly affect mitochondrial metabolisms and may lead to excessive production of reactive oxygen species, further enhancing the mutational burden and malignant adaptations of cancer cells [58]. High-fat diet fuels prostate cancer progression by enhancing the Myc transcriptional program through metabolic alterations and histone methylation [60]. Hyperglycemia and diabetes increase protein kinase C signaling and formation of advanced glycation end-products and activate NFκB. Activation of these and the hexosamine pathway is triggered by hyperglycemia-induced mitochondrial superoxide production and can be blocked through normalization of mitochondrial superoxide production [61, 62]. High-fat diet and obesity more generally alter lipid and cholesterol metabolism in adipocytes and cancer cells [63] and increase 27-hydroxycholesterol [64]. This metabolite is an endogenous estrogen receptor agonist and enhances metastasis in orthotopic breast and pancreatic cancer models, in part through its action on immune cells [64]. There is also evidence that obesity-associated pancreatic cancer is driven by metabolic alterations. A critical role of mitochondrial arginase, ARG2, and the urea cycle was identified using human pancreatic cancer cells and an orthotopic xenograft model of obesity-induced pancreatic ductal adenocarcinoma [65]. The data show a dependency on ARG2 for obesity-driven pancreatic cancer in this model. Lastly, colorectal and liver cancer patients are impacted by comorbidity-induced changes to the metabolism of the gut microbiome, as already discussed by us [45–47], highlighting the intertwined relationships between gut microflora metabolism and cancer. However, findings from these primarily animal-based investigations will need further validation with additional disease models and human data. Other poorly understood metabolic relationships that require further investigations include the crosstalk between tumor immune cell and adipocyte metabolism and the tumor microenvironment and cancer cell survival.
Concluding Remarks
Comorbidities should be considered throughout the cancer research process. The advent of COVID-19 infections is reinforcing the notion that diseases other than cancer decrease cancer survival [1]. Many chronic diseases have adverse effects on cancer outcomes, leading to excessive mortality among cancer patients. These deaths are preventable with treatment, diet and lifestyle changes, increased physical activity, and psychosocial support, among other intervention strategies that target these chronic diseases. Comorbidities influence tumor biology through various mechanisms (Figure 1). It is important that we understand these biology-based mechanisms to improve prevention and intervention as part of cancer care, specifically for patient populations that are at increased risk of multiple comorbidities like the elderly. There are many unanswered questions (see Outstanding Questions). For the most part, we have a limited understanding of how comorbidities affect therapy response in the elderly, underserved, and race/ethnic minorities, in part because clinical trials exclude those with common comorbidities (Figure 2). Certain comorbidities have been studied more extensively, such as obesity and diabetes, but others have been neglected, like liver and renal diseases, metabolic syndrome, neurological disorders, and autoimmune diseases. Lack of suitable animal cancer models to study comorbidities may have contributed to the dearth in mechanistic research. Likewise, funding opportunities for this research may not exist. We rarely have patient data that support our findings from in-vitro and animal studies. One reason could be the difficulty of obtaining biospecimens from cancer patients with known comorbidities. Comorbidities may affect younger subjects differently than elderly subjects and women differently than men. Age and gender are modifiers of health and disease and their influence remains to be understudied and underestimated in clinical research and medical practice, including the effects of comorbidities on cancer outcomes. Our understanding of tumor immune cell function and cancer metabolism is still in an evolving state. For example, we do not know how comorbidities affect tumor immune response through metabolic changes in immune cells. There are some data for adipocytes, endothelial cells, and tumor-associated fibroblasts, but they are generally sparse. Hence, more mechanistic research is needed into comorbidity-induced cancer progression and mortality for increased awareness among clinicians, and to develop novel prevention and intervention strategies.
Outstanding Questions.
Can we validate findings from animal models of comorbidities with patient data? Can we show that comorbidities in patients associate with a distinct tumor biology that is related to findings from experimental research? How do comorbidities relate to the development of an aggressive tumor biology in patients?
There is good evidence that obesity, diabetes, chronic infections, and stress exposures, can alter the tumor immune environment and gut microflora, but most other cancer-associated comorbidities have not been studied in this context. How do other cancer-associated comorbidities, like the metabolic syndrome, chronic cardiovascular disease, liver and renal disease, autoimmune diseases, or depression, affect the tumor immune environment and the gut microflora in cancer patients?
What is the effect of comorbidities on cancer therapy response in patient groups at high risk of multiple comorbidities, such as the elderly, underserved, and race/ethnic minorities?
Is the effect of comorbidities on tumor biology dependent on patients’ age or gender?
How do comorbidities affect the metabolism and function of all tumor-associated cells, including cancer cells, immune and endothelial cells, adipocytes, and tumor-associated fibroblasts?
Figure 2:
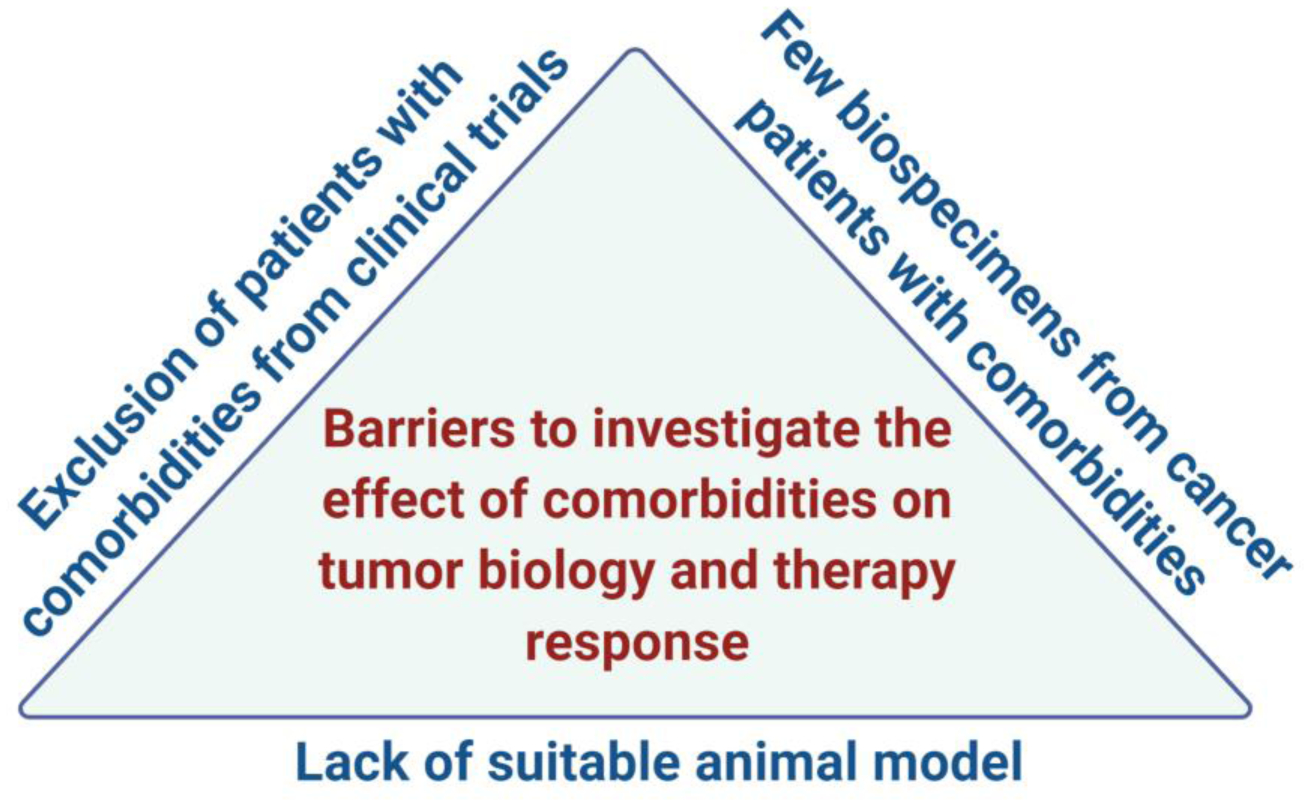
Barriers to studying the adverse effects of cancer-related comorbidities
Supplementary Material
Highlights.
The burden of comorbid chronic diseases is increasing as populations get older and is affecting underserved populations more so than the affluent
Comorbidities and cancer have common risk factors, but comorbidities and medications to treat them can directly or indirectly influence tumor biology
Comorbidities have adverse effects on cancer prevention and outcomes and lead to an excessive cancer mortality that is preventable
They exert their effects on tumor biology by altering the tumor microenvironment, cancer metabolism, and the gut microflora, and by affecting the cancer therapy response
They promote cancer progression by mechanisms including local and systemic inflammation, changes to the tumor immune environment, mesenchymal differentiation of cancer cells, and formation of a pro-metastatic niche at distant organ sites
Acknowledgement of funding:
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute (NCI), Center for Cancer Research (ZIA BC 010499, ZIA BC 010624, and ZIA BC 010887).
Glossary
- Hyperglycemia
Refers to a high blood sugar (glucose) level. Usually occurs when the body does not produce insulin or when insulin signaling is compromised. High blood sugar is an indicator of diabetes.
- PD-1 blockade
PD-1 or Programmed cell death protein 1 is a cell surface protein that regulates the immune system’s response to cells in the human body by down-regulating the immune response. PD-1 inhibitors cause a PD-1 signaling blockade and can activate the immune system to attack tumors.
- Gut microflora/microbiome
The microbiome represents all microbes - bacteria, fungi, protozoa and viruses - that live on or inside the human body. In our body, the gut microflora/microbiome is the largest entity. The microbiome consists of microbes that are both helpful and potentially harmful.
- Dysbiosis
Indicates a deleterious microbial imbalance in the body, usually an imbalance of the gut microflora.
- Epithelial to mesenchymal transition (EMT)
EMT is a reversible cellular program by which epithelial cells lose their cell polarity and cell-cell adhesion, and gain migratory and invasive properties to become mesenchymal type cells like fibroblasts. The mesenchymal differentiation allows cancer cells to become more metastatic.
- Reactive oxygen species
A type of unstable molecules that contain oxygen and easily react with other molecules in cells. They may cause damage to cell components such as DNA, RNA, lipids, and proteins and can cause mutations and cell death. However, reactive oxygen species can also participate in signal transduction.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement: None to declare.
References
- 1.Mehta V et al. (2020) Case Fatality Rate of Cancer Patients with COVID-19 in a New York Hospital System. Cancer Discov 10 (7), 935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renzi C et al. (2019) Comorbid chronic diseases and cancer diagnosis: disease-specific effects and underlying mechanisms. Nat Rev Clin Oncol 16 (12), 746–761. [DOI] [PubMed] [Google Scholar]
- 3.Sarfati D et al. (2016) The impact of comorbidity on cancer and its treatment. CA Cancer J Clin 66 (4), 337–50. [DOI] [PubMed] [Google Scholar]
- 4.Daw J (2017) Contribution of Four Comorbid Conditions to Racial/Ethnic Disparities in Mortality Risk. Am J Prev Med 52 (1S1), S95–S102. [DOI] [PubMed] [Google Scholar]
- 5.Coghill AE et al. (2019) HIV Infection, Cancer Treatment Regimens, and Cancer Outcomes Among Elderly Adults in the United States. JAMA Oncol 5, e191742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Unger JM et al. (2019) Association of Patient Comorbid Conditions With Cancer Clinical Trial Participation. JAMA Oncol 5 (3), 326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Louwman WJ et al. (2010) A 50% higher prevalence of life-shortening chronic conditions among cancer patients with low socioeconomic status. Br J Cancer 103 (11), 1742–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guilcher SJ et al. (2014) Level of disability, multi-morbidity and breast cancer screening: does severity matter? Prev Med 67, 193–8. [DOI] [PubMed] [Google Scholar]
- 9.Corrigan KL et al. (2019) Cancer disparities in people with HIV: A systematic review of screening for non-AIDS-defining malignancies. Cancer 125 (6), 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhatia D et al. (2020) Breast, cervical and colorectal cancer screening in adults with diabetes: a systematic review and meta-analysis. Diabetologia 63 (1), 34–48. [DOI] [PubMed] [Google Scholar]
- 11.Boakye D et al. (2018) Impact of comorbidity and frailty on prognosis in colorectal cancer patients: A systematic review and meta-analysis. Cancer Treat Rev 64, 30–39. [DOI] [PubMed] [Google Scholar]
- 12.Lee L et al. (2011) Impact of comorbidity on chemotherapy use and outcomes in solid tumors: a systematic review. J Clin Oncol 29 (1), 106–17. [DOI] [PubMed] [Google Scholar]
- 13.Hussain SP et al. (2003) Radical causes of cancer. Nat Rev Cancer 3 (4), 276–285. [DOI] [PubMed] [Google Scholar]
- 14.Tsilidis KK et al. (2015) Type 2 diabetes and cancer: umbrella review of meta-analyses of observational studies. BMJ 350, g7607. [DOI] [PubMed] [Google Scholar]
- 15.Klil-Drori AJ et al. (2017) Cancer, obesity, diabetes, and antidiabetic drugs: is the fog clearing? Nat Rev Clin Oncol 14 (2), 85–99. [DOI] [PubMed] [Google Scholar]
- 16.Giovannucci E et al. (2010) Diabetes and cancer: a consensus report. Diabetes Care 33 (7), 1674–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Novosyadlyy R et al. (2010) Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Res 70 (2), 741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu L et al. (2015) Pharmacologic Therapy of Diabetes and Overall Cancer Risk and Mortality: A Meta-Analysis of 265 Studies. Sci Rep 5, 10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Younossi ZM et al. (2019) The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J Hepatol 71 (4), 793–801. [DOI] [PubMed] [Google Scholar]
- 20.Wang P et al. (2012) Diabetes mellitus and risk of hepatocellular carcinoma: a systematic review and meta-analysis. Diabetes Metab Res Rev 28 (2), 109–22. [DOI] [PubMed] [Google Scholar]
- 21.Klein A et al. (2018) Rheumatoid arthritis and lymphoma: Incidence, pathogenesis, biology, and outcome. Hematol Oncol 36 (5), 733–739. [DOI] [PubMed] [Google Scholar]
- 22.Seretis A et al. (2019) Association between blood pressure and risk of cancer development: a systematic review and meta-analysis of observational studies. Sci Rep 9 (1), 8565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bousquenaud M et al. (2018) Obesity promotes the expansion of metastasis-initiating cells in breast cancer. Breast Cancer Res 20 (1), 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evangelista GCM et al. (2019) 4T1 Mammary Carcinoma Colonization of Metastatic Niches Is Accelerated by Obesity. Front Oncol 9, 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagahashi M et al. (2018) Targeting the SphK1/S1P/S1PR1 Axis That Links Obesity, Chronic Inflammation, and Breast Cancer Metastasis. Cancer Res 78 (7), 1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang X et al. (2017) Biologic Evaluation of Diabetes and Local Recurrence in Non-Small Cell Lung Cancer. Pathol Oncol Res 23 (1), 73–77. [DOI] [PubMed] [Google Scholar]
- 27.Fainsod-Levi T et al. (2017) Hyperglycemia Impairs Neutrophil Mobilization Leading to Enhanced Metastatic Seeding. Cell Rep 21 (9), 2384–2392. [DOI] [PubMed] [Google Scholar]
- 28.Jian Z et al. (2018) Glycemic Variability Promotes Both Local Invasion and Metastatic Colonization by Pancreatic Ductal Adenocarcinoma. Cell Mol Gastroenterol Hepatol 6 (4), 429–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Incio J et al. (2016) Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov 6 (8), 852–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meijers WC et al. (2018) Heart Failure Stimulates Tumor Growth by Circulating Factors. Circulation 138 (7), 678–691. [DOI] [PubMed] [Google Scholar]
- 31.Bertero E et al. (2018) Linking Heart Failure to Cancer: Background Evidence and Research Perspectives. Circulation 138 (7), 735–742. [DOI] [PubMed] [Google Scholar]
- 32.Chida Y et al. (2008) Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat.Clin Pract.Oncol 5, 466–475. [DOI] [PubMed] [Google Scholar]
- 33.Bower JE et al. (2018) Prometastatic Molecular Profiles in Breast Tumors From Socially Isolated Women. JNCI Cancer Spectr 2 (3), pky029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giese-Davis J et al. (2011) Decrease in depression symptoms is associated with longer survival in patients with metastatic breast cancer: a secondary analysis. J Clin Oncol 29 (4), 413–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sloan EK et al. (2010) The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res 70 (18), 7042–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le CP et al. (2016) Chronic stress in mice remodels lymph vasculature to promote tumour cell dissemination. Nat Commun 7, 10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z et al. (2019) Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat Med 25 (1), 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy KA et al. (2018) Cutting Edge: Elevated Leptin during Diet-Induced Obesity Reduces the Efficacy of Tumor Immunotherapy. J Immunol 201 (7), 1837–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Currier MB and Nemeroff CB (2014) Depression as a risk factor for cancer: from pathophysiological advances to treatment implications. Annu Rev Med 65, 203–21. [DOI] [PubMed] [Google Scholar]
- 40.Cole SW et al. (2015) Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer 15 (9), 563–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scott AJ et al. (2019) International Cancer Microbiome Consortium consensus statement on the role of the human microbiome in carcinogenesis. Gut 68 (9), 1624–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vujkovic-Cvijin I et al. (2020) Host variables confound gut microbiota studies of human disease. Nature 587 (7834), 448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Forslund K et al. (2015) Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528 (7581), 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thaiss CA et al. (2018) Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 359 (6382), 1376–1383. [DOI] [PubMed] [Google Scholar]
- 45.Schulz MD et al. (2014) High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 514 (7523), 508–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshimoto S et al. (2013) Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499 (7456), 97–101. [DOI] [PubMed] [Google Scholar]
- 47.Loo TM et al. (2017) Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity. Cancer Discov 7 (5), 522–538. [DOI] [PubMed] [Google Scholar]
- 48.Routy B et al. (2018) Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359 (6371), 91–97. [DOI] [PubMed] [Google Scholar]
- 49.Fuentes-Mattei E et al. (2014) Effects of obesity on transcriptomic changes and cancer hallmarks in estrogen receptor-positive breast cancer. J Natl Cancer Inst 106 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ebot EM et al. (2017) Gene expression profiling of prostate tissue identifies chromatin regulation as a potential link between obesity and lethal prostate cancer. Cancer 123 (21), 4130–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li R et al. (2018) Transcriptome and DNA Methylome Analysis in a Mouse Model of Diet-Induced Obesity Predicts Increased Risk of Colorectal Cancer. Cell Rep 22 (3), 624–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qin Y et al. (2018) An obesity-associated gut microbiome reprograms the intestinal epigenome and leads to altered colonic gene expression. Genome Biol 19 (1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu H et al. (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin. Invest 112 (12), 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park EJ et al. (2010) Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun H et al. (2018) An inflammatory-CCRK circuitry drives mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat Commun 9 (1), 5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kolb R et al. (2016) Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat Commun 7, 13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gunter MJ et al. (2015) Breast cancer risk in metabolically healthy but overweight postmenopausal women. Cancer Res. 75 (2), 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DeBerardinis RJ and Chandel NS (2016) Fundamentals of cancer metabolism. Sci Adv 2 (5), e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cui B et al. (2019) Stress-induced epinephrine enhances lactate dehydrogenase A and promotes breast cancer stem-like cells. J Clin Invest 129 (3), 1030–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Labbe DP et al. (2019) High-fat diet fuels prostate cancer progression by rewiring the metabolome and amplifying the MYC program. Nat Commun 10 (1), 4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Du XL et al. (2000) Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A 97 (22), 12222–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nishikawa T et al. (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404 (6779), 787–90. [DOI] [PubMed] [Google Scholar]
- 63.Louie SM et al. (2013) Mechanisms linking obesity and cancer. Biochim Biophys Acta 1831 (10), 1499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baek AE et al. (2017) The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun 8 (1), 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zaytouni T et al. (2017) Critical role for arginase 2 in obesity-associated pancreatic cancer. Nat Commun 8 (1), 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.